九州大学学術情報リポジトリ
Kyushu University Institutional Repository
ガス拡散型酸素電極に用いる窒素ドープカーボン複 合触媒に関する研究
立花, 直樹
https://doi.org/10.15017/4060203
出版情報:九州大学, 2019, 博士(工学), 課程博士 バージョン:
権利関係:
ガス拡散型酸素電極に用いる
窒素ドープカーボン複合触媒に関する研究
令和2年1月15日
九州大学大学院 総合理工学府
物質理工学専攻
立花 直樹
目次
第1章 緒論
1.1 緒言 2
1.2 金属空気電池
1.2.1 特徴 3
1.2.2 ガス拡散型電極 6
1.3 電極触媒
1.3.1 窒素ドープカーボン触媒 8
1.3.2 ペロブスカイト型酸化物触媒 14
1.4 触媒分散法 19
1.5 触媒担体 23
1.6 本研究の概要と目的 27
参考文献
第2章 窒素ドープポーラスカーボン触媒の調製条件の検討
2.1 緒言 35
2.2 実験方法
2.2.1 窒素ドープポーラスカーボンの調製 35
2.2.2 物性評価 36
2.3 電子顕微鏡観察および結晶構造解析 37
2.4 細孔構造解析 41
2.5 化学結合状態解析 46
2.6 本章のまとめ 49
参考文献
第3章 窒素ドープポーラスカーボン触媒の酸素還元活性とガス拡散型酸素 電極の特性
3.1 緒言 53
3.2 実験方法
3.2.1 回転ディスク電極法による評価 53
3.2.2 ガス拡散型酸素電極の作製 55
3.3 酸素還元活性 57
3.4 ガス拡散型酸素電極の分極特性 66
3.5 本章のまとめ 68
参考文献
第4章 ペロブスカイト型酸化物微粒子触媒の酸素還元活性
4.1 緒言 71
4.2 実験方法
4.2.1 逆ミセル法によるペロブスカイト型酸化物の合成 72
4.2.2 アモルファスリンゴ酸前駆体法によるペロブスカイト 75 型酸化物の合成およびビーズミル分散
4.3 結晶構造解析および電子顕微鏡観察 76
4.4 表面酸化状態の解析 82
4.5 酸素還元活性評価 90
4.6 本章のまとめ 93
参考文献
第5章 ペロブスカイト型酸化物微粒子/窒素ドープポーラスカーボン 複合触媒の酸素還元活性とガス拡散型酸素電極の特性
5.1 緒言 98
5.2 実験方法 98
5.3 電子顕微鏡観察および結晶構造解析 99
5.4 細孔構造および化学結合状態解析 104
5.5 酸素還元活性 107
5.6 ガス拡散型酸素電極の分極特性 110
5.7 本章のまとめ 111
参考文献
第6章 本研究の総括
6.1 総括 114
6.2 今後の展望 116
謝辞
1
第1章 緒論
1.1 緒言
1.2 金属空気電池 1.2.1 特徴
1.2.2 ガス拡散型電極 1.3 電極触媒
1.3.1 窒素ドープカーボン触媒 1.3.2 ペロブスカイト型酸化物触媒 1.4 触媒分散法
1.5 触媒担体
1.6 本研究の概要と目的 参考文献
2
1.1 緒言
金属空気電池は酸素を正極活物質とし,亜鉛,鉄等の金属を負極活物質として 用い,化学反応のエネルギーを電気エネルギーとして取り出す電気化学エネル ギーデバイスである.金属空気電池は電極内部に正極活物質を入れておく必要 がないため,正極にあたる空気極を非常に薄くすることができる.したがって,
セルに占める空気極の質量や体積は極めて小さい.これまでボタン型の亜鉛空 気電池が主に補聴器に使用され,近年では,非常用のマグネシウム空気電池が販 売されている.近い将来には自動車車載用電源,家庭や工場等の定置式分散電源 あるいは携帯電子機器の電源等への幅広い利用が期待されている.
リチウムイオン電池はこれまで使用されてきたニッケル水素電池や鉛蓄電池 と比較するとセル電圧が高く,質量当たりのエネルギー密度に優れるため携帯 電子機器や電気自動車に使用されている.しかし,これらの用途ではそのエネル ギー密度は満足できるものではない.また,可燃性の有機電解質を使用しており,
安全性に課題がある.原料となる炭酸リチウムの需要は2013—2017年において
年率 8%弱で成長しており,今後は年率 10%以上の伸びが予想され,需要の拡
大に伴い,2015年に約6,000$/tonであった炭酸リチウムの調達価格が,2018
年には約18,000$/tonまでに高騰した1).一方,金属空気電池は,亜鉛やアルミ
ニウム等の比較的,安価な金属を負極として使用することができる.また,Fig.
Fig. 1.1. Theoritical energy densities of metal-air batteries.
0 500 1000 1500 2000 2500 3000 3500
Theoritical energy density/ Wh kg-1
3
1.1に示すように,現行の電池の中で大きなエネルギー密度を持つリチウムイオ ン電池と比較しても,その理論エネルギー密度は極めて大きい2).しかし,金属 空気電池の課題の一つとして,その正極反応である酸素還元反応の進行が遅い 点が挙げられ,エネルギー変換効率を下げる主な要因となっている.白金あるい は白金合金の貴金属微粒子を用いた触媒は酸素還元反応に対して高い活性を示 すが,白金は資源が限られ,コストが高い.
本研究では,金属空気電池用酸素還元触媒として窒素ドープカーボン,ペロブ スカイト型酸化物およびこれらを複合した触媒を取り上げ,高活性で電池作動 条件下でも安定な触媒について検討した.本章では,金属空電池やガス拡散型電 極の基本的な特徴や特性について解説した後,本論文の目的を示す.
1.2 金属空気電池
金属空気電池は,負極活物質に亜鉛,アルミニウム,鉄等の金属,正極活物質 に酸素を用いた電池である.金属空気電池の放電時に負極の金属は酸化し,正極 である空気極において酸素が還元される.
1.2.1 特徴
Table 1.1に各種金属空気電池とリチウムイオン電池の理論セル電圧,理論エ
ネルギー密度および電解液を示す2).現行の電池の中で,大きなエネルギー密度 を持つリチウムイオン電池と比較しても,金属空気電池はエネルギー密度に優 れている.金属空気電池の負極活物質としては,様々な種類の金属が試されてき た.この中でアルカリ電解液中において使用できる亜鉛空気電池,鉄空気電池や アルミニウム空気電池は材料コストに優れ,長年にわたって研究が進められて いる.また,セル電圧が高く,エネルギー密度に優れたリチウム空気電池やマグ ネシウム空気電池を対象にした研究が近年,増加している3).さらに 2010 年代 に入って,ナトリウム空気電池4)やカリウム空気電池5)といった新しい金属空気 電池の研究が進められている.
4
Fig. 1.2に金属空気電池の一例として亜鉛空気電池の概略図を示す.亜鉛空気
電池の負極と正極および全反応は以下の通りである.
負極: Zn → Zn2+ + 2e− (E0 = −1.25 V vs. NHE) (R1.1) Zn2+ + 4OH− → Zn(OH)42− (R1.2)
Zn(OH)42− → ZnO + H2O + 2OH− (R1.3) 正極: O2 + 2H2O + 4e− → 4OH− (E0 = 0.40 V vs. NHE) (R1.4) 全反応: 2Zn + O2 → 2ZnO (R1.5)
これらの電池反応により,亜鉛空気電池の理論起電力は1.65 V となる.亜鉛空 気電池は補聴器や非常用の電池として使用され,実際の作動時には約 1.4 V の 電圧が出力されるが,その変動が小さくかつ長時間放電可能であることが特徴 である.負極では水素発生反応
Zn + 2H2O→ Zn(OH)2 + H2 (R1.6)
が併せて進行し,亜鉛が腐食する.正極で進行する酸素還元反応はアルカリ型燃 Table 1.1. Nominal cell voltage, theoretical energy density, and electrolyte of Li-ion battery and metal-air batteries.
Nominal cell voltage / V
Theoretical energy density* / Wh kg−1
Electrolyte Year
investigated
Li-ion 3.2−3.85 265 Aprotic 1980
Li-air 2.96 3458 Aprotic 1996
Na-air 2.27 1106 Aprotic 2012
Mg-air 3.09 2840 Saline 1966
Al-air 2.71 2796 Saline
/ alkaline
1962
K-air 2.48 935 Aprotic 2013
Zn-air 1.65 1086 Alkaline 1878
Fe-air 1.28 763 Alkaline 1968
* Energy densities are calculated including the contribution from O2.
