目次
頁
2.7.6 個々の試験のまとめ ... 4
臨床試験一覧表(表 2.7.6-1)... 5
2.7.6.1 バイオアベイラビリティ(BA)試験報告書 ... 6
2.7.6.1.1 N01377 試験(単回静脈内持続投与時及び単回経口投与時の薬物動態の比
較)(5.3.1.1.1:評価資料) ...
6
...
2.7.6.1.2 N01077 試験(単回静脈内持続投与時と単回経口投与時の薬物動態の比較及
び反復静脈内投与時の薬物動態の検討)(5.3.1.1.2:参考資料) ...
17
...
2.7.6.2 健康被験者における PK 及び初期忍容性試験報告書 ...
31
2.7.6.2.1 N01165 試験(健康成人におけるレベチラセタム用量漸増の 15 分間又は 5 分
間単回静脈内持続投与の安全性及び忍容性の検討)(5.3.3.1.1:参考資料) ...
31
...
2.7.6.3 内因性要因を検討した PK 試験報告書 ...
42
2.7.6.3.1 EP0038 試験(日本人と白人における静脈内持続投与時の薬物動態の比較)
(5.3.3.3.1:評価資料) ...
42
...
2.7.6.4 非対照試験報告書 ...
54
2.7.6.4.1 N01378 試験(成人てんかん患者における経口から静脈内への投与経路変更
時の安全性、薬物動態及び有効性の検討)(5.3.5.2.1:評価資料) ...
54
...
2.7.6.4.2 N01166 試験(成人てんかん患者における経口から静脈内への投与経路変更
時の安全性及び忍容性の検討)(
5.3.5.2.2:参考資料) ...
67
...
2.7.6.4.3 N01274 試験(4~16 歳の小児てんかん患者における静脈内持続投与時の安
全性及び薬物動態の検討)(5.3.5.2.3:参考資料) ...
79
...
略語及び名称一覧表
略語(略称)
内容
ALP
Alkaline phosphatase:アルカリホスファターゼ
ALT (GPT)
Alanine aminotransferase:アラニン・アミノトランスフェラーゼ
Glutamic pyruvic transaminase:グルタミン酸ピルビン酸トランスアミナーゼ
AST (GOT)
Aspartate aminotransferase:アスパラギン酸アミノトランスフェラーゼ
Glutamic oxaloacetic transaminase:グルタミン酸オキザロ酢酸トランスアミナー
ゼ
AUC
Area under the plasma concentration-time curve from zero to infinity:無限時間まで
の血漿中濃度-時間曲線下面積
AUC
day1Area under the plasma concentration-time curve after first dose:初回投与後の血漿中
濃度-時間曲線下面積
AUC(0-t)
Area under the plasma concentration-time curve from zero to the time of the last
quantifiable concentration:最終定量時点までの血漿中濃度-時間曲線下面積
AUCτ
Area under the plasma concentration-time curve over a dosing interval:投与間隔にお
ける血漿中濃度-時間曲線下面積
AUCτ
day1Area under the plasma concentration-time curve over a dosing interval after first dose:
初回投与後の投与間隔における血漿中濃度-時間曲線下面積
AUCτ
ssArea under the plasma concentration-time curve over a dosing interval at steady state:
定常状態の投与間隔における濃度-時間曲線下面積
AUMC
Area under the first moment curve from 0 to infinity:無制限時間までの 1 次モーメ
ント曲線下面積
2.7.6 個々の試験のまとめ
略語及び名称一覧表(続き)
略語(略称)
内容
BA
Bioavailability:バイオアベイラビリティ
BMI
Body mass index:体格指数
BSA
Body surface area:体表面積
β-hCG
Beta-human chorionic gonadotropin:β-ヒト絨毛性ゴナドトロピン
C
15’Plasma concentration at the end of the 15-minute infusion:15 分間静脈内投与終了
時の血漿中濃度
C
5’Plasma concentration at the end of the 5-minute infusion: 5 分間静脈内投与終了時
の血漿中濃度
CL
Total body clearance:全身クリアランス
C
lastConcentration at last time point measured:最終測定時の血漿中濃度
CL/F
Apparent total body clearance:みかけの全身クリアランス
CL
ssTotal body clearance at steady state:定常状態の全身クリアランス
C
maxMaximum plasma concentration:最高血漿中濃度
C
max day1Maximum plasma concentration after first dose:初回投与後の最高血漿中濃度
C
max,ssMaximum plasma concentration at steady state:定常状態の最高血漿中濃度
C
minMinimum plasma concentration:最低血漿中濃度
C
min,ssMinimum plasma concentration at steady state:定常状態の最低血漿中濃度
CPMP
Committee for Proprietary Medicinal Products:欧州医薬品委員会
CRP
Clinical Research Physician
CV
Coefficient of variation:変動係数
FAS
Full Analysis Set:最大の解析対象集団
FDA
Food and Drug Administration:米国食品医薬品局
γ-GTP
γ-glutamyl transpeptidase:γ グルタミルトランスペプチダーゼ
HBs 抗原
Hepatitis B surface antigen:B 型肝炎ウイルス表面抗原
HCV
Hepatitis C virus:C 型肝炎ウイルス
HIV
Human immunodeficiency virus:ヒト免疫不全ウイルス
IEC
Independent Ethics Committee:独立倫理委員会
ILAE
International League Against Epilepsy:国際抗てんかん連盟
ITT
Intention-to-Treat:治療を意図した解析対象集団
IV
Intravenous:静脈内投与
L057
ucb L057:レベチラセタムの主代謝物(カルボキシル体)
LDH
Lactate dehydrogenase:乳酸脱水素酵素
LEV
Levetiracetam:レベチラタセム
LF
Linearity factor:線形係数
MCH
Mean corpuscular hemoglobin:平均赤血球色素量
MCHC
Mean corpuscular hemoglobin concentration:平均赤血球血色素濃度
MCV
Mean corpuscular volume:平均赤血球容積
MedDRA
Medical Dictionary for Regulatory Activities:ICH 国際医薬用語集
MRT
Mean resident time:平均滞留時間
NA
Not applicable:該当せず
PCS
Possibly Clinically Significant:FDA Division of Neuropharmacologic Drug Products
ガイドラインをもとに
UCB 社が作成した基準
PK
Pharmacokinetics:薬物動態
PK-ITT
Pharmacokinetic ITT:治療を意図した薬物動態解析対象集団
PK-PPS
Pharmacokinetic Per Protocol Set:治験実施計画書に適合した薬物動態解析対象
集団
PMDA
Pharmaceuticals and Medical Devices Agency:医薬品医療機器総合機構
略語及び名称一覧表(続き)
略語(略称)
内容
