デュオトラバ配合点眼液
第 2 部 CTD の概要(サマリー)
2.7 臨床概要
2.7.6 個々の試験のまとめ
目 次 2.7.6 個々の試験のまとめ ··· 1 2.7.6.1 日本人健康被験者を対象とした薬物動態試験・・・5.3.3.1.1(C-12*) ··· 4 2.7.6.2 トラボプロスト 0.004%/チモロール 0.5%の配合点眼液に関する外国人健康被験者を 対象としたPK 試験・・・5.3.3.1.2(C-10*) ··· 20 2.7.6.3 日本人患者を対象とした第 III 相実薬対照(トラボプロスト単剤)比較試験・・・ 5.3.5.1.1(C-13*) ··· 34 2.7.6.4 日本人患者を対象とした第 III 相長期投与試験・・・5.3.5.2.2(C-14*) ··· 82 2.7.6.5 外国人患者を対象とした第 II 相比較試験・・・5.3.5.1.2(C-08*) ··· 140 2.7.6.6 外国人患者を対象とした第 III 相実薬対照(トラボプロスト単剤及びチモロール単 剤)比較試験・・・5.3.5.1.3(C-06*) ··· 159 2.7.6.7 外国人患者を対象とした第 III 相実薬対照(トラボプロスト/チモロール併用療法) 比較試験・・・5.3.5.1.4(C-07*) ··· 195 2.7.6.8 外国人患者を対象とした第 III 相実薬対照(トラボプロスト/チモロール併用療法、 チモロール単剤)比較試験・・・5.3.5.1.5(C-11*) ··· 228 2.7.6.9 外国人患者を対象とした第 III 相実薬対照(ラタノプロスト/チモロール配合剤)比 較試験・・・5.3.5.1.6(C-09*) ··· 269 2.7.6.10 外国人患者を対象とした第 III 相実薬対照(ラタノプロスト及びチモロール)比較 試験・・・5.3.5.4.1(C-03*) ··· 304 2.7.6.11 外国人患者及び日本人患者を対象とした第 III 相実薬対照(トラボプロスト 0.004%/ チモロール0.5%配合点眼液(BAC 含有製剤))比較試験・・・5.3.5.1.7(C-15*/C-16*) ··· 372 *:新薬承認審査情報提供時に置き換えた
略号(略称) 内 容 ACE 阻害薬 アンギオテンシン変換酵素阻害薬
AE 有害事象
Ae 薬物の尿中未変化体率
AGIS The Advanced Glaucoma Intervention Study AL-5848 トラボプロスト活性代謝物 Alb アルブミン Alb/Globulin Ratio アルブミン/グロブリン比 ALP アルカリフォスファターゼ ALT グルタミン酸ピルビン酸トランスアミナーゼ AST グルタミン酸オキサロ酢酸トランスアミナーゼ AUC 濃度-時間曲線下面積 AUC0-∞ 時間ゼロから無限までの濃度-時間曲線下面積 AUC0-t 時間ゼロからt までの濃度-時間曲線下面積 BAC 塩化ベンザルコニウム BID 用法;1 日 2 回 BLQ 定量限界未満 BPM 拍/分 BUN 尿素窒素 C/D 比 視神経乳頭陥凹比 CAI 炭酸脱水酵素阻害薬 Ca 拮抗薬 カルシウム拮抗薬 CI 信頼区間 CL 全身クリアランス CLcr クレアチニン・クリアランス CLr 腎クリアランス Cmax 薬物の最高血中濃度 Cmin 薬物の最低血中濃度
CNTGS Collaborative Normal-Tension Glaucoma Study
COPD 慢性閉塞性肺疾患
CPMP 医薬品委員会(欧州)
Cr クレアチニン
Css 定常状態の薬物血中濃度
CYP Cytochrome P450
CYP2D6 cytochrome P450, family 2, subfamily D, polypeptide 6
EU 欧州連合 F 女性 FDA 食品医薬品局(米国) Fe% 薬物の尿中排泄率(%) GTT 滴 h 時間 Hb ヘモグロビン HIV ヒト免疫不全ウイルス HPLC 高速液体クロマトグラフ HS 就寝時 Ht ヘマトクリット ICH 日米EU 医薬品規制調和国際会議
IND Investigational New Drug(臨床試験実施申請資料)
IOP 眼圧 ITT 治療を意図した割り付けに基づく対象集団 LAT ラタノプロスト0.005%(点眼液) LAT/TIM ラタノプロスト0.005%/チモロール 0.5%配合点眼液 LC 液体クロマトグラフ LDH 乳酸脱水素酵素
略号(略称) 内 容 LSMean 最小二乗平均 M 男性 Max 最大値 MCH 平均赤血球血色素量 MCHC 平均赤血球血色素濃度 MCV 平均赤血球容積
MedDRA Medical Dictionary for Regulatory Activities Terminology
Median 中央値 Min 最小値 MRM 複数反応モニター MS/MS タンデム質量分析 N 例数 N/A 該当なし ND 測定不能 NR 不明 NTG 正常眼圧緑内障 OC Observed Cases:観測例 OH 高眼圧症 OPHT 点眼 OTC 薬 一般用医薬品 OU 両眼 PD 薬力学 PDG 色素緑内障 PE 落屑緑内障 PG プロスタグランジン PK 薬物動態 PLT 血小板 POAG 原発開放隅角緑内障 PP 治験実施計画書に適合した対象集団 q.s. 十分な量 QC 品質管理 QD 用法;1 日 1 回 RA 規制当局 RBC 赤血球数 RDW 赤血球分布幅 RSD 相対標準偏差 SD 標準偏差 Sp 自発報告 STD, std 標準偏差 t1/2 消失半減期 T-Bill 総ビリルビン量 TIM チモロール0.5%(点眼液) Tmax 最高濃度到達時間 T-Pro 総タンパク質量 TRA トラボプロスト0.004%(点眼液) TRA15 トラボプロスト0.0015%(点眼液) TRA/TIM トラボプロスト0.004%/チモロール 0.5%(配合点眼液)
TRA/TIM BAC-free トラボプロスト0.004%/チモロール 0.5%(配合点眼液)BAC-free TRA/TIM AM トラボプロスト0.004%/チモロール 0.5%配合点眼液の朝点眼 TRA/TIM PM トラボプロスト0.004%/チモロール 0.5%配合点眼液の夜点眼 TRA+TIM トラボプロスト0.004%点眼液とチモロール 0.5%点眼液の併用療法 TRA15 トラボプロスト0.0015% (点眼液) UA 尿酸 UNOP イソプロピルウノプロストン upro 尿蛋白
uro ウロビリノーゲン
us 尿糖
VEH プラセボ
WBC 白血球
2.7.6 個々の試験のまとめ
1 2.7.6 個々の試験のまとめ
臨床データパッケージを構成する 11 試験(表 2.7.6)について、個々の試験の概要を以下に示 す。
2.7.6 個々の試験のまとめ 2 表 2.7.6 臨床試験一覧表 試験の種類 実施国 試験番号 報告書の 添付場所 試験の目的 試験デザイン 及び対照 試験薬 投与方法 被験 者数 対象 投与 期間 試験の進行状況 報告書の種類 評価資料又は 参考資料の区分 第 I 相 米国 5.3.3.1.1 C-12* 5.3.3.1 日本人健康被験者での 安全性/薬物動態の検討 二重遮蔽 並行群間比較 実薬及び プラセボ対照 TRA/TIM TRA TIM VEH いずれも 1 回 1 滴 1 日 1 回(6-9 時)両眼点眼 40 日本人 健康被験者 7 日 完了 完全な報告書 評価資料 第 I 相 米国 5.3.3.1.2 C-10* 5.3.3.1 外国人健康被験者での 安全性/薬物動態の検討 二重遮蔽 クロスオーバー 実薬対照 TRA/TIM TRA TIM いずれも 1 回 1 滴 1 日 1 回(6-9 時)両眼点眼 15 外国人 健康被験者 各薬剤 3 日 (計 9 日) 完了 完全な報告書 評価資料 第 III 相 日本 5.3.5.1.1 C-13* 5.3.5.1 日本人患者での有効性 及び安全性の検討 二重遮蔽 並行群間比較 実薬対照 TRA/TIM TRA いずれも 1 回 1 滴 1 日 1 回(8 時)両眼点眼 256 日本人 緑内障又は 高眼圧症患者 3 ヵ月 完了 完全な報告書 評価資料 第 III 相 日本 5.3.5.2.2 C-14* 5.3.5.2 日本人患者での有効性 及び安全性の検討 オープンラベル TRA/TIM 1 回 1 滴 1 日 1 回(朝)両眼点眼 141 日本人 緑内障又は 高眼圧症患者 12 ヵ月 完了 完全な報告書 評価資料 第 II 相 EU 5.3.5.1.2 C-08* 5.3.5.1 外国人患者での有効性 及び安全性の検討 