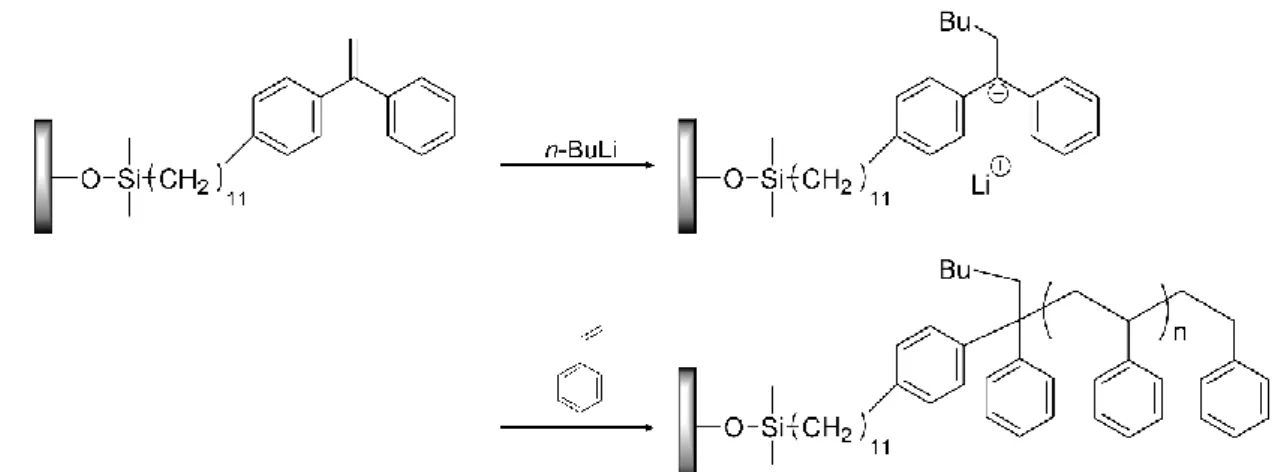
九州大学学術情報リポジトリ
Kyushu University Institutional Repository
精密重合法に基づくポリマーブラシの二次構造制御 と分子特性解析に関する研究
佐藤, 雅尚
http://hdl.handle.net/2324/1959097
出版情報:Kyushu University, 2018, 博士(工学), 課程博士 バージョン:
権利関係:
九州大学大学院 工学府 物質創造工学専攻
学位論文
精密重合法に基づくポリマーブラシの二次構造 制御と分子特性解析に関する研究
2018 年 5 月
佐藤 雅尚
第1章
序論
1-1 研究背景
材料同士が互いに触れ合う接点である界面の設計は,材料の諸物性を決めるうえで極 めて重要な役割を担っており,材料の微細・薄化が求められている現代産業において,
界面の設計および制御は必要不可欠である.材料表面を改質することにより,その材 料の性質を改変させることが可能であり,その簡便な手法として材料表面に高分子膜 を塗付する方法が挙げられる.高分子材料は,無機材料と比較して成形性,加工性に 富み,軽量であることから,近年様々な分野で研究が行われている.高分子膜による 表面改質手法として,スピンコート膜のような材料表面に高分子を塗布する手法と,
材料表面と高分子の相互作用により材料表面に高分子を固定化する手法が挙げられる.
前者は,簡便で均一な高分子膜を得ることが可能であるが,一方で材料と基板間の相 互作用が弱いため安定性に乏しい.後者の手法は,材料表面と高分子との物理的相互 作用を利用した物理的手法と,材料表面と高分子を化学結合で固定化する化学的手法 とに大別される.物理的な手法によって調製された高分子膜は,良溶媒中での剥離,
力学的に弱いなどいくつかの欠点を有している.化学的手法により調製された高分子 膜は,物理的相互作用と比較して強い化学結合を介して材料表面に固定化されている ため,耐溶剤性,熱的・機械的安定性を有しており,材料表面に恒久的な表面改質を 施す手法として多くの研究が成されてきた.その中でも高分子が共有結合あるいはイ オン結合を介して基材に高密度に結合したポリマーブラシは,優れた界面改質技術と して注目されている.
ポリマーブラシは,基材表面に高分子鎖の片側末端が高密度に固定化された高分子 膜であり,材料表面に対して垂直方向に高度に延伸・配向した構造を有する.これら に起因して濡れ性,摩擦特性および吸着特性などの特異的な表面物性を発現すること が知られており,ポリマーブラシは様々な分野への応用が期待されている.ポリマー ブラシの分子鎖形態はグラフト密度 (1 nm2あたりにグラフト鎖が占める割合.単位:
chain/nm2)によって大きく変化する.グラフト密度が高くなると,ポリマーと基材との 相互作用の他に高分子鎖同士で相互作用を生じ,より特異な性質を示す.ポリマーブ ラシの構造とグラフト密度の関係について Figure 1-1 に示す.グラフト密度が 0.01
chain/nm2 以下の場合,隣接するポリマー間の相互作用が働かず,高分子鎖は三次元的
な広がりを示す (マッシュルーム構造).一方でグラフト密度が高くなり準希薄溶液状 態にまで達すると,ポリマー間で相互作用を生じるようになり基材に対して鉛直方向 に伸長した構造を形成する (準希薄ポリマーブラシ).さらにグラフト密度が高くなる と,隣接ブラシ鎖間で斥力を生じ高分子鎖が材料表面に対して垂直方向に延伸した構 造を形成する (濃厚ポリマーブラシ).
従来報告されている多くのポリマーブラシはランダムコイル構造を形成しており,
その機能性はポリマーブラシ調製時に用いる重合性モノマーの化学構造によりほとん ど決定する 1-3.また,ポリマーブラシ調製後は,その密な分子鎖凝集構造に起因して 膜内部に新たに機能性を導入するような分子 (粒子)の導入が困難であることが問題点 として挙げられる(Figure 1-2a).この問題を解決するためには,ポリマーブラシ内部に 周期的に空間を導入するような分子設計の開発が必要不可欠である(Figure 1-2b).
Figure 1-1. Schematic illustration of molecular aggregation structure of polymer brushes with various graft densities.
らせん構造のような筒状の構造を有する高分子は,その空孔内部に分子を包接するこ とが可能である.らせん構造は DNA などの生体高分子で主に観測される代表的な構 造の一つであるが,合成高分子においてもらせん構造を形成するものが数多く報告さ れており,その一例として立体規則性高分子が挙げられる.立体規則性高分子とは,
側鎖のコンホメーションが規則的に配列した高分子のことであり,ビニル系高分子に おいては大別すると以下の四つに分類される(Figure 1-3).
側差が不規則に配列しているものはアタクチック (at-),側差が交互に配列している ものはシンジオタクチック (st-),側鎖がすべて同じ向きで配列しているものはイソタ
クチック (it-),側差が二つおきに交互に配列しているものはヘテロタクチック (ht-)で
ある.立体規則性高分子の発見は1947年にSchildknechtらによってなされ,ポリビニ ルエーテルの立体規則性に関する報告がなされている.しかし,立体規特異性重合の 研究が盛んに行われ始めたのは,Zieglerらが開発した新しい重合系を用いて,Nattaら Figure 1-2. Schematic illustration of introducing functional molecules into a) High-density polymer brush with random-coil structure and b) polymer brush with nano-pore.
Figure 1-3. Schematic illustration of stereoregularity of vinyl-type polymer.
が立体規則性ポリプロピレンの重合を報告してからのことである.立体規則性高分子 はガラス転移温度,融点,溶解性などの点においてアタクチックポリマーとは大きく 異なる性質を示すだけでなく,らせん構造に代表される特異的な構造を形成すること が知られている.汎用的な高分子材料として知られているポリメタクリル酸メチル
(PMMA)を例として挙げると,it-PMMA は二重らせん構造を形成して結晶化し 4,st-
PMMAは,トルエンやクロロアセトンなどの溶媒分子を包接したらせん構造を形成し ゲル化することが報告されている5-7.また,st-PMMAは包接する分子のサイズに合わ せて自身のらせんサイズを変化させ,it-PMMA,フラーレンおよびピレンなど,様々 な分子をらせん内部に包接することが可能である8-10.この概念をポリマーブラシに適 応することが可能となれば,ポリマーブラシ膜内部に新たに分子を包接可能な新規ポ リマーブラシの創製が期待される.しかしながら,これまでに立体規則性が精密に制 御された高密度ポリマーブラシの調製は成し遂げられていない.
