九州大学学術情報リポジトリ
Kyushu University Institutional Repository
カリウム選択性マンガン酸化物を用いた土壌のカリ ウム供給能評価法に関する研究
松田, 亜由美
https://doi.org/10.15017/1931968
出版情報:Kyushu University, 2017, 博士(農学), 課程博士 バージョン:
権利関係:
カリウム選択性マンガン酸化物を用いた 土壌のカリウム供給能評価法に関する研究
松田 亜由美
2018
目次
1 序論 ... 1
1.1 カリウムの地球化学 ... 1
1.2 Kの生物科学 ... 3
1.3 Kの土壌化学 ... 4
1.3.1 土壌中のKの存在形態 ... 4
1.4 土壌のK供給能の評価法 ... 8
1.4.1 植物の利用性からみた土壌のKの区分とそれらの評価法 ... 8
1.4.2 4区分との関連付けをしない土壌のK供給能評価 ... 10
1.4.3 既存のK供給能評価法の不十分な点 ... 10
1.5 この論文の構成 ... 11
2 カリウム選択的マンガン酸化物の調製に関する研究 ... 14
2.1 はじめに ... 14
2.2 実験方法 ... 15
2.2.1 K型マンガン酸化物の調製 ... 15
2.2.2 アンモニウム型マンガン酸化物の調製と酸洗浄方法の検討... 16
2.2.3 K吸着特性評価 ... 17
2.2.4 回収法の検討 ... 17
2.3 結果と考察 ... 18
2.3.1 K型マンガン酸化物の調製 ... 18
2.3.2 アンモニウム型マンガン酸化物の調製と酸洗浄方法の検討... 22
2.3.3 K吸着特性評価 ... 24
2.3.4 回収法の検討 ... 27
2.4 まとめ ... 28
3 土壌との接触試験条件に関する研究 ... 29
3.1 はじめに ... 29
3.2 試料と実験方法 ... 30
3.2.1 試料 ... 30
3.2.2 土壌充填量の決定 ... 31
3.2.3 固液比の決定 ... 32
3.3 結果と考察 ... 32
3.3.1 土壌充填量の決定 ... 32
3.3.2 固液比の決定 ... 33
3.4 まとめ ... 34
4 土壌との接触試験および粒径別試料を用いた接触試験に関する研究 ... 35
4.1 はじめに ... 35
4.2 試料と実験方法 ... 35
4.2.1 土壌試料の粒径分画 ... 35
4.2.2 土壌との接触試験 ... 36
4.2.3 粒径別試料を用いた接触試験 ... 37
4.3 結果と考察 ... 38
4.3.1 土壌との接触試験 ... 38
4.3.2 粒径別試料を用いた接触試験 ... 40
4.4 まとめ ... 42
5 総括 ... 43
1
1 序論
1.1 カリウムの地球化学
カリウム(K)は原子番号が19の元素で,第1族,第4周期元素に属する.元素周期表では,第 1族第3周期のナトリウム(Na)と第5周期のルビジウム(Rb)に挟まれた箇所に位置する.基底 状態の電子配置は,1s22s22p63s23p64s1であるが,閉殻構造は電子を1つ失った状態のものであるため,
自然界では1価の陽イオンとして存在する.Kは24種類の同位体を持つことでも知られているが,
中でも 39K, 40K, 41K の 3 種類は天然での存在割合が大きく,その割合はそれぞれ約 93.3%, 0.012%,
6.7%である(放射線医学総合研究所, 2002).39Kと41Kは安定同位体であるが,40Kは約1.277×109
年という半減期をもった放射性同位体である.約89%は安定なカルシウム-40(40Ca)にβ崩壊し,
約 11%は電子軌道捕獲され,安定なアルゴン-40(40Ar)に変換する(放射線医学総合研究所, 2002).
地殻構成主要元素の存在度を表 1.1に示す(Yaroshevsky, 2006).地殻の平均K 含量は質量ベー ス(mg kg-1)では25,000 mg kg-1であり,第1族のNaと等しい存在度である.第2族元素であるマ グネシウム(Mg)とカルシウム(Ca)の存在度もそれぞれ18,700 mg kg-1と29,600 mg kg-1であるた めKと近い存在度であるといえる.また,物質量ベース(mmol kg-1)でみた場合,Na, K, Mg, Caの 存在度はそれぞれ1,090, 831, 769, 738 mmol kg-1であるため,物質量ベースでも近い存在度であると いえる.地殻の元素組成はデーターベースによって大きく異なるが,上述した質量,物質量ベース の存在度はいずれのデーターベースでも同じ傾向がある.
表 1.1 地殻構成主要元素の存在度(Yaroshevsky, 2006)
原子番号 元素名 存在度(mg kg-1) 存在度(mmol kg-1) 3
6 7 8 11 12 13 14 15 19 20 26
リチウム (Li) 炭素 (C) 窒素 (N) 酸素 (O)
ナトリウム (Na) マグネシウム (Mg) アルミニウム (Al) ケイ素 (Si)
リン (P) カリウム (K) カルシウム (Ca) 鉄 (Fe)
32 230 19 470000 25,000 18,700 80,500 323,000 930 25,000 29,600 46,500
4.61 19.1 1.35 29,400 1,090 769 2,980 11,500 30.0 831 738 833
地殻に存在するKは造岩鉱物の構成元素として存在しており,その代表的なものとしてチョウセ キやウンモが挙げられる.チョウセキは地殻中で最も主要な造岩鉱物であり,複数の構造を持つ鉱
2
物である.チョウセキはアルカリチョウセキとシャチョウセキの 2 つの主要なサブグループに分か れている.アルカリチョウセキ(KAlSi3O8, NaAlSi3O8)のうち K 含量が最も高いものはカリチョウ セキと呼ばれる.シャチョウセキは固溶体の一つで,ソウチョウセキ(NaAlSi3O8)からカイチョウセ キ(CaAl2Si2O8)までの一連の鉱物の単成分で構成される.ウンモはK含量の高い鉱物であり,シロ ウ ン モ (KAl2AlSi3O10(OH)2) や ク ロ ウ ン モ な ど が 挙 げ ら れ る . ク ロ ウ ン モ は キ ン ウ ン モ
(KMg3AlSi3O10(OH)2)とテツウンモ(KFe3AlSi3O10(OH)2)の中間組成の固溶体である.
造岩鉱物が地球表層で降雨などの液体水にさらされると,溶解したり,液体水由来のプロトンと 反応して次第に変質する.この変質作用によってプロトンとの陽イオン交換が生じ,Na, Mg, K, Ca などのアルカリ金属イオンやアルカリ土類金属イオンが溶出する(Sposit, 1989; 和田,2017).陽イ オン交換によるイオンの溶出が継続すると,液体水にさらされた造岩鉱物は別の鉱物に構造変化す る.もとの造岩鉱物は 1 次鉱物と呼ばれ,2 次的に生成された鉱物は 2 次鉱物と呼ばれる(和田, 2017).