5
料電池と共通の反応であり,ともに電解液としてKOHを使用する.また,食塩 電解プロセスとして,水素発生極に代わって酸素ガス電極を用いることで大幅 な省エネルギー化が可能な酸素陰極法において,その陰極で酸素還元反応が進 行する.
アルカリ電解液中で使用する金属空気電池の共通の課題として,空気中の二 酸化炭素と電解液中の水酸化物イオンとの反応により生成する炭酸イオンおよ び炭酸塩に起因する性能低下が挙げられる.電解液として KOH を用いたとき は,以下のCO2との反応が進行する.
CO2 + 2OH− → CO32− + H2O (R1.7)
CO32− + 2K+ → K2CO3 (R1.8)
(R1.7)式により生成した炭酸イオンは電解液のイオン伝導度および酸素溶解 度を減少させる.また(R1.8)式により生成した炭酸カリウムは空気極中の電解 液や酸素ガスの拡散を阻害する.したがって,空気極に導入する空気中の二酸化 炭素を予めソーダ石灰により吸収させるシステムが提案されている6).また,ア ニオン交換膜を電解質として使用することで炭酸イオンの生成を抑制すること が可能である7).アニオン交換膜は主にアルカリ型燃料電池用の固体電解質とし
Fig. 1.2. Schematic illustration of zinc-air battery.
Electrolyte (KOH)
O2 (in air) OH-
e-
Zn2+
Anode e-
Cathode
6
て研究が進められてきた.耐久性に課題があるが,イオン伝導度は0.1 S cm−2を 超え,出力では1 A cm−2を超えるアルカリ膜型燃料電池が報告されている8). 1.2.2 ガス拡散型電極
金属空気電池の空気極には半疎水性の多孔質電極であるガス拡散型電極が使 用される(Fig. 1.3).ガス拡散型電極とは気体反応物質の電気化学的な酸化反応 あるいは還元反応を直接起こさせることができる電極である.この電極は通常,
ガス拡散層と触媒層からなる.ガス拡散層は ポリテトラフルオロエチレン
(PTFE)等の結着材と疎水性のカーボンからなる.カーボンとしては比較的,
粒子径の大きなアセチレンブラックが用いられることが多く,スムーズなガス 拡散パスを形成し,かつ電解液の漏出および空気側からの水分の混入を防ぐこ とができる9).また,触媒層は触媒,カーボン等の導電性の触媒担体およびPTFE バインダーからなる.触媒層にて進行する酸素還元反応は電極内部に三次元的 に存在する活物質(酸素)-電解液-触媒からなる三相界面近傍で反応が進行す るため,大きな電流密度を得るためには,反応サイトである三相界面を,より多 く形成させる必要がある.したがって,高性能なガス拡散型電極を作製するため には,活性の高い触媒を用いるだけでなく,触媒層の微細構造を制御する必要が ある.
Fig. 1.3. Schematic illustration of gas diffusion electrodes.
Gas diffusion
O2H2O
OH—
Catalyst layerGas diffusion layer Electrolyte
(KOH)
Current collector
7
Fig 1.4a にガス拡散型電極の放電特性の概略図を示す.電極電位は理想的に
は標準電極電位と一致するが,主に,次の 3 つの過電圧要因によって電位が低 下する.
・抵抗過電圧
・活性化過電圧
・濃度過電圧
抵抗過電圧とは電極内部での電荷自体の移動に必要なオーム損であり,電流 密度に比例する.活性化過電圧とは,電極での電気化学反応における電荷移動に 要する過電圧である.還元反応のとき,酸化体 Oxが還元体 Red に還元される 際の電荷移動に伴うエネルギー損失である(Fig. 1.4b).使用する電極触媒の活 性が高いほど電荷移動が速やかに進行する.濃度過電圧とは,電極反応における 物質輸送の遅れにより生じる過電圧である.還元反応であれば反応物である酸 化体 Oxの供給あるいは生成物である還元体 Red の散逸が遅いことに伴うエネ ルギー損失である.
(b) (a)
Fig. 1.4. (a) Polarization characteristics of gas diffusion electrodes and (b) schematic illustration of the pathways involved in solution phase.
Resistance overpotential Activation overpotential
Concentration overpotential
Potential / V
Current density / mA cm-2
Red Red
Ox Ox
Electrode
ne—
Electron transfer
Mass transfer
Electric double layer
8
1.3 電極触媒
常温で酸素還元反応を速やかに進行させるためには電極触媒が不可欠である.
これまで酸素還元触媒について数多くの報告がなされている2,3).貴金属触媒で ある白金やパラジウムは高い酸素還元活性を示すことが知られており,さらな る活性の向上のために合金化やコア―シェル構造型が検討されている10).しか し,これらの貴金属は資源が限られ,コストが高い.したがって,貴金属を使用 しない触媒の研究が進められており,金属酸化物,金属硫化物や金属窒化物等,
多岐にわたって検討されている3).充電を行う場合,空気極では酸素発生反応が 進行し,その反応を促進させるため,酸素発生触媒があわせて必要となる.酸化 イリジウムや酸化ルテニウムが高い酸素発生活性を示すことが知られている11). メカニカルチャージ方式の場合は放電済みの負極金属をプラントで再生するた め,充電時の電池内部の劣化を避けることができるが,再生プラントや回収シス テムを含めた社会的なインフラ整備が必要となる.
1.3.1 窒素ドープカーボン触媒 (a) 特徴
電気伝導に優れるカーボン材料はエネルギーデバイスの電極内で導電パスと なるため,白金や金属酸化物の微粒子といった電気化学触媒の担体としてこれ まで使用されてきた.2009年にDaiのグループは窒素ドープカーボンナノチュ ーブが酸素還元反応に対して高い触媒活性を示すことを報告し12),特にこの十 年間でカーボン系電極触媒は大いに発展した.ドーピング元素としては窒素だ けでなく,硫黄,ホウ素,リン等のヘテロアトムをドープしたカーボン触媒がピ ュアなカーボンと比較して優れた活性を示す.ヘテロアトムドープカーボンの 提案されているドープサイトを Fig. 1.5 に示す.ヘテロアトムをドープしたカ ーボン触媒は酸素発生反応や水素発生反応にも高い触媒活性を示す13,14).また,
色素増感型太陽電池の正極においてI3−イオンをI−イオンへ還元する触媒として 作用し,白金の代替材料として注目されている15).Table 1.2に提案されている 各触媒反応の記述子(デスクリプター)を示す.しかし,これらの反応に対する ヘテロアトムドープカーボンの触媒活性を支配する因子は未だ議論が多く,実 験的手法あるいはDFT等の計算化学的手法による検討が行われている.多孔性 の窒素ドープカーボンは二酸化炭素吸着材としても優れた性能を示し16),また,
9
二酸化炭素還元反応に対する触媒として作用することが報告されている17).Fig.
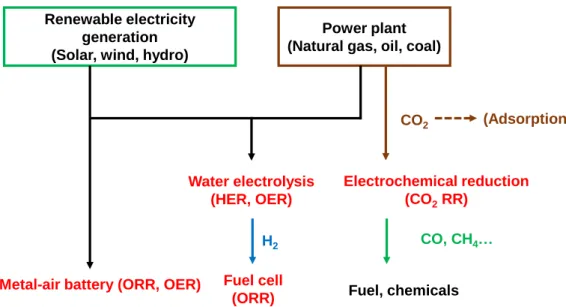
1.6にこれらのカーボン系触媒が活性を示す反応を,将来の二次エネルギー(電 気,水素)のフローに関連して示す.ヘテロアトムドープカーボン触媒は導電性 が高いことが最大の特徴であり,また,その微細構造はカーボン骨格の柔軟性を 利用した制御ができ,活性の向上が追求されている.
Table 1.2. Proposed intrinsic descriptors identified for carbon-based materials.
Descriptor Reaction Reference
Φ
(Φ = (Ex/Ec) × (Ax/Ac), electron affinity (A) and electronegativity (E))
Oxygen reduction reaction, oxygen evolution reaction, and
tri-iodine reduction reaction
18,19)
Free energy of OH adsorption, ΔGOH*
Oxygen reduction reaction and
oxygen evolution reaction 20) Free energy of I
adsorption, ΔGI*
Tri-iodine reduction reaction 19) Adsorption energy of H,
ΔGH*
Hydrogen evolution reaction 13) Halogens (F, Cl, Br, I)
Group IIIB, VB (B, N, P)
Bridge mode (S)
S-OH, P-OH S-O2, P-O2
Fig. 1.5. Schematic illustration of heteroatom-doped carbon.
