V/Ni コアに酸化物被覆層を形成した
CO 選択メタン化触媒に関する研究
山梨大学大学院医学工学総合教育部 博士課程学位論文 2016 年 3 月 林 克彦目次
第1 章 序論 ... 1 1.1 本研究の背景 ... 1 1.2 CO 選択メタン化触媒に求められる性能 ... 6 1.3 これまでの CO 選択メタン化触媒の研究と課題 ... 9 1.3.1 他の研究機関の研究 ... 9 1.3.2 本研究センターでのこれまでの研究 ... 12 1.4 本研究の目的 ... 13 1.5 本論文の構成 ... 14 参考文献 ... 16 第2 章メソポーラスシリカを被覆層とした Ni ナノ触媒の CO 選択メタン化活性とその発現機構 . 18 2.1 緒言 ... 18 2.2 実験方法 ... 19 2.2.1 触媒調製 ... 19 2.2.2 触媒活性評価 ... 24 2.2.3 触媒のキャラクタリゼージョン ... 27 2.3 酸化物被覆層による Ni 焼結抑制効果の検討 ... 29 2.3.1 V 担持効果 ... 29 2.3.2 V 担持及び MS 被覆層の効果 ... 40 2.4 MS/V/Ni 触媒の CO メタン化活性 ... 50 2.4.1 MS 被覆層形成による低温側 CO メタン化活性への効果 ... 50 2.4.2 MS 被覆層形成による長期連続耐久性への効果 ... 53 2.5 まとめ ... 59 参考文献 ... 59第3 章メソポーラスシリカ被覆コアシェル触媒の耐久性の向上 ... 61 3.1 緒言 ... 61 3.2 実験方法 ... 64 3.2.1 製造プロセスにおける触媒成分の溶出挙動解析 ... 64 3.2.2 触媒調製 ... 65 3.2.3 触媒活性評価 ... 67 3.3 各製造プロセスにおける触媒成分の溶出挙動 ... 67 3.3.1 MS/Ni/AlVOx 触媒からの成分溶出挙動 ... 67 3.3.2 MS/V/Ni 触媒からの成分溶出挙動 ... 72 3.4 Ni-Fe 含浸による MS 被覆コアシェル触媒の耐久性向上... 72 3.4.1 MS 含浸被覆層形成による低温側 CO メタン化活性への効果... 72 3.4.2 MS 含浸被覆層形成による長期連続耐久性への効果 ... 77 3.5 まとめ ... 79 参考文献 ... 80 第4 章シランカップリング剤によるシリカ薄層で被覆したNi ナノ触媒の CO 選択メタン化活性 . 81 4.1 緒言 ... 81 4.2 実験方法 ... 82 4.2.1 触媒調製... 82 4.2.2 触媒活性評価 ... 84 4.2.3 触媒のキャラクタリゼーション ... 86 4.3 SiO2被覆層の構造と機能 ... 87 4.3.1 SiO2被覆コアシェル触媒の構造 ... 87 4.3.2 CO メタン化活性に対する SiO2被覆量の最適化 ... 94 4.4 SiO2/V/Ni の CO 選択メタン化活性 ... 97 4.4.1 SiO2被覆層形成による低温活性向上への効果 ... 97 4.4.2 SiO2被覆層形成によるCO メタン化、CO2水素化反応への影響 ... 104
4.4.3 SiO2被覆層形成によるV 酸化状態の変化 ... 107 4.4.4 SiO2被覆層形成による長期連続耐久性への効果 ... 110 4.5 高温側でのCO2水素化反応の抑制 ... 113 4.5.1 高熱伝導度材料の添加による CO2水素化反応の抑制 ... 113 4.5.2 高熱伝導度材料添加触媒の CO 選択メタン化活性 ... 120 4.6 まとめ ... 123 参考文献 ... 124 第5 章総括 ... 125 研究論文 ... 128 学会発表 ... 129 謝辞 ... 130
第 1 章 序論
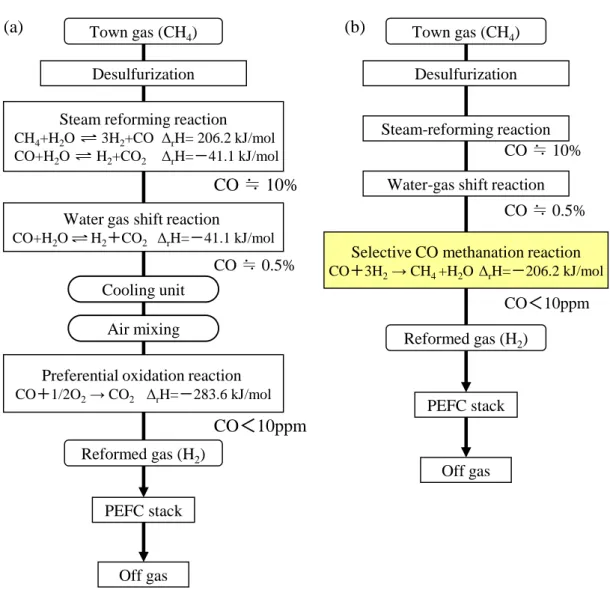
1.1 本研究の背景 現在、石油・石炭などの化石燃料の大量消費により、CO2の増大に伴う地球 温暖化の問題、資源の枯渇などのエネルギー問題が深刻化している。人類が持 続的に発展するためには、地球温暖化問題とエネルギー問題を同時に解決する ことが求められている。その解決手段の一つとして、水素エネルギーの利用に 大きな期待が寄せられている。 水素は、化石燃料、バイオマス、自然エネルギ ーなど様々なエネルギー源から製造することができる。このうち再生可能エネ ルギーから得られた水素を燃料として利用することで、CO2排出削減に大きく 貢献することができる。現在、我が国では水素の製造、輸送・貯蔵、利用まで を俯瞰した“水素社会”の実現に向けた研究開発が盛んに行われている。特に 水素利用技術として、家庭用燃料電池コージェネレーションシステムや燃料電 池車が実用化の段階まで進んでいる1)。 家庭用燃料電池コージェネレーションシステムは、我が国において 2009 年 に世界に先駆けて販売され、2015 年 9 月末の段階では累積販売台数が 14 万台 を突破している 2)。都市ガスを燃料として自宅で発電し、発電時に出る熱は給 湯・暖房に利用できることから、エネルギー効率の高い環境に優しいシステム として注目されている。また、省エネ・省 CO2への貢献が評価され、2014 年 に閣議決定された「エネルギー基本計画」において、2020 年に 140 万台、2030 年に530 万台(全世帯の 1 割)の普及が目標として掲げられている。家庭用燃料 電池コージェネレーションシステムには、固体高分子形燃料電池型(Polymer Electrolyte Fuel Cell:PEFC)と固体酸化物形燃料電池型(Solid Oxide Fuel Cell:SOFC)の 2 種類がある。現在は低温で扱いやすい PEFC 型が現在の主流 になっている3)。また、SOFC 型も発電効率の高さと低コスト化のポテンシャルにおいて関心が高まっている。今後は世帯人数が多く床暖房などを利用する 熱需要の大きい住宅向けではPEFC 型、一方高い発電効率をメリットとした熱 需要の少ない住宅向けでは SOFC 型と棲み分けが進んでいくと予想されてい る3)。 Fig.1-1 に家庭用 PEFC システムの概略図を示す。大きく分けて 3 ユニット から構成されており、それぞれ燃料電池ユニット、貯湯ユニット、バックアッ プ熱源機である。燃料電池ユニットでは、燃料である都市ガス等を燃料改質器 により水素に変換し、燃料電池スタックに供給する。発電された電気は直流で あるため、インバーターで交流に変換し、最大0.75kW の電力が家庭内に供給 される。燃料電池スタックや燃料改質器で排出された熱エネルギーは熱回収装 置(水循環)で回収し、貯湯ユニットでお湯として貯蔵される4)。 Fig.1-2(a)に現行の家庭用 PEFC システムに使用される燃料改質器のプロセ スフローを示す 5)。燃料改質器では水蒸気改質と水性ガスシフトの各反応によ り都市ガスを水素リッチガス(H2 70~80%)に改質している。水性ガスシフト工 程で生成した改質ガスをそのまま使用した場合、改質ガス中に含まれるCO が アノードで使用される白金系の電極触媒を被毒し、PEFC の性能を低下させる ため、最終段の CO 除去工程で出口 CO 濃度を 10ppm 以下まで低減させてい る。以下、現行の家庭用PEFC システムに用いられている燃料改質器の各工程 について更に詳細に説明する。 (1) 水蒸気改質工程 付臭剤成分を吸着剤や水素添加反応により除去した都市ガス(CH4)を水蒸気 改質反応(Steam Reforming Reaction:SR)により H2を製造する工程である。
CH4 + H2O → 3H2 + CO ΔrH= 206.2 kJ/mol (式 1-1) CO + H2O → H2 + CO2 ΔrH=-41.1 kJ/mol (式 1-2) 実機燃料改質器において水蒸気/カーボン比(S/C) を 2.8 とした場合、水蒸気
Fig.1-1 Schematic illustration of residential PEFC system. Hot water Water Fuel cell stack Inverter
Hot water storage unit
Backup heat
source equipment
Fuel Cell unit
Fuel reformer Electricity Heat exchanger Town gas (CH4) Hot water H2 HeatFig 1-2 Scheme of fuel processing for a residential fuel cell system : (a)Conventional fuel reformer using PROX, (b) fuel reformer using SCM.
