抗菌活性物質Amycolamicinの全合成研究
著者
目黒 康洋
学位授与機関
Tohoku University
学位授与番号
11301甲第19331号
抗菌活性物質
Amycolamicin の全合成研究
東北大学大学院農学研究科
生物産業創成科学専攻
目黒 康洋
指導教員
桑原 重文 教授
略 号 Ac acetyl AMM amycolamicin aq aqueous Ar aryl Bn benzyl
brsm based on recovered starting material Bu butyl
CAMHB cation-adjusted Mueller Hinton broth CBS Corey- Bakshi- Shibata
CD1d cluster of differentation 1d CSA 10-camphorsulfonic acid
DBU 1,8-diazabicyclo [5.4.0] undec-7-ene DDQ 2,3-dichloro-5,6-dicyano-p-benzoquinone DIAD diisopropyl azodicarboxylate
DIBAL-H diisobutylaluminium hydride DIPT diisopropyl tartrate
DMAP N,N-dimethyl-4-aminopyridine DMF N,N-dimethylformamide
DMP Dess- Martin periodinane DMSO dimethyl sulfoxide
DNA deoxyribonucleic acid dr diastereomer
EDC 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide ee enantiomeric excess
ent enantiomer EI electron impact Et ethyl
FAB fast atom bombardment Gyr gyrase
HOBt 1-hydroxybenzotriazole
HRMS high-resolution mass spectrometer HWE Horner- Wadsworth- Emmons LHMDS lithium bis(trimethylsilyl)amide Me methyl
MIC minimum inhibitory concentration
MRSA methicillin-resistant Staphylococcus aureus MS4A molecular sieve 4A
MSSA methicillin-susceptible Staphylococcus aureus MTPA α-methoxy-α-(trifluoromethyl)phenylacetyl NaHMDS sodium hexamethyldisilazide
Nc nosyl
NCS N-chlorosuccinimide NHK Nozaki- Hiyama- Kishi NMR nuclear magnetic resonance
NOESY nuclear Overhauser effect spectroscopy n.r. no reaction Nu nucleophile o- ortho- Ph phenyl Piv pivaloyl PMB p-methoxybenzyl ppm parts per million
PPTS pyridinium p-toluenesulfonate Pr propyl
RNA ribonucleic acid rt room temperature SM starting material sp species
TBAB tetrabutylammonium Bromide TBAF tetrabutylammonium Fluoride TBHP t-butyl hydroperoxide
TBS t-butyldimethylsilyl
TEMPO 2,2,6,6-tetramethylpiperidine 1-oxyl
t-, tert tertiary
Tf trifluoromethanesulfon TFA trifluoroacetic acid THF tetrahydrofuran TIPS triisopropylsilyl TLC thin-layer chromatography TMS trimethyl silyl TPP thiaminepyrophosphate Ts p-toluenesulfonyl
目 次 第 1 章 研 究 の 背 景 と 目 的 1 1.1 医 薬 品 の 創 製 1 1.2 有 機 合 成 化 学 を 基 盤 と す る 医 薬 品 開 発 2 1.2.1 有 機 化 合 物 の 合 成 2 1.2.2 グ リ コ シ ル 化 に 関 し て 3 1.2.3 合 成 創 薬 4 1.3 抗 生 物 質 5 1.3.1 抗 生 物 質 の 分 類 5 1.3.2 薬 剤 耐 性 菌 の 出 現 7 1.4 本 研 究 の 目 的 8 第 2 章 amycolamicin の 全 合 成 研 究 9 2.1 amycolamicin の 発 見 と 展 開 9 2.2 逆 合 成 解 析 12 2.3 DE ユ ニ ッ ト の 合 成 - Introduction 13 2.3.1 ピ ロ ー ル 環 部 分 の 合 成 15 2.3.2 DE ユ ニ ッ ト の 合 成 16 2.3.2.1 第 一 世 代 合 成 の 合 成 戦 略 16 2.3.2.2 DE ユ ニ ッ ト の 第 一 世 代 合 成 17 2.3.2.3 第 二 世 代 合 成 の 合 成 戦 略 21 2.3.2.4 DE ユ ニ ッ ト の 第 二 世 代 合 成 21 2.3.3 小 括 28 2.4 C ユ ニ ッ ト の 合 成 - Introduction 29 2.4.1 第 一 世 代 合 成 の 合 成 戦 略 30 2.4.2 C ユ ニ ッ ト の 第 一 世 代 合 成 31 2.4.3 第 二 世 代 合 成 の 合 成 戦 略 32 2.4.4 C ユ ニ ッ ト の 第 二 世 代 合 成 32 2.4.5 小 括 42 2.5 A ユ ニ ッ ト の 合 成 - Introduction 43 2.5.1 合 成 戦 略 44 2.5.2 A ユ ニ ッ ト の 合 成 45 2.5.3 合 成 中 間 体 159 の 立 体 化 学 49 2.5.4 A ユ ニ ッ ト の コ ン ホ メ ー シ ョ ン 50 2.5.5 小 括 51 2.6 各 ユ ニ ッ ト の 収 束 的 連 結 に よ る AMM の 合 成 に 向 け て 52 2.6.1 DE ユ ニ ッ ト の 改 良 合 成 52
2.6.2 C ユ ニ ッ ト の 二 炭 素 増 炭 53 2.6.3 A ユ ニ ッ ト の N-グ リ コ シ ル 化 54 2.6.4 187 と 16 の コ ン ホ メ ー シ ョ ン 57 2.6.5 AMM の 合 成 に 向 け た 各 ユ ニ ッ ト の 連 結 59 2.6.6 小 括 60 第 3 章 実 験 の 部 61 3.1 General information 61 3.1.1 2.3 に 関 す る 実 験 62 3.1.2 2.4 に 関 す る 実 験 76 3.1.3 2.5 に 関 す る 実 験 87 3.1.4 2.6 に 関 す る 実 験 90 3.2 NMR 97 第 4 章 総 括 151 4.1 参 考 文 献 151 4.2 結 言 156 4.3 研 究 生 活 を 振 り 返 っ て 157 4.4 謝 辞 158
1 第一章 研究の背景と目的 1.1 医薬品の創製 我々人類はその時代ごとに常に病の脅威にさらされてきた。それに対抗する ため、その時代の最高の技術と努力により薬を開発し、病を乗り越えてきた。「臨 床試験」による治療効果を評価する考え方が登場したのは18 世紀中ごろであり、 現在では動物試験などを経る厳密な指標が存在する1)。臨床試験という概念の登 場と時期を同じくして化合物による近代医薬として登場したのがモルヒネであ る。モルヒネは植物であるケシの実から抽出されるアヘンの有効成分であり、 強力な鎮痛剤としても利用されてきた。アヘンを数工程の化学的処理により精 製した後、結晶化によって純粋なモルヒネを得たことにより特定の化合物が薬 理活性を有することが実証された。そして、20 世紀になると微生物の二次代謝 産物を利用することで天然物由来の医薬品の開発が劇的に進歩した。二次代謝 産物は必ずしも生体活動には必要とされない化合物であり、なぜ生産されるの か本質的に不明な化合物が多い。抗生物質として利用される二次代謝産物にお いては、微生物が生存競争に勝ち残るため生産していると考えられている。一 方の微生物が他方の微生物に対して拮抗作用を示すことはL. Pasteur の時代に 観測されていた。そして、微生物由来として世界初の抗生物質であるペニシリ ンがA. Fleming によって発見された。その後、S. A. Waksman によって他方の 微生物の生育を阻害するような物質を「抗生物質」と呼ぶことが提唱された。 微生物の二次代謝産物を利用するメリットとして培養法による大量供給に加え、 ペニシリンのβ-ラクタム構造に見る新規構造およびこれまでに想像もしなかっ た作用機序の発見も挙げられる。さらに、β-ラクタム構造の発見により、新た な抗菌剤としての作用機序が見出されることになり、多様なβ-ラクタム系抗生 物質の開発に繋がった。このような作用機序の解明は医薬品開発において非常 に重要である。そもそも、ペニシリンのような低分子医薬品として機能する化 合物が標的とするのは特定のタンパク質である。つまり、「鍵穴」となる特定の タンパク質に作用することでそのタンパク質の機能を調整するような化合物 (鍵)が医薬品化合物となる。ペニシリンの場合は細菌のペプチドグリカン生合成 酵素に作用することで抗菌作用を示し、動物にはこの酵素が存在しないために 毒性が低い2)。
