博士論文
C 型肝炎ウイルスの新規耐性変異同 定法と B 型肝炎ウイルスの新規安定
発現細胞系の構築に関する研究
平成 30 年 9 月
小倉 直樹
岡山大学大学院
医歯薬学総合研究科
1
論文目次
1. 要旨 02
2. 略語表 04
3. 序論 05
3.1. 肝炎 05
3.2. C 型肝炎 06
3.3. B 型肝炎 11
4. 方法 16
4.1. C 型肝炎ウイルスの新規耐性変異同定法に関する研究
(第 1 章) 16
4.2. B 型肝炎ウイルスの新規安定発現細胞系の構築に関する研究
(第 2 章) 24
5. 結果,考察 33
5.1. C 型肝炎ウイルスの新規耐性変異同定法に関する研究
(第 1 章) 33
5.2. B 型肝炎ウイルスの新規安定発現細胞系の構築に関する研究
(第 2 章) 49
6. 総括 76
7. 引用文献 77
8. 論文目録 82
9. 謝辞 83
2
1. 要旨
肝臓が炎症を起こし,肝細胞が破壊される病気である肝炎が慢性化した慢性肝炎は長期 化すると肝硬変,肝がんへと進展する。慢性肝炎の主な原因は C型肝炎ウイルス(HCV), B型肝炎ウイルス(HBV)で,世界のピーク時のHCV持続感染者は1.7億人,HBV持続感 染者は3.5億人と推定されているが,治療薬やワクチンの普及により,現在のHCV及びHBV 持続感染者は減少傾向にある。現在の慢性 C 型肝炎治療薬の直接作用型抗ウイルス薬は,
薬剤耐性ウイルスが出現するため,耐性変異解析による薬効予測が必要である。また慢性B 型肝炎治療薬の免疫賦活薬や逆転写酵素阻害薬は休薬するとウイルスが再燃するため,新 規薬剤が必要である。私は新規肝炎治療薬を迅速に開発するためHCV及びHBV 領域の新 規評価系に関する研究を行った。
第 1 章では,HCV 領域に関する新規評価系の研究を実施した。HCV 非構造タンパクの NS5BタンパクがコードするRNA依存性RNAポリメラーゼのHCVポリメラーゼに結合す る非核酸系阻害剤のJTK-853は,Phase 1b試験でHCV感染患者に対して有効性が確認され ている。私は既存法とは異なる新規耐性変異同定法の確立を目的に,JTK-853投薬患者血清 を用いて耐性変異を解析した。Genotype 1a又は1bのHCV感染患者28例を4コホート(800,
1200,1600mg 1日2回又は1200mg 1日3回)に分けて,プラセボ投与群を各1例及びJTK-853 投与群を各6例で3日間経口投与した。JTK-853投薬患者の投薬前後の血清からHCV遺伝 子を単離後,HCVポリメラーゼ領域のpopulation sequencing解析により,in vitro耐性試験 で同定された耐性変異部位(C316,M414,C445,Y448,Y452,L466)のうち,患者0405 の投薬後にM414T変異が認められた。またclonal sequencing解析では,population sequencing 解析で認められた患者0405のM414T 変異以外に複数の患者の投薬後でM414T,C445R,
Y448H,Y448C,L466F変異が低頻度の割合で検出された。さらに患者由来のHCVポリメ
ラーゼ遺伝子を野生型(genotype 1b:Con1株)と組み換えたHCVレプリコン細胞に対する JTK-853の50%有効濃度(EC50)を検討したところ,M414T変異が検出された患者0405の 投薬後のJTK-853のEC50は,投薬前と比べて24倍減弱し,in vitro耐性試験のM414Tの感 受性低下と同等であった(44倍減弱)。一方,その他の患者の投薬後のJTK-853のEC50は,
投薬前と比べて著しい減弱は認められなかった。第 1 章の結果より,今回の耐性変異同定
法はJTK-853のin vitro耐性試験で検出された耐性変異が複数の患者で検出され,低頻度の
耐性変異も同定可能であったことから,耐性変異を確度高く検出可能であることを明らか にした。
第2章では,HBV領域に関する新規評価系の研究を実施した。既存のHBV安定発現細胞 系はHBV複製活性が低く,遺伝子型がgenotype Dのみのため,日米欧で患者が多いgenotype
A,B,Cを含めたpan-genotypicな治療薬の開発が困難である。私は化合物スクリーニング
3
に使用可能で,複数genotypeの新規HBV安定発現細胞系の構築を目的に,既存細胞(genotype D)及びHBV患者血清(genotype A,B,C)から新規細胞を構築した。最初にgenotype D の新規細胞としてテトラサイクリン誘導型の既存細胞であるHepAD38細胞からクローニン グしてHep38.7-Tet細胞を選抜した。Hep38.7-Tet細胞の細胞内HBV産生量及び細胞外HBV 分泌量は親株のHepAD38 細胞より高かった。またHep38.7-Tet 細胞に対して逆転写酵素阻 害薬のエンテカビル及びcccDNA形成阻害剤のCCC-0975は,細胞毒性を示さない濃度域で 濃度依存的にcccDNA産生量及びHBeAg分泌量を低下させた。さらにFDA approved drug screening libraryの414化合物から抗HBV剤を探索したところ,異なる作用機序を有する12 化合物(逆転写酵素阻害薬,HMG-CoA還元酵素阻害薬,ステロイドホルモン,免疫抑制薬,
テトラサイクリン誘導体)がHBeAg分泌量を低下させた。次にgenotype A,B,Cの新規細 胞としてHBV感染患者9人(genotype A:2人,genotype B:1人,genotype C:6人)の血 清からHBV遺伝子を単離後,clonal sequencing解析により全長HBV遺伝子配列を同定した。
同定した患者HBV 遺伝子を発現するプラスミドをヒト肝がん細胞株 HepG2細胞に導入し た一過性発現系により,各genotypeの患者HBV クローンからreferenceと比較して細胞内
core DNA産生量及び細胞外HBV分泌量が高いA-1,B-1,C-1クローンを選抜した。選抜
した患者 HBV クローンの遺伝子を発現するプラスミドを HepG2細胞に導入し,薬剤選択
によりA-1,B-1,C-1由来のHBV安定発現細胞を取得した。取得したA-1,B-1,C-1由来
のHBV安定発現細胞の細胞内HBV産生量及び細胞外HBV分泌量は経時的に増加し,エン テカビルは細胞毒性を示さない濃度域でcore DNA産生量及びextracellular HBV DNA分泌量 を濃度依存的に低下させ,エンテカビル以外の逆転写酵素阻害薬,免疫賦活薬,capsid形成 阻害剤,RNase H阻害剤はcore DNA産生量を低下させた。またA-1,B-1,C-1由来のHBV 安定発現細胞から産生されるウイルスを感染させた細胞ではextracellular HBV DNA分泌量
及び HBsAg 分泌量が経時的に増加し,エンテカビルは細胞毒性を示さない濃度域で core
DNA産生量を低下させた。第2章の結果より,genotype DはHep38.7-Tet細胞,genotype A, B,Cは患者由来のHBV安定発現細胞をそれぞれ取得し,様々なgenotypeの新規HBV安定 発現細胞を構築した。構築した安定発現細胞はHBV発現能を有しており,既存薬の阻害も 確認されたことから,新規抗HBV薬のスクリーニングに使用可能であることを明らかにし た。
本研究より,HCV領域では薬効予測に重要な耐性変異同定法を,HBV領域では新薬探索 に使用可能な細胞系を確立し,共に新薬創出における新規評価系を構築した。これら評価 系の確立により迅速な新規肝炎治療薬の創出につながると期待できる。
4
2. 略語表
論文中に以下の略語を用いた
ALT Alanine aminotransferase
BID Bis in die
BL Baseline
bps Base pairs
cccDNA Covalently closed circular DNA
DAA Direct acting antiviral
EC50 50% effective concentration
EOT End of treatment
FBS Fetal bovine serum
FDA Food and drug administration
HAV Hepatitis A virus
HBc Hepatitis B core protein
HBeAg Hepatitis B e antigen
HBeAb Hepatitis B e antibody
HBsAg Hepatitis B s antigen
HBV Hepatitis B virus
HCV Hepatitis C virus
HIV Human immunodeficiency virus
IFN Interferon
