トラゼンタ錠 5mg
CTD 第2部 資料概要
2.7
臨床概要
2.7.2 臨床薬理試験
2.7.2 臨床薬理の概要 ... 1 1. 背景及び概観 ... 6 1.1 薬物動態 ... 6 1.1.1 吸収および分布 ... 7 1.1.2 代謝および排泄 ... 8 1.1.3 用量比例性および累積 ... 8 1.1.4 2型糖尿病患者における薬物動態 ... 9 1.1.5 内因性要因および特別な集団 ... 9 1.1.6 外因性要因 ... 11 1.2 薬力学 ... 16 1.2.1 QT間隔に対する影響 ... 16 1.2.2 DPP-4阻害 ... 16 1.2.3 血糖およびGLP-1 ... 16 1.2.4 探索的に検討したバイオマーカー ... 17 1.3 患者における曝露-反応の評価 ... 18 2. 個々の試験結果の要約 ... 21 2.1 試験1218.1(単回-健康被験者) ... 23 2.2 試験1218.2(12日間反復-2型糖尿病患者) ... 27 2.3 試験1218.3(4週間反復-2型糖尿病患者) ... 30 2.4 試験1218.10(単回静脈内投与試験-健康被験者) ... 33 2.5 試験1218.7(14CヒトADME 静脈内および経口投与-健康被験者) ... 36 2.6 試験1218.26(腎機能障害患者) ... 38 Table of Contents
2.7 試験1218.27(肝機能障害患者) ... 45 2.8 試験1218.11(単回および12日間反復-日本人健康被験者) ... 48 2.9 試験1218.12(4週間反復-日本人2型糖尿病患者) ... 57 2.10 試験1218.23(第III相試験-日本人2型糖尿病患者) ... 62 2.11 試験1218.58(薬物動態-中国人健康被験者) ... 65 2.12 試験1218.31(DDI-リトナビル) ... 67 2.13 試験1218.67(DDI-リファンピシン) ... 71 2.14 試験1218.4(DDI-メトホルミン) ... 75 2.15 試験1218.13(DDI-ピオグリタゾン) ... 77 2.16 試験1218.30(DDI-グリブリド(グリベンクラミド)) ... 80 2.17 試験1218.9(DDI-シンバスタチン) ... 82 2.18 試験1218.28(DDI-ワルファリン) ... 84 2.19 試験1218.29(DDI-ジゴキシン) ... 86 2.20 試験1218.44(DDI-経口避妊薬) ... 88 2.21 試験1218.37(4週間薬力学) ... 90 2.22 試験1218.32(Thorough QT(TQT)) ... 93 2.23 母集団薬物動態解析 ... 97 2.24 母集団薬物動態/薬力学解析 ... 100 2.25 日本人患者における母集団薬物動態/薬力学解析 ... 103 3. 全試験を通しての結果の比較と解析 ... 105 3.1 ヒト試料を用いたin vitroデータおよび非臨床データ ... 105 3.2 健康被験者および2型糖尿病患者におけるリナグリプチンの基本的な薬物動態 ... 111 Table of Contents
3.2.1 健康被験者 ... 111 3.2.2 2型糖尿病患者 ... 111 3.2.3 要約および考察 ... 111 3.3 内因性要因および特別な集団 ... 124 3.3.1 年齢,BMI,体重および性別 ... 124 3.3.2 肝機能障害 ... 124 3.3.3 腎機能障害 ... 125 3.3.4 人種 ... 127 3.4 外因性要因 ... 129 3.4.1 薬物相互作用 ... 129 3.4.2 食事 ... 133 3.5 薬力学を検討した試験およびその結果 ... 134 3.5.1 QT間隔に対する影響 ... 134 3.5.2 DPP-4阻害 ... 135 3.5.3 インクレチン:GLP-1およびGIP ... 140 3.5.4 グルコース ... 143 3.5.5 インスリン ... 145 3.5.6 グルカゴン ... 146 3.5.7 フルクトサミン ... 147 3.5.8 1,5-アンヒドログルシトール ... 147 3.5.9 HbA1c ... 147 4 特別な試験 ... 150 Table of Contents
5. 付録 ... 151
5.1 本文中には含めないが引用する表 ... 151
5.2 本文中には含めないが引用する図 ... 214
Table of Contents
2.7.2 臨床薬理の概要
略号及び用語の定義一覧
ABC ATP binding casette
ACE Angiotensin converting
enzyme
アンジオテンシン変換酵素
1,5-AG 1,5-Anhydroglucitol 1,5-アンヒドログルシトール
ADME Absorption, Distribution,
Metabolism, Excretion
吸収,分布,代謝,排泄
ALT Alanine transaminase アラニンアミノトランスアミナーゼ
ANOVA Analysis of variance 分散分析
ANCOVA Analysis of covariance 共分散分析
AST Aspartate transaminase アスパラギン酸アミノトランスアミ
ナーゼ
AUC Area under the analyte plasma
concentration-time curve
血漿中濃度-時間曲線下面積
AUEC Area under the effect curve 効果-時間曲線下面積
AUC0-∞ Area under the analyte plasma
concentration-time curve from time point zero extrapolated to infinity
時間0 から無限大までの外挿した血
漿中濃度-時間曲線下面積
AUC0-tz Area under the
concentration-time curve of the analyte in plasma over the time interval from 0 to the last quantifiable analyte plasma concentration after single dose administration
定量可能最終時点tzまでの血漿中濃
度-時間曲線下面積
AUCτ,ss(,norm) Area under the analyte plasma concentration-time curve over a dosing interval at steady state (dose normalized)
(投与量補正した)定常状態におけ
る1 投与間隔 τ の血漿中濃度-時間曲
線下面積
BCRP Breast cancer resistance
protein
BI Boehringer Ingelheim ベーリンガーインゲルハイム社
BLQ Below limit of quantification 定量下限未満
BMI Body mass index 肥満度指数
°C Degrees Centigrade セルシウス温度
CD 1750 Racemate of linagliptin metabolite CD 1790
リナグリプチンの代謝物であるCD
1790 のラセミ体
CD 1790 Linagliptin metabolite formed
in vivo(S-enantiomer)
in vivo で生成されるリナグリプチン
代謝物(S-体)
CL Clearance クリアランス
CL/F,(ss) Apparent clearance of the
analyte in plasma following extravascular administration
(定常状態における)見かけのクリ アランス
CLR,t1_t2 Renal clearance of the analyte
from the time point t1 until the timepoint t2
時間t1から時間t2までの腎クリアラ
ンス Cmax(,norm) Maximum analyte plasma
concentration
(dose-normalized)
(投与量補正した)最高血漿中濃度
Cmax,ss(,norm) Maximum analyte plasma concentration at steady state (dose-normalized)
(投与量補正した)定常状態におけ る最高血漿中濃度
Cpre,ss Predose concentration at steady
state
定常状態における投与直前の濃度
CV Coefficient of variation 変動係数
DDI Drug-drug interaction 薬物相互作用
dL Decilitre デシリットル
DPP-4 Dipeptidyl peptidase-4 ジペプチジルペプチダーゼ-4
E24(,ss) Effect at time point 24 hours
after dosing(at steady state)
(定常状態での)投与24 時間後にお
ける効果
Eavg,ss Average effect at steady state 定常状態での平均効果
eCcr Estimated creatinine clearance 推定クレアチニンクリアランス
ECG Electrocardiogram 心電図
EE Ethinylestradiol エチニルエストラジオール
eGFR Estimated glomerular filtration
rate
推定糸球体ろ過率
Emax(,ss) Maximum effect at steady state (定常状態での)最大効果
ESRD End-stage renal-disease 末期腎障害
F Absolute bioavailability factor 絶対バイオアベイラビリティ
Fa Absorption 吸収率