10
ヘテロアトムドープカーボンの酸素還元活性はアルカリ溶液中では窒素ある いはホウ素をドープしたカーボンが特に高い酸素還元活性を示す21).ヘテロア トムドープカーボンの表面で進行する酸素還元反応の経路は
O2 + * → O2* (R1.9)
O2* + H2O + e— → HOO* + OH— (R1.10)
HOO* + e— → O* + OH— (R1.11)
O* + H2O + e— → HO* + OH— (R1.12)
HO* + e— →OH—+ * (R1.13)
と表される18).窒素をドープしたカーボンにはFig. 1.7 に示すピリジン型窒素
(pyridinic N),ピロール型窒素(pyrrolic N)あるいは四級窒素(quaternary
N)といった複数の窒素種が混在している.quaternary Nはグラファイト状窒
Renewable electricity generation (Solar, wind, hydro)
Power plant (Natural gas, oil, coal)
Metal-air battery (ORR, OER) Fuel cell (ORR) Water electrolysis
(HER, OER)
Electrochemical reduction (CO2RR)
Fuel, chemicals CO, CH4… CO2
H2
(Adsorption)
Fig. 1.6. Schematic illustration of a future secondary energy landscape (Oxygen reduction reaction, ORR; Oxygen evolution reaction, OER;
Hydrogen evolution reaction, HER; CO2 reduction reaction; CO2 RR).
11
素(graphitic N)とも表される.窒素種を選択的にドープすることは困難であ り,どの窒素種が活性をもたらし,どの部位が活性点であるかについては様々に 提案されている22).Guoらは酸性溶液中でpyridinic Nの隣の炭素原子に局在化 した電子準位が形成されてルイス塩基として機能して酸素還元反応の活性点と なることを報告している23).Jiao らはアルカリ溶液中で quaternary N に隣接 する炭素が酸素還元反応の活性点となることを,DFT 計算を用いたシミュレー ションにより報告している21).
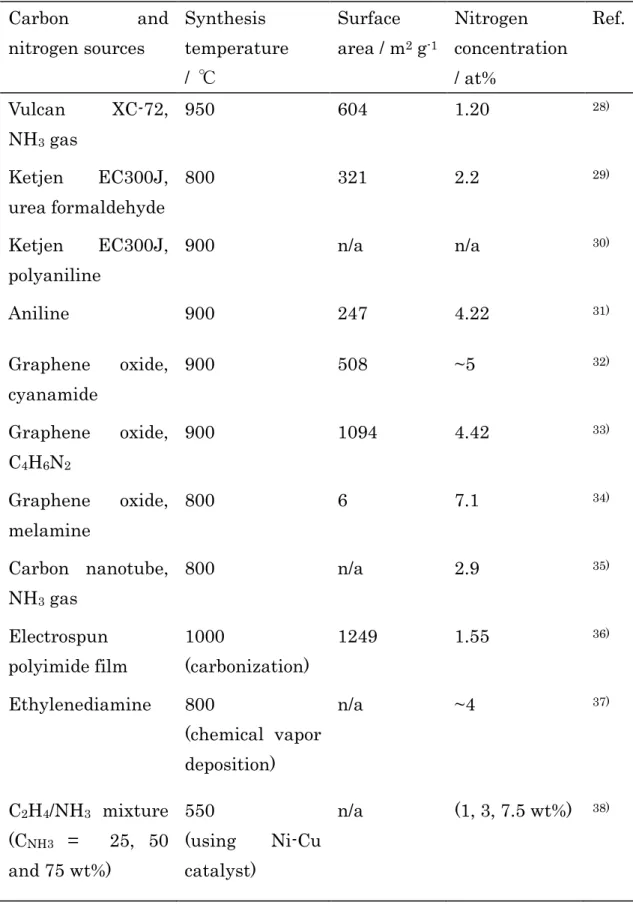
(b) 調製法
報告されている窒素ドープ炭素材料の炭素源,窒素源,合成温度,比表面積お よび窒素濃度をTable 1.3に示す.炭素源と窒素源とを含んだ前駆体を不活性ガ ス雰囲気下,もしくは炭素源をアンモニア雰囲気下で高温処理することで窒素 ドープ炭素材料を得ることができる.また,この前駆体に鉄源あるいはコバルト 源を加えて熱処理して得た触媒はそれぞれFe–C–N 触媒,Co–C–N触媒とも呼 ばれ,この種の触媒の歴史は古く,1964年にJasinskiがCoフタロシアニンの 酸素還元活性を報告した24).1976 年に Jahnkeらが Fe—N4や Co—N4の大環状
錯体を 700℃以上で熱処理することによって酸素還元活性が向上することを報
告し25),以来,様々な前駆体を用いて得たカーボン系触媒が報告されている.
1991 年にカーボンナノチューブ,2004 年にはグラフェンといった特徴的な形 態をもつ新しいナノカーボンが報告された.このカーボンナノチューブおよび グラフェンは,黒鉛と同様に炭素原子がsp2結合した六角格子から成るが,黒鉛 は炭素六角網面の積層体の集合からなる多結晶体であるのに対して,カーボン ナノチューブは炭素六角網面が同軸環状になり,グラフェンは炭素六角網面一 層のシートであり,特異な物性を示すことから注目されている.このカーボンナ
Pyrrolic N
Pyridinic N
Quaternary N
Fig. 1.7. Bonding configurations for nitrogen atoms in nitrogen-doped carbon.
12
ノチューブあるいはグラフェンを炭素源とし,窒素源を混合して不活性ガス雰 囲気下,あるいはアンモニア雰囲気下で 700—1100℃で熱処理することで,窒 素ドープカーボンナノチューブあるいは窒素ドープグラフェンを得ることがで きる.この熱処理法によって得られる窒素ドープ炭素材料は,出発原料や調製条 件によって比表面積や窒素濃度が大きく変化する.他の合成法としては化学気 相成長法,窒素プラズマ処理等が提案されている26).窒素ドープカーボン触媒は,
窒素原子に隣接した炭素原子に非対称な電荷分布を生じ,この特徴的な電荷分 布に起因してカーボンとは異なる高い酸素還元活性を示すと考えられているが,
ドープ量が多すぎると炭素六角網面を成すsp2ネットワークが壊れ,π電子モビ リィティが減少して電気伝導率が低下してしまうことが報告されている27).
13
Table 1.3. Carbon and nitrogen sources, synthesis temperature, surface area, and nitrogen concentration of nitrogen-doped carbon materials.
Carbon and
nitrogen sources
Synthesis temperature / ℃
Surface area / m2 g-1
Nitrogen concentration / at%
Ref.
Vulcan XC-72, NH3 gas
950 604 1.20 28)
Ketjen EC300J, urea formaldehyde
800 321 2.2 29)
Ketjen EC300J, polyaniline
900 n/a n/a 30)
Aniline 900 247 4.22 31)
Graphene oxide, cyanamide
900 508 ~5 32)
Graphene oxide, C4H6N2
900 1094 4.42 33)
Graphene oxide, melamine
800 6 7.1 34)
Carbon nanotube, NH3 gas
800 n/a 2.9 35)
Electrospun polyimide film
1000
(carbonization)
1249 1.55 36)
Ethylenediamine 800
(chemical vapor deposition)
n/a ~4 37)
C2Н4/NH3 mixture (CNH3 = 25, 50 and 75 wt%)
550
(using Ni-Cu catalyst)
n/a (1, 3, 7.5 wt%) 38)
14
1.3.2 ペロブスカイト型酸化物触媒 (a) 特徴
ペロブスカイト型酸化物(Fig. 1.8)はABO3で表され,Aサイトにはアルカ リ土類金属,アルカリ金属および希土類金属が占める.ペロブスカイト型酸化物 は構成元素が豊富であり,また,酸素欠陥の導入が比較的,容易である.ペロブ スカイト型酸化物は様々な機能性を有し,そのアプリケーションをTable 1.4に 示す.一例として,その高い触媒活性と熱的安定性を利用して自動車や種々の燃 焼器から出るCO,HC,NOx等を含んだ排ガスの浄化触媒として利用されてい る39).
Table 1.4. Applications of perovskite-type oxides.
Catalyst
・NOx decomposition
・Oxidation of CO, CH4, and methanol
・Oxidation of hydrocarbons
・VOCs combustion
Oxygen electrode
・Oxygen reduction and evolution in alkaline media
・Cathode material for solid oxide fuel cells
・Oxygen-permeable membranes
Gas sensor
・Alcohol sensor
・CO sensor
・NO2 sensor
・Lean-burn oxygen sensor A B O
Fig. 1.8. Crystal structure of perovskite-type oxides.