2 β-ラクタム構造の作用機序の解明によりセファロスポリン C などβ-ラクタ ム系抗生物質が多く開発されることになった(Figure 1-1)3-6)。医薬品の開発は 1) 標的タンパク質の同定、2) 薬剤候補物質の探索・選抜、3) 臨床試験を経て 行われることが多い。このようにして開発された医薬品が人類と感染症の戦い に大きく貢献してきた。その一因として,自然界に普通に存在し培養が容易な 微生物を取り尽くしつつあることが挙げられよう。さらに,培養が容易であり, かつ遺伝子にも化合物の情報がコードされているにも関わらず,何らかの理由 で微生物による化合物生産が行われなくなる場合もある。現在の培養法や遺伝 子工学の進展により,難培養微生物や化合物生産をやめてしまった微生物から も新たな医薬品候補化合物が発見される可能性はある。また,天然から微生物 を取り尽くしつつあるとは言え,実際のところ我々は足元に存在する微生物の 数さえ正確に把握できていない。そのため,天然には想像以上の種類の微生物 が存在しており,今後も医薬品化合物の宝庫となり続ける可能性はある。しか し,現実として発酵法による医薬品開発は減少傾向にあるため,微生物の発酵 法とは異なるアプローチを用いて医薬品開発を行う必要がある。そこで有用と なるのが有機合成化学を駆使した合成創薬である。最近は計算化学を駆使した 医薬品における最適構造の探索が行われるようになったが、依然として天然物 由来の医薬品は数多く存在する。そして、天然物の全合成研究は医薬品化合物 の供給、天然物の構造決定、生合成経路の解明や新規有機化学反応の発見に大 きく貢献しており、産業および学術の両面において重要な役割を果たしている。 1.2 有機合成化学を基盤とする医薬品開発 1.2.1 有機化合物の合成 以前の有機化合物の定義は生命活動によって生産される化合物であった。そ の後、F. Wöhler が尿素を合成したことで有機化合物の定義が改定され、炭素の 酸化物を除く、炭素を含む全ての化合物が有機化合物として定義された。それ から有機化合物を用いた多様な合成法が開発されてきた。天然物合成において -ラクタム系抗生物質 ベンジルペニシリン (1) セファロスポリンC (2) Figure 1-1 β-ラクタム系抗生物質
3 はこのように見出された反応のデータベースから最適な反応を探索して利用す る手法が一般的である。天然物合成ではより安価で単純な構造を有する出発原 料から合成を目指す。そのため、天然物の基本構造を構築するためには炭素- 炭素結合の形成が重要となる。炭素-炭素結合形成反応においてグリニャール 反応は代表的反応の一つであり、開発から一世紀経た現在でも利用例の多い重 要な反応である。 その後、グラブス触媒による炭素-炭素二重結合の連結や7)、高分子合成では チーグラー・ナッタ触媒による有用な重合反応が報告された(Figure 1-2)。そし て、多様な手法による炭素-炭素結合形成としてクロスカップリングが挙げら れる。これまでに多くの手法が開発されたがいくつか例を挙げて説明する。ま ず、有機亜鉛試薬と有機ハロゲン化物をパラジウム触媒もしくはニッケル触媒 存在下で反応させることにより炭素-炭素結合を形成する根岸カップリングが 初めて報告された。その後、パラジウム触媒存在下にてハロゲン化ビニルとオ レフィンを連結する溝呂木-ヘック反応や有機ホウ素化合物とハロゲン化アリ ールをカップリングさせる鈴木-宮浦反応などが登場した。通常、グリニャー ル試薬のようにカップリングに必要な有機金属化合物は水や空気に不安定であ る。一方、鈴木-宮浦反応では有機ホウ素化合物は水や空気に安定であり、取 り扱いが容易になっている。また、園頭カップリング反応のような末端アルキ ンとハロゲン化アリールを連結する有用な反応も開発されている。この他にも アルドール反応やウィッティヒ反応など様々な有機化合物の増炭方法が開発さ れることで以前では困難であった天然物合成を可能とした。 1.2.2 グリコシル化反応に関して 医薬品として利用される天然有機化合物にはオリゴ糖やグリコシド結合を有 する配糖体が多く存在する。このような糖含有化合物の生物活性ではグリコシ ド結合の立体選択性が重要となる場合がある。そのため、フィッシャーグリコ シル化が開発されてから現在に至るまで多くのグリコシル化反応が開発されて Grubbs 1stgeneration n TiCl3, Et3Al m a) b) Figure 1-2 a) グラブス触媒による閉環メタセシス反応、b) チーグラ ー・ナッタ触媒による重合反応
4 きた。最初のグリコシル化反応はH. E. Fischer により報告された手法で、塩酸 存在下にてアルコールと糖を加熱することによりグリコシド体を得る方である (Figure 1-3)。その後、中間体としてハロゲン化糖、トリクロロイミデート糖や アンヒドロ糖などを利用した様々なグリコシル化反応が報告された8-16)。長いグ リコシル化反応の開発を経て、グリコシル化反応は完成しつつあるが、まだデ オキシ糖への適応が困難であることや塩基性条件下での有用なグリコシル化が 少ないという課題も存在する。そのため、グリコシル化反応の開発を持続する 必要があり、今後の発展も期待したい。 1.2.3 合成創薬 アスピリン3 は医薬品開発において人類最大の成功例と言える(Figure 1-4)1)。 単純な構造で安価な原料から合成可能でありかつ約一世紀を経た現在でも鎮痛 剤としてベストセラーであり続けている。ヤナギの樹皮から単離されるアスピ リンの原料であるサリチル酸は副作用として胃腸障害を引き起こす。そこで、 バイエル社により無水酢酸を用いたサリチル酸のアセチル化という非常に容易 な変換によって得られるアスピリンが副作用を抑制した有効な鎮痛剤として見 出された。この合成によるアスピリンの開発が医薬品開発における有機合成化 学の有用性を示しており、現代合成創薬の扉を開いたと考える。その他の例と して、抗がん剤であるエクチナサイジン 743 はホヤから単離されるがごく微量 しか得られないため、微生物の培養から大量に得られるサフラシン B から半合 成によって標品供給されている17-18)。また、アベルメクチンB1aのC22 と C23 の炭素-炭素二重結合を還元することで得られるイベルメクチン B1a (15)がよ り有効な抗寄生虫作用を示すことも判明している。このように天然由来の化合 Figure 1-3 グリコシル化反応における様々な中間体 ROH, HCl Fischer glycosidation X = F, Cl, Br, I R’ = H, Ph X = F, Cl
Halide Thioglycoside Sulfoxide Sulfone Glycal
1,2-anhydro sugar Glycosyl Bunte salt Imidate Phosphate Acetate
5 物から分子構造を意図的に変換して理想的な構造へ最適化することで医薬品と しての有効性を向上している例も多く見られる。 1.3 抗生物質 1.3.1 抗生物質の分類 Fleming によって世界初の抗生物質であるペニシリンが見出され、Waksman によって抗生物質の定義が提唱されてから多様な抗生物質が開発されてきた。 以下のように抗生物質は作用機序により(a)から(d)の四種類に大分される19)。 (a) 細胞壁合成阻害 ペニシリンは細胞壁合成を阻害することで選択的に細菌に作用する。このよう な作用機序を示す化合物としてβ-ラクタム系抗生物質や MRSA を含む薬剤耐 性菌に対して有効性を示すバンコマイシンなどが挙げられる(Figure 1-5)。 Figure 1-5 バンコマイシンの構造 6 Figure 1-4 (左)アスピリンの構造、(中央)エクチナサイジン 743 の構造 (右)イベルメクチン B1aの構造 4 22 5 3
6 (b) 細胞膜機能阻害 細胞膜に作用することで細胞膜内のイオン濃度を撹乱することで細胞膜機能 の阻害もしくは障害を与えることにより抗菌作用を示す抗生物質の一群である。 モネンシンに代表されるイオノホア抗生物質は環状構造を形成しており、その 内部に金属イオンを取り込むことで金属イオン-イオノホア複合体を形成する (Figure 1-6)。この複合体が細胞膜での拡散や細胞膜に埋め込まれ通路を形成す ることで細胞内外のイオン濃度を撹乱して細菌を死に至らしめる。 (c) タンパク質合成阻害 タンパク質合成の生合成経路である開始、伸長、終了のいずれかの段階を阻害 することで抗菌作用を示す。選択性は真核生物と原核生物のリボソームの構造 の違いによって現れる。アミノグリコシド系抗生物質としてストレプトマイシ ンが代表例として存在する。ストレプトマイシンは Waksman によって放線菌 から単離され結核の治療薬として有効であることが判明した。その後、ストレ プトマイシンの副作用を改善したカナマイシン20 が発見された(Figure 1-7)。 (d) 核酸合成阻害 核酸合成の阻害活性を示す化合物群であり、核酸合成を阻害することから抗 がん剤として利用される化合物も多数く存在する。構造は多様でありペプチド 鎖を含むアクチノマシンD、アントラサイクリン系のアドリアマイシン(Figure 1-8)、糖ペプチド構造を有するブレオマイシン、ポリケチド生合成によって合成 されるアフラトキシンなどが挙げられる。それぞれ、作用機序は異なるがアド リアマイシンの場合では DNA の塩基対の間にインターカレーションすること でDNA に結合し、RNA ポリメラーゼの働きを阻害することで抗菌作用を示す。 8 Figure 1-7 カナマイシンの構造 Figure 1-6 モネンシンの構造 7