(a) CO ≒ 0.5% PEFC stack CO<10ppm Town gas (CH4) PEFC stack (b) Steam-reforming reaction Water-gas shift reaction
Desulfurization
Selective CO methanation reaction
CO+3H2→ CH4 +H2O ΔrH=-206.2 kJ/mol
CO ≒ 10%
CO ≒ 0.5%
CO<10ppm Steam reforming reaction
CH4+H2O 3H2+CO ΔrH= 206.2 kJ/mol
CO+H2O H2+CO2 ΔrH=-41.1 kJ/mol
Water gas shift reaction
CO+H2O H2+CO2 ΔrH=-41.1 kJ/mol
Desulfurization
Preferential oxidation reaction
CO+1/2O2→ CO2 ΔrH=-283.6 kJ/mol Town gas (CH4) Air mixing Cooling unit CO ≒ 10% Reformed gas (H2) Reformed gas (H2) Off gas Off gas
改質工程での出口CO 濃度は約 10%となる。 (2) 水性ガスシフト工程 改質ガス中に含まれる約 10%の CO を水性ガスシフト反応(Water-gas shft Reaction : WGS)により H2に変換する工程である。 CO + H2O
⇄
H2 + CO2 ΔrH=-41.1 kJ/mol (式 1-3) 水性ガスシフト反応は平衡反応であるため、反応温度を下げるほど出口の H2濃度を上げ、CO 濃度を低下することができる。これは使用する水性ガスシ フト触媒の性能に強く依存するものであり、現状のCu-Zn 系触媒を使った系で は反応温度として190~200℃が一般的である。水蒸気改質工程での水蒸気/カー ボンモル比(S/C)を 2.8 とすると、本工程出口での反応温度が 195℃とした場合、 平衡計算から出口CO 濃度は約 0.5%となる。 (3) CO 除去工程 水性ガスシフト工程では、使用する触媒性能の制約から出口 CO 濃度は約 0.5%までしか除去できない。そこで CO 選択酸化反応(Preferantial Oxidation Reaction :PROX)を用いて改質ガス中の CO 濃度を 10ppm まで低減している。 PROX で使用する酸化剤には空気を利用している。 CO + 1/2O2 → CO2 ΔrH=-283.6 kJ/mol (式 1-4) (副反応:H2 + 1/2O2 → H2O) しかしながら、PROX を採用した現行の燃料改質器には下記課題がある6),7)。 ① 空気を送り込むためのブロア、流量計及びバルブなどの補機が必要である。 ② 起動停止(Daily Start up and Stop:DSS)や負荷変動により、改質ガスの流 量やCO 濃度の変動に合わせて空気の供給を調節しているため複雑な制御が求 められる。③ PROX の操作温度領域は CO シフト工程の出口温度よりも 100℃低いこと から、CO シフト工程で処理した改質ガスを冷却する必要がある。
④ 低温で酸化反応を行うため、触媒には一般的に貴金属が使用されている。ま た遷移金属系の触媒を適用できないため、触媒の材料コストの低減が難しい。 そこで近年、PROX に代わる新しい CO 除去プロセスとして CO 選択メタン 化反応(Selective CO Methanation Reaction:SCM)が注目されている8)~16)。
CO 選択メタン化反応は、PROX と異なり外部から空気を導入する必要が無い ため、補機の削減や燃料改質器の構造・制御の簡略化による低コスト化と省ス ペース化が期待されている。 著者が所属する山梨大学 燃料電池ナノ材料研究センターでは、これまで CO 選択酸化プロセスに替わるCO 選択メタン化プロセス用触媒の開発に取り組ん でいる。 1.2 CO 選択メタン化触媒に求められる性能 CO 選択メタン化反応は主反応である CO メタン化反応(式 1-5)と副反応であ る CO2水素化反応(式 1-6,1-7)からなる。更に CO2水素化反応は式 1-6 の逆水 性ガスシフト反応(Reversible Water-gas shft Reaction :RWGS) と式 1-7 の CO2メタン化反応の2 種類に分けられる。 <主反応> CO メタン化反応: CO + 3H2 → CH4 + H2O ΔrH=-206.2 kJ/mol (式 1-5) <副反応> 逆水性ガスシフト反応: CO2 + H2 → CO + H2O ΔrH=41.1 kJ/mol (式 1-6) CO2メタン化反応: CO2 +4H2 → CH4 + 2H2O ΔrH=-165 kJ/mol (式 1-7) Fig.1-3 に CO 選択メタン化反応の温度依存性の概略図を示す。Fig.1-3(a)の
出口CO 濃度では、最初に反応温度を徐々に上昇させていくと、急激に CO 濃 度が低下していく。更に反応温度を上げていくと再び出口CO 濃度が上昇して いく。出口CO 濃度の変化は反応温度に対して極小値を持つ下に凸の曲線を描 き、この極小値は触媒種類に依存する。低温側の出口CO 濃度が急激に低下す る領域では、主に図中(1)の CO メタン化反応が発生している。一方高温側の出 口CO 濃度が上昇する領域では、主に図中(2)の逆水性ガスシフト反応が発生し ている。次に Fig.1-3(b)の出口 CH4濃度では、反応温度が上昇するのに伴い、 出口 CH4濃度は上昇する傾向となる。特に高温側になると CH4濃度は急激に 上昇する傾向となる。低温側での出口 CH4 濃度の上昇は、主に図中(1)の CO メタン化反応が原因である。高温側の CH4 濃度の急激な上昇は、主に図中(3) のCO2メタン化反応に由来する。 また、CO 選択メタン化触媒を家庭用 PEFC システムに適用するためには、 下記に示す性能が求められる。 (1) 高い CO メタン化活性 Fig.1-3 より CO メタン化反応を促進し、且つ逆水性ガスシフト反応を抑制し て改質ガス中の出口CO 濃度を 10ppm 以下にする。 (2) 高い反応選択性 CO シフト工程で処理した改質ガス中には CO 濃度が約 0.5%、CO2濃度が約 20%含まれる。CO 選択メタン化触媒では低濃度の CO のみを選択的に CO メ タン化反応で除去し、一方高濃度のCO2の水素化反応を抑制する反応選択性が 求められる。特にFig.1-3(b)より反応温度が高くなると CO2メタン化反応が発 生する。CO2メタン化反応で発生した反応熱は、高温側で熱暴走を引き起こす 可能性があるため、特にCO2メタン化反応の抑制は重要である。熱暴走が発生 すると燃料改質器の熱バランスが崩れ、燃料改質器の運転そのものに大きな影 響を及ぼす。
Fig.1-3 SCM temperature dependence images: (a)Outlet CO concentration, (b) Outlet CH4 concentration.
Reaction temperature (℃)
Ou
tle
t
CH
4co
n
ce
n
trat
io
n
(%
)
Reaction temperature (℃)
Ou
tle
t
C
O
co
n
ce
n
trat
io
n
(ppm
)
(a)
(b)
(1) CO Methanation : CO + 3H2→ CH4+ H2O ΔH = -206 kJ/mol(2) Reversible Water-gas shft : CO2 + H2→ CO + H2O ΔH = 41.2 kJ/mol (3) CO2Methanation : CO2+ 4H2→ CH4+ 2H2O ΔH = -165 kJ/mol
1
10
100
1000
140 150 160 170 180 190 200
0
0.5
1
1.5
2
140 150 160 170 180 190 200
5000
(1)
(1)
(2)
(3)
T
L(CO)
T
H(CO)
T
H(CH
4)
Window
width
(3) 広い操作温度領域 CO2メタン化反応により発生したCH4は改質ガス中のH2を過剰に消費する ことを意味する。H2の過剰な消費を回避するために CO2メタン化反応由来の CH4量は 0.5%以下(入口 CO 濃度が 0.5%である場合、50%の選択性に相当)に 抑える様に設定した。よってCO 選択メタン化触媒における CH4濃度の目標値 として、過剰に H2の消費をしない様に CH4濃度は 1%以下であることが望ま しい。ここでFig.1-3 中に示す様に、CO 濃度が 10ppm 以下で且つ CH4濃度が 1%以下となる触媒の温度範囲を操作温度領域(window width)として定義する。 この操作温度領域は安定運転のため30℃以上を目標とした。 (4) 高い触媒耐久性 システム搭載上 6~8 万時間の長期連続運転に対する耐久性が求められる。ま た、DSS 運転や負荷変動に伴う触媒の劣化(コーキング、酸化被膜の形成、活 性成分の焼結など)を抑制する必要がある。 (5) 触媒の低コスト化 貴金属の使用量削減及び触媒使用量の削減が求められる。そのため、遷移金 属系の触媒を適用し、且つ現行の触媒充填体積(約 500 mL)より 1/2 以下の量、 すなわち空間速度(Space velocity:SV)が 4800h-1以上でも高活性な触媒が望ま れる。 1.3 これまでの CO 選択メタン化触媒の研究と課題 1.3.1 他の研究機関の研究 CO 選択メタン化触媒の活性成分に関して、Panagiotopoulou らは、貴金属 成分を中心に反応活性を評価し、Pt、Pd に比較して Ru、Rh の方が触媒活性 が高いことを報告している20)。特にRu が触媒活性の点で有望な材料であると する報告が多い 21),22)。一方で、Ni の活性も高く且つ低コストであるため、Ni
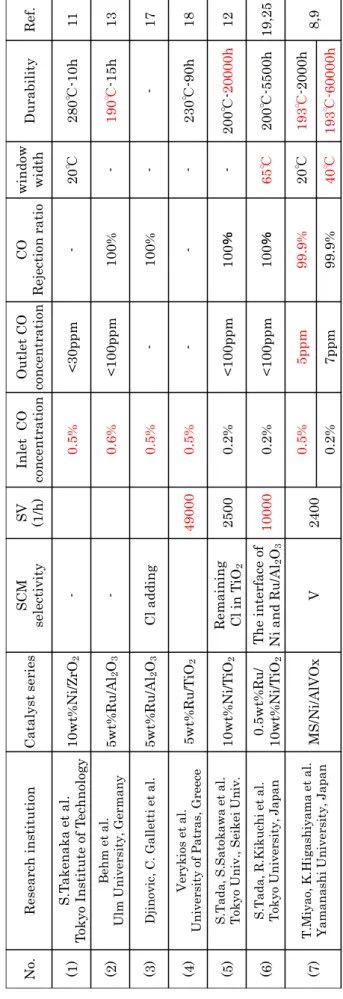