ポリマーブラシの調製法には,(ⅰ) grafting to法,(ⅱ) grafting from法の二通りの調製 法が存在する (Figure 1-4a,b).grafting to法は,あらかじめ調製したポリマーの末端と 基板との反応によってポリマーを基板上に固定化する方法である.この手法では精密 重合によってあらかじめ調製したポリマーを基材に固定化するため,ほとんどすべて の合成高分子に適応可能である.その一方で,固定化が進行するにつれて固定化され た高分子の立体障害が大きくなり,グラフト密度はある値以上には大きくならないこ とが問題として挙げられる (Figure 1-4a).grafting from法では,あらかじめ固体表面に 固定化した開始剤を用いて表面から重合を開始する手法である.この手法では反応点 に対する接近種が立体的にかさ高くないモノマーであり,立体障害による影響が小さ いため高密度領域での高分子の固定化が可能である.
ポリマーブラシの表面特性は分子量分布に大きく影響される 11.そのため,高密度で 均一なポリマーブラシを調製するためには,ポリマーブラシの分子量分布を精密に制 御する必要がある.近年,重合技術の発展により,表面開始重合において多種多様な 重合系が適用され,様々なポリマーブラシの調製が達成されている12-27.これらの重合 法の中でも,重合制御の簡便さおよび適用範囲の広さからラジカル重合法が最も汎用 Figure 1-4. Schematic illustration of preparation of polymer brush on the basis of a) grafting to and b) grafting from method.
a) b)
的なポリマーブラシの調製法として用いられている.ラジカル重合法は比較的容易な 条件で分子量,分子量分布の制御が可能であるが,一方,立体規則性制御が困難であ り,その制御度合いもイオン重合や配位重合などの重合と比較して低いことが問題と して挙げられる.表面開始重合法に基づくポリマーブラシの立体規則性制御の報告例 として,2008 年に Jiang らによるポリアクリルアミド (PAAM)ブラシの立体規則性制 御が挙げられる28.彼らは表面開始原子移動ラジカル重合 (SI-ATRP)系において,重合 系にルイス酸を添加することによって,PAAM の立体規則性を制御し,接触角測定法 に基づいて立体規則性の違いに起因した表面物性の変化を評価している.また,Idota らは,ポリ-N-イソプロピルアクリルアミド (PIPAAm)ブラシの重合に成功しており,
PIPAAm ブラシの表面濡れ性の温度依存性を評価し,立体規則性がポリマーブラシの
物性に及ぼす影響を報告している 29.これらのラジカル重合によって得られた立体規 則性高分子は,二連子でおよそ80 %程度のイソタクチック構造を有しているが,これ はイオン重合によって調製した高分子と比較して著しく低い値であり,ラジカル重合 法に基づくポリマーブラシの立体規則性制御には限界がある.
リビングアニオン重合はリビングラジカル重合と比較して生成するポリマーの分子 量・分子量分布および立体規則性を精密かつ同時に制御できるという点において大き なメリットを有する.さらに,ラジカル重合ではラジカル種同士が再結合や不均化を 起こして一部で停止反応が生じる一方で,アニオン重合の成長末端であるアニオン種 は静電反発によって反発しあうため,表面開始重合において成長末端基間での停止反 応が生じない.以上のことから,表面開始リビングアニオン重合はより均一に基材表 面を修飾する手法として期待される.しかしながら,アニオン重合では重合の制御が 困難であるため,表面開始重合に用いられた報告例はラジカル重合と比較して著しく
少ない 14-17, 30-31.表面開始リビングアニオン重合に用いる表面開始基の調製手法とし
て,基材表面に重合性官能基を固定化し重合開始剤と反応させる手法と,基材表面に
ハロゲン化アルキルを固定化しリチウム交換反応により開始基を調製する手法が挙げ られる.1988年に初めて報告された表面開始リビングアニオン重合は前者の手法に基 づいている.Schomakerらは,シリカ粒子表面にMMA誘導体を固定化し,Ggignard試 薬を用いてイソタクチック PMMAブラシの調製を行っている31.Oosterlingらは,ス チレン誘導体を基板表面に固定化し,アルキルリチウムを開始剤として重合を行って いる 30.これらの手法では,ポリマーブラシの成長反応と基材表面に固定化された重 合性官能基同士の反応が同時に進行するため,高密度ポリマーブラシの調製が困難で ある (Figure 1-5).
Hadjichristidis らは,1,1-ジフェニルエチレン誘導体を重合開始点として用いることに
よりこの問題を解決している (Figure 1-6)15.1,1-ジフェニルエチレンは,共鳴安定化お よび立体障害の影響から単独重合しないという性質を有しているため,従来の手法で 問題となった開始剤間の重合反応が進行しないという利点を有する.この 1,1-ジフェ ニルエチレン誘導体から表面開始リビングアニオン重合を行うことにより,これまで に様々なポリマーブラシの調製が達成されている.一方で,1,1-ジフェニルエチレン型 の表面開始剤を使用した場合,重合系中で生成するポリマーの分子量分布が広がる傾
向がある32-33.ポリマーブラシの一次構造 (分子量,分子量分布および立体規則性)は,
Figure 1-5. Schematic illustration of preparation of polymer brush on the basis of surface initiated living anionic polymerization method using styrene or methyl methacrylate derivative as a surface initiator.
基材表面からポリマーブラシを剥離し評価する手法,もしくは,より簡便な手法とし てポリマーブラシと同一重合系中で生成した基材に固定化されていないポリマー (フ リーポリマー)の一次構造から評価される.Zhouらは,1,1-ジフェニルエチレン型の表 面開始剤から調製したポリスチレンブラシを基材から剥離し,その一次構造をフリー ポリマーと比較することでポリマーブラシの分子量がフリーポリマーよりも低く,そ の分子量分布が広いことを報告している32.また,Kirらは,サイズ・形状の異なるシ リカおよびガラス微粒子表面に固定化した 1,1-ジフェニルエチレン型の表面開始剤を 用いて調製したポリイソプレンブラシの分子量がフリーポリマーの分子量と異なる値 を示し,その分子量分布が広いことを報告している33.これは,1,1-ジフェニルエチレ ン型の表面開始剤を活性化する際に用いた n-ブチルリチウムが系中に残存し重合を開 始する,フリーポリマーが重合過程で表面開始剤を攻撃することに起因していると考
えられ,1,1-ジフェニルエチレン型の表面開始剤を用いた表面開始重合では分子量分布
の制御は困難である.
Figure 1-6. Surface initiated living anionic polymerization using 1,1-diphenylethylene derivative as an initiator.
Min らは,基材表面に固定化したハロゲン化アルキル誘導体をハロゲン-リチウム 交換反応に基づき活性化し,調製した表面開始剤を用いて分子量分布の狭いポリスチ レンブラシを調製している (Figure 1-7)16.この知見から,ハロゲン-リチウム交換反応 に基づき調製した開始剤を用いた表面開始リビングアニオン重合法系では,分子量分 布の制御が可能であることを示唆している.以上のように,表面開始リビングアニオ ン重合系においても,様々な開始剤系が考案されることによりその適応範囲が広がり つつある.しかしながら,従来の重合法では今後要求されるであろう界面の構造・機 能の精密制御は困難であり,新たな表面開始重合技術の開発が必要不可欠である.
Figure 1-7. Surface initiated living anionic polymerization on the basis of halogen-lithium exchange reaction.
1-2 本研究の目的と本論文の構成
前節でも述べたように,材料の微細・薄化が求められている現代産業において,界面 の設計および制御は必要不可欠である.本論文では,高分子界面の中でも特異な分子 鎖凝集構造,表面特性を有するポリマーブラシを研究対象とし,新規表面開始リビン グアニオン重合系に基づいた合成的なアプローチからポリマーブラシの立体規則性を 制御し,ポリマーブラシが形成するらせん構造を用いた新規機能の創出を目的とした.
以下に本論文の構成を述べる.
第一章では,本研究の背景,目的および構成について記述した.
第二章では,新規表面開始リビングアニオン重合法を用いた立体規則性PMMAブラシ を合成し,その重合挙動を評価した.
第三章では,得られたst-PMMAブラシに対してフラーレンの導入を行い,st-PMMAブ ラシが形成するらせん構造を明らかにするとともに,立体規則性の違いによる分子鎖 凝集構造の変化を評価した.
第四章では,st-PMMAブラシのらせん空孔を用いたフラーレンの選択分離を行い,バ ルク高分子とポリマーブラシの分子包接挙動の違いを明らかにするとともに,らせん 状ポリマーブラシの分離剤への応用性を示した.
第五章では,らせん状ポリマーブラシの側鎖への機能性部位の導入を行う手法として,
分子間相互作用によるPMMAのらせん誘起に関する検討を行った.
第六章では,各章における結論を述べ,本研究を総括した.
1-3 参考文献
1.Hirai, T.; Kobayashi, M.; Takahara, A., Polym. Chem. 2017.
2.Higaki, Y.; Hatae, K.; Ishikawa, T.; Takanohashi, T.; Hayashi, J.; Takahara, A., ACS Appl.
Mater. Interfaces 2014, 6 (22), 20385-20389.
3.Higaki, Y.; Okazaki, R.; Takahara, A., ACS Macro Letters 2012, 1 (9), 1124-1127.