二重遮蔽 並行群間比較 実薬対照 TRA/TIM AM ・1 日 1 回(9 時) TRA/TIM PM ・1 日 1 回(21 時) いずれも1 回 1 滴両眼点眼 92 外国人 緑内障又は 高眼圧症患者 6 週間 完了 完全な報告書 安全性:評価資料 有効性:参考資料 第 III 相 米国 5.3.5.1.3 C-06* 5.3.5.1 外国人患者での有効性 及び安全性の検討 二重遮蔽 並行群間比較 実薬対照 TRA/TIM ・1 日 1 回(8 時) TRA ・1 日 1 回(20 時) TIM ・1 日 2 回(8、20 時) いずれも1 回 1 滴両眼点眼 263 外国人 緑内障又は 高眼圧症患者 6 ヵ月 完了 完全な報告書 安全性:評価資料 有効性:参考資料 第 III 相 米国 5.3.5.1.4 C-07* 5.3.5.1 外国人患者での有効性 及び安全性の検討 二重遮蔽 並行群間比較 実薬対照 TRA/TIM ・1 日 1 回(8 時) TRA+TIM ・TRA1 日 1 回(20 時) ・TIM 1 日 1 回(8 時) いずれも1 回 1 滴両眼点眼 316 外国人 緑内障又は 高眼圧症患者 6 ヵ月 完了 完全な報告書 安全性:評価資料 有効性:参考資料 第 III 相 米国 5.3.5.1.5 C-11* 5.3.5.1 外国人患者での有効性 及び安全性の検討 二重遮蔽 並行群間比較 実薬対照 TRA/TIM ・1 日 1 回(8 時) TRA+TIM ・TRA1 日 1 回(20 時) ・TIM 1 日 1 回(8 時) TIM ・1 日 2 回(8、20 時) 403 外国人 緑内障又は 高眼圧症患者 6 ヵ月 完了 完全な報告書 安全性:評価資料 有効性:参考資料
2.7.6 個々の試験のまとめ 3 試験の種類 実施国 試験番号 報告書の 添付場所 試験の目的 試験デザイン 及び対照 試験薬 投与方法 被験 者数 対象 投与 期間 試験の進行状況 報告書の種類 評価資料又は 参考資料の区分 第 III 相 EU 他 5.3.5.1.6 C-09* 5.3.5.1 外国人患者での有効性 及び安全性の検討 二重遮蔽 並行群間比較 実薬対照 TRA/TIM ・1 日 1 回(9 時) LAT/TIM ・1 日 1 回(9 時) いずれも1 回 1 滴両眼点眼 408 外国人 緑内障又は 高眼圧症患者 12 ヵ月 完了 完全な報告書 安全性:評価資料 有効性:参考資料 第 III 相 米国 5.3.5.4.1 C-03* 5.3.5.4 外国人患者での有効性 及び安全性の検討 二重遮蔽 並行群間比較 実薬対照 TRA TRA15 LAT ・1 日 1 回(20 時) TIM ・1 日 2 回(8、20 時) いずれも1 回 1 滴両眼点眼 801 外国人 緑内障又は 高眼圧症患者 12 ヵ月 完了 完全な報告書 参考資料 第Ⅲ相 米国 日本 5.3.5.1.7 C-15* C-16* 5.3.5.1 外 国 人 患 者 及 び日 本 人 患 者 で の 有 効 性及 び 安 全性の検討 二重遮蔽 並行群間比較 実薬対照 TRA/TIM BAC-free TRA/TIM いずれも 1 回 1 滴 1 日 1 回(9 時)、 両眼点眼 388 外国人:301 日本人:87 外国人及び日本人 緑内障又は 高眼圧症患者 6 週間 完了 完全な報告書 評価資料 本項で引用しない海外の臨床試験一覧については第 5.2 項に示す。 *:新薬承認審査情報提供時に置き換えた
2.7.6.1 日本人健康被験者を対象とした薬物動態試験・・・5.3.3.1.1(C-12*) 2.7.6.1.1 試験方法 試験方法の概略を表2.7.6.1.1-1 及び表 2.7.6.1.1-2 に示す。 表 2.7.6.1.1-1 試験方法の概略 項目 内容 目的 日本人健康被験者に対するトラボプロスト0.004%/チモロール 0.5%配合点眼液の安 全性、並びにトラボプロスト遊離酸代謝物(AL-5848)及びチモロールの血漿中濃度 について、トラボプロスト0.004%点眼液、チモロール 0.5%点眼液及びプラセボを単 独点眼した際の血漿中濃度と比較検討する。 デザイン 無作為化、二重遮蔽、並行群間比較、実薬及びプラセボ対照試験 治験対象 健康被験者 選択基準 対象:日本人(日系 3 世まで)の健康被験者 性別不問、年齢18 歳以上 除外基準 1. 妊娠、授乳婦、又は避妊不可の女性 2. 試験期間中コンタクトレンズの装用を中止できないもの 3. 片眼を摘出又は片眼の視力のないもの 4. 最高矯正視力が0.6logMARより悪いもの 5. PG製剤、β遮断薬又は本試験で使用する薬剤の成分に対するアレルギー又は過 敏症が疑われるもの 6. スクリーニング検査時及び点眼前日に、眼圧が10mmHg未満のもの 7. 慢性又は再発性の炎症性眼疾患の既往歴のあるもの(強膜炎、ぶどう膜炎、ヘ ルペス性角膜炎) 8. 過去6ヵ月以内に眼外傷の既往のあるもの 9. 過去3ヵ月以内に眼感染症又は眼内炎の既往のあるもの 10. 臨床上問題のある、又は進行性の網膜疾患(網膜変性、糖尿病網膜症、網膜剥 離など)の既往があるもの 11. 圧平眼圧計での眼圧測定に支障をきたすもの 12. 過去6ヵ月以内に内眼手術の既往のあるもの 13. 過去3ヵ月以内に眼科的レーザー手術の既往のあるもの 14. 処方薬又はOTC薬が投与されているもの(長期的に使用されているビタミン、 避妊薬あるいは類似薬(ホルモン補充療法)は除く) 15. スクリーニング検査日から14日前までに服薬されている全てのステロイド剤の 服薬を中止することのできないもの及び治験期間中にこれらの薬剤の服薬を止 められないもの 16. β遮断点眼液の点眼が容認できない重篤な慢性閉塞性肺疾患又は気管支喘息の 既往のあるもの 17. 眼又は鼻アレルギーを罹患しているもの 18. 身長を考慮した理想体重に対する体重比率が±25%を超えるもの 19. HIV陽性、B型肝炎、C型肝炎又はA型肝炎が陽性の既往のあるもの又はスクリ ーニング検査時に陽性となったもの 20. スクリーニング検査時に安静時脈拍が50/分未満又は110/分を超えるもの 21. スクリーニング検査時に安静時収縮期血圧が90~160mmHgの範囲外又は安静時 拡張期血圧が60~95mmHgの範囲外のもの 22. 臨床検査(血液、生化学、尿)において基準範囲を超える値につき治験責任医師が 臨床上問題と判断したもの
2.7.6 個々の試験のまとめ(5.3.3.1.1(C-12*)) 5 *:新薬承認審査情報提供時に置き換えた 表 2.7.6.1.1-1 試験方法の概略 項目 内容 24. 向精神薬、抗痙攣薬、アヘン性薬物を現在使用又は過去2年間において乱用歴の あるもの 25. 点眼前24時間以内及び治験期間中にアルコールを飲んだもの 26. 入院前30日以内に献血を行ったもの 27. スクリーニング検査日前72時間以内に血漿献血を行ったもの 28. 同意取得前30日以内に他の治験に参加したもの 29. 治験責任医師が本治験への参加が不適切又はリスクがあると判断したもの 投与群 及び 投与方法 1. トラボプロスト 0.004%/チモロール 0.5%配合点眼液:TRA/TIM 群 ・両眼に1 回 1 滴、1 日 1 回、午前(6-9 時)点眼 2. トラボプロスト 0.004%点眼液:TRA 群 ・両眼に1 回 1 滴、1 日 1 回、午前(6-9 時)点眼 3. チモロール 0.5%点眼液:TIM 群 ・両眼に1 回 1 滴、1 日 1 回、午前(6-9 時)点眼 4. プラセボ:VEH 群 ・両眼に1 回 1 滴、1 日 1 回、午前(6-9 時)点眼 (いずれの群も治験実施計画書にて鼻涙管圧迫の指示は行なわなかった) 投与期間 7 日間 観察項目 血漿中薬物濃度測定、眼圧、充血、フレア、前房細胞、最高矯正視力、瞳孔径、細 隙灯顕微鏡検査、眼底、脈拍、血圧、身体検査、臨床検査、有害事象 観察時期 表2.7.6.1.1-2 試験スケジュール参照 評価項目 薬物動態 単回投与と反復投与(7 日間投与)における AL-5848(トラボプロスト遊離酸代 謝物)及びチモロールの血漿中濃度並びに薬物動態パラメータ(Cmax、Cmin、Tmax、