PP
Per Protocol:治験実施計画書に適合した解析対象集団
Q1
1st quartile:第 1 四分位点
Q3
3rd quartile:第 3 四分位点
QTc
Corrected interval between Q and T waves on ECG:心拍数で補正した QT 間隔
R
AUCAccumulation ratio of AUC:AUC の累積係数
R
maxAccumulation ratio of C
max:C
maxの累積係数
RMSE
Root mean square error:二乗平均平方根誤差
SS
Safety Set:安全性解析対象集団
t
1/2Terminal elimination half-life:末端相の消失半減期
t
infInfusion duration:持続投与時間
t
maxTime to maximum plasma concentration:最高血漿中濃度到達時間
t
max,ssTime to maximum plasma concentration at steady state:定常状態の最高血漿中濃度
到達時間
USA
United States of America:アメリカ合衆国
V
zVolume of distribution during elimination phase:末端相の分布容積
V
z/F
Apparent volume of distribution during elimination phase:末端相のみかけの分布容
積
2.7.6 個々の試験のまとめ
2.7.6
個々の試験のまとめ
臨床試験一覧表を表
2.7.6-1 に示した。
Page 4
2 .7 .6 個々の試験のまと め
表 2.7.6-1 臨床試験一覧表
試験の種類 (資料区分) 実施国 試験番号 試験の目的 試験デザイン 治験薬、剤形、投与方法、投与経路 被験者数a) /完了時例数 被験者の種類 投与期間 試験報告書 添付場所 報告書の種類 バイオアベイラ ビリティ試験 (評価) 英国 N01377 薬物動態、安全 性及び忍容性 非盲検、無作為化、 2 期クロスオーバー、 単回投与 LEV 注射剤:単回静脈内投与 LEV 錠剤:単回経口投与 1 回 1500 mg×2 回 27/26 日本人健康成人 単回×2 期 5.3.1.1.1 完全な報告書 臨床薬理試験 (参考) ベルギー N01077 Part A 薬物動態、安全 性及び忍容性 非盲検、無作為化、 2 期クロスオーバー、 単回投与 LEV 注射剤:単回静脈内投与 LEV 錠剤:単回経口投与 1 回 1500 mg×2 回 18/18 外国人健康成人 単回×2 期 5.3.1.1.2 完全な報告書 Part B 薬物動態、安全 性及び忍容性 プラセボ対照、二重盲検、 無作為化、並行群間比較、 反復投与 LEV 又は PBO 注射剤 1 日 2 回反復静脈内投与 1 回 1500 mg LEV 群:12/12 PBO 群:6/6 外国人健康成人 4.5 日間 臨床薬理試験 (参考) ベルギー N01165 安全性、忍容性 及び薬物動態 プラセボ対照、単盲検、 無作為化、単回漸増投与 LEV 又は PBO 注射剤 単回静脈内投与、 2000、3000 及び 4000 mg を 15 分間投与 1500、2000 及び 2500 mg を 5 分間投与 LEV 群:36/36 PBO 群:12/12 外国人健康成人 単回 5.3.3.1.1 完全な報告書 臨床薬理試験 (評価) 日本 EP0038 薬物動態、安全 性及び忍容性 非盲検、反復投与 LEV 注射剤 単回静脈内投与 1 回 1500 mg 32/32 (日本人:16/16 白人:16/16) 日本人及び白人 健康成人男性 単回 5.3.3.3.1 完全な報告書 LEV 注射剤 1 日 2 回反復静脈内投与 1 回 1500 mg 4.5 日間 第 II 相試験 (評価) 日本 N01378 安全性、薬物動 態及び有効性 非対照、非盲検 LEV 注射剤 観察期間の LEV 経口製剤(1000~3000 mg/日) と同用量を 1 日 2 回反復静脈内投与 16/16 部分発作を有す る日本人成人て んかん患者(16 歳以上) 4 日間 5.3.5.2.1 完全な報告書 第 II 相試験 (参考) ドイツ、フラン ス、英国 N01166 安全性及び忍容 性 非対照、非盲検 LEV 注射剤 観察期間の LEV 経口製剤(1000~3000 mg/日) と同用量を 1 日 2 回反復静脈内投与 25/25 部分発作を有す る外国人成人て んかん患者(16 ~65 歳) 4 日間 5.3.5.2.2 完全な報告書 第 II 相試験 (参考) 米国、メキシ コ、トルコ、ベ ルギー、フラン ス、ドイツ N01274 安全性、忍容性 及び薬物動態 非対照、非盲検 LEV 注射剤 1 日 2 回反復静脈内投与 治験開始前から LEV 経口製剤を投与: LEV 経口製剤と同用量 治験開始前に LEV 経口製剤を投与せず: 体重 50 kg 未満は 20 mg/kg/日、体重 50 kg 以上は 1000 mg/日b) 33/33 外国人小児てん かん患者(4~ 16 歳) 最長 4 日間 5.3.5.2.3 完全な報告書 LEV:レベチラセタム、PBO:プラセボ a) 治験薬投与例数 b) 中等度腎機能不全(クレアチニンクリアランス 30~49 mL/分)の被験者は、体重 50 kg 未満は 10 mg/kg/日、体重 50 kg 以上は 500 mg/日を投与 2.7.6 個々の試験のまとめ Page 52.7.6 個々の試験のまとめ
2.7.6.1
バイオアベイラビリティ(
BA)試験報告書
2.7.6.1.1
N01377 試験(単回静脈内持続投与時及び単回経口投与時の薬物動態の比較)
(
5.3.1.1.1:評価資料)
治験方法の概略(表 2.7.6.1.1-1)及び結果の要約を示した。
表 2.7.6.1.1-1 治験方法の概略
項目 内容 標題 日本人健康成人被験者を対象として、レベチラセタムを経口投与及び静脈内持続投与 した時の生物学的同等性、安全性及び忍容性を検討する、単施設、非盲検、無作為 化、単回投与、2 期クロスオーバー試験 開発の相 第 I 相 目的 主要目的 日本人健康被験者を対象に、レベチラセタム 1500 mg を 15 分間単回静脈内持続投与 した時と、レベチラセタム 1500 mg(500 mg 錠 3 錠)を単回経口投与した時の薬物動 態を検討し、生物学的同等性を評価する。 副次目的 日本人健康被験者を対象に、レベチラセタム 1500 mg を 15 分間単回静脈内持続投与 した時と、レベチラセタム 1500 mg(500 mg 錠 3 錠)を単回経口投与した時の安全性 及び忍容性を検討する。 治験デザイン 単施設、非盲検、無作為化、単回投与、2 期クロスオーバー 治験方法 26 例の日本人健康成人被験者を 1 群 13 例ずつの 2 群(静脈内投与先行群及び経口投 与先行群)に無作為に割り付け、2 期クロスオーバー法により、投与第 1 期又は第 2 期にレベチラセタム 1500 mg を 15 分間静脈内又は経口のいずれかの投与経路で空腹 時単回投与することとした。いずれの投与期でも、投与前日から投与後 36 時間まで (必要な場合はそれ以降も)入院とした。なお、各投与の間は 7 日間以上の休薬期間 を設けた。 なお、第 2 期完了前に被験者が脱落した場合は、被験者を追加することとした。 被験者数 計画例数:26 例(男女各 10 例以上)、無作為化例数:27 例、完了例数:26 例 薬物動態解析対象例数:25 例、安全性解析対象例数:27 例 【被験者数設定の根拠】 先行試験(N01077 試験:5.3.1.1.2、Part A)の結果に基づき、被験者内変動係数 (CV)は、AUC(0-t)で 6.4%及び Cmaxで 20.8%と推定された。被験者数設定のための 被験者内 CV の推定値として、より高値である Cmaxの 20.8%を用いた。経口投与に対 する静脈内投与の Cmaxの真の比が 0.95~1.05 の範囲にあると仮定した。本治験では、 一般的な生物学的同等性の基準範囲である 0.8~1.25 を適用した。これらの仮定か ら、有意水準 0.05 で生物学的同等性を評価する際に 90%の検出力(第 2 種の過誤とな る確率 0.1)を確保するには、26 例が必要であると算出された。26 例を無作為化の計 画例数とした。 対象 健康成人 主要な選択/除外基準 主要な選択基準: 1) 20 歳以上、55 歳以下の男性又は女性 2) 祖父母の 4 名すべてが日本で生まれた日本人で、外国での生活が 10 年未満の者 3) BMI が 18 kg/m2以上、28 kg/m2以下の者 4) 体重が 45 kg 以上の者 5) 既往歴及び身体的所見より、身体的及び精神的に健康状態が良好である者 6) バイタルサインが以下の範囲の者 ・収縮期血圧:90 mmHg 以上、150 mmHg 以下 ・拡張期血圧:40 mmHg 以上、90 mmHg 以下 ・脈拍数:40 拍/分以上、100 拍/分以下 ・呼吸数:8 回/分以上、20 回/分以下 7) 心電図が正常、若しくは異常と判断されたが治験責任医師が臨床的に問題ない と判断した者 8) 臨床検査値が基準範囲内の者。検査値が基準範囲外であっても、治験マニュア ルに記載の臨床的に問題のないと合意した範囲にある者Page 6