FDA Food and Drug Administration 食品医薬品局(米国)
fe Fraction of drug eliminated in
urine
尿中排泄率
FPG Fasting plasma glucose 空腹時血糖
GGT Gamma-glutamyl transferase γ グルタミルトランスフェラーゼ
gCV Geometric coefficient of
variation
幾何変動係数
GFR Glomerular filtration rate 糸球体ろ過速度
GIP Glucose-dependent insulinotropic peptide
グルコース依存性インスリン分泌刺 激ペプチド
GLP-1 Glucagon-like peptide-1 グルカゴン様ペプチド-1
gMean Geometric mean 幾何平均値
h Hour 時間
HbA1c Glycosylated haemoglobin A1 糖化ヘモグロビン
(NGSP 基準で測定)
HI Hepatic impaired 肝機能障害
HV Healthy volunteer 健康被験者
HOMA Homeostasis Model
Assessment
HOMA-IS Homeostasis Model
Assessment-insulin secretion HPLC-MS/MS High-performance liquid chromatography-tandem mass spectrometry 高速液体クロマトグラフィー-タン デム質量分析法
IC50 Half maximal inhibitory
concentration
50%の阻害が得られる濃度
IC80 80% maximal inhibitory
concentration
80%の阻害が得られる濃度
iFF Intended final formulation 第III 相試験製剤
INR International normalized ratio
ISR Insulin secretion rate インスリン分泌
kg Kilogram キログラム
Ki Inhibition constant 阻害定数
L Litre リットル
LLOQ Lower limit of quantification 定量下限
ln Natural logarith 自然対数 ln2 Binary logarithm 底が2 の対数 LNG Levonorgestrel レボノルゲストレル MAO-B monoaminooxidase B モノアミンオキシダーゼB Max Maximum 最大値 MBq Megabecquerel メガベクレル MDR Multidrug resistance 多剤耐性 mg Milligram ミリグラム min Minute 分
Min Minimum 最小値 mL Milliliter ミリリットル
MRD Multiple rising dose 用量漸増反復投与
MRP Multidrug resistance-associated protein
MRT Mean residence time 平均滞留時間
ms Millisecond ミリ秒
µg Microgram マイクログラム
MTT Meal tolerance test 食事負荷試験
N Number 数
NC Not calculated 算出せず
ng Nanogram ナノグラム
nM Nanomole ナノモル
NONMEM Nonlinear mixed effect model 非線形混合効果モデル
µM Micromol マイクロモル
OATP Organic anion transporting
polypeptide
有機アニオントランスポートポリペ プチド
OCT Organic cation transporter 有機カチオントランスポーター
OGTT Oral glucose tolerance test 経口グルコース負荷試験
P-gp Permeability glycoprotein P-糖蛋白
PIB Powder in a bottle 瓶入り粉末製剤
PK Pharmacokinetics 薬物動態
p.o. Per os 経口
QTc Corrected QT interval QT 間隔の補正値
QTcB Heart rate-corrected QT
interval, using Bazzet’s method
QT 間隔の Bazett の方法を用いた心 拍数補正値
QTcF Heart rate-corrected QT
interval, using Fridericia’s method
QT 間隔の Fridericia の方法を用いた 心拍数補正値
QTcI Individually heart
rate-corrected QT interval
QT 間隔の被験者ごとの補正値
QTcN Heart rate-corrected QT
interval, using a study population-based approach
QT 間隔の試験対象集団に基づく方 法を用いた心拍数補正値
RA Accumulation ratio of the
analyte in plasma after multiple dose administration over a uniform dosing interval
一定の投与間隔で反復投与後の血漿 中における累積係数
RI Renal impaired 腎機能障害
SD Standard deviation 標準偏差
SLC Solute carrier
ss Steady state 定常状態
τ uniform dosing interval 一定の投与間隔
t1/2(,ss) Terminal elimination half-life
(at steady state)
(定常状態での)終末相における半 減期
T2DM Type 2 diabetes mellitus 2 型糖尿病
tmax(,ss) Time of maximum analyte
plasma concentration after administration(at steady state)
(定常状態における)最高血漿中濃 度到達時間
T/R Ratio test/reference Reference に対する test の比
tz(,ss) Time of last measurable
concentration of the analyte in plasma(at steady state)
(定常状態において)血漿中濃度が 測定可能な最終時間
U Units 単位
Vss Apparent volume of
distribution under steady state conditions
定常状態における見かけの分布容積
Vz/F(,ss) Apparent volume of
distribution during the terminal
phase λz following an
extravascular administration(at steady state)
(定常状態での)終末相λzにおける
見かけの分布容積
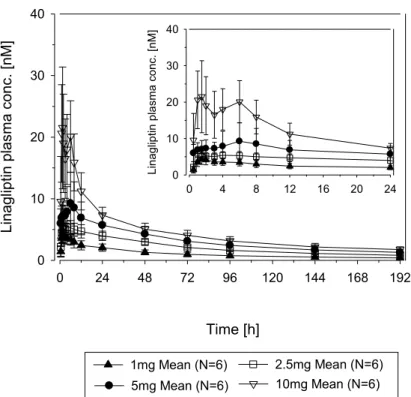
1. 背景及び概観 1.1 薬物動態 リナグリプチンは,2 型糖尿病の治療薬として開発されているジペプチジルペプチダーゼ 4 (DPP-4)阻害剤である。健康被験者および 2 型糖尿病患者におけるリナグリプチンの薬物動 態および薬力学を検討した。 リナグリプチンはDPP-4 に対して高親和性で飽和的に結合し,リナグリプチンと DPP-4 の複合 体を形成する。複合体からの解離は緩徐であるため,リナグリプチンは臨床用量の血漿中濃度 範囲で濃度依存的な蛋白結合を示し,総リナグリプチン濃度が増えると非結合型のリナグリプ チンが増加する。このためリナグリプチンは,経口投与および静脈内投与のいずれの場合にも 非線形の薬物動態を示し,臨床用量である5 mg を含む 1~10 mg の用量において,血漿中濃度 の上昇は用量比以下である。したがって薬物動態が線形な薬剤では用量に非依存的な薬物動態 パラメータ(例:クリアランス,分布容積および腎排泄率)が,リナグリプチンの場合には用 量の増加とともに増加する。薬物動態が線形および非線形な薬剤の血漿中濃度-時間曲線下面積 (AUC)と投与量の関係を以下の図 1.1: 1 に示す。リナグリプチンと同様の非線形な薬物動態 が,ACE 阻害剤であるトランドラプリルや ramipril の活性体であるトランドラプリラートや ramiprilat で報告されている[CTD 5.4-10,R05-0781;CTD 5.4-15,R07-4440]。リナグリプチン は主に未変化体のまま糞中に排泄される。臨床用量では,リナグリプチンの腎排泄は少ない。 図1.1: 1 線形および非線形な薬物動態を有する薬剤の血漿中濃度-時間曲線下面積 (AUC)と投与量の関係 非線形的薬剤 (用量比を上回る) 線形的薬剤 非線形的薬剤 (用量比を下回る) 用量