地球の海水は造岩鉱物から溶出したイオンが集積したものである.表 1.2 に海水中の主要溶質組 成を示す(Millero et al, 2008).Na, Mg, K, Caの各イオン濃度(mol L-1)をみると,最も高濃度で含 まれるのはナトリウムイオン(Na+)であり,その濃度は約0.480 mol L-1である.2番目に高い濃度 のイオンはマグネシウムイオン(Mg2+)であり約0.054 mol L-1である.カリウムイオン(K+)とカル シウムイオン(Ca2+)はこれら2つのイオンよりも濃度ははるかに低く,いずれも約0.010 mol L-1で ある.地殻内のNa, Mg, K, Caの含有量を表 1.1の物質量ベースで,Caを基準にみると,1.48:1.04:
1.12:1.00である.一方,海水内の濃度(mol L-1)を同じようにCaを基準にみると,45.58:5.13:
0.99:1.00である.これら2つの比を比較した場合,海水中のK, Mg, Caの存在割合はNaよりも低
いことが分かる.これは造岩鉱物から溶出したイオンのうち,Mg, K, Caは海水に流入する割合が低 く,海水に流入する前段階に除去されていることを示している.
これについては既往の研究で明らかにされている.K, Mg は造岩鉱物の風化によって 2 次鉱物に 取り込まれる割合が高く,Caは主に炭酸カルシウム(CaCO3)として除去されている(Kramer, 1965).
3
表 1.2 海水の主要溶質濃度 (Millero et al., 2008)
溶質 式量 濃度(g kg-1) 濃度(mol L-1) ナトリウムイオン(Na+) 22.9897 10.78 0.4796 マグネシウムイオン(Mg2+) 24.3050 1.283 0.0540 カリウムイオン(K+) 39.0983 0.3991 0.010447 カルシウムイオン(Ca2+) 40.0780 0.4120 0.010523 ストロンチウムイオン(Sr2+) 87.6200 0.0079 0.0000928 塩化物イオン(Cl-) 35.4530 19.35 0.5586 硫酸イオン(SO42-) 96.0626 2.712 0.02889 炭酸水素イオン(HCO3-) 61.0168 0.1048 0.001758 臭化物イオン(Br-) 79.9040 0.0672 0.0008618 炭酸イオン(CO32-) 60.0089 0.01434 0.0002446 ホウ酸イオン(B(OH)4-) 78.8403 0.00795 0.0001031
1.2 K の生物科学
K は植物にとっての必須多量元素である.表 1.3 に被子植物の平均必須元素含量と吸収形態を示 す(松本・三枝,1998).被子植物は乾物当たりで平均14,000 mg kg-1のKを含んでおり,これは炭 素(C),水素(H),酸素(O),窒素(N)を除けば,Ca に次いで高い含量である.植物によっ ては,CaよりもKを多く吸収するものもある(間藤ら,2001).
植物が吸収できるKの形態は,土壌の間隙水に溶存したK+としての形態であり,植物は全てのK+ を根から吸収する.Kは細胞質におけるpHや浸透圧の調節,多種の酵素の活性化に関与しており,
欠乏した場合は葉に褐色斑点が現れ下葉から枯れ上がる.また土壌内に過剰にカリ肥料が投入され た場合,マグネシウム欠乏を引き起こすこともある(日本作物学会,20100).
4
表 1.3 被子植物の平均必須元素含量(乾物当たり)と主な吸収形態(松本・三枝,1998)
元素名 元素記号 平均含量 (mg kg-1) 根からの吸収形態
炭素 C 454,000 CO2(葉から)
水素 H 55,000 H2O
酸素 O 410,000 H2O,O2(葉から)
窒素 N 30,000 NH4+,NO3-
リン P 2,300 H2PO4-,HPO42-
カリウム K 14,000 K+
硫黄 S 3,400 SO42-
カルシウム Ca 18,000 Ca2+
マグネシウム Mg 3,200 Mg2+
ホウ素 B 50 H3BO3
塩素 Cl 2,000 Cl-
鉄 Fe 140 Fe3+, Fe2+
マンガン Mn 630 Mn2+
銅 Cu 14 Cu2+
亜鉛 Zn 160 Zn2+
ニッケル Ni 3 Ni2+
モリブデン Mo 0.9 MoO42-
1.3 K の土壌化学
1.3.1 土壌中のKの存在形態
土壌は,造岩鉱物と造岩鉱物が構造変化した2 次鉱物と有機物の混合物である.間隙には2次鉱 物が生成される過程で液体水にアルカリ金属イオンやアルカリ土類金属イオンなどが溶け込んだ電 解質水溶液が保持される(和田,2017).この間隙水は,土壌水,土中水や土壌溶液などと呼ばれ,
K はK+としてこの土壌溶液中に溶存する.また,1.1 で述べたように造岩鉱物や 2 次鉱物の構成元 素として存在する.
表 1.4に地殻中および土壌中の主要元素含量と,土壌生成過程での富化率(土壌の含有量/地殻の 含有量)を示す.土壌中の主要元素含量はSposito (1989) の数値を引用し,地殻中の含有量は表 1.1 の存在度(mmol kg-1)と同じものを示している.土壌では炭素(C)と窒素(N)の富化率が非常に 高いことが分かる.最も土壌中の含有量の多い元素であるケイ素(Si)とアルミニウム(Al)の富化 率は1に近く,アルカリ金属元素やアルカリ土類金属元素の富化率は0.5程度である.
SiとAlの富化率が1に近いのは,多くの2次鉱物を構成する主要な構成元素がSiとAlであるた めである.アルカリ金属元素とアルカリ金属元素の富化率が0.5程度しかないのは,2次鉱物が生成 される過程でこれらの元素が溶脱するためである(和田,2017).
5
表 1.4 地殻中および土壌中の主要元素含量と土壌生成における富化率 原子番号 元素 地殻中の含有量 土壌中の含有量 土壌中の富化率*
---mmol kg-1---
3 Li 4.61 3.45 0.75
6 C 19.1 2,080 108
7 N 1.35 142 105
11 Na 1,090 522 0.47
12 Mg 769 370 0.48
13 Al 2,980 2,670 0.89
14 Si 11,500 11,000 0.96
15 P 30.0 13.9 0.46
19 K 831 384 0.46
20 Ca 738 599 0.81
26 Fe 833 465 0.55
*土壌中の含有量/地殻中の含有量
表 1.5 には土壌に含まれる代表的な2 次鉱物とその化学組成を示す.鉱物の産地や生成条件によ って組成に大きな幅があるため,この表の化学組成には平均的なものを示した.表から構造内にNa+ やCa2+を含む 2次鉱物がないことが分かる.これらの2 次鉱物のうち,土壌内の含量的に重要なも のは順に,層状ケイ酸塩鉱物,鉄の酸化物・水酸化物鉱物である.火山地帯に分布する火山灰由来の 土壌ではアロフェンおよびイモゴライトが土壌の主要2次鉱物であることもある.
層状ケイ酸塩鉱物は火成岩に含まれるケイ酸塩鉱物の不調和溶解によって生成され,1:1 型鉱物 は四面体層と八面体層が1層ずつの結晶単位層を形成する.2:1型鉱物は八面体層の両面を四面体 層で挟むような構造を形成し,2:1:1型鉱物は2:1型結晶単位層の間に八面体層が挟まった構造 を持つ.
2:1型鉱物は四面体層を構成するケイ素イオン(Si4+)がアルミニウムイオン(Al3+)と,また八 面体層を構成する Al3+が鉄イオン(Fe2+)や Mg2+と同形置換した場合,単位層は電気的に中性にな らず負電荷を帯びる.この負電荷を中和するため,単位層表面には何らかの陽イオンが静電気的に 吸着保持される(久馬,1997).表 1.5に示した2:1 型層状ケイ酸塩鉱物の組成式の太字のMは このような陽イオンを示している.スメクタイトグループやバーミキュライトグループの鉱物に吸 着する陽イオンの大部分はCa2+, Mg2+, K+, Na+である.水田のような湛水下の作土層では,還元的な 環境でもあることから,これらの陽イオンに加えてアンモニウムイオン(NH4+)や Fe2+が保持され ていることもある.一方,イライトグループの鉱物に吸着する陽イオンは全てK+である.植物のK の供給源として層状ケイ酸塩鉱物に保持されるKは最も重要なものであるといえる.