AUC0-、AUC0-12*) *投与 24 時間後のチモロール血漿中濃度は、ほとんどの症例で定量限界未満のた め、AUC0-24 に代えて AUC0-12 を算出した。 安全性 検査所見及び有害事象により評価を行なう。 データセット 薬物動態 Safety データセット 治験薬が投与された症例のデータ。 Intent-to-Treat(ITT)データセット 治験薬が投与され、点眼開始より後に少なくとも1 回以上の観察・検査データが存 在する症例のデータ。 Per Protocol(PP)データセット 治験薬が投与され、点眼開始より後に少なくとも1 回以上の観察・検査データが存 在し、選択基準と除外基準を満たした症例のデータをPP とする。PP においては、 個々の観察・検査データにおいても、治験実施計画書で定められた基準に違反した 場合は除外される。 薬物動態の解析にはITT と PP を用いた。安全性の解析には Safety を用いた。 薬物動態の 解析方法
AL-5848 及びチモロールの血漿中濃度並びに薬物動態パラメータ(Cmax、Cmin、Tmax、
AUC0-、AUC0-12)について、測定時点ごとに記述統計量(算術平均、標準偏差、例
数、最小値及び最大値)を算出した。 目標症例数 1 群 10 例
実施施設 米国1 施設
表 2.7.6.1.1-2 試験スケジュール スクリーニング (7 日前~3 日前) 入院 (前日) 1 日目 2 日目 3 日目 4 日目 5・6 日目 7 日目 終了時 (8 日目) 同意取得 ○ 被験者背景 ○ 治療歴の確認 ○ ○ 選択除外基準の確認 ○ ○ 身長・体重 ○ 血圧・脈拍数 ○ ○ ○ 身体検査 ○ ○ 臨床検査 (血液、生化学、尿) ○ ○ 尿薬物スクリーニング検査 ○ 最高矯正視力 ○ ○ ○ ○ 瞳孔径・充血 ○ ○ ○ ○ 細隙灯顕微鏡検査 ○ ○ ○ ○ 眼圧 ○ ○ ○ ○ 眼底 ○ ○ 治験薬投与 ○ ○ ○ ○ ○ ○ 血液採取 ○1 ○2 ○2 ○3 ○4 有害事象 ○ ○ ○ ○ ○ ○ ○ 血漿中薬物濃度測定用の血液採取: 1 点眼前、及び点眼後5、10、15、30、45分、1、2、3、4、6、8、12時間 2 点眼前(前日の点眼から24時間後) 3 点眼前、及び点眼後5、10、15、30、45分、1、2、3、4、6、8、12時間 4 前日の点眼から24時間後 2.7.6.1.2 試験成績 1)症例の内訳
本試験に組み入れられた40 例(TRA/TIM 群 10 例、TRA 群 10 例、TIM 群 10 例、VEH 群 10 例) は、全て試験を完了し、安全性(Safety)及び薬物動態(ITT、PP)の解析対象となった。
2)背景因子
背景因子を表2.7.6.1.2-1 に示す。群間に背景因子の大きな偏りはみられなかった。
表 2.7.6.1.2-1 背景因子
TRA/TIM TRA TIM VEH
N % N % N % N % p-value Total 10 100.0 10 100.0 10 100.0 10 100.0 Age <65 10 100.0 8 80.0 9 90.0 10 100.0 0.59511) 65 0 0.0 2 20.0 1 10.0 0 0.0 Mean SD 39.9 9.4 46.9 14.6 43.3 13.6 47.1 11.6 0.52952) Sex Male 5 50.0 5 50.0 4 40.0 6 60.0 0.84951) Female 5 50.0 5 50.0 6 60.0 4 40.0 1) 2検定又はFisher の正確確率検定 2) 分散分析 3)薬物動態 ①AL-5848(トラボプロスト遊離酸代謝物)
TRA/TIM 又は TRA 点眼投与(1 日 1 回)後の血漿中 AL-5848(トラボプロスト遊離酸代謝物) 濃度が測定された。血漿中AL-5848 濃度が定量限界(10pg/mL)以上であったサンプルは、20 例
2.7.6 個々の試験のまとめ(5.3.3.1.1(C-12*))
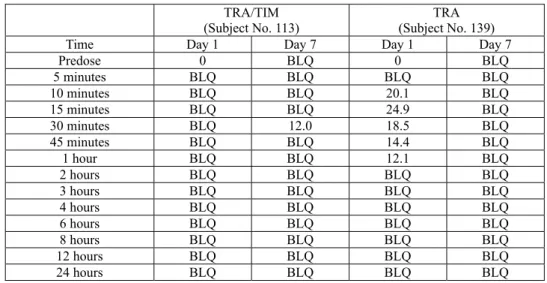
7 *:新薬承認審査情報提供時に置き換えた (TRA/TIM 群 10 例、TRA 群 10 例)中、TRA/TIM 群の 1 例(症例 113)1 サンプル及び TRA 群 の1 例(症例 139)5 サンプルのみであった(表 2.7.6.1.2-2)。TRA/TIM 群の 1 サンプルは 7 日目 (反復投与)の点眼30 分後のもので、血漿中濃度は 12pg/mL であった。他の 7 日目のサンプル及 び1 日目(単回投与)のサンプルは全て定量限界未満であった。TRA 群の 5 サンプルの血漿中濃 度は12.1~24.9pg/mL であり、いずれも 1 日目(単回投与)のものであった。Cmax(24.9pg/mL) は点眼15 分後であった。7 日目(反復投与)のサンプルについては、全て定量限界未満であった。 以上より、TRA/TIM 投与と TRA 投与の血漿中 AL-5848 濃度はともに極めて低く、蓄積性もみ られなかった。チモロールの配合は血漿中AL-5848 濃度に影響を及ぼさないことが示された。
表 2.7.6.1.2-2 血漿中 AL-5848 濃度(症例 113(TRA/TIM)及び症例 139(TRA))
TRA/TIM
(Subject No. 113) (Subject No. 139) TRA Time Day 1 Day 7 Day 1 Day 7
Predose 0 BLQ 0 BLQ 5 minutes BLQ BLQ BLQ BLQ 10 minutes BLQ BLQ 20.1 BLQ 15 minutes BLQ BLQ 24.9 BLQ 30 minutes BLQ 12.0 18.5 BLQ 45 minutes BLQ BLQ 14.4 BLQ 1 hour BLQ BLQ 12.1 BLQ 2 hours BLQ BLQ BLQ BLQ 3 hours BLQ BLQ BLQ BLQ 4 hours BLQ BLQ BLQ BLQ 6 hours BLQ BLQ BLQ BLQ 8 hours BLQ BLQ BLQ BLQ 12 hours BLQ BLQ BLQ BLQ 24 hours BLQ BLQ BLQ BLQ
BLQ=Less than the limit of quantitation (<LLOQ (10.0) pg/mL)
②チモロール TRA/TIM(10 例)又は TIM(10 例)点眼投与(1 日 1 回)後の血漿中チモロール濃度が測定さ れた。TIM 群の 1 例(症例 118)において、他の症例に比べ非常に高い血漿中チモロール濃度を 示した。その理由として、この症例の全身性のバイオアベイラビリティが他の症例に比べ著しく 高いことによるものと考えられたことから、TIM 群については症例 118 を含めた場合と除外した 場合の2 種類の分析が行なわれた。 <単回投与> TRA/TIM 又は TIM 単回投与後の血漿中チモロール濃度の推移を図 2.7.6.1.2-1 及び図 2.7.6.1.2-2 に、薬物動態パラメータを表2.7.6.1.2-3 に示す。 血漿中チモロール濃度は、TRA/TIM 群では 2 時間までに、TIM 群では 3 時間までにピークを迎 えた。
TIM 群(10 例)の平均 Cmaxは0.835 0.428 ng/mL であり、TRA/TIM 群(10 例)の 0.694 0.449
ng/mL に比べ 20%程度高かったが、症例 118 を除いて算出した TIM 群(9 例)の平均 Cmaxは0.722
0.250 ng/mL であり、TRA/TIM 群と大きな差はみられなかった。
かった。 図 2.7.6.1.2-1 単回投与時(1 日目)における血漿中チモロール濃度(平均値、SD)の推移 TRA/TIM(10 例)TIM(10 例:症例 118 含む)
0
6
12
18
24
Time (h)
0
0.2
0.4
0.6
0.8
1
1.2
1.4
1.6
T
im
olo
l C
on
ce
ntr
atio
n (
ng
/m
L
)
Timolol
Travoprost/Timolol
図 2.7.6.1.2-2 単回投与時(1 日目)における血漿中チモロール濃度(平均値、SD)の推移 TRA/TIM(10 例)TIM(9 例:症例 118 除外)0
6
12
18
24
Time (h)
0
0.2
0.4
0.6
0.8
1
1.2
1.4
1.6
T
im
ol
ol
Conc
en
tra
tion (ng/
m
L
)
Timolol
Travoprost/Timolol
2.7.6 個々の試験のまとめ(5.3.3.1.1(C-12*))
9 *:新薬承認審査情報提供時に置き換えた 表 2.7.6.1.2-3 単回投与時(1 日目)におけるチモロールの薬物動態パラメータ
投与群 Cmax
(ng/mL) Tmax (h) (ng*h/mL) AUC0-12 (ng*h/mL) AUC0-∞