IRES Internal ribosome entry site
mRNA Messenger RNA
NTCP Human sodium taurocholate cotransporting polypeptide
PCR Polymerase chain reaction
pgRNA Pregenome RNA
RT Reverse transcription
Tet Tetracycline
TID Ter in die
5
3. 序論
3.1. 肝炎
3.1.1. 肝炎の概要
肝炎は肝臓に炎症が起こった状態と定義され,原因の約80%がウイルス性肝炎1)であり,
その他に薬剤性肝炎2),アルコール性肝炎3),自己免疫性肝炎4)等がある。肝炎の症状は 発症の仕方で3種類に分類され,突発的に発症し,一過性の肝炎を起こす急性肝炎,6ヵ月 以上にわたり炎症が継続する慢性肝炎,急性肝炎を発症後,7~10日で死に至ることが多い 劇症肝炎がある。急性肝炎はウイルス感染後から数週間~数か月後又は薬剤投与後に発症 し,主な症状は黄疸,食欲不振,全身倦怠感である。慢性肝炎は急性肝炎が完治せず,肝 細胞の破壊と修復が長期間継続した状態で,主な症状は吐き気や食欲不振であるが,殆ど の場合で自覚症状がない。劇症肝炎は急性肝炎の約1%で発症し,肝性脳症という意識障害 の症状が出るのが特徴的である。また脳浮腫や感染症等の重い合併症を引き起こすことが 多く,死亡率も70~80%と高い。
ウイルス性肝炎の主な原因である肝炎ウイルスはA,B,C,D,E,G型の6種類が確認 されている。A型肝炎はA型肝炎ウイルス(Hepatitis A virus:HAV)が原因で起こる肝炎で,
急性肝炎の約 40%を占めている。A 型肝炎ウイルスは汚染された飲み水や魚介類を摂取す る経口感染で感染し,2~6 週間の潜伏期間を経て発症し,症状は一過性で慢性肝炎に移行 することはなく,劇症肝炎になることも稀(約5%)である。B型肝炎5)はB型肝炎ウイル ス(Hepatitis B virus:HBV)が原因で起こる肝炎で,出産時の母子感染,性行為,針刺し事 故による血液感染で感染する。成人が初感染すると殆どが一過性感染となり,一過性感染 の殆どが不顕性感染となり治癒するが,稀に1~6ヵ月間の潜伏期間を経て急性肝炎を発症 する。一方,幼少期に初感染するとウイルスが排除されずに体内に保有した状態(キャリ ア)の持続感染となり,持続感染の殆どが無症候性キャリアとなるが,稀に慢性肝炎に移 行する。C型肝炎6)はC型肝炎ウイルス(Hepatitis C virus:HCV)が原因で起こる肝炎で,
主に輸血による血液感染で感染し,HCV 感染が一旦成立すると,2~16 週間の潜伏期間を 経て急性肝炎を発症し,約 70%が慢性肝炎へと移行する。慢性化した場合,ウイルスの自 然排除は年率0.2%と稀であり,HCV感染による炎症の持続により10~30年の長期間を経 て慢性肝炎から肝硬変や肝細胞癌へと進展する。
6
3.2. C 型肝炎
3.2.1. C 型肝炎の概要
C型肝炎ウイルスは,1989年にChooらが輸血後の非A,非B型肝炎患者の血漿を接種し たチンパンジーの血漿中から,ウイルス遺伝子の一部を見出し,それにコードされる配列
(C100-3 抗原)を用いて血清中の抗体検査を行った。その結果,新たに発見された遺伝子 断片が輸血後の非A,非B型肝炎の主要な病原ウイルスであることが明らかになり,C型 肝炎ウイルスと命名された7)。ピーク時のHCVキャリアは全世界で1.7億人,本邦で150 万~200万人存在すると推定されているが,治療薬の普及により減少傾向にある。国内での 慢性肝疾患の70~80%はHCV感染に起因する。
3.2.2. C 型肝炎ウイルス
HCVはフラビウイルス科Flaviviridae,ヘパシウイルス属Hepacivirusに分類され,全長約 9600塩基よりなるプラス鎖1本鎖RNAをゲノムにもち,ウイルス粒子は直径55~65 nmの 球状粒子のウイルスである。HCV がレセプターを介して肝細胞に感染し,ウイルス粒子よ りウイルスゲノムRNAが放出される。そのゲノムRNAは5’と3’の非翻訳領域に挟まれた 1つのopen reading frameで構成されている。HCVのゲノムは真核細胞のmRNAと異なり,
CAP構造やポリA配列を持たず,5’領域にあるInternal ribosome entry site(IRES)を介して,
キャップ非依存的に翻訳され,約3000アミノ酸からなるポリプロテインとしてウイルスタ ンパクが発現する。そのポリプロテインは,宿主やウイルスのプロテアーゼによって10個 のウイルスタンパクに切断され,N末端側からウイルス粒子を構成する構造タンパク(コア タンパク,E1,E2エンベロープタンパク),残りがウイルス粒子には取り込まれない非構造 タンパク(NS2,NS3,NS4A,NS4B,NS5A,NS5B)が分布する。構造タンパクのコアタ ンパクはヌクレオキャプシドを構成する殻のタンパクで,エンベロープタンパクは受容体 と膜融合に関与する。一方,非構造タンパクはウイルスのゲノム複製機能を有する複合体 の成分であり,ウイルス粒子の組み立てにも関与している。NS2 は自身のC末端直後を切 断し,NS3 はそれ以外の非構造蛋白質領域の切断するプロテアーゼとヘリカーゼの機能を 併せ持つ。NS3 の下流に位置するNS4AはNS3 の共役因子として機能し,NS4Bはゲノム 複製に重要な膜のリモデリング(多重膜構造物形成)を誘導する。NS5Aの分子機能は正確 に分かっていないが,免疫回避やウイルス粒子形成に関与しており,その下流の NS5B は RNA 依存性 RNA ポリメラーゼとして機能する。これらのウイルスにコードされた酵素や 宿主因子によってゲノムRNAからマイナス鎖RNA が転写され,複製複合体が形成され,
プラス鎖RNAが合成される。ウイルスRNAがコアタンパクと結合してヌクレオキャプシ ドを形成し,ER膜で成熟後,トランスゴルジを通り感染性ウイルスとなって細胞外へ放出 される(図1)。
7
図 1 HCV ライフサイクル
*森石恆司「C型肝炎ウイルスの感染戦略と病原性発現機構」山梨医科学誌, 2012, 26, 1-8.
3.2.3. C 型肝炎ウイルスの遺伝子型
遺伝子型はHCV遺伝子配列の相同性に基づき分類されており,HCV 遺伝子配列の30~
35%の違いより,7種類の遺伝子型(genotype 1~7)に大別され,さらに60種類以上の亜
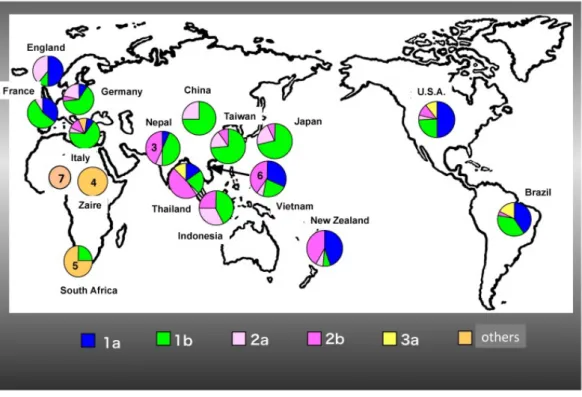
型(subtype a,b,cなど)に分類される8)。世界ではgenotype 1,2,3がアジア,アメリ カ,ヨーロッパで広く分布しており,その他の genotype は局地的な分布である。日本では genotype 1bが約70%,genotype 2aが約20%,genotype 2bが約10%の分布を示す。Genotype 1aはアメリカ,ヨーロッパで,genotype 1bは日本,アメリカ,ヨーロッパで広く分布して おり,genotype 1はウイルス量が多く,IFN治療抵抗性を示す。Genotype 2は日本,アメリ カ,ヨーロッパで,genotype 3はアメリカで分布しており,genotype 2,3はウイルス量が少 なく,IFN治療感受性を示す。一方,他のgenotypeとして,genotype 4は中央アフリカ,genotype 5が南アフリカ,genotype 6が東南アジア,genotype 7が中央アフリカで確認されている(図 2)。
8
図 2 HCV genotype の世界分布
*Tsukiyama-Kohara K「Hepatitis C Virus: Viral Quasispecies and Genotypes」 Int J Mol Sci, 2018, 19, 23.
3.2.4. C 型肝炎治療薬
C型肝炎治療には,インターフェロン(Interferon:IFN)とウイルスタンパクを標的とし た直接作用型抗ウイルス薬(Direct acting antiviral:DAA)が用いられいる。IFNは1992年 からC型肝炎に対する使用が保険適用になり,その後PEG化製剤であるPeg-IFNα-2aが保 険適用となった。IFNは抗ウイルス作用と免疫賦活作用を有している。一方,DAAはNS3/4A プロテアーゼ阻害薬の 6 種類(テラプレビル,シメプレビル,アスナプレビル,バニプレ ビル,パリタプレビル,グラゾプレビル),NS5A阻害薬の4種類(ダクラタスビル,レジ パスビル,オムビタスビル,エルバスビル),NS5B ポリメラーゼ阻害薬の核酸型のソホス ブビル,非核酸型のベクラブビルが認可され,バニプレビル以外が日常臨床に使用されて いる。NS3 には非構造タンパク領域のNS3-5 タンパクを切断する活性を有するセリンプロ テアーゼ領域を含んでおり,NS3/4Aプロテアーゼ阻害薬はこのセリンプロテアーゼを直接 阻害することでウイルス複製や粒子形成に必要なウイルスタンパクの産生を抑制し,ウイ ルス増殖を阻害する。NS3/4A プロテアーゼ阻害薬は分子構造の違いにより 2 種類存在し,