レベチラセタム 2.7.6 個々の試験のまとめ Page 6表 2.7.6.1.1-1 治験方法の概略(続き)
項目 内容 主要な選択/除外基準 (続き) 除外基準: 1) 本治験に以前に参加した者、又は 3 ヵ月以内に本治験で開発中の薬剤の別の治 験で投与を割り付けられたことがある者 2) 3 ヵ月以内に、他の治験薬又は医療機器の治験に参加した者、若しくは現在、他 の治験薬又は医療機器の治験に参加中の者 3) 被験者の医学的又は精神的状態が、本治験への参加に悪影響を及ぼす可能性が あると、治験責任医師が判断した者 4) 妊婦中又は授乳中の女性、若しくは妊娠の可能性のある女性では、医学的に認 められている受胎調節法を実施してない者 5) 薬剤の吸収、代謝又は排泄に影響を及ぼす可能性のある、若しくは治験薬の投 与に対する危険因子となる心血管系疾患、呼吸器疾患、肝疾患、腎疾患、消化 器系疾患、内分泌疾患又は神経系疾患を有する者、又は既往歴(6 ヵ月以内)の ある者 6) 薬物中毒者(薬物スクリーニング検査が陽性)又は薬物中毒の既往歴のある 者、若しくはアルコールを大量に摂取する者(週あたり 28 単位を超えるアルコ ールの摂取:1 単位は、ビール又はラガー1/2 パイント、ワイン 1 杯又はスピリ ッツ 1 杯)。治験への同意が無効となり得る又は治験実施計画書の遵守ができ ないような精神的又は他の情緒的な問題のある者[独立倫理委員会(IEC)のフ ィードバックに基づき、本基準は 2011 年 6 月 11 日に男性では週あたり 21 単 位、女性では週あたり 14 単位に変更となった] 7) スクリーニング時に症候性又は無症候性の起立性低血圧がある者。起立性低血 圧は、両腕を胸の高さに上げた状態で起立し 1 及び 3 分後に、収縮期血圧が 20 mmHg 以上、又は拡張期血圧が 10 mmHg 以上低下した場合と定義する(被験 者が起立した時点から時間を計測)。仰臥位で 5 分間安静にしたときの血圧を ベースラインとする 8) 治験薬投与前 6 ヵ月以内の喫煙量が、1 日あたり 5 本を超えるたばこ又はそれに 相当する者、また、治験期間中治験実施医療機関での入院中にニコチンの摂取 を控えることのできない者又は控える意思のない者 9) カフェイン含有飲料を大量に摂取する者(コーヒー又はお茶を 1 日あたり 5 杯 超) 10) 重度の頭痛を頻繁に発現する者 11) 治験薬初回投与前 14 日以内に、処方薬又は市販薬(女性でのエチニルエストラ ジオール 30 μg を超えない経口避妊薬、及び 1 日 2 g かつ 14 日間で 10 g を超えな いアセトアミノフェンを除く)を服用した者 12) 治験薬初回投与前 2 ヵ月以内に肝酵素誘導剤を使用した者 13) ウイルス性肝炎に罹患している者、又は HBs 抗原、HCV 抗体、HIV 抗体の陽性 者 14) 治験薬初回投与前 12 週間以内に、献血した者又はそれに相当する出血 (>400 mL)があった者 15) ピロリドン誘導体及び他の添加物(乳糖、コーンスターチ、セルロース等)に 対してアレルギー又は過敏症のある者 16) 治験責任医師が、臨床症状が本治験への参加に適切ではないと判断した者2.7.6 個々の試験のまとめ
表 2.7.6.1.1-1 治験方法の概略(続き)
項目 内容 治験薬、投与量及び 投与方法 1) 治験薬 レベチラセタム注射剤 5 mL バイアル(100 mg/mL) バッチ番号:BX1005881 レベチラセタム 500 mg 錠 バッチ番号:BX1005882 2) 投与量 レベチラセタム注射剤 1500 mg(15 mg/mL の希釈液を 100 mL) レベチラセタム錠 1500 mg(500 mg 錠 3 錠) 3) 投与方法 治験薬の投与は 10 時間以上の絶食の後とした。治験薬投与前後の 1 時間は水の 摂取を不可とした。治験薬投与後 4 時間は食事の摂取を不可とした。 治験薬は、各投与期の Day 1 の朝に治験責任医師又は治験責任医師が指名した者 の管理下で投与した。 レベチラセタム注射剤を 4 バイアルから 18 mL を採取し、生理食塩液バッグに 加え 15 mg/mL の持続投与用注射液とし、100 mL を投与した。静脈内投与時の体 位は臥位とし、その後 1 時間臥位を維持した。レベチラセタムは、較正した輸 液ポンプを用いて一定の速度で 15 分間静脈内投与した。 経口投与時の体位は座位とし、レベチラセタムは 200 mL の水と共に経口投与し た。被験者は、投与後 1 時間座位を維持した。 投与期間 単回投与:1 日(各投与の間は 1 週間の休薬期間を設けた) 評価項目 薬物動態 血漿中レベチラセタム濃度 測定時点 経口投与: 投与前、投与後 15、30、45 分、1、1.5、2、3、6、9、12、24 及び 36 時間 静脈内投与: 投与前、投与開始後 5、10、15、30、45 分、1、2、3、6、9、12、24 及び 36 時間 算出パラメータ 主要パラメータ:Cmax、AUC(0-t) 副次パラメータ:tmax、C15’、AUC、MRT、t1/2、λz、CL/F、CL、Vz/F、Vz 安全性 有害事象、臨床検査値(血液学的検査、血液生化学的検査、尿検査)、12 誘導心電 図、バイタルサイン(血圧、脈拍数、呼吸数)、身体的所見 統計解析方法 1) 解析対象集団Pharmacokinetic Per Protocol Set(PK-PPS)を薬物動態解析対象例、Safety Set (SS)を安全性解析対象例とした。 PK-PPS:SS のうち、薬物動態パラメータの算出に十分な血漿中レベチラセタム 濃度データが得られた被験者 SS:無作為化され、治験薬が 1 回以上投与されたすべての被験者
Page 8
レベチラセタム 2.7.6 個々の試験のまとめ Page 82.7.6 個々の試験のまとめ
表 2.7.6.1.1-2 被験者の内訳
静脈内投与先行群 経口投与先行群 全体 無作為化例数 13 (100) 14 (100) 27 (100) 完了例数 13 (100) 13 (92.9) 26 (96.3) 中止例数 0 1 (7.1) 1 (3.7) 中止理由数 その他a) 0 1 (7.1) 1 (3.7) 解析対象集団 Safety Set (SS) 13 (100) 14 (100) 27 (100)Pharmacokinetic Per Protocol Set (PK-PPS) 12 (92.3) 13 (92.9) 25 (92.6)
PK-PPS からの除外例数 1 (7.7) 1 (7.1) 2 (7.4) 除外理由 選択・除外基準不適合 (妊娠) 0 1 (7.1) 1 (3.7) 治験薬投与の不遵守 (静脈内投与量が過量又は不足) 1 (7.7) 0 1 (3.7) 例数 (%) a) 2 回目の投与(静脈内投与)前の妊娠検査が陽性であったため、静脈内投与前に治験を中止した。 N01377 試験総括報告書(5.3.1.1.1)Table 1.2、Table 2.1
(
2) 被験者背景
SS の 27 例の被験者背景を表 2.7.6.1.1-3 に示した。
すべての被験者は日本人であり、性別は男性 11 例及び女性 16 例であった。全体で年齢は 20~
50 歳(平均値 29.7 歳)、身長の平均値は 164.9 cm、体重の平均値は 59.94 kg、BMI の平均値は
22.01 kg/m
2であった。
表 2.7.6.1.1-3 被験者背景:SS
静脈内投与-経口投与 (N=13) 経口投与-静脈内投与 (N=14) 全体 (N=27) 年齢 (歳) 平均値 ± 標準偏差 27.5 ± 3.4 31.9 ± 8.3 29.7 ± 6.7 最小値 – 最大値 20 – 32 24 – 50 20 – 50 性別 男性 5 (38.5) 6 (42.9) 11 (40.7) n (%) 女性 8 (61.5) 8 (57.1) 16 (59.3) 民族 n (%) 日本人 13 (100) 14 (100) 27 (100) 身長 (cm) 平均値 ± 標準偏差 163.4 ± 9.6 166.4 ± 7.0 164.9 ± 8.3 最小値 – 最大値 147 – 183 154 – 177 147 – 183 体重 (kg) 平均値 ± 標準偏差 58.85 ± 7.71 60.95 ± 7.67 59.94 ± 7.62 最小値 – 最大値 45.9 – 71.6 52.7 – 79.7 45.9 – 79.7 BMI (kg/m2) 平均値 ± 標準偏差 22.02 ± 1.97 22.00 ± 2.15 22.01 ± 2.02 最小値 – 最大値 18.6 – 24.9 18.2 – 27.4 18.2 – 27.4 N:例数、n:層別例数 N01377 試験総括報告書(5.3.1.1.1)Table 3.1.1Page 10