t1/2 (h) TRA/TIM N=10 Mean 0.694 0.5 3.60 4.20 3.9 SD 0.449 0.6 2.03 2.45 1.5 Min 0.135 0.1 0.517 0.552 2.2 Max 1.71 2.0 6.15 7.90 6.9 TIM N=10 症例118 含む Mean 0.835 0.6 3.61 4.39 4.8 SD 0.428 0.9 1.71 2.20 1.9 Min 0.290 0.1 0.892 1.32 2.2 Max 1.85 3.0 6.02 8.16 7.6 TIM N=9 症例118 除外 Mean 0.722 0.6 3.35 4.18 5.0 SD 0.250 1.0 1.58 2.21 1.8 Min 0.290 0.1 0.892 1.32 2.2 Max 1.15 3.0 5.74 8.16 7.6 <反復投与> TRA/TIM 又は TIM 反復(7 日間)投与後の血漿中チモロール濃度の推移を図 2.7.6.1.2-3 及び図 2.7.6.1.2-4 に、薬物動態パラメータを表 2.7.6.1.2-4 に示す。 血漿中チモロール濃度は、両群ともに2 時間までにピークを迎えた。 TIM 群(10 例)の平均血漿中チモロール濃度は、TRA/TIM 群(10 例)のものと比べて高かっ たが、その差は症例 118 の血漿中チモロール濃度が他の症例に比べ著しく高いことによるもので あり、症例118 を除外して算出した TIM 群(9 例)の平均血漿中チモロール濃度は、TRA/TIM 群 と大きな差はみられなかった。
また、Cmax(平均値±SD)についても、TIM 群(10 例)の平均 Cmax (0.913 0.780 ng/mL)は
TRA/TIM 群(10 例)の平均 Cmax(0.704 0.407 ng/mL)に比べ 30%高かったが、症例 118 を除い
て算出したTIM 群(9 例)の平均 Cmaxは0.693 0.370 ng/mL であり、TRA/TIM 群と大きな差はみ
られなかった。AUC 及び t1/2についても、症例118 を除いて算出した場合には、TRA/TIM 群と TIM
群の間に大きな差はみられなかった。
Cmax及びAUC0-∞の7 日目/1 日目比を表 2.7.6.1.2-5 に示す。TRA/TIM 群及び TIM 群の Cmaxの7
日目/1 日目比(平均値SD)は、それぞれ 1.290.889 及び 1.080.473 であり、明らかな蓄積性は みられなかった。
以上より、TRA/TIM と TIM 反復投与時の血漿中のチモロール薬物動態に大きな差はみられな かった。
図 2.7.6.1.2-3 反復投与時(7 日目)における血漿中チモロール濃度(平均値、SD)の推移 TRA/TIM(10 例)TIM(10 例:症例 118 含む)
0
6
12
18
24
Time (h)
0
0.2
0.4
0.6
0.8
1
1.2
1.4
1.6
T
im
ol
ol
C
on
cen
trat
io
n (
ng
/m
L
)
Timolol
Travoprost/Timolol
図 2.7.6.1.2-4 反復投与時(7 日目)における血漿中チモロール濃度(平均値、SD)の推移 TRA/TIM(10 例)TIM(9 例:症例 118 除外)0
6
12
18
24
Time (h)
0
0.2
0.4
0.6
0.8
1
1.2
1.4
1.6
T
im
ol
ol
Conc
en
tra
tion (
ng/
m
L
)
Timolol
Travoprost/Timolol
2.7.6 個々の試験のまとめ(5.3.3.1.1(C-12*))
11 *:新薬承認審査情報提供時に置き換えた
表 2.7.6.1.2-4 反復投与時(7 日目)におけるチモロールの薬物動態パラメータ
投与群 Cmax
(ng/mL) Tmax (h) (ng*h/mL) AUC0-12 (ng*h/mL) AUC0-∞
t1/2 (h) TRA/TIM N=10 Mean 0.704 0.6 3.46 4.15 4.7 SD 0.407 0.8 2.56 3.03 1.3 Min 0.133 0.1 0.746 0.859 2.5 Max 1.62 2.0 8.16 9.10 6.7 TIM N=10 症例118 含む Mean 0.913 0.3 4.05 5.02 5.5 SD 0.780 0.2 2.85 3.21 1.7 Min 0.150 0.1 0.945 1.14 3.2 Max 2.90 0.5 10.9 12.4 8.9 TIM N=9 症例118 除外 Mean 0.693 0.3 3.29 4.20 5.5 SD 0.370 0.2 1.62 2.01 1.8 Min 0.150 0.1 0.945 1.14 3.2 Max 1.47 0.5 5.31 6.74 8.9 表 2.7.6.1.2-5 チモロールの Cmax及び AUC0-∞の 7 日目/1 日目比 1 日目 7 日目 7 日目/1 日目 TRA/TIM C max (ng/mL) Mean 0.694 0.704 1.29 SD 0.449 0.407 0.889 N 10 10 Min 0.135 0.133 Max 1.71 1.62 AUC0-∞ (ng*h/mL) Mean 4.20 4.15 1.14 SD 2.45 3.03 0.649 N 9 10 Min 0.552 0.859 Max 7.90 9.10 TIM C max (ng/mL) Mean 0.835 0.913 1.08 SD 0.428 0.780 0.473 N 10 10 Min 0.290 0.150 Max 1.85 2.90 AUC0-∞ (ng*h/mL) Mean 4.39 5.02 1.25 SD 2.20 3.21 0.660 N 10 10 Min 1.32 1.14 Max 8.16 12.4 ND = not determined 4)安全性 ①有害事象 程度別の有害事象発現率の一覧を本2.7.6.1 項末(表 2.7.6.1.2-7~表 2.7.6.1.2-10)に示す。 安全性解析対象40 例(TRA/TIM 群 10 例、TRA 群 10 例、TIM 群 10 例、VEH 群 10 例)のうち、 有害事象はTRA/TIM 群 3 例(30.0%)、TRA 群 5 例(50.0%)、TIM 群 3 例(30.0%)、VEH 群 1 例 (10.0%)にみられた。死亡例、重篤な有害事象及び有害事象による中止例はみられなかった。 治験薬との関連性を否定できない有害事象は、TRA/TIM 群 2 例(20.0%)、TRA 群 4 例(40.0%)、 TIM 群 1 例(10.0%)にみられた。VEH 群にはみられなかった。 眼局所の治験薬との関連性を否定できない有害事象のうち、最も多くみられたものは眼充血で あり、TRA/TIM 群 2 例(20.0%)、TRA 群 3 例(30.0%)であった。いずれも軽度で無治療にて消 失に至った。その他に、眼そう痒症がTRA 群で 2 例(20.0%)みられた(いずれも軽度で治療に
て消失)。これら以外には、複数例みられた有害事象はなかった。 眼局所以外の治験薬との関連性を否定できない有害事象は、TRA 群 1 例(γ-グルタミルトラ ンスフェラーゼ増加、アラニン・アミノトランスフェラーゼ増加)及びTIM 群 1 例(頭痛)であ った。いずれも軽度で無治療にて消失に至った。 ②PG 製剤に特徴的な所見(眼の充血) 眼充血は、ベースライン及び各検査日に、症状の程度順にスコア0~3 とグレーディングされた 4 枚の充血の標準写真と照合し、0.5 刻みの 7 段階のスコアで評価した。有害事象の判定基準は、 「充血により試験を中止した場合、又は患者から訴えが出た場合」とした。 充血スコアがベースラインから1 以上悪化した症例は、TRA/TIM 群 5 例(50.0%)、TRA 群 6 例(60.0%)であり、TIM 群と VEH 群ではみられなかった。TRA/TIM 群と TRA 群に大きな差は みられなかった。 表 2.7.6.1.2-6 眼充血スコアの 1 以上の悪化がみられた症例 Total 充血スコア1 以上悪化 N N % Total 40 11 27.5 TRA/TIM 10 5 50.0 TRA 10 6 60.0 TIM 10 0 0.0 VEH 10 0 0.0
p = 1.0 (TRA/TIM vs. TRA) Fisher の正確確率検定 p = 0.0325 (TRA/TIM vs. TIM) Fisher の正確確率検定 p = 0.0325 (TRA/TIM vs. VEH) Fisher の正確確率検定
