2.7.6.15 海外第 II 相継続試験[CS06A](添付資料番号 5.3.5.2-10)
2.7.6.15.1 試験方法の概略
治験の標題: 前立腺癌患者を対象とした FE200486 の反復投与時の長期安全性及び忍容性を検討する非盲 検,多施設共同,継続投与試験 治験責任医師名: 他 治験実施施設: 他 米国の計 12 施設 公表論文:なし 治験期間:3.1 年 治験開始日:2002 年 10 月 10 日(最初の患者の初回来院日) 治験終了日:2005 年 11 月 11 日(最後の患者の最終来院日) 開発のフェーズ:第 II 相試験 目的: 主要目的: ● 前立腺癌患者を対象に,ASP3550 反復投与時の長期安全性及び忍容性を検討する。 副次目的: ● 前立腺癌患者を対象に,ASP3550 反復投与時のテストステロン長期抑制効果を検討する。 ● ASP3550 反復投与時の血清 5α-dihydrotestosterone(DHT)値,血清黄体形成ホルモン(LH) 値,血清卵胞刺激ホルモン(FSH)値及び血清前立腺特異抗原(PSA)値の長期的変化を 検討する。 試験方法: 本試験は,前立腺癌患者を対象に ASP3550 長期反復投与時の安全性及び忍容性を検討する第 II 相,非盲検,多施設共同,継続投与試験であった。 海外第 II 相試験[CS06](以下[CS06]と略す)で ASP3550 の効果が認められた患者が,[CS06] の継続投与試験である本試験の対象となった。患者は[CS06]と同じ投与量を継続して投与さ れた。 本試験は,患者に合わせた投与間隔を確認する相(D-Phase)とその後の維持相(M-Phase) の 2 つの相にて実施した。D-Phase の最初の来院時(D-1 visit)に ASP3550 を初回投与,患者は測定を受けた。テストステロン抑制の持続時間により ASP3550 の投与間隔は異なっていた。D-1 visit 後 28 日以内にテストステロン抑制不十分の基準に合致した場合は試験を中止した。投与間 隔確定後 3 日以内に ASP3550 の 2 回目の投与を行い,M-Phase に移行した。M-Phase の間,患 者は投与レジメンに応じて 4~6 週間に 1 回来院することとした。投与間隔は,薬物動態/薬力 学的評価及び治験責任(分担)医師や治験依頼者の医学的判断により変更可能としたが,最低 間隔は 4 週間(28 日)とした。 被験者数(計画時及び解析時): 先行試験である[CS06]には 82 例(40 mg 群:10 例,80 mg 群:24 例,120 mg 群:24 例, 160 mg 群:24 例)が組入れられた。これらの患者のうち 37 例が本試験(継続投与試験)に組 入れられた(40 mg 群:1 例,80 mg 群:11 例,120 mg 群:16 例,160 mg 群:9 例)。 被験者数設定の根拠: 目標症例数の算出は実施しなかった。 診断及び主要な組入れ基準: 選択基準 [CS06]の選択基準 以下の基準を満たす場合,[CS06]の対象とした。 1. 18 歳以上の男性で,組織学的に前立腺癌(すべてのステージ)であることが確認されて おり,内分泌療法が必要な患者。ただし,ネオアジュバント内分泌療法は除く。前立腺癌 の転移が確認されていない場合は,スクリーニング前に 2 週以上の間隔で実施した 2 回の 連続した PSA 検査で PSA 値の上昇が認められた患者。
2. Eastern Cooperative Oncology Group(ECOG)の performance status(P.S.)が 2 以下の患者
3. ベースライン(Day 0)の血清テストステロン値が年齢別正常範囲内にある患者 4. 血清 PSA 値が 2 ng/mL 以上の患者 5. 少なくとも 6 カ月以上の生存が期待できる患者 [CS06A]の選択基準 以下の基準を満たす場合,本試験の対象とした。 1. 少なくとも 28 日間はテストステロン値の十分な抑制([CS06]の中止基準に規定した) が持続した患者 文書による同意は[CS06]又は[CS06A]の開始前に取得した。
除外基準 [CS06]の除外基準 1. 前立腺癌の内分泌療法[除睾術又は性腺刺激ホルモン放出ホルモン(GnRH)アゴニスト, GnRH アンタゴニスト,抗アンドロゲン剤,エストロゲン剤]を以前受けたことがある又 は現在受けている患者。ただし,以前受けたネオアジュバント内分泌療法が最大 6 カ月で あり,少なくともスクリーニングの 6 カ月以上前に終了している場合は除く。 2. ネオアジュバント内分泌療法が必要な患者 3. スクリーニング前の 12 週以内に血清テストステロン値又はテストステロンの機能に影響 を与える他の薬剤治療を受けた患者 4. スクリーニング後 6 カ月以内に根治的療法(根治的前立腺摘除術又は放射線療法)の対象 になると考えられる患者 5. 以下の既往歴を有する患者。 毎日吸入ステロイドの治療を必要とする重度の喘息,血管浮腫,アナフィラキシー反応。 6. 治験薬の成分(ASP3550 又はマンニトール)に対する過敏症を有する患者 7. 前立腺癌及び適切に処置された基底細胞癌又は表在性の扁平上皮癌以外の癌に過去 10 年 以内に罹患した患者 8. 治験責任(分担)医師により試験参加又は試験結果に影響を与える可能性があると判断さ れた,過度のアルコール又は薬物乱用を含む,臨床的に問題となる神経障害,胃腸障害, 腎障害,肝障害,循環器疾患,精神疾患,肺疾患,代謝異常,内分泌疾患,血液疾患,皮 膚疾患,感染性疾患又はその他の疾患を有する患者 9. 試験の参加又は試験の結果の評価の妨げになると治験責任(分担)医師が判断した,臨床 上問題となる臨床検査値異常を有する患者(肝機能の検査値は正常範囲内とする) 10. 適切な理解や協力を妨げる精神的障害又は言語障害のある患者 11. スクリーニング前 12 週以内に治験薬の投与を受けた患者 12. 過去に[CS06]に参加した患者 [CS06A]の除外基準 以下の基準のいずれかに該当する場合,本試験の対象としなかった。 1. [CS06]を以下の理由により中止した患者 ・有害事象 ・Day 28 にテストステロン抑制が達成されていなかった患者 ・[CS06]で規定したテストステロン値の上昇を伴わない PSA 値の低下不十分 ・治験実施計画書から逸脱した患者 2. 上記に示す[CS06]の除外基準の 2~10 に該当した患者
被験薬,用量及び投与方法,ロット番号: 被験薬: ASP3550 バイアル:1 バイアル中に ASP3550 の 20 mg 又は 40 mg を含有する注射用凍結乾燥 製剤 2.5%又は 5%マンニトール溶液で溶解し,それぞれ 10 mg/mL,20 mg/mL,30 mg/mL,40 mg/mL の投与液濃度とした。 用量及び投与方法: 10 mg/mL(40 mg),20 mg/mL(80 mg),30 mg/mL(120 mg)あるいは 40 mg/mL(160 mg) に調製した ASP3550 を,1 日量を 2 mL ずつ 2 回投与とし,腹壁の皮下に反復投与した。 なお,患者は[CS06]と同じ投与量をテストステロン抑制(テストステロン値が 0.5 ng/mL 以下)時間を基に,以下のような投与間隔で ASP3550 の投与を受けた。 ・テストステロン抑制時間が 28 日~41 日の場合,ASP3550 は 4 週ごとに投与 ・テストステロン抑制時間が 42 日~55 日の場合,ASP3550 は 6 週ごとに投与 ・テストステロン抑制時間が 56 日~69 日の場合,ASP3550 は 8 週ごとに投与 ・テストステロン抑制時間が 70 日~83 日の場合,ASP3550 は 10 週ごとに投与 また,14 週,18 週又は 22 週ごとの長期間隔の場合もあった。 ロット番号: ASP3550 20 mg:12605,22301,03F10-01,03K13-01,04C29-01 ASP3550 40 mg:13108,22601,03G07-01,03H04-01,03I11-01,04F21-01 治験実施計画書で規定された治験薬投与期間: ASP3550 が米国で上市される,ASP3550 の開発が中止又は個々の患者の中止基準に該当して 中止する(テストステロン又は PSA の中止基準値到達又はその他の理由による中止)までを投 与期間とした。190 週間後,7 例の患者を残すのみとなり,治験依頼者は管理上の理由で本試験 を打ち切った。 個々の患者の中止基準
・D-Phase での ASP3550 の単回投与後,nadir(最低値)後 Day 28 まで及びそれ以降,テス トステロン抑制(0.5 ng/mL 以下)が得られなかった場合
・M-Phase の Day 28 以降の来院日に 1 回でも,1.0 ng/mL を超える血清テストステロン値を 示した場合
・M-Phase の Day 28 以降の来院日に 2 回連続で,0.5 ng/mL 以上かつ 1.0 ng/mL 以下の血清 テストステロン値を示した場合
基準に該当した患者で,少なくとも 28 日間の抑制(0.5 ng/mL 以下)後のテストステロン 値増加が同時に起きた場合は,M-Phase へ移行してもよいこととした。 ・血清 PSA の投与前値([CS06]の Day 0 の値)から低下幅が 50%以下であった場合。ただ し,PSA 値の低下不十分が前立腺癌以外の理由によると判断された場合は,本試験を継続 してもよいこととした。 ・血清 PSA 値が nadir([CS06]又は[CS06A])と比べ 10 ng/mL 以上増加した場合。ただ し,[CS06A]の D-Phase にこれらの基準に該当した患者で,少なくとも 28 日間の抑制 (0.5 ng/mL 以下)後のテストステロン値の増加が同時に起きた場合は,M-Phase へ移行 してもよいこととした。 ・血清 ALT 値が参照範囲(10~45 U/L)上限の 5 倍以上であった場合 ・血清総ビリルビン値が参照範囲(0.1~1.0 mg/dL)上限の 2.5 倍以上であった場合 対照治療,用量及び投与方法,ロット番号: なし 併用治療: GnRH アゴニスト,GnRH アンタゴニスト,抗アンドロゲン剤又はエストロゲン剤との併用 療法は禁止とした。更に,血清テストステロン値又はテストステロンの機能に影響を与える他 の薬剤治療,例えばスピロノラクトン,ドンペリドン,シメチジン,アミトリプチリン,メチ ルドーパ,ミノキシジル,フィナステリド等を受けた患者は本試験から除外した。 ASP3550 以外の治験薬の投与は禁止とした。 評価スケジュール: [CS06]及び[CS06A]の評価スケジュールを表 2.7.6.15-1 及び表 2.7.6.15-2 に示した。