4.Kusanagi, H.; Chatani, Y.; Tadokoro, H., Polymer 1994, 35 (10), 2028-2039.
5.Kusuyama, H.; Takase, M.; Higashihata, Y.; Tseng, H.-T.; Chatani, Y.; Tadokoro, H., Polymer 1982, 23 (9), 1256-1258.
6.Kusuyama, H., Polym. Commun. 1983, 24, 119-122.
7.Berghmans, M.; Thijs, S.; Cornette, M.; Berghmans, H.; De Schryver, F. C.; Moldenaers, P.;
Mewis, J., Macromolecules 1994, 27 (26), 7669-7676.
8.Kawauchi, T.; Kumaki, J.; Kitaura, A.; Okoshi, K.; Kusanagi, H.; Kobayashi, K.; Sugai, T.;
Shinohara, H.; Yashima, E., Angew. Chem. Int. Ed. 2008, 47 (3), 515-519.
9.Kawauchi, T.; Kitaura, A.; Kumaki, J.; Kusanagi, H.; Yashima, E., J. Am. Chem. Soc. 2008, 130 (36), 11889-11891.
10.Kawauchi, T.; Kawauchi, M.; Kodama, Y.; Takeichi, T., Macromolecules 2011, 44 (9), 3452- 3457.
11.Yamaguchi, H.; Kikuchi, M.; Kobayashi, M.; Ogawa, H.; Masunaga, H.; Sakata, O.; Takahara, A., Macromolecules 2012, 45 (3), 1509-1516.
12.Barbey, R.; Lavanant, L.; Paripovic, D.; Schüwer, N.; Sugnaux, C.; Tugulu, S.; Klok, H.-A., Chemical Reviews 2009, 109 (11), 5437-5527.
13.Zoppe, J. O.; Ataman, N. C.; Mocny, P.; Wang, J.; Moraes, J.; Klok, H.-A., Chemical Reviews 2017.
14.Jordan, R.; Ulman, A.; Kang, J. F.; Rafailovich, M. H.; Sokolov, J., J. Am. Chem. Soc. 1999, 121 (5), 1016-1022.
15. Advincula, R.; Zhou, Q.; Park, M.; Wang, S.; Mays, J.; Sakellariou, G.; Pispas, S.;
Hadjichristidis, N., Langmuir 2002, 18 (22), 8672-8684.
16.Min, J.; Lin, Y.; Zheng, J.; Tang, T., Chemical Communications 2015, 51 (27), 5921-5924.
17.Sato, M.; Kato, T.; Ohishi, T.; Ishige, R.; Ohta, N.; White, K. L.; Hirai, T.; Takahara, A., Macromolecules 2016, 49 (6), 2071-2076.
18.Tsubokawa, N., Journal of Polymer Science: Polymer Chemistry Edition 1984, 22 (6), 1515- 1524.
19.Whitesell, J. K.; Chang, H. K., Science 1993, 261 (5117), 73-76.
20.Chang, Y.-C.; Frank, C. W., Langmuir 1998, 14 (2), 326-334.
21.Zhang, N.; Salzinger, S.; Deubel, F.; Jordan, R.; Rieger, B., J. Am. Chem. Soc. 2012, 134 (17), 7333-7336.
22.Prehn, F. C.; Boyes, S. G., Macromolecules 2015, 48 (13), 4269-4280.
23.Lim, E.; Tu, G.; Schwartz, E.; Cornelissen, J. J. L. M.; Rowan, A. E.; Nolte, R. J. M.; Huck, W. T. S., Macromolecules 2008, 41 (6), 1945-1951.
24.Chu, B.-F.; Chu, J.-H.; Zhao, S.-Q.; Liu, N.; Wu, Z.-Q., Polym. Chem. 2018, 9 (12), 1379- 1384.
25.Tkachov, R.; Senkovskyy, V.; Horecha, M.; Oertel, U.; Stamm, M.; Kiriy, A., Chemical Communications 2010, 46 (9), 1425-1427.
26. Senkovskyy, V.; Tkachov, R.; Beryozkina, T.; Komber, H.; Oertel, U.; Horecha, M.;
Bocharova, V.; Stamm, M.; Gevorgyan, S. A.; Krebs, F. C.; Kiriy, A., J. Am. Chem. Soc. 2009, 131 (45), 16445-16453.
27.Kang, S.; Ono, R. J.; Bielawski, C. W., J. Am. Chem. Soc. 2013, 135 (13), 4984-4987.
28.Jiang, J.; Wang, X.; Lu, X.; Lu, Y., Applied Surface Science 2008, 255 (5, Part 1), 1888-1893.
29.Idota, N.; Nagase, K.; Tanaka, K.; Okano, T.; Annaka, M., Langmuir 2010, 26 (23), 17781- 17784.
30.Oosterling, M. L. C. M.; Sein, A.; Schouten, A. J., Polymer 1992, 33 (20), 4394-4400.
31.Schomaker, E.; Zwarteveen, A.-J.; Challa, G.; Capka, M., Synthesis of isotactic poly(methyl methacrylate) covalently bound to microparticulate silica. 1988; Vol. 29, p 158.
32.Zhou, Q.; Wang, S.; Fan, X.; Advincula, R.; Mays, J., Langmuir 2002, 18 (8), 3324-3331.
33.Kir, O.; Binder, W. H., European Polymer Journal 2013, 49 (10), 3078-3088.
第 2 章
表面開始リビングアニオン重合法に基づく
st-PMMA ブラシの調製と一次構造解析
2-1. 緒言
第一章で述べたように,表面開始リビングアニオン重合法はその重合制御が困難であ るために報告例が限られている.本研究では,分子量,分子量分布および立体規則性 を精密に制御したPMMAブラシの創製を目指すため,得られるポリマーブラシの分子 量分布が広がる1,1-ジフェニルエチレン型ではなく,ハロゲン-リチウム交換反応に基 づく重合系を用いる必要がある.しかしながら,従来報告されているハロゲン-リチウ ム交換反応に基づく重合系は,開始剤効率が著しく低いもの1,反応性が高くメタクリ レート系高分子の極性基 (C=O基)を攻撃するもの2のみであり,立体規則性PMMAブ ラシを調製するには新たな表面開始剤系を提案する必要がある.
メタクリレート系モノマーの立体特異性重合は,2011年に北山らのグループにより 報告されており,アルキルアルミニウム存在下,三級のアルキルリチウムである tert- BuLiを開始剤として無極性溶媒中で重合を行うことによりシンジオタクチック構造を
90%程度有するPMMAの重合が報告されている.(Figure 2-1)3.
アルキルリチウムを用いてメタクリレート系モノマーの重合を行う場合,カルボニ ル部分への求核攻撃を抑制するために嵩高い三級のアルキルリチウムを反応に用いる 必要がある.また,表面開始リビングアニオン重合では,基板の表面電荷,基板表面 に付着した不純物等の影響を大きく受けるため,基材と重合開始基との距離が非常に 重要である.Kirらは,重合開始部と基材表面の間のスペーサー長x = 3, 5, 11の1,1-ジ フェニルエチレン誘導体から表面開始アニオン重合を行い,重合開始部と基材表面と
Figure 2-1. Stereospecific living anionic polymerization using alkyl aluminum regent.
の距離が重合に及ぼす影響に関して評価している4.その結果,スペーサー長が長くな るにしたがって,重合によって得られるポリマーの分子量分布が狭くなっていること が明らかとなった.この結果は,重合開始部が基板表面から離れるにしたがって重合 が制御されるということを示している.一方で,得られたポリマーブラシのグラフト 密度は,スペーサー長が長くなるにつれて低下した.これはスペーサー長が長くなる につれて開始剤誘導体の立体的な嵩高さが増し,開始剤誘導体の固定化率が低下した ことに起因する.以上の知見から,本研究では表面開始剤前駆体としてスペーサー長 が6の三級ハロゲン化アルキルを選択した.
本章では,三級のハロゲン化アルキルを用いたハロゲン-リチウム交換反応とアル キルアルミニウムを添加した立体特異性リビングアニオン重合を組み合わせた新規表 面開始リビングアニオン重合法を用いてPMMAブラシの立体規則性制御を試みた.
2-2. 実験
2-2-1. 使用試薬の合成と精製
1) Dichloro methane
市販品(Wako Pure Chemical Industry, 99.0%)をCaH2で脱水後,trap to trap法により 減圧蒸留することにより精製した.
2) Methyl methacrylate(MMA)
市販品(Wako Pure Chemical Industry, 99%)を中性アルミナカラムに通して重合禁止 剤を取り除き,CaH2存在下で減圧蒸留した.蒸留後,triethyl aluminum を用いた化 学処理に基づき脱水した後にtrap to trap法に基づき蒸留し,すぐに重合に用いた.