7 1.3.2 薬剤耐性菌の出現 これまで示したように抗生物質はペニシリンの発見からありとあらゆる種類 の抗生物質が見出された。副作用の改善やより高い抗菌作用を指向した改良を 目的として多くの抗生物質が開発された。そして、特に課題であるのが薬剤耐 性菌の出現である。ペニシリンが発見された当初から既に薬剤耐性菌の出現は 予測されていた1)。細菌の増殖は非常に速く、個体数が急激に増加するため、そ の中から突然変異による薬剤耐菌が現れやすい。また、最近の抗生物質の処方 として、使用頻度の急激な増加や単一の抗生物質の使用によっても薬剤耐性菌 の出現確立は高くなる。ペニシリンにおける細菌の薬剤耐性獲得のメカニズム は細菌のペニシリン結合部位の変異やペニシリンの分解酵素であるペニシリナ ーゼの発現である。そこで、ペニシリンの場合とは全く異なる作用機序を有す る抗生物質の開発が急務となった。この問題を解決する切り札として開発され たのがバンコマイシンであった。バンコマイシンの場合はペニシリンのような ペプチドグリカン生合成酵素の阻害ではなく、ペプチドグリカン前駆体の合成 を直接阻害することで抗菌作用を示す。ペプチドグリカンの合成を直接阻害で きるため、細菌は突然変異による薬剤耐性の獲得が困難となる。そのため、バ ンコマイシンの開発により感染症の恐怖を克服したかに思われたが、VRE や VRSA が出現し、現在に至るまで薬剤耐性菌と人類との戦いは続いている。薬 剤耐性菌が問題となるのは体力の低下した患者に対しては致死的な感染症を引 き起こす可能性があることである。そのため、新たな抗生物質の開発は緊喫の 課題である。 Figure 1-8 アドリアマイシンの構造 9
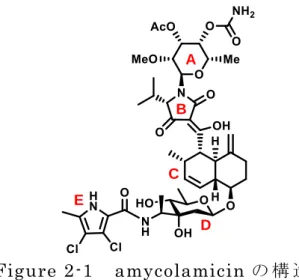
8 1.4 本研究の目的 本研究では,強力な抗菌活性と前例のない特異なハイブリッド型化学構造を 有 し , 新 し い カ テ ゴ リ ー の 抗 菌 薬 開 発 の 起 点 と な り 得 る 天 然 物 amycolamicin(AMM, 10)について,初の全合成と構造活性相関研究を実施し,画 期的な感染症治療薬の創製に繋げることを目指す。合成戦略としては,AMM の C ユニットと DE ユニットを調製した後に連結して CDE ユニットに導き,B ユ ニットを形成させながら別途調製した A ユニットと連結することで分子全体を 組み上げる収束的合成法を選択し,全合成の効率化を図る。 AMM の構造活性相関については,AMM の D 環部 2 級ヒドロキシ基のアセ チルエステル体と E 環部の 2 つの塩素の水素置換体について抗菌活性が調べら れたのみであるが(両誘導体ともに大幅な活性の減弱),本収束的合成法を用い れば,多彩な化合物ライブラリーの構築が可能となるだけでなく,全合成の過 程で得られる個々のユニットまたは複数ユニットの連結体について,抗菌活性 以外の未知の生理作用の探索も可能となる(実際に,構造決定の過程で得られ たDE ユニット単独では,細胞毒性を示すことが報告されている)。 Figure 1-9 amycolamicin の構造 10 B D E A C
9 第 2 章 amycolamicin の 全 合 成 研 究 2.1 amycolamicin の 発 見 と 展 開 感 染 症 治 療 の 研 究 現 場 で は 薬 剤 耐 性 菌 の 出 現 に よ り 、常 に 新 た な 脅 威 を 突 き 付 け ら れ て い る 。そ の た め 、交 差 耐 性 の 生 じ に く い 新 規 な 構 造 と 作 用 機 序 を 有 す る 、新 し い カ テ ゴ リ ー の 抗 生 物 質 の 起 点 と 成 り 得 る 薬 剤 の 開 発 が 急 務 と な っ て い る 。amycolamicin(AMM, 10)は 希 少 放 線 菌 Amycolatopsis sp. MK575-fF520-22)及 び Kibdelosporangium sp. MA739523)が 生 産 す る 抗 生 物 質 で あ る 。
AMM は 、 2 つ の 新 規 単 糖 ユ ニ ッ ト amykitanose(A)及 び amycolose(D)に 加 え
て 、テ ト ラ ミ ン 酸(B)、trans-デ カ リ ン (C)、ピ ロ ー ル カ ル ボ ン 酸 (D)が 連 結 し た 過 去 に 類 例 の な い 全 く 新 規 な ハ イ ブ リ ッ ド 型 化 学 構 造 を 有 す る と と も に 、 MRSA や VRE を 始 め と し た 極 め て 広 範 な グ ラ ム 陽 性 ・ 陰 性 の 薬 剤 耐 性 菌 に 対 し て 強 力 な 抗 菌 作 用 を 示 す(MICs 0.125- 1 μg/mL)。 Hydrophobic pocket D76/81 G80/85 T168/173 E53/58 R79/84 G103/109 H86/Q91 H101/107 amycolamicin H2O
Figure 2-2 S. aureus GyrB subunit と amycolamicin の
共 結 晶 X 線 結 晶 構 造 B D E A C Figure 2-1 amycolamicin の 構 造
10
AMM の 標 的 タ ン パ ク 質 は 細 菌 の DNA 合 成 必 須 酵 素 で あ る DNA gyrase の GyrB subunit 及 び topoisomerase IV の ParE subunit で あ る 。 共 結 晶 X 線
結 晶 構 造 か ら 、AMM の GyrB 及 び ParE に 対 す る 結 合 様 式 は ほ ぼ 同 じ で あ る
こ と が 判 明 し て い る(Figure 2-2)24)。 即 ち 、 各 ユ ニ ッ ト の 内 、 ピ ロ ー ル カ ル ボ
ン 酸 部 位(E ユ ニ ッ ト ) は 標 的 タ ン パ ク 質 の 疎 水 性 ポ ケ ッ ト に 深 く 入 り 込 む と
と も に 、N と O は 水 分 子 を 含 む 水 素 結 合 ネ ッ ト ワ ー ク を 形 成 し て い る 。ま た 、
テ ト ラ ミ ン 酸 部 位(B ユ ニ ッ ト ) の 2 つ の カ ル ボ ニ ル 基 は 親 水 性 ア ミ ノ 酸 残 基
に よ り 両 側 か ら 強 固 に 挟 み 込 ま れ て い る 。さ ら に 、AMM は 同 じ 標 的 を 持 つ 既
存 の ク マ リ ン 系 抗 生 物 質 と は 全 く 異 な る 新 規 な 結 合 様 式(dual arm, U-shaped
binding) を 持 つ こ と が 判 明 し た 。 特 に 、 ク マ リ ン 系 抗 生 物 質 で あ る novobiocin(11)や clorobiocin(12)の 場 合 は 、 GyrB subunit の D89 と の 結 合 が
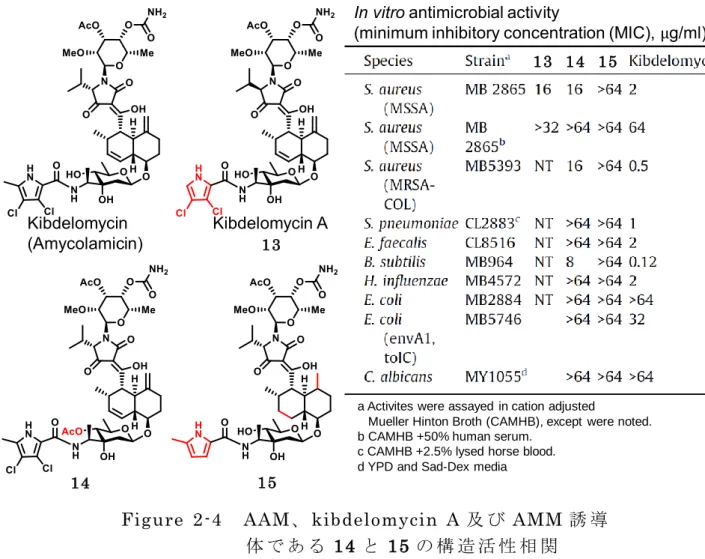
抗 菌 作 用 に 必 須 と な る の に 対 し 、AMM の 場 合 は D89 と の 結 合 は 形 成 さ れ な い (Figure 2-3)。 こ の よ う な 新 規 な 結 合 様 式 は 、 既 存 の 抗 菌 薬 に 交 差 耐 性 を 示 さ ず 、耐 性 発 現 頻 度 も 極 め て 低 い と い う AMM の 良 好 な 薬 理 特 性 の 根 拠 と な っ て い る 24)。 ま た 、AMM は ヒ ト の topoisomerase II を 阻 害 せ ず 、マ ウ ス に 対 す る 急 性 毒 性 も 無 い こ と か ら 、構 造 的 に も 作 用 機 序 的 に も 全 く 新 規 な 抗 生 物 質 リ ー ド と し て 注 目 を 集 め て い る 2 5-28)。こ れ ま で の AMM に 関 す る 研 究 は 構 造 解 析 、抗 菌 活 性 と タ ン パ ク 質 と の 相 互 作 用 に 関 し て 報 告 さ れ て い る 。AMM の 単 離 者 で あ る S.B.Singh ら に よ り AMM の 構 造 改 変 に よ る 構 造 活 性 相 関 研 究 が 実 施 さ れ た (Figure 2-4)29)。AMM の D ユ ニ ッ ト の ア セ チ ル 誘 導 体 14 及 び E ユ ニ ッ ト の Novobiocin Streptomyces caeruleus 1955-56, Merck Kibdelomycin (Amycolamicin) Novobiocin Clorobiocin Streptomyces roseochromogenes 1968, Rhone-Poulenc
Figure 2-3 S. aureus GyrB subunit と amycolamicin 及 び Novobiocin の 共 結 晶 X 線 結 晶 構 造
11
11 還 元 体 15 を そ れ ぞ れ 合 成 し 、天 然 物 由 来 で あ る AMM と kibdelomycin A (13) と の 活 性 評 価 の 比 較 が 行 わ れ た 。そ の 結 果 、合 成 化 合 物 で あ る 14 及 び 15 の 活 性 は AMM に 比 べ て 低 い 。つ ま り 、amycolose の ヒ ド ロ キ シ 基 は 無 保 護 で あ る こ と と ピ ロ ー ル 環 部 分 に 塩 素 置 換 基 が 存 在 す る 方 が 抗 菌 作 用 は 高 い こ と を 示 し て い る 。ま た 、13 に 関 し て も AMM よ り 抗 菌 活 性 が 低 い こ と か ら 、ピ ロ ー ル 環 部 分 に メ チ ル 基 が 必 要 で あ る こ と を 示 唆 し て い る 。現 在 の と こ ろ 構 造 活 性 相 関 研 究 は 以 上 で 記 述 し た 内 容 の み で あ り 、 各 ユ ニ ッ ト の 構 造 活 性 相 関 を 含 め 、 有 機 合 成 的 手 法 に よ る 新 薬 創 製 へ の ア プ ロ ー チ が 必 要 と な っ て い る 。 μ
a Activites were assayed in cation adjusted
Mueller Hinton Broth (CAMHB), except were noted. b CAMHB +50% human serum.