を 活 性 成 分 と し た 触 媒 の 研 究 が 多 く の 研 究 機 関 で 盛 ん に 行 わ れ て い る 11),15),23)~25)。 Table1-1 にこれまで行われた CO 選択メタン化触媒の研究の中から Ru 系及 びNi 系触媒の代表的なものを示した。実用化に近い結果は赤字で示す。Table 1-1(1)の竹中らは活性成分と担体との組み合わせに着目し、Ru/TiO2とNi/ZrO2 の優位性を報告している11)。特に10wt%Ni/ZrO2触媒では入口CO 濃度 0.5%、 反応温度280℃において 10 h 触媒活性を評価し、CO が 30ppm 以下に除去さ れていることを確認している。Table 1-1(2)の Abdel-Mageed らは 5wt% Ru/Al2O3触媒を検討し、入口CO 濃度 0.5%、反応温度 190℃で触媒活性を評 価し、CO は 100ppm 以下まで除去されることを確認した13)。またTable 1-1(3) のDjinovic らは 5wt%Ru/Al2O3触媒に塩素成分を添加することでCO 選択メ タン化触媒の反応選択性が向上することを指摘している17)。入口CO 濃度 0.5% で触媒活性を評価し、CO 除去性能が高いことを確認している17)。Table 1-1(4) のParaskevi らは 5wt%Ru/TiO2触媒についてSV49000 h-1の高いSV 条件下 で入口CO 濃度 0.5%に対する触媒活性を評価しており、反応温度 230℃で 90 h 経過後もCO 除去性能が維持されることを示した18)。Table 1-1(5) の多田、里 川らはTiO2及びAl2O3を担体としたRu 系、Ni 系触媒の検討から、著しい担 体効果が生じることを報告している12),26)。ここでは選択性低下の要因である副 反応のCO2メタン化反応は、担体と活性金属の界面で生じると述べている。界 面に吸着したCO2は、活性金属で解離吸着した水素が担体上にスピルオーバー することで、CO に還元される。この還元された CO が解離吸着した水素と反 応してCH4が生成される26)。この知見を基に10wt%Ni/TiO2触媒が活性及び 選択性に優れた触媒であることが見出された。また、TiO2に残留した塩素成分 も反応選択性に寄与し25)、10wt%Ni/TiO2触媒では入口CO 濃度 0.2%、反応 温度280℃で 20000 h 経過後も CO が 100ppm 以下に除去されていることを示
T
able
1-1
R
esearch
list
of
sele
ct
ive
C
O
met
h
an
ation
.
N o. Res ea rc h ins ti tuti on Cata ly st ser ies SCM selecti vi ty SV (1/h) Inl et CO conc entr ati on Outl et CO conc entr ati on CO Rejecti on ra ti o wi nd ow wi dth Durab il ity Ref . (1) S.T ak enaka et a l. Tokyo I ns ti tute of T ec hnol og y 10 wt%N i/ZrO 2 -0.5 % < 30 pp m -20 ℃ 280 ℃ -1 0h 11 (2) B ehm et al. Ulm Univ ers ity, G erm an y 5wt%Ru /Al 2 O3 -0.6 % < 10 0p pm 100% -190 ℃ -1 5h 13 (3) D jin ov ic , C . G allett iet al. 5wt%Ru /Al 2 O3 Cl a dd ing 0.5 % -100% -17 (4) V ery k ios et al . Univ ers ity of P atras , G reec e 5wt%Ru /T iO2 49000 0.5 % -230 ℃ -9 0h 18 (5) S. T ada , S. Satok aw a et al. T oky o Un iv ., Seikei Un iv . 10 wt%N i/T iO2 Rema ini ng Cl in T iO2 2500 0.2 % < 10 0p pm 100 % -200 ℃ -20 00 0h 12 (6) S. T ada ,R .K ik uchi et al. T ok yo Univ ers ity, Ja pan 0.5 wt%Ru / 10 wt%N i/T iO2 T he i nter face of Ni and Ru/Al 2 O3 10000 0.2 % < 10 0p pm 100 % 65 ℃ 200 ℃ -5 50 0h 19 ,2 5 (7) T .Miya o, K .H ig ashiy am a et al. Yam an ashi Univ ers ity, Ja pan M S/N i/ Al VOx V 2400 0.5 % 5p pm 99 .9 % 20 ℃ 193 ℃ -2 00 0h 8,9 0.2 % 7p pm 99 .9 % 40 ℃ 193 ℃ -6 00 00 hした。更に Table 1-1(6)では 10wt%Ni/TiO2 に Ru を担持した 0.5wt%Ru /10wt%Ni/TiO2触媒を検討した19)。この触媒はSV10000 h-1の高いSV 条件下 で入口 CO 濃度 0.2%に対して反応温度 280℃で 5500 h 経過後も、CO は 100ppm 以下に除去されていた。 以上より十分実用的な初期性能を有する触媒や、高い耐久性が期待される触 媒の報告はあるものの 26)、1.2 で述べた様に実際の燃料改質器に搭載する為に 必要な触媒性能を満足する触媒は現時点で開発されていない。 1.3.2 本研究センターでのこれまでの研究 本研究センターでは Ni/Al2O3系触媒を中心に研究を進め 8)~10),27),28)、当初は 塩化 Ru の添加によって優れた CO メタン化への選択性が得られた。塩化 Ru の添加による選択性の発現機構は不明であったが、選択性が水蒸気を含む改質 ガスに長時間暴露することで低下することや、種々の塩化物を添加すると選択 性が発現することから、選択性の発現にはNi/Al2O3触媒上の残留塩素に起因し ていることを突き止めた。またNi 上に吸着した塩素が、Ni 上の CO2の解離吸 着を抑制することで、CO2 の水素化反応の進行が抑制されることを CO2 吸着 FT-IR 等により明らかにした。しかしながら、塩素を添加した場合、水蒸気を 含む改質ガスに長時間曝される過程で塩素は脱離しやすく選択性が大幅に低下 することが確認された。そこで CO2 解離抑制効果を示し、反応条件下でも Ni/Al2O3系触媒中に安定に存在する塩素代替材料を CO2吸着 FT-IR を用いて 探索したところ、V 添加が反応選択性の向上に有効であることを見出した 8)。 V 添加 Ni/Al2O3系触媒(以下 Ni/AlVOx)の選択性は安定であり、長時間の反応 でも選択性の低下は見られなかったが、改質ガスに長時間曝されることによっ て触媒表面の炭素種の析出が進行し、CO 除去率が低下した。次に Ni/AlVOx をベースに改良を進め、炭素析出を抑制し耐久性を大幅に改善する手法として、
Ti 含有メソポーラスシリカ(以下 MS)で被覆した触媒(以下 MS/Ni/AlVOx)を見 出した。これはMS 被覆による著しい CO 除去性能と耐久性の向上は、MS 被 覆層中の活性金属種の存在、つまりメソ細孔中に新たな触媒活性サイトが出現 したことに由来している。以上より Table 1-1(7)に示した通り MS/Ni/AlVOx はCO 除去性能と耐久性能を著しく向上することができ、空間速度(SV)2400 h-1、 反応温度190 ℃以上で優れた触媒性能を示した8),9)。しかし、現行の改質器の 条件を大きく変更せずにPROX 触媒を CO 選択メタン化触媒に代替しようとし た場合、1.1 で述べた通り水性ガスシフト工程からの出口の CO 濃度が 0.5%を 下回る平衡温度は195℃以下であるため、現行の CO 選択メタン化触媒では操 作温度幅が190℃から 195℃間の 5℃しか取れない。本触媒は 190 ℃以上とい う現状の動作温度のままでは実機に適用するのは困難である。実機に適用する ためには、現状の動作温度を低温側に大きく拡張させることが望まれる。この 課題を解決する方策の一つとして Ni 担持量を増加することが考えらえるが、 MS/Ni/AlVOxのNi 担持量は現状 30 wt%で最適化しているため、Ni 担持量の 単純な増加ではNi 反応サイトの増加は期待できない。また SV を 2400 h-1よ り低くする(触媒量を増加する)ことで低温化は可能であるが、2400 h-1 は実機 で約500 mL の触媒充填体積に相当し、実機適用を考慮した場合、これ以上の SV 低減は難しくむしろ増加することが望まれている。 1.4 本研究の目的 本研究の目的は、家庭用 PEFC システムの燃料改質器の PROX 触媒の代替 としてCO 選択メタン化触媒を実機へ搭載するため、反応温度 195 ℃以下で広 い操作温度領域を有し、高いCO メタン化活性と高い反応選択性を両立させた CO 選択メタン化触媒を開発することである。 本研究では、CO 選択メタン化触媒の低温活性向上を図るため、次の様な共
通の戦略のもとで開発を進めることにした。つまり、従来 MS/Ni/AlVOx で使 用しているAl2O3担体の使用を止め、Ni ナノ粒子をコアとし、ガス透過が可能 な金属酸化物層をシェルとしたコアシェル構造の触媒を指向するものである。 単純に Al2O3等の担体を使用せずに Ni ナノ粒子のみから形成された触媒を 考えた場合、還元及び反応時に Ni ナノ粒子同士が焼結し、十分な触媒性能が 得られない。そこでその表面に金属酸化物のガス透過性の膜をシェルとして被 覆することで、Ni ナノ粒子の接触とそれに続く焼結を防ぐことが可能になると 考えた。この酸化物被覆層を形成したコアシェル触媒により、触媒単位体積当 たりの Ni 表面積が増え、反応サイトが増加するため低温側の CO メタン化反 応速度が向上することが期待される。 酸化物被覆層として、はじめにこれまで当研究センターで検討してきた MS 被覆層や V2O5の適用を試みる。次に MS 被覆層に代わる新たなシリカ被覆層 を検討する。併せてこれらの酸化物被覆層がCO 選択メタン化活性に与える影 響について考察することとした。 1.5 本論文の構成 本論文の構成は、本章の「序論」以下、次の構成で成る。 第 2 章「メソポーラスシリカを被覆層とした Ni ナノ触媒の CO 選択メタン 化活性とその発現機構」では、従来のMS/Ni/AlVOxの更なる低温領域でのCO メタン化活性を向上させる目的で、Al2O3 担体を使用せず、V を担持した Ni ナノ粒子に直接 MS 被覆層を形成したコアシェル構造を有する触媒(以下 MS 被覆コアシェル触媒) に着目し、実機搭載上必要とされる触媒活性の低温化と 長期耐久性を検証した。V 及び MS 被覆層が Ni 焼結抑制に与える効果を検討 し、低温側でのCO メタン化活性向上への効果を明らかにした。 第 3 章「MS 被覆コアシェル触媒の耐久性の向上」では、第 2 章で検討した