6
表 1.5 土壌の主要な2次鉱物とその化学組成
ケ イ 酸 塩 鉱 物
鉱物グループおよび鉱物名 化学組成
層状ケイ酸塩鉱物 1:1型鉱物 カオリナイト
ハロイサイト(10 Å)
ハロイサイト(7 Å)
2:1型鉱物 スメクタイト バーミキュライト イライト
2:1:1型
クロライト(2八面体)
クロライト(3八面体)
Si4Al4O10(OH)8
Si4Al4O10(OH)8·4H2O Si4Al4O10(OH)8
M0.66 Si8(Al3.34Mg0.66)O20(OH)4
M1.5(Si7Al)(Al3Fe0.5Mg0.5)O20(OH)4
K1.95(Si6.8Al1.2)(Al3Fe0.25Mg0.75)O20(OH)4
(Mg, Al)9.2~10(Si, Al)8O20(OH)16
(Mg10Al2)(Si6Al2)O20(OH)16
アロフェン・
イモゴライト
アロフェン イモゴライト
1.5SiO2·Al2O3
SiO2·Al2O3
酸 化
・ 水 酸 化 物 鉱 物
ケイ素の酸化物 酸化物 セキエイ 非晶質シリカ
SiO2
SiO2·nH2O アルミニウムの
水酸化物
水酸化物
ギブサイト Al(OH)3
鉄の酸化物・
水酸化物
酸化物 ヘマタイト 酸化水酸化物 ゲータイト
フェリハイドライト レピドクロサイト
Fe2O3
α-FeOOH 5Fe2O3・9H2O γ-FeOOH マンガンの酸化物 酸化物
パイロルーサイト バーネサイト
MnO2
MnO2
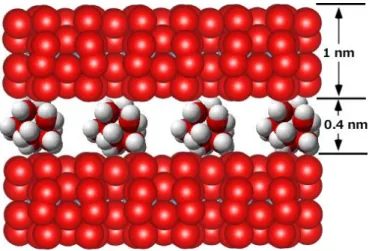
図 1.1 は層状ケイ酸塩鉱物であるスメクタイトグループとバーミキュライトグループの吸着陽イ オンの存在状態を模式的に示す(和田,2017).この 2 つのグループの鉱物は単位層の負電荷のバ ランスをとるため,水和陽イオンが保持される.図は 6 配位の水和陽イオンが保持されている様子 を示す.この場合には,厚さ1 nmの単位層が0.4 nmの水和イオン層を挟んで積層している.水和陽 イオンが層内に保持されている場合,単位層の負電荷と保持されている陽イオンは水分子によって 隔てられているため,作用する静電気力は弱く,層間は広くなる.そのため,鉱物粒子の外部から別
7
の水和陽イオンが層間に侵入し,もともと存在した陽イオンと交換するという現象が生じる.この 現象は陽イオン交換と呼ばれ,陽イオン交換される吸着イオンは交換性陽イオンと呼ばれる.
土壌の間隙水(土壌溶液)中のK+が植物根によって吸収されると,間隙水中のK+の相対的な濃度 は低下する.この結果,K+以外の陽イオンが層間に侵入する確率は高くなり,陽イオン交換によっ てK+が間隙水に溶出する.この陽イオン交換反応は,土壌の植物へのK供給に関わる反応として非 常に重要である.
図 1.1 スメクタイトやバーミキュライトグループの鉱物における吸着イオンの存在状態.
作図はVesta(Momma and Izumi, 2011)によった.
図 1.2にイライトグループの吸着陽イオンの存在状態を模式的に示す(和田,2017).イライトグ ループの鉱物では,単位層の負電荷を中和するために保持される陽イオンは全てK+である.スメク タイト,バーミキュライトグループとは異なり,水和イオンとしての陽イオンではなく,裸のK+と して単位層間に保持される.K+の大きさは,四面体層底面の酸素原子層のくぼみの大きさとほぼ等 しいため,K+はくぼみにぴったりと収まった状態で単位層間に挟まれることになる.イライトグル ープは,図 1.2の右図のように層間がほとんど閉じた状態となっており,保持されたK+は外部の陽 イオンとは非常に交換しにくいという特徴を持っている.
図 1.2はイライトグループの鉱物におけるKの存在様式を示したものではあるが,この構造は造 岩鉱物であるウンモにおいてもほとんど同様である.イライトとの違いは,ウンモの単位層の負電 荷量とK保持量がイライトよりも多いという点である.イライト,ウンモのような閉じた層間に保 持されたK+は陽イオン交換されにくい.しかし,全く陽イオン交換されないものではなく,層状鉱 物粒子の縁辺部に保持されたK+はある程度Ca2+などと交換し得ると考えられている.
8
図 1.2 イライトグループの鉱物におけるK+の存在状態.紫球はK+を示す.
左図は棒球モデル,右図は同じ構造の充填モデルを示す.
1.4 土壌の K 供給能の評価法
1.4.1 植物の利用性からみた土壌のKの区分とそれらの評価法
土壌のK は土壌分析においては,従来 4 種類の形態に分けられている(代表的なものは Helmke and Sparks, 1996).
1) 土壌溶液溶存態
2) 交換態
3) 非交換態(固定態)
4) 鉱物態(構造態)
これらのうち1)は最も植物に利用されやすい形態であり,次いで2), 3)が利用される.4)の利用性 は非常に低いと考えられている.従って,土壌のK供給能は上記の1)から3)の区分のK量を測定す ることによって評価されてきた.最も信頼性の高いK供給能評価法は,評価対象である植物を評価 対象の土壌で生育させ,実際に植物が吸収したK量を測定するという方法で栽培試験と呼ばれる方 法である.栽培試験の中でも,カリ肥料を無施用で行う三要素試験は土壌のK供給能の評価法とし て適している.
1)と 4)は存在形態にもとづいた区分であるが,3)と 4)は特定の条件下で陽イオン交換反応のしや
すさにもとづいた区分であるため,条件を厳密に指定しない限り,2)と3)の区分は曖昧である.土壌 の鉱物を原子間力顕微鏡のような高分解能で観察できたとしても,鉱物に含まれる陽イオンのどれ が交換反応するかを判別することはできない.また,ウンモやチョウセキのように,構造を構成す るKが部分的に陽イオン交換反応するものもある.これらのことから,2), 3)と4)を化学的,鉱物学 的にも明確かつ厳密な区分分けをすることは難しいといえる.
上記のK形態を厳密に定義づけて区分することはできないが,各形態のKを抽出する方法で区分 を定義することは可能である.抽出されたKで区分分けした場合の土壌Kは以下のようになる.
9 1) 土壌溶液溶存K
土壌から採取した土壌溶液中に存在するK.あるいは土壌から水で抽出できるK.
理論的には,水抽出を無限に繰り返した場合,土壌の全Kが抽出されるため,回数や用いる水 量を指定する必要がある.
2) 交換態K(交換性K)
指定された種類と濃度の塩溶液と指定された方法を用いて土壌から抽出されるK.