③脈拍、血圧 脈拍及び血圧は、ベースライン及び終了時に測定された。有害事象の判定基準は、「ベースライ ン検査時に比べ、臨床上重要な変化が認められたと治験責任医師等により判断された場合」とし た。臨床上意義のある変化は、脈拍及び血圧ともにみられなかった。 ④眼科検査所見 角膜所見の悪化がTRA 群 1 例(10.0%)にみられ、治験薬との関連性を否定できない有害事象 (点状角膜炎)として取り上げられた(軽度で無治療にて継続)。 その他、視力、虹彩/前房所見、水晶体、瞳孔径、眼底(網膜/黄斑/脈絡膜、視神経、硝子体) 所見、C/D 比、眼圧については、治験薬との関連性を否定できない悪化はいずれの投与群でもみ られなかった。 ⑤臨床検査 程度別の臨床検査値異常変動発現率の一覧を本2.7.6.1 項末(表 2.7.6.1.2-11)に示す。 有害事象の判定基準は、「点眼開始前検査時に比べ、臨床上重要な変化が認められたと治験責任 医師等が判断した場合」とした。
2.7.6 個々の試験のまとめ(5.3.3.1.1(C-12*)) 13 *:新薬承認審査情報提供時に置き換えた TRA/TIM 群の 1 例(10.0%)に、尿検査において尿中白血球エステラーゼ上昇、尿蛋白、尿潜 血及び尿細菌がみられ、尿路感染として有害事象に取り上げられた。軽度で無治療にて消失し、 治験薬との関連性は否定された。 TRA 群の 1 例(10.0%)に γ-グルタミルトランスフェラーゼとアラニン・アミノトランスフェ ラーゼの上昇がみられ、治験薬との関連性を否定できない有害事象として取り上げられた。軽度 で無治療にて消失に至った。 2.7.6.1.3 結論 血漿中のAL-5848(トラボプロスト遊離酸代謝物)濃度は、トラボプロスト 0.004%/チモロール 0.5%配合点眼液(1 日 1 回点眼)及びトラボプロスト 0.004%点眼液(1 日 1 回点眼)ともに極め て低かった。 血漿中のチモロール薬物動態は、トラボプロスト 0.004%/チモロール 0.5%配合点眼液(1 日 1 回点眼)とチモロール0.5%点眼液(1 日 1 回点眼)で大きな差はみられなかった。 トラボプロスト 0.004%/チモロール 0.5%配合点眼液(1 日 1 回点眼)の日本人健康被験者に対 する安全性が確認された。
14 2.7.6 個々の試験のまとめ( 5.3.5.1.1 ( C-1 2 *) ) 表 2.7.6.1.2-7 程度別有害事象一覧表 (全ての有害事象・眼局所) 全ての有害事象
安全性評価対象例:40 例 TRA/TIM (10) TRA (10) TIM (10) VEH (10)
有害事象 軽度 中等度 重度 計 軽度 中等度 重度 計 軽度 中等度 重度 計 軽度 中等度 重度 計 有害事象発現例数 3 3 4 1 5 3 3 1 1 有害事象発現率 30.0 30.0 40.0 10.0 50.0 30.0 30.0 10.0 10.0 有害事象発現件数 3 3 11 1 12 4 4 1 1 発現例数(眼局所) 2 2 4 4 3 3 発現率(眼局所) 20.0 20.0 40.0 40.0 30.0 30.0 発現件数(眼局所) 2 2 9 9 3 3 有害事象項目 N % N % N % N % N % N % N % N % N % N % N % N % N % N % N % N % 眼局所 眼充血 2 20.0 2 20.0 3 30.0 3 30.0 眼そう痒症 2 20.0 2 20.0 霧視 1 10.0 1 10.0 点状角膜炎 1 10.0 1 10.0 眼瞼そう痒症 1 10.0 1 10.0 眼刺激 1 10.0 1 10.0 硝子体浮遊物 1 10.0 1 10.0 皮質白内障 1 10.0 1 10.0 眼の異常感 1 10.0 1 10.0
15 2.7.6 個々の試験のまとめ( 5.3.5.1.1 ( C-1 2 *) ) *:新薬承認審査情報提供時に置き換えた 表 2.7.6.1.2-8 程度別有害事象一覧表 (全ての有害事象・眼局所以外) 全ての有害事象
安全性評価対象例:40 例 TRA/TIM (10) TRA (10) TIM (10) VEH (10)
有害事象 軽度 中等度 重度 計 軽度 中等度 重度 計 軽度 中等度 重度 計 軽度 中等度 重度 計 有害事象発現例数 3 3 4 1 5 3 3 1 1 有害事象発現率 30.0 30.0 40.0 10.0 50.0 30.0 30.0 10.0 10.0 有害事象発現件数 3 3 11 1 12 4 4 1 1 発現例数(眼局所以外) 1 1 1 1 2 1 1 1 1 発現率(眼局所以外) 10.0 10.0 10.0 10.0 20.0 10.0 10.0 10.0 10.0 発現件数(眼局所以外) 1 1 2 1 3 1 1 1 1 有害事象項目 N % N % N % N % N % N % N % N % N % N % N % N % N % N % N % N % 眼局所以外 感染症及び寄生虫症 尿路感染 1 10.0 1 10.0 神経系障害 頭痛 1 10.0 1 10.0 1 10.0 1 10.0 1 10.0 1 10.0 臨床検査 γ-グルタミルトランスフ ェラーゼ増加 1 10.0 1 10.0 アラニン・アミノトランス フェラーゼ増加 1 10.0 1 10.0
16 2.7.6 個々の試験のまとめ( 5.3.5.1.1 ( C-1 2 *) ) 表 2.7.6.1.2-9 程度別有害事象一覧表 (関連性の否定出来ない有害事象・眼局所) 関連性の否定出来ない有害事象
安全性評価対象例:40 例 TRA/TIM (10) TRA (10) TIM (10) VEH (10)
有害事象 軽度 中等度 重度 計 軽度 中等度 重度 計 軽度 中等度 重度 計 軽度 中等度 重度 計 有害事象発現例数 2 2 4 4 1 1 有害事象発現率 20.0 20.0 40.0 40.0 10.0 10.0 有害事象発現件数 2 2 11 11 2 2 発現例数(眼局所) 2 2 4 4 1 1 発現率(眼局所) 20.0 20.0 40.0 40.0 10.0 10.0 発現件数(眼局所) 2 2 9 9 1 1 有害事象項目 N % N % N % N % N % N % N % N % N % N % N % N % N % N % N % N % 眼局所 眼充血 2 20.0 2 20.0 3 30.0 3 30.0 眼そう痒症 2 20.0 2 20.0 霧視 1 10.0 1 10.0 点状角膜炎 1 10.0 1 10.0 眼瞼そう痒症 1 10.0 1 10.0 眼刺激 1 10.0 1 10.0 硝子体浮遊物 皮質白内障 眼の異常感 1 10.0 1 10.0
17 2.7.6 個々の試験のまとめ( 5.3.5.1.1 ( C-1 2 *) ) *:新薬承認審査情報提供時に置き換えた 表 2.7.6.1.2-10 程度別有害事象一覧表 (関連性の否定出来ない有害事象・眼局所以外) 関連性の否定出来ない有害事象
安全性評価対象例:40 例 TRA/TIM (10) TRA (10) TIM (10) VEH (10)
有害事象 軽度 中等度 重度 計 軽度 中等度 重度 計 軽度 中等度 重度 計 軽度 中等度 重度 計 有害事象発現例数 2 2 4 4 1 1 有害事象発現率 20.0 20.0 40.0 40.0 10.0 10.0 有害事象発現件数 2 2 11 11 2 2 発現例数(眼局所以外) 1 1 1 1 発現率(眼局所以外) 10.0 10.0 10.0 10.0 発現件数(眼局所以外) 2 2 1 1 有害事象項目 N % N % N % N % N % N % N % N % N % N % N % N % N % N % N % N % 眼局所以外 感染症及び寄生虫症 尿路感染 神経系障害 頭痛 1 10.0 1 10.0 臨床検査 γ-グルタミルトランスフェ ラーゼ増加 1 10.0 1 10.0 アラニン・アミノトランスフ ェラーゼ増加 1 10.0 1 10.0