MS 被覆コアシェル触媒の耐久性を向上させることを目的とした。従来の MS/Ni/AlVOxでは MS 被覆層中に析出した Ni、V が耐久性向上に寄与するこ とが既に確認されている。一方、MS 被覆コアシェル触媒では、MS 被覆層中 のNi、V の析出については不明であるため、MS 被覆層の形成過程における触 媒成分の溶出挙動の解析によりその存在の有無を明らかにした。次にMS 被覆 層へのNi-Fe、Ni-Fe-V を担持することにより更なる耐久性の向上を検討した。 第 4 章「シランカップリング剤によるシリカ薄層で被覆した Ni ナノ触媒の CO 選択メタン化活性」では、第 2 章の MS 被覆コアシェル触媒よりも Ni 含 有量を更に増し低温活性の向上を図ることを目的とした。SiO2薄層を形成する ことがよく知られているシランカップリング剤をSiO2の前駆体として選定し、 直接 Ni ナノ粒子と反応させることにより、Ni ナノ粒子全体を SiO2薄層で被 覆することを試みた。また、SiO2薄層がCO 選択メタン化反応に与える影響を 明らかにする為、SiO2とV との相互作用に着目し、XAFS、XPS、素反応評価 によりV の酸化状態と CO 選択メタン化反応の関係を解析した。 第 5 章 「総括」は、本研究から得られた知見をまとめた。
参考文献 1) 独立行政法人 新エネルギー・産業技術総合開発機構編, “NEDO 水素エネ ルギー白書”, 2015 2) 一般財団法人 コージェネ財団,“エネファームメーカー 販売台数(2015 年 9 月末現在)”,[オンライン]. [アクセス日:2015/12/17] Available:http://www.ace.or.jp/web/works/works_0090.html 3) 株式会社富士経済, “2015 年度版 燃料電池関連技術・市場の将来展望”,2015 4) 本間琢也,上松宏吉,“燃料電池のキホン”,ソフトバンククリエイティブ,2010 5) 宮尾敏広, 東山和寿, 山下寿生,渡辺政廣, 表面科学,Vol. 36(2015)pp.62-68. 6) E. Park, D. Lee, H. Lee, Catal. Today 139 (2009) 280–290.
7) 岩佐泰之, 松本隆也: 触媒 55, 27 (2013).
8) T. Miyao, S. Sakurabayashi, W. Shen, K. Higashiyama, M. Watanabe, Catal.Commun. 58 (2015) 93–96.
9) T. Miyao, J. Tanaka, W. Shen, K. Hayashi, K. Higashiyama, M. Watanabe, Catal.Today 251 (2015) 81–87.
10) T. Miyao, W. Shen, A. Chen, K. Higashiyama, M. Watanabe, Appl. Catal. A General,.486, 187,(2014).
11) S. Takenaka, T. Shimizu, K. Otsuka, Int. J. Hydrogen Energy 29 (2004)1065–1073.
12) S. Tada, R. Kikuchi, K. Urasaki, S. Satokawa, Appl. Catal. A: Gen. 404 (2011)149–154.
13) A. Abdel-Mageed, S. Eckle, H. Anfang, R. Behm, J. Catal. 298 (2013) 148–160.
14) P. Panagiotopoulou, D. Kondarides, X. Verykios, J. Phys. Chem. C 115 (2011)1220–1230.
15) R. Dagle, Y. Wang, G. Xia, J. Strohm, J. Holladay, D. Palo, Appl. Catal. A: Gen.326 (2007) 213–218.
16) F. Joensen, J. Rostrup-Nielsen, J. Power Sour. 105 (2002) 195–201. 17) P. Djinovic, C. Galletti, S. Specchis, V. Specchis, Top. Catal. 54
(2011)1042–1053.
18) Paraskevi Panagiotopoulou, Dimitris I. Kondarides, Xenophon E. Verykios, Catalysis Today 181 (2012) 138– 147
19) S. Tada, R. Kikuchi, K. Wada, K. Osada, K. Akiyama, S.
Satokawa and Y. Kawashima, J. Power Sources, 2014, 264, 59.
20) P. Panagiotopoulou, D. Kondarides, X. Verykios, Appl. Catal. A: Gen. 344(2008) 45–54.
21) C. Gallietti, S. Specchia, G. Saracco, V. Specchia, Chem. Eng. Sci. 65 (2010)590–596.
22) P. Panagiotopoulou, D. Kondarides, X. Verykios, Appl. Catal. B: Environ. 88(2009) 470–478.
23) K. Urasaki, Y. Tanpo, T. Takahiro, J. Christopher, R. Kikuchi, T. Kojima, S.Satokawa, Chem. Lett. 39 (2010) 972–973.
24) M. Krämer, M. Duisberg, K. Stöwe, W. Maier,J.Catal.251(2007)410–422. 25) N. Shimoda, D. Shoji, K. Tani, M. Fujiwara, K. Urasaki, R. Kikuchi, S.
Satokawa,Appl. Catal. B: Environ. 174 (2015) 486–495. 26) S. Tada, R. Kikuchi, Catal. Sci. Technol. 5 (2015) 3061–3070
27) M. Kimura, T. Miyao, S. Komori, A. Chen, K. Higashiyama, H. Yamashita, M.Watanabe, Appl. Catal. A: Gen. 379 (2010) 182–187.
28) A. Chen, T. Miyao, K. Higashiyama, H. Yamashita, M. Watanabe, Angew. Chem.Int. Ed. 49 (2010) 9895–9898.
第2章 メソポーラスシリカを被覆層とした Ni ナノ触媒の CO 選択メタン化
活性とその発現機構
2.1 緒言 山梨大学の当研究センターは、これまでにV を添加した Ni-Al 酸化物触媒を Ti 含有メソポーラスシリカ(以下 MS)で被覆することで、CO 除去性能と耐久 性能が大きく向上する事を見出している 1),2)。この触媒(以下 MS/Ni/AlVOx)で は、添加したV が反応選択性の向上に有効に作用する一方、MS 被覆層はその 細孔内部の高分散 Ni により高い CO 除去性能と耐久性を付与していると考え られる。これにより空間速度(SV)2400 h-1、反応温度190 ℃以上で優れた触媒 性能を示した。しかし実機水蒸気改質プロセスでの水蒸気/カーボン比 S/C を 2.8 とした場合、水性ガスシフトプロセス出口の CO 濃度が 0.5%を下回る平衡 温度は 195 ℃以下になるため、本触媒の 190 ℃以上という動作温度は実機に 十分適合しておらず、実機搭載には本触媒の動作温度を現状より低温側に大き くシフトさせる必要があった3)~7)。MS/Ni/AlVOxのNi 担持量は現状 30wt%で 最適化しているため、Ni 担持量の単純な増加では Ni 反応サイトの増加は期待 できない。また SV を 2400 h-1より低くする(触媒量を増加する)ことで低温化 は可能であるが、2400 h-1は実機で約500 mL の触媒充填体積に相当し、実機 適用を考慮した場合、これ以上の SV 低減は難しくむしろ増加することが望ま れている。 そこで本章では、従来のMS/Ni/AlVOx1),2)の更なる低温領域での CO メタン 化活性を向上させる目的で、Al2O3担体を使用せず、V を担持した Ni ナノ粒子 に直接MS 被覆層を形成するコアシェル構造に着目した3)。この構造では、触 媒単位体積当たりのNi 表面積が増え、反応サイトが増加するため低温側の CO メタン化反応速度が向上することが期待される。これまで逆ミセル法によりNi に SiO2を被覆したコアシェル触媒の研究例では、SiO2被覆層がNi の凝集・ 焼結を効果的に抑制していることが報告されている8),9)。 本章の研究ではMS/Ni/AlVOxで使用したMS 被覆層を Ni コアシェル構造の シェル材として適用し、低温側でのCO メタン化活性向上への効果を明らかに することを目的とした。2.3 節ではまず V 及び MS 被覆層が Ni 焼結抑制に与 える効果を明らかにすることにした。V の分散性向上の観点から Ni 原料とし てNi(OH)2を使用し、V イオンの吸着特性を解析した。V、Si は溶液中でポリ 陰イオンを形成することから、同様に溶液中でポリ陰イオンを形成する成分と して Mo、W を、ポリ陰イオンを形成しない成分として Fe を選定し、それぞ れの被覆材としての効果を比較検討した。2.4 節では凝集抑制効果と触媒活性 の観点からV 及び MS 被覆層の最適量を検討した。得られた最適構造の触媒に おいて、実機搭載上必要とされる触媒活性の低温化と長期耐久性を検証した。 2.2 実験方法 2.2.1 触媒調製 (a) Ni(OH)2コア粒子の作製
Fig.2-1 に Ni(OH)2コア粒子の製造フローを示す。Ni:NH3モル比が 1:6 となる様に5%硝酸 Ni 水溶液と 25 %アンモニア水を混合してヘキサアンミン ニッケル(Ⅱ)水溶液を調製した。この溶液に 5%KOH 水溶液を Ni:KOH モル 比が 1:2.5 となる様に加えて Ni(OH)2を沈殿させた。Ni(OH)2スラリー溶液 を5 時間撹拌後、この沈殿物をろ別し 50 ℃のイオン交換水で洗浄し、K+、NH4+、 NO3-の残留イオンを除去した。得られたNi(OH)2沈殿物を脱イオン水に懸濁し 回転円盤方式のスプレードライ装置(型式 L-8i, 大河原化工機)により 250 ℃で 乾燥してNi(OH)2粉末を得た。
Fig.2-1 Block diagram of preparation procedures for Ni(OH)2 powder.