3) 非交換態K(非交換性K)
1)と2)では抽出されないが,2)の塩溶液よりもやや抽出力の強い抽出剤と指定された方法を用い
て抽出されるK.抽出剤は酸溶液,テトラフェニルホウ酸ナトリウム溶液などがある.
4) 鉱物態K
フッ化水素酸溶解などの方法によって溶解させた土壌のKから1), 2)と3)のKを差し引いたK.
現在の土壌分析で扱われる土壌Kの区分は,基本的には上記のような抽出方法から定義されたも のに従っている.土壌の間隙水に溶存しているKは,遠心分離法や真空吸引法などによって間隙水 を採取し,そのK濃度を測定することで評価できる.しかし,土壌のK供給能評価のためのK分析 としてこの分析が行われることはほとんどないため,実際の土壌間隙水に溶存するKは無視されて いることになる.これは,土壌間隙水中に溶存しているK量は,植物のK要求量と比較すると非常 に少ないからである.
2)の交換性Kを定義する代表的な抽出法は,1.0 mol L-1, pH7の酢酸アンモニウムを用いた抽出法
である(Schollenberger, 1927).土壌試料をカラムに充填し,酢酸アンモニウム溶液を流下させるこ とによってK抽出を行うが,スケールダウンしたり(土壌養分分析法委員会,1970),カラムを用 いた方法を,繰り返し遠心分離法に置き換えた方法なども採用されている(村本ら,1992).この他 にも,チオ尿素銀(Wada, 1984),塩化ストロンチウム溶液(Simard and Zizka, 1994)などによる抽 出法も提案されている.いずれの方法も,Schollenberger法(Schollenberger, 1927)と同等の結果が得 られるように抽出条件が調節されているため,どの方法で抽出しても同じ結果を得る.
3)の非交換性Kを定義する抽出法は複数ある.しかし,交換性Kとは異なり,それぞれの抽出法
によって抽出されるK 量は大きく異なる.最もよく利用される方法は,1.0 mol L-1の硝酸を用いた 加熱抽出法(Wood and DeTurk, 1941)である.この他に,テトラフェニルホウ酸ナトリウム(NaTPB;
Scott et al., 1960; Scott and Smith, 1966)やCaまたは水素イオン飽和陽イオン交換樹脂による抽出法
(Arnold, 1958; Martin and Sparks, 1983)なども用いられる.それぞれの抽出法で抽出されたKから 酢酸アンモニウム溶液で抽出されたKを差し引いたものが非交換性Kと定義される.
鉱物態Kは土壌の全Kから土壌溶液溶存態,交換性,非交換性のKを差し引いたものと定義され る.土壌のK供給能評価のための調査や研究では,土壌の全K量の測定はほとんど行われていない ため,その数値が議論されることはほとんどない.
10
1.4.2 4区分との関連付けをしない土壌のK供給能評価
土壌のK供給能評価法は,1.4.1で述べた土壌Kの4つの区分と関連付けられることが多い.しか し,そのような関連付けのない評価法もある.0.05 mol L-1 硫酸-0.05 mol L-1 塩酸混合溶液による 抽出(Mehlich, 1953)や0.2 mol L-1酢酸-0.25 mol L-1硝酸アンモニウム-0.015 mol L-1フッ化アンモ ニウム-EDTA混合溶液による抽出(Mehlich, 1983)などがこれにあたる.これらの抽出法は土壌の K供給能評価を第一の目的とした評価法でなく,Ca, Mg, K, Pなどの複数の養分の供給能評価法とし て提案されたものである.
1.4.3 既存のK供給能評価法の不十分な点
これまで提案され,利用されてきた土壌のK供給能評価法は全て,土壌に抽出液を添加する液相 抽出法である.また,NaTPB法と陽イオン交換樹脂法を除けば,4区分それぞれのKあるいは植物 が利用できるKの全量を評価するものであるため,土壌のK供給速度を知ることはできない.水稲 など数種の例外を除けば,作物の大部分は不飽和状態の土壌で生育し,土壌の間隙に保持された土 壌溶液から水素イオンとの交換でKを吸収している(間藤ら,2001).より好ましい土壌のK供給 能評価法としては,このような土壌環境を模した条件下で土壌のK供給速度に関する情報を与えら れるものなのではないかと考える.
K 供給速度を得る方法としては,K に選択的な吸着材を不飽和の土壌と接触させ,土壌から吸着 材に溶出移行したK量を時間の関数として測定する方法が考えられる.この方法で必要なものは,
K に選択的な吸着材である.しかし,陽イオン交換樹脂はアルカリ金属イオンやアルカリ土類金属 イオンに対する選択性の差がほとんどなく(Helfferrich, 1962),水素イオン交換樹脂を用いた方法で は,土壌溶液中の陽イオンは全て水素イオンと交換される.また陽イオン交換樹脂法(Martin and Sparks, 1983)を不飽和土壌を用いた実験が出来るように変形することは難しいといった問題点があ る.これは,市販の陽イオン交換樹脂の粒度は大きいため,土壌と陽イオン交換樹脂層とを接触さ せると樹脂層も不飽和状態になる可能性があるためである.
不飽和土壌を用いて,土壌からのK放出と土壌内の移動の両方の過程を速度と共に評価するため には,不飽和土壌を接触させても吸着体層自体は飽和状態に保たれるような微粒子であり,K+に選 択的な吸着材である必要がある.
11
1.5 この論文の構成
この論文の第1章ではKの化学,鉱物学について基本的なことを述べ,土壌中のKの存在形態や その評価法と現在用いられている評価法の問題点や限界点について記述した。その中で,K 供給能 評価法としては,K+に対して選択的な吸着材を用いた方法が有用であることを述べた.
第 2 章では,クリプトメラン型マンガン酸化物が K+に対して非常に選択的であることを示した.
まず,既往の文献からその選択性の高さを示し,その合成法を示した.クリプトメラン型マンガン 酸化物はトンネル構造を持ち,そのトンネル内にK+やアンモニウムイオン(NH4+)が保持されてい る.この保持された陽イオンを水素イオン(H+)と交換したものは非常にK+に選択的な吸着材とな る.効率的な陽イオン交換についても述べる.
第3章では,Kに選択的なH+交換したクリプトメラン型マンガン酸化物を用いた土壌のK供給能 の評価試験方法を提示した.一定量のH+交換したクリプトメラン型マンガン酸化物をメンブランフ ィルターを介して土壌試料と一定時間接触させることで土壌から溶出したKが拡散移行する.その 量を測定する方法と土壌の充填量や土壌の湿潤状態といった接触条件などについて検討した結果を 述べる.
第4章では,H+交換したクリプトメラン型マンガン酸化物を4種類の土壌と2種類の土壌から分 離したシルトおよび砂画分のK供給能評価に応用した結果を述べる.
第5章では,この研究を総合して,土壌のK供給能評価法に関する今後の研究課題について概括 する.
14
2 カリウム選択的マンガン酸化物の調製に関する研究
2.1 はじめに
カリウム(K)は高等植物にとって窒素やリンに並ぶ多量必須元素の一つである.植物に供給され るKは伸長可能領域の土壌から溶出し,水溶態,交換態,非交換態といった多様な形態で存在する.
中でも水溶態や交換態のKは植物が利用しやすい画分であり,この画分の評価は施肥計画を立てる 上でも重要となる.
