CTD 第 2 部
2.5 臨床に関する概括評価
2.5 臨床に関する概括評価 - 1 -
目次
頁 表一覧 ... 4 図一覧 ... 5 略号及び用語の定義 ... 6 2.5 臨床に関する概括評価 ... 8 2.5.1 製品開発の根拠 ... 8 2.5.1.1 薬効分類 ... 8 2.5.1.2 化学的及び薬剤学的性質 ... 8 2.5.1.3 適応症 ... 9 2.5.1.4 科学的背景 ... 9 2.5.1.4.1 成人HSCT 患者におけるサイトメガロウイルス ... 9 2.5.1.4.2 予防及び先制治療:既存の抗CMV 治療の限界 ... 9 2.5.1.5 レテルモビルの開発背景及び作用機序 ... 11 2.5.1.6 臨床開発プログラムの概要 ... 11 2.5.1.6.1 臨床開発プログラムの概要 ... 11 2.5.1.6.2 日本での承認申請に用いる臨床データ ... 26 2.5.1.7 規制当局からの指針及び助言 ... 27 2.5.1.7.1 規制当局からの指針 ... 27 2.5.1.7.2 米国食品医薬品局 ... 27 2.5.1.7.3 欧州規制当局 ... 31 2.5.1.7.4 日本の規制当局 ... 32 2.5.1.8 医薬品の臨床試験の実施の基準(GCP)の遵守 ... 33 2.5.2 生物薬剤学に関する概括評価 ... 34 2.5.2.1 製剤開発 ... 34 2.5.2.1.1 レテルモビル経口剤の開発 ... 34 2.5.2.1.2 レテルモビル注射剤の開発 ... 35 2.5.2.2 バイオアベイラビリティ ... 35 2.5.2.3 食事の影響 ... 35 2.5.3 臨床薬理に関する概括評価 ... 36 2.5.3.1 臨床薬理試験の概括評価 ... 36 2.5.3.2 ヒトにおける薬物動態 ... 36 2.5.3.3 レテルモビルの用量設定及び変動許容範囲 ... 39 2.5.3.4 内因性要因 ... 41 2.5.3.5 外因性要因 ... 42 2.5.4 有効性の概括評価 ... 452.5 臨床に関する概括評価 - 2 - 2.5.4.1 第Ⅱ相試験 ... 45 2.5.4.1.1 前期第Ⅱ相試験(019試験) ... 45 2.5.4.1.2 後期第Ⅱ相試験(020試験) ... 46 2.5.4.2 第Ⅲ相試験(001試験) ... 47 2.5.4.2.1 試験デザイン(001試験) ... 47 2.5.4.2.2 有効性評価項目 ... 49 2.5.4.2.3 有効性評価方法 ... 50 2.5.4.2.4 患者の内訳及び背景 ... 51 2.5.4.3 第Ⅲ相試験の有効性の結果 ... 57 2.5.4.3.1 移植後24週以内に臨床的に意味のある CMV 感染がみられた患者の割合 (主要評価項目) ... 57 2.5.4.3.2 部分集団解析 ... 59 2.5.4.3.3 有効性の副次評価項目 ... 63 2.5.4.3.4 有効性の探索的評価項目 ... 67 2.5.4.3.5 有効性の追加解析 ... 73 2.5.4.3.6 薬物動態と有効性の関係 ... 73 2.5.4.3.7 耐性変異の解析 ... 74 2.5.4.4 全集団の結果を日本人患者に適用することの妥当性 ... 75 2.5.4.5 有効性の結論 ... 76 2.5.5 安全性の概括評価 ... 77 2.5.5.1 全体的な曝露状況 ... 77 2.5.5.2 第Ⅰ相試験(併合データ) ... 78 2.5.5.3 第Ⅱ相試験 ... 80 2.5.5.3.1 前期第Ⅱ相試験(019試験) ... 81 2.5.5.3.2 後期第Ⅱ相試験(020試験) ... 81 2.5.5.4 第Ⅲ相試験(001試験) ... 84 2.5.5.4.1 安全性評価計画及び試験対象集団 ... 84 2.5.5.4.2 有害事象の要約 ... 86 2.5.5.4.3 すべての有害事象の解析 ... 87 2.5.5.4.4 追加の安全性解析 ... 94 2.5.5.4.5 器官別又は症候群別の有害事象の解析 ... 96 2.5.5.4.6 臨床検査、バイタルサイン及び心電図所見 ... 98 2.5.5.5 特別な集団における安全性 ... 99 2.5.5.6 精巣毒性の可能性の評価 ... 100 2.5.5.7 薬物動態と安全性の関係 ... 101 2.5.5.8 安全性の結論 ... 101 2.5.6 ベネフィットとリスクに関する結論 ... 103
2.5 臨床に関する概括評価 - 3 -
2.5 臨床に関する概括評価 - 4 -
表一覧
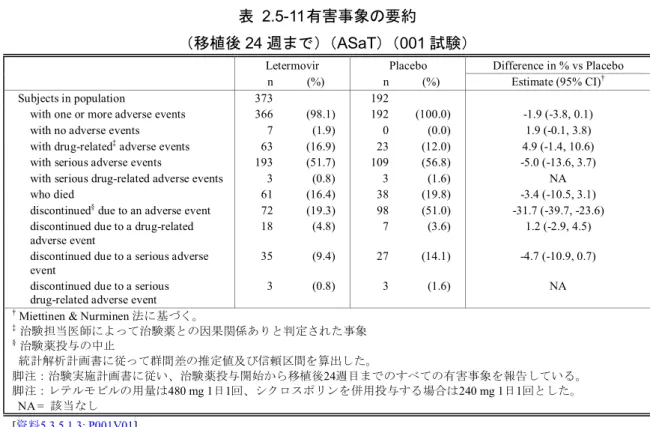
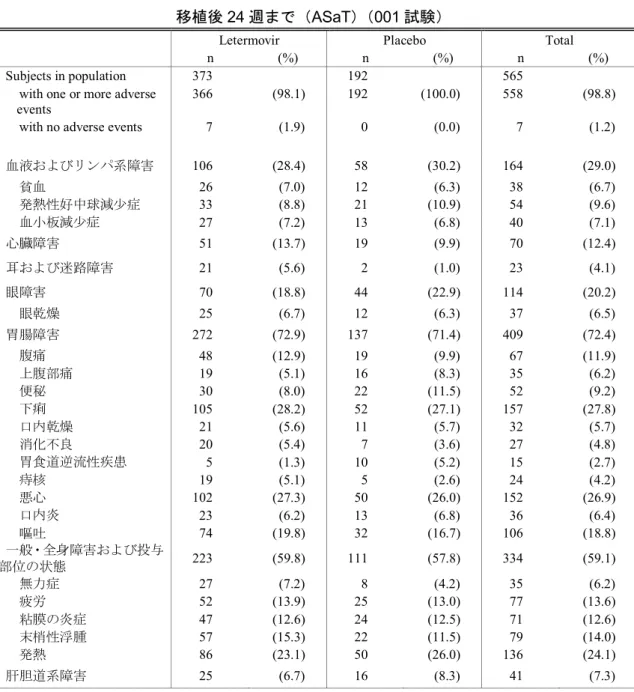
頁 表 2.5-1 レテルモビルの第Ⅰ相、第Ⅱ相及び第Ⅲ相試験の要約 ... 13 表 2.5-2 患者におけるAUC の概要 ... 38 表 2.5-3 患者の背景因子(ASaT、全集団)(001試験) ... 53 表 2.5-4 移植後24週以内に臨床的に意味のある CMV 感染がみられた患者の割合 (FAS、NC=F、全集団)(001試験) ... 58 表 2.5-5 移植後24週以内に臨床的に意味のある CMV 感染がみられた患者(予防不成 功)の割合に関する有効性解析の要約(FAS、全集団)(001試験) ... 59 表 2.5-6 有効性主要及び副次評価項目の要約(FAS、全集団)(001 試験) ... 64 表 2.5-7 探索的評価項目に関する有効性解析の要約(FAS、全集団)(001試験) ... 68 表 2.5-8 移植後24週以内に臨床的に意味のある CMV 感染がみられた患者又は 発症 しなかった患者での全死亡(移植後48週まで)(FAS、全集団)(001試験) ... 70 表 2.5-9 第Ⅰ相、第Ⅱ相及び第Ⅲ相試験におけるレテルモビル曝露状況の要約 ... 77 表 2.5-10 有害事象の要約(安全性解析対象集団)(020試験) ... 83 表 2.5-11 有害事象の要約 (移植後24週まで)(ASaT)(001試験) ... 87 表 2.5-12 有害事象発現例数(%)(いずれかの投与群で発現割合5%以上) 移植後24 週まで(ASaT)(001試験) ... 882.5 臨床に関する概括評価 - 5 -
図一覧
頁 図 2.5-1 試験デザインの概略(001試験)... 49 図 2.5-2 有効性解析対象集団の概略図(001試験) ... 51 図 2.5-3 移植後24週以内に臨床的に意味のある CMV 感染がみられた患者の割合に関 する フォレストプロット(リスク因子の部分集団別)(FAS、DAO、全集団) (001試験) ... 61 図 2.5-4 移植後24週以内に臨床的に意味のある CMV 感染がみられた患者の割合に関 する フォレストプロット(患者背景の部分集団別)(FAS、DAO、全集団) (001試験) ... 62 図 2.5-5 移植後24週以内に臨床的に意味のある CMV 感染がみられた患者の割合に関 する フォレストプロット(移植前処置レジメン及び併用免疫抑制レジメン の部分集団別) (FAS、DAO、全集団)(001試験) ... 63 図 2.5-6 移植後24週以内に臨床的に意味のある CMV 感染がみられるまでの期間 (Kaplan-Meier 曲線)(FAS、全集団)(001試験) ... 65 図 2.5-7 移植後48週以内の全死亡に関する、死亡までの期間(Kaplan-Meier 曲線) (FAS、全集団)(001試験) ... 69 図 2.5-8 移植後48週以内の非再発死亡に関する、死亡までの期間(Kaplan-Meier 曲線) (FAS、全集団)(001試験) ... 71 図 2.5-9 移植後48週以内の CMV 感染後の死亡に関する、死亡までの期間 (Kaplan-Meier 曲線)(FAS、全集団)(001試験) ... 72 図 2.5-10 Kaplan Meier 法による生着までの期間 無作為割付時に生着が認められなか った患者集団 (移植後24週まで)(ASaT)(001試験) ... 962.5 臨床に関する概括評価 - 6 -
略号及び用語の定義
略語 定義
AE Adverse event 有害事象
ARaT All Randomized and Treated
無作為割付け後に治験薬投与を1回以 上受けたすべての患者(無作為割付け された投与群)
ASaT All Subjects as Treated
無作為割付け後に治験薬投与を1回以 上受けたすべての患者(実際に投与さ れた治験薬に対応する投与群) AUC Area under the concentration-time curve 濃度-時間曲線下面積
AUC0-t Area under the concentration-time curve from time 0 to time t hours postdose 投与後0から t 時間までの濃度-時間曲線下面積 BCRP Breast cancer resistance protein 乳癌耐性蛋白質