Ni(OH)
2powder
Ni(NO
3)
2aqueous solution
25% NH
3aqueous solution
5% KOH aqueous solution
Filtration/Washing
Spray-drying
[Ni(NH
3)
6]
2+aqueous solution
Ni(OH)
2slurry
50℃ water
Stirring
Ni/KOH molar ratio 1/2.5
5h at 30℃
Ni(OH)
2slurry
Deionized water
Stirring
Ni/NH
3molar ratio 1/6
(b) V 担持 Ni(OH)2コア粒子の作製
Fig.2-2 に V 担持 Ni(OH)2コア粒子の製造フローを示す。Fig.2-1 で得られ た Ni(OH)2 粉末の所定量を 50 ℃に加温したバナジン酸アンモニウム水溶液 (0.079 mol/L)に 2 時間浸漬し、その後蒸発乾固することにより V 成分を Ni(OH)2粉末に担持した。V 成分の担持量の調節は Ni(OH)2の量を一定にして、 バナジン酸アンモニウム水溶液の仕込み量を変えることで対応した。V/Ni モル 比はそれぞれ0.010、0.025、0.039、0.046、0.048、0.097 とした。その後 120 ℃ -2 時間乾燥し V 担持 Ni(OH)2粉末を得た。V/Ni モル比が 0.039 以下の場合、 バナジン酸アンモニウム溶液に 2 時間浸漬した時点で、溶液中の V イオンは Ni(OH)2 に全量吸着していたことから、蒸発乾固は省略し、そのまま V 担持 Ni(OH)2をろ過、乾燥を行った。
また V、Si 以外に Mo、W、Fe の効果を検討する際は、Ni 1 mol に対して担 持成分が0.039 mol になる様に調製した。原料塩として、Mo はモリブデン酸 アンモニウム、W はパラタングステン酸アンモニウム、Fe は硝酸鉄をそれぞ れ使用した。Si 原料として、オルトケイ酸テトラエチルを使用した。各水溶液 濃度はV の場合と同様 0.079 mol/L に設定した。 (c) V 担持 Ni(OH)2コア粒子への MS 被覆層形成 Fig.2-3 に V 担持 Ni(OH)2コア粒子へのMS 被覆層の形成フローを示す。最 初にV 担持 Ni(OH)2コア粒子を NH3水溶液に添加してスラリーを作製した。 このときの仕込み量は、V 担持 Ni(OH)2コア粒子5 g、25%NH3水溶液2 g、 イオン交換水300 g とした。次に MS 被覆層を形成する際のテンプレートとし て、臭化ヘキサデシルトリメチルアンモニウム(CTAB, 99%, 和光純薬工業)を 用いた。エタノールとイオン交換水の混合液にCTAB を溶解し、CTAB 溶液を 調製した。このときの仕込み量は、CTAB 1.22 g、エタノール 96.8 g、イオン 交換水 10.6 g とした。V 担持 Ni(OH)2スラリーにCTAB 溶液を滴下し、V 担
Fig.2-2 Block diagram of preparation procedures for V-added Ni(OH)2 powder.
Ni(OH)
2powder
NH
4VO
3aqueous solution
(V 0.079 mol/L)
V Impregnation
Stirring
50℃-2h in water bath
Drying
V-added Ni(OH)
2powder
120℃-3h
Crushing
Fig.2-3 Block diagram of preparation procedures for MS/V/Ni powder after calcination. NH3 aqueous solution (2g) Stirring MS/V/Ni powder Room temperature- 15h Calcination
V-added Ni(OH)2powder (5g)
Deionized water (300g)
CTAB solution
(CTAB:Ethanol :Deionized water
=1.22g:96.8g:10.6g)
Stirring
TEOS solution @ Si/Ni=0.22
(TEOS:Ethanol : Isopropyl titanate : Acetylacetone =4.7g:20.1g:1.0g:0.2g) Stirring Filtration/Washing Drying 120℃-3h 550℃-3h Room temperature-Room temperature
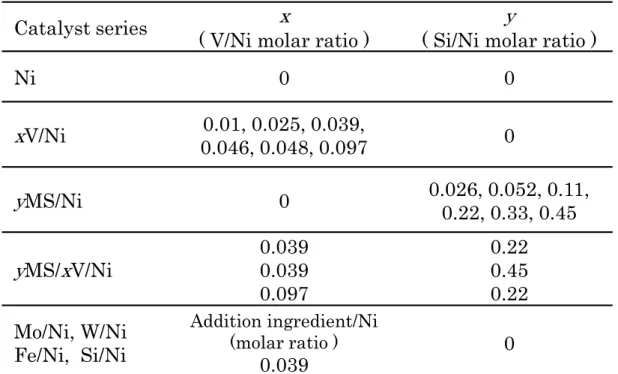
持Ni(OH)2表面にMS 被覆層のテンプレートを形成した。Si 源としてオルトケ イ酸テトラエチル(TEOS, 和光純薬工業)を用い、Ti 源としてチタン(IV)イソプ ロポキシド(Sigma-Aldrich)を用いた。例えば Si/Ni モル比 0.22 の場合、Ni(OH)2 5g に対して TEOS 4.7 g、エタノール 20.1 g、アセチルアセトン 1.0 g、チタン (IV)イソプロポキシド 0.2 g とした。Si/Ni モル比を変更する場合は、TEOS と チタン(IV)イソプロポキシドの添加量のみ変更した。CTAB を含んだ V 担持 Ni(OH)2スラリーにこのTEOS 溶液を添加し、室温で 15 時間撹拌した。その 後、ろ過して得られた固形物を洗浄し、120℃-3 時間で乾燥を行い、550℃- 3 時間で焼成を行った。 (d) 供試触媒 Table 2-1 に調製した全ての触媒試料を示した。ここで、yMS/xV/Ni の x は Ni に対する V のモル比(V/Ni)、y は Ni に対する Si のモル比(Si/Ni)である。こ れらのモル比は還元処理後の試料に対して高周波誘導結合プラズマ(ICP)発光 分析法により求めた。 2.2.2 触媒活性評価 (a) 触媒活性評価装置 Fig.2-4 に触媒活性評価装置の概略図を示す。CO 選択メタン化反応に対する 触媒特性は、非分散型赤外線吸収マルチガス分析計(VA-3000,堀場製作所製)を 装備した固定床常圧流通式反応装置で評価した。触媒活性評価装置はガス供給 系、反応系、分析系から構成されており、ガス供給系は市販の高圧ボンベから マスフローコントローラーで流量制御されたガスを反応系の反応管に導入する。 H2O はマイクロポンプでガス供給系の気化器に導入し、190℃で気化させた後 に N2 キャリアガスにより反応管へ導入した。反応管から排出された改質ガス は、冷却水サーキュレーターを通して水分を除去し、非分散型赤外線吸収マル
Table 2-1 List of catalyst samples.
Catalyst series ( V/Ni molar ratio )
x
( Si/Ni molar ratio )y
Ni 0 0
x
V/Ni 0.046, 0.048, 0.0970.01, 0.025, 0.039, 0y
MS/Ni 0 0.026, 0.052, 0.11,0.22, 0.33, 0.45y
MS/x
V/Ni 0.0390.039 0.097 0.22 0.45 0.22 Mo/Ni, W/Ni Fe/Ni, Si/Ni Addition ingredient/Ni (molar ratio ) 0.039 0Fig.2-4 Structure chart of fixed bed continuous flow reaction system.
