合成生物学的アプローチによる遺伝子発現システム の開発と組織工学への応用
小野, 章彦
https://doi.org/10.15017/1931747
出版情報:九州大学, 2017, 博士(工学), 課程博士 バージョン:
権利関係:
合成生物学的アプローチによる
遺伝子発現システムの開発と組織工学への応用
平成 30 年1月
小野 章彦
目次
第一章 序論 ... 1
合成生物学... 1
1.1.1 既往の生物学と比較した合成生物学 ... 2
1.1.2 微生物を利用した合成生物学 ... 2
1.1.3 動物細胞を利用した合成生物学 ... 3
誘導型遺伝子発現システム... 4
1.2.1 自然界でのストレス応答 ... 4
1.2.2 熱応答遺伝子発現システム ... 5
1.2.3 低酸素応答遺伝子発現システム ... 6
1.2.4 DNAダメージ応答遺伝子発現システム ... 8
1.2.5 光応答遺伝子発現システム ... 10
1.2.6 pH応答遺伝子発現システム ... 11
合成生物学の遺伝子治療、再生医療・組織工学への応用... 12
1.3.1 遺伝子治療への応用 ... 12
1.3.2 再生医療・組織工学への応用 ... 13
本研究の背景と目的... 14
本研究の方針... 16
本研究の構成... 17
第二章 温熱応答型遺伝子発現システムの組織工学への応用 ... 18
緒言... 18
温熱応答型遺伝子発現システムの構築... 19
本節の目的 ... 19
実験方法 ... 20
2.2.2.1 遺伝子発現システムの設計 ... 20
2.2.2.2 プラスミドベクターの作製 ... 23
2.2.2.3 レポーター遺伝子の検討 ... 33
2.2.2.4 細胞と培地の組成 ... 34
2.2.2.5 一過性遺伝子導入における経時的な GFPmodc 遺伝子発現挙動解析 ... 35
結果と考察 ... 36
2.2.3.1 レポーター遺伝子の検討 ... 36
2.2.3.2 各遺伝子発現システムの経時的GFPmodc発現挙動解析 ... 39
本節のまとめ ... 41
磁場誘導型遺伝子発現システムの機能評価... 42
本節の目的 ... 42
実験方法 ... 44
2.3.2.1 細胞と培地の組成 ... 44
2.3.2.2 Magnetite Cationic Liposome (MCL)の作製 ... 44
2.3.2.3 HepG2-HSP細胞の細胞特性評価 ... 45
2.3.2.4 HepG2-HSP 細胞の MCL 取り込み量評価および MCL 添加が生細胞 率に与える影響 ... 45
2.3.2.5 交番磁場照射によるHepG2-HSP細胞の遺伝子発現誘導 ... 46
2.3.2.6 HepG2-HSP細胞の三次元組織状態における遺伝子発現誘導 ... 47
結果と考察 ... 47
2.3.3.1 HepG2-HSP 細胞の温熱応答性および加温が生細胞率に与える影響 ... 47
2.3.3.2 HepG2-HSP 細胞の MCL 取り込み量評価および MCL 添加が生細胞 率に与える影響 ... 50
2.3.3.3 HepG2-HSP細胞の磁場照射による遺伝子発現誘導 ... 52
2.3.3.4 磁場照射時間を変えたときのHepG2-HSP細胞の遺伝子発現 ... 54
2.3.3.5 シート状組織での遺伝子発現誘導 ... 54
本節のまとめ ... 57
本章のまとめ... 57
第三章 低酸素応答型遺伝子発現システムの組織工学への応用 ... 58
3.1 緒言... 58
3.2 低酸素応答型遺伝子発現システムを利用した細胞センサー... 59
3.2.1 本節の目的 ... 59
3.2.2 実験方法 ... 59
3.2.2.1 HepG2 細胞由来ゲノム DNA からの RTP801 プロモーター配列のク ローニング ... 59
3.2.2.2 ODD遺伝子の人工遺伝子合成 ... 60
3.2.2.3 その他の遺伝子配列の取得 ... 60
3.2.2.4 プラスミドベクターの作製 ... 61
3.2.2.5 細胞と培地の組成 ... 69
3.2.2.6 蛍光タンパクEGFPを用いた検討 ... 69
3.2.2.7 一過性遺伝子発現解析 ... 69
3.2.2.8 安定発現株選抜のための遺伝子発現解析 ... 70
3.2.2.9 安定発現株の様々な酸素濃度に対する遺伝子発現解析 ... 70
3.2.2.10#24の三次元組織状態での低酸素応答性評価 ... 70
3.2.2.11#24の細胞センサーとしての機能評価 ... 71
3.2.3 結果と考察 ... 72
3.2.3.1 RTP801プロモーター配列のクローニング ... 72
3.2.3.2 作製した遺伝子発現システムの一過性発現解析 ... 73
3.2.3.3 EGFPをレポーターとして酸素濃度検出を行う細胞株の樹立 ... 75
3.2.3.4 様々な酸素濃度における低酸素応答細胞株の評価 ... 76
3.2.3.5 #24細胞の三次元組織状態での低酸素応答性 ... 78
3.2.4 本節のまとめ ... 80
3.3 高機能組織構築を目指した低酸素応答型遺伝子発現システムの応用 .... 80
本節の目的 ... 80
実験方法 ... 81
3.3.2.1 プラスミドベクターの作製 ... 81
3.3.2.2 安定発現株取得 ... 82
3.3.2.3 安定発現株の発現プロファイル解析 ... 82
3.3.2.4 安定発現株の自律的遺伝子発現抑制試験 ... 82
結果と考察 ... 83
3.3.3.1 安定発現株の発現プロファイル解析 ... 83
3.3.3.2 安定発現株の自律的遺伝子発現抑制試験 ... 85
本節のまとめ ... 86
3.4 本章のまとめ... 87
第四章 総括 ... 88
4.1 本論文のまとめ... 88
4.2 今後の展望... 90
参考文献 ... 92
謝辞 ... 101
1
第一章 序論
合成生物学
2000年にサンフランシスコで行われた米国化学会の年会において「合成生物 学」という用語が用いられた[1]。それ以前にも「合成生物学」という用語はあっ たが、これが「自然に存在しない分子を生きたシステムの中で機能させる」「生 物機能を人工的に再設計する」という意味での合成生物学の研究を加速させる 起点となった。上述の通り、合成生物学とは非常に広い意味をもつため、合成生 物学に関する様々な研究が報告されている。合成生物学が注目された初期のこ ろは、新規転写調節因子の開発が盛んに行われていた[2], [3]。これらの研究には 遺伝子工学研究のモデル生物である大腸菌が多く用いられてきた。研究が盛ん になるにつれ、合成生物学者は、遺伝子やタンパクを 1 つのユニットとして捉 えるようになり、各ユニットの組み合わせや設計によってコンピューターシス テム(入力→処理→出力)のように生物機能を制御するようになった[4]。特に遺 伝子に対しては、プロモーターをスイッチ、転写促進因子を増幅器などと見立て 電子回路のように扱い、設計された遺伝子を遺伝子回路と呼ぶようになった。生 物機能を人工的に制御していく中で、転写の促進・抑制のようなシンプルな制御 から、2細胞間でのシグナルのやり取りや、論理ゲートによる遺伝子発現の制御 といった、より複雑な機能を細胞に持たせることが可能になった[5], [6]。また近 年は、開発した遺伝子回路を細胞に導入することで細胞の高機能化が可能にな り、合成生物学を用いた有用物質生産や、様々な疾病に対する新規治療法の開発 も行われている[7]–[9]。
この節ではこうした合成生物学の歴史とともに合成生物学の研究対象につい て述べる。1.1.1 では既往の生物学と比較することで合成生物学の理解を深め、
1.1.2では微生物を対象とした合成生物学の報告例、1.1.3では動物細胞を対象と
2 した合成生物学の報告例について述べる。
1.1.1 既往の生物学と比較した合成生物学
既往の生物学では、自然に存在する生命現象を対象に研究が行われてきた。
例えば、有名なワトソンとクリックによるDNA二重らせん構造の決定にも見ら れるように、研究の目的は細胞が自然に有する分子の構造や機能を明らかにす ることであった。また遺伝子工学の発展に伴い、機能未知の遺伝子を欠損させた 後、表現型の変化を観察することで目的の遺伝子機能を調べる逆遺伝学的手法 も発達し、多くの遺伝子の機能同定が行われてきた。
一方で、機能の明らかになった遺伝子やタンパクの知見が蓄積されることで、
こうした機能既知の分子を人工的に組み合わせ、新たな生物機能を構築する試 みが行われるようになり、合成生物学という概念ができた。従来の生物学が自然 に存在する生命現象の理解のため、トップダウン(解析的)に機能同定していく のに対して、合成生物学は、ボトムアップ(構成的)に生物機能を構築していく 点に特徴を持つ。
しかしながら、合成生物学を用いた人工生物機能の構築は、倫理的な問題・