1.1.1 吸収および分布 リナグリプチン(分子量472.54 g/mol)5 mg は経口投与後速やかに吸収され,投与 1.5~2 時間 後(tmaxの中央値)に血漿中濃度がピークに達する。このことから本剤が主に腸管上部で吸収 されることが示唆される。外国人健康被験者に5 mg の単回経口投与を行った場合のリナグリ プチンのAUC0-24の幾何平均値は139 nM·h であり,最高血漿中濃度(Cmax)は8.90 nM であっ た[CTD 5.3.1.2-1,試験 1218.25]。日本人健康被験者に 5 mg の単回経口投与を行った場合のリ ナグリプチンのAUC0-24の幾何平均値は159 nM·h であり,最高血漿中濃度(Cmax)は9.00 nM であった[CTD 5.3.3.1-3,試験 1218.11]。 10 mg の経口投与後のリナグリプチンの絶対バイオアベイラビリティは約 30%である[CTD 5.3.1.1-2,試験 1218.10]。 非臨床試験[CTD 2.6.4,3.2.1.2 項および 6.1.2 項]およびリトナビルまたはリファンピシンと の薬物相互作用試験(試験1218.31[CTD 5.3.3.4-7]および試験 1218.67[CTD 5.3.3.4-9])のデ ータから,リナグリプチンの吸収にP-糖蛋白が影響することが示されている。 リナグリプチンを食後に投与すると吸収速度がわずかに低下する(tmaxの中央値が1.02 時間か ら2.99 時間へと延長し,Cmaxは約15%低下する[90%信頼区間:75.9~94.6%]が,吸収量に対 する食事の影響はみられない[AUC0-72 90%信頼区間:98.1~109.2%])[CTD 2.7.1,3.2 項参照]。 したがってリナグリプチンは食後または食間に投与することができると考えられる。 非臨床試験においてリナグリプチンと末梢組織との結合は強く,これはリナグリプチンが末梢 組織に分布するDPP-4 と結合しているためと推察された[CTD 2.6.4,4.1 項]。健康被験者に対 するリナグリプチン5 mg 単回静脈内投与後の定常状態における分布容積(Vss)は約1110 L で あり,これはヒトの全体積を大きく上回っており,リナグリプチンがヒト組織中へ広範囲に分 布することを示唆している[CTD 5.3.1.1-2,試験 1218.10]。 ヒト血漿中でのリナグリプチンの血漿蛋白結合は濃度依存的であり,リナグリプチン濃度が増 すにつれてDPP-4 との結合が飽和するため,蛋白結合率は 2 nM での 98.8%から 20 nM では 84% へと低下する。このため血漿中におけるリナグリプチンの蛋白非結合型分率は総血漿中濃度が 増すにつれて増加する。臨床用量である5 mg 経口投与後の血漿中濃度を上回る 100 nM より高 い濃度では,リナグリプチンの蛋白結合率は約70~80%で安定していた[CTD 2.6.4,4.1 項参 照]。 リナグリプチンの分布動態は,経口投与および静脈内投与のいずれの場合にも非線形である。 リナグリプチン単回経口投与後の見かけの分布容積(Vz/F)は,1 mg から 10 mg へと用量が増 加することによって約4 倍にしか上昇しない(1 mg の Vz/F:4510 L;5 mg の Vz/F:12700 L; 10 mg の Vz/F:20800 L)[CTD 5.3.3.2-1,試験 1218.2]。 血球への[14C]放射能の分布は濃度依存的であり,リナグリプチン濃度が増すにつれて上昇した。 これはリナグリプチンの血漿中DPP-4 への結合が強く,血球への移行は,血漿中 DPP-4 への結 合が飽和した後に始まるためと推察される。臨床用量の経口投与後のリナグリプチン濃度では,
リナグリプチンは主にDPP-4 に結合しており,血漿中の遊離型のリナグリプチンは少ない[CTD 2.6.4,4.1 項]。臨床用量である 5 mg の経口投与後の血球への移行は無視できる程度である(Cblood cells/Cplasma比の平均値の最大値は0.0668)[CTD 5.3.3.1-2,試験 1218.7]。 1.1.2 代謝および排泄 リナグリプチン5 mg 経口投与後の血漿中濃度は,終末相において長い半減期を示し,少なく とも2 相性に低下する。最初の速やかな血漿中濃度の低下は,主に大きな末梢コンパートメン トへの分布およびDPP-4 に結合していないリナグリプチンの速やかな排泄によるものである。 血漿中リナグリプチン濃度が血漿中DPP-4 濃度と同程度まで低下すると,長い半減期の終末相 (リナグリプチンの終末相における半減期は200 時間にまで及ぶ)が認められる。これは DPP-4 に対するリナグリプチンの結合が強く,リナグリプチンとDPP-4 の複合体の解離が緩徐である [CTD 4.3-1,P08-06359]ためである。したがって終末相における長い半減期は本剤の累積に は寄与しておらず,リナグリプチン5 mg 反復経口投与後の累積係数から求めた半減期は 11.4 時間である[CTD 5.4-7,P09-09363]。 リナグリプチンの定常状態における見かけのクリアランス(CL/F,ss)は用量依存性を示し,1 mg から10 mg にかけて用量の増加に伴って約 4 倍に上昇する(431 mL/min から 1850 mL/min へと 上昇;5 mg では 1120 mL/min)[CTD 5.3.3.2-1,試験 1218.2]。定常状態における 5 mg 経口投与 後の腎クリアランスは低かった(70.0 mL/min)[CTD 5.3.3.2-1,試験 1218.2]。 健康被験者に対する[14C]リナグリプチンの経口投与後,投与した放射能の約 90%が投与後 1 週 間以内に回収された。[14C]放射能は,投与後 96 時間以内に主に糞中(80%)に,少量が尿中(5%) に排泄された[CTD 5.3.3.1-2,試験 1218.7]。 リナグリプチンは主に未変化体のまま排泄され(静脈内および経口投与後に回収された放射能 の約76%および 90%は未変化体),リナグリプチンは代謝をあまり受けないと考えられる[CTD 5.3.2.3-6,U 1751]。リナグリプチン 5 mg 1 日 1 回反復経口投与後,CYP3A4 によって生成さ れる薬理活性をもたない代謝物CD 1790 の,定常状態での AUC のリナグリプチンに対する割 合は約13.3%であった[CTD 5.3.1.1-3,試験 1218.33]。ヒトで認められた CD 1790 以外の代謝 物は,リナグリプチンの血漿中濃度の10%未満であった[CTD 5.3.2.3-6,U -1751]。このこと からCD 1790 は主な代謝物であると考えられた。CD 1790 の生成は投与量依存的であり,リナ グリプチンの投与量が低いとリナグリプチンに対する相対曝露が低下する[CTD 5.3.1.1-3,試 験1218.33]。CD 1790 のヒト血漿蛋白結合率は 94.7%であり,濃度によらず一定の値であった [CTD 2.6.4,4.1 項]。 1.1.3 用量比例性および累積 健康被験者および患者における0.5~10 mg の単回経口投与後(試験 1218.1[CTD 5.3.3.1-1]; 試験1218.2[CTD 5.3.3.2-1];試験1218.3[CTD 5.3.5.1-1]),0.5~10 mg の単回静脈内投与後(試 験1218.10[CTD 5.3.1.1-2]),および 1~10 mg の 1 日 1 回反復経口投与後(試験 1218.2[CTD 5.3.3.2-1],試験 1218.3[CTD 5.3.5.1-1],試験 1218.33[CTD 5.3.1.1-3])のリナグリプチンの曝 露量の増加は,用量比以下であり,1 mg および 2.5 mg のリナグリプチンの 1 日 1 回投与によ
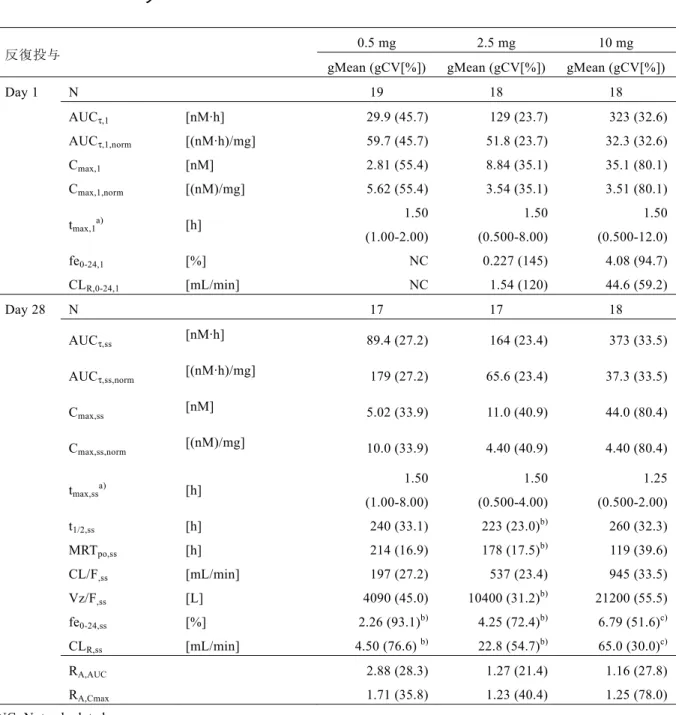
る曝露量は,臨床用量である5 mg での曝露量より 45%および 25%低下する程度であった[CTD 5.3.1.1-3,試験 1218.33]。 リナグリプチン5 mg を 1 日 1 回投与したところ,3 日目の投与までに定常状態に到達し,Cmax およびAUC の累積係数は約 1.3 であった。このため消失相における長い半減期は,リナグリプ チンの累積には寄与していないと考えられた。 リナグリプチンの薬物動態パラメータの個体間変動はほぼ低度から中程度であった(3.2.3 項参 照)。 1.1.4 2 型糖尿病患者における薬物動態 健康被験者および2 型糖尿病患者におけるリナグリプチンの薬物動態は同様であった(3.2 項 参照)。 1.1.5 内因性要因および特別な集団 年齢,体重,性別,BMI 年齢,体重,性別およびBMI の影響を母集団薬物動態解析において検討した。年齢,体重およ び性別を共変量として検討したところ,統計学的に有意な影響が認められた。これら個々の要 因によって,曝露量に−8.7%~6.8%の差が生じた。いずれの要因の影響も,±10%の範囲内であ り,臨床的に問題にはならないと考えられる。体重はBMI と相関が高いことから,体重でみら れた影響はBMI にも当てはまることが予想される。視覚的な判断から,リナグリプチンの曝露 量に対するBMI の明らかな影響は認められなかった。したがって年齢,体重,性別および BMI の内因性要因に基づく用量調節は必要ないと考えられる[CTD 5.3.3.5-1,U -1535-01]。 人種 非線形な薬物動態,終末相における半減期が長いが薬物動態的な特徴を表す半減期ではないこ と,および臨床用量ではリナグリプチンの尿中排泄率が低いことなどのリナグリプチンの薬物 動態的な特徴は,白人,日本人および中国人で同様であった。日本人および中国人における5 mg リナグリプチンの定常状態での曝露量は,白人と比較して約30%高かった(3.4.4 項参照)。し かし,5 mg リナグリプチン反復投与後のトラフ時における DPP-4 阻害率は全集団とも 80%以 上であったため,曝露量の差は臨床的に問題にならないと考えられる。日本人および白人にお いて,薬理活性をもたないリナグリプチンの代謝物であるCD 1790 の曝露量のリナグリプチン の曝露量に対する割合は同程度であった。黒人患者および非黒人患者間での曝露量の比較を行 うには例数が少なかった(黒人患者8 例に対し非黒人患者 454 例)が,探索的に行った比較の 結果に違いは認められなかった[CTD 5.3.3.5-1,U -1535-01]。