Gas Mixer
Furnace
Thermocouple
Water trap
Pump
H
2O
Vaporizer
Mass flow
controller
Catalyst
ND-IR
analyzer
CO CO 2 H 2 N 2チガス分析計で改質ガスを分析した。 触媒活性評価用サンプルは 2.2.1 で調製した粉末触媒試料を一軸プレスで加 圧成型後、粉砕・分級し1.2~2.0 mm の粒状触媒を使用した。内径 10 mm の 石英反応管にこの粒状触媒2.2 mL を充填し、活性評価前に 100%H2気流中、 500 ℃-1 時間還元処理を行った。 (b) 温度依存性評価 入口CO 濃度 0.5%、CO2濃度20%、H2(純度 99.99%)バランス、SV4800 h-1(ド ライベース)、steam/CO 比 34 のモデルガス条件下で 150~200℃の範囲内の各 温度での触媒性能を評価した。触媒層が各設定温度に達し、定常状態となった 1 時間後の出口ガス濃度を評価した。 (c) 長期連続耐久評価 後述する様に、初期性能が最も優れていた0.22MS/0.039V/Ni について、入 口CO 濃度 0.5%、CO2濃度20%、H2(純度 99.99%)バランス、SV4800 h-1(ド ライベース)、steam/CO 比 34 のガス条件下で反応温度 183 ℃の一定条件で連 続耐久評価を行った。反応温度183 ℃は温度依存性評価から求めた操作温度領 域の中央値である。 2.2.3 触媒のキャラクタリゼージョン (a) 触媒の分析 各触媒の微細構造観察には、透過型電子顕微鏡(TEM、加速電圧 200kV:日 立ハイテクノロジーズ、H-9500)、電解放射型走査電子顕微鏡(FE-SEM、加速 電 圧 5.0kV:FEI、XL-30 SFEG)および電界放射型走査透過電子顕微鏡 (FE-STEM、加速電圧 200kV : 日本電子、JEM-ARM200F)を用いた。各種触 媒の表面元素組成と化学結合状態の分析には X 線光電子分光装置(XPS、Al K α: 島津製作所、K-1)を使用し、XPS スペクトルの帰属には NIST X 線光電子
分光法データベースを利用した。触媒の結晶構造の分析には粉末X 線回折装置 (XRD、Cu Kα : リガク, RINT-TTR3)を用いた。NiO 及び Ni 結晶子径は、 Scherrer 式から算出した。長期連続耐久試験後のサンプルの炭素種の堆積状況 を確認する為に、TPO 分析(Temperature Programmed Oxidation)を行った。 評価装置は、BELCAT(マイクロトラックベル製)を用いた。評価サンプルは 2.2.2(b) の モ デ ル ガ ス 条 件 下 で 反 応 温 度 183 ℃ で 30 分 放 置 後 の 0.22MS/0.039V/Ni と 2.2.2(c)で 700 時間連続耐久評価後のサンプルを比較した。 温度を室温から700℃まで 10℃/min で昇温し、CO2発生量を比較した。 (b) 溶液中の V イオンの吸着挙動解析 最初に、溶液中での V イオンの存在形態を解析するために、熱力学的平衡計 算ソフト“OLI Stream Analyzer 3.1”(OLI system Inc.製)を使用した。解析 条件は、バナジン酸アンモニウム水溶液(濃度 0.079 mol/L, 溶液温度 50 ℃)、 圧力1 気圧、溶液の pH 変更は硝酸及びアンモニアで行うこととした。 次に、V イオンの Ni(OH)2 に対する吸着挙動の解析を行うため、平衡吸着量 を測定した。バナジン酸アンモニウム水溶液(濃度 0.079 mol/L, 溶液液温 50℃) を密閉容器に10mL 入れ、この中に Ni(OH)2粉(0.1~1.5g)を各量添加し、50℃ のウオーターバス中で定期的に振とうしながら 24 時間浸漬した。吸着時間が 24 時間の場合、V イオンの Ni(OH)2の吸着は平衡状態であることが確認され た。その後、ろ液の濃度をICP 発光分析法によって求めた。
2.3 酸化物被覆層による Ni 焼結抑制効果の検討 2.3.1 V 担持効果
(a) V 担持 Ni(OH)2の焼成時の NiO 焼結抑制
コアシェル構造において、V 成分の特徴である反応選択性を発揮させる為に は、微細なNi(OH)2表面に均一にV 成分を析出させることが重要である。本研 究では、V 成分を Ni(OH)2表面に析出させるプロセスとして、湿式条件下でV イオンを Ni(OH)2 表面に吸着させる担持法を採用した。本節では V 担持 Ni(OH)2 を焼成した際に V が Ni に与える影響を明らかにするため、始めに Ni(OH)2への V イオンの吸着挙動を検討し、次に焼成後の V/Ni のキャラクタ リゼーションを行った。 まず本題のV イオンの吸着挙動を検討する前に、Ni(OH)2粉末の結晶形態を 詳細に観察した。Fig.2-5 に Ni(OH)2粉末のFE-SEM,TEM の観察結果を示す。 Fig.2-5(a)の FE-SEM 像より Ni(OH)2粒子は表面に微細な凹凸が入った平均粒 径約3μm の球状の形態をしていた。Fig.2-5(b)の TEM 像より Ni(OH)2粒子は、 短軸3~5 nm、長軸 15~30 nm の針状一次粒子により構成されていることが 確認された。Ni(OH)2粉末のかさ密度は0.4 g/cm3、BET 比表面積は 120 m2/g となり、比表面積の高い微細なNi(OH)2粉末であった。この様に比表面積の高 いNi(OH)2が得られた要因として、以下の様な乾燥過程による10)。 ① 噴霧された Ni(OH)2スラリーの微細な液滴が空気中に飛ばされ、表面張力 により球状になる。 ② Ni(OH)2スラリー液滴の表面から熱風気流により水分が蒸発し始める。 ③ Ni(OH)2スラリー液滴の表面に乾燥したNi(OH)2被膜が形成される。 ④ Ni(OH)2乾燥被膜を通して水分が移動蒸発する。液滴中の Ni(OH)2は水分 に運ばれて表面に移動する。
Fig.2-5 FE-SEM image and TEM image of Ni(OH)2 : (a) FE-SEM image, (b) TEM image. 5μm
(a)
100 nm(b)
⑤ 乾燥した Ni(OH)2粒子内部のNi(OH)2スラリーの粘度が上昇する。 ⑥ 乾燥した Ni(OH)2粒子内に空隙が生じ、粒子内で水分が気化し続け、乾燥 収縮していく。先に乾燥した表面に圧縮力が残り、粒子中央付近には引っ 張り応力が発生し、部分的にクラックが形成される。 スプレードライ装置より得られた比表面積の高い微細なNi(OH)2の1 次粒子 表面に各種酸化物被覆層を形成し、焼成及び還元時に Ni コア材の焼結を抑制 させることが、Ni 金属表面積の増大と Ni 充填量の増大という、一般的に相い れない課題を解決するための重要な手法になると推測する。 次にこの微細な Ni(OH)2表面へのV イオンの吸着挙動を検討するため、最初 にバナジン酸アンモニウム溶液中のV イオンの存在形態を熱力学的平衡計算ソ フトで解析した。Fig.2-6 に OLI Stream Analyzer による解析結果を示す。V イオンは溶液中でバナジン酸イオンの状態で存在しており、このイオンの存在 形態は溶液のpH に大きく影響されることが推測された。Table 2-2 に V/Ni モ ル比0.039 で V イオンを Ni(OH)2に担持する際に使用した各溶液のpH を示す。 V 酸アンモニウム溶液は担持前に pH 6.9 であったが、V 担持後は pH 8.8 まで増加した。pH 6.9~8.8 の範囲では Fig.2-5 の結果から V イオンは H2VO4 - とHVO42-が混在した状態でNi(OH)2に吸着していることが考えられる。 次にNi(OH)2表面とV イオンとの反応について考察する。pH 8.8 において V イオンは溶液中でバナジン酸陰イオンとして存在していると推測されること から、Ni(OH)2 表面のヒドロキシル基とバナジン酸陰イオンは式(2・1)及び式 (2・2)に示すイオン交換反応により結合していると考えられる。 −𝑁𝑖 − 𝑂𝐻 𝐾↔ −𝑁𝑖1 + + 𝑂𝐻−
式(2・1)
−𝑁𝑖++ 𝑉𝑂 𝑥𝑛− 𝐾2 ↔ −𝑁𝑖 − 𝑉𝑂𝑥/𝑛
式(2・2)
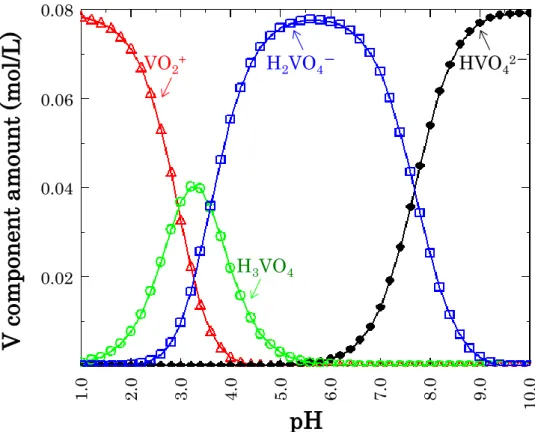
Fig.2-6 The result of OLI simulation about 0.079 mol/L NH4VO3 aqueous solution.
Table 2-2 pH data of V adsorbing process.