15
酸素還元触媒としては,Meadowcroft40)が1970年にペロブスカイト型酸化物 が高い活性を示すことを報告して以来,長年にわたって研究がなされている41). ペロブスカイト型酸化物の酸素還元活性は,主に遷移金属が占める B サイト元 素の性質に依存すると考えられている.この酸化物表面で進行する酸素還元反 応の経路は以下の通りである42).
Bm+—O2— + H2O + e— → B(m—1)+—OH— + OH— (R1.14)
O2 + e— → O2,ads— (R1.15)
B(m—1)+—OH— + O2,ads— → Bm+—O—O2— + OH— (R1.16) Bm+—O—O2— + H2O + e—→ B(m—1)+—O—OH—+ OH— (R1.17) B(m—1)+—O—OH—+ e—→ Bm+—O2—+ OH— (R1.18)
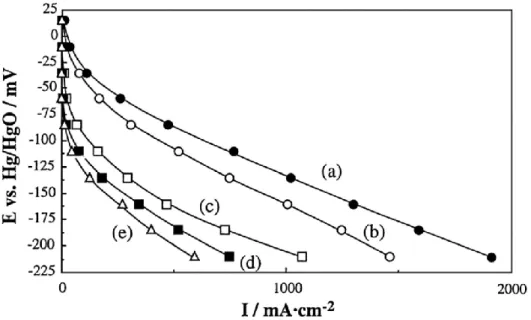
兵頭は,La系ペロブスカイト型酸化物を酸素還元触媒として使用したガス拡 散型電極を作製し,その分極特性を評価した(Fig. 1.9)43).LaMnO3 および LaCoO3を用いたガス拡散型電極が高い性能を示した.また,80 °C,9 M NaOH 水溶液中に浸漬したときの La 系ペロブスカイト型酸化物の安定性およびガス 拡散型電極をカソード分極したときの安定性を X 線回折測定によって評価した
(Table 1.5)43).LaMnO3はNaOH水溶液への浸漬後およびそれを用いたガス 拡散電極のカソード分極後において,その X 線回折ピークを確認できたが,
La(OH)3のピークがあわせて確認でき,一部,分解が起きていると考えられる.
LaCoO3は強アルカリ中でほとんど分解してしまうため強アルカリ電解液下で
の使用に適さないと考えられる.LaCrO3やLaFeO3は優れた安定性を示すが酸 素還元活性が低く,それらを用いたガス拡散型電極は性能が低い.LaNiO3はカ ソード分極に対する安定性が低く,分極後にはほとんど分解してしまっていた ことが報告されている.
16
Fig. 1.9. Polarization curves of gas diffusion electrodes using (a) LaMnO3,(b) LaCoO3,(c) LaNiO3,(d) LaCrO3,and (e) LaFeO3 at 80 °C in 9 M NaOH under oxygen flow.
Table 1.5. Chemical and electrochemical stability, and electrode performance of LaBO3 (B = Mn, Co, Ni, Cr, Fe).
(a) After immersion in 9 M NaOH at 80°C for 12 h.
(b) After cathodic polarization at −260 mV vs. Hg/HgO.
(c) Current density at −160 mV vs. Hg/HgO.
◎: Perovskite-type oxides only, ○: Perovskite-type oxides + lanthanum hydroxide, ×: Almost lanthanum hydroxide only.
17
A サイトおよびB サイトイオンの置換・欠陥の導入,また,酸素欠陥の導入 あるいは過剰酸素は,電気的中性条件を満足させるために反応サイトである B サイトイオンの異常原子価・混合原子価状態をつくり出し,間接的に触媒活性に 影響をもたらすものと考えられている.Matsumotoらは,ペロブスカイト型酸 化物の酸素還元活性は,この B サイトの遷移金属と表面吸着酸素との間で形成 する反結合性軌道σ*とその軌道の電子の充填状態が関与することを報告してい
る44, 45).Suntivichらはペロブスカイト型酸化物のBサイトの遷移金属のeg軌
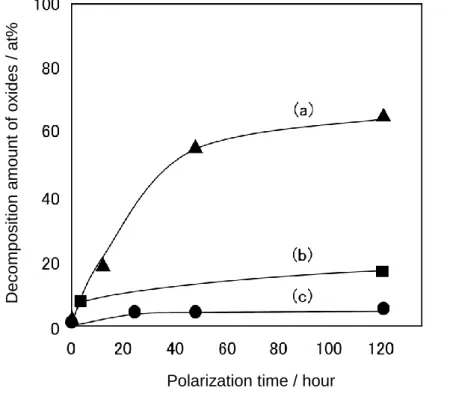
道に入る電子数と酸化物表面積で規格化した酸素還元電流値が25 µAのときの 電極電位との相関について調べ,eg が 0.5—1の間で最大の酸素還元活性を示す ことを報告している46).また,A,Bサイトの部分置換は触媒活性だけでなく安 定性にも影響を及ぼす.湯浅はLaMnO3のA,Bサイトをそれぞれ Ca,Feで 部分置換することで定電圧下での酸化物の分解量が減少し(Fig. 1.10),カソー ド分極に対する安定性が向上することを報告している47).
Fig. 1.10. Dependence of the decomposition amount of (a) LaMnO3, (b) LaMn0.6Fe0.4O3, and (c) La0.4Ca0.6Mn0.6Fe0.4O3 on the polarization time at —100 mV vs. Hg/HgO.
Polarization time / hour
Decomposition amount of oxides / at%
18
(b) 調製法
触媒として有利な高表面積酸化物を合成するためには,組成金属イオンの分 散性が高く,粒子径の小さな前駆体を用いることで,焼成温度を低くする必要が ある.
・金属塩分解法
水溶性の硝酸塩,酢酸塩等を所定のモル比で水に溶解させ,蒸発乾固すること によって得られる前駆体粉末を 800℃程度の温度で焼成してペロブスカイト型 酸化物を合成する方法である.しかし,混合水溶液中での金属塩の溶解度が著し く異なると,前駆体粉末が不均一な混合状態となって,高温焼成が必要となり,
不純物相が生成しやすくなる.
・共沈法
共沈法は,金属塩分解法と同様に水溶性の硝酸塩や酢酸塩の金属塩混合水溶 液を調製した後,水酸化物等の難溶性塩を同時に沈殿(共沈)させることで,均 一な前駆体粉末を得る方法である.しかし,共沈剤としてアルカリ水溶液を添加 すると溶液中で pH 値の偏りが生じるために,金属イオンの組合せによっては 均一な前駆体が得られないという課題がある.これを回避する方法として高pH の水溶液中に金属塩混合水溶液を滴下する方法(逆均一沈殿法)が提案されてい る48).
・逆ミセル法
ミセルは通常,20~100 個の界面活性剤から形成され,その直径は 50~100 Å程であり,親水基を外側に疎水基を内側に向けて配向している.したがって,
内部は疎水性となり,通常は可溶化しない疎水性溶媒を取り込んで,水中に可溶 化することが可能である.逆ミセルとは,ミセルとは逆に界面活性剤の疎水基を 外側に,親水基を内側に向けて配向したものであり,疎水性溶媒中に水を可溶化 させる.この逆ミセルは界面活性剤の親水・疎水バランス,溶液の温度,油相・
水溶液・界面活性剤の混合比,水溶液中のpH,含まれるイオン種やその濃度に より,安定性や内部に形成される水溶液相の大きさが変化する49).この水溶液相 を反応場として,酸化物前駆体を得る方法を逆ミセル法と呼び,ソフトテンプレ ート法の一つである(Fig. 1.11).ミセル内部の水溶液相は数ナノメートルサイ
19
ズに制御することができ,極めて微細な粒子を得ることができる50).
・有機酸錯体法
水酸基やカルボキシル基を含む有機酸は金属イオンと錯体を形成しやすく,
この錯体を利用することで均一性の良いアモルファス前駆体が得られる.クエ ン酸およびリンゴ酸は金属イオンの許容性が広く,それぞれ用いた方法をアモ ルファスクエン酸前駆体(Amorphous citrate precursor,ACP)法およびアモ ルファスリンゴ酸前駆体(Amorphous malate precursor,AMP)法と呼ぶ.
1.4 触媒分散法
ガス拡散型電極では活物質(酸素),電解液,触媒の三相界面近傍で反応が進 行する.しかし,触媒が凝集状態にあると,三相界面を有効に形成することがで きない.高分散状態にある触媒粒子は反応物である酸素分子が速やかに拡散し て供給されるが,凝集状態にあると酸素供給が速やかに行われず,電極性能が低 下する51).したがって,触媒の分散状態は電極性能を決める重要な因子となる.