テラプレビルを含む直鎖状構造を有する第一世代とシメプレビルを含む大環状構造を有す る第二世代に分けられる。また NS5A タンパクの機能は十分に知られていないが,酵素活 性を持たず,ウイルス複製で重要な働きを有すると考えられている。ダクラタスビルを含 むNS5A阻害薬は高選択的にNS5A複製複合体を阻害することでウイルス複製を阻害する。
さらにNS5Bはウイルス複製に必須なRNA依存性RNAポリメラーゼをコードしており,
9
NS5Bポリメラーゼ阻害薬はこのRNA依存性RNAポリメラーゼを直接阻害することでウイ ルス複製を阻害する。NS5Bポリメラーゼ阻害薬には作用形式の違いにより2 種類存在し,
ソホスブビルを含むウイルス遺伝子に直接取り込まれて,RNA 伸長反応を止める核酸型と ベクラブビルを含むポリメラーゼの酵素活性を阻害する非核酸型に分けられる(図3)。
テラプレビル シメプレビル ダクラタスビル
ソホスブビル ベクラブビル
図 3 主な抗 HCV 薬の構造式
3.2.5. C 型肝炎治療薬の薬剤耐性
種々の DAAの耐性変異として,NS3/4Aプロテアーゼ阻害薬の耐性変異は,NS3領域に 第一世代では36,54,155,156,168,170番目に,第二世代で80,155,156,168番目に アミノ酸変異が,またNS5A阻害薬の耐性変異は,NS5A領域に28,30,31,58,93番目 にアミノ酸変異が,さらにNS5Bポリメラーゼ阻害薬の耐性変異は,NS5B領域に核酸型で 282番目に,非核酸型ではThumb I系で495,496,499番目に,Thumb II系で392,419,
423,474,482,494,528番目に,Palm I系で414,451,558番目に,Palm II系で316,368 番目に,Palm-β hairpin系で316,445,448,452番目のアミノ酸変異が出現することが報告 されている9)。
3.2.6. C 型肝炎領域の課題と研究目的
現在まで慢性C型肝炎治療薬として複数のDAAが上市されているが,DAA投薬患者で は薬剤感受性を低下させる薬剤耐性ウイルスの出現が問題となっており,治療の中断や治 療薬の変更が必要となっている。しかし,薬剤耐性ウイルスの指標である耐性変異の同定 法はHCVレプリコン細胞を用いたin vitro耐性試験のみであり,患者サンプルを用いた耐性 変異同定法は確立されていない。特に早期に患者での薬効を予測するには,低頻度に出現
10
した耐性変異も検出する必要があるが,HCV領域では患者サンプルを用いた低頻度の耐性 変異を同定した報告がないのが現状である。一方,HIV領域ではclonal sequencing解析から 患者における低頻度の耐性変異も同定した報告があるが10, 11),HIVはレトロウイルスであ り遺伝子解析の基となるウイルスDNAがウイルスサイクルの逆転写反応により産生される ため,ウイルスサイクルで逆転写反応過程が存在しないHCVよりは技術的に容易と推測さ れる。
本論文の第1章では,患者サンプルで低頻度の耐性変異を同定するため,HCVポリメラ ーゼ阻害剤のJTK-853が投薬されたgenotype 1のHCV感染患者血清サンプルからHCVポ リメラーゼ領域の遺伝子増幅を試み,さらには増幅産物の遺伝子配列をpopulation
sequencing解析に加えてclonal sequencing解析も検討した。
11
3.3. B 型肝炎
3.3.1. B 型肝炎の概要
B型肝炎ウイルスは,1964年にBlumbergらオーストラリア抗原の同定からはじまり12),
1968年にPrinceら・大河内らにより,オーストラリア抗原が輸血後肝炎の一因となるHBV
の表面抗原であることが判明し,輸血後肝炎との関連が確認された13, 14)。1970年にはHBV の本態であるDane 粒子が同定され15),1979年にはウイルス粒子から HBVゲノムがクロ ーニングされ,ウイルスの全遺伝子が決定された16, 17, 18)。ピーク時のHBVキャリアは全 世界で3.5億人,本邦で150万~200万人存在すると推定されているが,ワクチンの普及に より減少傾向にある。国内での慢性肝疾患の15~20%がHBV感染に起因する。
3.3.2. B 型肝炎ウイルス(HBV)
HBVはヘパドナウイルス科Hepadnaviridae,オルソヘパドナウイルス属Orthohepadnavirus に分類され,長鎖の全長が約3200塩基からなり,全周の約15~50%が一本鎖構造を有する 不完全2本鎖DNAゲノムを有し,感染性のウイルス粒子は二重構造の直径約42 nmのDane 粒子である。HBVはS抗原のPre-S1を介して宿主の肝細胞のレセプターに結合し,感染を 引き起こす。肝細胞内に入る時にエンベロープが外れてコア粒子となり,コア粒子内のHBV ゲノムが核内に移行する。HBVゲノムの不完全二重鎖DNAはDNAポリメラーゼにより完 全二重鎖DNA(covalently closed circular DNA:cccDNA)となる。さらにcccDNAを鋳型と してRNAポリメラーゼにより,4種類(3.5kb,2.4kb,2.1kb,0.7kb)のmessenger RNA(mRNA)
が作成される。3.5kb mRNAはpregenome RNA(pgRNA)として転写産物の鋳型やcoreタ ンパクやポリメラーゼタンパク,hepatitis B e antigen(HBeAg)のmRNAとなる。また2.4kb mRNAはlarge hepatitis B s antigen(HBsAg)タンパクのmRNA,2.1kb mRNAはmiddle及び small HBsAgタンパクのmRNA,0.7kb mRNAはHBxタンパクのmRNAである。ウイルス 複製はε構造にポリメラーゼが結合したpgRNAとcoreタンパクによりヌクレオキャプシド が形成されることで開始される。ヌクレオキャプシド内のpgRNAを鋳型としてHBVポリ メラーゼの逆転写反応により,マイナス鎖のDNAが合成され,続いてプラス鎖DNAが合 成されるが,全てのプラス鎖DNA合成が終わる前に,ヌクレオキャプシドは小胞体でエン ベロープタンパクと相互作用して,感染性ウイルスとなり細胞外に放出される(図4)。
12
図 4 HBV ライフサイクル
*飯島沙幸「B型肝炎ウイルス(HBV)のリバースジェネティックス」ウイルス, 2013, 63, 23-32.
3.3.3. B 型肝炎ウイルスの遺伝子型
遺伝子型はHBV遺伝子配列の相同性に基づき分類されており,HBV遺伝子配列の8%以 上の違いより,10種類の遺伝子型(genotype A~J)に大別され,さらに30種類以上の亜型
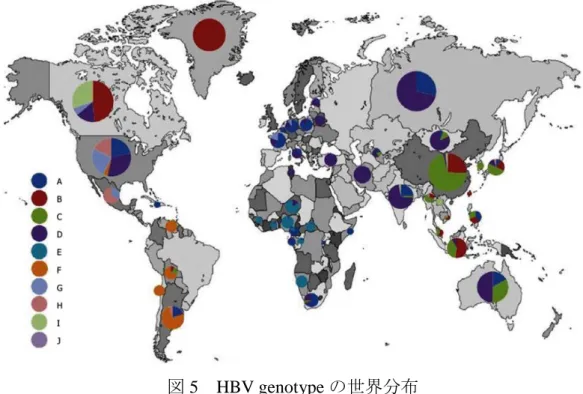
(sub-genotype)に分類される19)。世界ではgenotype AからDがアジア,アメリカ,ヨー ロッパ,アフリカで広く分布しており,その他の genotype は局地的な分布である。日本で はgenotype Cが約85%,genotype Bが約10%の分布を示す。Genotype Aはアメリカ,ヨー ロッパ,アジア,アフリカで広く分布しており,病態は急性肝炎後感染が遷延化する傾向 があり,キャリア化しやすいが,一般的に予後は良好である。Genotype B は主にアジアに 分布しており,病態は非常に穏やかで,殆どが無症候性キャリアとなり,肝細胞癌の発生 頻度は極めて低い。Genotype C は東南アジア,東アジアに分布しており,肝細胞癌を発症 しやすく,従来型のIFN治療に対して抵抗性を示す。Genotype Dは南ヨーロッパ,エジプ ト,インド等に分布しており,IFN治療抵抗性であり,予後不良である。一方,他のgenotype として,genotype Eは西アフリカ,genotype Fは主に中南米,genotype Gはフランス,ドイ ツ,北アメリカ等,genotype Hが主に中南米,genotype Iがベトナム及びラオス,genotype J がボルネオで確認されている(図5)。
13
図 5 HBV genotype の世界分布
*Sunbul M「Hepatitis B virus genotypes: Global distribution and clinical importance」World J Gastroenterol, 2014, 20, 5427-5434.