新種ウイルスの作製によるバイオテロへの悪用といった問題を抱えている。そ のため、合成生物学の健全な運用のための議論もなされており、研究者個人の責 任感はもちろん、社会科学・倫理学・工学・生物学の繋がりの重要性が提唱され ている[10], [11]。
1.1.2 微生物を利用した合成生物学
遺伝子工学のモデル生物である大腸菌や酵母は、遺伝子機能の知見が多く、
また研究手法も動物細胞に比べて容易であるため、合成生物学の多くは微生物
3
を対象に研究が行われてきた。また、一般的に微生物の取り扱いは動物細胞に比 べて時間・コストが少ないという点にもアドバンテージを有する。
合成生物学的手法によって設計・構築した遺伝子回路を微生物に導入するこ とで、任意の物質を検出するためのバイオセンサーとして利用する報告がある。
近年、薬剤耐性を獲得した薬剤耐性菌による感染症が問題となる中、この薬剤耐 性菌を標的としたバイオセンサーの報告もされている[12]。また、環境中の有害 物質である重金属を検出するバイオセンサーが開発されており、カドミウム、鉛、
アンチモンを数 nmol/L レベルで検出する Staphylococcus aureus RN4220 株と Bacillus subtilis BR151株も報告されている[13]。
さらに微生物は、バイオセンサーとしての利用だけでなく、代謝経路に影響 を与える遺伝子回路を導入することで、アミノ酸、アルコールなどの有用化合 物・バイオ燃料を生産させることも可能となっている[14]–[16]。
1.1.3 動物細胞を利用した合成生物学
微生物を対象とした合成生物学が盛んに研究される一方で、動物細胞を対象 とした合成生物学の研究も始まった。しかし、微生物と異なり、動物細胞を用い た合成生物学は実現困難だと考えられていた。その理由は、機能既知の遺伝子や タンパクの知見が少ない、遺伝子回路導入後の遺伝子・タンパクの挙動予測が難 しい、細胞内において遺伝子回路が分解される可能性があるなどの問題を抱え ていたためである[17]。しかし動物細胞に導入した遺伝子回路が設計通りに機能 したという報告[18]がなされると動物細胞を対象とした合成生物学の研究は加 速した。動物細胞を利用した合成生物学の目的の 1 つに人類の健康に貢献する ことが挙げられ、特にがん治療や糖尿病などの代謝性疾患治療の研究が報告さ れている。がん治療においては、腫瘍の特徴的な微小環境として挙げられる低酸 素環境に応答して自殺遺伝子を発現するよう設計した遺伝子回路を用いた遺伝
4
子治療の報告がされている[19]。代謝性疾患においては、高濃度のグルコースを 検出しインスリンを分泌するよう設計した細胞を移植する手法や、高レベルの 胆汁酸値を検出し、肝組織の再生を促進するhepatocyte growth factor (HGF)を発 現するよう設計した細胞を利用する治療法が報告されている[20], [21]。
誘導型遺伝子発現システム
合成生物学的手法によって細胞を高機能化する際、多くの場合は任意のタイ ミングで遺伝子発現することが求められる。この実現のために、細胞のストレ ス・刺激応答機構を利用することで、遺伝子発現を制御する研究が報告されてい る。そこで本節では、ストレス・刺激応答に着目し、それらを利用した遺伝子発 現システムについて述べる。1.2.1 では自然界における細胞のストレス応答につ いて述べ、1.2.2から1.2.6において、具体的な遺伝子発現システムの研究報告に ついて紹介する。
1.2.1 自然界でのストレス応答
細胞は自然界において体内で様々なストレスに曝されている。細胞外からの ストレスとしては、酸化ストレス、熱ストレス、薬剤によるストレス、重金属に よるストレスなどが挙げられる。また細胞内においてもミスフォールディング したタンパクが蓄積することによる小胞体ストレスが存在する[22]。こうした 種々のストレスに対して細胞は、恒常性を保つためのストレス応答を示す。しか しながら、過剰なストレスに対しては、細胞は逆に細胞死を誘導する方向へとス トレス応答を示す[23]。こうした細胞のストレス応答はタンパクレベルでの応答、
遺伝子レベルでの応答が存在する。本節ではこうしたストレス応答を利用した 遺伝子発現制御に関する研究報告を紹介する。
5
1.2.2 熱応答遺伝子発現システム
細胞は外部からのストレスに曝されると、生命を維持するために様々な応答 を示す。ストレスの 1 つとして熱ストレスが挙げられる。熱ショックタンパク
(heat shock protein; HSP)は熱ストレス応答の一端を担うタンパク質として知ら
れており[24]、ストレス条件下で発現量が上昇し、タンパク質の変性やミスフォ ールディングを防ぐ分子シャペロンの一群ということが報告されている[25]。 HSPの発現は、活性化した熱ショック転写因子(heat shock transcription factor-1;
HSF-1)がHSPプロモーター配列中に存在する熱ショック転写要素(heat-shock
element; HSE)に結合することによって開始される[26], [27]。ストレス下にない
通常の細胞内では、HSF-1は分裂促進因子活性化タンパク質キナーゼ(mitogen- activated protein kinase; MAPK)やグリコーゲン合成酵素キナーゼ 3(glycogen synthase kinase 3; GSK3)によってセリン残基Ser307、Ser303の構成的なリン酸 化の阻害を受けており[28]、HSPとのヘテロ複合体を形成することで細胞質内に 局在している。この際、RNA ポリメラーゼ II(Pol-II)は転写開始前複合体
(preinitiation complex; PIC)に捕捉されており、HSP遺伝子の転写は抑制状態に ある[29]。しかし熱ストレス条件下では、HSPがヘテロ複合体から外れ、HSF-1 の核内移行が起こる。核内移行した HSF-1 はホモ三量体を形成し、cAMP 依存 性タンパク質キナーゼ(cAMP-dependent protein kinase; PKA)やカルシウム/カル モジュリン依存性タンパク質キナーゼ(calcium/calmodulin-dependent protein kinase II; CaMKII)によってセリン残基Ser230、Ser326のリン酸化を受け活性化
する[30], [31]。活性化されたHSF-1がHSPプロモーター配列中のHSEに結合す
ることで、PICとPol-IIの結合が解かれ、HSP遺伝子の転写が誘導される[29]。 この転写機構によって、加温等のストレス下において、HSP 遺伝子の転写が促 進され、翻訳されたHSPは分子シャペロンとしてストレスに対処する。
HSP の転写機構を利用して、遺伝子発現を制御する際には、発現のリークが
6
少ない(加温時のみ遺伝子を発現する)ことが求められる。ヒトHSP70B’プロモ ーターは、加温をスイッチとして遺伝子発現を厳密に制御可能であることから in vitroおよびin vivoにおいて幅広く用いられている。
上述の通り、HSP プロモーターは細胞が熱ストレスにさらされたときに遺伝 子発現を誘導する。熱ストレスを与える方法としては、高温環境下(39.5 - 45℃)
に曝すという方法が一般的である[27]。一方で、熱ストレスを与えずともHSPプ ロモーターを駆動する薬剤も多数報告されている。例えば、胃潰瘍や胃粘膜障害 な ど の 治 療 薬 と し て 用 い ら れ る ゲ ラ ニ ル ゲ ラ ニ ル ア セ ト ン
(geranylgeranylacetone)、生薬として用いられるペオニフロリン(paeoniflorin)、 また生薬に含まれる成分であるカルベノキソロン(carbenoxolone)などである
[32]–[34]。細胞を高温環境下に曝す条件では、細胞へのダメージを制御すること
が難しいが、HSP 誘導剤の中には、細胞毒性は低いが、HSP プロモーターを駆 動するという利点を持つものある。また、細胞を高温環境下に曝す際の温度の調 節に比べて、薬剤濃度条件の調整は容易であり、その条件をより細かく詳細に設 定できるという利点も持つ。
1.2.3 低酸素応答遺伝子発現システム
酸素は多くの生物にとって生命維持に必須の分子である。細胞は、低酸素濃 度環境下に曝された際に、低酸素誘導因子(hypoxia inducible factor; HIF)の活性 化を通して様々な遺伝子の発現を促し、環境へ適応しようとする[35]。近年の報 告によると、HIFが幹細胞の維持、さらには狭心症や心筋梗塞といった虚血性疾 患に関与していることが明らかになった[36], [37]。また、がん細胞は腫瘍の増殖 速度に対し、血管新生が追いつかないため、局所的に低酸素環境であるというこ とが一般的に知られている。腫瘍組織では、HIFが活性化され、その結果として がんの転移・浸潤、治療抵抗性が引き起こされる。[38]–[42]。そのため、HIFを
7
ターゲットとした様々な治療法が開発されている[43], [44]。