レベチラセタム 2.7.6 個々の試験のまとめ Page 10(
3) 薬物動態
レベチラセタム 1500 mg を 15 分間単回静脈内投与及び単回経口投与した時の血漿中レベチラセ
タム濃度推移を図 1 に示した。また、レベチラセタムの薬物動態パラメータを表
2.7.6.1.1-4 に、主要薬物動態パラメータの比較を表 2.7.6.1.1-5 に示した。
静脈内投与開始後 1 時間までの血漿中レベチラセタム濃度は、経口投与に比べ高かったが、投与
開始後 2 時間以降の血漿中濃度は両投与経路間で類似した推移を示した。
C
maxの幾何平均値は静脈内投与時で 97.00 µg/mL と経口投与時の 58.94 µg/mL より大きく、t
maxの
中央値は静脈内投与時で 0.250 時間と経口投与時の 0.750 時間より短かった。静脈内投与時の t
maxの中央値は投与終了時(15 分)と一致していた。
静脈内投与時に対する経口投与時の AUC の比から、経口投与時の絶対的バイオアベイラビリテ
ィは 104%であった。
AUC(0-t)の静脈内投与 / 経口投与の幾何平均値の比の 90%信頼区間は 0.95~0.99 であり、生物学
的同等性の基準範囲である 0.8~1.25 の範囲内であったが、C
maxでは 1.47~1.83 と生物学的同等性
の基準範囲外であった。このため、本治験では日本人を対象として、レベチラセタム 1500 mg を
15 分間単回静脈内投与した時と単回経口投与した時に、生物学的に同等であると結論することは
できなかった。
幾何平均値 ± 95%信頼区間、例数=25 N01377 試験総括報告書(5.3.1.1.1)Figure 1.1、Figure 1.3図 2.7.6.1.1-1 レベチラセタム 1500 mg を 15 分間単回静脈内投与及び単回経口投与した時の
血漿中レベチラセタム濃度推移:PK-PPS
投与開始後の時間 (h) 血漿中レベチラセタム濃度 (μ g/ m L ) 静脈内投与 経口投与 静脈内投与経口投与 (投与開始後36時間までの推移) (投与開始後6時間までの推移) 血漿中レベチラセタム濃度 (μ g/ m L ) 投与開始後の時間 (h)2.7.6 個々の試験のまとめ
表 2.7.6.1.1-4 レベチラセタム 1500 mg を 15 分間単回静脈内投与及び単回経口投与した時の
レベチラセタムの薬物動態パラメータ:PK-PPS
薬物動態パラメータ 静脈内投与 (N=25) 経口投与 (N=25) Cmax (µg/mL) 97.00 (27.6) 58.94 (37.0) C15’ (µg/mL) 96.49 (27.7) NA AUC(0-t) (µgh/mL) 472.28 (15.4) 487.36 (15.9) AUC (µgh/mL) 486.22 (15.5) 503.51 (16.2) MRT (h) 9.349 (12.1) 10.273 (12.4) t1/2 (h) 7.106 (11.7) 7.230 (12.7) λZ (h-1) 0.098 (11.7) 0.096 (12.7) CL 又は CL/F a) (L/h) 3.055 (15.3) 2.979 (16.2) Vz又は Vz/F a) (L) 31.32 (18.0) 31.07 (18.8) tmax (h) 0.250 (0.17 – 0.27) 0.750 (0.50 – 3.00) 幾何平均値(変動係数%)、tmaxでは中央値 (最小値 – 最大値) NA:該当せず a) 静脈内投与では CL 及び Vz、経口投与では CL/F 及び Vz/F N01377 試験総括報告書(5.3.1.1.1)Table 8-1表 2.7.6.1.1-5 レベチラセタム 1500 mg を 15 分間単回静脈内投与及び単回経口投与した時の
レベチラセタムの主要薬物動態パラメータの解析結果:PK-PPS
薬物動態パラメータ 静脈内投与 / 経口投与 a) CV b) (%) 点推定値 90%信頼区間 Cmax 1.64 1.47, 1.83 22.8 AUC(0-t) 0.97 0.95, 0.99 3.6 a) 経口投与に対する静脈内投与の幾何平均値の比の点推定値及び 90%信頼区間(分散分析) b) 被験者内変動係数(分散分析) N01377 試験総括報告書(5.3.1.1.1)Table 8-2(
4) 安全性
1) 曝露状況
SS の 27 例中、26 例にレベチラセタム 1500 mg が静脈内及び経口にてそれぞれ単回投与され、1
例にレベチラセタム 1500 mg が単回経口投与された。経口投与のみの 1 例(投与順:経口-静脈内
投与)は、妊娠検査陽性のため静脈内投与の前日に治験を中止した。
2) 有害事象
有害事象は症例報告書の記載内容を MedDRA version 14.1 を用いて器官別大分類及び基本語に読
み替えた上で集計した。
因果関係の判定は「関連なし(Not related)」及び「関連あり(Related)」の 2 分類とした。こ
のうち治験責任医師が、治験薬との因果関係を「関連あり」と判断した有害事象を「因果関係が否
定できない有害事象」として取り扱った。
i) 有害事象発現例数の概要
治験薬投与開始後に認められた有害事象発現例数の概要を表 2.7.6.1.1-6 に示した。
Page 12
レベチラセタム 2.7.6 個々の試験のまとめ Page 12本治験では、有害事象は全体で 27 例中 23 例(85.2%)に認められた。投与経路別の有害事象の
発現率は同程度であり、静脈内投与で 26 例中 14 例(53.8%)及び経口投与で 27 例中 18 例(66.7%)
であった。因果関係が否定できない有害事象は全体で 22 例(81.5%)に認められ、静脈内投与で
12 例(46.2%)及び経口投与で 14 例(51.9%)であった。重症度が高度と判断された有害事象はな
かった。
死亡、重篤な有害事象及び治験薬の投与中止に至った有害事象は認められなかったが、本治験
のデータベースの固定後に、1 例の被験者で重篤な有害事象として治療的流産が報告された。本被
験者は、2 回目の投与(静脈内投与)前の妊娠検査が陽性であったため、静脈内投与前に治験を中
止し、治験期間終了後も本被験者のフォローアップを行い、データベース固定後に治療的流産の報
告を受けた。
表 2.7.6.1.1-6 有害事象発現例数の概要:SS
静脈内投与 (N=26) 経口投与 (N=27) 全体 (N=27) 有害事象発現例数 14 (53.8) 18 (66.7) 23 (85.2) 因果関係が否定できない有害事象 12 (46.2) 14 (51.9) 22 (81.5) 死亡 0 0 0 重篤な有害事象 0 0 0 因果関係が否定できない重篤な有害事象 0 0 0 治験薬の投与中止に至った有害事象 0 0 0 発現例数 (%)N01377 試験総括報告書(5.3.1.1.1)Table 9-1、Table 9-2、Table 6.2、Listing 7.2
ii) すべての有害事象
治験薬投与開始後に認められたすべての有害事象を表 2.7.6.1.1-7 に示した。
ほとんどの有害事象が、「神経系障害」に分類される有害事象(傾眠、浮動性めまい及び頭痛)
であった。最もよくみられた有害事象は傾眠であり、全体で 16 例(59.3%)に認められた。
注射部位に関連する有害事象が 2 例(各投与経路で 1 例ずつ)に認められたが、いずれも薬物動
態測定用検体採取時に発現した事象であり、治験薬との因果関係は「関連なし」と判断された。
2.7.6 個々の試験のまとめ
表 2.7.6.1.1-7 すべての有害事象:SS