粒子の分散状態はFig. 1.12に示すようにアグリゲーション,アグロメレーショ ン,フロキュレーションに分類することができる.アグリゲーション(凝結)は 一次粒子が面同士で結合して元の粒子の形状がほとんど確認できないほどに粗 大粒子化している状態である.アグロメレーションおよびフロキュレーション は凝集状態にあり,一次粒子のエッジ同士が点で結合している.フロキュレーシ ョンはアグロメレーションほど凝集が顕著でなく,軟凝集とも呼ばれる.
Precursor
Hydrophilic head Hydrophobic tail
Surfactant
Fig. 1.11. Schematic illustration of reverse micelles.
20
凝集体の分散には,ラボレベルのスケールでは超音波分散がよく行われる.超 音波振動子の振動が超音波ホモジナイザーのホーン先端あるいは超音波洗浄機 の洗浄槽に伝わって,接液面で圧縮,減圧が繰り返し発生する.この圧力差によ って微小な気泡の発生と消滅が起こり(キャビテーション),繰り返し凝集体に 衝撃を与える52).この衝撃により,液体中の凝集体が分散される.他のメディア レス分散法として,工業的にジェットミルがよく用いられる.ジェットミルは,
流体エネルギーを利用した粉砕機であり,ジェット気流で加速された粒子同士 を二方向または三方向から衝突させることにより粒子を粉砕する方法である.
しかし,ジェットミル等の乾式粉砕機では平均径でサブミクロンオーダーが粉 砕限界であると考えられている53).
メディアを使用した微粉砕法としてはビーズ(ボール)ミル法が挙げられる.
ボールおよびビーズはそれぞれの径で0.1—40 mm,0.015—0.5 mm とサイズに よって呼び方を分類することが多い54).この分散方法は,ジルコニア等の硬質の ビーズと,材料の粉体を円筒形の容器に入れて容器ごと回転あるいは攪拌する ことによって,ビーズに運動エネルギーを付与し,この運動エネルギーを持った ビーズに粉体を捕捉させて,衝突力,ずり応力,摩擦力およびせん断力によって 粉砕する方法である.Fig. 1.13にビーズによる凝集体の分散メカニズムを示す
54).Step (i) は初期状態であり,ビーズは回転しながら凝集体に近づく.Step (ii) においてビーズの間に凝集体が捕捉されて,衝突力やずり応力等が発生して凝 集体を分散する.Step (iii) でビーズと分散された粒子が離れていく.ビーズは 容器の回転や攪拌によって,減少した分の運動エネルギーを再び得て,また,
Stepを繰り返す.この手法はビーズに与えたエネルギーを直接,分散のための
Agglomeration Flocculation
Aggregation
Fig. 1.12. Dispersion states of particles.
21
エネルギーとして使用できるため効率が良く,また粉砕室の大きなビーズミル を使用することでラージスケールでの処理が可能であるため,工業的に広く利 用されている.
ビーズミル装置はメッシュあるいはスリットによって分散粒子とビーズの分 離を行うが,メッシュの目開きあるいはスリット幅をあまりにも小さくすると,
目詰まりや噛み込みによる閉塞を頻発してしまうため,通常,およそ0.1 mm以 上のサイズのビーズが使用される.この大きなビーズは凝集体に対して与える エネルギーが大きく,材料によっては結晶を破壊してしまう.新たに露出した表 面にはダングリングボンドがたくさんあるため化学的に不安定であり,容易に 再凝集してしまう(Fig. 1.14)55).これを過分散と呼ぶ.0.1 mm以下のビーズ に対応した遠心分離によるビーズ分離機構をもつビーズミルが開発され,この 十年で,酸化チタン56),酸化アルミナ57),カーボンナノチューブ58)等の小径ビー
Collision
Shear stress Step (i)
Step (ii)
Step (iii)
Fig. 1.13. Particle dispersion mechanism during bead milling.
22
ズによる分散が報告された.小径ビーズは質量が小さく,低周速で攪拌すると,
一つ一つのビーズが持つ運動エネルギーが極めて小さくなり,凝集体に与える ダメージが抑えられる.このビーズから分散系にもたらされるエネルギーを小 さくしたビーズミル法を,特に低エネルギービーズミル法と呼ぶ.ビーズの単位 容積当たりの数は径の三乗に逆比例するため,例えば0.3 mm のビーズ 1 個に
対して0.03 mmのビーズは1,000個となる.したがって,接触頻度が圧倒的に
多くなって分散が促進され,特に小さな粒子の分散には効果的である.また,弱 いエネルギーをもつビーズはビーズ同士の摩擦やずり応力による摩耗が減少し,
ビーズ由来のコンタミネーションが抑えられる.
Sufficient energy Agglomerated
particles
Dispersed particles Un-dispersed particles
Broken particles
Re-agglomerated particles Fig. 1.14. Morphological changes in particles during bead milling depending on supplied energy.
23
1.5 触媒担体
電気化学触媒に用いる担体は,電子の導電パスとなるため,高い電気伝導率を 示す材料である必要がある.また,大きな比表面積を持つ担体は触媒粒子を高分 散に担持し,その利用効率を高めることができる.したがって,電気伝導率が高 く,比表面積の大きなカーボンは電気化学触媒の担体としてよく使用される.
Table 1.6に主要なカーボン材料の比表面積および電気伝導率を示す.酸素還元
反応用触媒の担体の比表面積および電気伝導率はそれぞれ100 m2 g−1以上およ び0.1 S cm−1以上であることが望ましいとされる59).また,長期間使用するた めに,電解液中での高い安定性が求められる.ガス拡散型電極はFig. 1.3に示す ように,空気が拡散し,ガス拡散層を経て,触媒層に達し,活物質(酸素)—電 解液—触媒の三相界面近傍で反応が進行する.この触媒層において,担体はガス 拡散パスを形成する役割を担っており,三相界面を多数形成するために大きな 比表面積と細孔容積を持つ材料を用いる6).工業的にゴム製品の補強材や導電性 付与剤,また塗料等にも使用されているカーボンブラックは,これらの要件を満 たしているため,白金触媒や白金合金触媒の担体として用いられることが多い.
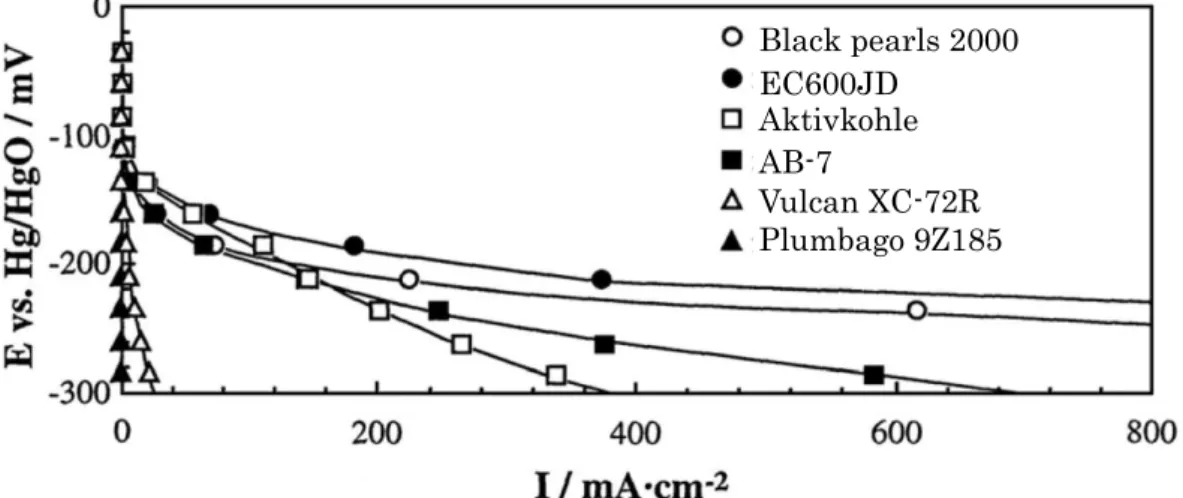
兵頭はカーボンブラックであるBlack pearls 2000,EC600JD,AB-7(Denka,
比表面積:49 m2 g−1)およびVulcan XC-72R(Cabot,254 m2 g−1),活性炭で あるAktivkohle(Merck,816 m2 g−1),グラファイトであるPlumbago 9Z185
(Ishizu,6 m2 g−1)のみを触媒層に用いたガス拡散型電極をそれぞれ作製し,
その分極特性を評価した(Fig. 1.15)43).カーボンはO2をHO2−に還元する触 媒として作用するため,比表面積が大きいEC600JDやBlack pearls 2000を用 いた電極が高い性能を示したと考えられる.また,濃度過電圧の影響が顕著であ ると考えられる卑な電位(−235 mV)における電流密度と細孔径0.004—0.2 µm の細孔容積との間に相関が確認できたことから,この径の細孔が反応物O2およ び生成物HO2−の拡散に寄与していることが示唆された43).