BCS Biopharmaceutics classification system 生物薬剤学分類システム
BID Twice daily 1日2回
BMI Body mass index 体格指数
BSEP Bile salt export pump 胆汁酸塩排出ポンプ
CAC clinical adjudication committee 臨床判定委員会
CAR Constitutive androstane receptor 構成的アンドロスタン受容体
CHMP Committee for Medicinal Products for Human Use 欧州医薬品委員会
CI Confidence interval 信頼区間
CL Clearance クリアランス
Cmax Maximum concentration 最高濃度
CMV Human cytomegalovirus ヒトサイトメガロウイルス
CsA Cyclosporin A シクロスポリン
CYP Cytochrome P450 チトクロムP450
DAO Data as Observed -
DNA Deoxyribonucleic acid デオキシリボ核酸
EE Ethinyl estradiol エチニルエストラジオール
EMA European Medicines Agency 欧州医薬品庁
FAS Full Analysis Set 最大の解析対象集団
FDA Food and Drug Administration 米国食品医薬品局
FFP Fit-for-purpose formulation 開発初期製剤
FMI Final market image 市販予定製剤
FSH Follicle-stimulating hormone 卵胞刺激ホルモン
GAP Genotyping Analysis Population 遺伝子解析対象集団
GVHD Graft-versus-host disease 移植片対宿主病
HLA Human leukocyte antigen ヒト白血球抗原
HMG-CoA 3-hydroxy-3-methylglutaryl-coenzyme A 3-ヒドロキシ-3-メチルグルタリル-CoA HPCD Hydroxypropyl-β-cyclodextrin ヒドロキシプロピル-β-シクロデキストリン HSCT Hematopoietic stem cell transplant 造血幹細胞移植
HSV Herpes simplex virus 単純ヘルペスウイルス
IgG Immunoglobulin G 免疫グロブリンG
INR International normalized ratio 国際標準比
LC-MS/MS Liquid chromatography/tandem mass spectrometory 液体クロマトグラフィー・タンデム質量分析法
2.5 臨床に関する概括評価 - 7 -
略号及び用語の定義(続き)
略語 定義
LNG Levonorgestrel レボノルゲストレル
MedDRA Medical dictionary for regulatory activities ICH 国際医薬用語集 MRP Multidrug resistance associated protein 多剤耐性関連蛋白質
NA Not applicable 該当なし
NC=F Non-Completer=Failure 非完了例=無効例
NNT Number Needed to Treat 治療必要数
OAT Organic anion transporter 有機アニオントランスポーター
OATP Organic anion transporting polypeptide 有機アニオン輸送ポリペプチド PBPK Physiologically-based pharmacokinetic 生理学的薬物動態
PCR Polymerase chain reaction ポリメラーゼ連鎖反応
P-gp P-glycoprotein P-糖蛋白質
PMF Preliminary market formulation 市販候補製剤
PMDA Pharmaceuticals and Medical Devices Agency 医薬品医療機器総合機構
POC Proof of concept -
PP Per-Protocol 治験実施計画書に適合した
pp65 low matrix phosphoprotein 65 (ウイルスの初期構造抗原)CMV が感染した白血球で発現する
PPK Population PK 母集団薬物動態
PT Preferred Term 基本語
PXR Pregnane X receptor プレグナンX 受容体
QD Once daily 1日1回
QOL Quality of life 生活の質
QTcF QT interval corrected for heart rate using Fridericia's formula Fridericia 補正法により心拍数で補正したQT 間隔
SOC System organ class 器官別大分類
t1/2 Elimination half-life 消失半減期
TEAE treatment-emergent adverse event 治療期に発現又は悪化した有害事象
TESAE treatment-emergent serious adverse event 治療期に発現又は悪化した重篤な有害事象 Tmax Time to reach maximum concentration 最高濃度到達時間
UGT Uridine 5'-diphospho-glucuronosyltransferase ウリジン酵素 5'-二リン酸グルクロン酸転移
VZV Varicella zoster virus 水痘帯状疱疹ウイルス
AIC001 Letermovir レテルモビル AIC090027 AIC246 BAY73-6327 MK-8228
2.5 臨床に関する概括評価 - 8 -
2.5
臨床に関する概括評価
2.5.1 製品開発の根拠 ヒトサイトメガロウイルス(CMV)は、ヘルペスウイルス科に属する二重鎖デオキシリボ核酸 (DNA)型ウイルスである。CMV は広く認められる一般的なウイルスであり、接触により容易 に感染するため、国内外を問わず成人の多くがCMV 既感染(抗体陽性)である。通常、CMV は 幼少期に不顕性感染の形で感染し、生涯その宿主に潜伏感染するが、免疫抑制状態下で再活性化 し、種々の病態を引き起こすおそれがある。特に同種造血幹細胞移植(HSCT)患者は重度の免疫 抑制状態にあるため、潜伏感染していたCMV の再活性化により活動性 CMV 感染(CMV 血症: 血液中より CMV が検出される状態)に至るリスクが高く、その結果、全身状態の悪化や高い死 亡率が懸念される。 成人HSCT 患者の65%~80%に CMV 感染歴があり、これらの患者では移植後の CMV 再活性化 及びCMV 感染症のリスクが懸念される[資料5.4: 2] [資料5.4: 3] [資料5.4: 4] [資料5.4: 5]。実際に、 移植前にCMV 抗体陽性であった HSCT 患者では、予防措置を実施しない場合、約80%で CMV が 再活性化し、20%~35%で CMV 感染症に進行することが報告されている[資料5.4: 2]。しかしなが ら、既存の抗CMV 薬では、骨髄抑制や腎毒性等の重大な毒性が認められていることから、HSCT 患者に対する予防投与は推奨されていない[資料5.4: 1]。そのため、HSCT 患者での CMV 感染予防 に有効かつ忍容性の良好な薬剤の開発が強く望まれている。 本項では、新規の強力な CMV ターミナーゼ阻害剤であるレテルモビル(同義語:MK-8228、 AIC246、AIC001、AIC090027及び BAY 73-6327)の開発のために実施した臨床試験の概要を示す。 2.5.1.1 薬効分類 レテルモビルは、CMV ターミナーゼ複合体に特異的に作用する新規の作用機序を有する。既存 の抗CMV 薬はいずれもウイルス DNA ポリメラーゼを阻害するため、交差耐性が懸念されるが、 レテルモビルは、既存の抗CMV 薬との交差耐性が認められていない。 2.5.1.2 化学的及び薬剤学的性質 レテルモビルは、錠剤、注射剤ともに240 mg 及び480 mg の2規格が製造されている[2.7.1 項]。 日本では、480 mg 錠のサイズが大きいことから、錠剤、注射剤ともにレテルモビルの240 mg 製剤 のみを製造販売承認申請の対象とした。 レテルモビル240 mg 錠剤の市販用製剤は速放性のフィルムコーティング錠であり、レテルモビ ルとして240 mg 及び標準的な添加剤を含有している。 レテルモビル240 mg 注射剤の市販用製剤は、Type I のガラスバイアル30 mL に20 mg/mL 無菌濃 縮注射液が充填されており、レテルモビルとして1バイアルあたり240 mg を含有する。バイアル には12.0 mL を抜き取るのに十分量の水溶液が充填されている。