3) Hydrofluoric acid
市販品(Wako Pure Chemical Industry, 48%)を蒸留水で10%に希釈したものを用い た.
4) Triethyl amine
市販品(Wako Pure Chemicals,)を水素化カルシウム存在下で蒸留し,すぐに用いた.
5) Ethyl-2-bromoisobutyrate (EBIB)
市販品 (Tokyo Chemical Inc., 98.0%) をCaH2存在下,減圧蒸留を行い精製した.精 製後はAr置換を行い冷蔵庫にて保管した。
以下の試薬は,購入したものをそのまま用いた.
5-hexen-1-ol (Tokyo Chemical Inc., 97.0%)
α- Bromoisobutyryl bromide (Sigma-Aldrich Co. LLC., 98.0%)
Sodium hydrogen carbonate (NaHCO3, Wako Pure Chemical Industry, 99.5%) Magnesium sulfate (Anhydrous) (MgSO4, Wako Pure Chemical Industry, 98.0%) Triethoxy silane (Tokyo Chemical Inc., 97.0%)
Platinum(0)-1,3-divinyl-1,1,3,3-tetramethyldisiloxane complex solution (Karstedt catalyst, Sigma-Aldrich Co. LLC., 2.0%, xylene)
Sulfuric acid (Nacalai Tesque, 98%)
Hydrogen peroxide (Wako Pure Chemical Industry, 30%) Ethanol (Wako Pure Chemical Industry, 99.5%)
Calcium hydrate (CaH2, Wako Pure Chemical Industry, 80.0%) Triethyl aluminum (Kanto Chemical, 1.04 M, n-toluene)
tert-Butyl lithium (tert-BuLi, Kanto Chemical, 1.60 M, n-pentane) Methanol (Wako Pure Chemical Industry, 99.5%)
Nano-size silica particles dispersed in water (SiP., Nissan Chemical Industry (MP2040), 40.7%, Diameter 130 ~ 230 nm)
Ammonia solution (NH3 aq., Wako Pure Chemical Industry, 28.0%) Chloroform (Wako Pure Chemical Industry, 99.5%)
n-Hexane (Wako Pure Chemical Industry, 95.0%)
Hydrochloric acid (HCl, Wako Pure Chemical Industry, 35.0%)
2-2-2. 1-(2-bromo-2-methyl)propyonyloxy-5-hexene (BPH)の合成
Ar雰囲気化,200 mLナスフラスコに5-hexen-1-ol (10.0 g, 99.9 mmol),triethyl amine (15.2 g, 150 mmol)お よ び dichloro methane (25 mL)を 収 め ,273 K に 冷 却 し た . α- bromoisobutyryl bromide (29.8 g, 130 mmol)をdichloro methane (15 mL)に溶解し,系中に ゆっくりと滴下し,12時間反応を行った.反応後,ろ過により生じたtriethyl amine塩 を除去した.その後,1 N HCl aq.,NaHCO3 aq.およびイオン交換水で洗浄し,有機層を 回収した.有機層をMgSO4で脱水後,dichloro methaneを減圧留去した.その後,減圧 蒸留により無色透明液体を得た.
2-2-3. (2-bromo-2-methyl)propyonyloxyhexyl triethoxysilane (BHE)の合成
Ar雰囲気化,200 mLナスフラスコにBPH (10 g, 40.2 mmol)を収め,313 Kで加熱した.
その後,triethoxy silane (19.8 g, 120 mmol),Karstedtcat.を順に加え,12時間反応を行っ た.反応追跡を1H NMRを用いて行い,BPHのvinyl 基に帰属されるシグナルが消失 するまで反応を行った.反応終了後,未反応のtriethoxy silaneを減圧留去し,減圧蒸留
Sheme 1-1. Synthesis of 1-(2-bromo-2-methyl)propyonyloxy-5-hexene (BPH).
α- bromoisobutyryl bromide
5-hexen-1-ol 1-(2-bromo-2-methyl)propyonyloxy-5-hexene
Scheme 2-2. Synthesis of (2-bromo-2-methyl)propyonyloxyhexyl triethoxysilane (BHE)
1-(2-bromo-2-methyl)propyonyloxy-5-hexene 2-bromo-2-methyl)propyonyloxyhexyl triethoxysilane
によりBHEを得た.得られた目的物の評価は,1H NMRおよび13C NMR測定により評 価した.
1H NMR (400 MHz, CDCl3) : 4.15 (t, 2H, CH2O), 3.80 (q, 6H, CH3CH2OSi), 1.91 (s, 6H, CCH3), 1.61-1.71 and 1.32-1.47 (m. 8H, CH2), 1.21 (t, 9H, CH3CH2OSi), 0.62 (t, 2H, SiCH2) ppm
13C NMR (100 MHz, CDCl3) : 172.0, 66.5, 58.7, 56.3, 32.9, 31.1, 28.6, 25.8, 23.0, 18.6, 10.7 ppm
2-2-4. シリコン基板表面へのBHEの固定化反応
シリコン基板は市販の片面鏡面シリコン基板(厚さ 0.5 mm, 結晶方位(111), SUMCO CORPORATION製)を用いた。シリコン基板はpiranha solution ( H2SO4 : H2O2 = 7 : 3 )を
用いて383 Kで1時間洗浄し,基板表面にSi – OH基を導入した.
ミクロチューブ (マルエム製)の中に BHE (0.07 mL)を入れ,toluene (0.3 mL)で希釈
し、BHEのtoluene希釈溶液を調製した。窒素置換したグローブボックス中で、セパラ
ブルフラスコに、piranha solutionを用いて洗浄した直後のシリコン基板およびBHE溶 液を入れて密閉し,428 Kで4時間反応を行った.反応後,シリコン基板を取り出し,
ethanolを用いて洗浄し,減圧乾燥した.固定化後の基板の表面組成を評価するために,
XPS測定を行った.
2-2-5. シリコン基板表面へのst-PMMAブラシの重合
Scheme 2-3. Surface initiated living anionic polymerization on Si substrate.
BHE を固定化したシリコン基板をフラスコに収め,373 Kで真空乾燥を行った.乾燥 後,dichloro methane (20 mL)を系中に加え,系を 195 K に冷却した.冷却後,系中に triethyl aluminum (0.45 mL, 0.50 mmol)およびtert-BuLi (0.063 mL, 0.10 mmol)を滴下し,
一時間撹拌した.撹拌後,系中にMMA (4 mL, 37.56 mmol)を加え12時間反応を行っ た.反応はmethanolで停止し,フリーポリマーはhexaneに再沈殿することにより回収 し,重合後の基板は chloroform を用いたソックスレー抽出により洗浄した.フリーポ リマーの分子量,分子量分布および立体規則性評価を,GPC 測定および NMR 測定に 基づき,重合後の基板表面の化学組成の評価を XPS 測定に基づきそれぞれ評価した.
2-2-6. シリカ微粒子表面へのBHEの固定化反応
500 mLのナスフラスコに平均粒形200 nmのSiP (13.03 g),ethanol (100 mL)を収め,
313 Kに加熱した.滴下漏斗にNH3 aq. (8.93 g)およびethanol (180 mL)を収め,系中に 6時間かけて滴下した.その後,滴下漏斗にBHE (1.90 g)およびethanol (10 mL)を収め,
系中に15 分かけて滴下した.滴下終了後,313 Kで 17 時間反応を行った.反応終了 後,反応溶液をテフロン製の遠心管に収め,室温,12,000 rpmで 10 分間遠心分離し,
上澄みを除去することにより反応溶液中の未反応のBHEおよびNH3 aq.を取り除いた.
その後,BHE固定化シリカ粒子 (BHE-g-SiP)を,ethanolに対して再分散した後,再び,
室温,12000 rpmで10分間遠心分離を行った.以上の操作を3回繰り返すことにより,
BHE-g-SiPの精製を行った.最後に,1,000 rpmで遠心分離を行いその上澄みを回収す
ることにより,反応時に凝集してしまったBHE-g-SiPを分離した.精製後,ethanolを 減圧留去し重合に用いた.
2-2-7. シリカ微粒子表面へのst-PMMAブラシの重合
重合フラスコにBHE-g-SiP (1 g)を収め,373 Kで12時間減圧乾燥を行った.乾燥後,
系中にdichloro methane (15 mL)を加え,超音波を用いてdichloro methaneに対してBHE- g-SiPを分散させた.分散後,重合系を195 Kに冷却し,triethyl aluminum (0.45 mL, 0.50 mmol)およびtert-BuLi (0.063 mL, 0.10 mmol)を用いて1時間Li交換反応を行った.反 応後,系中にMMA (4 mL, 37.56 mmol)を添加することにより重合を開始し,24時間反 応を行った.反応はmethanolで停止し,生成物はn-hexaneに再沈殿することにより回 収した.得られた生成物をchloroformに溶解しテフロン製の遠心管に収め,室温,6000 rpmで 10分間遠心分離し,上澄みを回収することによりフリーポリマーと PMMAグ ラフトシリカ微粒子 (st-PMMA-g-SiP)の分離を行った.その後,遠心管にchloroformを 加え沈殿物を再分散した後に,再び,室温,6000 rpmで10分間遠心分離し,上澄みを 回収した.回収した溶液のNMR測定を行い,PMMAのシグナルが存在しなくなるま でこの操作を繰り返し行った.また,回収した溶液を濃縮し,n-hexane に再沈殿する ことによりフリーポリマーを回収した.