c CAMHB +2.5% lysed horse blood. d YPD and Sad-Dex media
Figure 2-4 AAM、 kibdelomycin A 及 び AMM 誘 導
体 で あ る 14 と 15 の 構 造 活 性 相 関
13
15 14
12 2.2 逆 合 成 解 析 Scheme 2-1 に 示 す よ う に ,AMM(10)は A ユ ニ ッ ト 18 の L-バ リ ン 誘 導 体 に よ る N-グ リ コ シ ル 化 体 16 と CDE ユ ニ ッ ト 20 の 二 炭 素 増 炭 誘 導 体 17 と を ,B ユ ニ ッ ト を 形 成 さ せ な が ら 連 結 す る こ と で 合 成 可 能 で あ る と 考 え た 。18 は L-フ コ ー ス 19 か ら 2 位 の 立 体 化 学 を 反 転 さ せ る こ と で 調 製 で き る と 思 わ れ る 。CDE ユ ニ ッ ト 20 は DE ユ ニ ッ ト 21 と C ユ ニ ッ ト 22 の グ リ コ シ ル 化 で 得 る 。trans-デ カ リ ン 誘 導 体 22 は , 23 の 分 子 内 Diels–Alder 反 応 に よ り ジ ア ス テ レ オ 選 択 的 に 調 製 し よ う と 考 え た (23 の ヒ ド ロ キ シ 基 の 不 斉 に 起 因 す る 不 斉 誘 導 を 期 待 )。23 は 市 販 の 24 に 対 す る 増 炭 と 不 斉 還 元 に よ り 導 く 。 一 方 , DE ユ ニ ッ ト 21 は 25 の ア ジ ド 基 の 還 元 に よ る ア ミ ン へ の 変 換 と ピ ロ ー ル カ ル ボ ン 酸 誘 導 体 (E ユ ニ ッ ト ) と の 縮 合 , お よ び 2 つ の PMB の 除 去 に よ る 環 状 ヘ ミ ア セ タ ー ル の 形 成 に よ り 調 製 可 能 で あ る と 思 わ れ る 。四 置 換 不 斉 中 心 を 持 つ ア ル コ ー ル 25 は ,ケ ト ン 26 に 対 す る Cornforth–Evans 遷 移 状 態 モ デ ル( ま た は Felkin–Anh 遷 移 状 態 モ デ ル )に 基 づ く ビ ニ ル リ チ ウ ム 誘 導 体 の 求 核 付 加 に よ り ,ジ ア ス テ レ オ 選 択 的 に 得 ら れ る も の と 予 想 し た 。26 は エ ポ シ キ ド 27 の ア ジ ド ア ニ オ ン に よ る 開 環 に よ り ,27 は 28 の Sharpless 不 斉 エ ポ キ シ 化 に よ っ て 得 ら れ る は ず で あ る 。28 は (R)-乳 酸 メ チ ル か ら 4 工 程 で 得 ら れ る こ と が 知 ら れ て い る 。 Scheme 2-1 AAM の 逆 合 成 解 析
13 2.3 DE ユニットの合成-Introduction
AMM の DE ユニット(21)は新規糖骨格を有する amycolose 及びピロールカルボン酸 の縮合体である。DE ユニットは AMM の単離者である澤らによって AMM の分解物とし
て報告された(Figure 2-5)20, 21)。澤らは AMM を各ユニットに分解することでより単純な 部分構造にて構造解析を行い、AMM の構造を決定した。その際、天然物である AMM を酸性条件下にて加水分解することでグリコシド結合を開裂させ、DE ユニットの単離及び 構造決定を行っている。また、AMM を酸性条件下およびメタノール溶媒中にて加溶媒分 解することで DE ユニットのメチルグリコシル体(30,31)の単離および構造決定を行った。 21 および 30,31 の活性評価を実施した結果、大変興味深いことに細胞毒性を有するこ とが判明した(Figure 2-6)20)。AMM はマウスに対する急性毒性を示さない一方で部分 構造である 21 および 30,31 は細胞増殖抑制活性を有する。天然物とその部分構造にお いて生物活性が全く異なることは作用機序のさらなる解明が必要であり、構造活性相関研 究に大きく関心が寄せられる。 現在、DE ユニットの創薬に関連した作用機序などの報告 はない。 化合物 1 化合物 2 化合物 3 30 31 Figure 2-6 21 及 び 30,31 の 細 胞 毒 性 評 価 pyrrole amycolose HCl aq. THF HCl aq. MeOH
+
21 Figure 2-5 AMM の 加 溶 媒 分 解 に よ る DE ユ ニ ッ ト (21)及 び DE ユ ニ ッ ト(30-31)の メ チ ル グ リ コ シ ド 体 の 生 成 10 21 30 3114 amycolose 部 分 の 生 合 成 経 路 に 関 し て も 、 精 力 的 に 研 究 が 進 め ら れ た (Scheme 2-2)22)。同位体トレーサー法による分析から、生合成はまず糖 3 位のカルボニル誘導体 を基質として TPP 関連酵素によるピルビン酸の付加が進行することで枝分かれ型構造を 有するオキ ソエチ ル 糖誘導体となる 。続い て、アミ ノ基転位酵素 の 作用に よ り側鎖カルボ ニルに対してアミノ基が導入されることでamycolose が生合成される。また、オキソエチル 糖誘導体に対して還元酵素が作用した場合はヒドロキシエチル糖誘導体へと変換される。 先行研究によると本反応を触媒する酵素をコードする遺伝子配列が発見されていることか らも Scheme 2-2 の生合成経路が有力である。一方で糖部分に関しては、グリコーゲンな どのエネルギー貯蔵に関与する糖鎖 や核酸由来の糖鎖など生体内に 存在するどの 糖鎖 由来なのかは現在のところ不明である。ただし、枝分かれ構造を有する糖鎖は主に核酸か ら得られることが報告されている 30)。amycolose のような枝分かれ側鎖にアミノ基を有す る糖鎖は非常に稀であるが、側鎖にヒドロキシ基を有する糖鎖は核酸由来の糖鎖として数 多く存在する。これらの報告例からamycolose の糖骨格は核酸由来であると推測される。 また、E ユニットに相当するピロール環部分に関する生合成の報告はされていないが、ピ ロール構造は一般的にアミノ酸由来であること が知られているため、E ユニットはアミノ酸 由来であると考えられる。 D unit TPP (thiamine pyrophosphate) pyruvate reductase aminotransferase 3 Scheme 2-2 DE ユ ニ ッ ト の 生 合 成 経 路
15 2.3.1 ピロール環部分の合成 E ユニット部分は D ユニットとの縮合によって連結するため、まずピロールカルボン酸誘 導体の合成を計画した。ピロールカルボン酸は既知化合物 31)であるため、大量供給に最 適な合成経路を検討した。 まず初めに出発原料としてマロン酸ジエチル 32 を用いた(Scheme 2-3)。32 に対して 亜硝酸ナトリウムを酢酸条件下で作用させることでオキシム 33 へと変換した後、粉末亜鉛 による N-アセチル誘導体 34 の調製を行った。34 に対して別途 35 から調製した 36 とナ トリウムエトキシド存在下で反応させることによりピロール環 部分 37 を構築した。得られた 37 に対して、塩化スルフリルによるピロール環水素の塩素化により 38 を合成した。最後に 水酸化リチウムによるエチルエステル部分の加水分解によりピロールカルボン酸 39 の合 成を完了した。しかし、上記の合成に関して、収率および工程数において 課題を残してい た。まず一つ目として、37 から 38 の変換において収率 16%と低収率であった。低収率の 原因は 38 と同時にトリクロロ体 40 が副生するためである(Scheme 2-4)。そこで、37 から 直接 40 を合成した後に比較的温和な還元剤である NaCNBH3を用いることで工程数は 一工程増えるが収率の向上を可能とした(Scheme 2-5)。 NaNO2, AcOH 0 to 30 Ο C, 80 h 97% Zn, AcOH H2O, 40 Ο C, 2 h 25% NaOEt, EtOH H2O, 90 Ο C, 2 h 56% SOCl2, pyridine -40 Ο C to rt, 14 h 79% SOCl2, CCl4 0 Ο C, 22 h 16% LiOH THF/H2O, 75 ΟC, 20 h 82% SOCl2, CCl4 0 Ο C, 22 h 16%
+
by-product Scheme 2-3 ピ ロ ー ル 環 部 分 の 合 成 32 33 34 35 36 37 38 39 Scheme 2-4 ピ ロ ー ル 環 部 分 の 塩 素 化 反 応 37 38 4016 2 つ目の改善点として、34 から 37 の変換においてナトリウムエトキシドを大過剰使用す ることである。そのため、ピロール環構造の構築方法を変更し、出発原料として 41 と 42 を 用いた(Scheme 2-5)。これにより一工程にてピロール環の構築が可能であり、反応条件 の改善と工程数の削減を可能とした。以上によりグラムスケールの4 工程総収率 18%にて ピロールカルボン酸 E ユニットの合成を完了した。 2.3.2 DE ユニットの合成 2.3.2.1 第一世代合成の合成戦略 DE ユニットの第一世代合成の合成戦略として逆合成解析を示 した(Scheme 2-6)。 DE ユニット 21(cf. Scheme 2-1, 21: R = H)は 43 のオレフィン部位をオゾン分解する ことでアルデヒドへと変換し、糖骨格を構築することで合成できると考えた。43 は 44 の糖 1 位と 6 位のアセタール環の開環及び還元により得られると考えた。44 は 45 に対するグリ ニャール試薬の立体選択的求核付加により 四級不斉中心を構築することにした。45 は糖 1 位と 6 位のアセタール環の構築により導き、46 は出発原料であるD-グルコサミン塩酸塩 47 とピロールカルボン酸 39 との縮合により調製できると考えた(Scheme 2-6)。 Scheme 2-6 第 一 世 代 合 成 の 逆 合 成 解 析 NaNO2, Zn, AcOH/H2O 120 Ο C to rt, 2 h 33% SO2Cl2 CCl4, 0 ΟC, 2 h 80% NaCNBH3 NMP, 0 Ο C to rt, 20 h 82% LiOH THF/H2O, 75 Ο C, 20 h 82% 15 (E unit) gram scale 1.27 g 55 56 57 58 59 41 37 42 40 38 39 Scheme 2-5 改 良 し た ピ ロ ー ル 環 部 分 の 合 成 DE unit