表 2.7.6.1.2-11 程度別の臨床検査値異常変動発現率(TRA/TIM) TRA/TIM (10) 全ての臨床検査値異常変動 関連性の否定できない臨床検査値異常変動 臨床検査値異常変動 軽度 中等度 重度 計 軽度 中等度 重度 計 安全性評価対象例:40例 異常変動発現例数 1 1 異常変動発現率 10.0 10.0 異常変動発現件数 4 4 臨床検査項目 例数* 変動 N % N % N % N % N % N % N % N % 血液生化学的検査 ALT(GPT) 10 上昇 低下 γ-GTP 10 上昇 低下 尿検査 尿中白血球 10 上昇 1 10.0 1 10.0 エステラーゼ 低下 尿蛋白 10 上昇 1 10.0 1 10.0 低下 尿潜血 10 上昇 1 10.0 1 10.0 低下 尿細菌 10 上昇 1 10.0 1 10.0 低下 *安全性評価対象のうち、試験前後で検査を実施した症例 他の血液学的検査(白血球数、赤血球数、ヘモグロビン量、ヘマトクリット値、MCV、MCH、MCHC、RDW、血小板数、好中 球、リンパ球、単球、好酸球、好塩基球)、血液生化学的検査(血糖、BUN、クレアチニン、尿酸、ナトリウム、カリウム、, ク ロール、CO2、無機リン、AST(GOT)、LDH、ALT(GPT)、ALP、γ-GTP、ビリルビン、総蛋白、アルブミン、グロブリン、 A/G比、総コレステロール、中性脂肪、血清鉄)及び尿検査(尿色、尿外観、尿比重、尿pH、尿糖、尿ケトン体、尿中ウロビリ ノーゲン、尿中亜硝酸塩)に異常変動は認められなかった。 表 2.7.6.1.2-11 程度別の臨床検査値異常変動発現率(TRA) TRA (10) 全ての臨床検査値異常変動 関連性の否定できない臨床検査値異常変動 臨床検査値異常変動 軽度 中等度 重度 計 軽度 中等度 重度 計 安全性評価対象例:40例 異常変動発現例数 1 1 1 1 異常変動発現率 10.0 10.0 10.0 10.0 異常変動発現件数 2 2 2 2 臨床検査項目 例数* 変動 N % N % N % N % N % N % N % N % 血液生化学的検査 ALT(GPT) 10 上昇 1 10.0 1 10.0 1 10.0 1 10.0 低下 γ-GTP 10 上昇 1 10.0 1 10.0 1 10.0 1 10.0 低下 尿検査 尿中白血球 10 上昇 エステラーゼ 低下 尿蛋白 10 上昇 低下 尿潜血 10 上昇 低下 尿細菌 10 上昇 低下 *安全性評価対象のうち、試験前後で検査を実施した症例 他の血液学的検査(白血球数、赤血球数、ヘモグロビン量、ヘマトクリット値、MCV、MCH、MCHC、RDW、血小板数、好中 球、リンパ球、単球、好酸球、好塩基球)、血液生化学的検査(血糖、BUN、クレアチニン、尿酸、ナトリウム、カリウム、, ク ロール、CO2、無機リン、AST(GOT)、LDH、ALP、ビリルビン、総蛋白、アルブミン、グロブリン、A/G比、総コレステロー ル、中性脂肪、血清鉄)及び尿検査(尿色、尿外観、尿比重、尿pH、尿蛋白、尿糖、尿ケトン体、尿中ウロビリノーゲン、尿 潜血、尿中白血球エステラーゼ、尿中亜硝酸塩、尿細菌)に異常変動は認められなかった。
2.7.6 個々の試験のまとめ(5.3.3.1.1(C-12*)) 19 *:新薬承認審査情報提供時に置き換えた 表 2.7.6.1.2-11 程度別の臨床検査値異常変動発現率(TIM) TIM (10) 全ての臨床検査値異常変動 関連性の否定できない臨床検査値異常変動 臨床検査値異常変動 軽度 中等度 重度 計 軽度 中等度 重度 計 安全性評価対象例:40例 異常変動発現例数 異常変動発現率 異常変動発現件数 臨床検査項目 例数* 変動 N % N % N % N % N % N % N % N % 血液生化学的検査 ALT(GPT) 10 上昇 低下 γ-GTP 10 上昇 低下 尿検査 尿中白血球 10 上昇 エステラーゼ 低下 尿蛋白 10 上昇 低下 尿潜血 10 上昇 低下 尿細菌 10 上昇 低下 *安全性評価対象のうち、試験前後で検査を実施した症例 他の血液学的検査(白血球数、赤血球数、ヘモグロビン量、ヘマトクリット値、MCV、MCH、MCHC、RDW、血小板数、好中 球、リンパ球、単球、好酸球、好塩基球)、血液生化学的検査(血糖、BUN、クレアチニン、尿酸、ナトリウム、カリウム、, ク ロール、CO2、無機リン、AST(GOT)、LDH、ALT(GPT)、ALP、γ-GTP、ビリルビン、総蛋白、アルブミン、グロブリン、 A/G比、総コレステロール、中性脂肪、血清鉄)及び尿検査(尿色、尿外観、尿比重、尿pH、尿蛋白、尿糖、尿ケトン体、尿中 ウロビリノーゲン、尿潜血、尿中白血球エステラーゼ、尿中亜硝酸塩、尿細菌)に異常変動は認められなかった。 表 2.7.6.1.2-11 程度別の臨床検査値異常変動発現率(VEH) VEH (10) 全ての臨床検査値異常変動 関連性の否定できない臨床検査値異常変動 臨床検査値異常変動 軽度 中等度 重度 計 軽度 中等度 重度 計 安全性評価対象例:40例 異常変動発現例数 異常変動発現率 異常変動発現件数 臨床検査項目 例数* 変動 N % N % N % N % N % N % N % N % 血液生化学的検査 ALT(GPT) 10 上昇 低下 γ-GTP 10 上昇 低下 尿検査 尿中白血球 10 上昇 エステラーゼ 低下 尿蛋白 10 上昇 低下 尿潜血 10 上昇 低下 尿細菌 10 上昇 低下 *安全性評価対象のうち、試験前後で検査を実施した症例 他の血液学的検査(白血球数、赤血球数、ヘモグロビン量、ヘマトクリット値、MCV、MCH、MCHC、RDW、血小板数、好中 球、リンパ球、単球、好酸球、好塩基球)、血液生化学的検査(血糖、BUN、クレアチニン、尿酸、ナトリウム、カリウム、, ク ロール、CO2、無機リン、AST(GOT)、LDH、ALT(GPT)、ALP、γ-GTP、ビリルビン、総蛋白、アルブミン、グロブリン、 A/G比、総コレステロール、中性脂肪、血清鉄)及び尿検査(尿色、尿外観、尿比重、尿pH、尿蛋白、尿糖、尿ケトン体、尿中 ウロビリノーゲン、尿潜血、尿中白血球エステラーゼ、尿中亜硝酸塩、尿細菌)に異常変動は認められなかった。
2.7.6.2 トラボプロスト 0.004%/チモロール 0.5%の配合点眼液に関する外国人健康被験者を対象 とした PK 試験・・・・5.3.3.1.2(C-10*) 2.7.6.2.1 試験方法 試験方法の概略を表 2.7.6.2.1-1 及び表 2.7.6.2.1-2 に示す。 表 2.7.6.2.1-1 試験方法の概略 項目 内容 目的 健康被験者を対象に、トラボプロスト 0.004%/チモロール 0.5%の配合点眼液を 1 日 1 回、3 日間点眼した際のトラボプロスト遊離酸代謝物(AL-5848)又はチモロールの 血漿中濃度について、トラボプロスト 0.004%点眼液又はチモロール 0.5%点眼液を単 独点眼した際の血漿中濃度と比較検討する。 デザイン 無作為化二重遮蔽クロスオーバー(3 期 3 剤) 治験対象 健康被験者 選択基準 対象:健康被験者 年齢:18 歳以上 性別:不問 その他:点眼開始 3 日前からの禁酒、点眼開始前日からのカフェイン摂取の禁止及び 点眼開始前臨床検査の実施にあたり、検査 10 時間前からの絶食が可能なも の 除外基準 1. 妊婦、授乳婦、又は避妊不可の女性 2. 6 ヵ月以内に神経系疾患、心血管系疾患、免疫疾患、消化器疾患、血液疾患、肝 疾患、腎疾患の既往がある、又はスクリーニング時に実施する身体検査及び臨床 検査の結果から、重篤疾患を罹患し、治験責任医師が治験参加は容認できないと 判断したもの 3. 点眼開始 7 日前からコンタクトレンズの装用を中止できないもの(眼鏡は可) 4. 片眼を摘出した又は片眼の視力がないもの 5. 最高矯正視力が 0.6 logMAR より悪いもの 6. スクリーニング検査時眼圧が 10mmHg 未満のもの 7. PG 製剤、β遮断薬、又は本試験で使用する薬剤の含有成分に対するアレルギー 又は過敏症が疑われるもの 8. β遮断点眼液の投与が容認できない気管支喘息又は重篤な COPD の既往がある もの 9. 治験責任医師が治験参加は容認できないと判断した全身疾患又は眼疾患(肝炎、 急性又は慢性腎不全、ヘルペスウイルス感染症、アカントアメーバ感染症等)が あるもの 10. 身長を考慮した理想体重に対する体重比率が±25%を超えるもの 11. HIV 陽性、あるいはB型、C型もしくは A 型肝炎陽性の既往があるもの、又はス クリーニング検査時に実施する臨床検査において陽性であるもの 12. 点眼開始前 2 週間以内に処方薬又は OTC 薬を使用したもの(ホルモン避妊薬又 はこれに代わる避妊薬は除く) 13. 点眼開始前 30 日以内に全身ステロイド剤を使用したもの 14. 点眼開始前 90 日以内に免疫療法を行ったもの 15. スクリーニング検査時に眼又は鼻アレルギーを罹患しているもの 16. 薬物濫用歴がある、又はスクリーニング時及びベースライン時に実施する検査に て薬物濫用者であることが判明したもの 17. 過去 2 年間において向精神薬、抗痙攣薬の使用歴、あるいはアヘン性薬物の乱用