2-2-8. HF aq.処理に基づくst-PMMAブラシの剥離
重合により得られたst-PMMA-g-SiPをchloroformに分散し,テフロン製の遠沈管に収
めた.chloroformと等量の10% HF aq.を加え,室温で24時間反応を行った.反応終了
後,NaHCO3 aq.を用いて溶液を中和し,有機層のみを回収した.回収した溶液を濃縮
し,n-hexaneに再沈殿後,濾過によりポリマーを回収した.真空乾燥後,NMR測定お
よびGPC測定を行った.
Scheme 2-4. Surface initiated living anionic polymerization on Si particle.
2-3. 測定
Gel Permeation Chromatography (GPC)測定
得られたフリーポリマーの分子量および分子量分布を評価するために,GPC測定を行 った.装置はHLC-8120GPC (東ソー(株)製) を用い,送液速度は0.5 mL/min でカラム オーブンを40 ˚Cに設定して測定を行った.溶媒はTHF,分析カラムにTSK gel superH 2500とTSK gel superH 4000とTSK gel superH 6000 (東ソー(株)製) を直列に接続し,
カラムオーブンに CO-2065 plus(日本分光(株)製),RI 検出器に RI-2031 plus(日本分光
(株)製),ポンプにPU-2087 plus(日本分光(株)製)を用いた.
Nuclear Magnetic Resonance (1H-NMR)測定
得られたフリーポリマーの立体規則性を評価するため,1H-NMR 測定を行った.1H- NMR測定は,AVANCE-Ⅲ400 (Bruker Co., Ltd.)を用いて行った.測定周波数は400.13 MHz,サンプルはchloroform-d中で測定し,chemical shiftはchloroform (7.26 ppm)を基 準にした.測定モードはNONで行った.
Nuclear Magnetic Resonance (13C-NMR)測定
得られたフリーポリマーの立体規則性を評価するため,13C-NMR 測定を行った.13C- NMR測定は,AVANCE-Ⅲ400 (Bruker Co., Ltd.)を用いて行った.測定周波数は100.61 MHz,サンプルはchloroform-d中で測定し,chemical shiftはchloroform (77.3 ppm)を基 準にした.測定モードはNONで行った.
Thermogravimetric Analysis (TG)測定
BHE-g-SiPおよび st-PMMA-g-SiP の固定化量を評価するため,TG測定を行った.TG
測定には,セイコーインスツルメント(SII)製EXSTARを用い,窒素雰囲気化,298~800 Kの温度領域で測定を行い,10 K / minで熱走査した.
X-ray Photoelectron Spectroscopy (XPS) 測定
BHE 固定化シリコン基板および st-PMMA ブラシの表面化学組成を評価するために,
XPS測定を行った.測定はAPEX (アルバック・ファイ (株) 製) を用いて行った.X線 源に単色化Al K線を使用し,加速電圧kV (200 W),X線照射角45˚,測定室内の 圧力10-8 - 10-9 Torrにて測定を行った.全範囲測定はステップ1.0 eV,積算16回で行 い,高分解能測定はステップ0.10 eV,積算64回で行った.
Atomic Force Microscopy (AFM) 観察
BHE固定化シリコン基板およびst-PMMAブラシの表面形状をAFM観察により評価し た.形状像観察にはSPA400 (セイコーインスツル (株) 製) を用い,測定モードはDFM で行い,スキャナは20 µm PZT (SPA400-PZT (FS20N) ),カンチレバーはSI-DF20 (背面 Alコート) を用いた.
Spectroscopic Ellipsometry測定
開始剤固定化基板および st-PMMA ブラシの膜厚を分光エリプソメトリーにより評価 した.光源には150 WのHaランプを用い,入射角70 で測定を行った.シリコン基板 表面の酸化被膜およびPMMAブラシの屈折率をそれぞれ4.14,1.49,消光係数を0.045 として測定を行った.開始剤固定化基板の屈折率は1.49とした.
2-4. 結果および考察
2-4-1. 表面開始リビングアニオン重合法に基づくst-PMMAブラシの調製
本重合系では,基板表面に固定化する開始剤前駆体としてBHEと呼ばれる化合物を用 いた.BHEは表面開始原子移動ラジカル重合法で汎用的に用いられている化合物であ り,その合成,固定化条件が最適化されている.基板表面へのBHEの固定化を明らか にするために,XPS測定を行った.BHE 固定化反応前後の XPSスペクトルを Figure 2-1 に示す.BHE を固定化したシリコン基板からは,シリコン基板の化学組成には存 在しない炭素 (C)ならびに臭素 (Br)に帰属されるピークが観測された.また,C1sピー クの高分解能測定結果に着目すると,C1s領域においてピークが三つ観測された.XPS 測定では,測定元素の電子状態の違いに応じてピークのシフトが観測される.BHEの 分子内にはC=O結合,C-O結合ならびにC-C結合が存在しており,C1sピークの高分解 能測定において C=O 結合,C-O 結合,C-C 結合それぞれに起因したピークが観測され る.XPS測定の結果から,BHE のC=O結合,C-O 結合,C-C結合それぞれの結合に帰 属されるピークが観測されたことから,シリコン基板表面にBHEが固定化されたこと
Figure 2-1. XPS spectrum of BHE immobilized on Si substrate.
が示唆される.一方で,測定により得られた基板表面の原子組成比はBr 2.81%, Si 32.3%, C 22.6%, O 42.3%であり,これはBHEの原子組成比であるBr 5.9%, Si 5.9%, C 58.8%, O
29.4%とは一致しなかった.今回の系の場合,測定波長 0.154 nm,光電子角度が45 で
あり,BHEの分子鎖長がXPS測定の分析深さよりも短いためにシリコン基板由来のピ ークを検出している.また,炭素の存在比と比較して臭素の存在比が少なく観測され ている原因として,測定時に照射されるX線により比較的に弱い結合であるC – Br結 合が切断されているためだと考えられる.
次に,triethyl aluminum および tert-BuLi を用いて表面開始リビングアニオン重合を行 った基板のXPS測定を行った.Figure 2-2に表面開始リビングアニオン重合前後にお けるXPSスペクトルを示す.重合後の基板の XPSスペクトルからは,BHE の臭素に 帰属されるBr3dピークが消失した.これはリチウム交換反応の過程でBHE のBr基が Li に変換されたことに起因する.また,重合後の基板の XPS スペクトルからは Si2s, Si2pピークが完全に消失し,PMMAの化学組成に起因する O1s,C1sピークのみが観測 された.それぞれのピークの積分強度比から算出された重合後の基板表面における原 子組成比は C 71.9%, O 28.1%であり,これは PMMA の原子組成比の理論値である C
Figure 2-2. XPS spectra of a) PMMA brush and b) BHE immobilized Si substrate.
a) BHE substrate b) PMMA substrate
71.4%, O 21.6%とよく一致した.以上の結果は,リチウム交換反応により調製された基 板表面のアルキルリチウムからMMAの重合が進行し,XPSの分析深さ以上の膜厚を 有するPMMAブラシが調製されたことを示している.
得られたst-PMMAブラシの表面形態および膜厚の評価を AFMおよび分光エリプ
ソメトリー測定に基づき行った.Figure 2-3a, bにAFM測定から得られたst-PMMAブ ラシの形状像およびポリマーブラシをスクラッチ法に基づき剥離した際の形状像を示 す.得られた表面形状像から求めた平均二乗面粗さ(RMS ラフネス)は,5 × 5 m のスキャンエリアにおいて1 nm以下であった.この結果から,調製したPMMAブラ シ表面が比較的均一であることが明らかとなった.また,Figure 2-3bに示すポリマー ブラシをスクラッチ法に基づき剥離したサンプルの形状像からは,ポリマーブラシの 膜厚に関する情報が得られた.以上の結果は,シリコン基板表面で表面開始リビング アニオン重合が均一に進行し,目的とするポリマーブラシが得られたことを支持する 結果である.
一方,これまでに報告されている表面開始リビングアニオン重合の研究例では,シリ コン基板に調製したポリマーブラシの表面形態は不均一になるとされている5.このよ Figure 2-3. The topological image of a) obtained st-PMMA brush surface and b) scratched surface.