⇒
8 16 20 22 13⇒
⇒
⇒
ozonolysis⇒
⇒
⇒
⇒ ⇒ ⇒
⇒ ⇒ ⇒
D-glucosamine hydrochloride (14) condensation reductive ring opening stereoselective nucleophilic addition cyclic acetalization 21 45 46 39 47 43 4417 2.3.2.2 DE ユニットの第一世代合成 出発原料である D-グルコサミン塩酸塩 47 とピロールカルボン酸 39 に対して縮合剤と して EDC 塩酸塩を用いることにより 47 の 2 位アミノ基において選択的に縮合し、アミド 46 へと変換した(Scheme 2-7)。46 に対して TsCl とピリジン条件下にて反応させること により、6 位の水酸基を選択的に Ts 保護し、48 を合成した。続いて、48 に DBU を作用 させることで1,6-アンヒドロ糖 49 へと変換した後、PivCl を作用させることで 2 つある二級 水酸基のうち、より立体的に空いている4 位水酸基の Piv 保護を試みた。このとき、3 位の 水酸基もしくは 3 位と 4 位の両方の水酸基に Piv 保護された副生成物も同時に得られた が、検討の結果から収率 50%にて目的の 50 を得た。50 に対して、AZADOL 酸化により 3 位の水酸基をカルボニルへと変換して 45 とした後、鍵反応としてグリニャール試薬 51 による3 位の四級不斉中心の構築を試みた。反応条件として-78ºC の低温条件下にてグ リニャール試薬を作用させたところ、糖の convex 面から求核攻撃が進行した所望の立体 化学を有する 44 は収率 15%であった。一方、グリニャール試薬が糖の concave 面から 求核攻撃した望まない立体化学を有する52 は収率 59%であり、優先的に得られる結果と なった。そこでカルボニル体 45 の 1H NMR を精査したところ、糖 1 位がシングレットピー クでありカープラス則から考えると 45 の配座は想定していた 1C4型ではなく、実際は Bo, 3 型であると推定した。そこで、1,6-アンヒドロ糖の配座に関する文献を調査すると糖の置換 基が糖骨格の配座に影響を与えていることが示唆されている。 EDC·HCl, HOBt Na2CO3, THF, rt, 24 h 56% DBU EtOH, rt, 16 h 80% · TsCl, pyridine 0 ΟC to rt, 14 h 59% PivCl, pyridine 0 ΟC to rt, 15 h 50% AZADOL, KBr NaHCO3/NaClO aq.
CH2Cl2, 0 Ο C, 30 min THF, -78 Ο C, 1 h desired
+
D-glucosamine hydrochloride (14) 15 16 17 18 19 20 21 22 : 15% (2 steps) 23 : 59% (2 steps) Scheme 2-7 立 体 選 択 的 求 核 攻 撃 に よ る 44 の 合 成 48 50 49 35 47 46 39 52 45 51 4418 例とし て、無保 護の 1,6-アンヒドロ糖 54 は、室温で水もしくは DMSO 中において 15~20%程度は Bo, 3型(55)が存在することが報告されている(Figure 2-7)32) 。一方、3-アミノ-1,6-アンヒドロ糖 56 に関しては、DMSO 中において 1C4型と Bo, 3型(57)がおおよ そ 1:1 の割合で存在することが知られている 32)。さらに、1,6-アンヒドロ糖の 2 位及び 4 位水酸基が嵩高い置換基で保護されている場合は Bo ,3 型の方が優先的に存在 すること が報告されている。以上の知見を考慮すると本合成に用いた 45 の場合、2 位および 4 位 の水酸基に比較的嵩高い Piv 基とピロール環部位が存在するため、配座が Bo, 3 型(53) に変化したと考えられる。そのため、Bo ,3型の53 に対するグリニャール試薬の求核攻撃は より立体的に空いている糖のconcave 面から進行するため望まない立体化学を有する 52 が優先的に得られたと考えられる。また 、それぞれ 44 と 52 の立体化学は NOESY 測定 により決定した(Figure 2-8, 2-9)。 1C 4 Nu -Nu -BO,3 Chair-boat equilibrium conformational change 1C 4 BO,3 H2O or DMSO 15 ~ 20% DMSO 50% BO,3 O C3 H1 (singlet) O 2 H2 N 2 3 1 90Ο 1C 4 53 55 Figure 2-7 1,6- ア ン ヒ ド ロ 糖 の 配 座 変 換 56 57 54 45 concave
19 OH c-H 6-H NH 6 c Figure 2-8 44 の NOESY チ ャ ー ト (600 MHz, CDCl3) Figure 2-9 52 の NOESY チ ャ ー ト (600 MHz, CDCl3)
20 少量ではあったが得られた 44 を用いて糖アノマー位のジチアン化を試みた(Scheme 2-8)。反応条件は Lewis 酸として BF3·OEt2を用いてプロパンジチオール 58 を作用さ せ た 。1H NMR 測 定 に よ り 中 間 体 と し て 糖 オ キ サ ゾ リ ン 59 の 存 在 が 示 唆 さ れ た 。 BF3·OEt2によってアセタール環が活性化された後、糖 2 位のアミド基の隣接基関与によ ってオキ サゾリン環が 形成されたと考えら れる。 オキ サゾリン環の形成後、 反応を続ける こ とでモノチオアセタール体 60 を経由してジチアン 61 を得た。続いて、62 もしくは 63 を 用いて一級水酸基を選択的にチオカーボネートもしくはチオカーバメ ートへの変換を試み た。しかし、所望の 64 は得られず、2 級水酸基による求核攻撃が進行して環状チオカー ボネート 65 が得らた。64 が生成した場合、2 級水酸基の分子内求核攻撃が速やかに進 行するため、TLC による反応の追跡を行ったが単一生成物として 65 が得られた。そのた め、所望の 64 を選択的に得ることは困難であった。さらに、鍵反応となる四級不斉中心の 構築において条件検討を続けたが収率向上には至らなかった。上記の 2 点の問題により 新たな DE ユニットの合成戦略へと変更することにした。 THF, -78 Ο C, 1 h desired
+
BF3·OEt2, CH3CN rt to 40 Ο C, 40 h 70% desired 29 or 30, pyridine THF, 0 Ο C low yield overreaction 20 245) 1C 4 BO,3 22 : 15% (2 steps) 23 : 59% (2 steps) 21 26 27 28 29 30 31 32 25 Scheme 2-8 64 の 合 成 60 59 61 62 64 65 45 53 63 44 51 52 58 62 6321 2.3.2.3 第二世代合成の合成戦略 新たに提案する合成戦略を示した(Scheme 2-9)。DE ユニット 21 は 66 より二つの PMB 基の除去により糖骨格を構築することで合成できると考えた。66 はカルボニル 26 に対してビニルリチウム誘導体 67 を作用させることで本スキームの鍵となる四級不斉中心 の構築を達成できると考えた。26 はエポキシド 27 に対する位置選択的なアジド化によっ て導くことにした。27 はアリルアルコール 28 を Sharpless 不斉エポキシ化反応に付すこ とで変換できるとした。28 は出発原料である D-乳酸メチル 29 から調製可能であるとした。 2.3.2.4 DE ユニットの第二世代合成 Scheme 2-10 に実際の合成を示した。出発原料である D-乳酸メチル 29 に対して、酸 性条件下でトリクロロアセトイミデート 68 を用いることで PMB 保護(68→69)した後 33)、メ チルホスホン酸ジメチルを作用させることでホスホネート 70 を調製した。70 を Horner- Wadsworth-Emmons 反応(Roush-Masamune 法)に付すことでエノン 71 を導い た。本反応の E /Z 選択性は粗精製時において E /Z = 17:1 であった。続いて、71 に対 して Zn(BH4)2を用いた立体選択的還元により遷移状態 72 を経由してアリルアルコール 28 を得た34)。本反応でのジアステレオ選択性は粗精製時において dr = 11:1 であった。 また、合成初期の 69 から 70 の変換において強塩基によるカルボニルα位のメチル基の 異性化が懸念されるため、28 を得たところで Mosher 法により光学純度を測定した。その 結果、光学純度は 98% ee 以上であり 69 のメチル基の異性化はほぼ起こらないことを確 認した。28 を得た後、Sharpless 不斉エポキシ化反応によりエポキシド 73 へと変換し、 TBSCl により 2 級水酸基を保護することで 27 を導いた 35)。また、Sharpless 不斉エポ キシ化反応におけるジアステレオマー比は粗精製時において dr = 4.8:1 であった。