3.3.4. B 型肝炎治療薬
B型肝炎治療には,IFNと逆転写酵素阻害薬が用いられている。IFNの治療期間は24~48 週間と限定されており,また耐性ウイルスを生じないことが特徴であり,2011年にはPEG 化製剤であるPeg-IFNα-2aがHBe抗原の有無に関わりなくB型肝炎に保険適用となった。
一方,逆転写酵素阻害薬は,HBV増殖過程でのHBV自身がコードする逆転写酵素を特異的 に阻害し,HBV の生活環におけるマイナス鎖ならびにプラス鎖 DNA 合成を強力に抑制す る。継続して投与することで血中HBV DNA量は速やかに低下し,alanine aminotransferase
(ALT)値も改善するが,投与を中止すると高頻度に肝炎が再燃する。そのため,逆転写酵 素阻害薬の投与は原則的に中止せず,長期継続投与で持続的に HBV DNA量を抑制する必 要がある。本邦では2000年から2006年にかけて3種類の逆転写酵素阻害薬(ラミブジン,
アデホビル,エンテカビル)がB型慢性肝炎に対して保険適用となり,さらに2014年には テノホビル・ジソプロキシルフマル酸塩,2017 年にはテノホビル・アラフェナミドが保険 適用となった。ラミブジンはウイルスの逆転写酵素がRNAからDNA合成する際に必要な 基質であるデオキシシチジン 5’-三リン酸と競合的に逆転写酵素に結合し,ウイルス DNA 合成を阻害するが,耐性ウイルスが高率で出現する。アデホビルもラミブジンと同様な機 序でDNA 合成時の基質であるデオキシアデノシン5’-三リン酸と競合的に逆転写酵素に結 合し,ウイルスDNA合成を阻害する。エンテカビルは細胞内でリン酸化され,活性型のエ ンテカビル三リン酸となり,DNA合成時の基質であるデオキシグアニン5’-三リン酸と競合
14
的に逆転写酵素に結合し,阻害作用を示す。作用機序はプライミング,mRNA からのマイ ナス鎖DNA合成時の逆転写,HBV DNAのプラス鎖合成の3つの活性を阻害する。耐性ウ イルス出現率が低いため,第一選択薬として使用されている。テノホビル・ジソプロキシ ルフマル酸塩はテノホビルのプロドラッグであり,ジエステルの加水分解をうけてテノホ ビルへ変換後,細胞内酵素によりリン酸化を受けて活性型のテノホビル二リン酸となる。
このテノホビル二リン酸は DNA 合成時の基質であるデオキシアデノシン 5’-三リン酸と競 合的に逆転写酵素に結合し,ウイルスDNA合成を阻害する。テノホビル・アラフェナミド はテノホビルのプロドラッグであり,トランスポーターで肝細胞に取り込まれるとテノホ ビルに速やかに加水分解され,その後活性型のテノホビル二リン酸となる。テノホビル・
アラフェナミドの作用機序はテノホビル・ジソプロキシルフマル酸塩と同様であり,肝細 胞内に効率的に取り込まれることから必要量はテノホビル・ジソプロキシルフマル酸塩と 比べて少量である(図6)。
ラミブジン アデホビル エンテカビル テノホビル
図 6 主な抗 HBV 薬の構造式
3.3.5. B 型肝炎治療薬の薬剤耐性
B型肝炎ウイルスの逆転写酵素阻害薬はHBVポリメラーゼの逆転写領域に耐性変異が出 現する。ラミブジンでは 180,204 番目に出現し,治療には交叉耐性の点からアデホビル,
テノホビル・ジソプロキシルフマル酸塩,テノホビル・アラフェナミドが有効である。ア デホビルでは 181,233,236 番に出現し,テノホビル・ジソプロキシルフマル酸塩,テノ ホビル・アラフェナミドが有効である。エンテカビルではラミブジンの耐性変異である180,
204番目に184,202,250番目のいずれかが加わって出現する。エンテカビル耐性ウイルス
に対しては,ラミブジンは交叉耐性を有するが,アデホビル,テノホビル・ジソプロキシ ルフマル酸塩,テノホビル・アラフェナミドが有効である。テノホビル・ジソプロキシル フマル酸塩及びテノホビル・アラフェナミドでは 181,194,236 番目が耐性に関与すると の報告があるが,長期での耐性解析が必要である20)。
3.3.6. B 型肝炎領域の課題の研究目的
現在まで慢性 B 型肝炎治療薬として免疫治療薬や複数の逆転写酵素阻害薬が上市されて いるが,両薬剤とも治療を中断するとウイルスが再燃するため治療を長期で継続する必要
15
があり,HBVを根治することが困難なことが問題となっている。しかし既存薬とは異なる 作用機序を有する新規抗HBV薬の基となる化合物をスクリーニングする既存のHBV安定 発現細胞系は2種類のみである。特に既存細胞の2種類は異なるHBV誘導システムを有し ているだけで,両細胞共にgenotype DでありHBV複製能も低いことから,化合物のスク リーニングには長期間を要することや pan-genotypic な薬剤の選抜ができないのが現状で ある。また両既存細胞ともに取得には非常に多くの細胞クローンから選抜した経緯がある ことや他のgenotypeのHBV安定発現細胞系の報告がないことから,HBV領域で複製能を 有するHBV安定発現細胞系の構築は非常に難易度が高いと予想される。
本論文の第2章では,化合物スクリーニングに使用可能で複数genotypeの新規HBV安定 発現細胞系を構築するため,既存細胞(genotype D)から複製能の高いHBV安定発現細胞 を選抜又はHBV患者血清(genotype A,B,C)から複製能を有するHBV配列を選抜し,
HBV安定発現細胞の構築を試みた。
16
4. 方法
4.1. C 型肝炎ウイルスの新規耐性変異同定法に関する研究
(第 1 章)
4.1.1. 化合物
日本たばこ産業株式会社の医薬総合研究所でHCVポリメラーゼ阻害剤として合成された (2R)-4-(5-cyclopropylthiazolo[4,5-d]pyrimidin-2-yl)-N-[3-fluoro-4-(trifluoromethoxy)benzyl]-1-[4-(
trifluoromethyl)phenylsulfonyl]piperazine-2-carboxamideのJTK-853を使用した21)。
4.1.2. HCV 感染患者背景と臨床試験デザイン
Genotype 1a(23例)又は1b(5例)の慢性C型肝炎患者28例を対象に,プラセボ対照,
無作為化二重盲検比較試験を実施した22)。患者背景情報は,HCV RNA量が50000 IU/mL 以上,HBs抗原陰性,HAV抗体陰性,Human immunodeficiency virus(HIV)抗体陰性であ り,試験実施前にインフォームドコンセントを取得済みである。試験デザインはプラセボ 群:4例,800mg 1日2回(Bis in die:BID)群:6例,1200mg BID群:6例,1600mg BID 群:6例,1200mg 1日3回(Ter in die:TID)群:6例であり,投薬期間は3日間である。
各患者から投与前のDay 1(Baseline:BL),投与後のDay 4(End of treatment:EOT)の血 清を採取後,使用時まで-70℃に保管した。血清HCV RNA量はCobas TaqMan HCV Test(Roche Diagnostics)を用いて定量した。
4.1.3. Population sequencing 解析
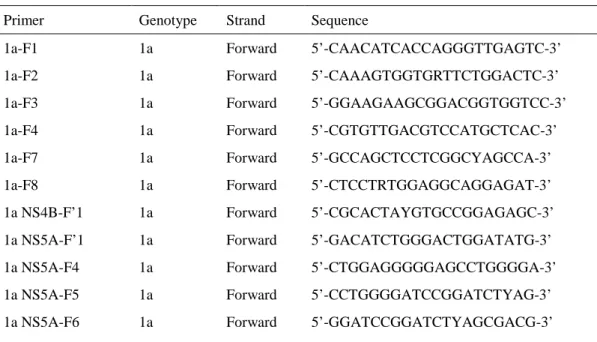
SepaGene RV-R kit(Eidia)又はHigh Pure viral RNA kit(Roche)で患者血清から抽出した total RNAから,各genotypeの特異的プライマー(表1),SuperScript III First-Strand Synthesis System(Invitrogen)を用いた逆転写(Reverse transcription:RT)反応によりHCV cDNAを 合成した。RT反応の条件は以下の通り行った。
<RT反応>
65℃,5分(変性) → 氷上,1分 → 50℃,50分(伸長) → 85℃,5分 → 37℃,20 分
17
表 1 RT 用増幅プライマー
Primer Genotype Strand Sequence
1a-1b 3UTR-R3 1a,1b Reverse 5’-TGCAGTCATGCGGCTCACGG-3’
1a 3UTR-R11 1a Reverse 5’-SCYGGAGTGKTTASCCCAA-3’
1a 3UTR-R12 1a Reverse 5’-SCYGGAGTGKTTASCCCAACCTTYA-3’
1b-RT 1b Reverse 5’-CCTGGAGTGKTTAGCTCCC-3’
*KはGとT,SはGとC,Y はCとTの混合塩基を示した。
各genotypeの特異的プライマー(表2)を用いたpolymerase chain reaction(PCR)により,
HCVのポリメラーゼ領域を増幅した。PCRの反応条件は以下の通り行った。
<First PCR>
Blend Taq-Plus polymerase(Toyobo)を用いて,94℃,2分 → 98℃,10秒(変性)→ 55℃,
30秒(アニーリング) → 72℃,2.5分(伸長)(下線部を30回反応を繰り返す)又は LA Taq polymerase(Takara)を用いて,94℃,3分 → 94℃,30秒 → 55℃,30秒 → 72℃,
2.5分 (下線部を30回反応を繰り返す)
<Second PCR>
KOD FX polymerase(Toyobo)を用いて,94℃,2分 → 98℃,10秒 → 55℃,30秒 → 68℃,
2分(下線部を40回反応を繰り返す)又はLA Taq polymerase を用いて,94℃,3分 → 94℃,
30秒 → 55℃,30秒 → 72℃,2分 (下線部を30回反応を繰り返す)
表 2 PCR 用増幅プライマー
Primer Genotype Strand Sequence
1a-F1 1a Forward 5’-CAACATCACCAGGGTTGAGTC-3’
1a-F2 1a Forward 5’-CAAAGTGGTGRTTCTGGACTC-3’
1a-F3 1a Forward 5’-GGAAGAAGCGGACGGTGGTCC-3’
1a-F4 1a Forward 5’-CGTGTTGACGTCCATGCTCAC-3’
1a-F7 1a Forward 5’-GCCAGCTCCTCGGCYAGCCA-3’
1a-F8 1a Forward 5’-CTCCTRTGGAGGCAGGAGAT-3’
1a NS4B-F’1 1a Forward 5’-CGCACTAYGTGCCGGAGAGC-3’
1a NS5A-F’1 1a Forward 5’-GACATCTGGGACTGGATATG-3’