表 2.7.6.15-1 [CS06]の評価スケジュール Visit 1 2 3 4 5 6a) 7a) 8a) 9b) 10b) 11b) 12b) 13b,c) → 最終 d) Weeks 0 − − 1 2 4 5 6 8 10 12 ..+2 Days 最大 −21 0 1 3 7 14 28 35 42 56 70 84 ..+14 同意取得 ● 選択基準/除外基 準 ● 既往歴及び合併症 ● 前立腺癌 組織学的診断/ ステージ ●e) ECOG P.S.スコア ● 患者背景 ● 喫煙歴及び飲酒歴 ● 身長 ● 体重 ● ● 身体所見 ● ● 心電図 ● ● ● ● ● バイタルサイン ● ● 血液学的検査 血液生化学的検査 尿検査 ● ●f) ● ● ● ● ● ● 注射部位反応の評 価 ●g) ● ● ● ● ● ● ●m) ●m) ●m) ●m) ●m) ● 有害事象 ● ● ● ● ● ● ● ● ● ● ● ● ● 併用薬 ● ● ● ● ● ● ● ● ● ● ● ● ● ● 治験薬の投与 ● バイタルサインを 含む臨床的観察 ●h) 採血(血液検体) 血漿中 ASP3550 及び代謝物 ●i) ● ● ● ● ● ● ● ● ● ● ● ● テストステロン DHT / LH / FSH SHBG ● ●j) ● ● ● ● ● ● ● ● ● ● ● ● PSA ● ●k) ● ● ● ● ● ● ● ● ● ● 抗 ASP3550 抗体 ● ● 採尿(尿検体) ● ● ASP3550 代謝物 中止基準の確認 ●l) ●l) ●l) ●l) ●l) ●l) ●l) ●l) a:Day 0 との関連で ± 2 日以内に実施してもよいこととした。 b:Day 0 との関連で ± 4 日以内に実施してもよいこととした。 c:追加の来院日(Day 98)は,中止基準に該当するまで 2 週間に 1 回とした。安全性評価のための臨床検査値の 測定は,最終来院日まで 4 週間に 1 回実施した。 d:いずれかの中止基準に該当した場合,最終の来院とした。安全性評価のための臨床検査値の測定が最終来院日 の 4 日以内に実施してあれば,繰り返し実施する必要はないこととした。 e:直腸診,CT 及び骨シンチグラフィが 12 週間以内に実施されていない場合は実施することとした。 f:Day 0 がスクリーニング後 7 日以上の場合,血液学的検査,血液生化学的検査及び尿検査は繰り返し実施する こととした。 g:注射部位反応は治験薬の投与前及び投与 6 時間後に評価することとした。 h:治験薬の投与後少なくとも 6 時間は臨床的観察(血圧及び脈拍数の測定も含む)を実施することとした。 i:治験薬投与前,投与 2 時間,4 時間及び 6 時間後に実施,j:治験薬投与前及び投与 6 時間後に実施 k:治験薬投与前に実施
表 2.7.6.15-2 [CS06A]の評価スケジュール
Phase D-Phase M-Phase
Visit D-1a) D-2,D-3,D-4...b) M-1c) その後の来院(M-2,M-3,M-4,...)d) 最終e) Weeks 0 D-1 から 2,4,6,... 0 (M-1 から開始して 4~6 週間間隔で来院) 同意取得 ● 選択基準/除外基準 ● 体重 ●g) ● ●j) ● 身体所見 ●g) ● ●j) ● 心電図 ●g) ● ●j) ● バイタルサイン ●g) ● ● ● ● 血液生化学的検査 ●g) ● ● ● 血液学的検査,尿検査 ●g) ● ●j) ● 有害事象 ●i) ●i) ●i) ●i) ●i) 併用薬 ●h) ● ● ● ● 治験薬の投与/投与後 1 時間観察 ●f, k) ... ●f, k) ... 注射部位反応の評価l) ● ● ● ● ● 投与前の採血(血液検体) 抗 ASP3550 抗体 ●g) ● ●j) ● 血漿中 ASP3550 ●g) ● ● ● テストステロン / DHT / LH / FSH / SHBG / PSA ●g) ● ● ● 中止基準の確認 テストステロン抑制 不十分 ● ● PSA 低下不十分 a:[CS06]の最終来院日とした。 b:Visit D-1 との関連で ± 2 日以内の来院とした。 c:D-Phase の最終来院から 2 日目の来院とした。 d:Visit M-1 との関連で ± 4 日以内の来院とした。 e:ASP3550 が上市されるか又は試験を中止した場合,可能な限り早急に最終来院とすることとした。 f:患者は,治験責任(分担)医師による血液生化学的検査結果のレビューの 2 日後に来院して治験薬の投与を受 けることとした。治験薬の投与後 1 時間は臨床的観察を実施した。 g:[CS06]の最終来院日に実施した。[CS06A]としての新たな検査・観察は実施せず,症例報告書にも記録しな かった。 h:[CS06]からの継続として記録した。 i:[CS06A]で新たに発現した有害事象と同様に,[CS06]からの継続の有害事象も追跡し,記録した。 j:M-Phase の Visit M-1 から開始して 12 ± 2 週間に 1 回の間隔で実施した。 k:本試験での治験薬の投与は,[CS06]に組入れられた投与量を基に実施した。M-Phase の投与間隔は D-Phase での患者の反応を基に,その後の薬物動態及び薬力学評価結果を基に調整した。投与後の観察は 1 時間とした。 l:注射部位反応は,すべての投与日の来院時に,治験責任(分担)医師(投与前後)及び患者(投与後)の両者 で評価した。投与日以外のすべての来院時にも治験責任(分担)医師及び患者の両者で評価した。 Source:CS06A 総括報告書,Table 5-6(5.3.5.2-10) 評価基準: 有効性: ● テストステロン値が 0.5 ng/mL を超えるまでの時間。[CS06A]の D-Phase での初回投与 から,テストステロン値が 0.5 ng/mL を超えた Day 28 又はそれ以降の日までの時間とし た。
● 血清テストステロン値,血清 DHT 値,血清 LH 値,血清 FSH 値及び血清 PSA 値の経時 変化 ● [CS06]及び[CS06A]の D-Phase でのテストステロン値が 0.5 ng/mL を超える直前の来 院時の血漿中 ASP3550 濃度 安全性: ● 有害事象/副作用の発現例数及び特性 ● 局所忍容性(注射部位反応) ・患者による評価(疼痛/圧痛,そう痒,発赤,腫脹,その他の反応) ・治験責任(分担)医師による評価(紅斑,硬結/結節/小結節,潰瘍/壊死) ● 臨床検査値(血液学的検査,血液生化学的検査,尿検査)の経時変化 ● 身体所見,バイタルサイン,体重及び心電図 有害事象の程度及び関連性を以下のように定義した。 程度 各有害事象の程度は,以下の 3 段階で評価した。 ● 軽度:兆候又は症状が認められるが,日常生活に支障なし ● 中等度:日常生活に影響を及ぼす事象(支障あり) ● 高度:仕事や日常生活が行えない(耐え難い) 関連性 有害事象と治験薬との関連性を,以下の 4 分類で判定した。 ● あり:治験薬投与と明確な時間的関連性がある。治験薬の投与中止又は投与量の減量に より改善がみられる。治験薬を再投与した場合に再発が認められる。既知の事象である。 ● 可能性あり:治験薬投与と妥当な時間的関係がある。患者の臨床状態あるいは他の治療 や環境因子による可能性もある。 ● 多分なし:治験薬投与と妥当な時間的関係がない。患者の臨床状態あるいは他の治療や 環境因子による可能性もある。 ● なし:明らかに治験薬投与とは別の原因によるものである。「多分なし」,「可能性あり」 又は「あり」の基準に合致しない。 統計手法: 1. 解析対象集団
2. 患者背景及びその他の基準値 本試験に組入れられた患者の患者背景及びその他の基準値は,[CS06]試験時に記録した。 患者背景,前立腺癌の病歴及びステージ,合併症及び治療歴,身長,体重,BMI 及び有効性 /PD パラメータを,CS06/CS06A 試験に組入れられたすべての患者を対象として,投与群ごと に要約統計量を用いて要約した。 カテゴリーデータは,カテゴリー別に患者数及び割合(率)を用いて要約した。連続変数は 平均値,標準偏差,中央値,最小値,最大値を用いて要約した。 3. 有効性 テストステロン値が 0.5 ng/mL を超えるまでの時間は,生命表法を用いて算出し,Log-rank 検定により投与群間の比較を行った。[CS06]及び[CS06A]の D-Phase で,テストステロン値 が 0.5 ng/mL を超える直前の来院時での血漿中 ASP3550 濃度は,要約統計量を用いて投与群ご とに要約した。 4. 安全性 安全性の解析は,[CS06]及び[CS06A]を併合して行った。 安全性の解析は,少なくとも 1 回は治験薬の投与を受け,有害事象発現の有無が確認された すべての患者を対象とした。 CS06/CS06A 試験の併合した安全性解析対象集団と ITT 解析対象集団は同一で,[CS06]に登 録したすべての患者 82 名より構成された。 発現した有害事象は ICH 国際医薬品用語集(MedDRA)version 8.1 を用い,器官別大分類と 基本語に従って分類した。 安全性に関するパラメータは,要約統計量を用いて要約した。 (1) 有害事象 [CS06]の最初の投与から[CS06A]の最終来院時までに発現した有害事象を,試験中に発 現した有害事象とした。有害事象は投与群ごとに,また,程度別及び治験薬との関連性別に要 約した。治験薬との関連性は,「あり」及び「可能性あり」を治験薬との関連性はありとし,「多 分なし」及び「なし」を治験薬との関連性はなしとした。治験薬との関連性に関するデータが 欠測している場合,治験薬との関連性はありとした。有害事象は器官別大分類別及び基本語別 に,発現頻度が高い順に集計した。 重篤な有害事象及び臨床上の重要性から特に注目すべきであると判断された他の重要な有害 事象を要約した。 (2) 臨床検査値 臨床検査値(血液学的検査,血液生化学的検査及び尿検査の測定値)を患者の安全性の評価
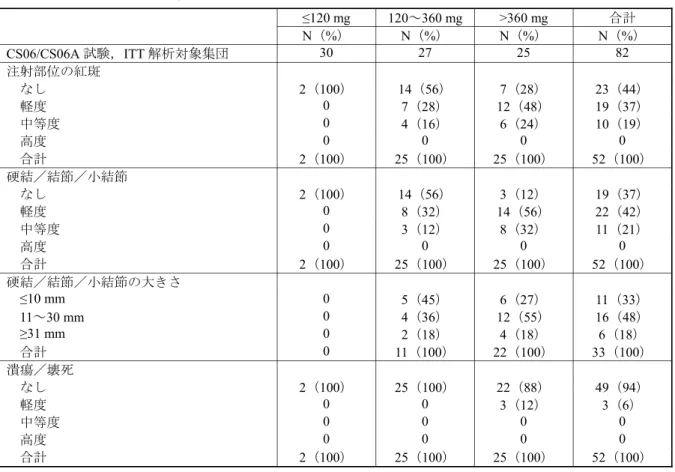
臨床検査値の変化は,試験終了時でのベースラインからの変化の平均値及び変化率の平均値 を,投与群ごとに要約した。臨床検査値の正常範囲との相対的な変動の発現率を,投与群ごと にシフトテーブルに要約した。シフトテーブルは,ベースライン値を「試験終了時」の値との 比較で「低い」,「正常内」又は「高い」に分類した。臨床検査値の測定値は来院時別でも要約 した。 (3) その他の安全性の評価項目:局所忍容性(注射部位反応) 認められたすべての注射部位反応の症状を要約統計量を用いて要約した。患者及び治験責任 (分担)医師の両者で,注射部位の評価を行った。 注射部位に症状(疼痛/圧痛,そう痒,発赤,腫脹/皮下の結節又は小結節及びその他の反 応)のいずれかを自覚した患者数を投与群,曝露量及び 1 日の投与量ごとに集計した。治験責 任(分担)医師による注射部位反応の評価も同様に発赤,腫脹及びその他の可視的変化を反応 なし,軽度,中等度,高度で評価し,集計した。皮下の腫脹/結節又は小結節は,その発現の 有無,有の場合はその大きさ(1~10 mm,11~30 mm 又は 31 mm を超える)別に集計した。