24
Table 1.6. Various carbon supports and their typical characteristics.
Carbon support Supplier Surface area / m2 g−1
Conductivity / S cm−1
Ref.
Carbon black Vulcan XC-72
Cabot 250 2.77 60,61)
Black pearls 2000
Cabot 1500 >1 59)
Ketjen EC300J
Lion Specialty Chemicals
800 4 59)
Ketjen EC600JD
Lion Specialty Chemicals
1270 10−100 59)
Denka black Denka 58 4 59)
Carbon nanotubes
200–900 0.3−104 60)
Graphene >2000
(theoretical value: 2630)
103−104 59)
Carbon nanofiber
10−300 102−104 59,60) Ordered
mesoporous carbon
600−2800 0.003−1.37 59)
Carbon nanocage
1276 813 59)
25
触媒担体は電子伝導のパスとなるだけでなく,担持された触媒と相互作用す ると考えられている.ナノ粒子触媒は接触界面において電荷移動が生じ,その触 媒活性および安定性の向上に寄与する.窒素ドープカーボン62),窒素およびリン ドープカーボンナノチューブ63)といったヘテロアトムをドープした炭素材料は 白金触媒との相乗効果が得られることが報告されている.また,カーボン材料だ けでなく様々な触媒担体が報告され,触媒活性および安定性の向上が図られて いる(Table 1.7).しかし,比表面積や電気伝導率はカーボン担体と比較すると 劣るものが多い.金属酸化物触媒においても,酸化マンガン/窒素ドープグラフ ェン64),Ba0.5Sr0.5Co0.8Fe0.2O3/カーボン65),酸化コバルト/窒素ドープカーボンナ ノチューブ66)は担持触媒と担体との間で相乗効果が得られて酸素還元活性が向 上することが報告されている.(ピュアな)カーボンは酸素を過酸化水素イオン に還元する二電子反応活性を有し,一部のペロブスカイト型酸化物担持カーボ ンでは,カーボン上で二電子還元反応が進行して生成した過酸化水素イオンを ペロブスカイト型酸化物が酸素および水酸化物イオンに接触分解する触媒とし て作用することが報告されている67,68,69).
Fig. 1.15. Polarization curves of gas diffusion electrodes using carbon materials in the catalyst layers at 80 °C in 9 M NaOH.
Black pearls 2000 EC600JD
Aktivkohle AB-7
Vulcan XC-72R Plumbago 9Z185
26
Table 1.7. Catalysts and supports for electrochemical reactions.
Catalyst Support Reaction Ref.
Pt TiN Oxygen reduction reaction
(0.1 M HClO4)
70)
Pt SiC Oxygen reduction reaction
(phosphoric acid)
71)
PtIr B4C Ammonia oxidation reaction 72)
Pt WC Hydrogen oxidation reaction 73)
Pt WC Methanol oxidation reaction 74)
Pt, PtRu W2C Methanol oxidation reaction 75) Pt Polyaniline Methanol oxidation reaction 76) Pt Polyaniline doped with
poly styrene sulfonic acid
Methanol oxidation reaction 77)
Pt Polyaniline/Nafion film Oxygen reduction reaction (0.5 M H2SO4)
78)
Pt Polypyrrole film Methanol oxidation reaction 79) Pt Polypyrrole nanowire Oxygen reduction reaction,
methanol oxidation reaction
80)
Pt Ti4O7 Hydrogen oxidation reaction, oxygen reduction reaction
81)
Pt TiO2 Oxygen reduction reaction 82)
Au SnO2 Oxygen reduction reaction 83)
PtRu WO3 Methanol oxidation reaction 84)
27
カーボン担体は化学的安定性に優れるが,アルカリ電解液中で炭素酸化反応 が進行し, (R1.19) 式と後続の(R1.20)式,もしくは(R1.21)式のように水酸化物 イオンと反応して二酸化炭素や炭酸イオンに酸化されてしまうことが知られて いる85).
C + 4OH− → CO2 + 2H2O + 4e− (R1.19)
CO2 + 2OH− → CO32− + H2O (R1.20)
C + 6OH− → CO32− + H2O + 4e− (R1.21)
その他にも,一酸化炭素やフミン酸等の有機物の生成も確認されている86).衣本 らは1.0 M KOH中で~0.65 V vs. Hg/HgOより貴な電位で炭素酸化反応による 酸化電流の増加およびカーボンの酸化による電極の重量減少を確認した87).し たがって空気極の電位を規制することは極めて重要である.
1.6 本研究の概要と目的
金属空気電池は質量エネルギー密度が極めて高いため,次世代の電源として 注目されているが,空気極に使用される酸素還元触媒として高い活性を示す白 金はコストが極めて高い.本研究では材料コストが低く,高い活性を示すことか ら注目されている窒素ドープカーボンとペロブスカイト型酸化物を取り上げ,
空気極における過電圧を低減させることを主な目的として,その調製条件や組 成を検討した.また,担体は電子伝導のパスとなるだけでなく,担持物との相乗 効果によって,その触媒活性の向上に寄与することから,窒素ドープカーボンを 担体としてペロブスカイト型酸化物を担持した複合触媒を合成し,その酸素還 元活性を評価した.
窒素ドープカーボンの合成方法としては,化学気相成長法,窒素プラズマ処理 等が提案されているが,シアナミドやメラミン等の窒素源を用いた熱処理法は 真空装置が不要であり,スケールアップに適した合成法である.しかし,この熱 処理法によって得られる窒素ドープカーボン材料は,出発原料や調製条件によ って細孔構造や表面の窒素種の濃度が大きく変化する.第 2 章では,酸素還元 触媒に最適な窒素ドープポーラスカーボンを得るために,炭素源として用いた カーボンブラックの前処理および熱処理温度が細孔構造や各窒素種の濃度に及 ぼす影響を評価した.第 3 章で,この窒素ドープポーラスカーボンの触媒活性
28
を回転ディスク電極法により評価した.物性評価と触媒活性評価の結果より,窒 素ドープカーボンの表面に形成される活性サイトについて考察した.第 4 章で は La 系ペロブスカイト型酸化物微粒子触媒について検討した.LaMnO3, LaFeO3 および La1-xCaxMn0.9Fe0.1O3 を逆ミセル法により合成した.また,
LaCoO3をAMP法により合成し,低エネルギービーズミルによって分散させた.
回転ディスク電極法により,これらのペロブスカイト型酸化物微粒子触媒の酸 素還元活性を評価した.第5章で,最適な組成であったLa0.4Ca0.6Mn0.9Fe0.1O3
と窒素ドープポーラスカーボンとを組み合わせた複合触媒を合成し,その酸素 還元活性を評価した.最後に,実際の金属空気電池やアルカリ型燃料電池の正極 として用いられているガス拡散型電極の触媒層にこの複合触媒を用い,その電 極特性を評価した.最後に,第6章で本研究で検討した内容を総括し,今後の展 望を述べた.
参考文献
1) 独立行政法人 石油天然ガス・金属鉱物資源機構,リチウム生産技術動向-
最 新 動 向 - , http://mric.jogmec.go.jp/wp-content/uploads/2018/12 /mrseminar2018_06_01.pdf.
2) X. Chen, Z. Zhou, H. E. Karahan, Q. Shao, L. Wei, and Y. Chen, Small, 14, 1–29 (2018).
3) F. Cheng and J. Chen, Chem. Soc. Rev., 41, 2172–2192 (2012).
4) P. Hartmann, C. L. Bender, M. Vra, A. K. Dürr, A. Garsuek, and P.
Adelhelm, Nat. Mater., 12, 228–232 (2012).
5) X. Ren and Y. Wu, J. Am. Chem. Soc., 135, 2923–2926 (2013).
6) F. Bidault, D. J. L. Brett, P. H. Middleton, and N. P. Brandon, J. Power Sources, 187, 39–48 (2009).
7) N. Fujiwara, M. Yao, Z. Siroma, H. Senoh, T. Ioroi, and K. Yasuda, J.
Power Sources, 196, 808–813 (2011).
8) D. R. Dekel, J. Power Sources, 375, 158–169 (2018).
9) M. Hayashi, and T. Shodai, Electrochemistry, 78, 529–539 (2010).
10) Y. J. Wang, N. Zhao, B. Fang, H. Li, X. T. Bi, and H. Wang, Chem. Rev.,
29
115, 3433–3467 (2015).
11) Y. Lee, J. Suntivich, K. J. May, E. E. Perry, and Y. Shao-Horn, J. Phys.