レテルモビル注射剤は、可溶化さ せるためヒドロキシプロピル-β-シクロデキストリン(HPCD)、pH 調整のため水酸化ナトリウム 及び等張化のため塩化ナトリウムを用いて製造されている。2.5 臨床に関する概括評価 - 9 - 2.5.1.3 適応症 日本におけるレテルモビルの予定される効能・効果は、「同種造血幹細胞移植患者におけるサイ トメガロウイルス感染又はサイトメガロウイルス感染症の予防」である。 2.5.1.4 科学的背景 本項では、HSCT 患者における CMV 感染の概要を示す。成人 HSCT 患者における CMV 関連の 罹患率及び死亡率については[2.5.1.4.1 項]に、予防又は先制治療を目的とした既存の抗 CMV 治療 の限界については[2.5.1.4.2 項]に記述する。 2.5.1.4.1 成人HSCT 患者におけるサイトメガロウイルス [2.5.1 項]のとおり、通常、CMV は潜伏感染しているが、HSCT(特に同種 HSCT)後などの極 端な免疫抑制状態の患者では、再活性化することがあり、その結果として全身状態の悪化や高い 死亡率が懸念される。全世界で年間約27,000件の同種 HSCT が実施されており[資料5.4: 11]、その うちの約8,000件は米国で実施されている[資料5.4: 12]。欧州47ヵ国では2014年に約16,000件[資料 5.4: 13]、東南アジア及び西太平洋諸国では、年間約6,400件の同種 HSCT が実施されている[資料 5.4: 11]。日本では2015年に3,724件の同種 HSCT が実施された[資料5.4: 14]。世界的に同種 HSCT の年間実施件数は増加している[資料5.4: 11]。 CMV の再活性化は、HSCT 患者における全身状態の悪化や高い死亡率と直接的、又は間接的に 関係している。「直接的」な影響として、CMV 感染症の発症率の増加[資料5.4: 15] [資料5.4: 16]、 また「間接的」な影響として、急性及び慢性移植片対宿主病(GVHD)、重篤な細菌感染症及び侵 襲性真菌日和見感染症、並びに非再発死亡など、CMV が免疫系に及ぼす作用によるもの[資料5.4: 17] [資料5.4: 19] [資料5.4: 18] [資料5.4: 20] [資料5.4: 21] [資料5.4: 22] [資料5.4: 23] [資料5.4: 24]が 挙げられる。移植後のHSCT 患者の CMV 再活性化の対策として先制治療が行われるようになっ てからは、CMV 感染症自体の発現率は著しく低下している。しかし、特に CMV 抗体陽性同種 HSCT 患者では CMV 血症が確認される可能性が高く[資料5.4: 24]、CMV 血症による全体的な死亡 のリスクは依然として高い[資料5.4: 25]。 2.5.1.4.2 予防及び先制治療:既存の抗CMV 治療の限界 HSCT 患者における CMV 感染症対策には、1)抗 CMV 薬の予防投与、2)先制治療(ウイルス の増殖を頻回にモニタリングし、CMV 血症が確認された場合、抗 CMV 薬の投与を開始する)[資 料5.4: 26] [資料5.4: 27]の2つの方法があるが、いずれの方法にも限界がある。 既存の抗 CMV 薬はいずれもヌクレオシド誘導体であり、骨髄抑制や腎毒性等の特徴的な毒性 を有するため予防薬として使用しにくいためあまり用いられない。最も広く使用されているガン シクロビル及びバルガンシクロビルでは、HSCT で特に問題となる骨髄抑制が副作用として認め られており、第二選択薬として用いられるホスカルネットでは腎毒性が認められることが多い。 通常、海外で第三選択薬として用いられる cidofovir(日本では未承認)では、骨髄抑制と腎機能
2.5 臨床に関する概括評価 - 10 - 障害が認められている。高用量のアシクロビル又はバラシクロビルの予防投与がこれまでに評価 されているが、CMV に対する有効性は確立しておらず、抗 CMV 薬としての適応はいずれも取得 していない[資料5.4: 26] [資料5.4: 27]。 このように、既存の抗 CMV 薬には毒性に関する懸念があることから、予防投与よりも先制治 療の方が望ましいとされており、国内外ともにHSCT を行う大多数の医療機関では、CMV 感染症 の発症抑制として特にCMV 感染のリスクが最も高い移植後100日間の CMV モニタリングに基づ く先制治療が推奨されている。しかし、先制治療は以下の理由により最適な方法とはいえない。 • 先制治療は患者にCMV 血症がみとめられた後に開始するため、CMV 感染自体は抑えられ ない。近年では、先制治療の有無にかかわらず、CMV 再活性化によりウイルス量依存的に 原因を問わない死亡(全死亡)が増加するとの報告がある[資料5.4: 25]。また、日本でも、 主に移植後早期のCMV 再活性化が全死亡及び原疾患以外の理由による死亡(非再発死亡) に対する有意な予後因子であるとの報告がある[資料5.4: 28]。 • 既存の抗CMV 薬では骨髄抑制や腎障害といった特徴的な毒性が報告されていることから、 過剰な先制治療は避けることが望ましい。特に、CMV 感染のリスクが最も高く、先制治療 が行われる可能性が高いのは移植後100日間であるが、移植後早期と定義されるこの期間は 好中球減少や粘膜障害、GVHD の発症、ステロイド投与等により HSCT 患者の免疫機能が 低下し、患者の状態管理に細心の注意を要するため、副作用の可能性がある過剰な先制治 療は避けることが望ましいと考えられる。しかしながら、CMV DNA 量の閾値は定められ ておらず、先制治療開始の判断にはばらつきがあるため、実際には不必要な先制治療が行 われている可能性がある[資料5.4: 29] [資料5.4: 1]。 • 先制治療ではCMV 血症の頻回なモニタリングが必要であることから、患者の負担と費用 がかかる[資料5.4: 30]。 これらのことから、CMV に対して有効かつ安全性の高い薬剤による予防投与は、CMV 感染症 対策として現在広く用いられている先制治療を上回るベネフィットが期待できる。そのため、 HSCT 患者での CMV 感染及び感染症の予防に有効かつ忍容性が良好な抗 CMV 薬の開発が望まれ ている[資料5.4: 30] [資料5.4: 31] [資料5.4: 3]。さらに、予防投与により先制治療よりも早い段階で CMV 感染自体を抑えることができれば、CMV 感染による「直接的」な影響(CMV 感染症の発症) のみならず「間接的」な影響(GVHD や死亡など、CMV が免疫系に及ぼす作用によるもの)への 予防効果も期待できる。レテルモビルは、新規の抗 CMV 薬であり、第Ⅰ相試験28試験、第Ⅱ相 試験(019試験及び020試験)及び第Ⅲ相試験(001試験)[表 2.5-1]において、概して良好な忍容 性を示した。前期第Ⅱ相試験(019試験)では Proof of Concept(POC)(抗ウイルス活性)が確認 され、後期第Ⅱ相試験(020試験)では HSCT 患者の CMV 感染予防においてレテルモビルの安全 性及び用量依存的な有効性が示された。第Ⅲ相試験(001試験)では、CMV 抗体陽性の成人同種 HSCT 患者の CMV 感染の予防においてレテルモビルの安全性及び有効性が確認されている。
2.5 臨床に関する概括評価 - 11 - 2.5.1.5 レテルモビルの開発背景及び作用機序 レテルモビルは新規の CMV ターミナーゼ阻害剤である。ウイルスターミナーゼは、ウイルス の子孫DNA を一単位長のゲノムへ切断し、空のウイルスカプシドに誘導する。感染症の細胞培養 モデルでは、レテルモビルは他の抗 CMV 薬に耐性を示す数種類の分離株を含め、実験室株及び CMV の臨床分離株を強力に阻害する。EC50値(増殖を50%抑制する濃度)は低く、nM レベルの 値であった。ウイルスのターミナーゼ複合体はヒトには存在しないことから、レテルモビルの作 用機序はウイルス特異的であり、作用機序に基づく副作用は発現しないと考えられる。 既存の抗CMV 薬はヌクレオシド誘導体である DNA ポリメラーゼ阻害剤であり、ポリメラーゼ
はUL54又は UL97遺伝子にマッピングされている。一方、レテルモビルは UL56及び UL89遺伝子
に由来する CMV のターミナーゼを標的とする。感染症の細胞培養モデルでは、レテルモビルと 既存の抗 CMV 薬との交差耐性は認められておらず、ポリメラーゼ阻害剤に対する耐性を示すウ イルスは、レテルモビルに対して感受性を示し、逆に、ターミナーゼに変異を有しレテルモビル に対する耐性を示すウイルスはポリメラーゼ阻害剤に対して十分な感受性を示す。これらのデー タから、レテルモビルに対する耐性が認められ、予防投与が無効であった患者でも、既存のDNA ポリメラーゼ阻害剤である抗CMV 薬に対する感受性は維持されることが示唆された。 2.5.1.6 臨床開発プログラムの概要 2.5.1.6.1 臨床開発プログラムの概要 レテルモビルの臨床開発プログラムの要約を[表 2.5-1]に示す。 健康成人被験者、肝及び腎機能障害者を対象として、第Ⅰ相試験を28試験実施した(日本人を