2.7.6.15.2 試験成績
1. 患者の内訳及び解析対象集団 CS06/CS06A 試験での患者の内訳を図 2.7.6.15-1 及び表 2.7.6.15-3 に示した。 [CS06]ではスクリーニングを受けた前立腺癌患者 150 例のうち,82 例が 4 つの投与群のいず れかに組入れられた。 [CS06]を完了した 39 例のうち,少なくとも 28 日間はテストステロン抑制がみられた 37 例 が[CS06A]に組入れられた。[CS06]と同じ投与量で ASP3550 の反復投与を受けた。 CS06/CS06A 試験の期間全体を通して,75 例が試験を中止したが,その内訳は 40 mg 群,80 mg 群,120 mg 群及び 160 mg 群でそれぞれ 10 例,20 例,24 例,21 例であった。[CS06]で 43 例, [CS06A]で 30 例が試験を中止した。[CS06]を完了した 80 mg 群の 2 例は,[CS06A]に組入れ られなかった。 [CS06]では,治験薬の投与開始後 3 カ月以内に 37 例(45%),3~6 カ月で 9 例(11%),6~ 12 カ月で 8 例(10%),12~18 カ月で 5 例(6%),18 カ月以降に 16 例(20%)が試験を中止した。 試験を中止した理由は「血清テストステロン値が 1 ng/mL を超えたため」が最も多く,32 例で あった。更に 6 例は「血清テストステロン値が 0.5~1 ng/mL であったため」に中止した。9 例は 「血清 PSA 値が 25%以上増加」により,2 例は「血清 PSA 値の 10 ng/mL 以上増加」,8 例は「血 清 PSA 値の低下が 50%以下」により中止した。 合計 5 例が有害事象により中止した。中止に至った有害事象は,両側性の亜急性及び慢性硬膜 下血腫,血中カリウム増加,脳血管発作,膀胱癌,扁桃癌が各 1 例であった。両側性の亜急性及現した患者 3 例は,中止理由を治験責任(分担)医師が誤って「その他」としたために,「その他」 の理由による中止として集計された。 4 例が治験実施計画書からの逸脱により中止した。3 例は[CS06A]の中止を選択した。2 例は 肝機能異常により中止した。 その他の理由により中止した例は,2 回来院しなかったために治験依頼者の要請により中止し た 1 例,血清テストステロン値及び血清 PSA 値ともに中止基準に該当した 1 例であった。 CS06/CS06A 試験を通して,合計 242 件の治験実施計画書からの逸脱が認められたが,大多数は 軽微な逸脱であった。 最も多く認められたのは,来院スケジュール又は ASP3550 の投与スケジュールの違反であった。 治験実施計画書からの重要な逸脱が,[CS06A]で 12 例に認められた。うち 9 例は選択基準違 反であった。これらの患者は[CS06]であらかじめ規定され PSA 値による中止基準に 12 週で該 当したが,[CS06A]に組入れられた。1 例は中止基準に該当したために[CS06]を終了したが, 治験責任(分担)医師が[CS06A]に組入れることを認めていた。1 例は試験期間中に併用禁止薬 の投与を受けており,もう 1 例は,同意文書の提出以前に Visit 1 に参加した。 CS06/CS06A の両試験ともに,ITT 解析対象集団及び安全性解析対象集団は同一で,[CS06]に 登録し治験薬の投与を 1 回以上受けたすべての患者 82 名により構成された。したがって,すべて の安全性に関する評価は,ITT 解析対象集団を対象に実施した。 スクリーニング (n=150) CS06 試験組入れ例 (n=82) CS06A 試験組入れ例 (n=37) A 群:40 mg B 群:80 mg C 群:120 mg D 群:160 mg (n=1) (n=11) (n=16) (n=9) 完了例:0 例 完了例:4 例 完了例:0 例 完了例:3 例 中止例:1 例 中止例:7 例 中止例:16 例 中止例:6 例 図 2.7.6.15-1 [CS06A]での患者の内訳
表 2.7.6.15-3 CS06/CS06A 試験での患者の内訳 A 群:40 mg B 群:80 mg C 群:120 mg D 群:160 mg 合計 N(%) N(%) N(%) N(%) N(%) CS06/CS06A 試験に組入れられた患者 10(100) 24(100) 24(100) 24(100) 82(100) ITT 解析対象集団 10(100) 24(100) 24(100) 24(100) 82(100) [CS06A]に組入れられた患者 1(10) 11( 46) 16(67) 9(38) 37(45) [CS06A]を完了した患者 0 4(17) 0 3(13) 7(9) 中止した患者 10(100) 20(83) 24(100) 21(88) 75(91) [CS06]を完了したが,[CS06A]に 組入れられなかった患者 0 2(8) 0 0 2(2) 中止理由 有害事象 0 2(8) 0 0 2(2) 肝機能異常 0 1(4) 0 1(4) 2(2) 患者の希望 0 0 2(8) 1(4) 3(4) 治験実施計画書からの逸脱 0 1(4) 3(13) 0 4(5) 血清 PSA 値が≥10 ng/mL 増加 0 1(4) 1(4) 0 2(2) 血清 PSA 値が≥25%増加 0 1(4) 5(21) 3(13) 9(11) 血清 PSA 値の低下が≤50% 0 2(8) 2(8) 4(17) 8(10) 血清テストステロン値>1.0 ng/mL 9(90) 9(38) 5(21) 9(38) 32(39) 血清テストステロン値 0.5~ 1.0 ng/mL 1(10) 0 3(13) 2(8) 6(7) その他a 0 1(4) 3(13) 1(4) 5(6) N:患者数,%:患者の割合 a:治験責任(分担)医師が誤って中止理由を「その他」としたが,実際は有害事象により中止した 3 例を含む。 Source:CS06A 総括報告書,Table 7-1(5.3.5.2-10) 2. 人口統計学的及び他の基準値の特性 主な患者背景を表 2.7.6.15-4 に,ベースライン時の前立腺癌のステージ及び罹病期間を表 2.7.6.15-5 及び表 2.7.6.15-6 に示した。 [CS06A]の患者集団の人口統計学的及び他の基準値は,[CS06]と同様であった。したがって, [CS06]と[CS06A]をプールしたデータを示す。患者の大多数(63 例,77%)が白人で,6 例 (7%)が黒人,13 例(16%)がその他の人種であった。全患者の平均年齢は 75.6 歳であった。年 齢,身長,体重及び BMI に,投与群間で差はなかった。 すべての患者は組織学的に確認された前立腺癌の患者であった。各投与群の約半数(42%~50%) の患者が限局性の前立腺癌であった。局所進行性前立腺癌が 80 mg 群,120 mg 群及び 160 mg 群 の 20%~25%であり,40 mg 群にはみられなかった。転移性前立腺癌と診断された患者の割合は, 120 mg 群及び 160 mg 群の各 8%に比べて,40 mg 群及び 80 mg 群で高かった(それぞれ 20%及び 17%)。 組織学的分類(Gleason スコア)は高分化が 3 例(4%),中分化が 42 例(52%),低分化が 36 例(44%)であった。他の投与群に比べて 40 mg 群で中分化が少なく,低分化が多かった。しか
CS06/CS06A 試験で,前立腺癌の罹病期間の中央値は 40 mg 群で 336 日,80 mg 群で 99.5 日, 120 mg 群で 1230 日,160 mg 群で 413 日であった。前治療歴は,40 mg 群で最終の直腸診からの 期間の中央値が長かった以外は,すべての投与群に差はみられなかった。多数の患者が前治療を 受けており,その内訳は放射線療法が 31 例(38%),根治的前立腺摘除術が 14 例(17%),ネオア ジュバント療法が 11 例(13%)であった。 ベースライン時の薬力学パラメータでは,血清 PSA の中央値(13.4 ng/mL)が 80 mg 群で他の 投与群(40 mg 群:9.30 ng/mL,120 mg 群:9.10 ng/mL,160 mg 群:6.40 ng/mL)に比べて高かっ た(表 2.7.6.15-7)。 既往歴で最も多かったのは筋骨格系および結合組織障害で,51 例(62%)であった。次いで胃 腸障害,外科および内科処置が各 48 例(59%),血管障害が 47 例(57%),代謝および栄養障害が 39 例(48%),生殖系および乳房障害が 38 例(46%),腎および尿路障害が 37 例(45%),眼障害 が 32 例(39%)の患者で報告された。事象名(基本語)では高血圧 NOS が最も多く,24 例(29%) であった。心臓障害は 23 例(28%)であった。既往歴は投与群間で同様であった。 表 2.7.6.15-4 主な患者背景(CS06/CS06A 試験,ITT 解析対象集団) A 群:40 mg B 群:80 mg C 群:120 mg D 群:160 mg 合計 N(%) N(%) N(%) N(%) N(%) CS06/CS06A 試験 ITT 解析対象集団 10 24 24 24 82 年齢(歳) N 10 24 24 24 82 平均値 ± SD 72.0 ± 5.72 77.5 ± 6.38 75.9 ± 5.79 74.7 ± 8.37 75.6 ± 6.90 中央値 73.0 78.5 78.0 75.5 76.0 最小値~最大値 64.0~81.0 64.0~86.0 62.0~88.0 59.0~88.0 59.0~88.0 人種,N(%) 黒人 2(20) 1(4) 2(8) 1(4) 6(7) 白人 5(50) 23(96) 15(63) 20(83) 63(77) 東洋人/アジア人 0 0 0 0 0 その他 3(30) 0 7(29) 3(13) 13(16) 合計 10(100) 24(100) 24(100) 24(100) 82(100) BMI(kg/m2) N 10 24 24 24 82 平均値 ± SD 26.7 ± 5.66 26.6 ± 3.54 27.2 ± 3.15 26.6 ± 3.39 26.8 ± 3.64 中央値 25.5 26.7 26.4 26.2 26.4 最小値~最大値 19.6~38.1 20.8~33.9 21.2~34.5 17.9~32.4 17.9~38.1 身長(m) N 10 24 24 24 82 平均値 ± SD 1.73 ± 0.122 1.75 ± 0.057 1.72 ± 0.094 1.74 ± 0.074 1.74 ± 0.082 中央値 1.75 1.75 1.74 1.75 1.75 最小値~最大値 1.50~1.93 1.65~1.85 1.50~1.88 1.57~1.88 1.50~1.93 体重(kg) N 10 24 24 24 82 平均値 ± SD 79.0 ± 11.7 81.7 ± 11.5 80.8 ± 12.8 81.4 ± 14.4 81.0 ± 12.6 中央値 82.4 82.6 78.5 77.6 79.8 最小値~最大値 63.5~99.3 56.7~102 58.7~109 53.1~109 53.1~109 N:患者数,%:患者の割合