従来,植物が利用可能な土壌のK供給能を評価する方法として中性1.0 mol L-1酢酸アンモニウム 抽出法が多く採用されてきたが,この方法で得られる量は水溶態や交換態K量であり,植物が吸収 することで土壌溶液中のKが枯渇した後に溶出する非交換態Kは評価していない.非交換態Kは雲 母,雲母の風化産物の層間や長石の結晶格子内に存在しており,熱硝酸抽出法など強酸を用いた抽 出法で評価されてきたが,植物の利用しやすさに応じた評価はほとんどされていない(Moritsuka, 2009).
化学抽出以外の土壌のK供給能評価方法として陽イオン交換樹脂を用いた方法が試行されてきた
(Martin and Sparks, 1983).これは土壌と吸着材を接触させることで目的元素を吸着材に拡散させ,
濃度勾配が生じることで溶出しにくい形態の元素も土壌から徐々に溶出し,吸着材に移行するとい うメカニズムを利用した方法である.吸着材に移行したKは熱硝酸抽出のように激しい条件で抽出 したものではないため,植物が吸収するような土壌Kに近いと考えられる.しかし,陽イオン交換 樹脂はK以外の共存する陽イオンも吸着するため,吸着容量やK選択性の低さの点で土壌のK供給 能評価のための吸着材としては適さない.
本研究ではK選択性が高く,吸着容量の大きいK型,アンモニウム(NH4)型のクリプトメラン 型マンガン酸化物を用いた評価法の開発を試みた.Tsuji and Abe(1985)はクリプトメラン型マンガ ン酸化物を合成し,構造内のKを硝酸で洗浄することで陽イオン交換体に調製した.調製した合成 物は,濃縮した海水から微量のKを除去するのに適用可能であることが確認された.また,リチウ ム(Li),ナトリウム(Na)やセシウム(Cs)よりもルビジウム(Rb)やK+を優先的に吸着するこ とを確認した(Tanaka and Tsuji, 1997).
この章では土壌のK分析のための,水素イオン交換したクリプトメラン型マンガン酸化物を調製 する方法を示す.合成物を陽イオン交換体に調製するために硝酸を用いた洗浄を行うが,洗浄期間 が長期化する点に問題があったため,その改善方法についても記す.
15
2.2 実験方法
2.2.1 K型マンガン酸化物の調製
2.2.1.1 K型マンガン酸化物の合成
構造内にK+を保持したクリプトメラン型マンガン酸化物(K-Cm)をTanaka and Tsuji(1994;
1997)による方法を基に合成した.44.55 gの炭酸マンガン(MnCO3)と5.45 gのカリウムブトキ
シド((CH3)3COK)をアルミナ乳鉢内で混合し,500℃の電気炉内で10時間加熱処理した.加熱 処理後はK-Cmをビーカーに移し,脱イオン水を添加,撹拌し,沈降後に上澄みを除去し,再び 脱イオン水を添加,撹拌,上澄み除去という操作を繰り返した.撹拌洗浄後は0.45 µmメンブラ ンフィルター(ADVANTEC C045A047A)でろ過し,回収したK-Cmをシャーレに広げて50℃で 乾燥した.
2.2.1.2 K+-H+交換反応時間の測定
K-Cm 1.0 gをトールビーカーに取り,脱イオン水を550 mL添加した.ガラス棒で撹拌後,上層部
分の懸濁がみられない状態になるまで静置し,5 mLの上澄み液をシリンジ(TERUMO SS-10Sz)で 採取した.採取後は13.14 mol L-1の硝酸(HNO3)を45 mL添加し,マグネチックスターラーで撹拌 した.0.5, 1.0, 2.0, 5.0, 10, 20時間後にスターラーを止め,上層部分に懸濁がみられない状態になるま で静置し,5 mL の上澄みをシリンジで採取した.各サンプルはシリンジフィルター(ADVANTEC
DISMIC-13c)でろ過し,K濃度を原子吸光光度計(Hitachi Z-2300)で測定した.
2.2.1.3 ガラスフィルターを用いた陽イオン交換
図 2.1のように底部にガラス繊維ろ紙(Whatman 1823-142)を敷いたφ1.47 cmのガラスフィルタ ー(HARIO 15AG1)内にK-Cm 10 gを充填し,1.0 mol L-1および6.0 mol L-1のHNO3で通液洗浄を行 った.適宜フィルター通過後の溶液を10 mL採取し,K濃度を原子吸光光度計で測定した.1.0 mol L-1 HNO3が9,700 mL, 6.0 molL-1 HNO3が3,500 mLそれぞれ通過した時点で洗浄溶液を脱イオン水に 換え,カラムから滴下する溶液がpH7 程度になるまで脱イオン水を流し続けた.酸処理と脱イオン 水による洗浄を行った調製物を以下K-CmHと略記する.K-CmHは懸濁液状態でポリエチレン容器 内に保存した.
高濃度の HNO3で長時間洗浄を行う場合,調製物の構造が破壊される可能性がある.破壊の有無 を確認するため,K-CmとK-CmHの一部を50℃の迅速乾燥機内で乾燥させ,X線回折分析(Rigaku Rint 2100V)とTG-DTA分析(Rigaku Thermo plus EVO TG 8120)を行った.
16
図 2.1 ガラスフィルターを用いた洗浄
2.2.2 アンモニウム型マンガン酸化物の調製と酸洗浄方法の検討
2.2.2.1 アンモニウム型マンガン酸化物の合成
構造内にアンモニウムイオン(NH4+)を保持したクリプトメラン型マンガン酸化物(NH4-Cm)を
Li et al.(2007)による方法を基に合成した.60.3 gの硫酸マンガン五水和物(MnSO4・5H2O),165
gの硫酸アンモニウム((NH4)2SO4)と57 gの過硫酸アンモニウム((NH4)2S2O8)を500 mLの脱イオ ン水に順に溶解し,50 mLずつテフロン容器に移して150℃のオーブンで24時間加熱した.水熱処 理後は懸濁液内の残ったNH4+を除去するため,NH4-Cmをビーカーに移し,脱イオン水を添加,撹 拌し,合成物が沈降後に上澄みを除去し,再び脱イオン水を添加,撹拌,上澄み除去という操作を繰 り返した.操作は 10 回繰り返し,余分な NH4+除去後は懸濁液状態でポリエチレン容器内に保存し た.
17 2.2.2.2 NH4+-H+交換処理の改善策
NH4-Cm懸濁液を250 mL容のポリカーボネート遠心分離管にとり,13分間遠心分離(2,500 rpm)
した後,上澄み液を除去した.再び懸濁液を添加し,遠心分離するという操作を繰り返し,全NH4- Cmを回収した.回収した NH4-Cmをガラスシャーレに広げ 50℃の迅速乾燥機内で乾燥した.乾燥 後は,NH4-Cmの凝集体を乳鉢に移し,0.5 mmふるいを通過する程度になるよう粗めに粉砕した.
粉砕したNH4-Cmをガラスフィルターに充填し,6.0 mol L-1のHNO3を用いてNH4+とH+の交換を行
った.約6.0 LのHNO3で洗浄が完了した時点で洗浄溶液を脱イオン水に換え,カラムから滴下する
溶液がpH7 程度になるまで脱イオン水を流し続けた.酸処理と脱イオン水による洗浄を行った調製 物を,以下NH4-CmHと略記する.NH4-CmHは懸濁液状態でポリエチレン容器内に保存した.
2.2.1.3と同様に調製物の構造破壊の有無を確認するため,NH4-Cmと NH4-CmH の一部を 50℃の
迅速乾燥機内で乾燥させ,X線回折分析(Rigaku Rint 2100V)とTG-DTA分析(Rigaku Thermo plus EVO TG 8120)を行った.