次に、HIFの特徴的な転写機構について述べる。HIFは二つのサブユニットか ら構成されており、βサブユニットは構成的に細胞内で発現している一方で、α サブユニットは主に低酸素環境下において発現がみられる。低酸素環境下で発 現されたαサブユニットは核内に移行し、βサブユニットと二量体を形成する ことでHIFとして働き始める[45]。様々な低酸素応答型プロモーター上の低酸素 応答領域(hypoxia response element; HRE)にHIFが結合することで、低酸素応答 型プロモーターが活性化され、低酸素応答型プロモーターの下流にある遺伝子 の転写誘導が起こる。低酸素応答型プロモーターは低酸素環境をスイッチとし て下流の遺伝子発現を迅速に誘導する。本研究の第四章では、低酸素応答型プロ モーターの中でもアポトーシス関連遺伝子として知られている RTP801 遺伝子 のプロモーター[44], [46], [47]を用いて低酸素応答型遺伝子発現システムの開発 を行った。
HIF-αの発現が低酸素環境下でのみ確認されるのは、通常酸素濃度下では、
HIF-αは速やかに分解されているためである。その分解制御機能を担うユニッ トが酸素依存性分解ドメイン(oxygen degradation domain; ODD)である。その作 用機序は以下のとおりである。通常酸素条件下において、HIFプロリン水酸化酵 素(prolyl hydroxylase domain; PHD)[48]はHIF-αのプロリン残基(ヒトHIF-1 αでは 402 番目と 564 番目)を水酸化する[49], [50]。この水酸化される位置が ODD領域である。水酸化されたHIF-αはvon Hippel-Lindau protein (pVHL) と結
合し、pVHL-Cullin2-elonginBC-Rbx1 が形成するユビキチンリガーゼ複合体[51]
によってユビキチン化され、プロテアソームによって分解される。この分解工程 の起点となる水酸化を担っているPHDは酸素濃度により活性が変化する。通常 酸素濃度下ではPHDは高い活性を示すため、HIF-αの水酸化が促進されるのに 対して、低酸素環境下ではその活性が低下し、HIF-αの水酸化が抑制されるため、
分解されずに発現がみられる。従って、このODD領域をタンパクに付加するこ
8
とで、通常酸素下でのODD付加タンパクの分解を促進することができる。実際 に、ルシフェラーゼにODD配列を付加することで、通常酸素下では分解される ルシフェラーゼの開発が行われており、低酸素環境である腫瘍のイメージング に利用するという報告がなされている[52]。
1.2.4 DNA ダメージ応答遺伝子発現システム
細胞は紫外線や放射線、化学物質などの外的因子によって損傷を受けている。
これらの損傷はDNAに直接作用し、細胞内のDNA分子に生じる構造を変化さ せることからDNAダメージと呼ばれており、主として、細胞の老化や死、細胞 のがん化などを引き起こす原因となっている。DNAダメージにはいくつかの種 類があり、それらは、アルキル化、微小管重合阻害、DNA二本鎖の切断、活性 酸素による酸化、紡錘体毒性、代謝拮抗等に分類される。このようにDNA及び 染色体に異常を引き起こすDNAダメージを検出する方法として、いくつかの遺 伝毒性検出法が存在するが、動物細胞を用いて、生きた状態で検出できる検出法 は少ない。また、DNA鎖の切断を高感度で検出するcomet assayのように、特定 の原因に起因する DNA ダメージの検出感度は高いが、DNA ダメージ全般を網 羅できない検出法が多い[53]。DNA ダメージを生体に近い形で広く検出するこ とが出来れば、化学物質の発がん性リスクの評価や医薬品開発段階における遺 伝毒性の一次スクリーニングへの応用が期待できる。
がん抑制機能を持つp53タンパクは、多種多様なDNA損傷ストレスから細胞 を守り、がんの発生を防ぐという働きから「ゲノムの守護神」と称される[54]。 事実、半分以上のがんにて p53 やそのシグナル伝達経路に関わる分子に変異が 見つかっている[55]。そのため、がん治療のターゲット遺伝子としても注目され ており、正常 p53 遺伝子をがん細胞に導入し、がんを治療しようとする試みが 続けられている。p53タンパクは多くの機能をもっており、特定の遺伝子の転写
9
を活性化する働きによりがんを抑制する作用がある[23]。p53タンパクによって 遺 伝 子 発 現 が 誘 導 さ れ る 遺 伝 子 上 流 の プ ロ モ ー タ ー に は 共 通 し て
PuPuPuC(A/T)(T/A)GPyPyPyという配列が存在し、p53タンパクは核内で4 量体
を形成してこの配列に結合する[56]。DNA 損傷を受けていない細胞では p53 は 転写・翻訳はされるが、主にMdm2を介したユビキチン-プロテアソーム系によ り速やかに分解されている。一方で、紫外線、放射線、化学物質、酸化ストレス などのストレスを受けた際には、様々なシグナル経路が活性化し、細胞内のリン 酸化 p53 タンパク量が上昇する。同時に転写活性化が誘導され,標的遺伝子群 の発現を調節し、多彩な生理作用を発揮する。
DNAダメージに応答する性質のあるp53タンパク同様、p53遺伝子のプロモ ーター領域もまたDNAダメージにより活性化する性質がある。p53遺伝子のプ ロモーター配列には様々な転写因子の結合モチーフが存在しており、中でもp53 プロモーター領域(-88 ~ +20)の内、-70 ~ -46の領域にはDNA損傷に 対する応答性の要となるcore promoter element(CPE)が存在する[57]。CPE配列 には NF-κBを含む重要な転写因子の結合モチーフがあり、DNA ダメージを受 けた際には、PKCδキナーゼ活性依存的に転写因子Btf(アポトーシス促進因子)
がp53プロモーター領域のCPE配列に結合することでp53の転写が上方調節さ れる。また、p53タンパクはp53プロモーター上の特定の結合部位に直接結合す る性質があり、細胞がDNAダメージに曝された際には内在性のp53タンパクの 発現誘導を活性化することが可能である[58]。
我々の研究室においてはp53プロモーターを利用することで、DNAダメージ に応答してLacZ遺伝子を発現するシステムを開発しており、これを細胞に導入 することで、DNAダメージを検出する細胞センサーの開発を行っている[59]。
10
1.2.5 光応答遺伝子発現システム
植物や細菌、シアノバクテリアなどの生物は、フィトクロムと呼ばれる光受 容タンパクを有する。こうしたタンパクは光受容ドメインを有しており、このド メインが特定の波長の光に応答して構造変化を起こすことでシグナル伝達が誘 導される[60]。こうした光受容タンパクを利用することで、光をスイッチとして 遺伝子発現誘導するシステムが開発されている。Kennedyらは、青色光照射によ ってヘテロダイマーを形成するCRY2タンパクとCIB1タンパクそれぞれに、組 換え酵素CreのN末側とC末側を二分割してリンカーによって繋いだ組換えタ ンパクを構築した。この組換えタンパクを利用することで、青色光下でヘテロダ イマーを形成したときのみ Cre 酵素が活性を持ち、目的遺伝子配列上流のスト ップコドン配列を削除することで、目的遺伝子が発現誘導される[61]。Polsteinら は上述のシステムを利用することで、in vitro 条件下において、青色光照射した 細胞がMyoD遺伝子(筋分化を誘導する)を発現し、筋分化が誘導されることを 示した。さらに、青色光照射に応答してVEGF遺伝子とAng1遺伝子を発現する 遺伝子回路を導入した細胞をヌードマウスに移植し、一定期間青色光照射下で 飼育すると、移植細胞組織の血管網領域が増加するというin vivo での光応答性 遺伝子発現システムの有用性も示した[62]。
光応答遺伝子発現システムのスイッチとして用いる光は、医療分野への応用 を考慮すると、人体に対してダメージの少ない長波長の光を用いることが求め られる。その報告例として遠赤色光に応答してインスリンを発現する遺伝子発 現システムの報告例がある[63]。さらにこのシステムの利点としては、遠赤色光 照射デバイスを遠隔操作できるようにしたことで、患者と医者が離れている場 合でも、患者の血中グルコース濃度データを受信した医者が、遠隔で遠赤色光照 射を施し、インスリンによる治療が可能になるという点である。
11
1.2.6 pH 応答遺伝子発現システム
pHは細胞を取り囲む環境における重要なファクターの1つである。細胞内に おいても、細胞質のpHが約7.2であり、リソソームでは5.5未満、初期エンド ソームでは約6.5 などと、それぞれ異なる pH環境を有している[64]。こうした 各オルガネラのpHの違い、特に酸性オルガネラによるpH勾配は、タンパク輸 送に重要な環境であることが報告されている[65]。また、細胞培養条件における pHは培養細胞の代謝に影響を与えることが報告されており、その制御が重要視 されている[66]。Fussenegger らは細胞膜タンパクである T cell death-associated
gene8 ( TDAG8 )を利用することで、細胞外低pHに応答して遺伝子発現誘導する