⇒
⇒
⇒
⇒
⇒
⇒
⇒
⇒ ⇒ ⇒
⇒ ⇒ ⇒
21 66 67 26 27 28 29 Scheme 2-9 改 良 し た DE ユ ニ ッ ト の 逆 合 成 解 析22 続いて、エポキシドの開環を伴う位置選択なアミノ基の導入を試みた(Scheme 2-11)。 27 のエナンチオマーである ent-27 をモデル基質として条件検討を行った。まず、アミン 74 を Lewis 酸存在下で導入を試みたが、目的物を得られず原料回収となった。そこで、 ピロールカルボン酸から導いたアミド 76 を上記と同様の Lewis 酸存在下で導入を試みた が原料回収となった。直接エポキシドに対してアミノ基を導入することが困難であったため、 合成戦略を変更し、エポキシドに対してアジド基を導入した後に還元することでアミノ基の 導入を試みた 36, 37 )。 ent-45 Lewis acid Lewis acid Lewis acid ・MgBr2・Mg(OTf)2 ・Cu(OTf)2・Yb(OTf)2 5 steps Methyl D-Lactate TfOH, Et2O, 16 h -78 Ο C to rt 70%
(MeO)2P(O)CH3, n-BuLi
THF, -78 ΟC to rt, 16 h 98%
LiCl, i-Pr2NEt, CH3CHO
THF, 0 ΟC, 24 h 89% Zn(BH4)2 THF, -20 ΟC, 2.5 h 77% (-)-DIPT, Ti(Oi-Pr)4, TBHP CH2Cl2, -20 Ο C, 36 h Horner-Wadsworth- Emmons olefination (crude dr = 11:1) 56% (crude dr = 4.8:1) (crude E/Z = 17:1) separable TBSCl, DMAP, imidazole CH2Cl2, 0 ΟC to rt, 4 h quant Sharpless epoxidation (>98% ee) 34 ent-57 76 29 28 Scheme 2-10 エ ポ キ シ ド 27 の 合 成 Scheme 2-11 ent- 27 に 対 す る 窒 素 官 能 基 の 導 入 75 ent- 27 74 76 77 42 68 69 70 71 72 73 27
23 エポキシドに対して赤矢印で示した位置でアジド化が進行すれば所望の 78 が得られる。 一方、青矢印の位置でアジド化が進行した場合は副 生成物である位置異性体 79 となる (Figure 2-10)。条件検討の結果、Entry1~6 で示したように酸および溶媒を検討したが 目的とする 78 を優先的に得ることはできなかった。そこで、Entry7 に示したように酸とし てトリメチルアミン塩酸塩を用いることで 78 の選択的が向上した。さらに、基質の検討とし
て Entry8 の TIPS 保護体 80 を Entry2 と同様の酸および溶媒にてアジド化を試みた
が アジド 基挿入の 位置選択性は向上し なかっ た 。以上の検討 結果よ り、本基質において Entry7 の反応条件を用いることにした。 acid, NaN3 desired
+
-undesired (regioisomer) a) Determined by 1H NMR. b) TBS → TIPS 78 : 79a) acid (eq.) NaN3(eq.)substrate entry solvent NH4Cl (2) 5 1.1 : 1.0 2 2-methoxyethanol/H2O = 8/1 3 NH4Cl (2) 5 DMSO/H2O = 8/1 1.2 : 1.0 NH4Cl (1) 7 2.5 : 1.0 1 EtOH/H2O = 3/1 15 4 PPTS (5) 2-methoxyethanol/H2O = 3/1 1.1 : 1.0 15 PPTS (5) 5 1,4-dioxane/H2O = 3/1 1.6 : 1.0 10 LiClO4 (3) 6 CH3CN 2.0 : 1.0 NH4Cl (2) 5 1.1 : 1.0b) 8 2-methoxyethanol/H2O = 8/1 solvent, 100 ΟC Me3N·HCl (3) 10 4.9 : 1.0 7 EtOH/H2O = 8/1 time 17 h 17 h 18 h 10 days 10 days 9 days 6 days 7 days Table 1 78 80 ent-27 79 ent-27 Figure 2-10 ア ジ ド 基 導 入 の 検 討
24 Figure 2-10(Entry 7)の条件にて、27 にトリメチルアミン塩酸塩とアジ化ナトリウムを 用いることで 81 を単離収率 57%にて得た(Scheme 2-12)。得られた 78 を Dess- Martin 酸化によりカルボニル 26 へと変換した後、tBuLi 存在下、ビニルブロマイド 82 を作用させることで所望の四級不斉中心を構築し、 単一立体異性体として25 を与えた。 また、本反応において用いた 82 はアニスアルコール 83 から既知化合物であるビニル エ ー テ ル 84 へ と変 換 し た 後 、 ジ ブ ロ モ 化 と 臭 化 水 素 の 脱 離 に よ り 合 成 し た (Scheme 2-13)38-40)。84 は実際に前述の反応に用いたように新規の増炭試薬としての利用が期待 される。
25 の四級不 斉中 心 の立 体選 択 性に つ いては polar Felkin - Anh model 及び Cornforth - Evans model が 最 適 モ デ ル で あ る と 考 え た 41, 42)。 す な わ ち 、polar
Felkin-Anh model の場合は 26 のカルボニルα位の TBSO と求核剤の間で双極子 の 反 発 が 起 こ る こ と で 反 対 側 に 配 向 す る(Scheme 2-14) 。一 方 、 Cornforth - Evans model においてはカルボニル酸素と TBSO の間で双極子反発により Scheme 2-14 で 示 し た 配 向 と な る 。 以 上 の こ と か ら 、polar Felkin -Anh model 及び Cornforth- Evans model の両モデルにおいて 25 の四級不斉中心の立体選択性を説明することが 可能である。 NaN3, Me3N·HCl aq. EtOH, 100 ΟC, 7 d 57% DMP, pyridine CH2Cl2, rt, 1 h 99% t-BuLi, Et2O, -78 ΟC, 30 min 84% “single diastereomer” 7 5 3 7 5 3
polar Felkin-Anh model
O OR C7 H C5 Nu -3 Cornforth-Evans model 3 O C5 C7 RO H Nu -3
New reagent for homologation reaction CaC2, KOH DMSO/H2O 120 ΟC, 15 h 75% 1) Br2, CH2Cl2 -78 Ο C, 2.5 h 2) Et3N, CH2Cl2 -78 to 0 Ο C, 4 h 54% (2 steps) 82 84 83 Scheme 2-13 ビ ニ ル ブ ロ マ イ ド 82 の 合 成 Scheme 2-12 四 級 不 斉 中 心 の 構 築 27 81 26 82 25
25 DE ユニット合成の鍵となる四級不斉中心の構築が完了した後、n-Bu3P によるアジド 基の還元によりアミン85 へと変換した(Scheme 2-15)。続いて、85 と予め合成したピロー ルカルボン酸 39 との縮合によりアミド 66 へと導いた。 41 57% 45 99% t-BuLi, Et2O, 30 min, 78 Ο C 84% “single diastereomer” 7 5 46 49 47 3 7 5 3
polar Felkin-Anh model7),8) O OSi C7 H C5 Nu -3 Cornforth-Evans model7),8) 3 O C5 C7 SiO H Nu -3 84% “single diastereomer” O RS R Nu -RM RL
Cram acyclic model
polar Felkin-Anh model O OTBS C7 H C5 3 Cornforth-Evans model 3 O C5 C7 TBSO H 3 3 7 5 7 5 3 ‐ ‐ Li + + Li Ο Ο Ο Ο n-Bu3P MeOH, rt, 12 h CH2Cl2, 0 Ο C to rt, 2 h 96% (2 steps) EDC·HCl HOBt, Et3N 25 26 Scheme 2-15 ア ミ ド 66 の 合 成 25 85 39 66 Scheme 2-14 四 級 不 斉 中 心 の 立 体 選 択 性