小児 小児患者を対象としたリナグリプチンの薬物動態を評価する試験は,まだ実施されていない。 腎機能障害 軽度(Cockroft-Gault 式に従って計算したクレアチニンクリアランス:>50~≤80 mL/min),中等 度(クレアチニンクリアランス:>30~≤50 mL/min),および高度(クレアチニンクリアランス: ≤30 mL/min)の腎機能障害患者,ならびに血液透析を必要とする末期腎障害(ESRD)患者に リナグリプチン5 mg を投与したところ,腎機能の低下がリナグリプチンの曝露量に大きな影 響を及ぼさないことが示された。単回経口投与後の曝露量は腎機能障害の程度によらず,いず れの腎機能障害患者群でも同程度であり,健康被験者対照群で認められた曝露量の126%~ 157%であった[CTD 5.3.3.3-1,試験 1218.26]。 定常状態での軽度腎機能障害患者における曝露量は,健康被験者と同程度であった。中等度腎 機能障害患者では,腎機能が正常な対照被験者と比較して約71%の曝露の上昇が認められたが, 薬物動態的な特徴を表す半減期の延長,終末相における半減期の延長,または累積係数の上昇 はみられなかった。中等度腎機能障害患者においてリナグリプチンのクリアランスに明確な変 化が生じたのであれば,これらは変化していたはずである[CTD 5.3.3.3-1,試験 1218.26]。 リナグリプチンの曝露に対する腎機能障害の影響を更に明らかにするために,腎機能正常(ク レアチニンクリアランス:>80 mL/min)および高度腎機能障害(クレアチニンクリアランス: ≤30 mL/min)を伴う 2 型糖尿病患者を対象として 10 日間の追加比較試験を実施した。リナグ リプチン5 mg の単回投与時の曝露は,高度腎機能障害を伴う 2 型糖尿病患者および腎機能正 常な2 型糖尿病患者間で同程度であった。高度腎機能障害を伴う 2 型糖尿病患者での定常状態 における曝露は,腎機能正常な2 型糖尿病患者と比較して約 1.4 倍上昇した。 ESRD 患者に対して単回投与のみを実施したため,定常状態の薬物動態パラメータを予測した ところ,ESRD 患者での定常状態の AUC は,中等度または高度腎機能障害患者での AUC と同
程度であると予測された。最もAUC が上昇すると考えられる ESRD 患者における定常状態で のAUC の予測値の上昇は,正常腎機能の 2 型糖尿病患者の 1.6 倍未満,および健康被験者の 1.9 倍未満と,いずれも 2 倍未満であった。 健康被験者,腎機能障害患者,腎機能正常な2 型糖尿病患者および高度腎機能障害を伴う 2 型 糖尿病患者における定常状態でのリナグリプチンの尿中排泄率は,いずれも投与量の7%未満 であった[CTD 5.3.3.3-1,試験 1218.26]。 リナグリプチンの蛋白結合率および血漿中DPP-4 濃度は腎機能の程度によらず同程度であった。
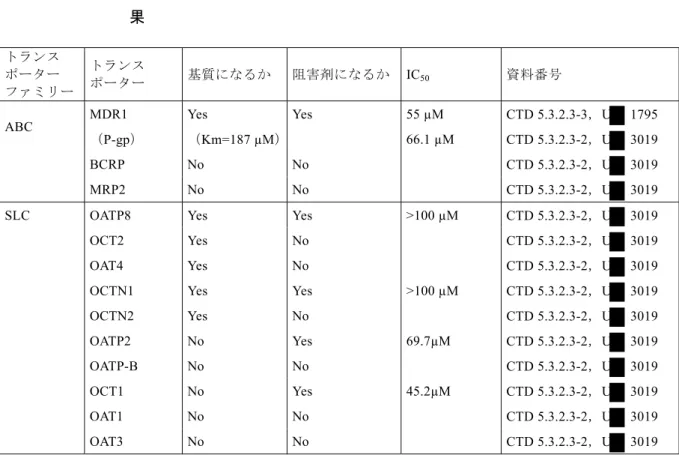
現在のFDA のガイダンス[CTD 5.4-14,R07-0393]に従い,非結合型のクリアランスが大きく, 非結合型の分布容積が大きいというリナグリプチンの薬物動態特性を考慮すると,透析により リナグリプチンが除去される可能性は低いと予測されることから,in vivo での薬物動態に対す る透析の影響は検討しなかった。 透析によって臨床的に意味のあるリナグリプチンの除去は起こらないと予想されることから, 透析の実施時期はリナグリプチンの薬物動態,安全性,有効性に影響しないと考えられる。 このほかにも試験1218.2,1218.3,1218.5,1218.6 のデータを用いた母集団薬物動態解析の結 果から,2 型糖尿病患者において軽度腎機能障害はリナグリプチンの薬物動態に影響を及ぼさ ないことが示された[CTD 5.3.3.5-1,U -1535-01]。第 III 相試験(試験 1218.16[CTD 5.3.5.1-6] および試験1218.23[CTD 5.3.5.1-9])における腎機能正常,軽度,中等度腎機能障害を有する 2 型糖尿病患者でのリナグリプチンのトラフ濃度は同程度であった。 以上より,いずれの程度の腎機能障害患者においてもリナグリプチンの用量調節は必要ないと 考えられる。 肝機能障害 軽度,中等度および高度の肝機能障害患者において,リナグリプチン5 mg の単回投与または 反復投与後のリナグリプチンのCmaxおよびAUC は,肝機能正常な対照被験者と同程度であっ た[CTD 5.3.3.3-2,試験 1218.27]。 したがって,いずれの程度の肝障害患者においてもリナグリプチンの用量調節は必要ないと考 えられる。 1.1.6 外因性要因 飲酒および喫煙 試験1218.2,1218.3,1218.5,1218.6 のデータを用いた母集団薬物動態解析の結果から,飲酒 または喫煙は,リナグリプチンの薬物動態に影響しないと考えられた[CTD 5.3.3.5-1, U -1535-01]。 薬物相互作用試験 In vitro 試験の結果から,リナグリプチンは CYP3A4 の弱~中程度の不可逆的阻害剤であり, Caco-2 細胞での P-糖蛋白を介したジゴキシン輸送に対し弱い阻害作用を示した。リナグリプチ
ンはCYP を誘導せず,CYP3A4 以外の CYP を阻害しない。
またリナグリプチンはP-糖蛋白の基質であり CYP3A4 の基質であることが in vitro で明らかに
されている(3.1 項参照)。しかしリナグリプチンは代謝が主な消失経路ではないことが示され ている(3.2.3 項参照)。
これらin vitro 試験の結果に基づき,リナグリプチンの薬物動態に対するリトナビル(CYP3A4 およびP-糖蛋白の強力な阻害剤)の影響およびリファンピシン(CYP3A4 および P-糖蛋白の誘 導剤)の影響,ならびにジゴキシン(P-糖蛋白の基質)およびシンバスタチン(CYP3A4 の基 質)に対するリナグリプチンの影響を検討した。 in vitro データに基づく相互作用試験のほかにも,FDA のドラフトガイドライン[CTD 5.4-19, R08-2669]の記載に従って,2 型糖尿病患者で一般的に併用される薬剤として,メトホルミン, ピオグリタゾン,グリブリド(グリベンクラミド),ワルファリンおよびエチニルエストラジオ ール-レボノルゲストレル配合剤との薬物相互作用試験を実施した。 リナグリプチンに対する他の薬剤の影響 リトナビル [CTD 5.3.3.4-7,試験 1218.31] P-糖蛋白および CYP3A4 の強力な阻害剤であるリトナビルの 200 mg 反復経口時にリナグリプ チン5 mg を単回併用投与すると,リナグリプチンの AUC および Cmaxはそれぞれ約2 倍および 3 倍に上昇した(幾何平均値の比[90%信頼区間]:AUC:201.4%[185.8~218.3%];Cmax:295.7% [252.0~347.0%])。 また,コンパートメントモデルに基づいてリトナビル併用投与時のリナグリプチンの定常状態 での曝露量を予測した結果,リトナビルを併用投与した場合のリナグリプチンの累積係数は低 く,AUCτ,ssは2 倍に上昇すると予測された。この影響はバイオアベイラビリティの上昇によっ て最も良く説明でき,これはP-糖蛋白の阻害に起因する可能性が最も高いと考えられた。 リナグリプチンの安全域は広く[CTD 2.6.6 参照],P-糖蛋白または CYP3A4 阻害剤の併用投与 による安全性プロファイルへの影響はないという結果が臨床試験から得られており[CTD 2.7.4, 5 項参照],リナグリプチンの曝露量の上昇は臨床的に問題にならないと考えられた。 リトナビルは最も強力なP-糖蛋白および CYP3A4 阻害剤のひとつであることから,本試験は P-糖蛋白および CYP3A4 が最大限に阻害された場合の結果であり,阻害作用のより弱いまたは 同程度の他のP-糖蛋白および CYP3A4 の阻害剤と併用した場合,臨床的に問題となる相互作用 はないと予想される。 リファンピシン [CTD 5.3.3.4-9,試験 1218.67] P-糖蛋白および CYP3A4 の誘導剤であるリファンピシンとリナグリプチンとの反復併用投与に よって,リナグリプチンの定常状態でのAUC および Cmaxは40%および 44%低下し(90%信頼 区間:AUCτ,ss 55.7~65.7%;Cmax,ss 47.8~66.0%),トラフ時の DPP-4 阻害率は約 35%低下した。 この影響はリファンピシンによるP-糖蛋白に対する誘導によると考えられた。リナグリプチン 5 mg とリファンピシンを併用投与した時と同程度の曝露量および DPP-4 阻害率が得られるリ ナグリプチン1 mg 投与時において,HbA1c は統計学的に有意に低下することが第 IIb 相試験の 結果から得られている[CTD 5.3.5.1-3,試験 1218.6]。したがって,リナグリプチンは強力な
P-糖蛋白誘導剤の併用時にも臨床的に有効であると予想されるが,最大の効果は得られない可 能性があると考えられる。 またリナグリプチンとリファンピシンの併用投与により,CYP3A4 で生成する代謝物の曝露は CYP3A4 の誘導により上昇するという予想に反し,CYP3A4 で生成する代謝物である CD 1790 のリナグリプチンに対する相対曝露が低下した。CD 1790 が投与量依存的に生成することを考 慮すると,リファンピシンの併用投与によるCD 1790 のリナグリプチンに対する相対曝露の低 下はP-糖蛋白の誘導および全身バイオアベイラビリティの低下による可能性が高いと考えら れる(3.5.1 項参照)。 グリブリド(グリベンクラミド) [CTD 5.3.3.4-6,試験 1218.30] グリベンクラミド1.75 mg 単回併用投与によるリナグリプチンの定常状態での薬物動態への影 響はみられなかった。 メトホルミン [CTD 5.3.3.4-1,試験 1218.4] 有機カチオントランスポーター(OCT)の基質であるメトホルミン[CTD 5.4-40,R10-2435; CTD 5.4-31,R09-6370]の 850 mg 1 日 3 回とリナグリプチン 10 mg 1 日 1 回の反復併用投与に より,リナグリプチンの定常状態でのAUC が 20%上昇したが(90%信頼区間:107.3~134.1%), リナグリプチンのCmaxに対する影響はみられなかった。これらの結果は母集団薬物動態解析の 結果とも一致しており,母集団薬物動態解析モデルからも同様にメトホルミンの併用時に 19.8%のリナグリプチン曝露の上昇が示された[CTD 5.3.3.5-1,U -1535-01]。リナグリプチン は安全域が広く,臨床試験でメトホルミン併用時の安全性データが得られている[CTD 2.7.4, 5 項]ことから,メトホルミン併用によるリナグリプチンの AUC の上昇に臨床的に意味のある 影響はないと考えられる。 ピオグリタゾン [CTD 5.3.3.4-3,試験 1218.13] リナグリプチン10 mg 1 日 1 回とピオグリタゾン 45 mg 1 日 1 回の反復併用投与時にリナグリ プチンのAUC および Cmaxの90%信頼区間はいずれも生物学的同等性の基準内(幾何平均値の 比[90%信頼区間]:AUCτ,ss 113.4%[103.0~124.9%];Cmax,ss 107.3%[92.3~124.8%])であり, リナグリプチンの薬物動態に対する影響はみられなかった。 P-糖蛋白/CYP3A4によるリナグリプチンの薬物動態への影響に関する結論 結論として,強力なP-糖蛋白および CYP3A4 の阻害剤であるリトナビルおよび P-糖蛋白および CYP3A4 誘導剤であるリファンピシンは,いずれもリナグリプチンの曝露に影響を与えた[CTD 5.3.3.4-7,試験 1218.31;CTD 5.3.3.4-9,試験 1218.67]。母集団薬物動態解析により,リトナビ ル併用投与時のリナグリプチンの曝露の上昇はバイオアベイラビリティの上昇により起こる可