1a NS5A-F4 1a Forward 5’-CTGGAGGGGGAGCCTGGGGA-3’
1a NS5A-F5 1a Forward 5’-CCTGGGGATCCGGATCTYAG-3’
1a NS5A-F6 1a Forward 5’-GGATCCGGATCTYAGCGACG-3’
18
1a NS5A-F7 1a Forward 5’-CGGATCTYAGCGACGGGTCA-3’
1a NS5A-F8 1a Forward 5’-CTYAGCGACGGGTCATGGTC-3’
1a 3UTR-R5 1a Reverse 5’-AATGGCCTAAGAGGCCGGAG-3’
1a 3UTR-R6 1a Reverse 5’-CCTAAGAGGCCGGAGTGTTT-3’
1a 3UTR-R7 1a Reverse 5’-GAGGCCGGAGTGTTTAYCCC-3’
1a 3UTR-R8 1a Reverse 5’-CGGAGTGTTTAYCCCAACCT-3’
1a 3UTR-R11 1a Reverse 5’-SCYGGAGTGKTTASCCCAA-3’
1a 3UTR-R12 1a Reverse 5’-SCYGGAGTGKTTASCCCAACCTTYA-3’
1b NS5A-F4 1b Forward 5’-CTTGAGGGRGAGCCGGGGGA-3’
1b NS5A-F5 1b Forward 5’-CCGGGGGAYCCCGATCTCAG-3’
1b NS5A-F6 1b Forward 5’-GGAYCCCGATCTCAGCGACG-3’
1b NS5A-F7 1b Forward 5’-CCGATCTCAGCGACGGGTCY-3’
1b NS5A-F8 1b Forward 5’-CTCAGCGACGGGTCYTGGTC-3’
1b NS5A-F10 1b Forward 5’-TCCGACGYTGAGTCGTACTC-3’
1b NS5A-F12 1b Forward 5’-ACGGTTGTCCTGACAGARTC-3’
1b NS5A-F13 1b Forward 5’-CCRGTGGTRCACGGGTGCCC-3’
1b-RT 1b Reverse 5’-CCTGGAGTGKTTAGCTCCC-3’
1b 3UTR-R5 1b Reverse 5’-RATGGCCTATTGGCCTGGAG-3’
1b 3UTR-R6 1b Reverse 5’-CCTATTGGCCTGGAGTGTTT-3’
1b 3UTR-R7 1b Reverse 5’-TGGCCTGGAGTGTTTAGCTC-3’
1b 3UTR-R8 1b Reverse 5’-TGGAGTGTTTAGCTCCCCGT-3’
1a-1b 3UTR-R3 1a, 1b Reverse 5’-TGCAGTCATGCGGCTCACGG-3’
1a-1b 3UTR-R5 1a, 1b Reverse 5’-CAGCTAGCCGTGACTAGGGC-3’
1a-1b 3UTR-R6 1a, 1b Reverse 5’-AGCCGTGACTAGGGCTAAGA-3’
1a-1b 3UTR-R7 1a, 1b Reverse 5’-TGACTAGGGCTAAGATGGAG-3’
*KはGとT,RはAとG,SはGとC,YはCとTの混合塩基を示した。
増幅したPCR産物を電気泳動後に精製し,各genotypeの特異的プライマー(表3)を用 いて塩基配列をABI Prism 3100 genetic analyzer(Applied Biosystems)で解析し,Vector NTI software(Invitrogen)で塩基配列を同定した。Reference株はgenotype 1aではH77株(GenBank accession number AF009606),genotype 1bではCon1株(GenBank accession number AJ238799)
を使用した。
19
表 3 Population sequencing 用シークエンスプライマー
Primer Genotype Strand Sequence
1a NS5A-F7 1a Forward 5’-CGGATCTYAGCGACGGGTCA-3’
1a NS5A-F8 1a Forward 5’-CTYAGCGACGGGTCATGGTC-3’
1a NS5B-Fa 1a Forward 5’-CCACATCAACTCCGTGTGGA-3’
1a NS5B-Fb 1a Forward 5’-TGTTGTGACCTGGACCCCCA-3’
1a NS5B-Fc 1a Forward 5’-AACGTGTCAGTCGCCCACGA-3’
1a NS5B-Fd 1a Forward 5’-GAGCTTGGAGACACCGGGCC-3’
1a NS5B-Ra 1a Reverse 5’-TTAACCTCCTTGAGCACGTC-3’
1a NS5B-Rb 1a Reverse 5’-ACGAGGAATTCAACCCGCTG-3’
1a NS5B-Rc 1a Reverse 5’-TGGTCATAGCCTCCGTGAAG-3’
1a NS5B-Rd 1a Reverse 5’-CCATGGAGTCTTTGAATGAT-3’
1a 3UTR-R7 1a Reverse 5’-GAGGCCGGAGTGTTTAYCCC-3’
1a 3UTR-R8 1a Reverse 5’-CGGAGTGTTTAYCCCAACCT-3’
1b NS5A-F7 1b Forward 5’-CCGATCTCAGCGACGGGTCY-3’
1b NS5A-F8 1b Forward 5’-CTCAGCGACGGGTCYTGGTC-3’
1b NS5B-Fa 1b Forward 5’-AAGGACGTCCGGAACCTATC-3’
1b NS5B-Fb 1b Forward 5’-TACCAATGTTGTGACTTGGC-3’
1b NS5B-Fc 1b Forward 5’-ATAACATCATGCTCCTCCAA-3’
1b NS5B-Fd 1b Forward 5’-CTGGAGACATCGGGCCAGAA-3’
1b NS5B-Ra 1b Reverse 5’-ACTGTGGACGCCTTCGCCTT-3’
1b NS5B-Rb 1b Reverse 5’-ACCAGGAACTCGACCCGCTG-3’
1b NS5B-Rc 1b Reverse 5’-TAGTCATAGCCTCCGTGAAG-3’
1b NS5B-Rd 1b Reverse 5’-TGGAGTGAAAATGCGCTAAG-3’
1b 3UTR-R7 1b Reverse 5’-TGGCCTGGAGTGTTTAGCTC-3’
1b 3UTR-R8 1b Reverse 5’-TGGAGTGTTTAGCTCCCCGT-3’
*YはCとTの混合塩基を示した。
4.1.4. Clonal sequencing 解析
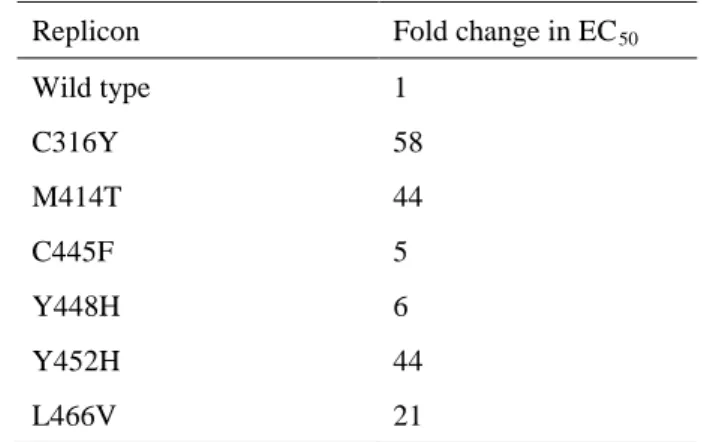
4.1.3.のPCR増幅産物を,Zero Blunt TOPO PCR cloning kit(Invitrogen)を用いてpCR Blunt II TOPO vector又はIn-Fusion HD Cloning Kit(Clontech)を用いてHCVレプリコンプラスミ ド(genotype 1b:Con1株)23)にクローンニングした。HCVレプリコンプラスミドはHCV 5’
非翻訳領域,ポリオウイルスIRES領域,ホタルルシフェラーゼ領域,脳心筋炎ウイルスIRES 領域,Con1株由来のNS3~3’XまでのHCV サブゲノム領域で構築されている(図7)。各
genotypeの特異的プライマー(表4)を用いた各患者のHCVポリメラーゼ領域の塩基配列
を10クローン以上,ABI Prism 3100 genetic analyzerで解析し,Vector NTI softwareで塩基配
20
列を同定した。各患者で同定した塩基配列で最も高頻度で検出された塩基配列をドミナン ト配列として選抜した。
図 7 HCV レプリコンプラスミドの模式図
*NS3~3’X領域はCon1株(genotype 1b)由来のHCV遺伝子配列
表 4 Clonal sequencing 用シークエンスプライマー
Primer Genotype Strand Sequence
3575FW 1a Forward 5’-CAACATGCACTACCCGTCAT-3’
M13 F-20 1a Forward 5’-GTAAAACGACGGCCAGT-3’
1a NS5A-F7 1a Forward 5’-CGGATCTYAGCGACGGGTCA-3’
1a NS5B-Fa 1a Forward 5’-CCACATCAACTCCGTGTGGA-3’
1a NS5B-Fb 1a Forward 5’-TGTTGTGACCTGGACCCCCA-3’
1a NS5B-Fc 1a Forward 5’-AACGTGTCAGTCGCCCACGA-3’
8183-R 1a Reverse 5’-CATTAACCTATAAAAATAGGC-3’
M13 R 1a Reverse 5’-AACAGCTATGACCATG-3’
3X-R1 1a Reverse 5’-AAAGTGCCACCTGACGTCTAAGAA-3’
3X-R2 1a Reverse 5’-GGCGTATCACGAGGCCCTTTCGTC-3’
3575FW 1b Forward 5’-CAACATGCACTACCCGTCAT-3’
M13 F-20 1b Forward 5’-GTAAAACGACGGCCAGT-3’
1b NS5B-F3 1b Forward 5’-CCCCCCTTGAGGGRGAGCCG-3’
1b NS5B-F7 1b Forward 5’-CCGATCTCAGCGACGGGTCY-3’
21
1b NS5B-Fa 1b Forward 5’-AAGGACGTCCGGAACCTATC-3’
1b NS5B-Fb 1b Forward 5’-TACCAATGTTGTGACTTGGC-3’
1b NS5B-Fc 1b Forward 5’-ATAACATCATGCTCCTCCAA-3’
8183-R 1b Reverse 5’-CATTAACCTATAAAAATAGGC-3’
M13 R 1b Reverse 5’-AACAGCTATGACCATG-3’
*RはAとG,YはCとTの混合塩基を示した。
4.1.5. 細胞
HCV レプリコン細胞は1999年にLohmannのグループが初めて樹立した細胞株で,ヒト 肝がん細胞株のHuh-7細胞中でHCV遺伝子を含むHCVレプリコンRNAが自律複製してい る細胞株である24)。
4.1.6. 細胞培養