うに得られたポリマーブラシの表面が不均一になる原因として,重合系中に残存する 不純物,重合開始剤の反応性などの要因が挙げられている1, 5.リビングアニオン重合 法は,リビングラジカル重合などの重合法と比較して不純物の影響を受けやすく重合 の制御が困難であることが知られている.従来の表面開始リビングアニオン重合系で は,基板表面に吸着した不純物の除去が困難であり,系中の不純物が調製された表面 開始剤を失活するため重合が均一に進行しないとされている.また,もう一つの要因 として溶媒効果による極性溶媒中でのアルキルリチウムの反応性の高さが挙げられる.
リビングアニオン重合系において,成長種であるカルバニオンは対イオンとの相互作 用の強さに依存して四つの状態をとる.この時,対イオンとカルバニオン間の相互作 用が弱いほど,両イオン間距離が大きくなりカルバニオンの反応性が増加する.THF などの極性溶媒を重合溶媒とした場合,溶媒が対イオンに配位することにより対イオ ンとカルバニオン間の相互作用が弱まり,カルバニオンの反応性が増す.これまでに 報告されているシリコン基板表面を用いた表面開始リビングアニオン重合において,
重合開始点となる開始剤誘導体は Si-O 結合により固定化されている.従来の系では,
極性溶媒によって活性化されたカルバニオンが開始剤誘導体のSi-O結合を攻撃するこ とにより開始剤誘導体が基板表面から剥離し,そのために基板表面において重合が均 一に進行しなかったと考えられる.
本研究で提案する重合系は,極性の低いジクロロメタンを溶媒として用いている ことからアルキルリチウムおよび成長末端カルバニオンによる基板表面のSi – O結合 への攻撃を抑制すると考えられる.さらに,立体規則性を制御するための添加剤とし て用いられている triethyl aluminum は,高い反応性を有する Lewis 酸としても知られ ている.本重合系では,重合系に重合開始剤である tert-BuLi を添加する前に triethyl
aluminum を添加しており,triethyl aluminum が重合系中の不純物を除去した後に表面
開始剤を調製するため,従来の重合系と比較して不純物の影響が抑制されたと考えら
れる.以上の理由から,本研究で提案する重合系を用いることにより,従来の表面開 始リビングアニオン重合法では調製困難であった均一なポリマーブラシの調製が達成 された.
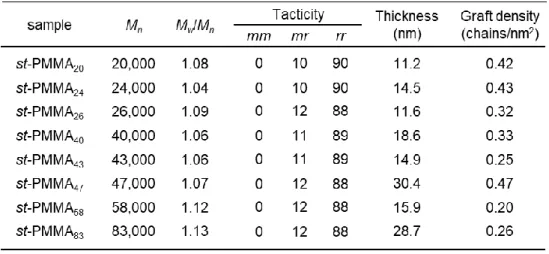
2-4-2. st-PMMAブラシの一次構造解析
表面開始重合により調製された高分子は,バルク状態と比較して運動性が低く,隣接 する分子間の立体障害などの影響から,バルクとは異なる分子量,分子量分布および 立体規則性を有している可能性がある 4, 6.そこで本重合系により調製した PMMA ブ ラシの一次構造を評価するために,ポリマーブラシを基材表面から剥離し,NMR測定 およびGPC測定を行った.Si基板表面に調製したポリマーブラシを用いた場合,Si基 板の表面積が小さく測定に十分な量のポリマーが得られないため,本実験ではSi基板 と比較して表面積の大きなシリカ微粒子(粒径:200 nm)上にポリマーブラシを調製し ポリマーブラシの剥離を行った.Si 基板と同様にシリカ微粒子表面にポリマーブラシ が調製されているかを評価するためにTGA測定を行った.Figure 2-4に表面開始リビ ングアニオン重合前後におけるシリカ微粒子のTGA曲線を示す.Figure 2-4に示すよ
Figure 2-4. XPS spectra of a) PMMA brush and b) BHE immobilized Si substrate.
うに BHE を固定化したシリカ微粒子からは,BHE の分解に基づく重量減少が観測さ れておりシリカ粒子表面にBHEが固定化されていることを示している.また,triethyl
aluminum および tert-BuLi を用いて表面開始リビングアニオン重合を行ったシリカ微
粒子の TGA 曲線では,340 ℃付近において顕著な重量減少が観測された.バルクの PMMAに関して同様の実験を行ったところ,同程度の温度領域において重量減少が観 測された.以上の結果から,シリカ微粒子表面にPMMAブラシが調製されたことが明 らかである.
得られた PMMA ブラシを基材から剥離するために,得られたポリマーブラシを
chloroformに分散し10 % HF水溶液で処理した.24時間後,有機相を回収し再沈殿お
よび乾燥した後,剥離したポリマーのNMR測定およびGPC測定を行った.本重合系 では,表面開始剤を調製するリチウム交換反応時に過剰量のtert-BuLiを反応に用いて おり,同一重合系中からポリマーブラシと同時に基材表面に固定化されていないポリ マー(以下,フリーポリマーと呼ぶ)が得られる.このフリーポリマーの一次構造評 価も同時に行い,ポリマーブラシの一次構造との比較を行うことで重合挙動の評価を 行った.Figure 2-5a,b にフリーポリマーおよび基材表面から剥離したポリマーブラシ のNMRスペクトルおよびGPC曲線を示す.フリーポリマーのNMR測定の結果から,
Figure 2-5. The results of a) 1H NMR and b) GPC measurement of polymer brush and free polymer.
Polymer brush
本重合系中でPMMAが得られたことを明らかにした.また,1.0 ppm近傍のPMMAの
-メチル基に帰属されるシグナルの積分強度比より得られた PMMA の立体規則性評 価を行った.その結果,得られたPMMAが90%程度のシンジオタクチック構造を有し ていることが明らかとなった.この値は,先行研究で報告されているジクロロメタン 中,triethyl aluminum/tert-BuLi を用いて調製したst-PMMAの値とよく一致している3. GPC測定の結果からも分子量分布の狭いポリマーが得られていることが明らかであり,
重合が制御されていることを示している.従来の表面開始リビングアニオン重合系で は,反応系中および基材に付着した不純物の影響により得られるポリマーの一次構造 の制御が困難であるとされているが,本重合系では,立体規則性の制御のために添加
した triethyl aluminum が重合系中でルイス酸として作用することにより重合系中の不
純物を失活し,重合の制御が達成されたと考えられる.また,Figure 2-5において剥離 したポリマーブラシのNMR スペクトル,GPC 曲線はフリーポリマーのものとよく一 致しており,同一重合系中で調製されたフリーポリマーとポリマーブラシの一次構造 が等しいことが明らかとなった(以下,ポリマーブラシの一次構造は,フリーポリマ ーの一次構造から評価する).Table 2-1 に測定により得られたフリーポリマーおよび ポリマーブラシの一次構造を示す.
Table 2-1. The Results of the Polymerization.
2-4-3. st-PMMAブラシのグラフト密度評価
表面開始リビングアニオン重合により調製した st-PMMA ブラシのグラフト密度を評 価した.シリコン基板に調製したst-PMMAブラシのグラフト密度は,分光エリプソメ ーターより得られた膜厚を用いて,Eq. 2-1から算出した.
Eq. 2-1
Eq. 4-1において,, , h, NA, Mnは,それぞれグラフト密度 (chains/nm2),バルク状態 におけるポリマー密度 (g/cm3),ポリマーブラシの乾燥膜厚 (nm),アボガドロ数 (mol-
1 ),フリーポリマーの数平均分子量 (g/mol)である.Table 2-2にst-PMMAブラシの一
次構造および得られたポリマーブラシの膜厚,グラフト密度を示す.Eq. 4-1から算出
されたst-PMMAブラシのグラフト密度は最大で0.63 chains / nm2であった.すべての
st-PMMAブラシにおいてグラフト密度 > 0.1を満たしていることから,本重合系で調
製した st-PMMA ブラシは濃厚ブラシ構造を形成している.また,Table 2-2 に示すよ
うに,重合条件を変更することにより幅広い範囲の分子量を有する高密度st-PMMAブ ラシを調製できることが明らかとなった.一方で,ポリマーブラシのグラフト密度は,
ポリマーブラシの分子量が大きくなるにつれて低下するという傾向が観測された.本 重合系では,リチウム交換反応に用いるtert-BuLiの添加量を変更することによりポリ マーブラシの分子量を調整している.リチウム交換反応時のtert-BuLi濃度は交換反応 の反応率に影響をおよぼすと考えられ,高分子量体を調製する場合,添加しているtert-
BuLi量が低分子量体調製時よりも少ないためリチウム交換反応の反応率が低下しグラ フト密度が低下したと考えられる.