器官別大分類 基本語 静脈内投与 (N=26) 経口投与 (N=27) 全体 (N=27) 有害事象発現例数 14 (53.8) 18 (66.7) 23 (85.2) 神経系障害 14 (53.8) 15 (55.6) 23 (85.2) 傾眠 10 (38.5) 9 (33.3) 16 (59.3) 浮動性めまい 5 (19.2) 7 (25.9) 10 (37.0) 頭痛 3 (11.5) 3 (11.1) 5 (18.5) 胃腸障害 1 (3.8) 2 (7.4) 3 (11.1) 悪心 1 (3.8) 1 (3.7) 2 (7.4) 腹痛 0 1 (3.7) 1 (3.7) 上腹部痛 0 1 (3.7) 1 (3.7) 一般・全身障害および投与部位の状態 1 (3.8) 2 (7.4) 3 (11.1) カテーテル留置部位関連反応 1 (3.8) 0 1 (3.7) カテーテル留置部位紅斑 0 1 (3.7) 1 (3.7) カテーテル留置部位そう痒感 0 1 (3.7) 1 (3.7) 疲労 0 1 (3.7) 1 (3.7) 感染症および寄生虫症 0 1 (3.7) 1 (3.7) 鼻咽頭炎 0 1 (3.7) 1 (3.7) 生殖系および乳房障害 0 1 (3.7) 1 (3.7) 月経困難症 0 1 (3.7) 1 (3.7) 呼吸器、胸郭および縦隔障害 0 1 (3.7) 1 (3.7) 口腔咽頭痛 0 1 (3.7) 1 (3.7) 発現例数 (%) 事象名:MedDRA ver. 14.1 N01377 試験総括報告書(5.3.1.1.1)Table 9-1iii) 因果関係が否定できない有害事象
治験薬投与開始後に認められた因果関係が否定できない有害事象を表 2.7.6.1.1-8 に示した。
ほとんどの有害事象が治験薬との因果関係を「関連あり」と判断された。「神経系障害」に分
類される有害事象(傾眠、浮動性めまい及び頭痛)が最もよくみられた。
表 2.7.6.1.1-8 因果関係が否定できない有害事象:SS
器官別大分類 基本語 静脈内投与 (N=26) 経口投与 (N=27) 全体 (N=27) 因果関係が否定できない有害事象発現例数 12 (46.2) 14 (51.9) 22 (81.5) 神経系障害 12 (46.2) 13 (48.1) 22 (81.5) 傾眠 10 (38.5) 9 (33.3) 16 (59.3) 浮動性めまい 5 (19.2) 6 (22.2) 9 (33.3) 頭痛 1 (3.8) 1 (3.7) 2 (7.4) 胃腸障害 1 (3.8) 1 (3.7) 2 (7.4) 悪心 1 (3.8) 1 (3.7) 2 (7.4) 腹痛 0 1 (3.7) 1 (3.7) 一般・全身障害および投与部位の状態 0 1 (3.7) 1 (3.7) 疲労 0 1 (3.7) 1 (3.7) 発現例数 (%) 事象名:MedDRA ver. 14.1 N01377 試験総括報告書(5.3.1.1.1)Table 9-2Page 14
レベチラセタム 2.7.6 個々の試験のまとめ Page 14iv) 重症度別有害事象
治験薬投与開始後に認められた有害事象を、重症度別に表 2.7.6.1.1-9 に示した。
すべての有害事象の重症度は軽度又は中等度と判断され、中等度と判断された有害事象は全体
で 4 例(14.8%)に認められた。中等度の有害事象は、静脈内投与では浮動性めまい 1 例(3.8%)、
経口投与では頭痛 3 例(11.1%)及び月経困難症 1 例(3.7%)であった。
表 2.7.6.1.1-9 重症度別有害事象:SS
器官別大分類 基本語 静脈内投与 (N=26) 経口投与 (N=27) 全体 (N=27) 軽度 中等度 軽度 中等度 軽度 中等度 有害事象発現例数 14 (53.8) 1 (3.8) 17 (63.0) 4 (14.8) 23 (85.2) 4 (14.8) 神経系障害 14 (53.8) 1 (3.8) 14 (51.9) 3 (11.1) 23 (85.2) 4 (14.8) 傾眠 10 (38.5) 0 9 (33.3) 0 16 (59.3) 0 浮動性めまい 5 (19.2) 1 (3.8) 7 (25.9) 0 10 (37.0) 1 (3.7) 頭痛 3 (11.5) 0 1 (3.7) 3 (11.1) 4 (14.8) 3 (11.1) 胃腸障害 1 (3.8) 0 2 (7.4) 0 3 (11.1) 0 悪心 1 (3.8) 0 1 (3.7) 0 2 (7.4) 0 腹痛 0 0 1 (3.7) 0 1 (3.7) 0 上腹部痛 0 0 1 (3.7) 0 1 (3.7) 0 一般・全身障害および投与部位の状態 1 (3.8) 0 2 (7.4) 0 3 (11.1) 0 カテーテル留置部位関連反応 1 (3.8) 0 0 0 1 (3.7) 0 カテーテル留置部位紅斑 0 0 1 (3.7) 0 1 (3.7) 0 カテーテル留置部位そう痒感 0 0 1 (3.7) 0 1 (3.7) 0 疲労 0 0 1 (3.7) 0 1 (3.7) 0 感染症および寄生虫症 0 0 1 (3.7) 0 1 (3.7) 0 鼻咽頭炎 0 0 1 (3.7) 0 1 (3.7) 0 生殖系および乳房障害 0 0 0 1 (3.7) 0 1 (3.7) 月経困難症 0 0 0 1 (3.7) 0 1 (3.7) 呼吸器、胸郭および縦隔障害 0 0 1 (3.7) 0 1 (3.7) 0 口腔咽頭痛 0 0 1 (3.7) 0 1 (3.7) 0 発現例数 (%) 事象名:MedDRA ver. 14.1 N01377 試験総括報告書(5.3.1.1.1)Table 6.4v) 死亡及びその他の重篤な有害事象
本治験期間中に、死亡及びその他の重篤な有害事象は認められなかったが、データベースの固
定後に、1 例の被験者で重篤な有害事象として治療的流産が報告された。
vi) 治験薬の投与中止に至った有害事象
本治験では、治験薬の投与中止に至った有害事象は認められなかった。
3) 臨床検査値及びその他の安全性評価項目
臨床検査値、バイタルサイン、12 誘導心電図及び身体的所見に、臨床的に重要な所見は認めら
れなかった。
2.7.6 個々の試験のまとめ
(
5) 結論
日本人健康成人を対象として、レベチラセタム 1500 mg を 15 分間単回静脈内投与及び単回経口
投与した時の生物学的同等性及び安全性を検討した。
レベチラセタム 1500 mg を 15 分間静脈内投与した時の血漿中レベチラセタム濃度は、経口投与
に比べ、投与開始後 1 時間までは高く推移したが、投与開始後 2 時間以降は類似した推移を示した。
AUC(0-t)の静脈内投与 / 経口投与の幾何平均値の比の 90%信頼区間は生物学的同等性の基準範囲で
ある 0.8~1.25 の範囲内であったが、C
maxでは生物学的同等性の基準範囲外であった。このため、
本治験では日本人を対象として、レベチラセタム 1500 mg を 15 分間単回静脈内投与した時と単回
経口投与した時に、生物学的に同等であると結論することはできなかった。
日本人健康成人男女を対象として、レベチラセタム 1500 mg を 15 分間静脈内投与した時の忍容
性は C
maxが経口投与した時に比べ高かったものの、経口投与した時と同様に良好であった。静脈
内投与時の安全性プロファイルは経口投与時と類似し、本治験で報告された有害事象はレベチラセ
タム投与で既知のものであった。
Page 16
レベチラセタム 2.7.6 個々の試験のまとめ Page 162.7.6.1.2
N01077 試験(単回静脈内持続投与時と単回経口投与時の薬物動態の比較及び反
復静脈内投与時の薬物動態の検討)(
5.3.1.1.2:参考資料)
治験方法の概略(表 2.7.6.1.2-1)及び結果の要約を示した。
表 2.7.6.1.2-1 治験方法の概略
項目 内容 標題 健康成人を対象とした、 Part A:無作為化、非盲検、2 期クロスオーバー法による、レベチラセタム 1500 mg の 15 分間単回静脈内持続投与時と、レベチラセタム 500 mg 錠 3 錠の単回経口投与時のバ イオアベイラビリティの比較 Part B:無作為化、二重盲検、プラセボ対照による、レベチラセタム 1500 mg を 15 分間 静脈内持続投与で、1 日 2 回 4.5 日間反復投与した時の安全性、忍容性及び薬物動態の検 討 開発の相 第 I 相 目的 主要目的 Part A:レベチラセタム 1500 mg を 15 分間単回静脈内持続投与した時と、レベチラセタ ム 1500 mg(500 mg 錠 3 錠)を単回経口投与した時のバイオアベイラビリティを比較す る。 Part B:レベチラセタム 1500 mg を 15 分間静脈内持続投与で 1 日 2 回 4.5 日間反復投与 した時の薬物動態を検討する。 副次目的 レベチラセタム 1500 mg を 15 分間静脈内持続投与で 1 日 2 回 4.5 日間反復投与した時の 安全性及び忍容性を検討する。 治験デザイン Part A:単施設、非盲検、無作為化、単回投与、2 期クロスオーバー Part B:単施設、二重盲検、無作為化、プラセボ対照、反復投与、並行群間比較 治験方法 健康成人男性及び女性各 9 例の合計 18 例を対象に、 Part A:クロスオーバー法にて、レベチラセタム 1500 mg を 15 分間単回静脈内投与 (500 mg/5 mL アンプル)及び単回経口投与(500 mg 錠 3 錠)した。各投与の間は 1 週 間の休薬期間を設けた。 