2.2.3 K吸着特性評価
85 mL 容ポリカーボネート遠心分離管に,懸濁液をろ過して乾燥させたK-CmH,NH4-CmH をそ
れぞれ0.1 gずつ取り入れ,50 mmol L-1の塩化カルシウム(CaCl2)と50 mmol L-1の塩化マグネシウ
ム(MgCl2)を共存させた1.0から10 mmol L-1の塩化カリウム溶液(KCl)を10 mLずつ添加し,24 時間振とうした.振とう後は遠心分離し,上澄み液をシリンジフィルターでろ過した.採取した上 澄み液は適宜希釈した後,K濃度を原子吸光光度計で測定して,K-CmH, NH4-CmHそれぞれのK吸 着量を算出した.
2.2.4 回収法の検討
調製したK-CmH,NH4-CmHを土壌との接触試験に用いた場合,土壌から溶出して吸着材に移行
したKを定量的に回収する必要がある. MnO2は還元させることで溶解させることができるた め,吸着材を溶解させる溶解試薬として硫酸酸性シュウ酸アンモニウムと硫酸酸性シュウ酸ナトリ ウムを検討した.
硫酸酸性シュウ酸アンモニウムは0.1 molのシュウ酸アンモニウム((NH4)2C2O4)をビーカーに 取り,脱イオン水を約500 mL添加して溶解させた.撹拌しながら濃硫酸(H2SO4)を55.5 mL添加 し,放冷した後に1 Lに定容した.硫酸酸性シュウ酸ナトリウムも同様に,0.1 molのシュウ酸ナト リウム(Na2C2O4)をビーカーに取り,脱イオン水を約500 mL添加して溶解させた.撹拌しながら
濃硫酸を55.5 mL添加し,放冷した後に1 Lに定容した.2.2.1,2.2.2にて得た懸濁液をろ過して乾
燥させた0.1 gのK-Cm, K-CmHには0.1 mol L-1の硫酸酸性シュウ酸アンモニウムを50 mL添加し,
0.1 gのNH4-CmHには0.1 mol L-1の硫酸酸性シュウ酸ナトリウムを50 mL添加した.各溶液を添加
後は室温下で時間を計りながら溶解反応を観察した.溶解後は適宜希釈して原子吸光光度計でK濃 度を測定した.
18
2.3 結果と考察
2.3.1 K型マンガン酸化物の調製
2.3.1.1 K+-H+交換処理
図 2.2にK-CmのK+-H+交換反応の予備試験結果を示す.K-Cmに脱イオン水を添加直後のK-Cm
からのK溶出量は96.3 mmol kg-1であった.HNO3添加後はビーカー内のHNO3濃度が1.0 mol L-1と
なるため K+と H+が陽イオン交換しやすい状態となる.1 時間後の採取サンプルの K 濃度から,約
400 mmol kg-1のKがK-Cmから溶出したことを確認した.20時間後のK溶出量も約400 mmol kg-1
であったため,K-CmにおけるK+とH+の交換反応に要する時間は1時間以内であり,陽イオン交換 速度は速いといえる.
2.3.1.2 ガラスフィルターを用いた陽イオン交換
図 2.3に1.0 mol L-1および6.0 mol L-1のHNO3洗浄量に対するK-CmからのK溶出量を示す.1
mol L-1のHNO3を用いた場合,約4.5 L通液させると511 mmol kg-1のKが溶出したが,その後も継
続して約 9.0 L 通液させてもK 溶出量は 550 mmol kg-1程度で,大きな変化がみられなかった.6.0
mol L-1のHNO3を用いた場合,少量の洗浄液量で1.0 mol L-1のHNO3以上のK溶出量を確認した.
約260 mL通液させた場合は669 mmol kg-1のKが溶出し,約3.6 L通液させた場合は1,011 mmol kg-
1のKがK-Cmから溶出した.K-Cm内に保持されるK量をできる限り少なくし,使用するHNO3量 を少なくするためには6.0 mol L-1のHNO3を用いてK+とH+交換するのが最適である.
K-Cm, K-CmHのDTA-TG分析結果を図 2.4に示す.K-Cmの吸熱ピークは30℃と891℃で現れ,
0-600℃,600-870℃,870-899℃の 3 段階の減量がみられた.一方,K-CmH の吸熱ピークは 30℃,
590℃と892℃でK-Cmよりも鋭いピークとして現れ,0-520℃,520-625℃,625-870,870-899℃の4
段階の減量がみられた.
K-Cm, K-CmHのX線回折図を図 2.5に示す.K-Cmは32°から33°にみられる微小なピークを
除いて,クリプトメラン型マンガン酸化物のX線回折パターンと同様の特徴を示していた(Li et al.,
2007).K-CmHでも同じピークの傾向がみられたことから,6.0 mol L-1のHNO3を使用してK-Cm内
のK+とH+交換を行っても,構造が破壊されることはないことを確認した.
19
図 2.2 K-CmからのK溶出量の経時変化(バッチ試験)
図 2.3 K-Cmからの積算K溶出量の経時変化(カラム試験)
2 4 6 8 10 12 14 16 18 20 22 100
200 300 400 500
0
Reaction time (h)
M igr at ed K ( m m ol /kg)
2000 4000 6000 8000 10000 12000 200
400 600 800 1000 1200
0
Cumulative eluent volume (mL)
C um ul at ive a m ou nt of l ea che d K ( m m ol /kg )
6 mol/L
1 mol/L
20
図 2.4 DTA-TG分析結果(a: K-Cm, b: K-CmH)
0 200 400 600 800 1000
-100 -80 -60 -40 -20 0 20 40 60 80 100
-1 -0.8 -0.6 -0.4 -0.2 0
Temperature ( ℃ )
D T A ( μ V) T G ( m g)
DTA TG
(a)
0 200 400 600 800 1000
-100 -80 -60 -40 -20 0 20 40 60 80 100
-1 -0.8 -0.6 -0.4 -0.2 0
Temperature ( ℃)
D T A ( μ V) T G ( m g)
DTA TG
(b)
21
図 2.5 K-CmとK-CmHのX線回折図を用いた構造比較
10 20 30 40 50 60
500 1000 1500 2000 2500 3000
0
2θ (CuKα)
In te ns it y (c ps )
K-Cm
K-CmH
22
2.3.2 アンモニウム型マンガン酸化物の調製と酸洗浄方法の検討
2.3.2.1 NH4+-H+交換処理
合成したNH4-Cmをそのままカラムに充填して6.0 mol L-1のHNO3を約6 L通液させた場合の溶 液通過速度は0.075 mL min-1,通液を完了させるまでの期間は4か月近くを要した.一方,一度乾燥 させて粗めに粉砕したNH4-Cmをカラムに充填して6.0 mol L-1のHNO3を約6 L通液させた場合の 溶液通過速度は2.4 mL min-1,通液を完了させるまでの期間は9 日間であった.0.1 gの乾燥させた
NH4-CmHに10 mmol L-1のKCl溶液を10 mL添加してK吸着量を測定したが,洗浄期間に影響され
ることなく75 cmolc kg-1のKを吸着した.
合成直後の NH4-Cm の粒子は非常に小さく,土壌の単粒構造のように緻密に詰まった状態になり やすい傾向がある.粒子間の隙間が非常に小さくなってしまったため,HNO3の通液速度が遅くなっ てしまったと考えられた.一方,一度乾燥させて粗く粉砕した場合,粒子同士は土壌の団粒構造の ようにくっつき合った状態であるため,緻密に詰まることはないと考えられた.団粒間の隙間は粒 子間の隙間よりはるかに大きいため,HNO3の溶液通過速度を維持することが可能である.