対象とした2試験を含む)。このうち、14試験は AiCuris GmbH & Co. KG(以下、AiCuris)又は Bayer Healthcare AG が実施し、14試験は MSD 株式会社(申請者)及び Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc.(以下、米国本社)が実施した。第Ⅰ相試験には、安全性及び忍容
性、薬物動態、外因性(薬物相互作用)及び内因性要因、食事の影響並びにレテルモビルが QTc 間隔に及ぼす影響の評価が含まれ、いずれの試験でも概して良好な忍容性が示されている。 第Ⅱ相試験2試験(019試験及び020試験)は AiCuris が実施した。前期第Ⅱ相試験(019試験、 AiCuris AIC001-2-001)では腎又は腎/膵臓移植患者17例及び HSCT 患者1例を対象にレテルモビ ルを2つの用法で14日間投与し、実薬対照(実施医療機関の標準治療)と比較した結果、レテルモ ビルのPOC(抗ウイルス活性)が確認された。後期第Ⅱ相試験(020試験、AiCuris AIC246-01-II-02) では、同種HSCT 患者に84日間3用量のレテルモビルを投与し、CMV 感染予防において、プラセ ボと比較したレテルモビルの安全性及び用量依存的な有効性が確認された。 第Ⅱ相試験の完了後、レテルモビルの開発及び製造販売権を米国本社が取得し、第Ⅲ相試験(001 試験)を計画及び実施した。001試験は国際共同試験として実施し、日本からも参加した。001試 験では、CMV 抗体陽性の同種 HSCT 患者570例を2:1の比でレテルモビル群又はプラセボ群に無 作為割付けし、移植後14週まで投与した。無作為割付けした570例中、565例に治験薬を投与した。 日本人としては、無作為割付けした36例中、35例に治験薬を投与した。001試験の有効性の主要評
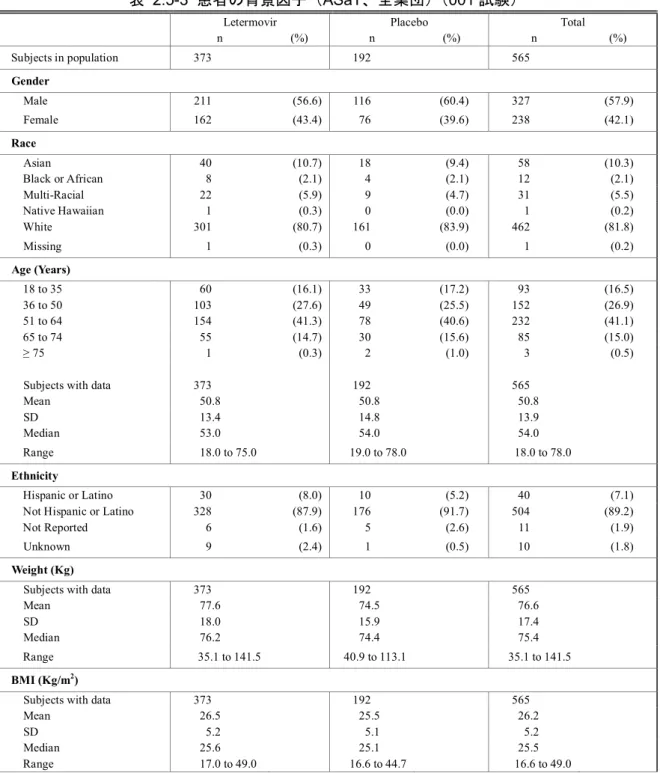
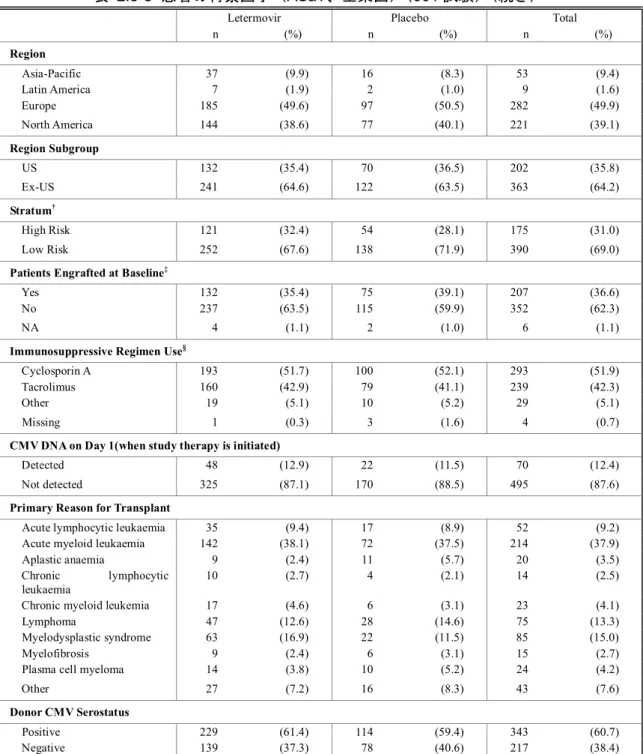
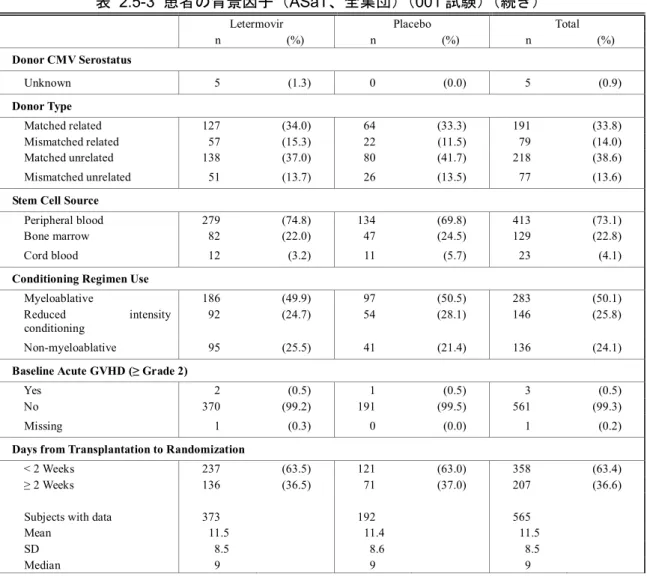
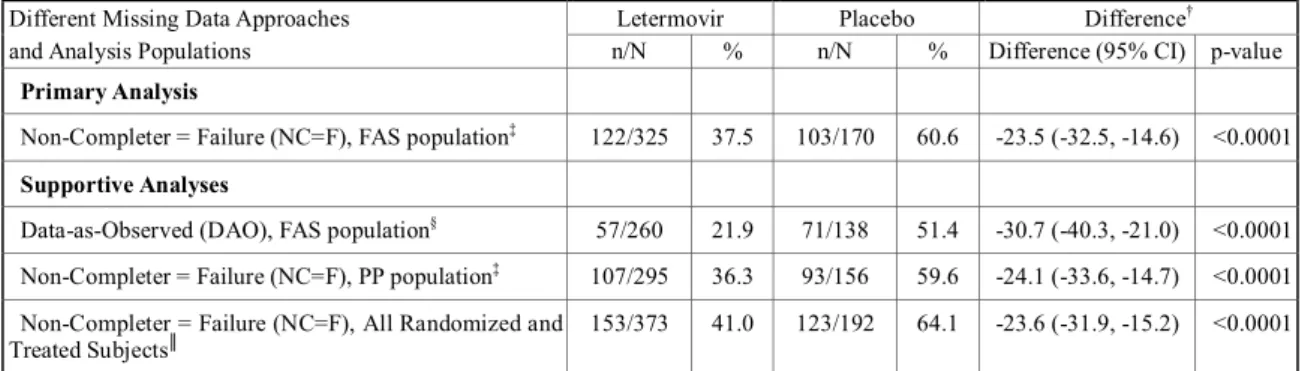
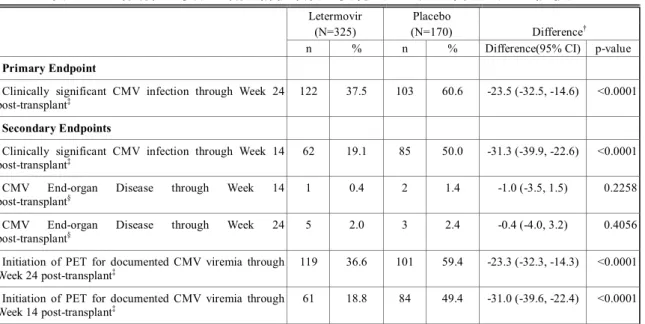
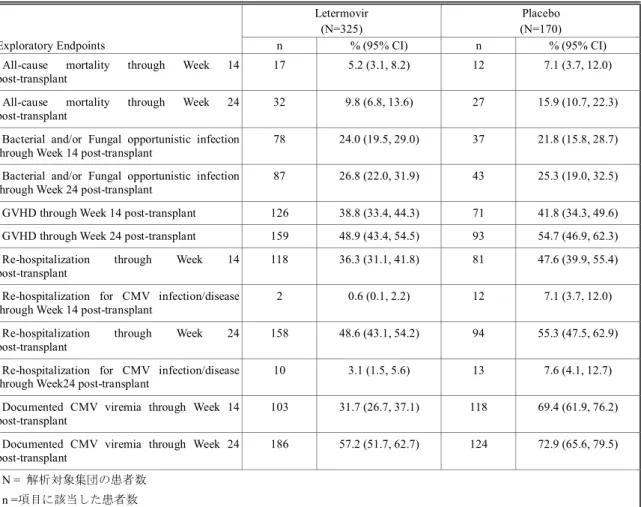
2.5 臨床に関する概括評価 - 12 - 価項目は移植後24週時(約6ヵ月)に設定しており、製造販売後承認申請時点の本申請資料では、 この時点までに収集可能であったデータに基づきレテルモビルの安全性を評価した。有効性の主 要評価項目は、移植後24週以内に臨床的に意味のある CMV 感染がみられた患者の割合とした。 以下のいずれかを認めた場合を、臨床的に意味のあるCMV 感染と定義した。 • 終末器官におけるCMV 感染症の発症 又は • CMV 血症の確認(中央検査機関による測定)及び臨床状態に基づいた、抗 CMV 薬による 先制治療の開始。本治験における先制治療の開始とは、CMV の活発なウイルス複製が確認 された場合に以下の抗CMV 薬のいずれかの投与を開始することである:ガンシクロビル、 バルガンシクロビル、ホスカルネット又はcidofovir(日本では未承認)。 001試験には、日本を含む20ヵ国67施設が参加した。レテルモビルの投与量は480 mg 1日1回と した(シクロスポリンを併用投与する場合は、レテルモビルの用量を240 mg 1日1回に調整)[2.5.3.3 項]。注射剤及び錠剤の両製剤が使用されたが、いずれの製剤でも同一用量を投与した。 本申請資料において001試験の結果は、主に治験総括報告書[資料5.3.5.1.3: P001V01]を基に作成 した。治験総括報告書[資料5.3.5.1.3: P001V01]は、有効性の主要評価項目である移植後24週の評価 がすべての症例で完了した時点の固定データに基づくものである。001試験では、無作為割付けし た570例中565例に治験薬が投与され、347例(60.9%)が移植後14週までの治験薬投与を完了した。 431例(75.6%)が移植後24週の来院を完了し、134例(23.5%)が移植後24週の来院前に治験を中 止した。移植後24週の来院を完了した431例は後観察期(追跡期間)(移植後24週~48週)に移行 した。有効性の主要解析のためのデータカットオフ時点で、302例(無作為割付したすべての患者 の53.0%)が移植後48週の来院を完了し、64例(11.2%)が治験を継続中であった[2.7.3.2.2.3.2 項]。 その後得られた探索的な目的である移植後48週までの評価については、移植後48週の治験総括報 告書[資料5.3.5.1.5: P001V02]に示し、本項には結果の概要を示す。なお、主要解析のデータ固定後、 移植後48週までの追跡調査の過程で得られた新たな情報に基づき、移植後24週までのデータの一 部も更新されている。移植後48週の治験総括報告書[資料5.3.5.1.5: P001V02]は、48週間を通して更 新されたデータに基づいている。 第Ⅲ相試験(001試験)の部分集団である日本人集団の結果及び全体集団との比較の詳細につ いては、[2.7.6.3.3 項]にまとめて記載した。
2.5 臨床に関する概括評価 - 13 - 試験の種類 資料の 分類 試験番号† (海外/国内) 略称/デザイン 主要目的 レテルモビルを 投与された 被験者/患者数 第Ⅰ相 健康被験者 を対象とし たPK 及び初 期忍容性試 験 評価資料 027試験 (国内) 日本人健康女性被験者を対象とした安全性、忍 容性及びPK を評価する単回投与試験 二重盲検、無作為化、プラセボ対照、2パート、 投与順序固定、用量漸増、単回経口及び静脈内 投与試験 日本人健康女性被験者に用量漸増 単回経口投与及び静脈内投与した 際の安全性及び忍容性の評価 パート1: 8例 パート2: 8例 評価資料 032試験 (海外) 日本人健康女性被験者を対象とした反復投与 及びシクロスポリンとの薬物相互作用を検討 する反復投与試験 二重盲検、無作為化、単施設、プラセボ対照、 2期、投与順序固定、反復経口投与試験 -日本人健康女性被験者に反復経口 投与した際のPK パラメータを非日 本人被験者のPK パラメータと比較 -空腹時反復経口投与後、シクロスポ リンを併用投与した際のレテルモ ビルのPK の評価 14例 参考資料 007試験 (AIC001-1-001/ BAY-73-6327-011 696試験) (海外) 初回ヒト投与 健康男性被験者に単回経口投与した際の安全 性、忍容性及びPK を評価する試験 単盲検、無作為化、単施設、プラセボ対照、並 行群間、用量漸増、単回経口投与試験 健康男性被験者に液剤(FFP1製剤) 及び錠剤(FFP2製剤)を単回経口投 与した際の安全性及び忍容性の評 価 40例 参考資料 011試験 (AIC001-1-005 試験) (海外) 健康男性被験者を対象とした安全性、忍容性及 びPK を評価する用量漸増単回経口投与試験 二重盲検(群内)、無作為化、プラセボ対照、 並行群間、用量漸増、単回経口投与試験 用量依存的な有害事象プロファイ ルの評価、用量制限有害事象の特定 及び最大耐用量の決定を目的とし て、健康男性被験者に用量漸増単回 経口投与した際の安全性及び忍容 性の確認 36例
2.5 臨床に関する概括評価 - 14 - 試験の種類 資料の 分類 試験番号† (海外/国内) 略称/デザイン 主要目的 レテルモビルを 投与された 被験者/患者数 第Ⅰ相 健康被験者 を対象とし たPK 及び初 期忍容性試 験(続き) 参考資料 021試験 (AIC246-01-Ⅰ -08試験) (海外) 健康被験者に単回及び用量漸増反復経口投与 した際の安全性、忍容性及びPK を評価する試 験、並びに健康男性被験者に反復経口投与した 際のマスバランス及び代謝プロファイルを検 討する試験 パート1: 二重盲検、無作為化、単施設、プラセボ対照、 単回経口投与試験 パート2: 二重盲検、無作為化、単施設、プラセボ対照、 用量漸増、反復経口投与試験 パート3: 非盲検、単施設、ADME 試験 パート1: 健康女性被験者に単回経口投与し た際の安全性、忍容性及びPK の評 価 パート2: -健康被験者に用量漸増反復経口投 与した際の安全性、忍容性及びPK の評価 -有害事象の発現に用量依存的な傾 向があるか、及び最大耐用量の確認 -ある一定の用量以上で発現する有 害事象の特定 パート3: -マスバランスの評価 -血漿中及び排泄物中の代謝物並び に排泄経路の同定 パート1: 6例 パート2: 36例 パート3: 8例
2.5 臨床に関する概括評価 - 15 - 試験の種類 資料の 分類 試験番号† (海外/国内) 略称/デザイン 主要目的 レテルモビルを 投与された 被験者/患者数 第Ⅰ相 健康被験者 を対象とし たPK 及び初 期忍容性試 験(続き) 参考資料 009試験 (AIC001-1-003/ BAY-73-6327-011 698試験) (海外) 健康男性被験者を対象とした安全性、忍容性及 びPK を検討する、用量漸増反復投与試験、並 びにミダゾラムのPK に及ぼす影響を評価する 試験 ステージ1: 単盲検、無作為化、単施設、プラセボ対照、群 間比較、用量漸増、反復経口投与試験 ステージ2: 非盲検、非無作為化、単施設、非プラセボ対照、 群間比較試験 ステージ1: 健康男性被験者に反復経口投与し た際の安全性及び忍容性の評価 ステージ2: ミダゾラムのPK に及ぼす影響の評 価 23例