Huh-7.5細胞25)及びHCVレプリコンRNAを導入したHCVレプリコン細胞は,10% ウ シ胎児血清(Fetal bovine serum:FBS),0.1 mM非必須アミノ酸,100 U/mL ペニシリン,100 mg/mL ストレプトマイシン,2 mM L-グルタミンを含む high glucose Eagle 培地(Life Technologies)用いて培養した。
4.1.7. 感受性評価
4.1.4.のclonal sequencing解析より選抜した各患者のドミナント配列を有するHCVレプリ コンプラスミドを構築するため,各genotypeの特異的プライマー(表5)を用いてPCRで HCVポリメラーゼ領域を増幅した。PCRの反応条件は以下の通り行った。
<PCR>
KOD FX polymeraseを用いて,94℃,2分 → 98℃,10秒(変性) → 68℃,2分(伸長)
(下線部を40回反応を繰り返す)
表 5 HCV レプリコンプラスミド構築用増幅プライマー
Primer Genotype Strand Sequence
1a NS5B-FW1 1a Forward 5’-GACGTCGTCTGCTGCTCAATGTCTTATTCC-3’
1a NS5B-FW4 1a Forward 5’-GACGTCGTCTGCTGCTCAATGTCCTATTCC-3’
1a NS5B-FW5 1a Forward 5’-GACGTCGTCTGCTGCTCAATGTCTTATTCT-3’
1a NS5B-FW6 1a Forward 5’-GACGTCGTCTGCTGCTCAATGTCYTATTCC-3’
1a NS5B-FW7 1a Forward 5’-GACGTCGTCTGCTGCTCAATGTCTTATTCY-3’
1a NS5B-FW8 1a Forward 5’-GACGTCGTCTGCTGCTCYATGTCTTATTCC-3’
22
1a NS5B-FW9 1a Forward 5’-GACGTCGTCTGCTGCTCAATGTCCTATTCY-3’
1a NS5B-FW10 1a Forward 5’-GACGTCGTCTGCTGCTCRATGTCTTATTCC-3’
1a NS5B-FW11 1a Forward 5’-GACGTCGTCTGCTGCTCGATGTCTTATTCC-3’
1a NS5B-FW12 1a Forward 5’-GACGTCGTCTGCTGCTCCATGTCCTACTCC-3’
1a NS5B-FW13 1a Forward 5’-GACGTCGTCTGCTGCTCAATGTCCTATWCC-3’
1a NS5B-FW14 1a Forward 5’-GACGTCGTCTGCTGCTCGATGTCCTATTCC-3’
1a NS5B-FW15 1a Forward 5’-GACGTCGTCTGCTGCTCAATGTCTTACTCT-3’
1a NS5B-FW16 1a Forward 5’-GACGTCGTCTGCTGCTCGATGTCTTACTCT-3’
1b NS5B-FW1 1b Forward 5’-GACGTCGTCTGCTGCTCGATGTCCTACACA-3’
1a NS5B-RV1 1a Reverse 5’-TGTTTAGCTCCCCGTTCACCGGTTGGGGAG-3’
1a NS5B-RV2 1a Reverse 5’-TGTTTAGCTCCCCGTTCATCGGTTGGGGAG-3’
1a NS5B-RV3 1a Reverse 5’-TGTTTAGCTCCCCGTTCAYCGGTTGGGGAG-3’
1a NS5B-RV4 1a Reverse 5’-TGTTTAGCTCCCCGTTCACCGGTYGSGGAG-3’
1a NS5B-RV5 1a Reverse 5’-TGTTTAGCTCCCCGTTCATCGGTYGSGGAG-3’
1a NS5B-RV6 1a Reverse 5’-TGTTTAGCTCCCCGTTTAYCGGTTGGGGAG-3’
1a NS5B-RV7 1a Reverse 5’-TGTTTAGCTCCCCGTTTATCGGTYGSGGAG-3’
1a NS5B-RV8 1a Reverse 5’-TGTTTAGCTCCCCGTTTATCGGTTGGGGAG-3’
1b NS5B-RV1 1a Reverse 5’-TGTTTAGCTCCCCGTTCATCGGTTGGGGAG-3’
1a-1b NS5B-RV1 1a, 1b Reverse 5’-TGTTTAGCTCCCCGTTTACCGGTTGGGGAG-3’
*RはAとG,SはGとC,W はAとT,YはCとTの混合塩基を示した。
増幅したPCR産物を電気泳動後に精製して,In-Fusion HD Cloning Kitを用いて,Xho I
(Takara)及びSpe I(Takara)処理したHCVレプリコンプラスミドに各患者のドミナント 配列の精製産物を導入した。導入後のHCV レプリコンプラスミドのHCVポリメラーゼ領 域がドミナント配列と同一であることをシークエンス解析で確認後,制限酵素のSpe I処理 したHCVレプリコンプラスミドからMEGAscript T7 kit(Ambion)を用いて,HCVレプリ コンRNAを作成した。Gene Pulser II(Bio-Rad)を用いて,960 µF,270 Vの条件下で,8×106 cellsのHuh-7.5細胞にHCVレプリコンRNA(5 µg又は15 µg)を導入した。導入後の細胞 を96 wellプレートに2×104 cells/well で播種し,37℃,5% CO2の条件で48時間培養後,
JTK-853を含むジメチルスルホキシド(Dimethyl sulfoxide:DMSO)溶液を添加し,さらに
37℃,5% CO2の条件で48時間培養した。JTK-853のHCVに対する複製阻害はSteady-Glo luciferase assay system(Promega)を用いたルシフェラーゼ活性26)をMicroplate Luminescence CounterのTopCount(Packard)で測定し,RNA導入96時間後のルシフェラーゼ活性から下 記の方法で阻害率を算出した。ルシフェラーゼ活性による阻害率から算出される 50%効果 濃度(50% effective concentration:EC50)を指標に複製阻害を評価した21)。
23
<ルシフェラーゼ活性による阻害率の算出法>
阻害率(%) = 100 – [100 × (Ls) / (Lc)]
Ls:JTK-853処置群のルシフェラーゼ活性
Lc:DMSO処置群のルシフェラーゼ活性
24
4.2. B 型肝炎ウイルスの新規安定発現細胞系の構築に関する研究
(第 2 章)
4.2.1. 化合物
エンテカビル,テノホビル,ラミブジンは和光純薬工業より購入した。Bay41-4109 27), HBF-0259 28),β-Thujaplicinol 29),CCC-0975 30)は日本たばこ産業株式会社,医薬総合研究 所で合成した。Food and drug administration(FDA)approval drug screening libraryの414化合 物はSelleck chemicalより購入した。
4.2.2. 細胞
HepG2.2.15細胞31)は1987年にSellsのグループが樹立した細胞株で,ヒト肝がん細胞株
HepG2細胞のゲノム中にHBV遺伝子が組み込まれており,細胞の増殖と共にHBV複製が
起こる細胞株である。またHepAD38細胞32)は 1997年にLadnerのグループが樹立した細 胞株で,HepG2 細胞のゲノム中にテトラサイクリン(Tetracycline:Tet)プロモーターを有 するHBV遺伝子が組み込まれており,TetによりHBV複製が調節可能な細胞株である。
4.2.3. 細胞培養
HepG2細胞は10% FBS,100 U/mL ペニシリン,100 µg/mL ストレプトマイシン,5 µg/mL インスリンを含む DMEM/F12 培地(Life Technologies)用いて培養した。また HepG2.2.15 細胞及びHepG2.2.15.7細胞は,HepG2細胞の培地に400 µg/mL ジェネティシンを添加して,
HepAD38細胞及びHep38.7-Tet細胞はHepG2細胞の培地に400 µg/mL ジェネティシン,400
ng/mL Tetを添加して培養した。さらに,新規HBV安定発現細胞はHepG2細胞の培地に400
µg/mL ジェネティシン,250 µg/mL ハイグロマイシンを添加して培養した。
4.2.4. 細胞播種
Hep38.7-Tet細胞を6 cmディッシュにTet添加培地を用いて,8×105 cells/wellで播種し,
37℃,5% CO2の条件で24時間培養後,Tet非添加培地に交換して,37℃,5% CO2の条件で 培養を開始した。6 日間培養後,Tet 添加培地に交換し,さらに 37℃,5% CO2の条件で 6 日間培養後の細胞のcore DNA量,cccDNA量,HBV RNA量及び培養上清中のHBV DNA 量をreal time PCR法で,培養上清中のHBeAg量及びHBsAg量をELISA法で測定した。
4.2.5. 化合物スクリーニング
Hep38.7-Tet細胞を96 wellプレートにTet添加培地を用いて,3×104 cells/wellで播種し,
37℃,5% CO2の条件で24時間培養後,化合物を含むTet非添加培地に交換して,37℃,5%
CO2の条件で培養を開始した。6日間培養後,Tet添加培地に交換し,さらに37℃,5% CO2 の条件で6日間培養した。化合物のHBVに対する複製阻害は細胞内のcccDNA量をreal time
25
PCR 法で,培養上清中の HBeAg 量を ELISA 法で測定し,宿主細胞に対する細胞毒性は CellTiter 96 AQueous One Solution Cell Proliferation Assayを用いてEnVision(PerkinElmer)で
490 nmの吸光度を測定し,それぞれ下記の方法で% of controlを算出した。
<Real time PCRによる% of controlの算出法>
% of control = 100 × (Rs) / (Rc) Rs:化合物処置群の増幅量
Rc:DMSO処置群の増幅量
<吸光度による% of controlの算出法>
% of control = 100 × (As – Ab) / (Ac – Ab) As:化合物処置群の吸光度
Ac:DMSO処置群の吸光度
Ab:Blankの吸光度