表 2.7.6.15-5 ベースライン時の前立腺癌のステージ(CS06/CS06A 試験,ITT 解析対象集団) A 群:40 mg B 群:80 mg C 群:120 mg D 群:160 mg 合計 N(%) N(%) N(%) N(%) N(%) CS06/CS06A 試験 ITT 解析対象集団 10 24 24 24 82 ステージa 限局性 5(50) 11(46) 12(50) 10(42) 38(46) 局所進行性 0 6(25) 5(21) 5(21) 16(20) 転移性 2(20) 4(17) 2(8) 2(8) 10(12) 分類不能 3(30) 3(13) 5(21) 7(29) 18(22) 合計 10(100) 24(100) 24(100) 24(100) 82(100) 根治治療b あり 6(60) 8(33) 14(58) 10(42) 38(46) なし 4(40) 16(67) 10(42) 14(58) 44(54) 合計 10(100) 24(100) 24(100) 24(100) 82(100) 組織学的分類 (Gleason スコア) 高分化(2~4) 1(10) 1(4) 1(4) 0 3(4) 中分化(5~6) 3(30) 14(61) 12(50) 13(54) 42(52) 低分化(7~10) 6(60) 8(35) 11(46) 11(46) 36(44) 合計 10(100) 23(100) 24(100) 24(100) 81(100) N:患者数,%:患者の割合 a:限局性;T1/2・(NX 又は N0)・M0,局所進行性;[T3/4・(NX 又は N0)・M0]又は[N1・M0],転移性;M1 b:根治的前立腺摘除術又は放射線療法 Source:CS06A 総括報告書,Table 7-3(5.3.5.2-10) 表 2.7.6.15-6 前立腺癌の罹病期間(CS06/CS06A 試験,ITT 解析対象集団) A 群:40 mg B 群:80 mg C 群:120 mg D 群:160 mg 合計 CS06/CS06A 試験 ITT 解析対象集団 10 24 24 24 82 前立腺癌の罹病期間(日) N 10 22 24 23 79 平均値 ± SD 851 ± 1077 954 ± 1329 1609 ± 1836 1095 ± 1385 1181 ± 1494 中央値 336 99.5 1230 413 231 最小値~最大値 19.0~2932 14.0~4268 26.0~6674 25.0~3915 14.0~6674 N:患者数 Source:CS06A 総括報告書,Table 7-4(5.3.5.2-10)
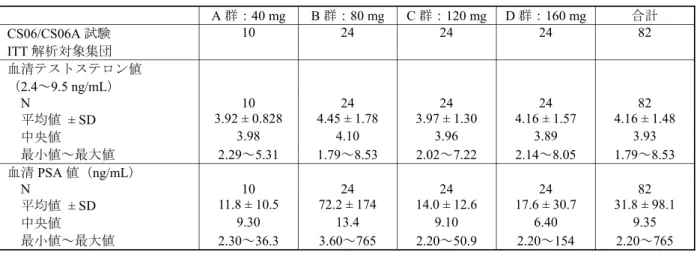
表 2.7.6.15-7 ベースラインの血清テストステロン値及び血清 PSA 値(CS06/CS06A 試験,ITT 解析対象集団) A 群:40 mg B 群:80 mg C 群:120 mg D 群:160 mg 合計 CS06/CS06A 試験 ITT 解析対象集団 10 24 24 24 82 血清テストステロン値 (2.4~9.5 ng/mL) N 10 24 24 24 82 平均値 ± SD 3.92 ± 0.828 4.45 ± 1.78 3.97 ± 1.30 4.16 ± 1.57 4.16 ± 1.48 中央値 3.98 4.10 3.96 3.89 3.93 最小値~最大値 2.29~5.31 1.79~8.53 2.02~7.22 2.14~8.05 1.79~8.53 血清 PSA 値(ng/mL) N 10 24 24 24 82 平均値 ± SD 11.8 ± 10.5 72.2 ± 174 14.0 ± 12.6 17.6 ± 30.7 31.8 ± 98.1 中央値 9.30 13.4 9.10 6.40 9.35 最小値~最大値 2.30~36.3 3.60~765 2.20~50.9 2.20~154 2.20~765 N:患者数
Source:CS06A 総括報告書,EOT Table 13(5.3.5.2-10)
3. 治験薬の曝露
ASP3550 の投与量を表 2.7.6.15-8 に示した。
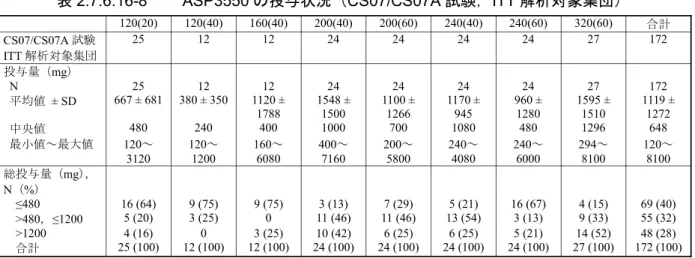
CS06/CS06A 試験期間で,ASP3550 の投与量の平均値は 472 mg であった。2 試験を通じて,30% の患者が 360 mg を超える ASP3550 を投与された。
表 2.7.6.15-8 ASP3550 の投与量(CS06/CS06A 試験,ITT 解析対象集団)
A 群:40 mg B 群:80 mg C 群:120 mg D 群:160 mg 合計 CS06/CS06A 試験 ITT 解析対象集団 10 24 24 24 82 投与量(mg) N 10 24 24 24 82 平均値 ± SD 72.0 ± 101 467 ± 722 457 ± 370 660 ± 1056 472 ± 732 中央値 40 80 360 160 160 最小値~最大値 40~360 80~2800 120~1680 160~4640 40~4640 総投与量(mg),N(%) ≤120 9(90) 13(54) 8(33) 0 30(37) >120,≤360 1(10) 5(21) 5(21) 16(67) 27(33) >360 0 6(25) 11(46) 8(33) 25(30) 合計 10(100) 24(100) 24(100) 24(100) 82(100) N:患者数,%:患者の割合 Source:CS06A 総括報告書,Table 8-1(5.3.5.2-10) 4. 有効性 本試験の主要目的は,前立腺癌患者を対象に ASP3550 を長期反復投与したときの安全性及び忍 容性を検討することであった。この継続投与試験に組入れられた患者は 4 群で 37 例であり,各投
方法(投与間隔)は,[CS06A]の最初の投与でもたらされたテストステロン抑制時間を基に患者 ごとに決定したので,種々の投与レジメンの有効性に関する結論を導き出すことは困難であった。 1) テストステロン値が 0.5 ng/mL を超えるまでの時間 [CS06A]でのテストステロン値が 0.5 ng/mL を超えるまでの時間を表 2.7.6.15-9 に示した。 生命表法により推定したテストステロン値が 0.5 ng/mL を超えるまでの時間の中央値[95%信頼 区間(CI)]は,80 mg 群,120 mg 群及び 160 mg 群でそれぞれ 70 日(42~140 日),98 日(70~ 126 日),112 日(28~126 日)であった。40 mg 群の中央値は,患者が 1 例のみであったため推定 できなかった。テストステロン値が 0.5 ng/mL を超えるまでの時間の中央値に,投与群間で統計的 に有意な差はみられなかった(Log-rank 検定,P=0.8950)。 患者内でテストステロン値が 0.5 ng/mL を超えるまでの時間を[CS06]と[CS06A]の D-Phase とで比較した。両試験での時間の差は,26 例(70%)で ± 28 日以内であった。これは,[CS06] と[CS06A]の D-Phase での ASP3550 の一貫した反応を示唆している(表 2.7.6.15-10)。
表 2.7.6.15-9 [CS06A]の D-Phase での Day 28 及びそれ以降にテストステロン値が
0.5 ng/mL を超えるまでの時間([CS06A],ITT 解析対象集団) A 群:40 mg B 群:80 mg C 群:120 mg D 群:160 mg [CS06A],ITT 解析対象集団 1 11 16 9 テストステロン値が Day 28 及びそれ以降に 0.5 ng/mL を超えるまでの時間(日) N 1 11 16 9 中央値 98.0 70.0 105 58.0 最小値~最大値 98.0~98.0 30.0~154 42.0~154 28.0~140 生命表法による推定値 中央値 − 70.0 98.0 112 95%CI − 42.0~140 70.0~126 28.0~126 Log-rank 検定 0.8950 N:患者数 Source:CS06A 総括報告書,Table 9-1(5.3.5.2-10) 表 2.7.6.15-10 [CS06]と[CS06A]の D-Phase でのテストステロン値が 0.5 ng/mL を超える までの時間の患者内比較(CS06/CS06A 試験,ITT 解析対象集団) N % テストステロン値が 0.5 ng/mL を超えるまでの時間の差 CS06 < CS06A − ASP3550 の投与 −28 日 5 14 CS06 = CS06A − ASP3550 の投与(±28 日) 26 70 CS06 > CS06A − ASP3550 の投与 +28 日 6 16 合計 37 100 N:患者数,%:患者の割合 Source:CS06A 総括報告書,Table 9-2(5.3.5.2-10)
2) 血清テストステロン値,血清 DHT 値,血清 LH 値,血清 FSH 値及び血清 PSA 値の経時変化 [CS06A]では,ASP3550 の単回投与後,すべての薬力学パラメータ(血清テストステロン値, 血清 DHT 値,血清 LH 値,血清 FSH 値,血清 PSA 値)はベースライン値から低下し,その後徐々 に増加した。一方,これらの薬力学パラメータの値は,維持相を通して比較的安定していた。 投与スケジュールが患者で異なっていたために,平均値又は中央値を経時的に図示することは できなかった。
薬力学パラメータに関し,[CS06A]の D-Phase 及び M-Phase のいずれでも投与量に関連した明