26 66 の エ ノ ー ル エ ー テ ル 部 分 の 立 体 化 学 を 決 定 す る た め NOESY 測 定 を 行 っ た (Figure 2-11)。1 位と 2 位のプロトン間において明確な相関が確認されたため、エノール エーテル部分の幾何異性は Z 体であると決定した 43, 44)。 ア ミ ド 66 の PMB 基 の 除 去 に 関 し て 、 ま ず 初 め に 酸 化 剤 で あ る DDQ を 用 い た (Scheme 2-16)。その結果、2 級水酸基上の PMB 基は除去された(66→86)一方で反応 を 36 時間続けたが、エノールエーテル上の PMB 基は全く脱離せず、目的とする 87 を 得ることはできなかった。そこで、酸としてTFA を用いて 2 つの PMB 基の除去を試みた。 反応を TLC で追跡すると TFA により 2 級水酸基上の PMB 基が脱離して糖骨格を構築 した単一異性体である中間体88 が生成した後、アセタール上の PMB 基が脱離して所望 の 87(cf. Scheme 2-1, 21: R = TBS)を単離収率 83%で得ることに成功した。また、88 に関して、反応を途中でクエンチしてシリカゲルカラムクロマトグラフィーにより単離可能で あった。酸として TFA を用いた際には良好な収率で 87 を得たが BF3·OEt2を用いた場 合には反応系中が複雑化し、87 は 16%の低収率となった。おそらく 66 の三級アルコー ルの脱離が原因であると考えられる。最後に 87 の糖 4 位の TBS 基を TBAF により除去 Figure 2-11 66 の NOESY チ ャ ー ト (600 MHz, CDCl3) 1 2
27 することで DE ユニット 21 の合成を完了した。出発原料である D-乳酸メチルから総工程 数 13 工程及び総収率 9.2%にて DE ユニットの合成を達成した。比旋光度に関しては文 献値と近い値を示した。さらに、87 を共通中間体としてメタノール溶媒中で TMSCl を作 用させることで、メチルグリコシル化と TBS 基の除去を一挙に行い、DE ユニットのメチル グリコシド体(30,31)の合成にも成功した 45)。このとき溶媒として塩化メチレンや THF を 用いた場合には TBS 基の脱離は全く進行しなかったがメタノール溶媒では TBS 基の脱 離が良好に進行した。30 の X 線結晶構造解析が既に報告されていたため、単離 NMR と の比較によって目的物であることを確認した 21)。本合成の鍵であった四級不斉中心の立 体選択性に関しては25 を合成したときには不明であったが DE ユニットの合成により所望 の立体選択性で反応が進行していたことを確認した。 以上で DE ユニットの合成を完了と した。 66 86 88 87 21 30 31 Scheme 2-16 DE ユ ニ ッ ト の 合 成 2 5 - Ο one pot TBAF, THF rt, 12 h 94%
DE-a-OMe : 19% DE-b-OMe : 50% X-ray21)
2 5 [a]27 D+19.2 (c 0.225, CHCl3) lit. [a]23 D+15 (c 0.51, CHCl3) B) BF3·OEt2, CH2Cl2 -20 to 0 Ο C, 1 h
28 2.3.3 小括 D-乳酸メチルを出発原料に用い、鍵反応である四級不斉中心の構築を経て、 21 及び 30、31 の合成を達成した。以下に DE ユニット全体の合成を示した(Scheme 2-17)。 29 69 68 70 Zn(BH4)2 THF, -20 Ο C, 2.5 h 77% (crude dr = 11:1) LiCl, i-Pr2NEt, CH3CHO
THF, 0 ΟC, 24 h 89% (crude E/Z = 17:1) (-)-DIPT, Ti(Oi-Pr)4, TBHP CH2Cl2, -20 Ο C, 36 h 56% (crude dr = 4.8:1) (>98% ee) Methyl D-Lactate TfOH, Et2O, 16 h -78 Ο C to rt 70%
(MeO)2P(O)CH3, n-BuLi
THF, -78 Ο C to rt, 16 h 98% TBSCl, DMAP, imidazole CH2Cl2, 0 Ο C to rt, 4 h quant NaN3, Me3N·HCl aq. EtOH, 100 ΟC, 7 d 57% DMP, pyridine CH2Cl2, rt, 1 h 99% t-BuLi, Et2O, -78 Ο C, 30 min 84% “single diastereomer” n-Bu3P MeOH, rt, 12 h CH2Cl2, 0 Ο C to rt, 2 h 96% (2 steps) EDC·HCl HOBt, Et3N TFA, CH2Cl2 -20 to 0 Ο C, 3 h 83% TBAF, THF rt, 12 h 94% DE unit TMSCl, MeOH rt, 62 h
+
DE-a-OMe : 19% DE-b-OMe : 50% 71 28 Scheme 2-17 DE ユ ニ ッ ト の 合 成 73 27 81 26 82 25 66 39 85 87 21 31 3029 2.4 C ユニットの合成-Introduction
Amycolamicin の C ユニットはトランスデカリン構造を主骨格としてヒドロキシ基を含む
官能基化によって構築されている。C ユニットの構造決定は AMM を ABC ユニットの
amykitanoseーテトラミン酸-trans-デカリン環複合体 89 に分解し、新 Mosher 法にて
決定された(Scheme 2-18)21)。 C ユニット部分の活性評価に関する報告例は無いがテトラミン酸-デカリン環に相当す る類縁体の単離、合成及び活性評価に関する報告例は多数存在する。これまでの報告に よると、テトラミン酸-デカリン環類縁体の一部では S. aureus や C. difficile に対して 抗菌作用を示すことが知られている(Figure 2-12)46-51)。AMM が抗菌作用を示すことや、 細菌のGyrB において AMM のテトラミン酸部分との強固な相互作用が確認されているこ とを踏まえると、AMM の抗菌活性発現の中心はテトラミン酸-デカリン環であると推測さ れる。さらに 、天然物ライブラリーからの抗菌活性物質の探索の結果もテトラミン酸-デカ リン環部分が抗菌作用を示す本体であることを示唆している 51)。
Scheme 2-18 AMM の ABC ユ ニ ッ ト の 構 造 決 定
(R)-MTPA-Cl, Et3N DMAP, CH2Cl2 (S)-MTPA-Cl, Et3N DMAP, CH2Cl2 (S) (R) 10 89 90 91 A B C D E
30 2.4.1 第一世代合成の合成戦略 C ユニットの第一世代合成の逆合成解析を示した(Scheme 2-19)。まず、C ユニット 111 は 112 に対する分子内 Diels-Alder 反応によって得られると考えた。112 は出発 原料であるジヒドロキシアセトン 113 から Horner-Wadsworth-Emmons 反応及び Johnson-Claisen 転位反応により導く。 Figure 2-12 テトラミン酸-デカリン環類縁体の構造 Scheme 2-19 C ユ ニ ッ ト の 逆 合 成 解 析 111 112 113 n n = 0 : signemycin D (92) n = 1 : signemycin A (93) n = 2 : signemycin B (94) n = 3 : signemycin C (95) R3= OH, R4= CO2H Sch 210971 (96) R3= CO2H, R4= OH Sch 210972 (97) JBIR-22 (98) zopfiellamide (99) R1= R2= Me : equisetin (100) R1= H, R2= Me : trichosetin (101) R1= Me, R2= CH2OH : ophiosetin (102)
cryptocin (103) paecilosetin (104) phomasetin (105)
streptosetin (106) R5= H, R6= Me : coniosetin (107) R5= R6= H : altersetin (108)
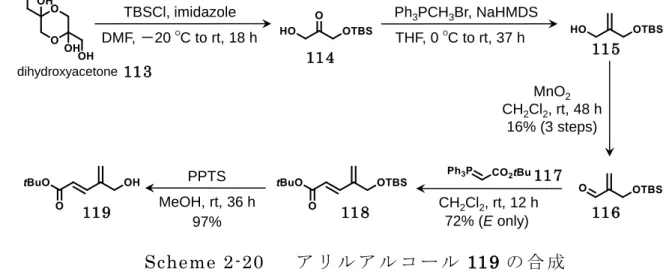
31 2.4.2 C ユニットの第一世代合成 出発原料であるジヒドロキシアセトン 113 に TBSCl を作用させることで TBS 保護体 114 へと変換した後、Wittig 反応によりオレフィン部位を導入することでアリルアルコール 115 を調製した 52, 53)。115 を二酸化マンガンによる酸化反応によってエノン 116 へと導いた。 続いて 116 に Wittig 試薬 117 を用いることで単一異性体にて 118 を得た。PPTS によ り 118 を処理することで TBS 基を除去してアリルアルコール 119 へと変換した(Scheme 2-20)。 続いて、Johnson-Claisen 転位反応により二炭素増炭したメチルエステル 120 の合 成を試みた 54,55)。用いた酸の検討結果を以下に示した(Figure 2-13)。過去の報告例を 参考に比較的温和な条件を選択して本反応を試みたが、Entry 1~ 3 で示したように 120 の収率は 14~27%の低収率であった。また、Scheme 2-10 の合成初期の 113 から 116 の 3 工程の変換に関しても、3 工程収率 16%の低収率であり、検討を重ねたが改善が見 い出せないことから合成戦略を見直すことにした。 TBSCl, imidazole DMF, -20 ΟC to rt, 18 h Ph3PCH3Br, NaHMDS THF, 0 ΟC to rt, 37 h MnO2 CH2Cl2, rt, 48 h 16% (3 steps) CH2Cl2, rt, 12 h PPTS MeOH, rt, 36 h 72% (E only) 97% dihydroxyacetone Scheme 2-20 ア リ ル ア ル コ ー ル 119 の 合 成 113 114 115 116 117 118 119 entry 3 2 1 CH3C(OMe)3(eq.) 5 5 20 acid (eq.)