2-4-4. ハロゲン-リチウム交換反応に基づく表面開始リビングアニオン重合の開始剤
効率評価
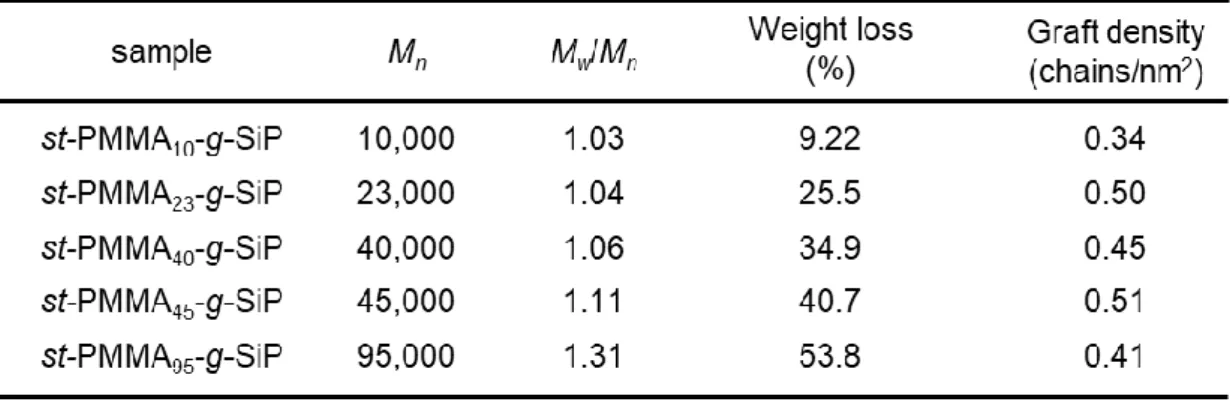
表面開始リビングアニオン重合の開始剤効率を評価するために,シリカ粒子表面に固 定化された BHE およびst-PMMAブラシのグラフト密度を TGA 測定に基づき算出し た.それぞれのグラフト密度,TGA測定より得られた重量減少の値を用いて以下に示
すEq. 2-2を用いて算出した.
Eq. 2-2
Table 2-2. The Results of the Polymerization.
, m, d, M, NAは,それぞれ,グラフト密度 (chains/nm2),一粒子あたりに固定化されて いるポリマー重量 (g),シリカ粒子の粒径 ( 200 nm),BHE の分子量 (g/mol),アボガ ドロ数 (chains/mol)である., wは実験に用いたシリカ微粒子の密度 (1.6 g/nm3)および TGA測定により得られた重量減少の値である.Figure 2-4に示すBHE-g-SiPのTGA測 定の結果を用いて,Eq. 2-2から算出されたBHEの固定化量は,3.5 molecules/nm2であ った.先行研究において,シリカ微粒子上にSi-OHグループが1 nm2あたりおよそ5- 6分子存在しているとされており,本系では比較的高密度にBHEが固定化されている と考えられる.また,この固定化密度は辻井らによって報告されている先行研究の値 ともよく一致している7.
次に表面開始リビングアニオン重合により調製した st-PMMA-g-SiP の TGA 測定を 行った.Figure 2-6に様々な分子量を有するst-PMMA-g-SiPのTGA曲線を,Table 2-3 にその分子量,分子量分布およびグラフト密度を示す.Figure 2-6に示すように,全て
の st-PMMA-g-SiP ブラシにおいて 380 ℃付近に顕著な重量減少が観測されている.
得られた重量減少からst-PMMA-g-SiPのグラフト密度を算出した結果,グラフト密度
Figure 2-6. TGA curves of st-PMMA grafted of silica particles.
は0.34 ~0.51 chains/nm2の値を示した.シリカ微粒子に固定化されたBHEのグラフト
密度が 3.5 molecules/nm2であることから,本重合系で用いている表面開始剤の開始剤
効率は最大でおよそ14.6 %である.これは先行研究で報告されている表面開始リビン グアニオン重合法で用いられている他の表面開始剤の開始剤効率(25%)と比較すると 若干低い値である2.
Table 2-3. The Results of Surface Initiated Living Anionic Polymerization on Silica Particles.
2-5. 結論
本研究では,立体規則性を制御するための新規表面開始リビングアニオン重合系を提 案し,シンジオタクチックPMMAブラシの調製を行った.はじめに基材表面で重合さ れたポリマーの一次構造を評価するために,シリカ粒子表面に調製したポリマーブラ シを粒子表面から剥離し,一次構造解析を行った.剥離したポリマーは高いシンジオ タクチック構造および狭い分子量分布を有しており,本研究で提案する重合系を用い ることによってポリマーブラシの立体規則性を制御できることが明らかとなった.ま た,ポリマーブラシの一次構造は同一重合系から得られたフリーポリマーの一次構造 とよく一致していた.次に平滑なシリコン基板表面にポリマーブラシを調製し,得ら れたポリマーブラシの表面形態を評価した.従来報告されている表面開始リビングア ニオン重合系で得られたポリマーブラシは,重合系中の不純物の影響によりポリマー ブラシ表面が不均一になると報告されているが,本重合系で調製したポリマーブラシ が均一な表面を有していることが明らかとなった.これは,本重合系で立体規則性制 御のために添加したトリエチルアルミニウムが重合系中でルイス酸としても作用し,
重合系中の不純物を失活したためであると考えられる.
2-6. 参考文献
1. Jordan, R.; Ulman, A.; Kang, J. F.; Rafailovich, M. H.; Sokolov, J., Surface-Initiated Anionic Polymerization of Styrene by Means of Self-Assembled Monolayers. J. Am. Chem. Soc.
1999, 121 (5), 1016-1022.
2. Min, J.; Lin, Y.; Zheng, J.; Tang, T., A novel strategy to synthesize well-defined PS brushes on silica particles by combination of lithium-iodine exchange (LIE) and surface-initiated living anionic polymerization (SI-LAP). Chemical Communications 2015, 51 (27), 5921-5924.
3. Nishiura, T.; Abe, Y.; Kitayama, T., Syndiotactic-specific polymerization of methyl methacrylate with tert-butyllithium/trialkylaluminum in dichloromethane. Polymer Bulletin 2011, 66 (7), 917-923.
4. Kir, O.; Binder, W. H., Living anionic surface initiated polymerization (LASIP) of isoprene from silica nano- and glass particles. European Polymer Journal 2013, 49 (10), 3078- 3088.
5. Advincula, R.; Zhou, Q.; Park, M.; Wang, S.; Mays, J.; Sakellariou, G.; Pispas, S.;
Hadjichristidis, N., Polymer Brushes by Living Anionic Surface Initiated Polymerization on Flat Silicon (SiOx) and Gold Surfaces: Homopolymers and Block Copolymers. Langmuir 2002, 18 (22), 8672-8684.
6. Zhou, Q.; Wang, S.; Fan, X.; Advincula, R.; Mays, J., Living anionic surface-initiated polymerization (LASIP) of a polymer on silica nanoparticles. Langmuir 2002, 18 (8), 3324-3331.
7. Ohno, K.; Morinaga, T.; Koh, K.; Tsujii, Y.; Fukuda, T., Synthesis of Monodisperse Silica Particles Coated with Well-Defined, High-Density Polymer Brushes by Surface-Initiated Atom Transfer Radical Polymerization. Macromolecules 2005, 38 (6), 2137-2142.
第 3 章
st-PMMA ブラシのフラーレン包接挙動の
評価および分子鎖凝集構造解析
3-1. 緒言
ポリマーブラシは高分子鎖片側末端が基材に対して密に固定化されている高分子膜で あり,隣接する分子鎖間の立体障害に基づき,基材に対して垂直方向に伸長した構造 を有する.1-6 濃厚ポリマーブラシは密な分子鎖凝集構造に基づき,ブラシ膜内部への 分子の侵入を阻害する性質を有するため,調製されたポリマーブラシ内部に新たに分 子を導入することは困難である.
らせん構造のような二次構造を形成するポリマーブラシの例として,ポリペプチド 系やポリイソシアニド系のポリマーブラシが報告されている.7-10ペプチド系ポリマー ブラシは基材表面に固定化されたアミノ基を開始剤としてN- carboxyanhydride(NCA)
の開環重合を行うことにより調製されるが,NCAモノマーの合成,精製の観点から分 子設計が困難であることが問題として挙げられる.また,ペプチド系ポリマーブラシ は分子内の水素結合に基づき-helix構造を形成するが,-helix構造のらせん空孔径が 小さいことや形成するらせん構造が比較的剛直であるためらせん内部に分子の導入が 困難であることが問題として挙げられる.ポリイソシアニド系ポリマーブラシも分子 設計,形成するらせん構造の剛直さの観点かららせん内部への分子の導入が困難であ る.