VO2+ H 2VO4- HVO42- H3VO4 0.02 0.04 0.06 0.08 1.0 2.0 3.0 4.0 5.0 6.0 7.0 8.0 9.0 10.0
V
c
ompo
n
en
t
am
ou
n
t
(m
ol
/L)
pH
pH@50℃
a
ammonium vanadate solution
(0.079 mol/L )
6.9
b
15% Ni(OH)
2slurry
7.0
イオン交換反応が生じた場合、(2・1)式より Ni(OH)2より排出されたOH-によ りpH は上昇すると推測される。このことは Table 2-2 の V 担持時のスラリー のpH は 8.8 まで上昇していることから裏付けられる。ただし、V 担持時の pH の上昇は遊離アンモニウムイオンの影響も考えられることから、バナジン酸ナ トリウム溶液についてTable2-2 と同様の試験を実施したところ、混合後のスラ リー溶液はpH 8.6 まで上昇することが確認された。 これまで得られた結果を整理すると、Ni(OH)2表面へのV イオンの吸着挙動 は下記の通りLangmuir 吸着等温モデルの前提条件が考えられる11)。 <Langmuir 吸着等温モデルの前提条件> ① V イオンが吸着する Ni(OH)2表面は単一成分で一定数の吸着点を有する。 ② V イオン(バナジン酸イオン)と Ni(OH)2がイオン交換反応と想定した場合、 Ni(OH)2の吸着点に1 つの V イオンが吸着する。 ③ Ni(OH)2表面に吸着したV イオン同士の相互作用はないと推測される。 ここでV イオンの Ni(OH)2への吸着挙動を解析するために、Langmuir 吸着 等温モデルが成り立つか検討する。V イオン分子を V、Ni(OH)2の吸着点をσ とした場合、次式(2・3)の平衡状態を考える。 V + σ
⇋
V・σ 式(2・3) V・σは V イオンが吸着されている Ni(OH)2の吸着点を表す。V・σの割合を θとし、未反応の溶液中のV の濃度を c、V が吸着されていない Ni(OH)2の吸 着点σの割合を 1‐θとした場合、吸着速度( )は次式(2・4)で表すことがで きる。 ① 吸着速度式 式(2・4) ここで、k は吸着速度定数、m は担体 1 mol あたりの V イオン吸着量、A は Ni(OH)2 1 mol あたりの V イオン最大吸着量と定義する。m は V イオンの仕込
A
m
kc
kc
v
1
1
v
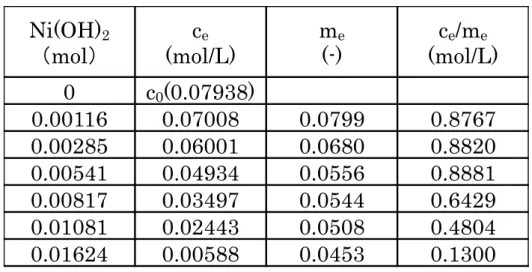
み濃度c0 (mol/L)、仕込み液量 L (L)、Ni(OH)2仕込み量M (mol)とすると、 m=(c0-c)L/M から算出される。 一方、式(2・4)の逆反応である脱着速度( )は V・σの濃度すなわちθに比 例することから、式(2・5)で表される。 ② 脱着速度式 式(2・5) k'は脱着速度定数と定義する。 式(2・3)の速度式は式(2・4)と式(2・5)を合わせた式になることから、吸着/脱 着を合わせた速度式は式(2・6)で表される。 ③ 吸着/脱着を合わせた速度式 式(2・6) ここでV イオンの Ni(OH)2への吸着が平衡状態に達すると となり、v=0 となることから、式(2・6)を変形していくと、式(2・7)の Langmuir 式が得ら れた。 として更に整理すると、 式(2・7) b は吸着平衡定数となる。 Table 2-3 に V イオンを Ni(OH)2に担持させたときの平衡吸着試験の結果を 示す。Fig.2-7 には Table 2-3 について式(2・7)の Langmuir 式に従い ceとce/me をプロットした結果を示す。ce が 0.05 以下で直線関係が成り立つことから、 この領域の V イオンの吸着は Langmuir 型の吸着であると推測できる。 Langmuir プロットから直線の傾き(1/A)は 17.4 であることから、A は 0.0575
A
m
k
k
v
'
' A m k A m kc v v dt dc v 1 ' v vA
m
k
A
m
kc
e e e 1 '
e
e eA
m
m
c
k
k
' k' k b bA
c
A
m
c
e e e1
1
v
Table 2-3 The result of V adsorbing test .
Fig.2-7 Langmuir plot of V adsorbing process.
y =
17.4
x + 0.0377
R² = 0.9984
0
0.2
0.4
0.6
0.8
1
0
0.02
0.04
0.06
0.08
c
e(mol/L)
c
e/m
e(m
ol
/L)
Ni(OH)
2(mol)
(mol/L)
c
em
(-)
e(mol/L)
c
e/m
e0
c
0(0.07938)
0.00116
0.07008
0.0799
0.8767
0.00285
0.06001
0.0680
0.8820
0.00541
0.04934
0.0556
0.8881
0.00817
0.03497
0.0544
0.6429
0.01081
0.02443
0.0508
0.4804
0.01624
0.00588
0.0453
0.1300
mol が得られた。切片(1/bA)は 0.0377 となり、吸着平衡定数 b を計算すると 460.5 となった。本試験で実施した V イオンの Ni(OH)2への吸着条件では、V/Ni モル比0.039 が上限であり、理論値の 6 割程度の V イオンしか担持されていな い。V/Ni モル比が 0.039 以下ならば V イオンは Langumuir モデルに従い、V イオンは単分子層でNi(OH)2に吸着していると考えられる。
次にV 担持 Ni(OH)2を焼成したyV/Ni について検討する。Fig.2-8 に横軸に 触媒調製時の仕込みV/Ni モル比、縦軸には XPS から測定した V/Ni モル比の 関係を示す。V/Ni モル比が 0.039 までは直線関係が成り立つことから、V は NiO 表面に均一に分散していると推測される。Fig.2-9 に焼成後の 0.039V/Ni のSTEM-EDS 観察結果を示す。Fig.2-9(b)は Fig.2-9(a)中に示された点線の四 角の箇所を拡大したSTEM 像である。Fig.2-9(a)より焼成後の 0.039V/Ni は針 状結晶の凝集粒子で構成されていた。Fig.2-9(b)の点線で示した針状粒子が、こ の凝集粒子中の一次粒子に該当する。Fig.2-9(a) 中に挿入した 0.039V/Ni の制 限視野電子線回折パターンを解析した結果、NiO の面心立方格子に帰属された。 Fig.2-9(c, V)及び Fig.2-9(d, Ni)の EDS マッピング像から V 成分は NiO 一次粒 子の表面に均一に分散していた。しかしながら、V 成分に関係する結晶粒子な どはSTEM 像より確認されなかった。Fig.2-10 に焼成後の 0.039V/Ni の XRD 及びXPS 測定結果を示す。Fig.2-10(a)では焼成後の 0.039V/Ni の X 線回折ピ ークからは、V 成分に帰属される結晶相は確認されなかった。一方で、 Fig.2-10(b)に示した焼成後の 0.039V/Ni の X 線光電子スペクトルを解析したと
ころ、V2p3/2結合エネルギーが517 eV で V2O5に帰属されるピークが確認され
た。Fig.2-10(b)の 525eV および 531eV 付近で確認された他の XPS ピークは、
それぞれ V2p1/2と O1sに帰属することができた。以上より焼成後の 0.039V/Ni
について、V 成分は NiO 一次粒子の表面で均一に分散しており、その析出形態 はアモルファス状あるいはV2O5の極めて微細な結晶として存在していると推
Fig.2-8 V/Ni molar ratio of the charge composition by ICP and observed by XPS of xV/Ni samples.
0
0.01
0.02
0.03
0.04
0.05
0.06
0
0.01
0.02
0.03
0.04
V/Ni molar ratio by ICP ( -)
V
/N
i m
olar
ra
tio
by
X
P
S
(-
)
Fig.2-9 FE-STEM images and EDS mappings of 0.039V/Ni after calcination: (a),(b) HAADF-STEM images, (c) V mapping, (d) Ni mapping. Inset of (a) is the electron diffraction pattern.
10nm
10nm
10nm
(a)
(b)
(c)
(d)
100nm
Fig.2-10 XRD patterns and XPS spectrums of 0.039V/Ni: (a) XRD and (b) XPS after calcination.
535 530 525 520 515 510
Binding energy (eV)
3000 2500 2000 1500 1000
In
te
nsi
ty
(
cps
)
(b)
0 0.2 0.4 0.6 0.8 1 10 20 30 40 50 60 70 80 NiO (20 0) NiO (1 11 ) N iO (22 0) NiO (31 1) N IO (22 2)(a)
2θ (deg.)