4.2.6. 核酸精製と Southern blot 法及び Northern blot 法
細胞内core DNAはGuoらの論文33)を参考に,細胞内cccDNAはHirtらの論文34)を参 考に調製した。抽出したDNA(15 µg)を1.2%アガロース電気泳動で分離し,20×Saline sodium citrate buffer(Nacalai Tesque)でHybond-XL membrane(GE Healthcare)に転写した。また TRIzol reagents(Invitrogen)で精製したtotal RNA(10 µg)は2.2 Mホルムアルデヒドを含
む 1.2%アガロース電気泳動で分離し,Hybond-XL membrane に転写した。メンブラン上の
DNA及びRNAはAlkPhos direct labeling reagents(GE Healthcare)でラベル化された全長HBV DNAをプローブに用いて,65℃,6時間ハイブリダイズし,LAS-4000(GE Healthcare)で 検出した。
4.2.7. ウイルス精製
培養上清中のウイルスはLenhoffらの論文35)を参考に調製した。10% PEG8000を添加し て,氷上で1時間静置し,8000 rpm,4℃,10分間で遠心して調製した。遠心後の沈殿物を TNE bufferに溶解させた。調製後のウイルス溶液中のHBV DNAは,QIAamp DNA Mini Kit
(Qiagen)を用いて精製した。
4.2.8. Real time PCR 法
ウイルスDNAをEXPRESS SYBR GreenER qPCR Supermix(Life Technologies)で定量し た。以下に使用したプライマーとPCR の反応条件を示しており,Applied Biosystems 7500 sequence detection system(Life Technologies)で検出した。
26
<coreDNA,extracellular HBV DNA用プライマー>
5’-CTCGTGGTGGACTTCTCTC-3’(Forward)
5’-AAGATGAGGCATAGCAGCA-3’ (Reverse)
<cccDNA用プライマー>
5’-CGTCTGTGCCTTCTCATCTGC-3’(Forward)
5’-GCACAGCTTGGAGGCTTGAA-3’ (Reverse)
<PCR>
50℃,2分 → 95℃,2分 → 95℃,15秒 → 60℃,1分(下線部を45回反応を繰り返す)
4.2.9. RT-real time PCR 法
TRIzol reagentsで精製したtotal RNA(5 µg)を5 unitsのRQ1 RNase-free DNases (Promega)
で処理後,RNeasy mini kit(Qiagen)で精製した。精製したtotal RNA(1 µL)からSuperScript III First-Strand Synthesis Systemを用いたRT反応によりcDNAを合成した。合成したDNA
を4.2.8.と同様な試薬,反応条件及び検出器で定量した。以下に使用したプライマーを示し
た。
<プライマー>
5’-GACCACCAAATGCCCCTATC-3’(Forward)
5’-GATTGAGATCTTCTGCGACGC-3’(Reverse)
4.2.10. RT-PCR 法
Total RNAからSuperScript III First-Strand Synthesis Systemを用いたRT反応により,合成 したcDNAをPrimeSTAR Max DNA polymerase(Takara)で増幅し,1%アガロース電気泳動 により増幅産物を分離した。以下に使用したプライマーとPCRの反応条件を示した。
<プライマー>
5’-TAGGCATAAATTGGTCTG-3’(Forward)
5’-GATTGAGATCTTCTGCGACGC-3’(Reverse)
<PCR>
94℃,1分 → 98℃,10秒 → 55℃,5秒 → 72℃,1分(下線部を45回反応を繰り返す)
27
4.2.11. 免疫染色
4%パラホルムアルデヒドで細胞をスライドガラス上に固定して,0.3% Triton-X-100で処
理後,抗hepatitis B core protein(HBc)抗体(Dako)で染色した36, 37)。
4.2.12. ELISA 法
培養上清中のHBeAg量はEnzygnost HBe monoclonal kit (Siemens),HBsAg量はEnzygnost HBsAg 6.0 kit(Siemens)を用いて,EnVisionで450 nmの吸光度を測定した。吸光度による%
of controlは4.2.5.と同様に算出した。
4.2.13. HBV 感染患者の背景情報
インフォームドコンセントは取得済みの 9 人の慢性 B 型肝炎患者(genotype A:2 人,
genotype B:1人,genotype C:6人)から血清を取得した。全患者の血清中HBV DNA量は 高値を示し,Patient C-2を除く8人の血清中HBeAgは陽性,血清中hepatitis B e antibody
(HBeAb)は陰性を示した(表6)。
表 6 HBV 感染患者の背景情報
Patient ID
Country of origin
Age (years)
Sex ALT (IU/L)
Genotype HBeAg HBeAb HBV DNA (log10 copies/mL) A-1 Japan 39 Male 317 A >1250 (-) 9.0
A-2 Philippines 23 Female 26 A 1166.6 (-) 9.7 B-1 China 26 Female 18 B 1371.4 (-) 8.4
C-1 Japan 34 Female 20 C 839.4 0.0 8.8
C-2 Japan 35 Male 157 C NT NT 9.1
C-3 Japan 26 Male 33 C 1604.8 (-) 9.4
C-4 China 26 Female 16 C 1357.8 (-) 8.8 C-5 Thailand 33 Female 33 C 1448.3 (-) 9.3 C-6 Japan 26 Female 39 C 1368.9 (-) 9.1 NT:未実施
4.2.14. 患者由来の HBV 配列の同定
HBV DNAはQIAmp DNA blood kit(Qiagen)を用いて,HBV感染患者血清から精製した。
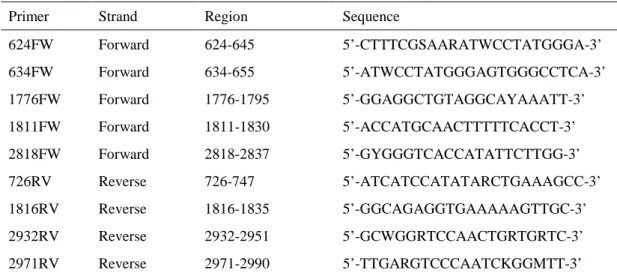
全長のHBV遺伝子を3領域に分けて増幅用プライマー(表7)を用いたPCRにより,HBV 遺伝子の3領域を増幅した。PCRの反応条件は以下の通り行った。
<PCR>
PrimeSTAR HS DNA Polymerase(Toyobo)を用いて,94℃,10秒 → 98℃,10秒 → 55℃,
10秒 → 72℃,2分 (下線部を40回反応を繰り返す)
28
表 7 増幅用プライマー
Primer Strand Region Sequence
624FW 634FW 1776FW
Forward Forward Forward
624-645 634-655 1776-1795
5’-CTTTCGSAARATWCCTATGGGA-3’
5’-ATWCCTATGGGAGTGGGCCTCA-3’
5’-GGAGGCTGTAGGCAYAAATT-3’
1811FW Forward 1811-1830 5’-ACCATGCAACTTTTTCACCT-3’
2818FW Forward 2818-2837 5’-GYGGGTCACCATATTCTTGG-3’
726RV Reverse 726-747 5’-ATCATCCATATARCTGAAAGCC-3’
1816RV 2932RV
Reverse Reverse
1816-1835 2932-2951
5’-GGCAGAGGTGAAAAAGTTGC-3’
5’-GCWGGRTCCAACTGRTGRTC-3’
2971RV Reverse 2971-2990 5’-TTGARGTCCCAATCKGGMTT-3’
*塩基番号は全長HBVゲノムのEcoR I サイトを開始点とした。KはGとT,MはAとC,RはAとG,S はGとC,WはAとT,YはCとTの混合塩基を示した。
増幅したPCR産物を電気泳動後に精製し,Zero Blunt TOPO PCR Cloning Kitでクローニン グ後,シークエンスプライマー(表8)で各患者の領域毎に4クローン以上,3130xl Genetic Analyzer(Applied Biosystems)で塩基配列を解析し,GENETYX ver.9 (GENETYX)で塩基 配列を同定した。各患者で同定した塩基配列で最も高頻度で検出された塩基配列をドミナ ント配列として選抜した。Reference配列38)はgenotype A ではAe株(GenBank accession number AB246337),genotype BではBj株(GenBank accession number AB246341),genotype C ではC株(GenBank accession number AB246345)を使用した。
表 8 シークエンスプライマー
Primer Strand Region Sequence
M13FW 192FW 1174FW
Forward Forward Forward
Vector 192-213 1174-1195
5’-GTAAAACGACGGCCAG-3’
5’-TCGTGTTACAGGCGGKGTTTTT-3’
5’-TGYCAAGTRTTTGCTGACGCAA-3’
2374FW Forward 2374-2395 5’-TAGAAGAAGAACTCCCTCGCCT-3’
M13RW Reverse Vector 5’-CAGGAAACAGCTATGAC-3’
243RV 1264RV
Reverse Reverse
243-263 1264-1284
5’-TCCACCACGAGTCTAGACTCT-3’
5’-TARGAGTTCCGCWGTATGGAT-3’
2426RV Reverse 2426-2445 5’-TCCCGAGATTGAGATCTTCT-3’
*塩基番号は全長HBVゲノムのEcoR I サイトを開始点とした。KはGとT,RはAとG,WはAとT, YはCとTの混合塩基を示した。
29
4.2.15. 一過性発現系用の HBV 発現プラスミドの構築