システムを開発している[6]。 TDAG8 は細胞外ドメインのヒスチジン残基によ ってプロトンを検出可能であり、pH7.2を下回ると下流のシグナル伝達を駆動し、
cAMPの蓄積を誘導することが報告されている[67]。このcAMPがProtein Kinase
A ( PKA ) を活性化し、サブユニットの核内移行を誘導する。核内のPKAサブ
ユニットはcAMP-responsive element-binding protein 1 ( CREB1 ) をリン酸化し、
合成プロモーター上のcAMP-response elementsへの結合・下流の遺伝子発現誘導 をおこなう。上述のシステムを導入した細胞を培養する際、CO2濃度を変化させ ることで培地中の pH を制御可能であるため、特別な装置や試薬は不要であり、
インキュベーターの設定を変更するだけで培養細胞の遺伝子発現制御が可能と なる。pHに応答してバイオ医薬品であるリツキシマブを発現するシステムを導
入したFreeStyle 293F細胞を培養した結果、CO2濃度の上昇に応答してリツキシ
マブ生産量が上昇した。このようにpH応答遺伝子発現システムは、バイオ医薬 品生産への応用が可能である。
12
合成生物学の遺伝子治療、再生医療・組織工学への応 用
次に、合成生物学の医療・組織工学分野への応用例について紹介する。1.3.1 では、遺伝子治療への応用、1.3.2 では組織工学としての三次元組織構築への応 用について述べる。
1.3.1 遺伝子治療への応用
遺伝子治療は大きく 2 つに分類される。1 つは治療用遺伝子をそのまま患者 に導入する方法であり、もう1つは、治療遺伝子を細胞に導入後、治療遺伝子を 搭載した細胞を患者に導入する細胞ベースの手法である[68], [69]。合成生物学の 研究においては、設計した遺伝子回路が細胞内で目的の挙動を示すかを評価す ることが多いため、本節では、後者の細胞ベースの遺伝子治療について報告例を 紹介する。細胞ベースの治療として注目されているのがchimeric antigen receptors
( CARs )-T細胞を用いたがん治療である[70]。CARsとはキメラ抗原受容体のこ
とであり、CARs遺伝子を導入したT細胞では、その細胞膜上にCARsが局在す ることで、がん抗原を認識し、細胞内に存在するCARsのシグナリングドメイン によって T 細胞が活性化され腫瘍特異的な治療を可能にする。また、T 細胞の 活性化のみならず、シグナル伝達によって、がんの免疫療法に用いられるサイト カインであるinterleukin 12 (IL-12)を分泌させる試みも行われている[71]。このシ ステムでは、IL-12はNFAT6 minimalプロモーターの制御下にある。この合成プ ロモーターはnuclear factor of activated T cells (NFAT) binding siteの6回繰り返し
配列とinterleukin 2 minimalプロモーターから構成されているため、T 細胞が活
性化した条件でのみ駆動される[72]。さらに、腫瘍特異性を向上させるための試 みも行われており、CARs が 2 種類の腫瘍抗原を認識したときのみ T 細胞の活
13
性化が誘導されるシステムや、腫瘍以外の細胞を認識した条件下では T 細胞の 活性化を抑制するシステムなどが報告されている[73], [74]。
1.3.2 再生医療・組織工学への応用
再生医療における移植組織構築において、組織工学的に生体外で三次元組織 を構築する研究がなされている。かつての研究では、生体の失った機能を取り戻 すことを目的に研究が行われていたが、合成生物学的手法によって高機能化し た細胞を用いて三次元組織を構築することで、移植組織に付加価値を与える研 究も注目されている。Yi らは加齢などによって減少していく軟骨組織の再生医 療に合成生物学的手法を用いた[75]。この手法では、軟骨細胞特異的マトリック スタンパクの転写因子であるSox9をドキシサイクリン存在下で遺伝子発現する システムを構築している。このシステムを軟骨細胞に導入しラットへと移植後、
移植組織におけるSox9下流遺伝子の発現量を調べている。その結果、ドキシサ イクリン投与群では、ドキシサイクリン非投与群に比べて、タイプ II コラーゲ ンやアグリカンといったSox9下流遺伝子の発現量が有意に増加した。さらにこ のシステムでは、ドキシサイクリン投与時にのみSox9が発現するので、Sox9の 過剰発現による副作用のリスクが低減される。このように合成生物学的手法を 用いることで、治療遺伝子の発現を時間的に制御可能となる。また三次元組織構 築法そのものに合成生物学的手法を用いる研究も報告されている[76], [77]。
Cachatらは、組織構築に合成生物学的手法を応用するにあたって、細胞間接着タ
ンパクであるカドヘリンに着目した。T-Rex-293細胞にテトラサイクリン存在下 でE-カドヘリンを発現するシステムを導入したE-cellsと、同条件下で、E-カド ヘリンより結合力の弱いP-カドヘリンを発現するシステムを導入したP-cellsを 樹立し、1:1 の細胞数で共培養しスフェロイドを形成させた。その結果、E-cells
とP-cellsはスフェロイド中で分離した。より強い結合能を有するE-cellsがスフ
14
ェロイドの中央を帯状に占め、P-cellsはスフェロイドの端に局在した。このよう に異なる細胞間接着タンパクを発現するよう細胞を設計することで、三次元組 織構築時に、細胞のパターニングが可能となる。
本研究の背景と目的
上述のように近年、個々の遺伝子やタンパクの生物機能を明らかにする従来 の分子生物学による解析的なアプローチに対して、機能の明らかな遺伝子やタ ンパクを組み合わせることで、ボトムアップ的に人工の生物機能を構築する合 成生物学という新しい概念の構成的なアプローチによる研究が盛んになってい
る[1], [78]。合成生物学的手法によって細胞を高機能化する際、多くの場合は任
意のタイミングで目的遺伝子を発現する人工遺伝子発現システムを細胞に導入 する手法が採られる。こうした遺伝子発現システムを設計する際は、遺伝子発現 のスイッチとなるプロモーター配列や、転写調節因子を自在に組み合わせ、あら かじめ設計した発現挙動を実現する。特に遺伝子発現のスイッチは、発現挙動の 制御において重要となるため、細胞のストレス・刺激応答機構を利用して遺伝子 発現タイミングを制御する研究が多数報告されている[79], [80]。こうした遺伝子 発現システムは、がん治療や代謝性疾患治療、バイオ医薬品生産分野といった医 療分野への応用が報告されており、その応用性の高さから、遺伝子発現システム の開発研究は合成生物学において重要な位置を占めている[6], [81], [82]。
人工多能性幹細胞(induced pluripotent stem cell; iPS cell)が報告されて以降、
再生医療分野の研究が加速している。再生医療分野において、組織工学的に生体 外で作製した三次元組織を患者に移植するといったアプローチが採られている。
しかし、移植組織の「乏血管性」と、移植部位での「組織形成能の低さ」によっ て移植組織の生着率が低いという問題を抱えている。特に、「乏血管性」は、組 織内部まで栄養・酸素が行き届かず、組織内部の細胞が死んでしまうという深刻
15
な問題を引き起こすため、作製する三次元組織の厚さを制限する要因となって いる[83]。三次元組織の乏血管性を解決するには、作製した移植組織内で血管内 皮増殖因子(vascular endothelial growth factor ; VEGF)を遺伝子発現させ、組織内 部に血管網を構築させる方法が考えられる。しかし VEGF の長時間の発現は血 管の膜透過性を過剰にしてしまい、水腫の原因となるため[84]、血管網構造の安 定化に働く Angiopoietin-1 と共発現させるなどの方法が採られている[85], [86]。
一方で「組織形成能の低さ」の解決には、生体外で三次元組織を予め作製する 方法がある。三次元組織構築法としては、培養担体を足場材料として三次元組織 を構築する方法や、バイオプリンターを用いる方法、培養液の液滴中で球状組織 を構築する方法などが報告されている[87]–[89]。我々の研究室においては、機能 性磁性ナノ粒子(Magnetite Cationic Liposome; MCL)を細胞に取り込ませた後、
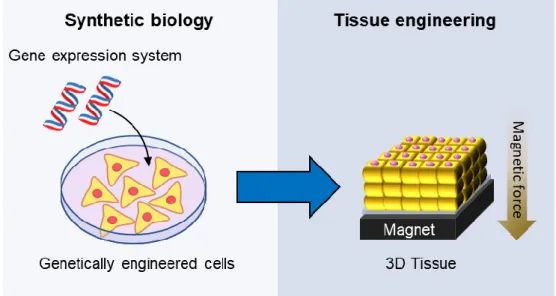
磁力によって細胞を積層させる三次元組織構築法が開発されている[90]。 本研究では、近年注目されている合成生物学的アプローチによって、組織工学 分野に応用可能な遺伝子発現挙動を制御するシステムの開発を目指した。この 遺伝子発現システムを導入した細胞を用いて組織工学的に三次元組織を作製す ることで、遺伝子発現制御可能な高機能三次元組織として再生医療への応用の 可能性を示すことを目的とした(Fig. 1)。
Fig. 1 A research strategy for combination of synthetic biology and tissue engineering.