2.5 臨床に関する概括評価 - 16 - 試験の種類 資料の 分類 試験番号† (海外/国内) 略称/デザイン 主要目的 レテルモビルを 投与された 被験者/患者数 第Ⅰ相 健康被験者 を対象とし たPK 及び初 期忍容性試 験(続き) 参考資料 018試験 (AIC246-01-Ⅰ -13試験) (海外) 健康女性被験者を対象とした用量漸増単回及 び反復経口投与した際の安全性、忍容性及び PK を検討する試験、 並びに 製剤を単回及び静脈内投与 した際の安全性、忍容性及びPK を検討する試 験、 並びに健康女性被験者を対象としたジゴキシ ン経口投与後のPK に及ぼす影響を検討する試 験 パートA(コホート1): 非盲検、単施設、非プラセボ対照、反復経口投 与試験 パートA(コホート2及び7): 二重盲検、無作為化、単施設、プラセボ対照、 用量漸増、単回及び反復経口投与試験 パートB: 二重盲検、無作為化、単施設、プラセボ対照、 用量漸増、単回及び反復静脈内投与試験 パートC: 非盲検、単施設、2期、2投与順序、クロスオー バー試験 パートA: -健康女性被験者に投与した際の安 全性及び忍容性の評価 -最大耐用量の確認 -単回投与時及び定常状態での曝露 量の評価 -高用量の定常状態で QT/QTc 間隔に 影響を及ぼす可能性の評価 パートB: 単回及び反復静脈内投与した際の 定常状態のPK の評価 パートC: 定常状態のレテルモビルがジゴキ シン単回経口投与後の血漿中ジゴ キシンのPK に及ぼす影響の評価 パートA: 28例 パートB: 16例‡ パートC: 24例
2.5 臨床に関する概括評価 - 17 - 試験の種類 資料の 分類 試験番号† (海外/国内) 略称/デザイン 主要目的 レテルモビルを 投与された 被験者/患者数 第Ⅰ相 健康被験者 を対象とし たPK 及び初 期忍容性試 験(続き) 参考資料 026試験 (海外) 健康女性被験者を対象とした高用量反復経口 投与及びHPCD 製剤を反復静脈内投与した際 の安全性、忍容性及びPK を評価する試験 パート1: 二重盲検、無作為化、単施設、プラセボ対照、 反復経口投与試験 パート2: 二重盲検、無作為化、単施設、プラセボ対照、 反復静脈内投与試験 パート1: 健康女性被験者に高用量を反復経 口投与した際の安全性及び忍容性 の評価 パート2: 健康女性被験者にHPCD 製剤を反復 静脈内投与した際の安全性及び忍 容性(局所忍容性を含む)の評価 パート1: 18例 パート2: 9例 参考資料 005試験 (AIC246-01-Ⅰ -14試験) (海外) 用量漸増単回静脈内投与及び反復静脈内投与 した際の安全性、忍容性及びPK を評価する試 験 パートA: 二重盲検、無作為化、単施設、プラセボ対照、 用量漸増、単回静脈内投与試験 パートB: 二重盲検、無作為化、単施設、プラセボ対照、 反復静脈内投与試験 パートA: 健康女性被験者に用量漸増単回静 脈内投与した際の安全性及び忍容 性の評価 パートB: 健康女性被験者に反復静脈内投与 した際の安全性及び忍容性の検討 パートA: 30例 パートB: 8例
2.5 臨床に関する概括評価 - 18 - 試験の種類 資料の 分類 試験番号† (海外/国内) 略称/デザイン 主要目的 レテルモビルを 投与された 被験者/患者数 第Ⅰ相 BA 試験 参考資料 008試験 (AIC001-1-002/ BAY-73-6327-011 697試験) (海外) 健康男性被験者を対象とした液剤及び錠剤の 安全性、忍容性、PK 及び液剤経口投与に対す る錠剤経口投与の相対的BA の評価、並びに錠 剤経口投与のBA に対する高脂肪・高カロリー 食の影響を評価する試験 非盲検、無作為化、単施設、非プラセボ対照、 4期クロスオーバー、単回経口投与試験 -錠剤(FFP2製剤)及び液剤(FFP1 製剤)を経口投与した際の相対的 BA の評価 -錠剤(FFP2製剤)の BA に対する高 脂肪・高カロリー食の影響の評価 11例 参考資料 017試験 (AIC246-01-Ⅰ -12試験) (海外) 健康女性被験者を対象とした経口及び静脈内 投与した際の相対的曝露量及び絶対的BA を評 価する試験、並びに用量漸増単回静脈内投与し た際の安全性、忍容性及びPK を評価する試験 コホート1: 非盲検、無作為化(投与順序に対して無作為に 割り付ける)、単施設、2期クロスオーバー、単 回経口及び静脈内投与試験 コホート2~5: 二重盲検、無作為化、並行群間、プラセボ対照、 用量漸増、単回静脈内投与試験 コホート1: 経口(PMF1製剤)及び静脈内( 製剤)投与した際の相対的 曝露量の評価 コホート2~5: 静脈内( 製剤)投与した 際の用量依存的な有害事象プロフ ァイル、ホルター心電図を用いた QT 間隔への影響、PK 及び最大耐用 量の評価、安全性及び忍容性の確認 34例§ 評価資料 029試験 (海外) 健康被験者に食後及び空腹時に経口投与した 際のBA を評価する試験 非盲検、無作為化、2期、2処置、2投与順序、 クロスオーバー、単回経口投与試験 健康女性被験者に錠剤(PMF3製剤) を食後及び空腹時に単回経口投与 した際の相対的BA の評価 14例
2.5 臨床に関する概括評価 - 19 - 試験の種類 資料の 分類 試験番号† (海外/国内) 略称/デザイン 主要目的 レテルモビルを 投与された 被験者/患者数 第Ⅰ相 比較BA 及び BE 試験 参考資料 014試験 (AIC246-01-Ⅰ -09試験) (海外) 健康女性被験者に経口投与した際の錠剤間の 相対的BA を評価する試験 非盲検、無作為化、単施設、5期、5処置、クロ スオーバー、単回経口投与試験 健康女性被験者にPMF1製剤(4種類 の力価)とFFP2製剤を空腹時投与し た際の製剤間の相対的BA の評価 15例 参考資料 028試験 (海外) 健康女性被験者を対象に含量の異なる錠剤を 空腹時投与した際の相対的BA を評価する試験 非盲検、無作為化、単施設、2期、クロスオー バー、単回経口投与試験 錠剤(PMF3製剤、2種類の力価)を 空腹時単回経口投与した際の血漿 中の主要PK パラメータの比較 14例 内因性要因 を検討した PK 試験 参考資料 015試験 (AIC246-01-Ⅰ -10試験) (海外) 肝機能障害者及び健康被験者に反復経口投与 した際のPK、安全性及び忍容性に対する肝機 能障害の影響を検討する試験 非盲検、並行群間、反復経口投与試験 反復経口投与した際のPK に対する 肝機能障害の影響の評価 33例 参考資料 006試験 (AIC246-01-Ⅰ -16試験) (海外) 健康被験者、中等度及び重度腎機能障害者を対 象とした反復経口投与した際のPK、安全性及 び忍容性を検討する試験 非盲検、非無作為化、単施設、反復経口投与試 験 反復経口投与した際のPK に対する 腎機能障害の影響の評価 24例
2.5 臨床に関する概括評価 - 20 - 試験の種類 資料の 分類 試験番号† (海外/国内) 略称/デザイン 主要目的 レテルモビルを 投与された 被験者/患者数 第Ⅰ相 外因性要因 を検討した PK 試験 参考資料 016試験 (AIC246-01-Ⅰ -11試験) (海外) 健康女性被験者を対象とした反復経口投与が ミダゾラムを単回経口及び静脈内投与した際 のPK に及ぼす影響を検討する試験 非盲検、単施設、反復経口投与試験 反復経口投与時(定常状態)に、単 回静脈内投与したミダゾラム(2種 類の力価)のPK に及ぼす影響の検 討 16例 参考資料 010試験 (AIC001-1-004 試験) (海外) 健康男性被験者を対象とした安全性及び忍容 性、並びにシクロスポリンとの薬物相互作用を 検討する反復投与試験 パート1: 非盲検、非無作為化、単施設、非プラセボ対照、 反復経口投与試験 パート2: 非盲検、部分無作為化(シクロスポリン投与順 序)、2投与順序、3処置、クロスオーバー、反 復経口投与試験 パート1: 反復経口投与時に、単回経口投与し たシクロスポリンのPK に及ぼす影 響の検討 パート2: 用量の異なるシクロスポリンの単 回経口投与が、反復経口投与したレ テルモビルのPK に及ぼす用量依存 的な影響の検討 パート1: 8例 パート2: 12例