NaHSO4-SiO2(0.1 g/mmol) 2-nitrophenol (0.1) propionic acid (0.4) yield 26% 14% 27% Table 2 Johnson-Claisen rearrangment 100 ΟC, 24 h Table 2 119 120 Figure 2-13 Johnson-Claisen 転位反応による 120 の合成
32 2.4.3 第二世代合成の合成戦略 以下に、新たに提案する C ユニットの合成戦略を示した(Scheme 2-21)。trans-デカ リン構造の 構築は第 一世代合成 の 逆合成 解析 と同様に し て、112 から分子内 Diels- Aldar 反応によって導く。112 は新たな出発原料である 2,3-ジブロモプロペン 24 から Horner-Wadsworth-Emmons 反応及び Mizoroki-Heck 反応によって合成でき ると考えた。24 を出発原料とすることでエキソメチレンを起点に増炭して、第一世代合成よ りも効率的に Diels-Aldar 反応の前駆体まで導けることを期待した。 2.4.4 C ユニットの第二世代合成 24 をヨウ化銅存在下で酢酸メチルとのアルキル化により、メチルエステル 121 へと変換 した。この変換においてヨウ化銅を添加しないと 121 が全く得られないことから、ヨウ化銅 の添加は必須である(Scheme 2-22)56)。
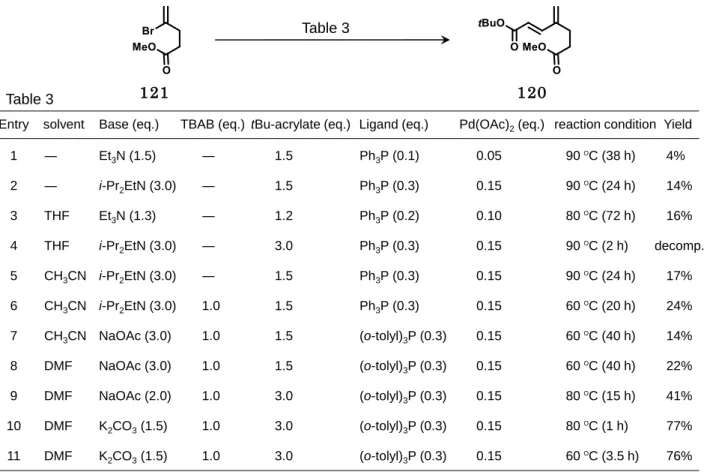
続いて、アクリル酸t-Bu との Mizoroki-Heck 反応を試みた(Figure2-14)。反応条
件 の 検 討 結 果 を 次 項 に 示 し た 。Entry 1~2 の よ う に 無 溶 媒 条 件 に お い て 、 120 は 4~14%の低収率であった。そこで、Entry 3~8 において溶媒、塩基及び配位子の変換を 試みたが大きな収率の向上は見られなかった。しかし、Entry 9 では 120 の収率が 41% に向上したことから、溶媒はDMF および配位子は(o-tolyl)3P が最適であるとして塩基の 検討を行った。酢酸ナトリウムから炭酸カリウムに塩基を変更したところ、Entry 10,11 に 示したように 120 の収率が 70%以上まで改善された。特に Entry 11 の条件を用いてグ ラムスケールにて反応を行ったところ、24 から 2 工程収率 63%にて 120 を得ることに成 功した(Scheme 2-23)。 MeOAc, NaHMDS, CuI, THF -78 to -20 Ο C, 17 h 2,3-dibromopropene 24 121 Scheme 2-22 メ チ ル エ ス テ ル 121 の 合 成 Scheme 2-21 C ユ ニ ッ ト の 第 二 世 代 合 成 の 逆 合 成 解 析 111 112 24
33 また、120 はE 体の単一異性体で得られた。120 は室温で放置すると重合反応が進行 する不安定な化合物であるため 、Entry 1~9 のように原料消失までに長時間を要すると 120 の生成と重合が同時に進行し、低収率になったと考える。一方で Entry 10~11 では 反応時間が 1~3.5 時間であり比較的速やかに原料が消失したため収率改善を可能とした。 配位子に関しては、(o-tolyl)3P と酢酸パラジウムの組み合わせにより安定なパラダサイク ル触媒が形成され触媒回転数が向上するため速やかな反応完結の一因になったと考えら れる。 Yield solvent Entry THF decomp. 4 5 CH3CN 17% THF 16% 3 6 CH3CN 24% DMF 8 22% Base (eq.) i-Pr2EtN (3.0) i-Pr2EtN (3.0) Et3N (1.3) i-Pr2EtN (3.0) NaOAc (3.0) NaOAc (2.0) DMF 9 41% reaction condition 90ΟC (2 h) 90 ΟC (24 h) 80 ΟC (72 h) 60 ΟC (20 h) 60ΟC (40 h) 80 ΟC (15 h) ― 4% 1 Et3N (1.5) 90 ΟC (38 h) CH3CN 7 NaOAc (3.0) 60 ΟC (40 h) 14% K2CO3(1.5) DMF 10 77% tBu-acrylate (eq.) 3.0 1.5 1.2 1.5 1.5 3.0 1.5 1.5 3.0 80ΟC (1 h) Ligand (eq.) Ph3P (0.3) Ph3P (0.3) Ph3P (0.2) Ph3P (0.3) (o-tolyl)3P (0.3) (o-tolyl)3P (0.3) Ph3P (0.1) (o-tolyl)3P (0.3) (o-tolyl)3P (0.3) Pd(OAc)2(eq.) 0.15 0.15 0.10 0.15 0.15 0.15 0.05 0.15 0.15 ― 14% 2 i-Pr2EtN (3.0) 1.5 Ph 90 ΟC (24 h) 3P (0.3) 0.15 TBAB (eq.) ― ― ― 1.0 1.0 1.0 ― 1.0 1.0 ― K2CO3(1.5) DMF 11 3.0 (o-tolyl) 60ΟC (3.5 h) 76% 3P (0.3) 0.15 1.0 Table 3 Table 3 121 120 Figure 2-14 Mizoroki-Heck 反応による 117 の合成検討 MeOAc, NaHMDS, CuI, THF -78 to -20 Ο C, 17 h tBu-acrylate, Pd(OAc)2, (o-tolyl)3P, K2CO3, TBAB DMF, 80 ΟC, 1 h 63% (E only : 2 steps) 2,3-dibromopropene 121 120 24 Scheme 2-23 Mizoroki-Heck 反応による 120 の 合 成
34
続いて、120 のメチルエステル部分を選択的にホスホネートへと変換することで 122 へ
と導いた(Figure 2-15)57)。本反応において Entry1~2 に示すように n-BuLi 及びメチ
ルホスホン酸ジメチルの当量を 2.4~2.6 当量用いた場合、t-Bu エステルもホスホネート へと変換されるため中程度の収率になった。そこで 、各試薬の当量を 1.7 当量まで減少さ せたところ単離収率 92%にて 122 を得た。 得られた 122 をクロトンアルデヒド 123 との Horner-Wadsworth-Emmons 反応 に付すことでE /Z = 9.8:1.0 にてジエン部分を構築し、124 へと導いた(Scheme 2-24)。 124 に対して CBS 試薬による不斉還元でアリルアルコール 112 の合成を試みた (Figure 2-16)58, 59)。条件検討の結果、Entry 1 では CBS 試薬を 0.1 当量の触媒量に て反応させたところ、単離収率は 80%と良好であったが光学純度は 60% ee であった。そ こで、Entry 2 において CBS 試薬を 1 当量まで増加させることにより、光学純度は 80% ee まで向上した。最後に、Entry 3 で示すように CBS 試薬を 2 当量、BH3·THF を 3 当量用いることで定量的に反応は進行し、光学純度を 88% ee にて 112 を得た。 Yield Entry 3 92% 45% 2 n-BuLi (eq.) 1.7 2.4 phosphonate (eq.) 1.7 2.4 temp. (time) -78ΟC (2 h) to rt (20 h) -78 ΟC (2 h) to -20 ΟC (3 h) 41% 1 2.4 2.6 -78 ΟC (1 h) Table 4
(MeO)2P(O)CH3, n-BuLi THF Table 4 120 122 Figure 2-15 ホスホネート 122 の合成 LiBr, Et3N, THF rt, 12 h 79% (E/Z = 9.8/1.0) 122 Scheme 2-24 Horner-Wadsworth-Emmons 反応による 124 の 合 成 124 123
35 これまでの C ユニットの合成を Scheme 2-25 にまとめた。最後に、112 のヒドロキシ基 の不斉に起因する分子内 Diels-Alder 反応により C ユニットの全ての不斉炭素の構築 を試みた。しかし、各種の Lewis 酸条件、加熱条件およびマイクロウェーブなどの反応条 件を試みたが分子内 Diels-Alder 反応は全く進行せず、目的とする trans-デカリン体 125 を得ることはできなかった。この一因として、比較的嵩高いt-Bu 基によりジエンとジエ ノフィルが接近しにくいために Diels-Alder 反応が進行しなかったと考えられる。また、 反応を活性化させるために BF3·OEt2 などの比較的強い Lewis 酸を用いた場合、112 が不安定化合物であるため徐々に重合等が進行し反応系中が複雑化した 。そこで、t-Bu 基をアルデヒドに変換した後に Diels-Alder 反応を試みた。 - - Ο Ο - Ο · - - Ο 121 120 24 123 Scheme 2-25 分子内 Diels-Alder 反応による 125 の 合 成 124 122 112 125 entry 3 2 temp. (time) -78 Ο C (2 h) to -40 ΟC (8 h) -78 Ο C (4 h) 1 -40 Ο C (3 days) (S)-CBS (eq.) 2.0 1.0 0.1 BH3·THF (eq.) 3.0 2.0 1.2 yield quant 77% 80% b ’ ee1) 88% 80% 60% Table 5 (S)-CBS, BH3·THF THF Table 5 124 Figure 2-16 不斉還元による 112 の合成 112