一般的なビニル系ポリマーブラシは,分子鎖がコンフォメーション的に無秩序なラ ンダムコイル構造を有しており,膜内部に他分子を取り込むことが困難であるが,立 体規則性を制御したビニル系高分子は,アタクチックの高分子と異なりらせん構造の ような二次構造を形成することが知られている.11-16例えば,汎用的な高分子材料とし て知られているポリメタクリル酸メチル(PMMA)は, 立体規則性がアタクチック(at-
PMMA)の時にランダムコイル構造を,立体規則性がイソタクチック(it-PMMA),シン
ジオタクチック(st-PMMA)の時にらせん構造を形成する.特に,st-PMMA はその内部 に様々な種類の分子を包接することが知られており,内部に包接する分子のサイズに
合わせてらせんサイズが変化する柔軟ならせん構造を形成することが明らかにされて いる(induced fit 型)12-13, 15, 17.この柔軟ならせん構造がポリマーブラシにおいても形成 可能であるとするならば,導入分子サイズによりその微細構造を変化させる機能性界 面の創製が期待される.
また,ポリマーブラシの特異な分子鎖凝集構造は,形成するらせん,包接錯体の構 造にも影響をおよぼすと考えられる.らせん構造のような筒状高分子が分子を包接す る場合,包接された分子は一軸方向に配向し18,st-PMMA/フラーレン包接錯体におい ても内包されたフラーレンはらせん軸に沿って配向する.フラーレンの配向制御は電 気デバイスなどへの応用を考えた際に非常に重要である一方で,従来のst-PMMA/フラ ーレン包接錯体では包接錯体自身の配向を制御することは困難である.前述したよう に,ポリマーブラシは隣接する分子鎖間の立体的な反発に基づき分子鎖が基材に対し て垂直方向に伸長した構造を形成する.ポリマーブラシを構成するグラフト鎖が包接 錯体を形成した場合,ポリマーブラシ特有の分子配向が包接錯体の配向にも影響を及 ぼすと考えられ,内包したフラーレンを基材に対して垂直方向に配向させることが可 能になると期待される.
本章では,st-PMMAブラシがフラーレンを包接した際に形成する構造および立体規 則性がポリマーブラシの構造におよぼす影響を評価した.
3-2. 実験
3-2-1. 使用試薬の合成と精製
1) Methyl methacrylate(MMA)
市販品(Wako Pure Chemical Industry, 99%)を中性アルミナカラムに通して重合禁止 剤を取り除き,水素化カルシウム存在下で減圧蒸留した.
2) CuBr (Ⅰ)
市販品 (Wako Pure Chemicals, 99.9%) を試験管に加え,Acetic acid中での撹拌 (撹拌 後,上澄み溶液を除去) を10回,その後,Ethanol中での撹拌 (撹拌後,上澄み溶液 を除去) を10回行った後,室温で減圧乾燥を行うことで精製した.
3) Ethyl-2-bromo isobtylate
市販品 (Tokyo Chemical Inc., 98.0%) を水素化カルシウム存在下,減圧蒸留を行い精 製した.精製後はアルゴン置換を行い冷蔵庫にて保管した。
4) バルクのst-PMMAおよびst-PMMAブラシは,第二章で調製したものを用いた.
以下の試薬は,購入したものをそのまま用いた.
Acetic acid (Wako Pure Chemical Industry, 99%) Fullerene C60 (Tokyo Chemical Industry, 99.5%) Fullerene C70 (Tokyo Chemical Industry, 98.0%) Toluene (Wako Pure Chemical Industry, 99.5%)
Calcium hydride (CaH2, Wako Pure Chemical Industry, 80.0%) Methanol (Wako Pure Chemical Industry, 99.5%)
Chloroform (Wako Pure Chemical Industry, 99.5%) Anisole (Wako Pure Chemical Industry, 99.0%)
4,4’-dinonyl-2,2’-bipyridyl (Tokyo Chemical Inc., 98.0%)
3-2-2. フリーポリマーへのfullerene導入13
Fullerene (2 mg)およびtoluene (1 mL)を2 mLサンプル瓶に加え,5分間撹拌した.撹拌 後,フリーポリマー50 mgを加え,110 Cで10分間撹拌した.撹拌後,ゆっくりと室 温まで冷却し,ゲル化後,2000 rpmで10分間遠心分離を行った.遠心分離後,上澄み を捨て再びtolueneを加えて室温で撹拌した.撹拌後,2000 rpmで10分間遠心分離を 行った後,上澄みを回収した.以上の操作を繰り返し行い,系中の余剰な fullerene を 取り除いた.その後,真空乾燥を行い,XRD測定を行った.
3-2-3. ATRP法に基づくポリマーブラシの調製
重合管にBHE固定化シリコン基板,MMA(5.00 mL),EBIB anisole溶液(0.100 M, 0.600 mL, 0.0600 mmol)およびanisole (3.1 mL) を収め,30分間アルゴンバブリングを行っ た.また,別の容器にanisole(1 mL)および4,4’-dinonyl-2,2’-bipyridyl(144 mg, 0.353 mmol)を収め,30分間Arバブリングを行った後,CuBr (Ⅰ) (17.4 mg, 0.176 mmol)を加 えて再び30分間Arバブリングを行った.重合管に調製した溶液を加え,333 Kで24 時間反応を行った.重合後,tolueneを用いたソックスレー抽出により基板を洗浄した.
Scheme 3-1. Surface initiated living radical polymerization.
また,重合溶液をアルミナカラムにより精製した後,methanol に再沈殿することでフ リーポリマーを得た.
3-2-4. ポリマーブラシへのfullerene導入
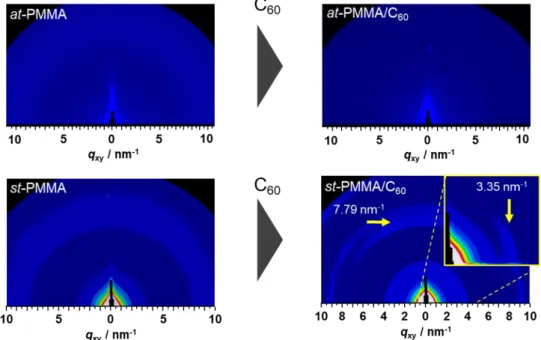
Fullerene (2 mg)およびtoluene (1 mL)を2 mLサンプル瓶に加え,5分間撹拌した.撹拌 後,調製したポリマーブラシを溶液に浸漬し,383 Kで10分間撹拌した.撹拌後,ゆ っくりと室温まで冷却し,溶液から取り出した.乾燥後,GIWAXD測定を行った.
3-3. 測定
Differential Scanning Calorimetry (DSC) 測定
St-PMMAブラシがfullerene と包接錯体を形成していることを評価するために DSC 測
定を行った.DSC測定には,セイコーインスツルメント(SII)製EXSTAR 6000を用 い,窒素雰囲気化,273~553 Kの温度領域で測定を行い,10 K / minで熱走査した.
Powder X-ray diffraction (XRD) 測定
St-PMMAブラシがfullereneと包接錯体を形成していることを評価するために XRD測
定を行った.XRD測定には,Rigaku Co., Ltd.製Smartlabを用い,測定波長0.154 nmで 行った.
Spectroscopic Ellipsometry測定
at-PMMA ブラシの膜厚を分光エリプソメトリーにより評価した.光源には 150 W の
Ha ランプを用い,入射角 70 で測定を行った.シリコン基板表面の酸化被膜および PMMAブラシの屈折率をそれぞれ4.14,1.49,消光係数を0.045として測定を行った.
開始剤固定化基板の屈折率は1.49とした.
Atomic Force Microscopy (AFM) 観察
at-PMMAブラシの表面形状をAFM観察により評価した.
形状像観察にはSPA400 (セイコーインスツル (株) 製) を用い,測定モードはDFMで 行い,スキャナは20 µm PZT (SPA400-PZT (FS20N) ),カンチレバーはSI-DF20 (背面Al コート) を用いた.
Grazing incidence wide angle X-ray diffraction (GIWAXD) 測定
調製したポリマーブラシの分子鎖凝集構造を評価するために,GIWAXD測定を行った.
測定は,大型放射光施設SPring-8 BL40B2で行い,測定波長0.1 nm,検出器はIP(IP、
R-AXIS VII、Rigaku Co., Ltd.)を用い,カメラ長833 mmで測定を行った.カメラ長及
びビーム中心はベヘン酸銀を標準試料として算出し,視斜角を 0.12 で測定した.ま た,二次元像から回折ピークの配向が観測されたサンプルに関して,回折ピークの方 位角プロットを作成し,Eq. (1)に示すガウス関数を用いてFittingを行った後,Eq. (2)に
示すHermanの配向関数を用いて配向度を算出した.
Eq.
(1)
Eq.
(2)
Hermanの配向関数から算出された配向度Fに関して,F = 1, F = -1の場合,それぞれ
in-plane方向,out-of plane方向に完全配向,F = 0の場合,完全無配向を意味している.
また,本論文では散乱ベクトルq = (4/)sinと定義する.