Intens
ity
(cps
)
×
10
5測される。これは V イオンが Ni(OH)2に吸着する際の Langmuir 型の吸着挙 動に起因していると考えられる。
(b) V 担持 NiO の還元時の Ni 焼結抑制
Fig.2-11 に FE-SEM で観察した還元後の V/Ni の結晶形態を示す。NiO から Ni への還元に伴い、結晶形態は針状結晶(Fig.2-9 の数 nm 程度)から粒状結晶 (100~200nm 程度)へ大きく変化した。V 含有量の増加に伴い、V/Ni 粒子は微 細化していく傾向となった。Fig.2-12 に還元後の 0.039V/Ni の XRD 及び XPS 測定結果を示す。Fig.2-12(a)に示した還元後の 0.039V/Ni の X 線回折パターン で確認された鋭い回折ピークは Ni 金属(PDF 071-3740)に帰属された。 Fig.2-12(b)に示した還元後の 0.039V/Ni の X 線光電子スペクトルを解析したと ころ、V2p3/2結合エネルギーが516 eV で V2O3に帰属されるピークが確認され
た。Fig.2-12(b)の 525eV および 531eV 付近で確認された他の XPS ピークは、
それぞれ V2p1/2と O1sに帰属することができた。Fig.2-13 に還元後の V/Ni サ
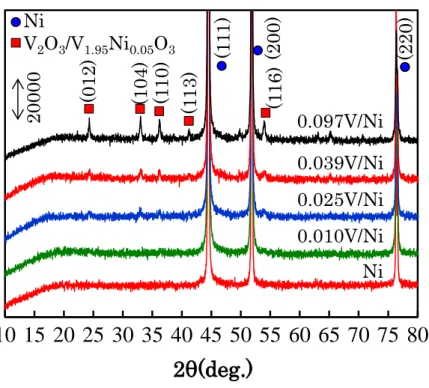
ン プ ル の X 線 回 折 像 を 示 す 。 V/Ni モ ル 比 が 0.025 を 超過 し た 場 合 、 V2O3(PDF085-1411)あるいは V1.95Ni0.05O3(PDF 072-5931)に帰属される回折 ピークが確認された。
2.3.2 V 担持及び MS 被覆層の効果
Fig.2-14 に Ni、V/Ni および MS/V/Ni について還元前後の NiO と Ni の結晶 子径を示す。Ni のみの場合、NiO(焼成後)および Ni(還元後)の結晶子径はそれ ぞれ27nm および 78nm であった。一方 0.039V/Ni では結晶子径の減少が確認 され、焼成後のNiO 結晶子径は 8.4nm となり、還元後の Ni 結晶子径は 49.9nm となった。V の添加は還元時に NiO から Ni へ相転移する際の Ni の焼結抑制 に対して、多少の効果を有していると言える。次に0.22MS/Ni では NiO の結 晶子径はほとんどV/Ni と同等レベルであったが、還元後の Ni 結晶子径は
Fig.2-11 FE-SEM images of the series of xV/Ni sample after reduction: (a) Ni, (b) 0.010V/Ni, (c) 0.039V/Ni, (d) 0.097V/Ni.
200nm
(a)
(b)
Fig.2-12 XRD patterns and XPS spectrums of 0.039V/Ni: (a) XRD and (b) XPS after reduction.
In
te
n
si
ty
(c
ps
)
535 530 525 520 515 510Binding energy (eV)
3000 2500 2000 1500 1000
(b)
0 1 2 3 4 10 20 30 40 50 60 70 80 N i(20 0) N i(1 11 ) N i(22 0)(a)
In
te
n
si
ty
(c
ps)
×
10
52θ (deg.)
Fig. 2-13 XRD patterns of V/Ni samples after reduction.
Fig.2-14 NiO and Ni crystallite size of Ni and V/Ni, MS/V/Ni after reduction.
10 15 20 25 30 35 40 45 50 55 60 65 70 75 80
2θ(deg.)
(01 2) (10 4) (1 10 ) (1 13 ) (1 16 ) (1 11 ) (20 0) (22 0) 0.097V/Ni 0.039V/Ni 0.025V/Ni 0.010V/Ni Ni V2O3/V1.95Ni0.05O3 Ni 20000 0 10 20 30 40 50 60 70 80 0 10 20 30 40 50 60 70 80N
iO
cr
ysta
ll
it
e
si
ze
(n
m)
N
i c
ryst
al
li
te
s
ize
(n
m)
after reduction after calcination Ni xV/Ni yMS/xV/Ni x 0 0.039 0.097 0 0.039 0.097 0.039 y 0 0 0 0.22 0.22 0.22 0.4515.1nm であった。MS 被覆層は Ni の焼結を著しく抑制していた。MS/V/Ni である0.22MS/0.39V/Ni では Ni 結晶子径は 10.6nm となり、0.22MS/Ni と比 較すると若干ではあるがNi 結晶子径の更なる減少が確認された。この現象は V 添加とMS 被覆層による相乗効果を示している。V や MS 被覆層を更に増やし た 場 合(0.22MS/0.097/Ni, 0.45MS/0.039/Ni)、Ni 及び NiO の結晶子径は 0.22MS/0.039V/Ni と殆ど変らなかった。 ここで V および MS 被覆層により NiO 及び Ni の焼結抑制効果が得られた要 因を考察する。V 及び MS 被覆層の共通点として、Ni(OH)2に被覆する際にV 及び Si は溶液中でオキソ陰イオンとして存在している点が挙げられる 12)。溶 液中でNi(OH)2表面に担持されたこれらのオキソ陰イオンが乾燥時に脱水縮合 反応により酸素原子が基本単位間を架橋することで、多彩な構造の酸化物被覆 層をNi(OH)2表面に形成していると推測される。この様にNi(OH)2の粒界に酸 化物被覆層が形成されることで、焼成時及び還元時に Ni 同士の接触を防止す る仕切り材として働き、その結果焼結が抑制されたと考えられる。そこで、 Fig.2-15 に Ni(OH)2表面に各種成分を担持した際の還元前後のNiO と Ni 結晶 子径を示す。溶液中でポリ陰イオンを形成する成分を添加した V/Ni、Mo/Ni、 W/Ni、Si/Ni は還元前後で NiO 及び Ni 結晶子径は無担持の Ni のみと比較し て大幅に低減した。一方、ポリ陰イオンを形成しない成分を添加したFe/Ni は 無担持のNi のみより NiO 及び Ni の結晶子径の低下が若干確認できたものの、 他の成分と比べて焼結抑制は明らかに低い。V/Ni、Mo/Ni、W/Ni、Si/Ni を比 較すると、焼成後のNiO 結晶子径は殆ど変らないが、還元後では Ni 結晶子径 の序列がSi/Ni>W/Ni>Mo/Ni>V/Ni の順になった。この序列は溶液中でのポ リ陰イオンの形成しやすさの傾向に一致していた12)。 Fig.2-16 に V 及び SiO2含有量に対する還元後のNi の結晶子径との関係を示 す。コア材がNi のみで構成されている場合、Ni の結晶子径は 78nm であった
Fig.2-15 NiO and Ni crystallite size of various component added Ni after reduction of after reduction.
0 10 20 30 40 50 60 70 80 0 10 20 30 40 50 60 70 80 Ni only V -added Ni
N
iO
cr
ysta
ll
it
e
si
ze
(n
m)
N
i c
rystal
li
te
s
iz
e
(n
m)
after reduction after calcination Mo -added Ni W -added Ni Si -added Ni Fe -added NiFig.2-16 Change in Ni crystallite size of Ni, V/Ni , MS/Ni and MS/Ni/AlVOx samples with V/Ni or Si/Ni values.
0
10
20
30
40
50
60
70
80
90
0
0.1
0.2
0.3
0.4
0.5
N
i c
ry
sta
lli
te
si
ze
(n
m)
V/Ni or Si/Ni (molar ratio)
Ni
V/Ni
MS/Ni
(y 軸上の○)。V 含有量が V/Ni モル比 0.097(■)まで増加するに伴い、Ni 結晶 子径は連続的に減少した。MS 被覆層(△)は V 添加と比較して Ni の焼結抑制に 対して更に有効であることが明らかとなった。MS 被覆量が Si/Ni モル比 0.22 まで増加するに従い、Ni 結晶子径は連続的に減少した。MS 被覆量が Si/Ni モ ル比0.22 を超過すると、Ni 結晶子径はほとんど一定になった。この結果は、 特に MS 被覆層によって、還元中に Ni の焼結が有効に抑制されていることを 示す。またMS/Ni-AlVOx(●)は還元後の Ni 結晶子径が 40nm となり、MS 被 覆層(△)よりの 2 倍の Ni 結晶子径であった。これは微細な Ni ナノ粒子に直接 MS 被覆層を形成しているため、還元後も Ni 結晶子径が小さく維持されたと推 測する。CO メタン化反応は構造敏感反応と知られており13),14)、微細なNi 粒 子である程、より高い触媒活性を示す傾向となる。今回の研究で確認された MS 被覆層による著しい Ni の焼結抑制は、Ni 表面積だけでなく構造敏感性に よる触媒活性の改良に対して明確な効果を有すると考えられる。
Fig.2-17 に還元後の 0.22MS/0.039V/Ni の TEM 観察結果を示す。Fig.2-17 よりV/Ni コア粒子の表面は MS 層で均一に被覆されていた。この MS 被覆層 はコア材の表面上に垂直で隙間のない規律正しいメゾポア孔を形成し、MS 被 覆層の厚みは平均30nm であった。Fig.2-18 に還元後の 0.22MS/0.039V/Ni の 高解像度SEM 像および EDS マッピング像を示す。Fig.2-11(c)で示した MS 被 覆 層 を 形 成 し て い な い 0.039V/Ni と 比 較 し て 、 MS 被 覆 層 を 形 成 し た 0.22MS/0.039V/Ni の方は非常に微細な Ni 粒子を有していると言える。また Fig.2-18(c),(d)より、Si 及び V 成分は Ni 表面上で均一に分散していた。 以上より、MS/V/Ni はコアシェル構造を有していると考えられる。また、CO パルス吸着測定によってMS 被覆層のガス透過性を確認することができた。さ らに、0.22MS/0.039V/Ni の BET 比表面積は 307 m2/g と比較的高く、MS 被 覆層は高い多孔質構造を有していることが言える。
Fig.2-17 TEM image of 0.22MS/0.039V/Ni sample after reduction.
100 nm
0.039V/Ni core
Fig.2-18 SEM images of 0.22MS/0.039V/Ni after reduction: (a) SEM image and EDS mappings (b) Ni, (c) Si, (d) V .