Part B:Part A に参加した被験者 18 例を無作為にレベチラセタム群(12 例)又はプラセ ボ群(6 例)に割り付け、Part A の第 2 期の投与 3 日目から、レベチラセタム 1500 mg 又はプラセボを 15 分間静脈内投与で 1 日 2 回 4.5 日間(Part B の投与 5 日目は朝のみ投 与)反復投与した。 被験者数 計画例数:18 例、完了例数:18 例 薬物動態解析対象例数:Part A 17 例、Part B 12 例 安全性解析対象例数:Part A 18 例、Part B 18 例 【被験者数設定の根拠】 2 剤 2 期クロスオーバー法による Part A は、レベチラセタム 1500 mg(500 mg/5 mL アン プル)の 15 分間静脈内投与とレベチラセタム 500 mg 錠 3 錠の経口投与の生物学的同等 性を評価するためにデザインした。 被験者内変動係数(CV)は、過去の試験(N01002 試験及び N01072 試験)から AUC で は 7.1%、Cmaxでは 9.7%と推定された。例数設定は、被験者内 CV を 9.7%として算定し た。 真の幾何平均値の比が 90~111%の範囲であるとした時、第 I 種の過誤による危険率 5% で 90%の検出力を得るには、2×2 クロスオーバー法にて 14 例の例数が必要と算出された。 少数例の中止の可能性を考慮し、本治験では 18 例の被験者を組み入れ、無作為化した。 この被験者数と上述の仮説のもとでの検出力は 96%となった。 対象 健康成人2.7.6 個々の試験のまとめ
表 2.7.6.1.2-1 治験方法の概略(続き)
項目 内容 主要な選択/除外基準 主要な選択基準: 1) 18 歳以上、55 歳以下の男性又は女性(各 9 例)。人種は問わない 2) BMI が 19 kg/m2以上、28 kg/m2以下の者 3) 既往歴及び一般身体所見により、身体的及び精神的に健康状態が良好である者 4) 臥位血圧、脈拍数及び呼吸数が以下の範囲の者 ・収縮期血圧:100 mmHg 以上、150 mmHg 以下 ・拡張期血圧:50 mmHg 以上、90 mmHg 以下 ・脈拍数:45 拍/分以上、90 拍/分以下 ・呼吸数:10 回/分以上、18 回/分以下 5) 心電図が正常、又は異常と判断されたが治験責任医師が臨床的に問題ないと判断し た者 6) ドップラー心エコー検査において正常、又は異常と判断されたが治験責任医師が臨 床的に問題ないと判断した者 7) 臨床検査値が基準範囲内の者。検査値が基準範囲外であるが治験責任医師が臨床的 に問題ないと判断した場合は、治験依頼者の文書による承認を得たうえで、組み入 れることを可能とする 除外基準: 1) 妊娠中又は授乳中の女性、若しくは妊娠する可能性のある女性では医学的に認めら れている避妊法を実施していない女性。治験薬初回投与前の 48 時間以内の検査で β-hCG 検査が陰性であり、医学的に認められている避妊手段[ホルモン避妊薬、避 妊リング、埋め込み型避妊薬、殺精子剤付ペッサリー、両側卵管結紮、精管切除し ているパートナーとの関係(3 ヵ月間以上)又は殺精子ジェル入りコンドームの使 用、禁欲等]を治験期間中に講じる場合は除く 2) 薬剤の吸収、代謝又は排泄に影響を及ぼす可能性のある、若しくは治験薬の投与に 対する危険因子となる心血管系疾患、呼吸器疾患、肝疾患、腎疾患、消化器系疾患、 内分泌疾患又は神経系疾患を有する者又は既往歴のある者 3) 薬物中毒者(薬物スクリーニング検査が陽性)又は薬物中毒の既往歴のある者、若 しくはアルコールを大量に摂取する者(週あたり 28 単位を超えるアルコールの摂 取:1 単位は、ビール又はラガー1/2 パイント、ワイン 1 杯又はスピリッツ 1 杯)。 または、治験への同意が無効となり得る又は治験実施計画書の遵守ができないよう な精神的又は他の情緒的な問題のある者 4) スクリーニング時に症候性又は無症候性の起立性低血圧がある者。起立性低血圧は、 両腕を力を抜いて下した状態で起立し 1 及び 3 分後に、収縮期血圧が 20 mmHg 以上、 又は拡張期血圧が 10 mmHg 以上低下した場合と定義する(被験者が起立した時点か ら時間を計測)。仰臥位で 5 分間安静にしたときの血圧をベースラインとする 5) 喫煙者及びたばこを止めてから本治験薬初回投与まで 6 ヵ月を経過していない者 6) カフェイン含有飲料を大量に摂取する者(コーヒーやお茶等を 1 日あたり 5 杯超) 7) 重度の頭痛を頻繁に発現する者 8) 治験薬初回投与前 14 日以内に、処方薬又は市販薬(女性でのエチニルエストラジオ ール 30 μg を超えないホルモン避妊薬、及び 1 日 2 g かつ 14 日間で 10 g を超えない アセトアミノフェンを除く)を服用した者 9) 治験薬初回投与前 2 ヵ月以内に、肝酵素を誘導することが知られている薬剤(例え ば、糖質コルチコイド、フェノバルビタール、イソニアジド)を服用した者 10) 血清肝炎に罹患している者、又は HBs 抗原、HCV 抗体、HIV 抗体の陽性者 11) 治験薬初回投与前 12 週間以内に、他の治験に参加した者、献血した者又は異常出血 があった者 12) ピロリドン誘導体及び他の添加物(乳糖、コーンスターチ、セルロース等)に対し てアレルギー又は過敏症のある者 13) 治験責任医師が、被験者の健康状態が本治験への参加に適切でないと判断した者Page 18
レベチラセタム 2.7.6 個々の試験のまとめ Page 18表 2.7.6.1.2-1 治験方法の概略(続き)
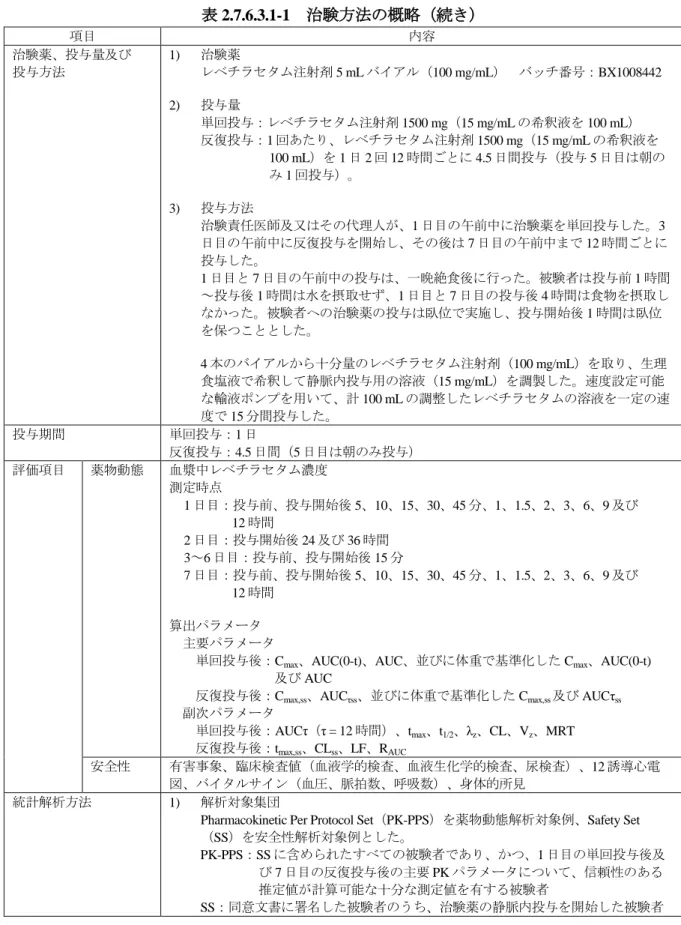
項目 内容 治験薬、投与量及び投 与方法 1) 治験薬 レベチラセタム 500 mg 錠 バッチ番号:11716 レベチラセタム注射剤 5 mL アンプル(100 mg/mL) バッチ番号:11989 プラセボ注射剤 5 mL アンプル(生理食塩液) バッチ番号:11987 2) 投与量 Part A:単回投与 レベチラセタム錠 1500 mg(500 mg 錠 3 錠) レベチラセタム注射剤 1500 mg(100 mg/mL の 5 mL アンプルを 3 アンプル) Part B:反復投与 1 回あたり、レベチラセタム注射剤 1500 mg(100 mg/mL の 5 mL アンプルを 3 アン プル)又はプラセボ注射剤 3 アンプルを 1 日 2 回 12 時間ごとに 4.5 日間投与した(Part B の投与 5 日目は朝のみ 1 回投与した)。 3) 投与方法 治験薬は、治験責任医師又は治験責任医師が指名した者の管理下で投与した。治験 薬投与前後の 1 時間は、経口投与時に用いる水(240 mL)を除き水の摂取を不可と した。経口投与時の体位は座位とした。静脈内投与時は臥位とした(ドップラー心 エコー検査の実施時には検査終了まで臥位とした)。その後は座位とし、1 時間は座 位を維持した。 Part A: 各投与期の 1 日目の朝の空腹時に以下のいずれかの治験薬を投与した。各投与の間 は 1 週間の休薬期間を設けた。 レベチラセタム 500 mg 錠 3 錠を水 240 mL と共に単回経口投与した。 レベチラセタム注射液 15 mL(1500 mg)を生理食塩液 100 mL で希釈し、15 分間静 脈内投与した。 Part B: Part A の第 2 期の投与 3 日目から 4.5 日間、朝及びその 12 時間後の 1 日 2 回(Part B の投与 5 日目は朝のみ)、レベチラセタム又はプラセボのいずれかを静脈内投与し た。治験薬は生理食塩液 100 mL で希釈し 15 分間静脈内投与した。 投与期間 Part A:単回投与;1 日(各投与の間は 1 週間の休薬期間を設けた) Part B:4.5 日間(5 日目は朝のみ投与) 評価項目 薬物動態 レベチラセタム及び L057 の血漿中濃度 測定時点 Part A: 経口投与: 投与前、投与後 15、30、45 分、1、1.5、2、3、6、9、12、24 及び 36 時間 静脈内投与: 投与前、投与開始後 5、10、15、30 分、1、2、3、6、9、12、24 及び 36 時間 Part B: 1~4 日目:投与前及び投与開始後 15 分 5 日目:投与前、投与開始後 5、10、15、30 分、1、2、3、6、9 及び 12 時間 算出パラメータ Part A: レベチラセタム:AUC、AUC(0-t)、AUCτ、λz、C15’(15 分間静脈内投与終了時の 血漿中濃度)、Cmax、tmax、t1/2、CL/F 又は CL、Vz/F 又は Vz L057:AUC、AUC(0-t)、λz、Cmax、tmax、t1/2Part B:
レベチラセタム:AUCτss、Cmax,ss、tmax,ss、Cmin,ss、Rmax(Cmaxの累積係数)、RAUC (AUC の累積係数)、LF(線形係数)
L057:Cmax,ss、tmax,ss、Cmin,ss
安全性 有害事象、身体的所見、バイタルサイン(血圧、脈拍数、呼吸数)、臨床検査(血液学