らかな差はみられなかった。 3) [CS06]及び[CS06A]の D-Phase での,テストステロン値が 0.5 ng/mL を超える直前の来院 時の血漿中 ASP3550 濃度 [CS06]では,テストステロン値が 0.5 ng/mL を超える直前の来院時の血漿中 ASP3550 血漿中 濃度の平均値は,40 mg 群,80 mg 群,120 mg 群及び 160 mg 群でそれぞれ 1.71 ng/mL,2.47 ng/mL, 2.14 ng/mL 及び 2.69 ng/mL であった。 [CS06A]の D-Phase では,テストステロン値が 0.5 ng/mL を超える直前の来院時の血漿中 ASP3550 濃度の平均値は,40 mg 群,80 mg 群,120 mg 群及び 160 mg 群でそれぞれ 1.90 ng/mL, 3.32 ng/mL,4.01 ng/mL 及び 3.44 ng/mL であった。 テストステロン値が 0.5 ng/mL を超える直前の来院時の血漿中 ASP3550 濃度は,80 mg 群,120 mg 群及び 160 mg 群の投与群間では同様であった。40 mg 群での血漿中 ASP3550 濃度は低かったが, [CS06A]の D-Phase に組入れられた 40 mg 群の患者は 1 例のみである。 5. 安全性 (1) 有害事象 CS06/CS06A 試験で発現した有害事象の要約を総投与量別に表 2.7.6.15-11 に示した。 ASP3550 を投与された 82 例中 62 例(76%)に 460 件の有害事象が発現した。副作用は 46 例(56%) に 107 件認められた。総投与量別の発現率は,120 mg 以下の群で 70%,120~360 mg 群で 63%, 360 mg を超える群で 96%であった。 重篤な有害事象が,合計で 14 例(17%)に 22 件認められた。試験期間中に死亡した患者が 1 例あり,本患者では 2 件の重篤な有害事象が認められたが,いずれも ASP3550 との関連性は「な し」と判断された。5 例が重篤な有害事象により試験を中止した。
表 2.7.6.15-11 総投与量別有害事象(CS06/CS06A 試験,ITT 解析対象集団) ASP3550 の総投与量 ≤120 mg 120~360 mg >360 mg 合計 N(%) 件数 N(%) 件数 N(%) 件数 N(%) 件数 CS06/CS06A 試験 ITT 解析対象集団 30(100) - 27(100) - 25(100) - 82(100) - すべての有害事象 21(70) 60 17(63) 82 24(96) 318 62(76) 460 死亡 0 0 0 0 1(4) 2 1(1) 2 重篤な有害事象 1(3) 1 3(11) 6 10(40) 15 14(17) 22 中止に至った有害事象a 0 0 1(4) 1 1(4) 1 2(2) 2 高度の有害事象 1(3) 1 3(11) 6 10(40) 21 14(17) 28 副作用 14(47) 25 12(44) 19 20(80) 63 46(56) 107 N:患者数,%:患者の割合 副作用:治験薬との関連性が「あり」又は「可能性あり」と判断された有害事象 a:中止に至った有害事象を報告された 2 例に加えて,「その他」の理由で中止したと報告された 3 例は,実際は 有害事象により中止した。 Source:CS06A 総括報告書,Table 10-1(5.3.5.2-10) 1) すべての有害事象 [CS06]とその継続試験である本試験を通して,合計 460 件の有害事象が 62 例(76%)に認め られた。投与群別の発現状況は,40 mg 群で 6 例(60%)に 10 件,80 mg 群で 19 例(79%)に 221 件,120 mg 群で 21 例(88%)に 156 件,160 mg 群で 16 例(67%)に 73 件であった。 CS06/CS06A 試験で発現した有害事象の要約を投与群別及び器官別大分類別に表 2.7.6.15-12 に 示した。 有害事象の発現率が最も高かった器官別大分類は「血管障害」で,発現率は 41%(34/82 例)で あった。その他,発現率が 20%以上の器官別大分類は,「全身障害および投与局所様態」が 33% (27/82 例),「感染症および寄生虫症」が 26%(21/82 例),「胃腸障害」が 24%(20/82 例),「神 経系障害」が 23%(19/82 例),「筋骨格系および結合組織障害」及び「生殖系および乳房障害」が 各 20%(16/82 例)であった。
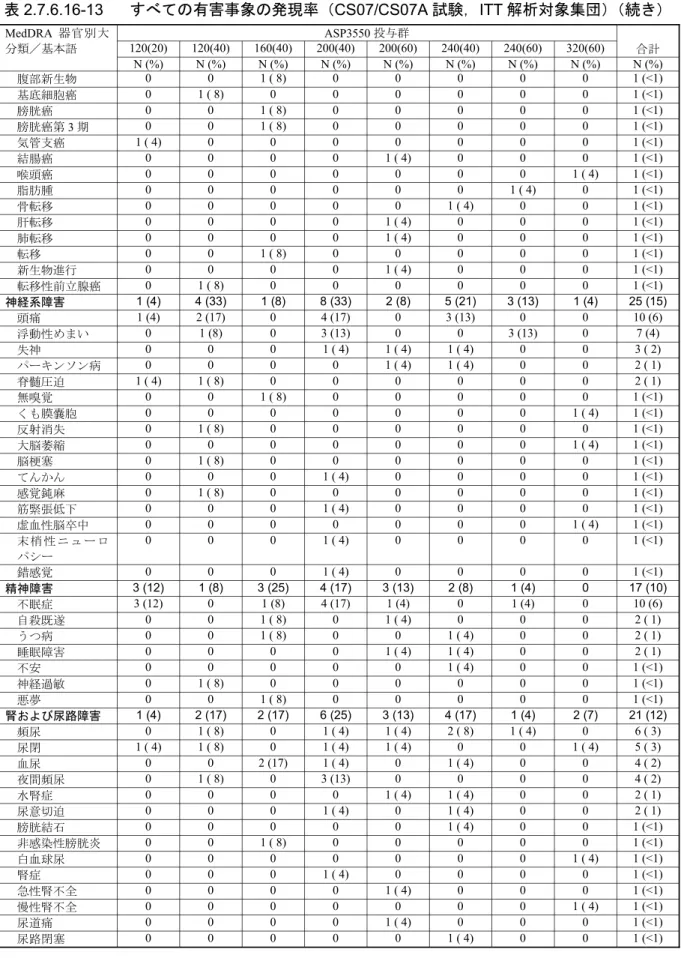
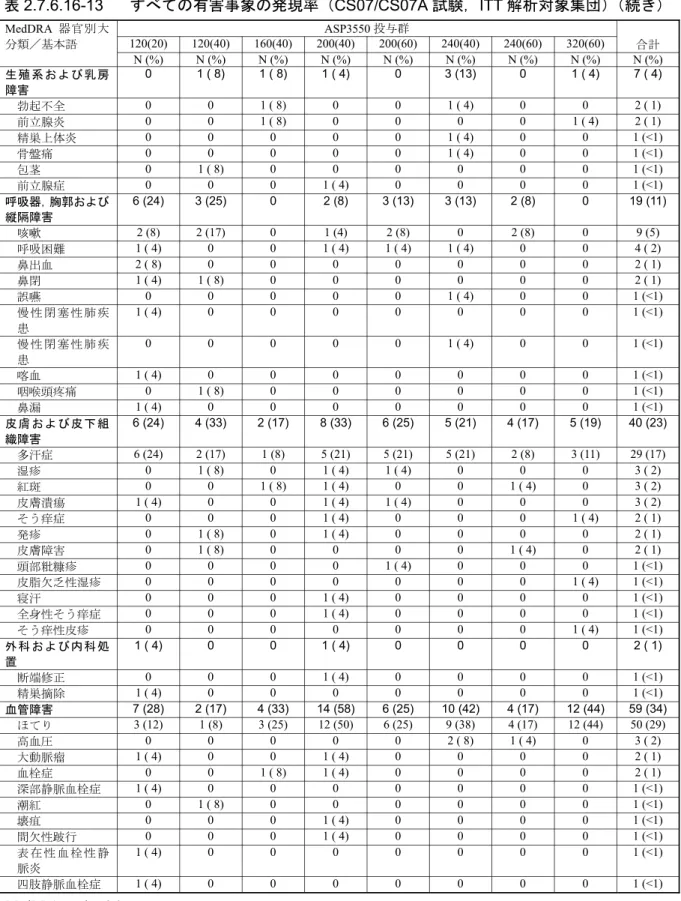
表 2.7.6.15-12 器官別大分類別有害事象(CS06/CS06A 試験,ITT 解析対象集団) MedDRA 器官別大分類 A 群:40 mg B 群:80 mg C 群:120 mg D 群:160 mg 合計 N(%) N(%) N(%) N(%) N(%) CS06/CS06A 試験,ITT 解析対象集団 10(100) 24(100) 24(100) 24(100) 82(100) 有害事象発現例数 6(60) 19(79) 21(88) 16(67) 62(76) 血液およびリンパ系障害 0 3(13) 5(21) 1(4) 9(11) 心臓障害 0 3(13) 5(21) 0 8(10) 先天性,家族性および遺伝性障害 0 0 2(8) 0 2(2) 耳および迷路障害 0 1(4) 0 0 1(1) 眼障害 1(10) 4(17) 2(8) 1(4) 8(10) 胃腸障害 0 9(38) 9(38) 2(8) 20(24) 全身障害および投与局所様態 1(10) 11(46) 9(38) 6(25) 27(33) 免疫系障害 0 0 3(13) 0 3(4) 感染症および寄生虫症 0 7(29) 8(33) 6(25) 21(26) 傷害,中毒および処置合併症 0 6(25) 1(4) 2(8) 9(11) 臨床検査 0 6(25) 6(25) 3(13) 15(18) 代謝および栄養障害 0 3(13) 3(13) 1(4) 7(9) 筋骨格系および結合組織障害 2(20) 6(25) 4(17) 4(17) 16(20) 良性,悪性および詳細不明の新生物 (嚢胞およびポリープを含む) 0 2(8) 2(8) 0 4(5) 神経系障害 2(20) 7(29) 7(29) 3(13) 19(23) 精神障害 0 1(4) 2(8) 3(13) 6(7) 腎および尿路障害 1(10) 6(25) 3(13) 2(8) 12(15) 生殖系および乳房障害 0 6(25) 6(25) 4(17) 16(20) 呼吸器,胸郭および縦隔障害 1(10) 6(25) 4(17) 2(8) 13(16) 皮膚および皮下組織障害 0 7(29) 5(21) 1(4) 13(16) 外科および内科処置 0 0 0 1(4) 1(1) 血管障害 2(20) 16(67) 10(42) 6(25) 34(41) MedDRA version 8.1 N:患者数,%:患者の割合 Source:CS06A 総括報告書,Table 10-2(5.3.5.2-10) すべての有害事象を,器官別大分類別及び基本語別に表 2.7.6.15-13 に示した。 発現率が 10%以上の比較的よくみられた有害事象で発現率が最も高かったのは,ほてりや潮紅 で,発現率は 34%(28/82 例)であった。ほてり関連事象の発現率が最も高かったのは 80 mg 群で, 58%(14/24 例)であった。他の投与群での発現率は 40 mg 群,120 mg 群及び 160 mg 群でそれぞ れ 20%(2/10 例),29%(7/24 例)及び 21%(5/24 例)であった。これらの事象のうち,2 例以外 は,ASP3550 との関連性が「可能性あり」又は「あり」と判断された。 その他のよくみられた有害事象の発現率は鼻咽頭炎が 11%(9/82 例),便秘,疲労及び浮動性め まいが各 10%(8/82 例)であった。本長期試験では,その他の有害事象の発現率はすべて 10%未 満であった。 ASP3550 の長期投与によりほてり,リビドー消失,インポテンス及び不妊,多汗等のテストス テロン抑制に関連した有害事象が生じることが予測された。しかしながら,安全性に関する併合 データでは,性機能不全に関連した有害事象はごくわずかしか報告されていない。「生殖系および 乳房障害」の有害事象は,合計で 16 例(20%)に認められた。その内訳は女性化乳房が 6 例(7%),
精巣萎縮症が 4 例(5%),勃起不全が 2 例(2%)であった。その他の「生殖系および乳房障害」 の有害事象はいずれも 1 例(1%)ずつの発現だった。寝汗が 2 例(2%)に認められた。 「心臓障害」の有害事象は 8 例(10%)に認められた。そのうち狭心症が最も発現率が高く,3 例(4%)に認められた。その他の心臓障害の有害事象はいずれも 1 例(1%)ずつの発現だった。 心臓障害の有害事象が認められた患者の中で 5 例は心臓障害の既往歴があった。 損傷が 4 例(5%)に認められたが,いずれも ASP3550 との関連性は「なし」又は「多分なし」 と判断された。
表 2.7.6.15-13 すべての有害事象の発現率(CS06/CS06A 試験,ITT 解析対象集団) MedDRA 器官別大分類/基本語 A 群:40 mg B 群:80 mg C 群:120 mg D 群:160 mg 合計 N(%) N(%) N(%) N(%) N(%) CS06/CS06A 試験,ITT 解析対象集団 10 (100) 24 (100) 24 (100) 24 (100) 82 (100) 有害事象発現例数 6 (60) 19 (79) 21 (88) 16 (67) 62 (76) 血液およびリンパ系障害 0 3 (13) 5 (21) 1 (4) 9 (11) 貧血 0 2 (8) 5 (21) 0 7 (9) 貧血 0 0 1 (4) 0 1 (1) 貧血 0 0 1 (4) 0 1 (1) 白血球増加症 0 1 (4) 0 0 1 (1) 白血球減少症 0 0 1 (4) 0 1 (1) 悪性貧血 0 0 0 1 (4) 1 (1) 心臓障害 0 3 (13) 5 (21) 0 8 (10) 狭心症 0 2 (8) 1 (4) 0 3 (4) 大動脈弁閉鎖不全症 0 0 1 (4) 0 1 (1) 心房細動 0 1 (4) 0 0 1 (1) 徐脈 0 0 1 (4) 0 1 (1) 左脚ブロック 0 0 1 (4) 0 1 (1) 心停止 0 0 1 (4) 0 1 (1) 冠状動脈粥状硬化症 0 0 1 (4) 0 1 (1) 冠動脈疾患 0 1 (4) 0 0 1 (1) 僧帽弁閉鎖不全症 0 1 (4) 0 0 1 (1) 上室性期外収縮 0 0 1 (4) 0 1 (1) 三尖弁閉鎖不全症 0 1 (4) 0 0 1 (1) 先天性,家族性および遺伝性障害 0 0 2 (8) 0 2 (2) 表皮母斑 0 0 1 (4) 0 1 (1) 陰嚢水腫 0 0 1 (4) 0 1 (1) 耳および迷路障害 0 1 (4) 0 0 1 (1) 耳痛 0 1 (4) 0 0 1 (1) 眼障害 1 (10) 4 (17) 2 (8) 1 (4) 8 (10) 白内障 0 1 (4) 1 (4) 0 2 (2) 霧視 0 0 1 (4) 1 (4) 2 (2) 糖尿病性網膜症 1 (10) 0 0 0 1 (1) 複視 0 1 (4) 0 0 1 (1) 眼乾燥 0 1 (4) 0 0 1 (1) 瞳孔障害 0 1 (4) 0 0 1 (1) 網膜剥離 0 1 (4) 0 0 1 (1) 胃腸障害 0 9 (38) 9 (38) 2 (8) 20 (24) 便秘 0 4 (17) 2 (8) 2 (8) 8 (10) 鼡径ヘルニア 0 3 (13) 2 (8) 0 5 (6) 消化不良 0 1 (4) 2 (8) 1 (4) 4 (5) 下痢 0 1 (4) 2 (8) 0 3 (4) 下痢 0 1 (4) 2 (8) 0 3 (4) 鼓腸 0 1 (4) 1 (4) 1 (4) 3 (4) 上腹部痛 0 1 (4) 1 (4) 0 2 (2) 結腸ポリープ 0 1 (4) 1 (4) 0 2 (2) 痔核 0 0 2 (8) 0 2 (2) 腹部ヘルニア 0 1 (4) 0 0 1 (1) 腹痛 0 1 (4) 0 0 1 (1)