2.5 臨床に関する概括評価 - 21 - 試験の種類 資料の 分類 試験番号† (海外/国内) 略称/デザイン 主要目的 レテルモビルを 投与された 被験者/患者数 第Ⅰ相 外因性要因 を検討した PK 試験(続 き) 参考資料 003試験 (海外) 健康女性被験者を対象とした反復経口投与が シクロスポリン及びタクロリムスのPK に及ぼ す影響を評価する薬物相互作用試験 非盲検、2パート、反復経口投与試験 パート1: 健康女性被験者を対象として反復 経口投与時(定常状態)に、単回で 併用経口投与したシクロスポリン のPK に及ぼす影響の評価 パート2: 健康女性被験者を対象として、反復 経口投与時(定常状態)に、単回で 併用経口投与したタクロリムスの PK に及ぼす影響の評価 パート1: 14例 パート2: 14例 参考資料 013試験 (AIC001-1-007 試験) (海外) 健康男性被験者を対象とした反復経口投与と タクロリムスの単回経口投与の薬物相互作用 試験 非盲検、非無作為化、単施設、1投与順序、2処 置、反復経口投与試験 反復経口投与が、単回経口投与した タクロリムスのPK に及ぼす影響の 評価 16例 参考資料 036試験 (海外) 健康女性被験者を対象とした反復経口投与が シロリムスの単回投与のPK に及ぼす影響を評 価する薬物相互作用試験 非盲検、2期、投与順序固定、反復経口投与試 験 健康女性被験者を対象として反復 経口投与時(定常状態)に、単回投 与したシロリムスのPK に及ぼす影 響の評価 14例
2.5 臨床に関する概括評価 - 22 - 試験の種類 資料の 分類 試験番号† (海外/国内) 略称/デザイン 主要目的 レテルモビルを 投与された 被験者/患者数 第Ⅰ相 外因性要因 を検討した PK 試験(続 き) 参考資料 022試験 (海外) 健康女性被験者を対象としたミコフェノール 酸モフェチルの活性代謝物ミコフェノール酸 との薬物相互作用試験 非盲検、単施設、投与順序固定、単回及び反復 経口投与試験 健康女性被験者を対象として反復 経口投与時(定常状態)に、ミコフ ェノール酸のPK に及ぼす影響の評 価 14例 参考資料 034試験 (海外) 健康女性被験者を対象としたアシクロビルと の薬物相互作用試験 非盲検、1期、投与順序固定、反復経口投与試 験 健康女性被験者を対象として反復 経口投与時(定常状態)に、単回併 用経口投与したアシクロビルのPK に及ぼす影響の評価 16例 参考資料 025試験 (海外) 健康女性被験者を対象としたボリコナゾール のPK に及ぼす影響を評価する試験 非盲検、単施設、投与順序固定、反復経口投与 試験 健康女性被験者を対象として反復 経口投与時(定常状態)に、併用経 口投与したボリコナゾールのPK に 及ぼす影響の評価 14例 参考資料 033試験 (海外) 健康女性被験者を対象としたポサコナゾール との薬物相互作用試験 非盲検、2期、2処置、投与順序固定、反復経口 投与試験 健康女性被験者を対象として反復 経口投与時(定常状態)に、単回併 用経口投与したポサコナゾールの PK に及ぼす影響の評価 13例 参考資料 023試験 (海外) 健康女性被験者を対象としたアトルバスタチ ンとの薬物相互作用試験 非盲検、2期、投与順序固定、反復経口投与試 験 健康女性被験者を対象として反復 経口投与時(定常状態)に、単回併 用経口投与したアトルバスタチン のPK に及ぼす影響の評価 13例
2.5 臨床に関する概括評価 - 23 - 試験の種類 資料の 分類 試験番号† (海外/国内) 略称/デザイン 主要目的 レテルモビルを 投与された 被験者/患者数 第Ⅰ相 外因性要因 を検討した PK 試験(続 き) 参考資料 035試験 (海外) 妊娠する可能性のない健康女性被験者を対象 とした反復投与時に、経口避妊薬(エチニルエ ストラジオール及びレボノルゲストレル)の単 回投与時のPK に及ぼす影響を評価する試験 非盲検、2期、投与順序固定、反復経口投与試 験 反復経口投与時に、単回併用経口投 与したエチニルエストラジオール 及びレボノルゲストレルのPK に及 ぼす影響の評価 22例 健康被験者 におけるPD 試験及び PK/PD 試験 評価資料 004試験 (海外) 健康女性被験者を対象としたQTc 間隔に及ぼ す影響を検討する試験 無作為化、単施設、4期、8投与順序、クロスオ ーバー、単回投与試験 予定臨床用量を超える高用量及び 予定臨床用量がQTc 間隔に及ぼす 影響の評価 37例 第Ⅱ相 申請する適 応症に関す る比較対照 試験 参考資料 019試験 (AIC001-2-001 試験) (海外) CMV 血症患者を対象に先制治療下で投与した 際の安全性、忍容性及び抗ウイルス活性を評価 する試験 非盲検、無作為化、多施設共同、実薬対照、用 量反応、POC、前期第Ⅱ相試験 腎移植又は腎及び膵臓移植後の CMV 血症患者を対象とした14日間 の投与(2用量)後における CMV DNA 量の減少の検討、実薬対照群と の比較 18例 評価資料 020試験 (AIC246-01-Ⅱ -02試験) (海外) 再感染又は再活性化によるCMV の増殖抑制を 検討するために投与した際の安全性、忍容性及 び抗ウイルス活性を評価する試験 二重盲検、無作為化、多施設共同、プラセボ対 照、用量反応、後期第Ⅱ相試験 CMV 抗体陽性の同種造血幹細胞移 植患者を対象として投与(3用量) した期間のCMV の増殖抑制効果及 び安全性の検討 98例
2.5 臨床に関する概括評価 - 24 - 試験の種類 資料の 分類 試験番号† (海外/国内) 略称/デザイン 主要目的 レテルモビルを 投与された 被験者/患者数 第Ⅲ相 申請する適 応症に関す る比較対照 試験 評価資料 001試験 (国内及び海 外) CMV 抗体陽性の成人同種造血幹細胞移植患者 を対象に臨床的に意味のあるCMV 感染の予防 を目的として投与した際の安全性及び有効性 を評価する試験 二重盲検、無作為化、多施設共同、プラセボ対 照、第Ⅲ相試験 CMV 抗体陽性の成人同種造血幹細 胞移植患者を対象として投与した 際の、移植後24週(約6ヵ月)以内 の臨床的に意味のあるCMV 感染の 予防に対する有効性の評価 373例 (日本人:25例) PK=薬物動態、FFP =開発初期製剤、ADME=吸収、分布、代謝及び排泄、HPCD=ヒドロキシプロピル-β-シクロデキストリン、BA=バイオアベイラビリティ、PMF=市販候補製剤、BE=生物 学的同等性、PD=薬力学、CMV=ヒトサイトメガロウイルス、POC= Proof of concept
† 他の試験番号がある場合はその番号(AIC001、AIC246、BAY73-6327はレテルモビルと同義語) ‡ 018試験のパート B:16例の被験者はいずれもレテルモビルの 製剤(注射剤)を投与された。
2.5 臨床に関する概括評価 - 25 - 臨床薬理プログラムの概要 レテルモビルの臨床開発プログラムには、健康成人被験者並びに肝又は腎機能障害者を対象に レテルモビルの安全性及び薬物動態を評価した第Ⅰ相試験28試験が含まれた([2.7.1 項] [2.7.2 項])。これらの第Ⅰ相試験には、日本人健康被験者33例を含む766例の被験者が組み入れられた。 766例のうち、668例がレテルモビルを単独又は他剤との併用(薬物相互作用試験として)で投与 された。レテルモビルの経口投与の用量は、単回投与が5~720 mg、反復投与が40 mg 1日1回(QD) 投与~720 mg 1日2回(BID)投与であった。レテルモビルの静脈内投与の用量は、単回投与が30 ~960 mg、反復投与が120 mg QD 投与~480 mg QD 投与であった。 本臨床開発プログラムでは、2種類の注射剤(HPCD 製剤及び 製剤)を使用した。第 Ⅰ相試験2試験(017試験及び018試験)でのみ、錠剤とともに注射剤として 製剤が用い られ、組み入れた110例のうち50例に 製剤を静脈内投与した。 製剤は、安全 性の問題(軽度から中等度の注射部位刺激感及び血栓性静脈炎)により開発を中止したため、本 申請資料には含めない[2.7.1.1.2.2.1 項]。また、以降の開発は注射剤として HPCD 製剤を用いて行 った。 レテルモビルの薬物動態の評価には、第Ⅰ相、第Ⅱ相及び第Ⅲ相試験(001試験)から得られた 併合データを用いた母集団薬物動態解析も含めた。母集団薬物動態解析の目的は、健康被験者及 び同種HSCT 患者におけるレテルモビル薬物動態特性、有機アニオン輸送ポリペプチド(OATP) 1B1及びウリジン5’-二リン酸グルクロン酸転移酵素(UGT)の遺伝子多型がレテルモビルの曝露 量に及ぼす影響、並びに内因性及び外因性要因がレテルモビルの薬物動態に及ぼす影響を評価す ることであった[2.7.2.2.6.2 項]。OATP1B1及び UGT の遺伝子多型の影響については、線形混合効 果モデルによる解析でも検討した[2.7.2.2.6.4 項]。さらに、生理学的薬物動態(PBPK)モデルに より、レテルモビルの薬物動態の非線形性に寄与する因子や、日本人及び非日本人健康被験者間 で認められたレテルモビルの曝露量の差を、メカニズムに基づいて説明することが可能であった。 第Ⅰ相、第Ⅱ相及び第Ⅲ相試験(001試験)から得られた併合データを用いて、母集団薬物動態 解析、曝露-応答解析及び薬理遺伝学的解析を実施し、レテルモビルの薬物動態、有効性及び安 全性評価項目との曝露-応答関係を明らかにし、レテルモビルの曝露量及び応答に及ぼす特定の 内因性及び外因性要因の影響を評価した。第Ⅰ相試験、母集団薬物動態解析及び曝露-応答解析 の結果を[2.7.2.2 項]に要約する。 臨床開発プログラムの概要 第Ⅱ相及び第Ⅲ相臨床開発プログラムには、第Ⅱ相試験2試験(019試験及び020試験)及び第Ⅲ 相試験1試験(001試験)が含まれた。 019試験は、27例の患者(腎又は腎/膵臓移植患者26例及び HSCT 患者1例)を対象として用法 が異なるレテルモビル(40 mg BID 及び80 mg QD)を14日間投与した際の安全性、忍容性及び抗 ウイルス活性を実薬対照[実施医療機関の標準治療(バルガンシクロビル)]と比較した前期第Ⅱ 相非盲検 POC 試験であった[資料5.3.5.1.1: P019]。組み入れられた27例の患者は、レテルモビル 40 mg BID 群、80 mg QD 群又は実薬対照(バルガンシクロビル)群に各9例が割り付けられた。