2.7.6 個々の試験のまとめ(5.3.3.1.2(C-10*)) 21 *:新薬承認審査情報提供時に置き換えた 表 2.7.6.2.1-1 試験方法の概略 項目 内容 18. 点眼開始前 30 日以内に献血をしたもの 19. スクリーニング検査前 3 日以内に血漿献血を行ったもの 20. 同意取得前 30 日以内に他の治験に参加したもの 21. スクリーニング検査 30 日以内に煙草を服用したもの 投与群 及び 投与方法 ・トラボプロスト 0.004%/チモロール 0.5%配合点眼液 ・トラボプロスト 0.004%点眼液 ・チモロール 0.5%点眼液 1 日 1 回、両眼に 1 回 1 滴、午前 6 時~9 時に両眼に点眼する。 (点眼時刻は各治療期を通じ同一とする) (いずれの群も治験実施計画書にて鼻涙管圧迫の指示は行なわなかった) 投与期間 1 剤あたり 3 日間、計 9 日間 第Ⅰ期~第Ⅲ期の 3 治療期を設け、上記 3 薬剤を各治療期に 1 剤ずつ割付ける。 なお、各治療期間で 5~10 日の休薬期間を設ける。 観察項目 血漿中薬物濃度測定、最高矯正視力、眼底、細隙灯顕微鏡検査、眼圧、有害事象 観察時期 表 2.7.6.2.1-2 試験スケジュール参照 評価項目 薬物動態 トラボプロスト 0.004%/チモロール 0.5%の配合点眼液の投与時における各薬物 動態パラメータ(血漿中濃度及び Cmax、Tmax、t1/2、AUC0-12、AUC0-)について、
トラボプロスト 0.004%点眼液又はチモロール 0.5%点眼液を単独点眼した際の各 薬物動態パラメータと比較検討する。 安全性 検査所見及び有害事象により評価を行う。 データセット 薬物動態 ITT 治験薬が投与され、点眼開始より後に少なくとも 1 回以上の採血が実施され、 適切な PK データが存在する症例のデータ。 PP 治験薬が投与され、点眼開始より後に少なくとも 1 回以上の採血が実施され、 適切な PK データが存在し、治験実施計画書にて定めた基準を満たした症例の データを Per Protocol とする。Per Protocol においては、個々の観察・検査デー タにおいても、治験実施計画書で定められた基準に違反した場合(例:併用禁 止薬の使用)除外される。 安全性 治験薬が投与された症例のデータ。 有効性の 解析方法 各薬物動態パラメータについて、対応のある t-検定により投与群間の比較を行った (有意水準:両側 5%)。また、AUC と Cmaxについて、投与群ごとに幾何平均を算出 し、トラボプロスト 0.004%/チモロール 0.5%配合点眼液群とチモロール 0.5%点眼 液群の幾何平均の比の 90%信頼区間を算出した。 目標症例数 15 例(トラボプロスト 0.004%/チモロール 0.5%配合点眼液、トラボプロスト 0.004% 点眼液、チモロール 0.5%点眼液より成る 3 つの投与順序に 5 例ずつ割り付けた) 実施施設 米国 1 施設 治験期間 20xx 年 xx 月 xx 日~20xx 年 xx 月 xx 日
表 2.7.6.2.1-2 試験スケジュール スクリーニング 入院 Ⅰ期・Ⅱ期・Ⅲ期 5~10 日 休薬後 終了時 21 日前~ 3 日前 前日 1・9・17 日目 2・10・18 日目 3・11・19 日目 8・16 日目 入院 19 日目又 は中止時 同意取得 ○ 被験者背景 ○ 治療歴の確認 ○ ○ 選択除外基準の確認 ○ ○ 身長・体重 ○ 身体検査 ○ 血圧・脈拍数 ○ 肝炎ウイルス及び HIV 検査 ○ 臨床検査 (血液、生化学、尿) ○ 尿薬物スクリーニング検査 ○ ○ ○ 最高矯正視力 ○ ○ ○ ○ ○ 眼底 ○ ○ 細隙灯顕微鏡検査 ○ ○ ○ ○ ○ 眼圧 ○ ○ ○ ○ ○ 血液採取 ○1) ○2) ○1) 有害事象 ○ ○ ○ ○ ○ 治験薬投与 ○ ○ ○ 1)血漿中薬物濃度測定用の血液採取は、1、3、9、11、17、19 日目の点眼前、点眼後 5、10、15、30、45 分後、 1、2、3、4、6、8、12 時間後に行う 2)2 日目、10 日目、18 日目の血液採取は点眼前のみ実施 2.7.6.2.2 試験成績 1)症例の内訳 本試験に組み入れられた 15 例は、全例が薬物動態及び安全性の解析対象となったが、1 例の被 験者は、TRA 及び TRA/TIM の投与を完了した試験開始 16 日目での薬物スクリーニングテストに おいて、陽性反応が認められたために試験中止となり、TIM は投与されなかった。このため、こ の 1 例は、TIM 群の各データセットから除外された。
2.7.6 個々の試験のまとめ(5.3.3.1.2(C-10*)) 23 *:新薬承認審査情報提供時に置き換えた 2)背景因子 解析対象となった 15 例の背景因子を表 2.7.6.2.2-1 に示す。 表 2.7.6.2.2-1 解析対象例の背景因子 被験者数 15 年齢 <65 12 65 3 平均年齢 38.9 標準偏差 20.2 性別 男 8 女 7 人種 白人 13 黒人 1 ヒスパニック 1 虹彩色 茶 7 ヘーゼル 2 緑 1 青 3 灰 2 3)薬物動態 ①AL-5848(トラボプロスト遊離酸代謝物) トラボプロストは点眼後に角膜で加水分解され、活性本体はその遊離酸代謝物(AL-5848)と なるため、血漿中 AL-5848 濃度が測定された。
TRA/TIM 又は TRA の投与後における AL-5848 の血漿中濃度を表 2.7.6.2.2-2 に示す。
試験期間を通じて、AL-5848 の血漿中濃度が定量限界(10pg/mL)以上であった血漿サンプル は、4 例から採取された 18 サンプルのみであり、これら 18 サンプルの血漿中濃度も 10~20pg/mL と低かった。
TRA/TIM 群と TRA 群との間で、血漿中 AL-5848 濃度に違いはみられず、TIM の併用は、AL-5848 の全身血中移行性に影響を及ぼさないことが示唆された。 単回投与時(投与 1 日目)と反復投与時(投与 3 日目)との間で、血漿中 AL-5848 濃度に差は みられず、また、投与 1 時間後の血漿中濃度は、ほとんど定量限界未満と速やかに消失しており、 AL-5848 の蓄積性はみられなかった。 なお、ほとんどのサンプルにおいて、AL-5848 濃度が定量限界未満であったことから、各種薬 物動態パラメータに関する幾何平均値解析は行わなかった。
表 2.7.6.2.2-2 血漿中 AL-5848 濃度(pg/mL)1) 症例番号 血液 採取日 採取時間 (点眼後経過時間) TRA/TIM TRA 103 3 日目 30 分 BLQ 12 45 分 BLQ 10 106 1 日目 10 分 BLQ 17 15 分 BLQ 19 30 分 11 20 45 分 BLQ 16 60 分 BLQ 11 3 日目 10 分 11 BLQ 15 分 16 15 30 分 11 10 45 分 11 BLQ 108 1 日目 10 分 11 BLQ 109 1 日目 15 分 12 BLQ 30 分 11 BLQ 3 日目 30 分 11 BLQ BLQ =定量限界未満 (10pg/mL) 1) 10pg/mL 未満の症例については記載せず ②チモロール TRA/TIM 又は TIM が投与された全例の血漿中よりチモロール濃度が検出定量された。 単回投与時(投与 1 日目)及び反復投与時(投与 3 日目)におけるチモロールの血漿中濃度の 推移を図 2.7.6.2.2-1 及び図 2.7.6.2.2-2 にそれぞれ示す。また、投与 1 日目及び 3 日目のチモロール に関する各種薬物動態パラメータ(Cmax、Tmax、AUC0-12、AUC0-∞、t1/2)を表 2.7.6.2.2-3 に、さ
らに、これらの薬物動態パラメータに関する配合剤群と単剤群との比較について表 2.7.6.2.2-4 に 示す。 <単回投与> 血漿中のチモロール濃度は、TRA/TIM 群及び TIM 群ともに速やかに上昇し、投与 2 時間後ま でにピークに達し、その後単相性に消失した。平均 Cmax(平均値±SD)は、TRA/TIM 群 0.552 ± 0.297 ng/mL、TIM 群 0.482 ± 0.244 ng/mL であった。両群間に統計的有意差はみられなかった。 <反復投与> 血漿中のチモロール濃度は、TRA/TIM 群及び TIM 群ともに速やかに上昇し、投与 2 時間後ま でにピークに達し、その後単相性に消失した。平均 Cmax(平均値±SD)は、TRA/TIM 群 0.692± 0.384 ng/mL、TIM 群 0.613±0.281 ng/mL であり、TRA/TIM 群の値が若干高かったが、統計的有意 差はなく、また臨床上意義ある差ではないと考えられた。 t1/2は、投与 3 日目では両群間に有意差がみられたが、その差は 30 分であり、臨床上意義ある 差ではないと考えられた。