16
本研究の方針
本研究では、まず、環境刺激により制御可能な遺伝子発現システムとして、
加温をスイッチとする遺伝子発現システムを構築した。遺伝子発現挙動として 持続的発現と一過性発現の2種類の異なった挙動を示す発現システムを設計し、
これらの遺伝子発現挙動を比較・解析することで、遺伝子発現システムの評価を 行った。次に、設計した温熱応答型遺伝子発現システムのうち持続的発現を示す システムを用いて、磁場照射による加温について検討した。機能性磁性ナノ粒子 MCL を細胞に取り込ませた後に、細胞に交番磁場を照射することで、細胞内 MCLの発熱を誘導し、細胞内局所加温による温熱応答型遺伝子発現システムの 発現誘導が可能かどうか調べた。その際、生細胞率測定を行うことで、磁場照射 による遺伝子発現誘導法の細胞傷害性を評価した。さらに、温熱応答型遺伝子発 現システムの、組織工学への応用可能性を調べるために、三次元組織(細胞シー ト)状態での磁場照射による遺伝子発現誘導が可能かを検討した。
次に、自律的に細胞外環境を検出し遺伝子発現誘導するシステムとして低酸 素環境下で緑色蛍光タンパク(Enhanced green fluorescent protein; EGFP)を発現 する「低酸素応答型遺伝子発現システム」を開発した。開発したシステムをヒト 子宮頸がん細胞株(HeLa 細胞)に導入し、低酸素環境下で培養した際の EGFP 蛍光を観察することで、低酸素環境を検出する細胞センサーとしての有用性を 評価した。構築した細胞センサーの二次元培養状態での低酸素応答性を評価し た後、細胞シートを構築し低酸素応答性を調べることで、三次元組織状態におけ る細胞センサーとしての機能評価を行った。
さらに、三次元組織構築の問題点である乏血管性による組織内部の低酸素環 境を自律的に克服することを目指し、低酸素環境に応答して血管内皮増殖因子
(VEGF)とEGFPを共発現するようなシステムを構築した。このシステムをゲ ノム上に組込んだマウス筋芽細胞株(C2C12細胞)を樹立し、低酸素環境に応答
17
したVEGF発現量の増加がみられるか、また通常酸素条件下に戻すことでVEGF 発現が抑制されるかを調べた。
本研究の構成
第1章では、本研究の関連分野の既往の研究を紹介し、本研究の背景、目的、
意義を示し、研究方針について説明した。
第 2 章では、加温をスイッチとして遺伝子発現する温熱応答型遺伝子発現シ ステムの発現挙動解析を行った。さらに、本システムと「磁場照射による磁性ナ ノ粒子の発熱」を利用した磁場誘導型遺伝子発現システムの遺伝子発現誘導能 とその際の細胞傷害性を評価し、移植用三次元組織の遺伝子発現システムとし ての有用性を示した。
第 3 章では、低酸素ストレスをスイッチとして遺伝子発現する低酸素応答型 遺伝子発現システムを設計・構築し、細胞に導入することで、低酸素下でEGFP 蛍光を示す、低酸素状態を検出する生きた細胞センサーを開発した。さらに、本 システムを移植用三次元組織の高機能化に応用した。具体的には低酸素環境下 において VEGF を発現する遺伝子発現システムを細胞に導入することで、移植 後に自律的に血管網を構築し、低酸素状態を克服するシステムの構築を行った。
第 4 章では、本論文の総括を行い、本研究の成果を踏まえた今後の展望につ いて述べた。
18
第二章 温熱応答型遺伝子発現システムの組 織工学への応用
緒言
合成生物学的アプローチによる遺伝子発現システム構築にあたって、効率的 に遺伝子発現を制御可能なため、すでに幅広く用いられているHSP プロモータ ーを利用し、加温を遺伝子発現のトリガーとした。本章で用いる HSP70B’プロ モーターは加温に厳密に応答するプロモーターであるが、一方でサイトメガロ ウイルス(cytomegalovirus; CMV)などの構成的に下流の遺伝子発現を誘導する ウイルスプロモーターと比較して、その発現量が低いという欠点を有する。我々 の研究室では、人工の大量遺伝子発現システムであるTet-offシステムとHSP70B’
プロモーターを組み合わせることで HSP70B’プロモーターの発現量を増幅する ことに成功している[91]。本章でもこの Tet-off システムと HSP70B’プロモータ ーの組み合わせによって、遺伝子発現挙動の異なる遺伝子発現システムを設計
した。Tet-offシステムとは遺伝子上の配列であるTRE配列とtTAタンパクから
なるシステムである。TRE認識ドメインであるtetRと転写活性ドメインである vp16からなるtTAタンパクがTRE配列に結合することで下流の遺伝子発現を促 進するシステムである。
また、HSP プロモーターをトリガーとすることで、ウォーターバスによる加 温、磁場照射による磁性ナノ粒子の発熱を利用した加温が発現誘導のスイッチ として利用可能となる。我々の研究室では、機能性磁性ナノ粒子MCLを取り込 ませた細胞に交番磁場を照射することで、HSP70B’プロモーターを駆動する研究 を行っており、遺伝子療法と温熱療法を組み合わせたがん治療法の開発にも成 功している[92]。
本章では合成生物学的手法によって環境刺激をスイッチとして遺伝子発現を
19
駆動できる遺伝子発現システム(温熱応答型遺伝子発現システムと磁場誘導型 遺伝子発現システム)の発現挙動解析と組織工学分野への応用についての成果 を示す。
温熱応答型遺伝子発現システムの構築 本節の目的
本節では、HSP70B’プロモーターと Tet-off システムを組み合わせることで2 種類の温熱応答型遺伝子発現システム(遺伝子持続的発現と遺伝子一過性発現)
を構築し、それらの遺伝子発現挙動を解析することを目的とした。まず、EGFP 遺伝子と、遺伝子工学的改変によって半減期を短くしたEGFP(GFPmodc)の蛍 光持続時間を比較することで、遺伝子発現挙動の解析に適当なレポーター遺伝 子の検討を行った。次に、GFPmodc を発現する温熱応答型遺伝子発現システム を、持続型と一過性型の2種類構築した。この際、持続型・一過性型それぞれに 対して、1つのベクター上に必要な遺伝子発現カセットが存在するOne-Packシ ステムと、遺伝子発現カセットを2つのベクターに分割した Two-Plasmids シス テムを設計し計4種類の遺伝子発現システムを構築することで、より詳細な遺 伝子発現挙動の解析を目指した(Figs. 2 and 3)。これら4種類の遺伝子発現シス テムをマウス繊維芽細胞株 NIH3T3 細胞に一過的に遺伝子導入し、加温後、
GFPmodc 蛍光を経時的に観察・解析することで、作製した遺伝子発現システム
の発現挙動解析を行った。
20
実験方法
2.2.2.1 遺伝子発現システムの設計
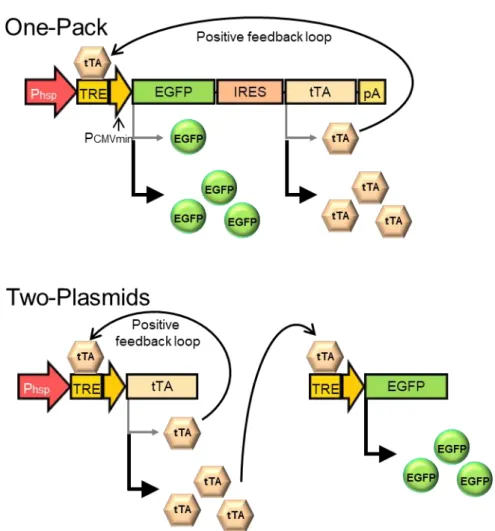
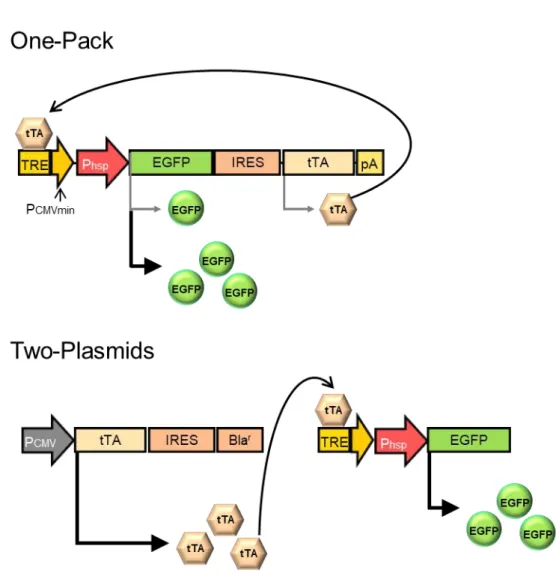
本研究で設計した持続型遺伝子発現システムでは、まず、加温をスイッチと してプロモーター下流の遺伝子配列がmRNAに転写された後、目的遺伝子と共 にtTAが翻訳される。発現したtTAはHSPプロモーター下流のTRE配列に結合 することで、目的遺伝子が、ポジティブフィードバックを伴って大量生産される
(Fig. 2)。一方、一過性型遺伝子発現システムでは、目的遺伝子の下流にあるtTA
が,HSPプロモーターの上流にあるTRE配列に結合することで、一過的な発現 増強が起こる(Fig. 3)。また、どちらのシステムも、テトラサイクリンの誘導体 であるドキシサイクリン(Dox)の添加によって、tTAの活性を抑制することが できる。
21
Fig. 2 Schematic drawing of the transgene expression strategy for heat-inducible transgene expression with transcriptional positive feedback loop. Phsp, HSP70B’ promoter; TRE, Tet-responsive element; EGFP, enhanced green fluorescent protein; IRES, internal ribosomal entry site;
tTA, Tet-responsive transactivator gene; pA, poly-A tail.
A
22
Fig. 3 Schematic drawing of the transgene expression strategy for heat-inducible transgene expression without transcriptional positive feedback loop. Phsp, HSP70B’ promoter; TRE, Tet-responsive element;
PCMVmin, cytomegarovirus minimum promoter; EGFP, enhanced green fluorescent protein; IRES, internal ribosomal entry site; tTA, Tet-responsive transactivator gene; pA, poly-A tail; Blar, blasticidin resistance gene.