2.5 臨床に関する概括評価 - 26 - 019試験では、治験開始時に CMV 血症が認められ、先制治療の開始基準を満たした患者における CMV DNA 量の減少を評価した。 020試験は、移植前に CMV 免疫グロブリン G(IgG)抗体陽性で、治験薬投与開始前5日以内に CMV DNA が検出されなかった同種 HSCT 患者133例を対象としてレテルモビルの安全性及び抗ウ イルス活性をプラセボと比較した後期第Ⅱ相無作為化、二重盲検、プラセボ対照試験であった。 患者は3用量のレテルモビル群(60、120又は240 mg 1日1回)又はプラセボ群のいずれかに無作為 割付され、治験薬を84日間経口投与した。無作為割付した133例中131例に治験薬を投与し、内訳 は、レテルモビル60 mg 群33例、120 mg 群31例、240 mg 群34例及びプラセボ群が33例であった[資 料5.3.5.1.2: P020]。020試験では、CMV 再活性化の予防(CMV 感染又は感染症の予防)における レテルモビルの有効性をプラセボと比較して評価した。 001試験は、CMV 抗体陽性の成人同種 HSCT 患者を対象にレテルモビル480 mg(シクロスポリ ンを併用投与する場合はレテルモビル240 mg)を1日1回投与した際の安全性及び有効性をプラセ ボと比較して評価する無作為化、二重盲検、プラセボ対照第Ⅲ相試験であった。投与期間は移植 後14週(約100日)までとした。レテルモビル群376例及びプラセボ群194例の計570例を無作為割 付した。日本人は36例が無作為割付けされた。001試験では、CMV 再活性化の予防(臨床的に意 味のある CMV 感染又は感染症の予防)におけるレテルモビルの有効性をプラセボと比較して評 価した。 レテルモビルの全体的な曝露状況 臨床開発プログラムにおけるレテルモビル、プラセボ及び実薬対照の全体的な曝露状況の要約 を[2.5.5.1 項] [2.7.4.1.2 項] 及び[表 2.7.4-5]に示す。これらのデータには、健康成人被験者及び腎 又は肝機能障害者を対象とした第Ⅰ相試験、移植後の患者を対象とした第Ⅱ相試験(019試験及び 020試験)及び第Ⅲ相試験(001試験)が含まれる。レテルモビルを投与されたのは全試験を通じ て1157例(第Ⅰ相試験の健康被験者並びに肝又は腎機能障害者668例、第Ⅱ相及び第Ⅲ相試験の患 者489例)であった。レテルモビルを投与された日本人は全試験を通じて54例(第Ⅰ相試験の健康 被験者30例、第Ⅲ相試験の患者24例)であった。 第Ⅲ相試験(001試験)でのレテルモビル投与例は373例であったが、001試験と同一用量以上の レテルモビルの投与例は、第Ⅰ相試験で317例(レテルモビル240 mg 1日1回とシクロスポリンの 併用投与26例を含む)、第Ⅱ相試験(020試験)で18例(レテルモビル240 mg 1日1回 とシクロス ポリンの併用投与)であった。 2.5.1.6.2 日本での承認申請に用いる臨床データ 本製造販売承認申請では、20 年 月の医薬品医療機器総合機構(PMDA)との医薬品 相談での臨床データパッケージに対する助言[2.5.1.7.4 項]を踏まえ、臨床データパッケ ージを作成した。 [2.5.1.4.2 項]で示したように、HSCT 後の CMV 感染症対策は、海外では CMV のモニタリング
2.5 臨床に関する概括評価 - 27 - に主に用いられているPCR 法が日本では保険適応外であり、代わりに抗原血症検査が主流である という違いはあるものの、両検査法の相関性は報告されており[資料5.4: 32]、国内外ともに先制治 療が主流である。また、先制治療開始の判断基準については、現時点で国際的に標準化された開 始基準は存在しないものの、国内外共通して施設及び担当医の判断が重視されているという点で、 地域による大きな違いはない。また、海外で市販されている抗 CMV 薬のうち、cidofovir のみ日 本では未承認であるが、国内外共にガンシクロビルが第一選択薬であり、その他にバルガンシク ロビルやホスカルネットが用いられる。いずれの抗 CMV 薬も、国内外で用いられている用法・ 用量はほぼ同様である。 母集団薬物動態解析により詳細な検討を行った結果、大多数が日本人(日本人30例及び日本人 以外のアジア人3例)で構成されたアジア人の曝露量のベイズ推定値は、非アジア人と比較してわ ずかに(33.2%)高かったものの、日本人 HSCT 患者におけるレテルモビルの曝露量の分布は、非 日本人HSCT 患者の分布と大部分が重なった[図 2.7.2-4] [表 2.7.2-6]。なお、レテルモビルの曝露 量でみられた人種間の差は、大部分が体重の違いで説明可能であった。さらに、アジア人と白人 の薬物動態の違いに寄与することが報告[資料5.4: 8] [資料5.4: 9] [資料5.4: 10]されている OATP1B1 及び UGT1A1の遺伝子多型に関して、それらのレテルモビルの薬物動態における潜在的な影響に ついても検討したが、レテルモビルの曝露量に臨床的に意味のある影響を及ぼさなかった [2.7.2.3.1.1.3 項]。これらの結果から、日本人と非日本人の曝露量に明らかな違いは認められない と判断した。 以上より、日本人及び非日本人の間で薬効評価上問題となる外因性民族的要因及び内因性民族 的要因の違いは存在しないと考えられる。したがって、日本人被験者の参加した第Ⅲ相国際共同 試験成績を主に用いて日本人での本剤の有効性及び安全性を評価することが可能と判断した。な お、日本での臨床データパッケージは、日本人を対象とした臨床試験(027試験、032試験)及び 第Ⅲ相国際共同試験(001試験)に加え、非日本人を対象とした第Ⅰ相試験(QT/QTc 評価試験: 004試験及び食事の影響試験:029試験)及び後期第Ⅱ相試験(020試験)を評価資料、非日本人を 対象として実施したその他の海外臨床試験を参考資料とした[表 2.5-1]。 2.5.1.7 規制当局からの指針及び助言 2.5.1.7.1 規制当局からの指針 本剤の開発は、その過程において、各国の規制当局からの助言を随時得ながら進めている。米 国食品医薬品局(FDA)、欧州医薬品庁(EMA)及び PMDA から入手した正式な指針を以下に要 約する。 2.5.1.7.2 米国食品医薬品局 20 年 月 日、FDA は、 することで、 と結論付けた。
2.5 臨床に関する概括評価 - 28 - 2011年5月25日、FDA から「移植患者における CMV 感染症の予防」に対してファストトラック 指定を受けた。 2011年12月12日、「リスク集団での CMV 血症及び感染症の予防」に対してオーファン・ドラッ グ指定を受けた。 20 年 月 日、 を実施した。FDA との主な合意事項及び新薬承認申請に関する FDA からの助言を以下に示す。 非臨床開発 以下の点についてFDA と合意した。 • 及び を支持するための の充足性 • 必要はないこと • に対する懸念に対し、 にて説明を行 った結果、 こと • に対する懸念に対し、 を測定することは、FDA は受け入れ可能とした。 臨床薬理 以下の点についてFDA と合意した。 • 決定すること • する必要はないこと • すること • する必要はないこと • 評価すること また、以下の点についてFDA から見解を得た。 • FDA は、 することを推奨した。 • FDA は、 する必要があるとした。
2.5 臨床に関する概括評価 - 29 - 臨床的有効性及び安全性 以下の点についてFDA から見解を得た。 • FDA は 受け入れ可能とし た。また、 が必要であるとした。 • FDA は、 よいこととした。FDA は、 、この助言を再検討 するとした。 20 年 月 日、FDA と米国本社との間で が実施された。この相談の目 的は、 に関しFDA の合意を得ることであった。以下に検討さ れた事項の要約を示す。 品質 • FDA は受け入れ可能とした。 臨床薬理 以下の点についてFDA と合意した。 • を支持すること • について また、以下の点についてFDA から見解を得た。 • を評価するこ と。 臨床的有効性及び安全性 以下の点についてFDA と合意した。 • • と