以上のことから,一度乾燥させてNH4-Cm に団粒を形成させた後に HNO3で洗浄する方法は,洗 浄期間を大幅に短縮することが出来ることから,効率面の改善が出来たといえる.
NH4-Cm, NH4-CmHのDTA-TG分析結果を図 2.6に示す.NH4-Cmの吸熱ピークは38℃と553℃で現 れ,0-430℃,430-567℃,567-899℃の3段階の減量がみられた.一方,NH4-CmHの吸熱ピークは35℃,
536℃で現れ,0-440℃,440-560℃,560-899℃の3段階の減量がみられた.NH4-Cm, NH4-CmHの500℃付 近の減量は脱水によるものである可能性が考えられるが,NH4-Cm, NH4-CmHにおける200℃付近のDTA 測定結果の傾向は異なっていた.
NH4-Cm, NH4-CmHのX線回折図を図 2.7に示す.NH4-CmはK-CmHはクリプトメラン型マンガン酸 化物のX線回折パターンと同様の特徴を示していた(Li et al.,2007).NH4-CmHでも同じピークの傾向 がみられたことから,6.0 mol L-1のHNO3を使用してNH4-Cm内のNH4+とH+交換を行っても,構造が破 壊されることはないことを確認した.
23
図 2.6 DTA-TG分析結果(a: NH4-Cm, b: NH4-CmH)
0 200 400 600 800 1000
-30 -20 -10 0 10
-3 -2 -1 0
Temperature ( ℃)
DTA ( μ V) TG (mg)
DTA TG
(a)
0 200 400 600 800 1000
-30 -20 -10 0 10
-3 -2 -1 0
Temperature ( ℃)
DTA ( μ V) TG (mg)
DTA TG
(b)
24
図 2.7 NH4-CmとNH4-CmHのX線回折図を用いた構造比較
2.3.3 K吸着特性評価
図 2.8にK-CmH, NH4-CmHの吸着等温線を示す.50 mmol L-1のCaやMgが共存していても,溶 存陽イオンがKのみの時の吸着等温線とほぼ同じであるため,K吸着にほとんど影響を与えていな いことが分かる.K-CmHは約50 cmolc kg-1で吸着平衡量に達し,40 cmolc kg-1のKを吸着した場合 の平衡濃度は6.0×10-3 mmol L-1であった.一方,NH4-CmHは約60 cmolc kg-1で吸着平衡量に達し,
53 cmolc kg-1のKを吸着した場合の平衡濃度は1.4×10-3 mmol L-1であった.
図 2.9 に調製法を改善して調製した吸着材の吸着等温線を示す.約 75 cmolc kg-1で吸着平衡に達 し,吸着容量が50 cmolc kg-1の時,平衡濃度は1.0×10-3 mol L-1であった.調製法改善前と同等の吸 着能であることを確認できた.
本研究で用いる供試土壌の交換性K量の最大値は1.8 cmolc kg-1,交換性K+非交換性K量の最大
値は1.9 cmolc kg-1である.この土壌1.0 gと調製したK-CmH,NH4-CmHを0.1 g用いて接触試験を
行った場合,交換性K+非交換性K全量とそれ以上のKを吸着することが出来るため,K-CmH, NH4- CmHいずれの場合でも0.1 gを用いれば吸着容量としては十分である.
NH4-Cm内のNH4+とH+を交換する際に用いたHNO3の積算通液量と各NH4-CmHの吸着等温線を 図 2.10 に示す.HNO3 の積算通液量を増加した場合のそれぞれの最大吸着容量と平衡濃度は,250 mLでは38 cmolc kg-1で6.89 mmol L-1,500 mLでは44 cmolc kg-1で6.42 mmol L-1,1,000 mLでは76 cmolc kg-1で3.86 mmol L-1,1,500 mLでは88 cmolc kg-1で2.90 mmol L-1であった.このことから,NH4-
10 20 30 40 50 60
500 1000 1500 2000
0
2 θ (CuK α )
In ten si ty (cp s)
NH
4-Cm
NH
4-CmH
25
CmHの吸着能はHNO3の積算通液量に依存しており,通液量が多いほど最大吸着容量は大きくなり,
平衡濃度を低く抑える吸着材に調製されることが分かった.HNO3の通液量によって吸着量は変化す るため,異なるNH4-CmHを吸着材として使用する際は,供試土壌の交換性K+非交換性K量とNH4- CmHのK吸着容量を比較し,土壌のK量がNH4-CmHの吸着容量を超過しないように使用する必要 がある.
図 2.8 調製した吸着材の吸着等温線(a: K-CmH, b: NH4-CmH)
0.5 1.0 1.5 2.0
20 40 60
0
Equilibrium K concentration (mmol/L) K ad sor pti on (c mo l
c/kg )
KCl
KCl in 50 mmol/L of CaCl
2KCl in 50 mmol/L of MgCl
2(b)
1 2 3 4 5 6
20 40 60
0
Equilibrium K concentration (mmol/L) KCl
KCl in 50 mmol/L of CaCl
2KCl in 50 mmol/L of MgCl
2K ad sor pti on (c mo l
c/kg )
(a)
26
図 2.9 調製法を改善して調製したNH4-CmHの吸着材の吸着等温線
図 2.10 NH4+-H+交換時の6.0 mol L-1 HNO3の積算通液量とNH4-CmHの吸着等温線の関係
1.0 2.0 3.0
20 40 60 80 100
0
Equilibrium K concentration (mmol/L) K ads orpt ion (c mol
c/kg)
2 4 6 8
20 40 60 80 100
0
Equilibrium K Concentration (mmol/L) K adsor pti on (c mo l
c/kg)
1500 mL 1000 mL
500 mL
250 mL
27 2.3.4 回収法の検討
図 2.11 は,K-Cm に硫酸酸性シュウ酸アンモニウムを添加した直後の溶液と 30 分経過後の溶液 の比較を示す.K-Cm, K-CmH には硫酸酸性シュウ酸アンモニウムを,NH4-CmH には硫酸酸性シュ ウ酸ナトリウム溶液を添加したが,K-Cm, K-CmH, NH4-CmH はいずれも室温下で 30 分間程度反応 させることで完全溶解することを確認した.
各試料のK含有量を表 2.1に示す.硝酸を用いたK+とH+交換前のK-Cmには159 cmolc kg-1, 洗
浄後のK-CmHには73 cmolc kg-1のKが含まれていた.K型マンガン酸化物は,6.0 mol L-1のHNO3
を用いて構造内の K+とH+を交換しても,初期含有量の半量程度の K+が残存していたため,完全に K+を除去することは難しいと考えられる.NH4型マンガン酸化物は合成する際に使用した試薬に K が含まれていないことから,NH4-CmH内に含まれるK量も0 cmolc kg-1であった.