的検査、血液生化学的検査、尿検査)、12 誘導心電図、ドップラー心エコー検査(Part B のみ)
表 2.7.6.1.2-2 被験者の内訳
Part 無作為化例 治験薬投与例 完了例 PP 集団:薬物動態解析対象例 ITT 集団:安全性解析対象例 A 18 18 18 17 18 B 18 18 18 12 18 例数 N01077 試験総括報告書(5.3.1.1.2)10.1 項、11.1 項(
2) 被験者背景
ITT 集団 18 例の被験者背景を表 2.7.6.1.2-3 に示した。
すべての被験者は白人であった。全体で年齢は 19.3~52.9 歳(平均値 35.00 歳)、身長の平均値
は 174.3 cm、体重の平均値は 73.3 kg、BMI の平均値は 23.88 kg/m
2、BSA の平均値は 1.875 m
2であ
った。
表 2.7.6.1.2-3 被験者背景:ITT
項目 全体 (N=18) 年齢a) (歳) 平均値 ± 標準偏差 35.00 ± 9.28 最小値 – 最大値 19.3 – 52.9 性別 男性 9 (50.0) n (%) 女性 9 (50.0) 人種 n (%) 白人 18 (100) 身長 (cm) 平均値 ± 標準偏差 174.3 ± 10.2 最小値 – 最大値 157 – 189 体重 (kg) 平均値 ± 標準偏差 73.3 ± 14.2 最小値 – 最大値 50 – 94 BMI (kg/m2) 平均値 ± 標準偏差 23.88 ± 2.47 最小値 – 最大値 19.7 – 27.3 BSA (m2) 平均値 ± 標準偏差 1.875 ± 0.230 最小値 – 最大値 1.48 – 2.21 N:例数、n:層別例数 a) 治験薬初回投与時の年齢 N01077 試験総括報告書(5.3.1.1.2)Table 14.1.2:1(
3) 薬物動態
1) Part A(単回投与)
レベチラセタム 1500 mg を単回経口投与した時及び単回静脈内投与した時の血漿中レベチラセタ
ム濃度推移を図 2.7.6.1.2-1 に、レベチラセタムの薬物動態パラメータ及び投与経路間の比較をそれ
ぞれ表 2.7.6.1.2-4 及び表 2.7.6.1.2-5 に示した。
レベチラセタムの AUC(0-t)、AUC 及び C
maxの幾何平均値は、経口投与時と静脈内投与時でほぼ
同程度であった。静脈内投与時に対する経口投与時の AUC の比から、経口投与の絶対的バイオア
ベイラビリティは 109%であった。経口投与時に対する静脈内投与時の幾何平均値の比の 90%信頼
区間は、AUC(0-t)では 88.3%~95.3%、AUC では 89.0%~95.6%、C
maxでは 91.6%~117.4%と、いず
2.7.6 個々の試験のまとめ
t
maxの中央値は経口投与時で 0.75 時間、静脈内投与時で 0.25 時間であった。t
1/2の幾何平均値は、
経口投与時で 7.13 時間、静脈内投与時で 7.07 時間と、投与経路による差はなく、また、全身クリア
ランスと分布容積の幾何平均値も両投与経路でほぼ同程度の値であった。
平均値 ± 標準偏差、例数=17 N01077 試験総括報告書(5.3.1.1.2)Figure 11:2図 2.7.6.1.2-1 レベチラセタム 1500 mg を単回経口投与及び 15 分間単回静脈内投与した時の
血漿中レベチラセタム濃度推移:PP
表 2.7.6.1.2-4 レベチラセタム 1500 mg を単回経口投与及び 15 分間単回静脈内投与した時の
レベチラセタムの薬物動態パラメータ:PP
薬物動態パラメータ 経口投与 (N=17) 静脈内投与 (N=17) Cmax (µg/mL) 45.87 (29.8) 47.50 (36.9) C15’ (µg/mL) NA 44.14 (43.1) AUC(0-t) (µgh/mL) 405.92 (21.7) 371.75 (20.0) AUC (µgh/mL) 419.21 (21.2) 386.27 (18.7) AUCτ (µgh/mL) NA 260.90 (22.6) tmax (h) 0.75 (0.5 – 2.0) 0.25 (0.2 – 2.0) t1/2 (h) 7.13 (16.7) 7.07 (16.0) CL/F 又は CL a) (L/h) 3.58 (21.2) 3.88 (18.7) Vz/F 又は Vz a) (L) 36.78 (30.1) 39.63 (28.8) 幾何平均値(変動係数%)、tmaxでは中央値 (最小値 – 最大値) NA:該当せず a) 静脈内投与では CL 及び Vz、経口投与では CL/F 及び Vz/FN01077 試験総括報告書(5.3.1.1.2)Table 14.2.1:2、Table 14.2.1:3、Table 14.2.1:4 一部改変(追加解析:5.4.21)
血漿中レベチラセタム濃度 (μg /m L ) 投与開始後の時間 (h) 経口投与 静脈内投与 投与開始後の時間 (h) 投与開始後4時間までの推移
Page 22
レベチラセタム 2.7.6 個々の試験のまとめ Page 22表 2.7.6.1.2-5 レベチラセタム 1500 mg を単回経口投与及び 15 分間単回静脈内投与した時の
レベチラセタムの薬物動態パラメータの解析結果:PP
薬物動態パラメータ 静脈内投与 / 経口投与 a) CV b) (%) 点推定値 90%信頼区間 Cmax 103.7 91.6, 117.4 20.8 AUC(0-t) 91.7 88.3, 95.3 6.4 AUC 92.2 89.0, 95.6 5.9 a) 経口投与時に対する静脈内投与時の幾何平均値の比の点推定値及び 90%信頼区間(分散分析) b) 被験者内変動係数(分散分析) N01077 試験総括報告書(5.3.1.1.2)Table 11:5レベチラセタム 1500 mg を単回経口投与した時及び単回静脈内投与した時の血漿中 L057 濃度推
移を図 2.7.6.1.2-2 に、L057 の薬物動態パラメータを表 2.7.6.1.2-6 に示した。
主要代謝物である L057 の AUC(0-t)、AUC 及び C
maxの幾何平均値は、レベチラセタムと同様に経
口投与時と静脈内投与時でほぼ同程度であり、それぞれ、18.36 及び 16.66 µg eq. LEV·h/mL(レベチ
ラセタム当量に換算した濃度として表示、以下同様)、18.67 及び 17.55 µg eq. LEV·h/mL、並びに
1.01 及び 0.93 µg eq. LEV/mL であった。
t
maxの中央値も経口投与時と静脈内投与時でそれぞれ 6.05 及び 6.00 時間と違いは認められず、t
1/2の幾何平均値も 8.43 及び 8.31 時間と同程度であった。
平均値 ± 標準偏差、例数=17 ng eq. LEV/mL:レベチラセタム当量に換算した血漿中 L057 濃度 N01077 試験総括報告書(5.3.1.1.2)Figure 11:4図 2.7.6.1.2-2 レベチラセタム 1500 mg を単回経口投与及び 15 分間単回静脈内投与した時の
血漿中 L057 濃度推移:PP
血漿中 L 0 5 7 濃度 (n g e q . L E V /m L ) 投与開始後の時間 (h) 経口投与 静脈内投与2.7.6 個々の試験のまとめ
表 2.7.6.1.2-6 レベチラセタム 1500 mg を単回経口投与及び 15 分間単回静脈内投与した時の
L057 の薬物動態パラメータ:PP
薬物動態パラメータ 経口投与 (N=17) 静脈内投与 (N=17)Cmax (µg eq. LEV/mL) 1.01 (17.8) 0.93 (18.7)
AUC(0-t) (µg eq. LEVh/mL) 18.36 (23.5) 16.66 (26.1)
AUC a) (µg eq. LEVh/mL) 18.67 (24.0) 17.55 (30.6 )
tmax (h) 6.05 (6.0 – 12.0) 6.00 (3.0 – 9.0)
t1/2 a) (h) 8.43 (13.4) 8.31 (13.0)
幾何平均値(変動係数%)、tmaxでは中央値 (最小値–最大値)
µg eq. LEVh/mL:レベチラセタム当量に換算した血漿中 L057 の AUC
µg eq. LEV/mL:レベチラセタム当量に換算した血漿中 L057 濃度 a) N=13 N01077 試験総括報告書(5.3.1.1.2)Table 14.2.1:8、Table 14.2.1:9 一部改変(追加解析:5.4.21)
2) Part B(反復投与)
レベチラセタム 1500 mg を反復静脈内投与した時の血漿中レベチラセタム濃度推移を図
2.7.6.1.2-3 に、レベチラセタムの薬物動態パラメータを表 2.7.6.1.2-7 に示した。
レベチラセタムの血漿中濃度は、Part B の投与 2 日目に定常状態に達した。単回投与時の