Chem. Lett., 3, 399–404 (2012).
12) K. Gong, F. Du, Z. Xia, M. Durstock, and L. Dai, Science, 323, 760–764 (2009).
13) Y. Zheng, Y. Jiao, L. H. Li, T. Xing, Y. Chen, M. Jaroniec, and S. Z. Qiao, ACS Nano, 8, 5290–5296 (2014).
14) L. Zhang, J. Xiao, H. Wang, and M. Shao, ACS Catal., 7, 7855–7865 (2017).
15) Y. Xue, J. Liu, H. Chen, R. Wang, D. Li, J. Qu, and L. Dai, Angew.
Chemie Int. Ed., 51, 12124–12127 (2012).
16) G. P. Hao, W. C. Li, D. Qian, and A. H. Lu, Adv. Mater., 22, 853–857 (2010).
17) G. L. Chai, and Z. X. Guo, Chem. Sci., 7, 1268–1275 (2016).
18) Z. Zhao, M. Li, L. Zhang, L. Dai, and Z. Xia, Adv. Mater., 27, 6834–6840 (2015).
19) Z. Zhao, C. Lin, J. Tang, and Z. Xia, Nano Energy, 49, 193–199 (2018).
20) J. Zhang, Z. Zhao, Z. Xia, and L. Dai, Nat. Nanotechnol., 10, 444–452 (2015).
21) Y. Jiao, Y. Zheng, M. Jaroniec, and S. Z. Qiao, J. Am. Chem. Soc., 136, 4394–4403 (2014).
22) K. H. Wu, D. W. Wang, D. S. Su, and I. R. Gentle, ChemSusChem, 8, 2772–2788 (2015).
23) D. Guo, R. Shibuya, C. Akiba, S. Saji, T. Kondo, and J. Nakamura, Science, 351, 361–365 (2016).
24) R. Jasinski, Nature, 201, 1212–1213 (1964).
25) H. Jahnke, M. Schönborn, and G. Zimmermann, Top. Curr. Chem., 61, 133–181 (1976).
26) H. Wang, T. Maiyalagan, and X. Wang, ACS Catal., 2, 781–794 (2012).
27) Z. R. Ismagilov, A. E. Shalagina, O. Y. Podyacheva, A. V. Ischenko, L. S.
30
Kibis, A. I. Boronin, Y. A. Chesalov, D. I. Kochubey, A. I. Romanenko, O. B.
Anikeeva, T. I. Buryakov, and E. N. Tkachev, Carbon, 47, 1922–1929 (2009).
28) M. Chisaka, T. Iijima, A. Tomita, T. Yaguchi, and Y. Sakurai, J.
Electrochem. Soc., 157, B1701–B1706 (2010).
29) N. P. Subramanian, X. Li, V. Nallathambi, S. P. Kumaraguru, H. Colon- Mercado, G. Wu, J. W. Lee, and B. N. Popov, J. Power Sources, 188, 9–44 (2009).
30) G. Wu, K. L. More, C. M. Johnston, and P. Zelenay, Science, 332, 443–
447 (2011).
31) W. Ding, Z. Wei, S. Chen, X. Qi, T. Yang, J. Hu, D. Wang, L. J. Wan, S. F.
Alvi, and L. Li, Angew. Chemie - Int. Ed., 52, 11755–11759 (2013).
32) K. Parvez, S. Yang, Y. Hernandez, A. Winter, A. Turchanin, X. Feng, and K. Müllen, ACS Nano, 6, 9541–9550 (2012).
33) S. Liu, H. Zhang, Q. Zhao, X. Zhang, R. Liu, X. Ge, G. Wang, H. Zhao, and W. Cai, Carbon, 106, 74–83 (2016).
34) Z. H. Sheng, L. Shao, J. J. Chen, W. J. Bao, F. Bin Wang, and X. H. Xia, ACS Nano, 5, 4350–4358 (2011).
35) T. C. Nagaiah, S. Kundu, M. Bron, M. Muhler, and W. Schuhmann, Electrochem. commun., 12, 338–341 (2010).
36) Q. Liu, Y. Wang, L. Dai, and J. Yao, Adv. Mater., 28, 3000–3006 (2016).
37) S. Zhu, Z. Chen, B. Li, D. Higgins, H. Wang, H. Li, and Z. Chen, Electrochim. Acta, 56, 5080–5084 (2011).
38) O. Y. Podyacheva, A. S. Lisitsyn, L. S. Kibis, A. I. Stadnichenko, A. I.
Boronin, E. M. Slavinskaya, O. A. Stonkus, S. A. Yashnik, and Z. R.
Ismagilov, Catal. Today, 301, 125–133 (2018).
39) J. Zhu, H. Li, L. Zhong, P. Xiao, X. Xu, X. Yang, Z. Zhao, and J. Li, ACS Catal., 4, 2917–2940 (2014).
40) D. B. Meadowcroft, Nature, 226, 847 (1970).
41) D. Chen, C. Chen, Z. M. Baiyee, Z. Shao, and F. Ciucci, Chem. Rev., 115, 9869–9921 (2015).
31
42) X. Ge, A. Sumboja, D. Wuu, T. An, B. Li, F. W. T. Goh, T. S. A. Hor, Y.
Zong, and Z. Liu, ACS Catal., 5, 4643–4667 (2015).
43) 兵頭健生,博士論文,ペロブスカイト型酸化物の酸素還元活性とガス拡散
型酸素電極への応用,九州大学 (1997).
44) Y. Matsumoto, H. Yoneyama and H. Tamura, J. Electroanal. Chem., 83, 237–243 (1977).
45) Y. Matsumoto, H. Yoneyama and H. Tamura, J. Electroanal. Chem., 79, 319–326 (1977).
46) J. Suntivich, H. A. Gasteiger, N. Yabuuchi, H. Nakanishi, J. B.
Goodenough, and Y. Shao-Horn, Nat. Chem., 3, 546–550 (2011).
47) 湯浅雅賀,博士論文,ランタン―遷移金属系ペロブスカイト型酸化物微 粒子を用いたガス拡散型酸素電極の酸素還元・発生特性に関する研究,九州大 学 (2011).
48) Y. Teraoka, S. Nanri, I. Moriguchi, S. Kagawa, K. Shimanoe, and N.
Yamazoe, Chem. Lett., 29, 1202–1203 (2005).
49) M. Yuasa, G. Sakai, K. Shimanoe, Y. Teraoka, and N. Yamazoe, J.
Electrochem. Soc., 151, A1477–A1482 (2004).
50) M. Yuasa, K. Shimanoe, Y. Teraoka, and N. Yamazoe, Catal. Today, 126, 313–319 (2007).
51) M. Watanabe, H. Sei, and P. Stonehart, J. Electroanal. Chem., 261, 375–
387 (1989).
52) R. R. Retamal Marín, F. Babick, and M. Stintz, Powder Technol., 318, 451–458 (2017).
53) 猪ノ木 雅裕,粉砕,57,66–73 (2014).
54) T. Ogi, R. Zulhijah, T. Iwaki, and K. Okuyama, KONA Powder Part. J., 2017, 3–23 (2017).
55) R. Zulhijah, A. Suhendi, K. Yoshimi, C. W. Kartikowati, T. Ogi, T. Iwaki, and K. Okuyama, Langmuir, 31, 6011–6019 (2015).
56) M. Inkyo, Y. Tokunaga, T. Tahara, T. Iwaki, F. Iskandar, C. J. Hogan, and K. Okuyama, Ind. Eng. Chem. Res., 47, 2597–2604 (2008).
57) C. Schilde, C. Mages-Sauter, A. Kwade, and H. P. Schuchmann, Powder
32
Technol., 207, 353–361 (2011).
58) M. Matsuoka, J. Tatami, T. Wakihara, K. Komeya, and T. Meguro, J.
Asian Ceram. Soc., 2, 199–203 (2014).
59) Y. J. Wang, B. Fang, H. Li, X. T. Bi, and H. Wang, Prog. Mater. Sci., 82, 445–498 (2016).
60) Antolini, E. Appl. Catal. B Environ. 88, 1–24 (2008).
61) S. Tang, G. Sun, J. Qi, S. Sun, J. Guo, Q. Xin, and G. M. Haarberg, Chinese J. Catal., 31, 12–17 (2010).
62) S. Zhang, S. Chen, J. Power Sources, 240, 60–65 (2013).
63) J. Zhu, G. He, L. Liangshu, Q. Wan, P. Shen, Electrochim. Acta, 158, 374–382 (2015).
64) R. Chen, J. Yan, Y. Liu, and J. Li, J. Phys. Chem. C, 119, 8032–8037 (2015).