表 2.7.6.15-13 すべての有害事象の発現率(CS06/CS06A 試験,ITT 解析対象集団)(続き) MedDRA 器官別大分類/基本語 A 群:40 mg B 群:80 mg C 群:120 mg D 群:160 mg 合計 N(%) N(%) N(%) N(%) N(%) 憩室 0 1 (4) 0 0 1 (1) 出血性腸憩室 0 0 0 1 (4) 1 (1) 嚥下障害 0 1 (4) 0 0 1 (1) 歯肉痛 0 1 (4) 0 0 1 (1) 舌痛 0 1 (4) 0 0 1 (1) 鼡径ヘルニア 0 1 (4) 0 0 1 (1) 嚥下痛 0 0 1 (4) 0 1 (1) 肛門周囲痛 0 1 (4) 0 0 1 (1) 小腸閉塞 0 0 1 (4) 0 1 (1) 歯の障害 0 0 0 1 (4) 1 (1) 歯痛 0 1 (4) 0 0 1 (1) 嘔吐 0 0 1 (4) 0 1 (1) 全身障害および投与局所様態 1 (10) 11 (46) 9 (38) 6 (25) 27 (33) 疲労 1 (10) 3 (13) 1 (4) 3 (13) 8 (10) 注射部位疼痛 0 3 (13) 3 (13) 1 (4) 7 (9) 注射部位そう痒感 0 4 (17) 2 (8) 0 6 (7) 末梢性浮腫 0 1 (4) 3 (13) 0 4 (5) 胸痛 0 3 (13) 0 0 3 (4) 無力症 0 2 (8) 0 0 2 (2) 発熱 0 0 1 (4) 1 (4) 2 (2) カテーテル留置部位関連反応 0 1 (4) 0 0 1 (1) 胸痛 0 1 (4) 0 0 1 (1) 悪寒 0 1 (4) 0 0 1 (1) 脂肪織増加 0 1 (4) 0 0 1 (1) インフルエンザ様疾患 0 1 (4) 0 0 1 (1) 注射部位内出血 0 1 (4) 0 0 1 (1) 注射部位刺激感 0 0 1 (4) 0 1 (1) 注射部位紅斑 0 1 (4) 0 0 1 (1) 注射部位硬結 0 1 (4) 0 0 1 (1) 注射部位結節 0 0 0 1 (4) 1 (1) 注射部位反応 0 1 (4) 0 0 1 (1) 小結節 0 1 (4) 0 0 1 (1) 疼痛 0 0 1 (4) 0 1 (1) 潰瘍 0 0 0 1 (4) 1 (1) 無力症 0 0 1 (4) 0 1 (1) 免疫系障害 0 0 3 (13) 0 3 (4) 季節性アレルギー 0 0 3 (13) 0 3 (4) 感染症および寄生虫症 0 7 (29) 8 (33) 6 (25) 21 (26) 鼻咽頭炎 0 3 (13) 3 (13) 3 (13) 9 (11) インフルエンザ 0 1 (4) 2 (8) 0 3 (4) 尿路感染 0 2 (8) 0 1 (4) 3 (4) 精巣上体炎 0 1 (4) 1 (4) 0 2 (2) 咽頭炎 0 2 (8) 0 0 2 (2) 上気道感染 0 1 (4) 0 1 (4) 2 (2) 尿路感染 0 1 (4) 1 (4) 0 2 (2)
表 2.7.6.15-13 すべての有害事象の発現率(CS06/CS06A 試験,ITT 解析対象集団)(続き) MedDRA 器官別大分類/基本語 A 群:40 mg B 群:80 mg C 群:120 mg D 群:160 mg 合計 N(%) N(%) N(%) N(%) N(%) 気管支炎 0 1 (4) 0 0 1 (1) 蜂巣炎 0 0 0 1 (4) 1 (1) 齲歯 0 0 0 1 (4) 1 (1) 耳感染 0 0 1 (4) 0 1 (1) 胃腸感染 0 0 1 (4) 0 1 (1) 歯肉感染 0 0 0 1 (4) 1 (1) 単純ヘルペス 0 0 1 (4) 0 1 (1) 感染性皮脂嚢腫 0 1 (4) 0 0 1 (1) 大葉性肺炎 0 0 1 (4) 0 1 (1) 肺炎 0 1 (4) 0 0 1 (1) 肺炎 0 0 1 (4) 0 1 (1) 敗血症 0 0 1 (4) 0 1 (1) 副鼻腔炎 0 0 1 (4) 0 1 (1) 股部白癬 0 1 (4) 0 0 1 (1) 傷害,中毒および処置合併症 0 6 (25) 1 (4) 2 (8) 9 (11) 損傷 0 3 (13) 0 1 (4) 4 (5) 処置による疼痛 0 3 (13) 0 0 3 (4) 事故 0 2 (8) 0 0 2 (2) 節足動物刺傷 0 0 0 1 (4) 1 (1) カテーテル合併症 0 1 (4) 0 0 1 (1) 挫傷 0 1 (4) 0 0 1 (1) 焼痂 0 1 (4) 0 0 1 (1) 擦過傷 0 0 0 1 (4) 1 (1) 関節損傷 0 0 1 (4) 0 1 (1) 裂傷 0 1 (4) 0 0 1 (1) 引っかき傷 0 1 (4) 0 0 1 (1) 硬膜下血腫 0 0 1 (4) 0 1 (1) 臨床検査 0 6 (25) 6 (25) 3 (13) 15 (18) 体重増加 0 1 (4) 2 (8) 0 3 (4) アラニン・アミノトランスフェラー ゼ増加 0 2 (8) 0 0 2 (2) アスパラギン酸アミノトランス フェラーゼ増加 0 2 (8) 0 0 2 (2) 血中カリウム増加 0 1 (4) 1 (4) 0 2 (2) 心拍数不整 0 1 (4) 0 1 (4) 2 (2) 肝機能検査値異常 0 1 (4) 0 1 (4) 2 (2) 体重減少 0 1 (4) 1 (4) 0 2 (2) 血中クレアチニン増加 0 0 1 (4) 0 1 (1) 血中カリウム減少 0 0 1 (4) 0 1 (1) 頚動脈雑音 0 0 1 (4) 0 1 (1) 心電図異常 0 0 1 (4) 0 1 (1) ヘマトクリット減少 0 1 (4) 0 0 1 (1) ヘモグロビン減少 0 1 (4) 0 0 1 (1) 心拍数減少 0 0 0 1 (4) 1 (1)
表 2.7.6.15-13 すべての有害事象の発現率(CS06/CS06A 試験,ITT 解析対象集団)(続き) MedDRA 器官別大分類/基本語 A 群:40 mg B 群:80 mg C 群:120 mg D 群:160 mg 合計 N(%) N(%) N(%) N(%) N(%) 代謝および栄養障害 0 3 (13) 3 (13) 1 (4) 7 (9) 高コレステロール血症 0 2 (8) 0 0 2 (2) 食欲減退 0 0 1 (4) 0 1 (1) 糖尿病 0 0 1 (4) 0 1 (1) 高脂血症 0 0 1 (4) 0 1 (1) 高脂血症 0 0 1 (4) 0 1 (1) 高ナトリウム血症 0 0 0 1 (4) 1 (1) 低血糖症 0 1 (4) 0 0 1 (1) 低カリウム血症 0 1 (4) 0 0 1 (1) 筋骨格系および結合組織障害 2 (20) 6 (25) 4 (17) 4 (17) 16 (20) 関節痛 0 2 (8) 1 (4) 1 (4) 4 (5) 背部痛 1 (10) 1 (4) 0 1 (4) 3 (4) 関節炎 0 0 1 (4) 0 1 (1) 骨痛 0 1 (4) 0 0 1 (1) 滑液包炎 0 1 (4) 0 0 1 (1) 殿部痛 0 0 0 1 (4) 1 (1) 肋軟骨炎 0 0 1 (4) 0 1 (1) 鼡径部痛 0 0 0 1 (4) 1 (1) 筋痙縮 0 1 (4) 0 0 1 (1) 筋痙縮 0 0 0 1 (4) 1 (1) 骨関節炎 0 0 0 1 (4) 1 (1) 骨関節炎 1 (10) 0 0 0 1 (1) 骨粗鬆症 0 0 0 1 (4) 1 (1) 四肢痛 0 1 (4) 0 0 1 (1) 足底筋膜炎 0 0 1 (4) 0 1 (1) 多発性関節炎 0 1 (4) 0 0 1 (1) 肩部痛 0 0 1 (4) 0 1 (1) 腱炎 0 1 (4) 0 0 1 (1) 良性,悪性および詳細不明の新生物 (嚢胞およびポリープを含む) 0 2 (8) 2 (8) 0 4 (5) 基底細胞癌 0 1 (4) 0 0 1 (1) 膀胱癌 0 0 1 (4) 0 1 (1) 肺腺癌 0 0 1 (4) 0 1 (1) 扁桃癌 0 1 (4) 0 0 1 (1) 神経系障害 2 (20) 7 (29) 7 (29) 3 (13) 19 (23) 浮動性めまい 1 (10) 3 (13) 3 (13) 1 (4) 8 (10) 頭痛 0 2 (8) 1 (4) 0 3 (4) 頭痛 1 (10) 0 1 (4) 0 2 (2) 頭痛 0 1 (4) 1 (4) 0 2 (2) 坐骨神経痛 0 2 (8) 0 0 2 (2) 失語症 0 1 (4) 0 0 1 (1) 頚動脈閉塞 0 1 (4) 0 0 1 (1) 頚動脈狭窄 0 0 1 (4) 0 1 (1) 脳出血 0 0 1 (4) 0 1 (1) 脳血管発作 0 1 (4) 0 0 1 (1) 痙攣 0 0 1 (4) 0 1 (1)