各患者で同定したドミナント配列を用いて,Sugiyamaらの論文38)を参考に1.24倍長(約 4000 base pairs:bps)のHBV配列を組み込んだHBV発現プラスミド(図8)を構築するた め,増幅用プライマー(表9)を用いたPCRでHBV遺伝子を増幅した。PCRの反応条件は 以下の通り行った。
<First PCR>
PrimeSTAR MAX polymerase(Toyobo)を用いて,94℃,10秒 → 98℃,10秒 → 55℃,
10秒 → 72℃,2分 (下線部を35回反応を繰り返す)
<Second PCR>
PrimeSTAR MAX polymeraseを用いて,94℃,10秒 → 98℃,10秒 → 55℃,10秒 → 72℃,
3.5分 (下線部を40回反応を繰り返す)
図 8 HBV 発現プラスミドの模式図(一過性発現用)
30
表 9 HBV 発現プラスミド構築用増幅プライマー(一過性発現用)
Primer Step Strand Sequence
1814FWi First Forward 5’-ATGCAACTTTTTCACCTCTGC-3’
2824FWi First Forward 5’-ACCATATTCTTGGGAACAAGA-3’
676FWi First Forward 5’-TTTACTAGTGCCATTTGTTCA-3’
1413HFW1 Second Forward 5’-TGATTACGCCAAGCTTGACGTCCTTTGTCTACGTC CCGT-3’
1413HFW2 Second Forward 5’-TGATTACGCCAAGCTTGACGTCCTTTGTTTACGTC CCGT-3’
3200FW Second Forward 5’-ATCCTCAGGCCATGCAGTGGA-3’
2820RVi First Reverse 5’-TCCCAAGAATATGGTGACCC-3’
670RVi First Reverse 5’-AATGGCACTAGTAAACTGAGC-3’
1808RVi First Reverse 5’-GTGAAAAAGTTGCATGGTGCT-3’
2164SRV1 Second Reverse 5’-TGATTAAGCTGTCGACTTTAAACCCATGTTAGTAT TAACA-3’
2164SRV2 Second Reverse 5’-TGATTAAGCTGTCGACTTTAGGCCCATGTTAACAT TGACA-3’
2164SRV3 Second Reverse 5’-TGATTAAGCTGTCGACTTTAGGCCCATATTAATGT TGACA-3’
2164SRV4 Second Reverse 5’-TGATTAAGCTGTCGACTTTAGGCCCATATTAACAT TGACA-3’
2164SRV5 Second Reverse 5’-TGATTAAGCTGTCGACTTCAGGCCCATATTAACAT TGACA-3’
2164SRV6 Second Reverse 5’-TGATTAAGCTGTCGACTTTAGGCCCATGTTAGTAT TAACA-3’
243RV Second Reverse 5’-TCCACCACGAGTCTAGACTCT-3’
増幅したPCR産物を電気泳動後に精製して,Hind III(Takara)及びXba I(Takara)又は EcoR I(Takara),Sal I(Takara)及びXba I又はEcoR I処理した。DNA Ligation Kit Ver.2.1
(Takara)を用いて,Hind III及びSal I処理したpUC19に各患者のドミナント配列を制限酵 素処理した産物を導入した。導入後のHBV発現プラスミドのHBV領域がドミナント配列 と同一であることをシークエンス解析で確認した。
4.2.16. 一過性発現系
HepG2細胞を24 wellプレートに3×105 cells/wellで播種し,37℃,5% CO2の条件で24時 間培養後,Lipofectamine 3000 transfection reagent(Invitrogen)を用いて4.2.15.で構築した1.24
31
倍長のHBV発現プラスミド(0.5 µg)を細胞内に導入した。さらに37℃,5% CO2の条件で 48時間培養後に細胞のcore DNA量及び培養上清中のHBV DNA量をreal time PCR法で,
培養上清中のHBeAg量及びHBsAg量をELISA法で測定した。
4.2.17. HBV 安定発現細胞用の HBV 発現プラスミドの構築
Genotype A,B,Cの1.24倍長のHBV発現プラスミドを用いて,全長のHBV遺伝子を増 幅し,In-Fusion HD Cloning Kitを用いて2倍長(約6400 bps)のHBV配列を組み込んだHBV 発現プラスミドを構築した(図9)。
図 9 HBV 発現プラスミドの模式図(HBV 安定発現細胞用)
4.2.18. HBV 安定発現細胞の選抜
HepG2細胞を10 cmディッシュに3×106 cellsで播種し,37℃,5% CO2の条件で24時間 培養した。Lipofectamine 3000 transfection reagentを用いて,4.2.17.で構築した2倍長のHBV 発現プラスミド(5 µg)とハイグロマイシン耐性遺伝子を発現するプラスミド(250 ng)で 共導入した。さらに37℃,5% CO2の条件で96時間培養後にハイグロマイシンを250 µg/mL 添加して細胞を薬剤選択した。選抜後の細胞を限界希釈法でクローニングし,クローニン グ後の培養上清中のHBeAg量及びHBsAg量をELISA法で測定し,分泌能が確認できた細 胞をHBV安定発現細胞として選抜した。
32
4.2.19. 細胞ゲノム DNA の精製と確認
ゲノムDNAはWaltersらの論文39)を参考に調製した。HBV安定発現細胞を8.0×106 cells で回収し,細胞中のゲノムDNAを抽出した。ゲノムDNAをHBV遺伝子配列中のみに制限 酵素サイトを有する制限酵素EcoT22 I(Takara)で処理後,電気泳動によりDNAを分離し,
AlkPhos direct labeling reagentsでラベル化した全長のHBV DNAプローブで検出した。
4.2.20. HBV 安定発現細胞の複製活性
HBV安定発現細胞を24 wellプレートに2×105 cells/wellで播種し,細胞播種1,4,7日後 の細胞中のcore DNA量,HBV RNA量及び培養上清中のHBV DNA量をreal time PCR法で,
培養上清中のHBeAg量及びHBsAg量をELISA法で測定した。抗HBV 剤の複製阻害は細 胞播種24時間後に化合物を添加し,6日間培養後の細胞のcore DNA量,HBV RNA量及び 培養上清中のHBV DNA量をreal time PCR法で測定した。同時に宿主細胞に対する細胞毒 性をCellTiter 96 AQueous One Solution Cell Proliferation Assayを用いてEnVisionで490 nmの 吸光度を測定した。Real time PCR及び吸光度からの% of controlは4.2.5.と同様に算出し,
エンテカビルのEC50はreal time PCRの% of controlから算出した。
4.2.21. ウイルス調製と感染性評価
ウイルスはWatashiらの論文37)を参考に調製した。HepG2細胞にhuman sodium taurocholate cotransporting polypeptide(NTCP)を安定発現させたHepG2/NTCP細胞40)に,HBV安定発 現細胞から調製したウイルスを3×102 genome euivalents/cellで感染させ,ウイルス感染6時 間後及び4日後に培地を交換した。ウイルス感染7,11,14,18日後の培養上清中のHBV DNA 量をreal time PCR法で,HBsAg量をELISA法で測定した。抗HBV剤の複製阻害はウイル ス感染6時間後に化合物を添加し,3又は4日毎に化合物添加培地に交換し,ウイルス感染 18日後の細胞のcore DNA量をreal time PCR法で測定した。Real time PCRの% of controlは 4.2.5.と同様に算出した。
33
5. 結果,考察
5.1. C 型肝炎ウイルスの新規耐性変異同定法に関する研究
(第 1 章)
HCV非構造タンパクのNS5BタンパクがコードするHCVポリメラーゼは,RNA依存性 RNAポリメラーゼの機能を有しており,HCV複製過程に必須なウイルスタンパクである41,
42)。HCVポリメラーゼは,ヒトDNA及びRNAポリメラーゼとは塩基配列及びタンパク構 造が異なっているため,複数の抗HCV治療薬のターゲット分子となっている。NS5Bポリ メラーゼ阻害剤は核酸型又は非核酸型の2種類に大別され,核酸型はHCV複製時に基質の 核酸に代わり取り込まれることでRNA伸長反応を停止させる。一方,非核酸型は HCVポ リメラーゼの活性中心とは異なる5つのアロステリック部位に相互作用し,HCVポリメラ ーゼ活性を阻害することが知られている43)。日本たばこ産業株式会社が開発したJTK-853 は,HCV ポリメラーゼに結合する非核酸型のアロステリック阻害剤であり,ハイスループ ットスクリーニングから見出された21, 44)。JTK-853の構造を図10に示した。JTK-853はin
vitro酵素評価系で複数のgenotypeのHCVポリメラーゼ酵素活性を阻害し,in vitro細胞評
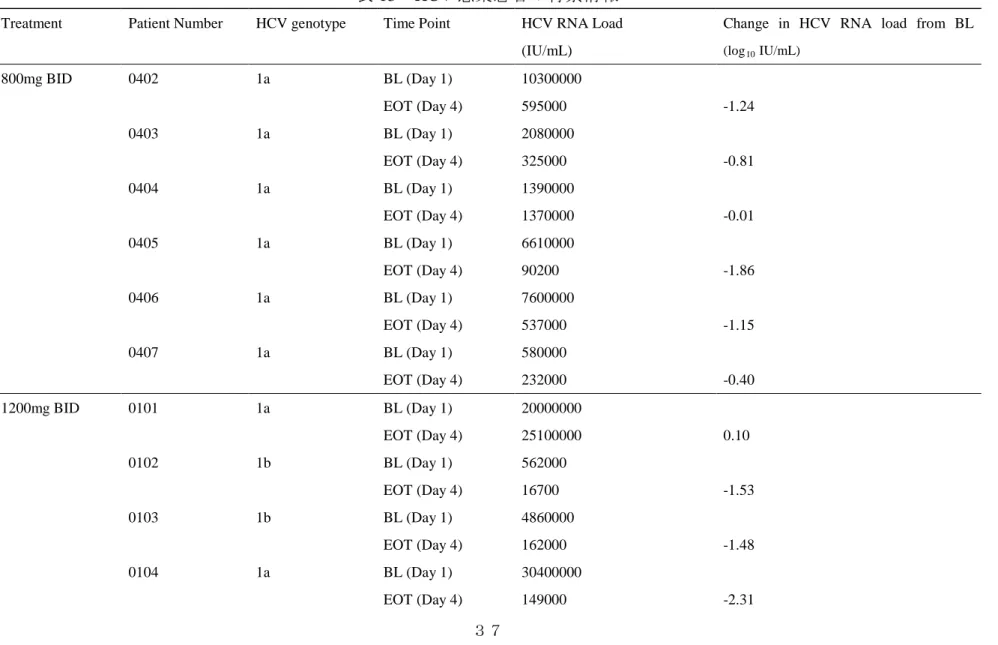
価系でgenotype 1a及び1bのHCVレプリコン細胞の複製活性を阻害した(表10, 11)。また 他の抗HCV治療薬(NS3/4Aプロテアーゼ阻害剤,NS5A阻害剤,核酸型のNS5Bポリメラ ーゼ阻害剤等)との併用では相加作用を示すことが報告されている21)。またin vitro耐性試 験においてHCVレプリコン細胞にJTK-853を長期処置して誘導される耐性変異には,HCV ポリメラーゼ領域のC316Y,M414T,C445F,Y448H,Y452H,L466Vがあり,各耐性変異 を導入したレプリコン細胞に対するJTK-853の感受性は,野生型レプリコン細胞に比べて5
~58倍低下した(表12)。またHCVポリメラーゼとJTK-853の共結晶構造解析から,確認 されたこれらの耐性変異部位はHCVポリメラーゼとJTK-853の結合部位の周辺であること が報告されている(図11)21)。現在まで抗HCV薬に対する確立された耐性変異同定法は,
HCVレプリコン細胞を用いたin vitro耐性試験のみであり,臨床サンプルを用いた耐性変異 同定法で確立された方法はない。