能性が高いことが示された。一方でリナグリプチンの排泄に対するリトナビルの影響はほとん どなかった。 またリナグリプチンとリファンピシンの併用投与により,CYP3A4 で生成する代謝物の曝露は CYP3A4 の誘導により上昇するという予想に反し,CYP3A4 で生成する代謝物である CD 1790 のリナグリプチンに対する相対曝露が低下した。CD 1790 が投与量依存的に生成することを考 慮すると,リファンピシンの併用投与によるCD 1790 のリナグリプチンに対する相対曝露の低 下はP-糖蛋白の誘導および全身バイオアベイラビリティの低下による可能性が高いと考えら れる(3.5.1 項参照)。 この結果は,[14C]リナグリプチンを用いた ADME 試験(試験 1218.7)で示されたように,リナ グリプチンは代謝をあまり受けないという結果とも一致する[CTD 5.3.3.1-2,試験 1218.7]。 リトナビルおよびリファンピシンでみられたリナグリプチンの曝露に対する影響は,P-糖蛋白 の強い阻害剤および誘導剤を併用した場合のみで生じるものと予想される。 その他の薬剤に対するリナグリプチンの影響 シンバスタチン [CTD 5.3.3.4-2,試験 1218.9] 健康被験者におけるCYP3A4 の基質であるシンバスタチンの薬物動態に対するリナグリプチン 反復併用投与の影響はごくわずかであった。シンバスタチン40 mg とリナグリプチン 10 mg を 6 日間反復併用投与すると,シンバスタチンの AUC は 34%上昇し,Cmaxは10%上昇した。した がってin vivo でのリナグリプチンによる CYP3A4 阻害は弱いと考えられ,リナグリプチンと併 用されるCYP3A4 で代謝される薬剤の用量調節は不要と考えられる。なお本試験は申請するリ ナグリプチンの臨床用量である5 mg よりも高用量の 10 mg で実施されている。5 mg 投与時か ら10 mg 投与時にかけて非結合型のリナグリプチン濃度が用量比を上回って上昇することを考 慮すると,シンバスタチンの薬物動態に対するリナグリプチン5 mg 併用の影響は小さいと予 想される。 ジゴキシン [CTD 5.3.3.4-5,試験 1218.29] ジゴキシン0.25 mg とリナグリプチン 5 mg を 6 日間反復併用投与した結果,ジゴキシンの薬物 動態に対するリナグリプチンの影響はみられなかった。したがってリナグリプチンは,in vivo でP-糖蛋白またはジゴキシンの薬物動態に関与する他のトランスポーターを阻害しないと考 えられる。 メトホルミン [CTD 5.3.3.4-1,試験 1218.4] リナグリプチンの併用投与によるメトホルミンの曝露に対する臨床的に意味のある影響はみら れなかった。リナグリプチンの併用投与により,メトホルミンの定常状態でのAUC は影響を
受けず,定常状態でのCmaxの低下は11%であった(90%信頼区間:78.2~100.4%)ことから,
リナグリプチンはin vivo において臨床的に意味のある OCT の阻害剤ではないと考えられた。
ピオグリタゾン [CTD 5.3.3.4-3,試験 1218.13]
CYP2C8 および CYP3A4 の基質であるピオグリタゾン 45 mg 1 日 1 回とリナグリプチン 5 mg 1
日1 回の反復併用投与による,ピオグリタゾンの定常状態での AUC やピオグリタゾンの活性
型代謝物であるM III および M IV の定常状態での AUC および Cmaxへの影響はなく,リナグリ
プチンはCYP2C8 を阻害しないこと,ならびにリナグリプチンによる CYP3A4 の阻害はほとん どないことが裏付けられた。ピオグリタゾンのCmax,ssは14%低下(90%信頼区間:78.1~93.8%) したが,ピオグリタゾンのように長期投与を行う薬剤では有効性はCmaxよりもAUC に関連す ると推察され,本試験でみられた併用投与によるCmaxの低下に臨床的に意味のある影響はない ものと考えられる。 ワルファリン [CTD 5.3.3.4-4,試験 1218.28] リナグリプチン5 mg の併用投与によって,CYP2C9 の基質である S(−)および R(+)ワルフ ァリンの薬物動態への影響はみられず,リナグリプチンがin vivo において CYP2C9 の阻害剤で ないことが示された。 グリブリド(グリベンクラミド) [CTD 5.3.3.4-6,試験 1218.30] 主にCYP2C9 で代謝されるグリベンクラミドの 1.75 mg 単回投与とリナグリプチン経口投与の 併用投与によって,グリベンクラミドのAUC および Cmaxがいずれも14%低下した(90%信頼 区間:AUC 79.8~92.1%,Cmax 79.6~93.3%)。グリベンクラミドを 1 年間投与した 2 型糖尿病 患者での試験結果から投与量と血清中薬物濃度間に明確な相関がみられなかったこと[CTD 5.4-35,R10-2430],およびグリベンクラミドの曝露の低下により低血糖のリスクは増大しない と考えられることから,リナグリプチンの併用によるグリベンクラミドの曝露の低下は臨床的 に問題になるものではないと考えられる。グリベンクラミドと同様に主にCYP2C9 で代謝され る他のスルホニル尿素系の薬剤(例:グリピジド,トルブタミド,グリメピリド)についても, リナグリプチンの併用による臨床的に意味のある影響はないものと予想される。 エチニルエストラジオール-レボノルゲストレル配合剤 [CTD 5.3.3.4-8,試験 1218.44] リナグリプチン5 mg 1 日 1 回反復併用投与を行っても,エチニルエストラジオールまたはレボ ノルゲストレルの定常状態の曝露に影響はみられなかった。 In vitro データからは,CYP450 またはトランスポーターを介するリナグリプチンとの相互作用 のリスクは示唆されていない(3.1 項参照)。さらに,臨床試験において,リナグリプチンの併 用により,メトホルミン,グリベンクラミド,ピオグリタゾン,ワルファリン,シンバスタチ
ン,ジゴキシンおよび経口避妊薬の薬物動態に対する臨床的に問題になる影響がみられなかっ たことから,リナグリプチンの併用によりCYP3A4,CYP2C9,CYP2C8,P-糖蛋白および OCT の基質との薬物相互作用を引き起こす可能性は低いと考えられた。 1.2 薬力学 以下に,Thorough QT 試験(試験 1218.32)の結果および主に投与期間が 4 週間以下の臨床試験 におけるバイオマーカーの結果について記載する。実施期間が4 週間を上回る主な臨床試験に おけるバイオマーカーの結果については,CTD 2.7.3,臨床的有効性の概要に記載されている。 1.2.1 QT 間隔に対する影響 Thorough QT 試験である試験 1218.32[CTD 5.3.4.1-1]でリナグリプチンによる心臓への影響を 検討した。臨床用量である5 mg の単回投与または臨床用量を上回る用量である 100 mg の単回 投与を行った健康被験者において,心拍数で補正したQT 間隔のベースラインからの変化量の 両側90%信頼区間の上限はいずれの用量レベルにおいても全時点で 10 ms 未満であったことか ら,リナグリプチンは臨床的に問題となるQT 間隔の延長を起こさないと考えられた。さらに, 心拍数,補正なしのQT 間隔またはいずれの方法の心拍数で補正した QT 間隔にも臨床的に問 題となる変化はなかった。リナグリプチンおよびCD 1790 の最高血漿中濃度到達時間付近でも 心電図を測定しており,リナグリプチン投与時のQT 間隔に対する影響の検出感度は十分であ ると考えられた。試験1218.32 の結果は,単回投与試験である試験 1218.1[CTD 5.3.3.1-1]お よび反復投与試験である試験1218.2[CTD 5.3.3.2-1]で探索的に検討した結果を裏付けるもの であった。 1.2.2 DPP-4 阻害 リナグリプチン投与後,血漿中DPP-4 は速やかに阻害され,阻害は強力かつ長時間持続した。 リナグリプチンの単回投与後の最大DPP-4 阻害率は,2.5 mg で 72%,5 mg で 88.5%,25 mg 以 上で95%を上回っていた[CTD 5.3.3.1-1,試験 1218.1]。5 mg または 10 mg のリナグリプチン を1 日 1 回反復投与したとき,定常状態での血漿中 DPP-4 阻害率は投与後 24 時間を通して 80% を上回っていた[CTD 5.3.3.2-1,試験 1218.2;CTD 5.3.5.1-1,試験 1218.3]が,投与量が 2.5 mg 以下の場合,大部分の患者で投与24 時間後の阻害率が 80%に到達することはなかった。 日本人患者においてリナグリプチン5 mg 投与によりトラフ時に 80%を上回る DPP-4 阻害率が みられ,白人および日本人の健康被験者も同様であった(表3.5.2: 1 および 2 参照)。 1.2.3 血糖およびGLP-1 2 型糖尿病患者におけるインクレチンおよび血糖値に対するリナグリプチンの影響を検討した [CTD 5.3.4.2-1,試験 1218.37]。リナグリプチン 5 mg 28 日間投与によって,プラセボと比較 して食事負荷試験(MTT)後のグルカゴン様ペプチド-1(GLP-1)の AUEC0-2は18.1 pmol·h/L 上昇し,この上昇は統計学的に有意であった(p<0.0001)。MTT 後の血糖の加重平均値および