AUC(0-12)に対する反復投与時の AUCτ
ssの比(AUC の累積係数:R
AUC)は 1.41 であり、反復投与
時の C
max,ssも AUCτ
ssと同様に、単回投与時に比べて増加した。t
maxの中央値は反復又は単回投与時
のいずれも 0.25 時間であった。単回投与時の AUC に対する反復投与時の AUCτ
ssの比(線形係数:
LF)は 0.95 であり、反復静脈内投与時のレベチラセタムは線形の薬物動態プロファイルを示すと考
えられた。
平均値 + 標準偏差、例数=12 N01077 試験総括報告書(5.3.1.1.2)Figure 11:6図 2.7.6.1.2-3 レベチラセタム 1500 mg を 1 日 2 回 12 時間間隔で 4.5 日間反復静脈内投与した
時の血漿中レベチラセタム濃度推移:PP
血漿中レベチラセタム濃度 (μg /m L ) Part Bの初回投与開始後の時間 (h)Page 24
レベチラセタム 2.7.6 個々の試験のまとめ Page 24表 2.7.6.1.2-7 レベチラセタム 1500 mg を反復静脈内投与した時のレベチラセタムの薬物動態
パラメータ:PP
薬物動態パラメータ 反復静脈内投与 (N=12) Cmax,ss (µg/mL) 68.97 (29.9) AUCτss (µgh/mL) 364.09 (21.6) tmax,ss (h) 0.25 (0.2 – 0.3) Cmin,ss (µg/mL) 13.61 (26.9) Rmax 1.43 (36.7) RAUC 1.41 (15.9) LF 0.95 (11.7) 幾何平均値(変動係数%)、tmaxでは中央値 (最小値 – 最大値) N01077 試験総括報告書(5.3.1.1.2)Table 14.2.1:11 一部改変(追加解析:5.4.21)レベチラセタム 1500 mg を反復静脈内投与した時の血漿中 L057 濃度推移を図 2.7.6.1.2-4 に、L057
の薬物動態パラメータを表 2.7.6.1.2-8 に示した。
反復投与時の t
max,ssの中央値は 3.00 時間であり、C
max,ssの幾何平均値は 1.59 µg eq. LEV/mL(レベ
チラセタム当量に換算した濃度として表示、以下同様)であった。図 2.7.6.1.2-4 に示すように、C
minは Part B の初回投与後 24 時間以降一定であり、C
min,ssの幾何平均値は 0.82 µg eq. LEV/mL であった。
平均値 + 標準偏差、例数=12 ng eq. LEV/mL:レベチラセタム当量に換算した血漿中 L057 濃度 N01077 試験総括報告書(5.3.1.1.2)Figure 11:7
図 2.7.6.1.2-4 レベチラセタム 1500 mg を 1 日 2 回 12 時間間隔で 4.5 日間反復静脈内投与した
時の血漿中 L057 濃度推移:PP
血漿中 L 0 5 7 濃度 (n g e q . L E V /m L ) Part Bの初回投与開始後の時間 (h)2.7.6 個々の試験のまとめ
表
2.7.6.1.2-8 レベチラセタム 1500 mg を反復静脈内投与した時の
L057 の薬物動態パラメータ:PP
薬物動態パラメータ 反復静脈内投与
(N=12)
Cmax,ss (µg eq. LEV/mL) 1.59 (33.2)
tmax,ss (h) 3.00 (2.0 – 3.0)
Cmin,ss (µg eq. LEV/mL) 0.82 (45.7)
幾何平均値(変動係数%)、tmaxでは中央値 (最小値 – 最大値) µg eq. LEV/mL:レベチラセタム当量に換算した血漿中 L057 濃度 N01077 試験総括報告書(5.3.1.1.2)Table 14.2.1:14 一部改変(追加解析:5.4.21)
(
4) 安全性
1) 曝露状況
Part A では、18 例にレベチラセタム注射剤 1500 mg を単回静脈内投与し、18 例にレベチラセタム
500 mg 錠 3 錠を経口投与した。このうち、1 例の被験者では、レベチラセタム 1500 mg を静脈内投
与すべきところを
1200 mg しか投与しなかった。
Part B では、12 例にレベチラセタム 1500 mg を 1 日 2 回 4.5 日間反復静脈内投与し、6 例にプラ
セボを同様に投与した。
2) 有害事象
有害事象は症例報告書の記載内容を
MedDRA version 6.0 を用いて器官別大分類及び基本語に読み
替えた上で集計した。
因果関係の判定は「関連なし(
None)」、「おそらく関連なし(Unlikely)」、「どちらともい
えない(
Possible)」、「おそらく関連あり(Probable)」、「関連あり(Highly probable)」の 5 分
類とした。このうち、治験責任医師が、治験薬との因果関係を「関連あり」、
「おそらく関連あり」、
「どちらともいえない」と判断した有害事象を「因果関係が否定できない有害事象」として取り扱
った。なお、重症度別の集計では、同一被験者に重症度が異なる同一事象が複数件発現した場合は、
より重い重症度で集計した。
i) 有害事象発現例数の概要
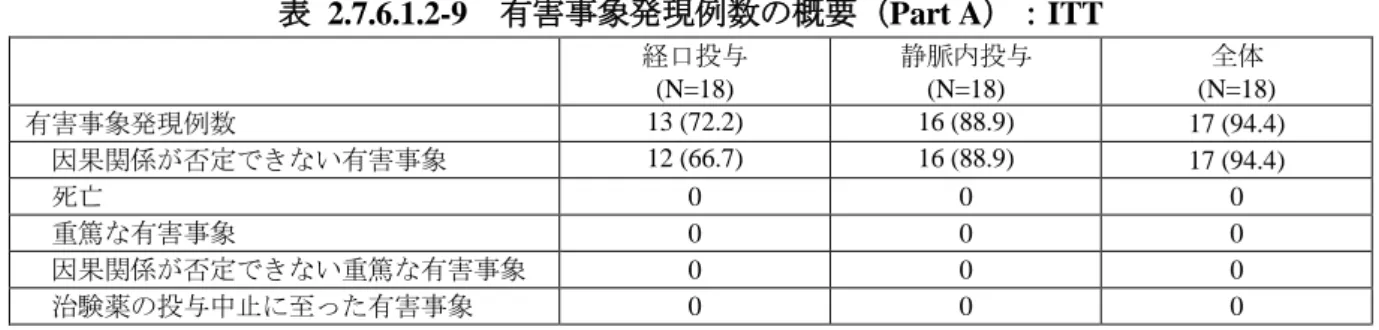
治験薬投与開始後に認められた有害事象発現例数の概要を、
Part A 及び Part B の別にそれぞれ表
2.7.6.1.2-9 及び表 2.7.6.1.2-10 に示した。
Part A では、有害事象は全体で 18 例中 17 例(94.4 %)に認められ、経口投与で 13 例(72.2%)、
静脈内投与で
16 例(88.9%)であった。因果関係が否定できない有害事象は全体で 17 例(94.4%)
に認められ、経口投与で
12 例(66.7%)、静脈内投与で 16 例(88.9%)であった。
Part B では、有害事象及び因果関係が否定できない有害事象は、18 例中 10 例(55.6%)に認めら
れ、レベチラセタム群の発現率は
66.7%(12 例中 8 例)であり、プラセボ群 33.3%(6 例中 2 例)
より高かった。
本治験では、死亡、重篤な有害事象及び治験薬の投与中止に至った有害事象は認められなかった。
また、
重症度が高度と判断された有害事象はなく、
いずれの有害事象も治験終了時までに消失した。
レベチラセタム 2.7.6 個々の試験のまとめ Page 26表 2.7.6.1.2-9 有害事象発現例数の概要(Part A):ITT
経口投与 (N=18) 静脈内投与 (N=18) 全体 (N=18) 有害事象発現例数 13 (72.2) 16 (88.9) 17 (94.4) 因果関係が否定できない有害事象 12 (66.7) 16 (88.9) 17 (94.4) 死亡 0 0 0 重篤な有害事象 0 0 0 因果関係が否定できない重篤な有害事象 0 0 0 治験薬の投与中止に至った有害事象 0 0 0 発現例数 (%) N01077 試験総括報告書(5.3.1.1.2)Table 14.3.1:1、Table 14.3.2:1表 2.7.6.1.2-10 有害事象発現例数の概要(Part B):ITT
プラセボ群 (N=6) レベチラセタム群 (N=12) 全体 (N=18) 有害事象発現例数 2 (33.3) 8 (66.7) 10 (55.6) 因果関係が否定できない有害事象 2 (33.3) 8 (66.7) 10 (55.6) 死亡 0 0 0 重篤な有害事象 0 0 0 因果関係が否定できない重篤な有害事象 0 0 0 治験薬の投与中止に至った有害事象 0 0 0 発現例数 (%) N01077 試験総括報告書(5.3.1.1.2)Table 14.3.1:5、Table 14.3.2:2ii) すべての有害事象
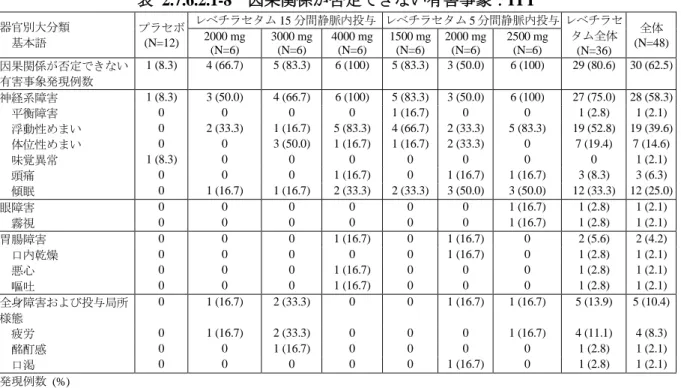
治験薬投与開始後に認められたすべての有害事象を、Part A 及び Part B の別にそれぞれ表
2.7.6.1.2-11 及び表 2.7.6.1.2-12 に示した。
Part A の全体で最もよくみられた有害事象は、
「神経系障害」
(16 例 88.9%)
[傾眠(11 例 61.1%)、
体位性めまい(8 例 44.4%)、浮動性めまい(4 例 22.2%)及び頭痛(4 例 22.2%)]であった。ま
た、静脈内投与で 2 例(11.1%)に注射部位そう痒感が認められた。
Part B でプラセボ群と比べてレベチラセタム群で発現率が高かった有害事象のうち、2 例以上の
被験者に認められた有害事象は、傾眠[プラセボ群、レベチラセタム群(以下同様):1 例 16.7%、
4 例 33.3%]、体位性めまい(0 例、3 例 25.0%)、頭痛(0 例、3 例 25.0%)及び血圧低下(0 例、
2 例 16.7%)であった。反復投与では、注射部位そう痒感は認められなかった。
2.7.6 個々の試験のまとめ