A
23
2.2.2.2 プラスミドベクターの作製
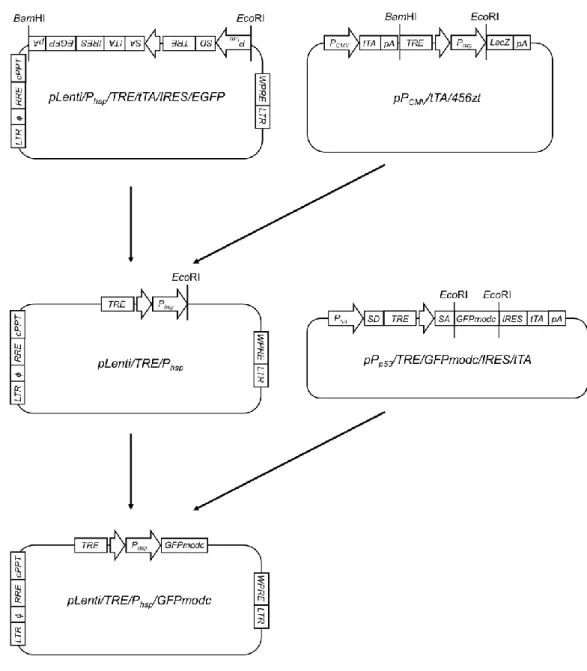
設計したプラスミドベクターの作製法を以下に示す。
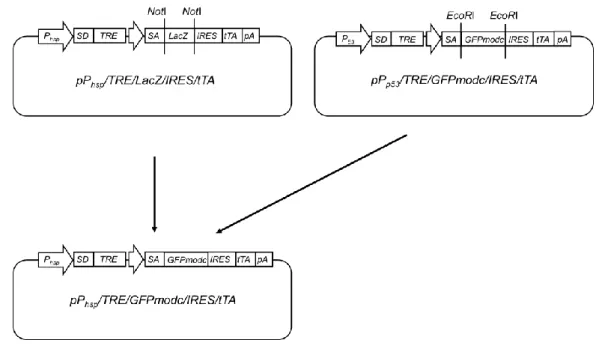
<pPhsp/ TRE/GFPmodc/IRES/tTA の作製>
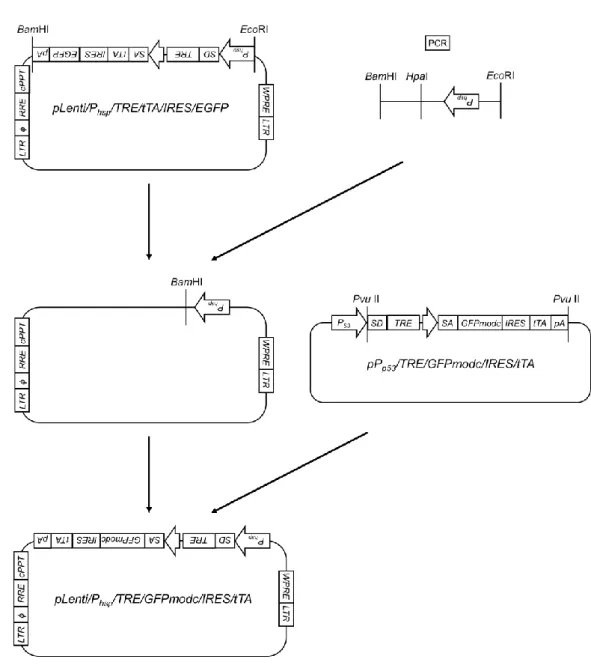
pPhsp/TRE/GFPmodc/IRES/tTAのコンストラクトとその作製手順をFig. 4に示
す。まず、すでに構築されたプラスミドpPhsp/TRE/LacZ/IRES/tTA(山口雅紀氏 よ り 供 与 ) を 制 限 酵 素 NotI に よ っ て 切 断 し 、 同 様 に
pPp53/TRE/GFPmodc/IRES/tTA(鈴木大雅氏より供与)を制限酵素EcoRIによっ
て切断した。得られた2つの遺伝子断片についてBlanting処理(DNA断片末端 の平滑化処理)を、その後 pPhsp/TRE/LacZ/IRES/tTA由来のDNA断片のみCIAP 処理(DNA5’末端の脱リン酸化処理)を行い、LigaFast™ Rapid DNA Ligation
System ( Promega ) を 用 い て ラ イ ゲ ー シ ョ ン 反 応 を 行 い 、
pPhsp/TRE/GFPmodc/IRES/tTA を作製した。ライゲーション反応後の DNA 溶液
は、大腸菌 DH5αに形質転換し、1 時間 LB 液体培地(1% ポリペプトン、1%
NaCl、0.5% 酵母エキス、pH 7.0)により培養後、最終濃度100 µg/mLアンピシ
リンを含む LB 寒天培地上に塗布することで目的プラスミドをもつ形質転換体 をスクリーニングした。その後アルカリ法によるプラスミド抽出を行い、
pPhsp/TRE/GFPmodc/IRES/tTA を 作 製 し た 。 作 製 し た
pPhsp/TRE/GFPmodc/IRES/tTAは、QIAfilter Plasmid Midi Kit(Qiagen)を用いた カラム抽出により精製した。
24
Fig. 4 Flowchart for the construction of pPhsp/ TRE/GFPmodc/IRES/tTA plasmid.
<pLenti/Phsp/TRE/GFPmodc/IRES/tTAの作製>
pLenti/Phsp/TRE/GFPmodc/IRES/tTAのコンストラクトとその作製手順をFig. 5 に示す。まず、Phsp 遺伝子配列をクローニングするためのプライマー(Fw:5’- GGAATTCGCCTCTAAAGTTGCTGCTTTTGC-3’, Rev:5’-aaGGATCCGTTAACCT
TGTCGGATGCTGGAGGC-3’)を設計し、シグマジェノシス社に合成を委託した。
なお、このプライマーの各端には、それぞれ制限酵素EcoRIとBamHI-HpaIの認 識サイトを付加した。鋳型 DNA として、pPhsp/TRE/GFPmodc/IRES/tTA を用い た。PCR反応は、94℃で2分間処理した後、98℃で 10秒間、66℃で30秒間、
68℃で14秒間を1サイクルとし、これを30サイクル繰り返した。得られたPCR 産物について、アガロースゲル電気泳動を行い、目的のDNA断片が得られてい るかどうかを確認した後、pBluescript II SK(-)上の制限酵素EcoRVの認識サイト に挿入した。このEcoRVサイトにPCR産物を持つpBluescript II SK(-)をBamHI
と EcoRI によって制限酵素処理し、アガロースゲル電気泳動後のバンドプレッ
プ に よ り 目 的 遺 伝 子 断 片 を 回 収 し た 。 次 に 、 プ ラ ス ミ ド
25
pLenti/Phsp/TRE/tTA/IRES/EGFP(薗田裕人氏より供与)を制限酵素BamHI およ
び EcoRI で切断した。得られた 2 断片を用いてライゲーション反応を行った。
得られたプラスミドをBamHIによって切断し、Blanting処理、CIAP処理した後、
Mag 精 製 を 行 い DNA 断 片 を 得 た 。 こ の DNA 断 片 と 、
pPp53/TRE/GFPmodc/IRES/tTAを制限酵素PvuIIによって切断して得られたDNA
断 片 を 用 い て ラ イ ゲ ー シ ョ ン 反 応 を 行 い 、 目 的 プ ラ ス ミ ド pLenti/pPhsp /TRE/GFPmodc/IRES/tTAを作製した。
Fig. 5 Flowchart for the construction of pLenti/Phsp/TRE/GFPmodc/IRES/tTA plasmid.
26
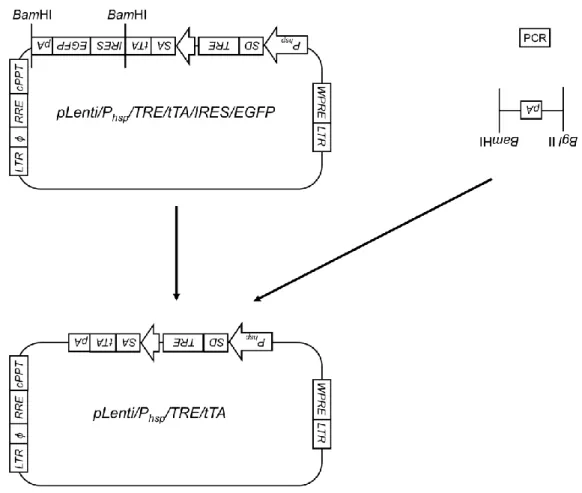
<pLenti/Phsp/TRE/tTAの作製>
pLenti/Phsp/TRE/tTAのコンストラクトとその作製手順をFig. 6に示す。まず、
polyA 遺 伝 子 配 列 を ク ロ ー ニ ン グ す る た め の プ ラ イ マ ー (Fw:5’- AAAGATCTCTGTGCCTTCTAGTTGCCAGC-3’, Rev:5’-aaGGATCCCCATAGAGC
CCACCGCATC-3’)を設計し、シグマジェノシス社に合成を委託した。なお、こ
のプライマーの各端には、それぞれ制限酵素 BglII と BamHI の認識サイトを付 加した。鋳型DNAとして、pcDNA4/TO/myc-His/A(Invitrogen)を用いた。PCR 反応は、94℃で2分間処理した後、98℃で10秒間、66℃で30秒間、68℃で10 秒間を1サイクルとし、これを30サイクル繰り返した。得られたPCR産物につ いて、アガロースゲル電気泳動を行い、目的のDNA断片が得られているかどう かを確認した後、pBluescript II SK(-)上の制限酵素EcoRVの認識サイトに挿入し た。EcoRVサイトにPCR産物を持つpBluescript II SK(-)をBglIIとBamHIによっ て 制 限 酵 素 処 理 し 、 目 的 遺 伝 子 断 片 を 回 収 し た 。 次 に 、 プ ラ ス ミ ド pLenti/Phsp/TRE/tTA/IRES/EGFP を制限酵素 BamHI を用いて切断し、目的 DNA 断 片 を 得 た 。 以 上 の 2 断 片 を 用 い て ラ イ ゲ ー シ ョ ン 反 応 を 行 い 、 pLenti/Phsp/TRE/tTAを作製した。
27
Fig. 6 Flowchart for the construction of pLenti/Phsp/TRE/tTA plasmid.