65) E. Fabbri, R. Mohamed, P. Levecque, O. Conrad, R. Kötz, and T. J.
Schmidt, ACS Catal., 4, 1061–1070 (2014).
66) I. A. Khan, A. Badshah, and M. A. Nadeem, Catal. Commun., 99, 10–14 (2017).
67) 三浦 則雄, 清水 陽一, 山添 昇, 清山 哲郎,日本化学会誌,4,644–650 (1985).
68) T. Poux, F. S. Napolskiy, T. Dintzer, G. Kéranguéven, S. Y. Istomin, G. A.
Tsirlina, E. V. Antipov, and E. R. Savinova, Catal. Today, 189, 83–92 (2012).
69) T. Poux, A. Bonnefont, G. Kéranguéven, G. A. Tsirlina, and E. R.
Savinova, ChemPhysChem, 15, 2108–2120 (2014).
70) B. Avasarala, T. Murray, W. Li, and P. Haldar, J. Mater. Chem., 19, 1803–1805 (2009)
71) A. Honji, T. Marl, Y. Hishinuma, and K. Kurita, J. Electrochem. Soc., 135, 917–918 (1988).
72) D. W. McKee, A. J. Scarpellino, I. F. Danzig, and M. S. Pak, J.
Electrochem. Soc., 116, 562–568 (1969).
73) Y. Hara, N. Minami, H. Matsumoto, and H. Itagaki, Appl. Catal. A Gen., 332, 289–296 (2007).
74) M. K. Jeon, H. Daimon, K. R. Lee, A. Nakahara, and S. I. Woo,
33
Electrochem. commun., 9, 2692–2695 (2007).
75) R. Ganesan and J. S. Lee, Angew. Chemie Int. Ed., 44, 6557–6560 (2005).
76) H. Laborde, J. M. Léger, and C. Lamy, J. Appl. Electrochem., 24, 1019–
1027 (1994).
77) L. M. Huang, W. R. Tang, and T. C. Wen, J. Power Sources, 164, 519–526 (2007).
78) Q. Yang, Y. Wang, H. Nakano, and S. Kuwabata, Polym. Adv. Technol., 16, 759–763 (2005).
79) K. H. Xue, C. X. Cai, H. Yang, Y. M. Zhou, S. G. Sun, S. P. Chen, and G.
Xu, J. Power Sources, 75, 207–213 (1998).
80) J. Li and X. Lin, J. Electrochem. Soc., 154, 1074–1079 (2007).
81) T. Ioroi, Z. Siroma, N. Fujiwara, S. I. Yamazaki, and K. Yasuda, Electrochem. commun., 7, 183–188 (2005).
82) S. Y. Huang, P. Ganesan, S. Park, and B. N. Popov, J. Am. Chem. Soc., 131, 13898–13899 (2009).
83) W. S. Baker, J. J. Pietron, M. E. Teliska, P. J. Bouwman, D. E. Ramaker, and K. E. Swider-Lyons, J. Electrochem. Soc., 153, 1702–1707 (2006).
84) K. W. Park, J. H. Choi, K. S. Ahn, and Y. E. Sung, J. Phys. Chem. B, 108, 5989–5994 (2004).
85) R. Mohamed, E. Fabbri, P. Levecque, R. Kötz, T. J. Schmidt, and O.
Conrad, ECS Trans., 58, 9–18 (2014).
86) N. Staud, H. Sokol, and P. N. Ross, J. Electrochem. Soc., 136, 3570–3576 (1989).
87) 衣本 太郎, 江藤 誠, 小野 晃平, 松岡 美紀, 津村 朋樹, 豊田 昌宏, 炭素, 280, 207–210 (2017).
34
第 2 章 窒素ドープポーラスカーボン触媒の調製条件の検討
2.1 緒言 2.2 実験方法
2.2.1 窒素ドープポーラスカーボンの調製 2.2.2 物性評価
2.3 電子顕微鏡観察および結晶構造解析 2.4 細孔構造解析
2.5 化学結合状態解析 2.6 本章のまとめ 参考文献
35
2.1 緒言
窒素ドープカーボンは酸素還元反応,酸素発生反応や二酸化炭素還元反応を 促進させる電気化学触媒として作用する.また,窒素ドープポーラスカーボンは 大きな静電容量をもつことから,電気二重層キャパシタとしての応用が期待さ れている1,2).調製条件が窒素ドープカーボンに与える影響を明らかにすること は,これらの用途において最適な材料を得るためには欠かせない.本研究では熱 処理法により,カーボン材料表面に窒素をドープした.この表面修飾的アプロー チによる手法は,簡便であり,スケールアップも容易である.しかし,特定の窒 素種のドーピングおよびその量を制御することは困難であり,また,熱処理は形 態にも大きな影響を与え,比表面積や細孔容積を変化させる.本章では,炭素源 として使用したカーボンブラックの前処理および熱処理が,得られる窒素ドー プポーラスカーボンの窒素種や細孔構造に与える影響を評価した.
2.2 実験方法
2.2.1 窒素ドープポーラスカーボンの調製
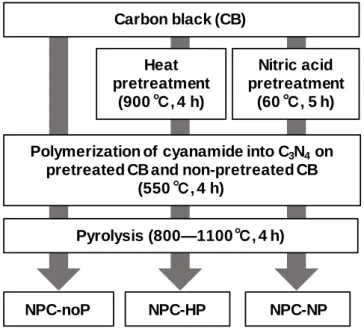
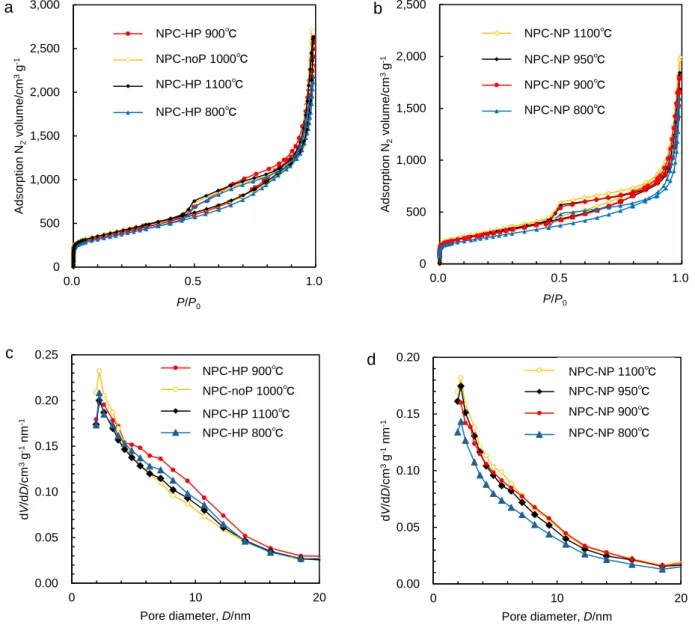
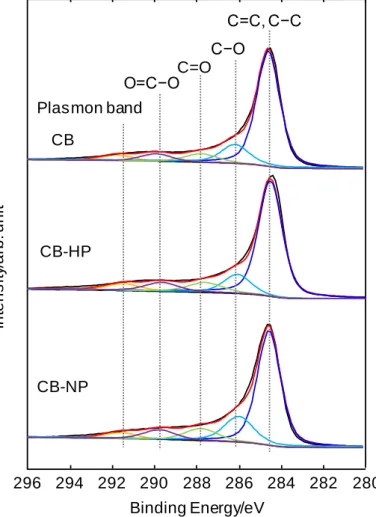
窒素ドープポーラスカーボンの合成フローチャートを Fig. 2.1 に示す.市販 のカーボンブラック(Carbon black,CB;EC600JD,ライオン)に,窒素フロ ー中での熱処理(900℃,4時間)もしくは70 wt%濃硝酸(和光純薬)中で60℃,
5時間の酸処理の二種類の異なる前処理を施し,それぞれ前処理を施したCBを
CB-HP,CB-NPと示す.前処理をしたCBもしくは無処理のCBを炭素源とし,
シアナミド水溶液に含浸させた後,蒸発乾固して得た前駆体を窒素流通下で,ま
ず550℃で 4時間保持してカーボンナイトライド(C3N4)をCB 表面で重合さ
せ3,4),続けて800—1100℃で4時間,熱処理を行ってC3N4を完全に分解させて 窒素ドープポーラスカーボンを得た.以後,無処理のCB,CB-HP,CB-NPを 炭素源として合成した窒素ドープポーラスカーボンをそれぞれ NPC-noP,
NPC-HP,NPC-NPと表す.熱処理は管状炉を使用して石英管(内径:19.5 mm,
外径:22.5 mm)中で窒素を300 mL min−1でフローさせながら行った.