図 2.11 K-Cmに硫酸酸性シュウ酸アンモニウム溶液を添加した際の反応性の確認
(左:添加直後,右:30分後)
表 2.1 K-Cm, K-CmH, NH4-CmHのK含有量
試料 K含量
(cmolc kg-1) K-Cm
K-CmH NH4-CmH
159 73
0
28
2.4 まとめ
調製したK-CmH, NH4-CmHはK吸着容量が大きく,K選択性がかなり高いことを確認した.吸着
容量は陽イオン交換の際に用いる硝酸の濃度と積算通液量に依存するが,高濃度の硝酸に長期間さ らされてもクリプトメランの構造に影響はなかった.しかし,構造内の K+や NH4+をH+に交換する ための調製は非常に長期の洗浄期間を要するため,効率面で問題があった.合成物は非常に小さい 粒径であるため高密度化を生じ,合成物をそのまま充填する方法では水位差を大きくしても溶液通 過速度の改善は確認できなかった.合成物を一度乾燥させた後,0.5 mmふるいを通過するサイズに 粉砕したものをカラムに充填する方法を採用した洗浄では,洗浄期間を 3 か月以上短くすることが でき,その吸着容量は改善前と同程度のものであることを確認した.効率面での問題点を改善でき たことから,一度乾燥させた後に洗浄をする方法は洗浄方法として最適であるといえる.
NH4-CmHの吸着能はNH4+とH+を交換する際に用いるHNO3の積算通液量に依存することを確認 した.通液量が多いほど最大吸着容量は大きくなり,溶液中のK濃度をより低くすることが出来る.
調製する NH4-CmH は必ずしも吸着容量が等量になるとはいえないため,異なった調製物を土壌と の接触試験で吸着材として使用する場合は,吸着容量が同じになるように使用量を変化させる必要 がある.
K-CmHは構造内に残存するKが存在するため,土壌との接触試験に使用する際には,硫酸酸性シ
ュウ酸アンモニウムで接触後の吸着材を溶解させた溶液の測定値から残存分のK量を差し引く必要 がある.一方,NH4-CmHを用いた接触試験では,硫酸酸性シュウ酸ナトリウムで溶解後の測定値を 土壌から吸着材への移行量としてそのまま使用できる.
29
3 土壌との接触試験条件に関する研究
3.1 はじめに
第2 章で調製した吸着材であるK-CmHとNH4-CmHを使用して土壌との接触予備試験を行った.
K-CmHを使用する際は,0.1 gの吸着材と3.0 gの土壌試料をメンブランフィルターを介して接触さ
せ,0.01 mol L-1のCaCl2溶液を3.0 mL添加した.NH4-CmHを使用する際は,0.1 gの吸着材と2.0 g の土壌試料をメンブランフィルターを介して接触させ0.01 mol L-1のCaCl2溶液を2.0 mL添加した.
各接触時間の土壌から各吸着材に移行したK量を図 3.1に示す.左図からアンモニウム型のクリプ トメラン型マンガン酸化物であるNH4-CmH は供試土壌から吸着材に移行するKの経時変化を精度 よく測定することができることが分かる.一方,右図から,K型であるK-CmHを使用して接触試験 を行った場合, 2~3連の試験で測定値が大きく異なることを確認した.K-CmHを使用する際は,
硫酸酸性シュウ酸アンモニウムで接触試験後の K-CmHを溶解させた溶液の K 量から K-CmH 内に 残存するK量を差し引いた値を供試土壌からK-CmHに移行したK量と考えたが,K-CmHの構造内 に残存するKは不均一に存在する可能性があるため,このような測定値にばらつきのある結果にな った可能性があると考えた.今後の土壌との接触試験では,精度の高い測定値を得るため,NH4-CmH を吸着材として用いた試験を行った.
NH4-CmHを使用した予備試験では,供試土壌2.0 gに0.01 mol L-1 CaCl2溶液を2.0 mL添加して吸 着材と接触させたが,接触条件である供試土壌の充填量や CaCl2溶液の添加量については未検討で あった.土壌充填量を0.5,2.0 g,CaCl2溶液添加量を0.5,2.0 mLとして接触試験を行った場合,単 位質量換算のK溶出量に違いが生じることを確認した.第3章では接触試験の際の供試土壌の充填 量とCaCl2溶液添加量について検討した.
図 3.1 土壌から吸着材に溶出移行するK量の経時変化(プロットは平均値を示す)
30
3.2 試料と実験方法
3.2.1 試料
本実験では福岡県三潴郡大木町の福岡県農林業総合試験場筑後分場(筑後),福岡市筑紫野市吉 木の福岡県農林業総合試験場(吉木),大韓民国京畿道鳥山市内の混合二次林(オサン),熊本県 合志市の九州沖縄農業研究センター(合志)で採取し,風乾した4種類の土壌試料を用いた.充填 量,CaCl2溶液添加量を検討する際には,粒径によるK溶出の影響も考えられるため212 µmふる い通過分を使用した.土壌試料の理化学性として交換性K,非交換性Kを測定した.交換性Kは 酢酸アンモニウムを用いた振とう浸出法(Muramoto et al., 1992)で抽出し,非交換性Kは熱硝酸抽 出法(Haylock, 1956)で抽出した.抽出溶液中のK濃度は原子吸光光度計で測定した.
供試土壌の交換性K, 非交換性K 含量,主要粘土鉱物を表 3.1 に示す.粘土鉱物組成については Mori et al. (2012),Yoshinaga et al. (1986)から引用した.筑後試料の主要粘土鉱物はスメクタイト,吉 木試料はバーミキュライトとクロライト,オサン試料は雲母,合志試料はアロフェンが主要であっ た.
表 3.1 供試土壌の交換性K, 非交換性K含量と粘土鉱物組成(筑後,吉木,合志試料の主要粘土 鉱物はMori et al. (2012),オサン試料の主要粘土鉱物はYoshinaga et al. (1986)から引用)
交換性K (cmolc kg-1)
非交換性K (cmolc kg-1)
主要粘土鉱物
筑後 1.071 1.925 Sm>K>Mc
吉木 0.096 0.087 Vm>Ch-Vm
オサン 0.190 0.950 Mc>K
合志 1.386 1.779 Allo
Allo:アロフェン,Ch:クロライト,K:カオリン鉱物,Mc:雲母,Sm:スメクタイト Vm:バーミキュライト,Ch-Vm:クロライト-バーミキュライト中間種鉱物
31
3.2.2 土壌充填量の決定
接触試験の際に用いた器具を図 3.2に示す.プラスチックホルダー(ADVANTEC PPO-47)のベー
スに直径 47 mm のポリエチレンフィルムを置き,吸着容量が 0.1 mmol 相当量になるように NH4-
CmH(0.25 g)を3枚のセルロースアセテートタイプの0.45 mmメンブランフィルター(ADVANTEC
C045A047A)でパックしたものをのせ,PTFE製Oリングとキャップで固定した.
NH4-CmHをパックしたメンブランフィルター上に,212 µmふるい通過分の供試土壌を0.2, 0.5, 1.0 g充填し,0.01 mol L-1 CaCl2溶液0.2, 0.5, 1.0 mLを固液比が1:1になるようにそれぞれ添加した.
開口部からの蒸発を防ぐためパラフィルムをかぶせ,プラスチックホルダーを密閉可能なポリエチ レン袋内に入れて24時間から90時間接触させた.接触期間中は25℃の恒温室で静置した.
各回収時間に供試土壌を脱イオン水で洗浄して除去し,NH4-CmHをパックしたメンブランフィル ターを回収した.回収したNH4-CmHは0.1 mol L-1硫酸酸性シュウ酸ナトリウムを30 mL添加して 完全に溶解させた.溶解後は適宜希釈し,原子吸光光度計を用いてK濃度を測定し,供試土壌から 吸着材に溶出移行したK量を算出した.
図 3.2 接触試験に用いた試験器具