食後血糖のAUEC0-3は,それぞれ−19.9 mg/dL および−106.5 mg·h/dL 低下し,統計学的に有意で あった(p<0.0001)。 また,リナグリプチン28 日間投与によって,プラセボと比較して空腹時血糖値は−10.8 mg/dL 低下した[CTD 5.3.4.2-1,試験 1218.37]。 1.2.4 探索的に検討したバイオマーカー このほかにもグルコースの恒常性に対する影響を検討するために,バイオマーカーとして,グ ルカゴン,C-ペプチド,インスリン,フルクトサミン,1,5-アンヒドログルシトール(1,5-AG) およびHbA1c を測定した。それぞれのマーカーで効果がみられるまでの期間は,グルカゴン, インスリンおよびC-ペプチドについては投与数日後,フルクトサミンおよび 1,5-AG について 2~4 週間であり,HbA1c については 12 週間以上を要すると考えられている。したがって,以 下の記載では,グルカゴン,C-ペプチドおよびインスリンを「短期のバイオマーカー」,フルク トサミンおよび1,5-AG を「中期のバイオマーカー」,HbA1c を「長期のバイオマーカー」と分 類する。 短期のバイオマーカー リナグリプチンがDPP-4 を阻害することから予想されるように,リナグリプチンの反復投与に よって,空腹時および食後のインスリンおよびC-ペプチド濃度は上昇した。 5 mg リナグリプチン投与によってインスリン分泌は増加した。このことはベースラインからの HOMA-IS がプラセボと比較して 24.2%上昇したこと,および 28 日間投与後のグルコースの曝 露量に対するインスリンの曝露量の変化がプラセボと比較して28.7 pM/(mg/dL·h)上昇し,統計 学的に有意(p=0.0004)であることから裏付けられた。 また,リナグリプチン4 週間投与後のグルカゴンのピーク濃度も,プラセボと比較して 16.8 pg/mL 低下した(95%信頼区間:−28.7~−4.9;p=0.0064)[CTD 5.3.4.2-1,試験 1218.37]。 中期のバイオマーカー 2~3 週間の血糖コントロールを反映するマーカーとして,フルクトサミンおよび 1,5-AG を測 定した。フルクトサミンはグルコースと蛋白質の反応によって生成されるが,1,5-AG は食物に 含まれる天然の単糖類であり,血液および組織中に一定量存在している。1,5-AG は腎臓でろ過 されるが,グルコーストランスポーターによってほぼ完全に再吸収される。したがって血糖値 が高値になるとグルコースと1,5-AG の再吸収が競合し,1,5-AG の再吸収が抑制されるため, 高血糖時には1,5-AG の血中濃度が低下する[CTD 5.4-42,R10-2438]。 血糖コントロールに対する中期の効果は,フルクトサミンおよび1,5-AG に対する変化で評価 できる。 リナグリプチン5 mg を 4 週間投与すると,プラセボと比較して 1,5-AG は 1.8 µg/mL 上昇し, これは統計学的に有意(p<0.0001)であった。リナグリプチンの投与により,グルコース濃度 の変動が低下することが示唆された[CTD 5.3.4.2-1,試験 1218.37]。リナグリプチン投与 4 週
間後の濃度は,グルコースが180 mg/dL を上回ることがない場合の 1,5-AG の濃度と報告されて いる10 µg/mL であった[CTD 5.4-41,R10-2437;CTD 5.4-42,R10-2438]。 長期のバイオマーカー HbA1c は,長期間の平均血糖値を確認するのに最も多く用いられるヘモグロビンの非酵素的な 不可逆的糖化産物である。したがってHbA1c は,赤血球が曝露された平均グルコース濃度を反 映する。赤血球の平均寿命は約120 日間なので,HbA1c は 3 カ月間までの平均血糖濃度を反映 すると考えられる。 HbA1c は投与期間が 4 週間の臨床試験においても測定した[CTD 5.3.5.1-1,試験 1218.3;CTD 5.3.5.1-4,試験 1218.12;CTD 5.3.4.2-1,試験 1218.37]。投与期間が 4 週間と短期間であったも のの,リナグリプチンの投与により,HbA1c はプラセボに比べて最大 0.48%低下し,統計学的 に有意であった[CTD 5.3.5.1-4,試験 1218.12]。白人患者および日本人患者とも HbA1c の低下 の程度は同程度であった。投与期間が4 週間を超える試験では,HbA1c は主要評価項目として 測定された。(試験1218.15~18[CTD 5.3.5.1-5~5.3.5.1-8]および試験 1218.23[CTD 5.3.5.1-9])。 これらの試験でリナグリプチンは,プラセボに対しHbA1c を統計学的に有意に低下させた(詳 細はCTD 2.7.3 を参照)。 1.3 患者における曝露-反応の評価 図1.3: 1 に示すように,リナグリプチンの血漿中濃度は血漿中 DPP-4 阻害率と良好に相関した。
Linagliptin plasma conc. [nmol/L]
0.1
1
10
100
DPP-4 i
nhi
bi
tion [%
]
0
20
40
60
80
100
120
Cmax,ss Cpre,ss 破線の横線はDPP-4 阻害率 50%および 80%を示し,実線の縦線は試験 1218.2 における Cpre,ssおよびCmax,ssの幾 何平均値を示す引用元:CTD 5.3.3.2-1,試験 1218.2,Table 15.5.1.1: 7 to 10 and Table 15.6.1: 18 to 21 より作成
図1.3: 1 血漿中DPP-4 阻害率とリナグリプチン血漿中濃度の関係(試験 1218.2) 試験1218.2,1218.3,1218.5,1218.6 のデータを用いた母集団薬物動態解析の結果から,DPP-4 を50%および 80%阻害するには,それぞれ 2.97 nM および 5.30 nM のリナグリプチンの血漿中 濃度が必要と考えられた[CTD 5.3.3.5-2,U -1554-01]。 メトホルミンの併用投与,腎機能障害または肝機能障害による,定常状態DPP-4 阻害率に対す る大きな違いはみられなかった(3.7.2 項参照)。母集団薬物動態/薬力学解析において,年齢, 体重,BMI,性別,アラニンアミノトランスフェラーゼ(ALT),アスパラギン酸アミノトラン スフェラーゼ(AST),空腹時血糖(FPG),飲酒,喫煙およびクレアチニンクリアランスを含 む様々な共変量による影響を検討した。共変量のうち,統計学的に有意であったいずれの共変 量も,50%または 80% DPP-4 阻害を生じる濃度に対して±20%を超える変化を与えないことか ら,いずれの共変量による影響も臨床的に問題にならないと考えられた[CTD 5.3.3.5-2, U -1554-01]。 DP P-4 阻害率[ % ] リナグリプチン血漿中濃度[nM]
リナグリプチン以外のDPP-4 阻害剤を用いた非臨床試験から,80%以上の DPP-4 阻害が 24 時 間持続することで,最大のインクレチン効果および血糖低下が得られることが報告されている ([CTD 5.4-29,R09-6021;CTD 5.4-25,R09-4256]および 2.24 項参照)。大部分の試験で,リ ナグリプチン5 mg の反復投与後のトラフ時に DPP-4 活性は 80%阻害されたが,投与量が 2.5 mg ではDPP-4 阻害率は 80%未満であった。試験 1218.2,1218.3,1218.5,1218.6 のデータから, 2.5 mg および 5 mg のリナグリプチンの投与によって,それぞれトラフ時の DPP-4 阻害率が 77.0%および 84.8%となることが示された[CTD 5.3.3.5-2,U -1554-01]。5 mg より投与量を増 やしても,トラフ時のDPP-4 阻害率の中央値が大きく上昇することはなかった。したがって DPP-4 阻害率に基づくと,臨床用量としては 5 mg が適切であると考えられた。リナグリプチン 1 mg,5 mg および 10 mg を 12 週間投与した試験 1218.6[CTD 5.3.5.1-3]の結果,HbA1c が 10 mg 投与によって 5 mg 投与よりさらに低下することはなかった。また,日本人を対象にリナグ リプチン5 mg および 10 mg を 12 週間投与した試験 1218.23[CTD 5.3.5.1-9]においても,5 mg および10 mg 投与時の HbA1c の低下および DPP-4 阻害は同程度であった。4 週間を上回る臨床 試験の主要評価項目の結果については,CTD 2.7.3,臨床的有効性の概要に記載されている。