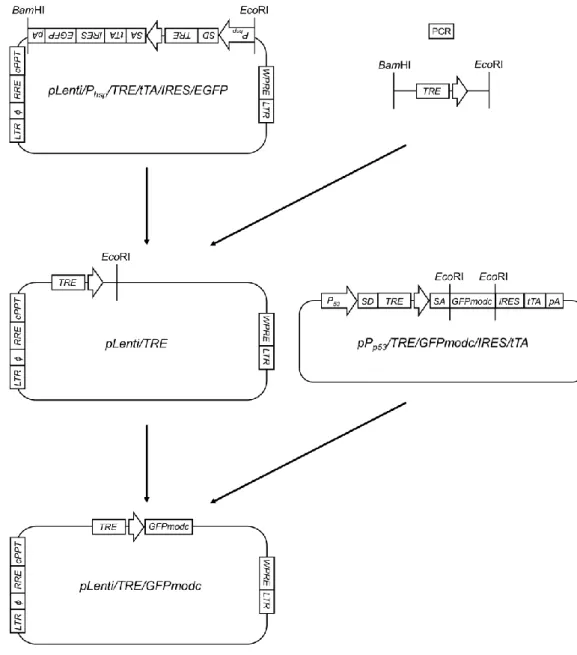
<pLenti/TRE/GFPmodcの作製>
pLenti/TRE/GFPmodcのコンストラクトとその作製手順をFig. 7に示す。まず、
TRE 遺 伝 子 配 列 を ク ロ ー ニ ン グ す る た め の プ ラ イ マ ー (Fw:5’- ttggcgcgcGGATCCGCCCTTTCGTCTTCAgTCGA-3’, Rev:5’-aagaattcGGCGATCTG ACGGTTCACTAAAC-3’)を設計し、シグマジェノシス社に合成を委託した。な お、このプライマーの各端には、それぞれ制限酵素BssHII- BamHIとEcoRIの認 識サイトを付加した。鋳型DNAとして、pTRE-tight(Clontech)を用いた。PCR 反応は、94℃で2分間処理した後、98℃で10秒間、66℃で30秒間、68℃で10 秒間を 1 サイクルとし、これを 30 サイクル繰り返した。得られた PCR 産物を
pBluescript II SK(-)上の制限酵素EcoRVの認識サイトに挿入した。EcoRVサイト
28
にPCR産物を持つpBluescript II SK(-)をBamHIとEcoRIによって制限酵素処理 し 、 目 的 遺 伝 子 断 片 を 回 収 し た 。 次 に 、 プ ラ ス ミ ド pLenti/Phsp/TRE/tTA/IRES/EGFPを制限酵素BamHIとEcoRIを用いて切断し、目 的DNA断片を回収した。以上の2断片を用いてライゲーション反応を行い、組 み 換 え 中 途 産 物 pLenti/TRE を 作 製 し た 。 次 に 、 作 製 し た pLenti/TRE、
pPp53/TRE/GFPmodc/IRES/tTAそれぞれを制限酵素EcoRIによって切断し、目的
遺伝子断片をアガロースゲル電気泳動後のバンドプレップにより回収した。
pLenti/TRE より得られた遺伝子断片を CIAP 処理した後、Mag 精製を行い、
pPp53/TRE/GFPmodc/IRES/tTA由来の DNA 断片とライゲーション反応を行い、
目的プラスミドpLenti/TRE/GFPmodcを作製した。
29
Fig. 7 Flowchart for the construction of pLenti/TRE/GFPmodc plasmid.
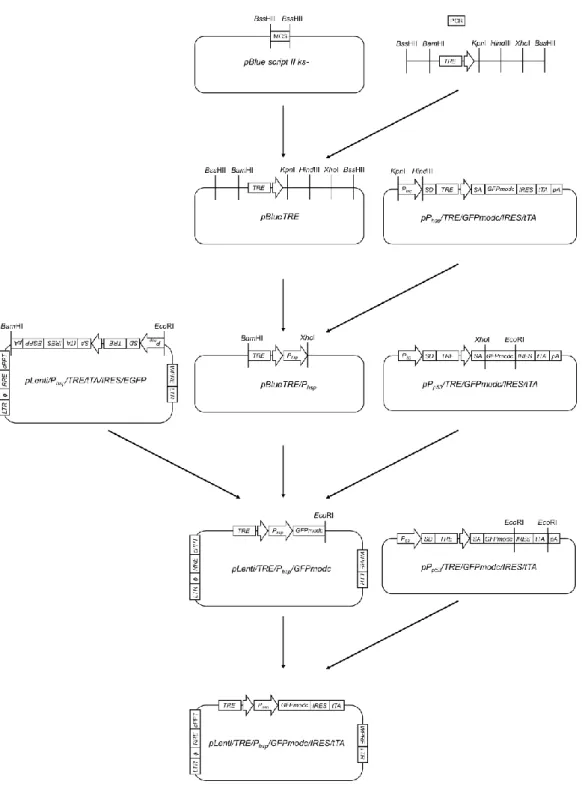
<pLenti/TRE/Phsp/GFPmodc/IRES/tTAの作製>
pLenti/TRE/Phsp/GFPmodc/IRES/tTAのコンストラクトとその作製手順をFig. 8 に示す。まず、TRE 遺伝子配列をクローニングするためのプライマー(Fw:5’- ttggcgcgcGGATCCGCCCTTTCGTCTTCAgTCGA-3’, Rv:5’-ttgGCGCGCCTCGAGA AGCTTaaGGTACCGGCGATCTGACGGTTCACTAAAC-3’)を設計し、シグマジェ
30
ノシス社に合成を委託した。なお、このプライマーの各端には、それぞれ制限酵 素BssHII - BamHIとBssHII - XhoI - HindIII - KpnIの認識サイトを付加した。鋳型 DNAとして、pTRE-tightを用いた。PCR反応は、94℃で2分間処理した後、98℃ で10秒間、66℃で30 秒間、68℃で10秒間を 1サイクルとし、これを30 サイ クル繰り返した。得られた PCR 産物を pBluescript II SK(-)上の制限酵素 EcoRV の認識サイトに挿入した。EcoRVサイトにPCR産物を持つpBluescript II SK(-)と
pBluescript II SK(-)をBssHIIによって制限酵素処理し、目的遺伝子断片を回収し
た。以上の 2 断片を用いてライゲーション反応を行い、組み換え中途産物
pBlueTRE を 作 製 し た 。 次 に 、 プ ラ ス ミ ド pBlueTRE と
pPhsp/TRE/GFPmodc/IRES/tTAを制限酵素KpnIとHindIIIを用いて切断し、目的 DNA断片を回収した。得られた2つの遺伝子断片を用いてライゲーション反応 を行い組換え中途産物 pBlueTRE/Phsp を作製した。続いて pBlueTRE/Phsp を BamHI、XhoIによって、pLenti/Phsp/TRE/tTA/IRES/EGFP をBamHI、EcoRI によ って、pPp53/TRE/GFPmodc/IRES/tTA を XhoI と EcoRIによってそれぞれ制限酵 素処理を行い得られた3つの遺伝子断片を用いて3断片ライゲーションを行い、
組 換え 中途産物 pLenti/TRE/Phsp/GFPmodc を作製し た。 最後 に 、得ら れた pLenti/TRE/Phsp/GFPmodcとpPp53/TRE/GFPmodc/IRES/tTAをそれぞれ制限酵素 EcoRI で切断し、2つの目的 DNA 断片を回収した。pLenti/TRE/Phsp/GFPmodc 由来の DNA断片については、CIAP処理した後、Mag精製を行った。以上の 2 断片を用いてライゲーション反応を行い、pLenti/TRE/Phsp/GFPmodc/IRES/tTAを 作製した。
31
Fig. 8 Flowchart for the construction of pLenti/TRE/Phsp/GFPmodc/IRES/tTA plasmid.
32
<pLenti/Pcmv/tTA/IRES/Blar>
pLenti/Pcmv/tTA/IRES/BlarのコンストラクトをFig. 9に示す。このコンストラ クトは、藤原昇氏により既に作製されたものを用いた。
Fig. 9 Construction of pLenti//Pcmv/tTA/IRES/Blar plasmid.
<pLenti/TRE/Phsp/GFPmodcの作製>
pLenti/TRE/Phsp/GFPmodcのコンストラクトとその作製手順をFig. 10に示す。
ま ず 、pLenti/Phsp/TRE/tTA/IRES/EGFP と 既 に 作 製 さ れ た プ ラ ス ミ ド
pPCMV/tTA/456zt(岡本憲明氏作製)をそれぞれ BamHI と EcoRI によって制限
酵素処理し、2つの目的遺伝子断片を回収した。得られた 2 断片を用いてライ ゲーション反応を行い、組換え中途産物 pLenti/TRE/Phsp を作製した。次に、
pLenti/TRE/Phspと pPp53/TRE/GFPmodc/IRES/tTA それぞれを EcoRI によって制 限酵素処理し、2つの目的遺伝子断片を回収した。その後pLenti/TRE/Phsp由来 のDNA断片のみCIAP処理、Mag精製を行った。これら2断片を用いてライゲ ーション反応を行うことで、pLenti/TRE/Phsp/GFPmodcを作製した。
33
Fig. 10 Flowchart for the construction of pLenti//TRE/Phsp/GFPmodc plasmid.