表 2.7.6.15-13 すべての有害事象の発現率(CS06/CS06A 試験,ITT 解析対象集団)(続き) MedDRA 器官別大分類/基本語 A 群:40 mg B 群:80 mg C 群:120 mg D 群:160 mg 合計 N(%) N(%) N(%) N(%) N(%) 体位性めまい 0 0 1 (4) 0 1 (1) 顔面神経麻痺 0 0 1 (4) 0 1 (1) 不全片麻痺 0 1 (4) 0 0 1 (1) 感覚減退 0 0 0 1 (4) 1 (1) 筋緊張低下 0 1 (4) 0 0 1 (1) 思考散乱 0 0 1 (4) 0 1 (1) 錯感覚 0 1 (4) 0 0 1 (1) 副鼻腔炎に伴う頭痛 0 0 0 1 (4) 1 (1) 失神 0 0 1 (4) 0 1 (1) 精神障害 0 1 (4) 2 (8) 3 (13) 6 (7) 不眠症 0 0 1 (4) 1 (4) 2 (2) リビドー減退 0 0 0 2 (8) 2 (2) 不安 0 1 (4) 0 0 1 (1) うつ病 0 0 1 (4) 0 1 (1) 妄想症 0 0 0 1 (4) 1 (1) 腎および尿路障害 1 (10) 6 (25) 3 (13) 2 (8) 12 (15) 排尿困難 0 1 (4) 1 (4) 2 (8) 4 (5) 尿閉 0 2 (8) 1 (4) 1 (4) 4 (5) 血尿 0 1 (4) 1 (4) 1 (4) 3 (4) 尿失禁 0 1 (4) 1 (4) 0 2 (2) 排尿困難 0 1 (4) 0 0 1 (1) 水腎症 0 0 1 (4) 0 1 (1) 排尿異常 0 1 (4) 0 0 1 (1) 夜間頻尿 0 1 (4) 0 0 1 (1) 頻尿 0 0 0 1 (4) 1 (1) 尿道狭窄 0 0 1 (4) 0 1 (1) 尿流量減少 1 (10) 0 0 0 1 (1) 生殖系および乳房障害 0 6 (25) 6 (25) 4 (17) 16 (20) 女性化乳房 0 4 (17) 1 (4) 1 (4) 6 (7) 精巣萎縮症 0 2 (8) 1 (4) 1 (4) 4 (5) 勃起不全 0 1 (4) 0 1 (4) 2 (2) 亀頭炎 0 1 (4) 0 0 1 (1) 精巣上体障害 0 0 1 (4) 0 1 (1) 前立腺萎縮 0 1 (4) 0 0 1 (1) 前立腺炎 0 0 1 (4) 0 1 (1) 性機能不全 0 0 1 (4) 0 1 (1) 精巣嚢胞 0 0 1 (4) 0 1 (1) 精巣痛 0 0 0 1 (4) 1 (1) 呼吸器,胸郭および縦隔障害 1 (10) 6 (25) 4 (17) 2 (8) 13 (16) 咳嗽 0 2 (8) 2 (8) 1 (4) 5 (6) 呼吸困難 0 1 (4) 1 (4) 0 2 (2) 鼻閉 0 1 (4) 1 (4) 0 2 (2) 副鼻腔うっ血 0 1 (4) 0 1 (4) 2 (2) アレルギー性咳嗽 0 0 1 (4) 0 1 (1) 呼吸音異常 1 (10) 0 0 0 1 (1)
表 2.7.6.15-13 すべての有害事象の発現率(CS06/CS06A 試験,ITT 解析対象集団)(続き) MedDRA 器官別大分類/基本語 A 群:40 mg B 群:80 mg C 群:120 mg D 群:160 mg 合計 N(%) N(%) N(%) N(%) N(%) 慢性閉塞性肺疾患 0 0 1 (4) 0 1 (1) 呼吸困難 0 1 (4) 0 0 1 (1) 鼻出血 0 0 1 (4) 0 1 (1) 咽喉頭疼痛 0 0 1 (4) 0 1 (1) 嚥下性肺炎 0 1 (4) 0 0 1 (1) 肺高血圧症 0 1 (4) 0 0 1 (1) 皮膚および皮下組織障害 0 7 (29) 5 (21) 1 (4) 13 (16) 斑状出血 0 1 (4) 2 (8) 0 3 (4) 寝汗 0 1 (4) 1 (4) 0 2 (2) そう痒症 0 2 (8) 0 0 2 (2) 発疹 0 1 (4) 0 1 (4) 2 (2) 臍紅斑 0 1 (4) 1 (4) 0 2 (2) 色素沈着障害 0 1 (4) 0 0 1 (1) そう痒症 0 1 (4) 0 0 1 (1) 乾癬 0 1 (4) 0 0 1 (1) 発疹 0 0 1 (4) 0 1 (1) 皮膚刺激 0 1 (4) 0 0 1 (1) 皮膚病変 0 1 (4) 0 0 1 (1) 皮下結節 0 0 1 (4) 0 1 (1) 外科および内科処置 0 0 0 1 (4) 1 (1) 嚢胞切除 0 0 0 1 (4) 1 (1) 血管障害 2 (20) 16 (67) 10 (42) 6 (25) 34 (41) ほてり 2 (20) 8 (33) 4 (17) 2 (8) 16 (20) ほてり 0 6 (25) 3 (13) 3 (13) 12 (15) 高血圧 0 0 3 (13) 0 3 (4) 大動脈粥状硬化症 0 1 (4) 0 0 1 (1) 潮紅 0 1 (4) 0 0 1 (1) 血腫 0 1 (4) 0 0 1 (1) 高血圧 0 0 1 (4) 0 1 (1) 低血圧 0 0 0 1 (4) 1 (1) MedDRA version 8.1 N:有害事象を発現した患者数,%:有害事象を発現した患者の割合 Source:CS06A 総括報告書,EOT Table 27(5.3.5.2-10)
2) 有害事象の程度及び ASP3550 との関連性 ASP3550 との関連性が「可能性あり」又は「あり」と判断された有害事象(副作用)が 46 例(56%) に 107 件認められた。副作用と判断された事象の数が,報告されたすべての有害事象の 4 分の 1 よりも少なかった(107/460 件)ことは注目すべきである。 すべての副作用の発現率を表 2.7.6.15-14 に示した。 投与群別の副作用発現率は,他の投与群と比べて 80 mg 群が最も高かった。各投与群での副作 用の発現状況は 40 mg 群,80 mg 群,120 mg 群及び 160 mg 群でそれぞれ 3 例(30%)に 3 件,17 例(71%)に 61 件,14 例(58%)に 26 件,12 例(50%)に 17 件であった。
他の投与群での発現率は 40 mg 群,120 mg 群及び 160 mg 群でそれぞれ 20%(2/10 例),25%(6/24 例)及び 21%(5/24 例)であった。 女性化乳房が 6 例(7%),精巣萎縮症が 4 例(5%)に認められた。注射部位疼痛,注射部位そ う痒感,疲労がそれぞれ 7 例(9%),6 例(7%),5 例(6%)に認められた。副作用の発現パター ンに投与群間による大きな差はなかった。ほてり及び潮紅,精巣萎縮症は,テストステロン抑制 に関連してよくみられる有害事象である。 CS06/CS06A 試験での副作用発現数は,[CS06]での発現数と一致しており,ASP3550 の長期反 復投与は副作用の著しい増加又は新たな予期せぬ副作用の発現を引き起こさなかったことを示唆 している。 [CS06]では,41 例(50%)に認められた 72 件の副作用のうち,160 mg 群で高度の疲労 1 件 が報告された以外は,すべて軽度又は中等度であった。発現率が最も高かったのはほてりで,発 現率は 29%(24/82 例)であった。 [CS06]では注射部位疼痛,疲労,注射部位そう痒感がそれぞれ 6 例(7%),4 例(5%),3 例 (4%)に認められた。女性化乳房,精巣萎縮症が各 2 例(2%)に認められた。全体的に,有害事 象の発現パターンに投与群間による大きな差はなかった。 表 2.7.6.15-14 副作用の発現率(CS06/CS06A 試験,ITT 解析対象集団) MedDRA 器官別大分類/基本語 A 群:40 mg B 群:80 mg C 群:120 mg D 群:160 mg 合計 N(%) N(%) N(%) N(%) N(%) CS06/CS06A 試験,ITT 解析対象集団 10 (100) 24 (100) 24 (100) 24 (100) 82 (100) 副作用発現例数 3 (30) 17 (71) 14 (58) 12 (50) 46 (56) 血液およびリンパ系障害 0 0 1 (4) 0 1 (1) 貧血 0 0 1 (4) 0 1 (1) 貧血 0 0 1 (4) 0 1 (1) 眼障害 0 0 0 1 (4) 1 (1) 霧視 0 0 0 1 (4) 1 (1) 全身障害および投与局所様態 0 10 (42) 6 (25) 4 (17) 20 (24) 注射部位疼痛 0 3 (13) 3 (13) 1 (4) 7 (9) 注射部位そう痒感 0 4 (17) 2 (8) 0 6 (7) 疲労 0 3 (13) 0 2 (8) 5 (6) 無力症 0 1 (4) 0 0 1 (1) 脂肪織増加 0 1 (4) 0 0 1 (1) 注射部位内出血 0 1 (4) 0 0 1 (1) 注射部位刺激感 0 0 1 (4) 0 1 (1) 注射部位紅斑 0 1 (4) 0 0 1 (1) 注射部位硬結 0 1 (4) 0 0 1 (1) 注射部位結節 0 0 0 1 (4) 1 (1) 注射部位反応 0 1 (4) 0 0 1 (1) 発熱 0 0 1 (4) 0 1 (1) 感染症および寄生虫症 0 0 0 1 (4) 1 (1) 蜂巣炎 0 0 0 1 (4) 1 (1)
表 2.7.6.15-14 副作用の発現率(CS06/CS06A 試験,ITT 解析対象集団)(続き) MedDRA 器官別大分類/基本語 A 群:40 mg B 群:80 mg C 群:120 mg D 群:160 mg 合計 N(%) N(%) N(%) N(%) N(%) 臨床検査 0 3 (13) 0 0 3 (4) アラニン・アミノトランスフェラー ゼ増加 0 2 (8) 0 0 2 (2) ア ス パ ラ ギ ン 酸 ア ミ ノ ト ラ ン ス フェラーゼ増加 0 2 (8) 0 0 2 (2) 肝機能検査値異常 0 1 (4) 0 0 1 (1) 筋骨格系および結合組織障害 0 1 (4) 0 0 1 (1) 四肢痛 0 1 (4) 0 0 1 (1) 神経系障害 1 (10) 1 (4) 0 0 2 (2) 浮動性めまい 1 (10) 0 0 0 1 (1) 筋緊張低下 0 1 (4) 0 0 1 (1) 精神障害 0 0 0 2 (8) 2 (2) リビドー減退 0 0 0 2 (8) 2 (2) 腎および尿路障害 0 2 (8) 0 0 2 (2) 排尿困難 0 1 (4) 0 0 1 (1) 排尿異常 0 1 (4) 0 0 1 (1) 尿閉 0 1 (4) 0 0 1 (1) 生殖系および乳房障害 0 6 (25) 3 (13) 3 (13) 12 (15) 女性化乳房 0 4 (17) 1 (4) 1 (4) 6 (7) 精巣萎縮症 0 2 (8) 1 (4) 1 (4) 4 (5) 勃起不全 0 1 (4) 0 0 1 (1) 性機能不全 0 0 1 (4) 0 1 (1) 精巣痛 0 0 0 1 (4) 1 (1) 呼吸器,胸郭および縦隔障害 0 1 (4) 0 0 1 (1) 呼吸困難 0 1 (4) 0 0 1 (1) 皮膚および皮下組織障害 0 3 (13) 3 (13) 0 6 (7) 寝汗 0 1 (4) 1 (4) 0 2 (2) 臍紅斑 0 1 (4) 1 (4) 0 2 (2) 発疹 0 1 (4) 0 0 1 (1) 皮下結節 0 0 1 (4) 0 1 (1) 血管障害 2 (20) 14 (58) 7 (29) 6 (25) 29 (35) ほてり 2 (20) 8 (33) 4 (17) 2 (8) 16 (20) ほてり 0 6 (25) 2 (8) 3 (13) 11 (13) 高血圧 0 0 1 (4) 0 1 (1) 低血圧 0 0 0 1 (4) 1 (1) MedDRA version 8.1 N:副作用を発現した患者数,%:副作用を発現した患者の割合 Source:CS06A 総括報告書,EOT Table 29(5.3.5.2-10)