2. 個々の試験結果の要約 本項の個々の試験結果の要約には,CTD 2.7.2 に関連する以下のリナグリプチンの臨床試験の 主に薬物動態と薬力学の結果について記載した。これらの試験の被験者背景および安全性に関 する結果についてはCTD 2.7.4 に記載されている。 基本的な薬物動態を検討した試験: 試験1218.1(単回-健康被験者[CTD 5.3.3.1-1]), 試験1218.2(12 日間反復-2 型糖尿病患者[CTD 5.3.3.2-1]), 試験1218.3(4 週間反復-2 型糖尿病患者[CTD 5.3.5.1-1]), 試験1218.10(単回静脈内投与試験-健康被験者[CTD 5.3.1.1-2]), 試験1218.7(14C ヒト ADME 静脈内および経口投与-健康被験者[CTD 5.3.3.1-2]), 試験1218.58(薬物動態-中国人健康被験者[CTD 5.3.3.1-4]) 特別な集団における試験: 試験1218.26(腎機能障害患者[CTD 5.3.3.3-1]), 試験1218.27(肝機能障害患者[CTD 5.3.3.3-2]) 日本人を対象とした試験 試験1218.11(単回および 12 日間反復-日本人健康被験者[CTD 5.3.3.1-3]), 試験1218.12(4 週間反復-日本人 2 型糖尿病患者[CTD 5.3.5.1-4]), 試験1218.23(第 III 相試験-日本人 2 型糖尿病患者[CTD 5.3.5.1-9]) 薬物相互作用(DDI)試験: 試験1218.31(DDI-リトナビル[CTD 5.3.3.4-7]), 試験1218.67(DDI-リファンピシン[CTD 5.3.3.4-9]), 試験1218.4(DDI-メトホルミン[CTD 5.3.3.4-1]), 試験1218.13(DDI-ピオグリタゾン[CTD 5.3.3.4-3]), 試験1218.30(DDI–グリブリド(グリベンクラミド)[CTD 5.3.3.4-6]), 試験1218.9(DDI-シンバスタチン[CTD 5.3.3.4-2]), 試験1218.28(DDI-ワルファリン[CTD 5.3.3.4-4]), 試験1218.29(DDI-ジゴキシン[CTD 5.3.3.4-5]), 試験1218.44(DDI-経口避妊薬[CTD 5.3.3.4-8]) 薬力学試験: 試験1218.37(4 週間薬力学[CTD 5.3.4.2-1]), 試験1218.32(Thorough QT[CTD 5.3.4.1-1]) さらに,母集団薬物動態解析の結果[CTD 5.3.3.5-1,U -1535-01;CTD 5.3.3.5-2,U -1554-01; CTD 5.3.3.5-3,U -1849-01]についても記載した。 バイオアベイラビリティ関連および食事の影響試験の結果はCTD 2.7.1,生物薬剤学及び関連 する分析法の概要に記載した。また,CTD 2.7.1 には用量比例性検討試験である試験 1218.33 [CTD 5.3.1.1-3]の結果も記載した。ただし,本試験は市販予定製剤を用いて行われたので, 本試験の結果はCTD 2.7.2 でも参照する。
全臨床試験において,リナグリプチンおよびその代謝物CD 1790(薬理活性はない)の血漿中 および尿中濃度は,内標準物質に[13C3]リナグリプチンおよび[13C3]CD 1750 を使用し,バリデ ートされた高速液体クロマトグラフィー-タンデム質量分析法(HPLC-MS/MS)を用いて測定さ れた[CTD 2.7.1,1.4.1.1 項参照]。なお CD 1790 の定量には,検量線にラセミ体の CD 1750 を 用いた。ただし,検討を行ったすべての動物種およびヒトでは,S-体である CD 1790 のみの生 成がみとめられた[CTD 5.3.2.3-5,U -2471;CTD 5.3.1.4-9,U -1831-01]。したがって,臨 床試験の報告書中で,リナグリプチンの主な代謝物は"CD 1750"と記載されている場合があるが, 本CTD では"CD 1790"と統一して記載した。 また,開発の初期段階ではリナグリプチン濃度をng/mL 単位で測定したが,その後はモル単位 を使用した。本CTD では変換係数 2.116 を使用して ng/mL 単位をモル単位に変換した(リナグ リプチンの分子量:472.54 g/mol)値に統一して記載した。適宜,元の単位の値を括弧内に示し た。 さらに,開発の初期段階では,報告書ではジペプチジルペプチダーゼ4 の略語として DPP-4 で はなくDPP-IV を使用していたが,本 CTD では略語を DPP-4 に統一して記載した。 また,開発の初期段階の報告書ではDPP-4 活性[%]を算出したが,その後は DPP-4 阻害率[%] を用いた。DPP-4 活性[%]および DPP-4 阻害率[%]は以下のように算出した。 DPP-4 活性[%]=100×(薬剤投与後のある時点での DPP-4 活性[RFU]/ベースラインの DPP-4 活性[RFU]) DPP-4 阻害率[%]=100 − 100 ×(薬剤投与後のある時点の DPP-4 活性[RFU]/ベースライン のDPP-4 活性[RFU]) 定常状態での投与24 時間後の DPP-4 阻害率を E24,ss,定常状態での平均DPP-4 阻害率を Eavg,ss として算出した。 本CTD では DPP-4 阻害率に記載を統一するために,報告書で DPP-4 活性[%]が算出されて いる場合は,次の式を使用して変換を行った: DPP-4 阻害率[%]=100 − DPP 活性[%] ただし添付資料の表(CTD 2.7.2.5)では,適宜括弧中に元の値を示す。 またDPP-4 阻害率の記述統計量として,個々の報告書では平均値または中央値のいずれかを用 いたが,CTD 中では中央値を用いた。DPP-4 阻害率の値は非対称に分布するので,DPP-4 阻害 率の記述統計量として中央値は適切であると考えられる。 本CTD 中では各パラメータの数値は有効数字 3 桁で示す。幾何平均値の比およびその 90%信 頼区間は小数点以下1 桁まで示した。ただし CTD 2.7.2 の 5.1 項の表には,各報告書に示され ている元の値を示した。
2.1 試験1218.1(単回-健康被験者) 外国人健康男性被験者に対して液剤としてリナグリプチン2.5 mg,5 mg および 100 mg,なら びに錠剤としてリナグリプチン25~600 mg を用量漸増単回経口投与後の安全性,忍容性,薬 物動態および薬力学検討するための,ランダム化,二重盲検,プラセボ対照試験(錠剤および 液剤としてリナグリプチン100 mg 投与時の被験者内バイオアベイラビリティ比較を含む)。 参照先: 試験 1218.1[CTD 5.3.3.1-1] 目的: 用量漸増単回経口投与後のリナグリプチンの安全性,忍容性,薬物動態および薬力学を検討す ること。 方法: 健康男性被験者を対象として,単施設で,ランダム化,用量群内二重盲検,プラセボ対照,用 量漸増単回投与試験を実施した。100 mg の用量で,錠剤および液剤の被験者内バイオアベイラ ビリティ比較を行った。リナグリプチンは2.5 mg,5 mg および 100 mg を液剤として,25 mg, 50 mg,100 mg,200 mg,400 mg,600 mg を錠剤として単回投与した。各投与群は 8 例(リナ グリプチン投与6 例,プラセボ投与 2 例)で実施し,100 mg のバイオアベイラビリティ比較群 のみ10 例(リナグリプチン投与 8 例,プラセボ投与 2 例)で実施した。 投与後最大192 時間までリナグリプチン濃度および投与後 96 時間まで DPP-4 活性の測定用に 採血を行った。投与後120 時間にわたってリナグリプチン濃度測定用に尿を採取した。回帰モ デルを使用してリナグリプチンの用量比例性の探索的検討を行った。100 mg の用量で錠剤およ
び液剤投与後のAUC0-∞およびCmaxの被験者内比に対して分散分析(ANOVA)を行い,100 mg
のリナグリプチンの錠剤投与時に対する液剤投与時の相対バイオアベイラビリティを検討した。 その他の薬物動態および薬力学パラメータについて記述統計量を算出した。 結果: 薬物動態:リナグリプチン投与後0.733~3 時間以内に最高血漿中濃度(Cmax)に達し,いずれ の投与量でも吸収は中程度から速やかであることが示された。液剤(瓶入り粉末製剤:PIB) または錠剤として投与した各用量で最高血漿中濃度到達時間(tmax)の中央値に大きな違いはな かった。Cmaxの上昇は2.5 mg および 5 mg の間では用量比以下であり,25 mg から 100 mg の間 では用量比以上であり,100 mg(錠剤)から 600 mg では用量比例的であった。リナグリプチ ンの薬物動態プロファイルはいずれの投与量も二相性の消失を示した。錠剤として高用量投与 後に2 つの吸収ピークがみられた。曝露量(AUC0-∞)については,2.5 mg から 25 mg の間では 用量比以下の上昇を示し,100 mg(錠剤)から 600 mg では統計学的に用量に比例した上昇が 認められた。見かけのクリアランス(CL/F)の値はほぼ中程度の値であり,2.5 mg から 25 mg の間では用量の増加に伴って上昇し,100 mg(錠剤)から 600 mg の間ではほぼ一定であった。 2.5 mg から 50 mg での終末相における半減期(t1/2)は69.7~79.9 時間であり,100 mg から 600 mg では 128~184 時間であった。リナグリプチンの見かけの分布容積(Vz/F)は,2.5~5 mg
の用量範囲で2100~2490 L,25~600 mg では 5490~10700 L と大きかった。t1/2の値はサンプ リング期間に明らかに依存しており,100 mg 以上の用量ではより長時間にわたってサンプリン グが行われているため,Vz/F 値の比較には注意する必要がある。また絶対バイオアベイラビリ ティが不明なので,CL/F および Vz/F の値も注意して解釈しなければならない。CL/F および Vz/F の値から,非線形の分布および(または)排泄過程が示唆されている。尿中排泄率は用量 依存性を示し,2.5 mg では濃度が定量下限未満のため算出不可(0%)であり,600 mg では 32.7% まで上昇した。リナグリプチンの腎排泄においても非線形性が示され,少なくともリナグリプ チン低用量投与では腎排泄の役割は小さいことが示唆された。 薬物動態パラメータの要約を表2.1: 1 および 2 に示す。 表2.1: 1 リナグリプチン2.5~50 mg の単回経口投与後のリナグリプチンの主な薬物 動態パラメータ リナグリプチン 2.5 mg 液剤 (N=5) 5 mg 液剤 (N=6) 25 mg 錠剤 (N=6) 50 mg 錠剤 (N=5) パラメータ 単位 gMean(gCV%) gMean(gCV%) gMean(gCV%) gMean(gCV%)