2.2.2.3 レポーター遺伝子の検討
本節では持続的遺伝子発現と一過性遺伝子発現の発現挙動解析を行う。その ため、レポーター遺伝子から転写・翻訳されて合成されたタンパク質の半減期が 長いと、遺伝子発現レベルでの解析に支障が出てしまう。そこで本節ではレポー ター遺伝子として通常の EGFP よりも半減期の短い GFPmodc を使用した。
34
GFPmodcはEGFP配列の終止コドンを欠損させた 3’側に不安定化配列マウスオ
ルニチンデカルボキシラーゼ配列(modc配列)を付加させることによって半減 期を短くしたものである[93]。
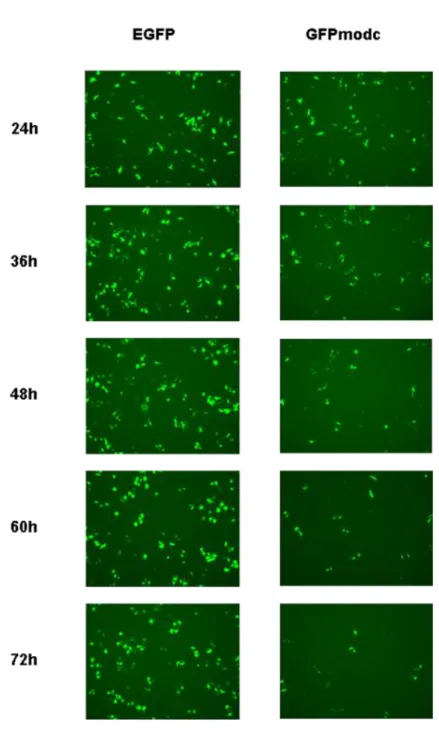
まず、予備検討としてEGFPとGFPmodcの半減期を比較した。プラスミドベ ク タ ーpPhsp/TRE/EGFP/IRES/tTA ( 山 口 雅 紀 氏 よ り 供 与 ) も し く は pPhsp/TRE/GFPmodc/IRES/tTAをLipofectamine 2000(Invitrogen)によるリポフェ クション法により、HeLa細胞に一過性で遺伝子導入した。前日に細胞を1.0×106 cells/dishで播種し、翌日に各ディッシュにプラスミド8 µgをLipofectamine 2000 を用いて遺伝子導入した。遺伝子導入から 24 時間後、遺伝子導入細胞に EGFP
もしくはGFPmodcの発現誘導を行うために、細胞に熱ストレスを与えた。熱に
よるストレス誘導はディッシュをパラフィルムで覆いディッシュごと 43℃に設 定した恒温槽に 1 時間沈めることで行った。熱ストレス処理後にトリプシン処 理により 60 mm ディッシュから剥がした細胞を 2.5×105 cells/well になるよう に 6 well plateへと播種し直した。熱ストレス後の時刻を0 hとし、24, 36, 48, 60, 72時間後の細胞の蛍光写真を撮影し、画像解析ソフトBZ-II Analyzer(Keyence) により蛍光強度を測定した。
2.2.2.4 細胞と培地の組成
ヒト子宮頸がん細胞株(HeLa細胞)、マウス繊維芽細胞株(NIH3T3細胞)の
培地は、10 % 牛胎児血清、0.1 mg/mL ストレプトマイシン、100 U/mL ペニシリ
ンを添加した基本培地DMEM(high)(Invitrogen)を、pH 7.2に調製して用いた。
35
2.2.2.5 一過性遺伝子導入における経時的な GFPmodc 遺伝子発現挙動
解析
今 回 作 製 し た プ ラ ス ミ ド ベ ク タ ー pPhsp/TRE/GFPmodc/IRES/tTA 、 pLenti/Phsp/TRE/GFPmodc/IRES/tTA、pLenti/Phsp/TRE/tTA、pLenti/TRE/GFPmodc、 pLenti/TRE/Phsp/GFPmodc/IRES/tTA 、 pLenti/Pcmv/tTA/IRES/Blar 、 pLenti/TRE/Phsp/GFPmodcをLipofectamine 2000(Invitrogen)を用いた60 mmス ケールにおけるリポフェクション法により、NIH3T3細胞に一過性で遺伝子導入 した。前日に細胞を5.0×105 cells/wellで播種し、翌日に各ディッシュに各プラス ミ ド 当 た り 5 µg を Lipofectamine 2000 を 用 い て 遺 伝 子 導 入 し た 。 pLenti/Phsp/TRE/GFPmodc/IRES/tTA と pLenti/TRE/Phsp/GFPmodc/IRES/tTA は pBluescript II SK(-)と共導入し、pLenti/Phsp/TRE/tTAはpLenti/TRE/GFPmodcと、
pLenti/Pcmv/tTA/IRES/BlarはpLenti/TRE/Phsp/GFPmodc と共導入した。遺伝子導 入から 24 時間後、遺伝子導入細胞内で GFPmodc の発現誘導を行うために、細 胞に熱ストレスを与えた。熱によるストレス誘導はディッシュ側面をパラフィ ルムで覆いディッシュごと43℃に設定した恒温槽に1時間沈めることで行った。
熱ストレスの終了時刻を0 hとし、トリプシン処理により剥がした細胞を2.0× 105 cells/wellで6 well plateに再播種した。その後、経時的な発現挙動解析のため
に24, 36, 48, 60時間後の蛍光写真を蛍光顕微鏡(Keyence)によって撮影した。
撮影した蛍光写真に対して、画像解析ソフトを用いてGFPmodcの蛍光強度を測 定することで、遺伝子発現挙動解析を行った。
36
結果と考察
2.2.3.1 レポーター遺伝子の検討
は じ め に 、EGFP と GFPmodc の 半 減 期 を 比 較 し た 。 プ ラ ス ミ ド pPhsp/TRE/EGFP/IRES/tTAもしくはpPhsp/TRE/GFPmodc/IRES/tTAを HeLa細胞 へ一過性で遺伝子導入し、熱ストレス後の時間を0 hとし、24, 36, 48, 60, 72 hに おける各レポーター遺伝子(EGFP, GFPmodc)の発現解析を行った。また、この 際の熱ストレスは、温度と時間を変化させた際のレポーター遺伝子解析におい て細胞へのダメージが少なく、かつ高く遺伝子発現が誘導された 43℃、1 時間 の条件にて行った[29]。Fig. 11はEGFP、GFPmodcそれぞれの各タイムポイント における蛍光写真である。今回の実験では、遺伝子非導入HeLa細胞の蛍光写真 における蛍光強度をバックグラウンドとして測定し、それよりも高い強度を示 す細胞を蛍光細胞として扱った。蛍光写真より、EGFPと比較して、GFPmodcの 蛍光は48時間と早い段階で消光し始めていることが示された。また、Fig. 12は 各タイムポイントでの相対蛍光強度を表したグラフである。各条件に対して3視 野×3ウェル分の写真を撮影し、蛍光写真1視野中から細胞を5つ選び、その相 対強度を測定し、平均値を算出した。Fig. 12AはDox非添加条件の結果を示し、
Fig. 12B は Dox 添加条件の結果を示している。両図とも縦軸は蛍光細胞の相対
蛍光強度を、横軸は熱ストレス処理後の経過時間を示している。Dox非添加条件 のEGFPにおいて、熱ストレス処理24時間後の蛍光強度が3.14, 72時間後の蛍 光強度が2.55と、72時間後まで著しい蛍光強度の減少は観測されなかった。こ れはポジティブフィードバックの効果によるものだと考えられる。一方 Dox 非 添加条件のGFPmodcにおいて、24時間後の蛍光強度は2.90、48時間後には2.22 と、EGFPにおける最低蛍光強度を下回り、72 時間後には 1.39と、ほとんどバ ックグラウンドの値と同等まで減少した。これらの結果から、GFPmodcはEGFP と比較して半減期が短く、遺伝子発現解析のレポーター遺伝子として適してい
37
るということが示された。この際、EGFP、GFPmodcともにポジティブフィード バック効果があるにも関わらず蛍光強度が減少しているのは、GFP タンパクの 分解と、細胞分裂によるプラスミドの希釈が原因だと考えられる。また、Dox非 添加条件と比較して Dox 添加条件の相対蛍光強度は低かった(Dox 非添加条件 では24時間後の相対蛍光強度が EGFP、GFPmodc それぞれ3.14, 2.90 であった のに対して、Dox添加条件ではそれぞれ2.21, 1.36)。この結果より、いずれのレ ポーター遺伝子においても Dox 添加による遺伝子発現増幅の抑制が起きている ということが明らかになった。また、Dox添加条件でのGFPmodcの強度は24時 間後の時点で 1.36 と著しく低い。これは、GFP と modc 配列を結合させた
GFPmodcの半減期が2 時間であり[26]、かつDox による発現量増幅の抑制が起
こっているため、一過的発現した GFPmodc は 24 時間後の時点でほとんど分解 されているからだと考えられる。
38
Fig. 11 Fluorescent images. Left; time course images of EGFP expressing HeLa cells. Right; time course images of GFPmodc expressing cells.
39
Fig. 12 Comparison of relative fluorescence intensity between EGFP and GFPmodc. Time course analysis for relative fluorescence intensity for EGFP expressing HeLa cells and GFPmodc expressing cells without (A) or with (B) Dox addition.
2.2.3.2 各遺伝子発現システムの経時的GFPmodc発現挙動解析
次に、作製した各遺伝子発現システムが熱ストレスによって応答し、どのよ うな発現挙動を示すのかを調べるため、一過性遺伝子導入条件での経時的な
GFPmodc 遺伝子発現挙動解析を行った。各遺伝子発現システムを一過性で遺伝
子導入した3T3細胞に熱ストレスを与え終えた時間を0 hとし、24, 36, 48, 60 h におけるレポーター遺伝子GFPmodcの発現解析を行った(Fig. 13)。また、この 際の熱ストレスは、43℃、1時間の条件にて行い、相対蛍光強度は、遺伝子非導
入 NIH3T3 細胞の強度を 1 として測定した。各条件に対して 3 視野分の写真を
撮影し、蛍光写真1視野中から細胞を5つ選び、その相対蛍光強度を測定し、平 均値を算出した。その結果、持続型 One-Pack, 持続型 Two-Plasmids, 一過性型
One-Pack, 一過性型Two-Plasmidsのいずれも熱ストレスを与えた24時間後には
それぞれ4.13, 3.98, 3.94, 3.71を示し、遺伝子非導入の相対蛍光強度1と比較し
てGFPmodcの相対強度が高くなった(Fig. 13)。また、持続型遺伝子発現システ
Relative fluorescence intensity Relative fluorescence intensity