2.5 臨床に関する概括評価 - 30 - することについて • 計画 また、以下の点はFDA から推奨され、米国本社が合意した事項である。 • を用いること • すること その他、FDA からの見解を以下に示す。 • 受け入れは可能であること。 する必要があるとした。 • FDA は、レテルモビルがオーファン・ドラッグ指定を受けていることから、 することとした。 20 年 月 日、FDA との 相談を実施した。 に関連する協議事 項、合意事項及び要求事項の要約を以下に示す。 • FDA は、 について合意した。 • FDA は、 について合意した。 • FDA は米国本社に対し、 推奨した。 • FDA は、 と述べた。FDA は、 ることで合意した。 • FDA は、 と合意した。 • FDA は、 あるとした
2.5 臨床に関する概括評価 - 31 - • FDA は、 ことについて合意した。 • FDA の提言を受けて、 ことに合意した。 • 2017年1月9日、レテルモビルの Breakthrough therapy の指定を申請し、2017年2月27日に指 定された。 2.5.1.7.3 欧州規制当局 2011年4月15日、「細胞性免疫障害のリスク患者における CMV 感染症の予防」、2012年6月6日、 「細胞性免疫障害のリスク患者における CMV 感染症の治療」に関するオーファン・ドラッグ指 定を受けた。 20 年 月 日、 に対して欧州医薬品委員会(CHMP)から助言を得た。 CHMP は、 について確認し た。 に関連するCHMP からの助言及び合意事項の要約を以下に示す。 品質 • 、受け入れ可能とした。 非臨床開発 • 米国本社が提示した は不要である。 臨床薬理 • が推奨された。 臨床的有効性及び安全性 • に関して受 け入れ可能と判断された。 するこ とは妥当であると判断され、 ことが推奨され た。
2.5 臨床に関する概括評価 - 32 - • する必要が ある。 • であると考えられた。 20 年 月 日、 からは肯定的な意見が得られた。 20 年 月 日、 医薬品局 との 相談を実施し、 された につ いて受け入れ可能である旨コメントがあった。 20 年 月 日、 医薬品局 との 相談を実施し、 された。 について合意に達した。また、 とコメントがあった。 20 年 月 日、EMA との 相談を実施した。 が検討された。 について詳細な議論 を行った。 20 年 月 日、CHMP に対して迅速審査の申請書を提出した。 2.5.1.7.4 日本の規制当局 20 年 月 日に、日本での第 相試験実施に先立ち について、PMDA と医薬品 相談を実施した。 を相談した。 • 機構は、 は受け入れ可能であるとした。 •
2.5 臨床に関する概括評価 - 33 - ことは可能とした。 • また、日本では新規添加物となる本薬注射剤の添加剤HPCD の安全性を、 機構は、製剤開発 の経緯及び臨床試験データに基づく安全性等を考察することで受け入れ可能とした。 • 機構は、 受け入れ可能とした。 20 年 月 日までに書面にて、医薬品 相談を実施した。欧米では240 mg 錠に加えて480 mg 錠の承認取得を計画しているが、日本では240 mg 錠のみの承認取得を計画して いるため、 について 相談し、PMDA は受け入れ可能とした。 20 年 月 日に、医薬品 相談を実施した についてPMDA は受け入れ可能とした。 なお、本剤は、2016年2月25日に、厚生労働大臣より希少疾病用医薬品として指定を受けた[指 定番号(28薬)第374号]。 2.5.1.8 医薬品の臨床試験の実施の基準(GCP)の遵守 日本で実施した国内第Ⅰ相臨床試験(027試験)及び第Ⅲ相臨床試験(001試験)、並びに本申請 で利用した臨床試験は、必須文書の保管を含めた試験のデザイン、実施及び解析に関する既存の 標準的な研究方法に従って実施した。すべての試験は医薬品の臨床試験の実施の基準(GCP)に 従い、試験実施時に適用されていたヒト被験者に対する倫理的治療を考慮して実施した。 。
2.5 臨床に関する概括評価 - 34 - 2.5.2 生物薬剤学に関する概括評価 製剤開発、各製剤の挙動、バイオアベイラビリティ及び食事の影響について明らかにするため に実施した生物薬剤学試験の要約を[2.7.1 項]に示す。すべての生物薬剤学試験は健康被験者を対 象に実施し、各試験の結果を[2.7.1.2 項]に示す。レテルモビルの臨床開発のために、液体クロマ トグラフィー・タンデム質量分析法(LC-MS/MS)を用いてヒトの血漿、尿及び唾液中のレテル モビル測定のため特異的で感度の高い分析法を開発した。さらに、レテルモビル投与後の循環血 中のレテルモビルグルクロン酸抱合体の濃度を測定するため、アルカリ処理したヒト血漿中のレ テルモビル測定法を開発した[2.7.1.1.4.1 項]。 生物薬剤学分類システム(BCS)では、レテルモビルは240 mg 及び480 mg の用量でクラスⅡ: 低溶解性-高膜透過性医薬品に該当する。レテルモビルは pH ~ の範囲でわずかに溶解し(約 mg/mL)、pH 未満及び pH を超えると溶解度が急速に上昇する[2.7.1.1.1 項]。 レテルモビルの製剤開発については、[2.5.2.1 項]に示す。レテルモビルのバイオアベイラビリ ティ及び臨床的に意味のある食事の影響はないことを示す生物薬剤学試験の概括評価を、それぞ れ[2.5.2.2 項]及び[2.5.2.3 項]に示す。 2.5.2.1 製剤開発 レテルモビルには経口剤(液剤及び錠剤)及び注射剤がある。レテルモビルの臨床開発では、 経口剤6種類及び注射剤2種類を開発し、臨床試験で評価した[表 2.7.1-3] [表 2.7.1-4]。海外市場向 けには、錠剤及び注射剤のいずれも2種類の含量の規格があり、経口剤は240 mg 錠及び480 mg 錠、 注射剤は無菌溶液の240 mg バイアル及び480 mg バイアルである[2.5.2.1.1 項] [2.5.2.1.2 項]。日本 では、240 mg 錠及び240 mg バイアルのみを承認申請し、市販する予定である[2.7.1.1.2 項]。 2.5.2.1.1 レテルモビル経口剤の開発 本臨床開発プログラムでは、6種類の主要な経口剤を開発し、臨床試験で評価した。これらの内 訳は、開発初期製剤として液剤(FFP1製剤)及び を用いた錠剤(FFP2製剤)の2種類、市 販候補製剤として を用いた錠剤(PMF1製剤)、 及び を用いた錠剤(PMF2製剤)及び を用いた製剤(PMF3製剤)の3 種類、並びに市販予定製剤(FMI 製剤)が1種類である。FMI 製剤は、最終的なフィルムコーティ ング錠に を施していることを除き、PMF3製剤と同一の製剤である。第Ⅲ相試験(001試験) ではPMF3製剤及び FMI 製剤の240 mg 錠及び480 mg 錠を使用した。なお、日本では経口剤に関し てPMF3製剤及び FMI 製剤の240 mg 錠のみを001試験で使用した。レテルモビル480 mg 錠はサイ ズが大きく、日本の市場には適切ではないと考えられたため、日本では、レテルモビル群の患者 に480 mg を投与する場合、480 mg 錠1錠の代わりに、240 mg 錠2錠を投与した[2.7.1.1.2.1 項]。 レテルモビルの主要な経口剤の開発経緯及び製剤間の類似性を評価した溶出性/相対的バイオ アベイラビリティの概略を[図 2.7.1-1]に示す。レテルモビルの開発に使用された経口剤は、相対 的バイオアベイラビリティ試験及び溶出性データによりほぼ生物学的に同等であることが示され
2.5 臨床に関する概括評価 - 35 - ている[2.7.1.3.3.1 項]。第Ⅰ相試験(028試験)の結果から、PMF3製剤の240 mg 錠2錠と480 mg 錠 1錠の薬物動態は生物学的同等性基準の範囲内であった[2.7.1.2.2.2 項]。 2.5.2.1.2 レテルモビル注射剤の開発 本臨床開発プログラムでは、2種類の注射剤[ヒドロキシプロピル-β-シクロデキストリン (HPCD)製剤及び 製剤]が開発された。 製剤は、安全性の理由により開発 が中止され、本承認申請の対象ではないため詳述しない[2.7.1.1.2.2.1 項]。注射剤の市販予定製剤 (HPCD 製剤)は、レテルモビルを溶解補助剤である HPCD を用いて水に溶解した、無菌の濃縮 水溶液であり、以後の第Ⅰ相試験で使用された。市販予定製剤は第Ⅲ相試験(001試験)でも使用 された[2.7.1.1.2.2.2 項]。レテルモビル注射剤の開発経緯を[図 2.7.1-2]に示す。 2.5.2.2 バイオアベイラビリティ 第Ⅰ相試験の統合データを用いて実施した母集団薬物動態(PPK)解析に基づくと、レテルモ ビルの絶対的バイオアベイラビリティは高く、健康被験者で94%であった[2.7.1.3.2 項]。 2.5.2.3 食事の影響 PMF3製剤を用いて、単回投与時のレテルモビルの薬物動態に及ぼす標準的高脂肪・高カロリー 食の影響を評価した。 標準的高脂肪・高カロリー食摂取後にレテルモビル480 mg を単回経口投与した際のレテルモビ ルのAUC に食事の影響は見られず、最高濃度(Cmax)は空腹時と比較して約30%増加した[2.7.1.2.3.2 項]。これらの結果から、レテルモビルの血漿中濃度に臨床的に意味のある食事の影響はなかった。 よって、レテルモビルは食事に関係なく投与することできる[2.7.1.3.4 項]。また、第Ⅲ相試験(001 試験)では、食事に関係なく治験薬を投与し、安全性及び有効性が確認された。