Cmax {ng/mL} [nM] 4.41 (19.1) {2.08} 5.72 (19.4) {2.70} 72.3 (40.2) {34.2} 250 (47.0) {118} Cmax,norm {(ng/mL)/mg} [nM/mg] 1.76 (19.1) {0.833} 1.14 (19.4) {0.540} 2.89 (40.2) {1.37} 5.01 (47.0) {2.37} tmaxa) [h] 2.05 (1.48-3.05) 1.47 (1.02-5.95) 2.97 (0.700-4.02) (0.450-1.48) 0.733 AUC0-∞ {ng h/mL} [nM·h] 291 (33.8) {137} 427 (33.0) {202} 1110 (15.8) {525} 1930 (25.7) {913} AUC0-∞,norm {(ng·h/mL)/mg} [nM·h/mg] 116 (33.8) {55.0} 85.4 (33.0) {40.4} 44.4 (15.8) {21.0} 38.7 (25.7) {18.3} t1/2 [h] 79.9 (34.7) 69.7 (17.2) 79.9 (24.6) 75.9 (5.60) CL/F [mL/min] 303 (33.8) 413 (33.0) 794 (15.8) 912 (25.7) Vz/F [L] 2100 (12.9) 2490 (26.7) 5490 (37.5) 5990 (26.4) fe0-tz [%] NC 0.958 (69.6) 6.81 (49.1) 9.36 (43.9) a)tmaxについては中央値および範囲(最小値-最大値)を示す。 引用元:CTD 5.3.3.1-1,試験 1218.1,Table 15.5.2.1: 1-4 より作成
表2.1: 2 リナグリプチン 100~600 mg の単回経口投与後のリナグリプチンの主な薬 物動態パラメータ リナグリプチン 100 mg 錠剤 (N=8) 100 mg 液剤 (N=8) 200 mg 錠剤 (N=6) 400 mg 錠剤 (N=5) 600 mg 錠剤 (N=6) パラメータ 単位 (gMeangCV%) (gMeangCV%) (gMeangCV%) (gMeangCV%) (gMeangCV%)
Cmax {ng/mL} [nM] 757 (38.8) {358} 310 (57.9) {147} 1440 (25.9) {682} 3270 (36.7) {1550} 4340 (32.1) {2050} Cmax,norm {(ng/mL)/mg} [nM/mg] 7.57 (38.8) {3.58} 3.10 (57.9) {1.47} 7.22 (25.9) {3.41} 8.18 (36.7) {3.87} 7.24 (32.1) {3.42} tmaxa) [h] (0.517-3.03) 1.73 (0.483-5.97) 2.49 (0.467-2.03) 1.13 (0.683-4.00) 3.00 (0.700-3.02) 2.21 AUC0-∞ {ng·h/mL} [nM·h] 5690 (21.0) {2690} 3760 (29.0) {1780} 10700 (16.8){5060} 27700 (35.7) {13100} 39500 (19.6){18700} AUC0-∞,norm {(ng·h/mL)/mg} [nM·h/mg] 56.9 (21.0) {26.9} 37.6 (29.0) {17.8} 53.5 (16.8) {25.3} 69.3 (35.7) {32.8} 65.9 (19.6) {31.1} t1/2 [h] 143 (19.8) 132 (28.5) 172 (43.2) 184 (50.9) 128 (41.3) CL/F [mL/min] 620 (21.0) 938 (29.0) 659 (16.8) 509 (35.7) 535 (19.6) Vz/F [L] 7670 (17.5) 10700 (44.8) 9830 (52.0) 8090 (44.9) 5920 (57.5) fe0-tz [%] 18.2 (26.2) 13.2 (48.2) 21.1 (23.4) 30.4 (19.7) 32.7 (13.4) a)tmaxについては中央値および範囲(最小値-最大値)を示す。 引用元:CTD 5.3.3.1-1,試験 1218.1,Table 15.5.2.1: 5-9 より作成 錠剤および液剤(100 mg)の被験者内比較の結果,8 例中 7 例の被験者において錠剤に対する 液剤のバイオアベイラビリティは約半分(Cmax:幾何平均値の比41.0%;90%信頼区間 28.5~ 59.0%,AUC0-∞:幾何平均値の比66.1%;90%信頼区間 54.9~79.5%)であった。 薬力学:リナグリプチンのすべての投与量群で,血漿中のDPP-4 活性の阻害がみられた。2.5 mg および5 mg のリナグリプチン投与においても,その投与後の最大 DPP-4 阻害率(中央値)は, それぞれ72%および 88.5%であった。25 mg 以上の用量では,95%を上回る最大 DPP-4 阻害率 (中央値)がみられた。最大阻害率に達するまでの時間は,2.5 mg 群での 3 時間から 200 mg 以上の用量群での0.7 時間未満へと,用量が増加するにつれて短縮した。血漿中 DPP-4 活性に 対するリナグリプチンによる阻害は長時間にわたって持続し,本剤の投与から96 時間後でも, DPP-4 活性はベースラインレベルに戻らなかった。 DPP-4 阻害率と血漿中リナグリプチン濃度には良好な相関性が認められ,約 4~5 nM(2~2.5 ng/mL)の濃度によって 50%の阻害が生じ,約 10.6 nM(5 ng/mL)を上回る濃度では DPP-4 活 性は95%以上阻害された。 結論: リナグリプチンは100 mg までの用量において非線形の分布および(または)排泄過程を示し た。Cmaxの上昇は2.5~5 mg では用量比を下回り,100 mg までは用量比を上回った。100~600 mg の用量範囲では CmaxおよびAUC は,いずれも用量に比例して上昇した。尿中排泄率は,最
低用量群(2.5 mg)の 0%から最高用量群(600 mg)の 32.7%へと,投与量の上昇に伴って増加 した。 投与した全用量群でDPP-4 活性の阻害がみられ,25 mg 以上の用量群で 95%を上回る最高阻害 率がみとめられた。全用量群とも投与96 時間後でも DPP-4 活性はベースラインレベルに戻ら なかった。リナグリプチンの血漿中濃度とDPP-4 阻害率の間に良好な相関が認められ,約 4~5 nM の濃度によって 50%の阻害が生じ,約 10.6 nM(5 ng/mL)を上回る濃度では DPP-4 活性は 95%以上阻害された。
![図 1.3: 1 血漿中 DPP-4 阻害率とリナグリプチン血漿中濃度の関係(試験 1218.2) 試験 1218.2,1218.3,1218.5,1218.6 のデータを用いた母集団薬物動態解析の結果から,DPP-4 を 50%および 80%阻害するには,それぞれ 2.97 nM および 5.30 nM のリナグリプチンの血漿中 濃度が必要と考えられた[CTD 5.3.3.5-2,U -1554-01]。 メトホルミンの併用投与,腎機能障害または肝機能障害による,定常状態 DPP-4 阻害率に対](https://thumb-ap.123doks.com/thumbv2/123deta/6515336.663687/25.892.137.736.200.671/リナグリプチンおよびそれぞれおよびリナグリプチンメトホルミン.webp)

![表 2.13: 1 リナグリプチン単独またはリファンピシン併用下でのリナグリプチンの主 な薬物動態パラメータ リナグリプチン 5 mg 1 日 1 回 ( N=16) リナグリプチン 5 mg 1 日 1 回および リファンピシン600 mg1 日 1 回併用 ( N=16) リナグリプチン gMean gCV [%] gMean gCV [%] AUC τ,ss [nM·h] 145 25.4 87.6 16.8 C max,ss [nM] 9.84 38.6 5.53](https://thumb-ap.123doks.com/thumbv2/123deta/6515336.663687/79.892.107.781.204.602/リナグリプチンリナグリプチンリファンピシンリナグリプチン.webp)
![表 2.22: 1 リナグリプチン( 5 mg もしくは 100 mg)またはモキシフロキサシン(400 mg)投与後の主要および副次的 QT 評価項目の統計解析 評価項目 間隔/時点 [時間] プラセボに対する変化 [SE] 両側 90%信頼区間 [ms] 下限 [ms] 上限 [ms] リナグリプチン 5 mg QTcI N=43 間隔 1~4 −1.12 (0.96) −2.72 0.48 QTcI N=43 間隔 0.5~24 −0.91 (0.81) −2.26](https://thumb-ap.123doks.com/thumbv2/123deta/6515336.663687/101.892.105.786.216.596/リナグリプチンモキシフロキサシンに対するリナグリプチン.webp)
![表 3.3.4: 2 外国人 2 型糖尿病患者および日本人 2 型糖尿病患者にリナグリプチン 5 mg を反復投与時の定常状態でのトラフ時の血漿中濃度の比較 トラフ時の 血漿中濃度 [nM] 外国人(試験 1218.16) 日本人(試験 1218.23) 日本人 / N gMean (gCV [%]) N gMean (gCV [%]) 外国人 Week 12 293 6.38 (63.3) 153 7.15 (30.5) 1.12 Week 24 / 26 24](https://thumb-ap.123doks.com/thumbv2/123deta/6515336.663687/134.892.94.796.194.326/外国人型糖および日本人リナグリプチントラフトラフ日本人日本人.webp)

![図 3.5.2: 2 血漿中 DPP-4 阻害率とリナグリプチン血漿中濃度の関係(試験 1218.2) 薬物相互作用試験である試験 1218.4[CTD 5.3.3.4-1,メトホルミン]および試験 1218.67 [CTD 5.3.3.4-9,リファンピシン]においても,リナグリプチンの血漿中濃度と DPP-4 阻害率 の関係を検討した。これらの試験でもリナグリプチンの血漿中濃度と DPP-4 阻害率の相関は良 好であり,メトホルミンまたはリファンピシンの併用による影響を受けなかった。同様に,日](https://thumb-ap.123doks.com/thumbv2/123deta/6515336.663687/145.892.116.673.161.597/リナグリプチンリファンピシンリナグリプチンリファンピシン.webp)