CS06/CS06A 試験で発現した有害事象を程度別及び投与群別に表 2.7.6.15-15 に示した。合計 62 例(76%)に発現した有害事象のうち,53 例(65%)が軽度で,42 例(51%)が中等度で,14 例 (17%)が高度であった。
中等度の有害事象では,便秘及び鼻咽頭炎の発現率が高く,それぞれ 6%(5/82 例),5%(4/82 例)であった。次いで貧血,下痢,消化不良,血尿,尿閉,女性化乳房,咳嗽,ほてりが各 4%(3/82 例)であった。 高度の有害事象は,器官別大分類では「全身障害および投与局所様態」及び「胃腸障害」が各 5%(4/82 例),「傷害,中毒および処置合併症」が 4%(3/82 例)であった。高度の疲労及び損傷 が 2 例(2%)に認められた。その他の高度の有害事象はいずれも 1 例(1%)ずつに発現した。投 与群別の高度の有害事象発現患者数は,80 mg 群,120 mg 群及び 160 mg 群でそれぞれ 5 例(21%), 6 例(25%),3 例(13%)であった。 160 mg 群の 1 例は,[CS06]で高度の疲労を発現し,ASP3550 との関連性は「可能性あり」と 判断された。[CS06A]では高度の副作用の報告はなかった。 表 2.7.6.15-15 程度別の有害事象(CS06/CS06A 試験,ITT 解析対象集団) A 群:40 mg B 群:80 mg C 群:120 mg D 群:160 mg 合計 N(%) N(%) N(%) N(%) N(%) CS06/CS06A 試験,ITT 解析対象集団 10(100) 24(100) 24(100) 24(100) 82(100) 有害事象の程度 軽度 3(30) 19(79) 18(75) 13(54) 53(65) 中等度 4(40) 12(50) 15(63) 11(46) 42(51) 高度 0 5(21) 6(25) 3(13) 14(17) N:患者数,%:患者の割合 Source:CS06A 総括報告書,Table 10-4(5.3.5.2-10) 3) 死亡及びその他の重篤な有害事象 CS06/CS06A 試験で発現した死亡例を含む重篤な有害事象を器官別大分類別及び基本語別に表 2.7.6.15-16 に示した。 [CS06]及びその継続試験である本試験を通して,重篤な有害事象が 14 例(17%)に 22 件認 められた。[CS06]では重篤な有害事象を発現したのは 2 例であった。[CS06]でも[CS06]と [CS06A]を併合した ITT 解析対象集団でも,重篤な有害事象に明らかな傾向はなかった。 CS06/CS06A 試験では,80 mg 群で 5 例(21%)に 7 件,120 mg 群で 7 例(29%)に 13 件,160 mg 群で 2 例(8%)に 2 件の重篤な有害事象が認められた。重篤な有害事象の発現パターンに投与量 による明らかな差はなかった。40 mg 群では重篤な有害事象は認められなかった。 最も多い器官別大分類別の重篤な有害事象は「心臓障害」で,4 例(5%)に認められた。損傷 が 2 例に認められた。その他の重篤な有害事象はいずれも 1 例に 1 件ずつの発現だった。 「心臓障害」に分類された重篤な有害事象を発現したのは 4 例で,徐脈,狭心症,心停止,大 動脈弁閉鎖不全症が各 1 例であった。これらの重篤な有害事象は,いずれも ASP3550 との関連性 は「なし」と判断された。心停止を発現した 1 例は死亡したが,その他の 3 例は回復した。3 例 に心臓障害関連の既往歴があった。 [CS06A]試験中に死亡した 1 例は 120 mg 群で,ASP3550 の投与開始から約 24 カ月後に,硬
以下に死亡例の経過を簡単に叙述した。 被験者番号 18C026(120 mg 投与群)は 81 歳の白人男性で,硬膜下血腫により心停止を起こし 2005 年 2 月 3 日に死亡した。本事象は[CS06A]試験中に起こった。本患者は自宅で崩れるよう に倒れた後,2005 年 1 月 31 日に入院した。CT 検査の結果では両側性の慢性血腫及び小さい領域 に亜急性血腫が認められた。2005 年 2 月 2 日に硬膜下血腫を抜くための手術を行った。しかしな がら,術後の改善が得られず,その後昏睡状態になり次の日に死亡した。症状発現時までに ASP3550 の投与を 5 回受けており最終投与は 2004 年 10 月 22 日であった。本事象は ASP3550 と の因果関係は「なし」と判断された。 表 2.7.6.15-16 重篤な有害事象(死亡に至った有害事象も含む)(CS06/CS06A 試験,ITT 解析 対象集団) MedDRA 器官別大分類/基本語 A 群:40 mg B 群:80 mg C 群:120 mg D 群:160 mg 合計 N(%) N(%) N(%) N(%) N(%) CS06/CS06A 試験,ITT 解析対象集団 10(100) 24(100) 24(100) 24(100) 82(100) 重篤の有害事象発現例数 0 5(21) 7(29) 2(8) 14(17) 心臓障害 0 1(4) 3(13) 0 4(5) 大動脈弁閉鎖不全症 0 0 1(4) 0 1(1) 徐脈 0 0 1(4) 0 1(1) 心停止 0 0 1(4) 0 1(1) 冠動脈疾患 0 1(4) 0 0 1(1) 胃腸障害 0 0 2(8) 1(4) 3(4) 出血性腸憩室 0 0 0 1(4) 1(1) 鼡径ヘルニア 0 0 1(4) 0 1(1) 小腸閉塞a 0 0 1a(4) 0 1(1) 全身障害および投与局所様態 0 1(4) 0 0 1(1) 胸痛a 0 1a(4) 0 0 1(1) 感染症および寄生虫症 0 0 1(4) 0 1(1) 大葉性肺炎 0 0 1(4) 0 1(1) 傷害,中毒および処置合併症 0 1(4) 1(4) 1(4) 3(4) 損傷 0 1(4) 0 1(4) 2(2) 関節損傷 0 0 1(4) 0 1(1) 硬膜下血腫 0 0 1(4) 0 1(1) 臨床検査 0 1(4) 0 0 1(1) 血中カリウム増加 0 1(4) 0 0 1(1) 良性,悪性および詳細不明の新生物 (嚢胞およびポリープを含む) 0 1(4) 2(8) 0 3(4) 膀胱癌 0 0 1(4) 0 1(1) 肺腺癌 0 0 1(4) 0 1(1) 扁桃癌 0 1(4) 0 0 1(1) 神経系障害 0 0 2(8) 0 2(2) 頚動脈狭窄 0 0 1(4) 0 1(1) 脳血管発作 0 1(4) 0 0 1(1) 失神 0 0 1(4) 0 1(1) 腎および尿路障害 0 1(4) 0 0 1(1) 血尿 0 1(4) 0 0 1(1)
4) 中止に至った有害事象 [CS06]及びその継続試験である本試験を通して,80 mg 群の 2 例が重篤な有害事象により投 与を中止した。投与中止に至った有害事象は,血中カリウム増加と脳血管発作であった。いずれ の重篤な有害事象も程度は高度で,ASP3550 との関連性は「なし」と判断された。 更に,他の 3 例が「その他の理由」により投与を中止したと報告されたが,これら 3 例は,実 際は扁桃癌,膀胱癌及び硬膜下血腫のいずれも重篤な有害事象と判断された有害事象による中止 であった。 治験責任(分担)医師が誤って「その他」としたために中止理由は有害事象と報告されなかっ た。 5) その他の重要な有害事象 多くの臨床検査値異常が臨床的に重要と判断され,有害事象として報告された。有害事象とさ れた臨床検査値異常を表 2.7.6.15-17 に示した。 貧血が 80 mg 群の 2 例及び 120 mg 群の 5 例の合計 7 例(9%)に認められた。これらのうち 3 例は,それぞれ貧血,貧血増悪,白血球減少症の事象も発現していた。更に 160 mg 群の 1 例は悪 性貧血を発現した。 血尿が 3 例(4%),アラニン・アミノトランスフェラーゼ増加,アスパラギン酸アミノトラン スフェラーゼ増加,血中カリウム増加,肝機能検査 NOS 異常及び高コレステロール血症が各 2 例 (2%)に認められた。その他の重要な有害事象はいずれも 1 例(1%)ずつの発現であった。 80 mg 群の 1 例に認められた高度の血中カリウム増加は重篤な有害事象と報告された。ASP3550 との関連性は「なし」と判断されたが,投与中止に至った。 臨床的に重要な臨床検査値異常のうち,4 例に認められた事象が副作用と判断された。内訳は ALT 増加及び AST 増加が 2 例,貧血,肝機能検査 NOS 異常が各 1 例であった。
(2) 臨床検査値 ALP 及び AST のベースラインからの平均変化率に,投与群間で統計的に有意な差が認められた (P<0.05)。総投与量が,ヘマトクリット,ヘモグロビン,赤血球数及び好酸球(%)の平均変化 率に重大な影響を及ぼすことが示唆された(P<0.05)。ヘマトクリット,ヘモグロビン及び赤血球 数の値は,ベースライン後総投与量の増加に伴い減少した。しかし,好酸球のベースラインから の平均変化率と総投与量の間には明らかな関連性はみられなかった。 1 回投与量が ALP,ヘマトクリット,ヘモグロビン及び赤血球数に重大な影響を及ぼすことが 示された(P<0.05)。しかし,ベースラインからの変化に 1 回投与量が影響するかどうかの明確な 傾向は認められなかった。臨床検査値には,ベースラインと比較して低下したものもあれば,増 加がみられたものもあった。各投与群の患者数が少ないため,結果の解釈は困難であった。
![表 2.7.6.15-3 CS06/CS06A 試験での患者の内訳 A 群:40 mg B 群:80 mg C 群: 120 mg D 群: 160 mg 合計 N(%) N(%) N(%) N(%) N(%) CS06/CS06A 試験に組入れられた患者 10(100) 24(100) 24(100) 24(100) 82(100) ITT 解析対象集団 10(100) 24(100) 24(100) 24(100) 82(100) [CS06A]に組入れられた患者 1(10) 11( 46) 16(67](https://thumb-ap.123doks.com/thumbv2/123deta/8591734.936072/12.892.111.796.215.592/A試験患者内訳合計CSA試験組入ITT解析対象集団CSA組入られ患者.webp)