2.7.6 個々の試験のまとめ(5.3.3.1.2(C-10*)) 25 *:新薬承認審査情報提供時に置き換えた <考察> 薬物相互作用に関する検討に関するガイドライン(「薬物相互作用の検討方法について」)では、 「一般に、薬物動態パラメータ(Cmax及び AUC)の比の 90%信頼区間が 80-125%の区間に収まる なら、当該薬物間の薬物動態学的な相互作用がないと判断できる」と示されている。 本試験でも、ガイドラインに従い、トラボプロストがチモロールに及ぼす薬物相互作用の有無 についての判定を行うため、TRA/TIM 群及び TIM 群におけるチモロールに関する各種薬物動態 パラメータ(Cmax、AUC0-12、AUC0-∞)の幾何平均値を求め、さらに、その幾何平均値の TRA/TIM
群/TIM 群間対比の 90%信頼区間を求めた。 これらの解析結果を表 2.7.6.2.2-5 に示す。 Cmax及び AUC の両群間対比の 90%信頼区間は 1 を跨いでおり、統計的有意差はみられなかった が、上限値は 125%以上とガイドラインで示された範囲内には収まらなかった。 しかしながら、Cmax及び AUC の両群間差自体は、投与 1 日目及び投与 3 日目ともに小さく、臨 床上意義ある差とは考えにくいことから、本試験では、各被験者の体内変動に加えて、症例数が 少ないことにより、信頼区間幅に多少の広がりがみられたと考えられた。 以上より、TRA/TIM(1 日 1 回点眼)と TIM(1 日 1 回点眼)の血漿中のチモロール薬物動態 パラメータに、大きな差はないと考えられた。 0 0.2 0.4 0.6 0.8 0 2 4 6 8 10 12 Time (h) Timolol Concentration (ng/mL) Travoprost 0.004%/Timolol 0.5% Timolol 0.5% 図 2.7.6.2.2-1 単回投与時(1 日目)におけるチモロール血漿中濃度(平均値、SD)の推移
0 0.2 0.4 0.6 0.8 1 0 2 4 6 8 10 12 Time (h) Timolol Concentration (ng/mL) Travoprost 0.004%/Timolol 0.5% Timolol 0.5% 図 2.7.6.2.2-2 反復投与時(3 日目)におけるチモロール血漿中濃度(平均値、SD)の推移 表 2.7.6.2.2-3 チモロールに関する各種薬物動態パラメータ 薬物動態パラメータ TRA/TIM (N=15) TIM (N=14) 1 日目 Cmax (ng/mL) 0.552 0.297 (0.196 - 1.02) 0.482 0.244 (0.144 - 0.890) Tmax (h) 0.47 0.66 (0.08 - 2.00) 0.54 0.58 (0.08 - 2.00) AUC0-12 (ngh/mL) (0.864 - 5.44) 2.53 1.32 2.15 1.04 (0.830 - 4.16) AUC0- (ngh/mL) (0.991 - 5.74) 2.99 1.35 2.70 1.26 (1.27 - 5.45) t1/2 (h) 4.6 2.2 (2.6 - 11.4) 5.4 2.9 (2.4 - 13.3) 3 日目 Cmax (ng/mL) 0.692 0.384 (0.244 - 1.38) 0.613 0.281 (0.172 - 1.23) Tmax (h) 0.56 0.38 (0.08 - 1.00) 0.57 0.55 (0.08 - 2.00) AUC0-12 (ngh/mL) (1.10 - 6.45) 3.25 1.70 2.91 1.26 (0.978 - 4.89) AUC0- (ngh/mL) (1.26 - 7.29) 3.84 2.02 3.51 1.56 (1.22 - 6.66) t1/2 (h) 4.2 1.6 (2.6 - 8.6) 4.7 1.4 (2.7 - 7.3) 数値は平均値±SD(範囲)
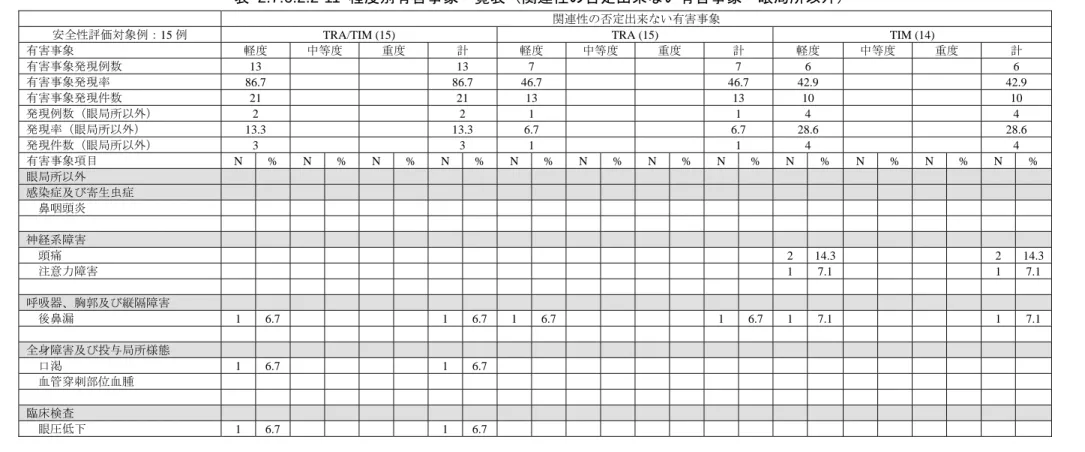
2.7.6 個々の試験のまとめ(5.3.3.1.2(C-10*)) 27 *:新薬承認審査情報提供時に置き換えた 表 2.7.6.2.2-4 チモロールに関する薬物動態パラメータの比較(TRA/TIM vs TIM) 薬物動態パラメータ 群間差±SD (TRA/TIM-TIM)(N=14) 対応のある t 検定 (p 値) 1 日目 Cmax (ng/mL) 0.083 0.314 0.3387 Tmax (h) -0.18 0.63 0.3145 AUC0-12 (ngh/mL) 0.380 1.26 0.2804 AUC0- (ngh/mL) 0.282 1.33 0.4413 t1/2 (h) -0.7 2.7 0.3547 3 日目 Cmax (ng/mL) 0.087 0.340 0.3557 Tmax (h) -0.05 0.48 0.7255 AUC0-12 (ngh/mL) 0.314 1.23 0.3547 AUC0- (ngh/mL) 0.304 1.32 0.4033 t1/2 (h) -0.5 0.8 0.0418 表 2.7.6.2.2-5 チモロールに関する各種薬物動態パラメータの幾何平均値解析 Cmax (ng/mL) AUC(0-12) (ngh/mL) (ngh/mL) AUC(0-) 1 日目 TRA/TIM 幾何平均値(N=15) 0.474 2.21 2.68 TIM 幾何平均値(N=14) 0.421 1.90 2.44 群間対比(T/R)1) 1.13 1.16 1.10 群間対比の 90%信頼区間上限 1.49 1.52 1.39 群間対比の 90%信頼区間下限 0.849 0.893 0.863 3 日目 TRA/TIM 幾何平均値(N=15) 0.607 2.84 3.33 TIM 幾何平均値(N=14) 0.549 2.62 3.17 群間対比(T/R)1) 1.11 1.09 1.05 群間対比の 90%信頼区間上限 1.43 1.40 1.36 群間対比の 90%信頼区間下限 0.858 0.839 0.812 1)群間対比(T/R)は配合剤群(T)の単剤群(R)に対する幾何平均値の対比を表す。 4)安全性 ①有害事象 程度別因果関係別の有害事象発現率の一覧を 2.7.6.2 項末(表 2.7.6.2.2-8~表 2.7.6.2.2-11)に示 す。 安全性解析対象 15 例(クロスオーバー試験:各群 15 例)のうち、有害事象は TRA/TIM 群 14 例(93.3%)、TRA 群 9 例(60.0%)及び TIM 群 6 例(42.9%)にみられた。死亡例、重篤な有害 事象及び有害事象による中止例はみられなかった。 治験薬との関連性を否定できない有害事象は、TRA/TIM 群 13 例(86.7%)、TRA 群 7 例(46.7%) 及び TIM 群 6 例(42.9%)にみられた。 眼局所の治験薬との関連性を否定できない有害事象の一覧を表 2.7.6.2.2-6 に示す。 最も多くみられたものは各群とも眼の充血(眼充血及び結膜充血)であり、TRA/TIM 群 13 例 (眼充血 1 例 6.7%、結膜充血 12 例 80.0%)、TRA 群 9 例(眼充血 2 例 13.3%、結膜充血 7 例 46.7% 及び TIM 群 4 例(結膜充血 4 例 28.6%)であった(いずれも軽度)。その他に、眼刺激が TRA/TIM 群 2 例(13.3%)及び TRA 群 1 例(6.7%)にみられた(いずれも軽度)。これら以外には、複数例 みられたものはなかった。
表 2.7.6.2.2-6 眼局所の治験薬との関連性を否定できない有害事象 TRA/TIM N= 15 TRA N= 15 TIM N= 14 有害事象名 (MedDRA Term) N % N % N % 結膜充血 12 80.0 7 46.7 4 28.6 眼刺激 2 13.3 1 6.7 - - 眼充血 1 6.7 2 13.3 - - 眼部不快感 - - 1 6.7 1 7.1 霧視 1 6.7 - - 1 7.1 眼痛 - - 1 6.7 - - 眼そう痒症 1 6.7 - - - - 眼瞼そう痒症 1 6.7 - - - - 眼局所以外の治験薬との関連性を否定できない有害事象の一覧を表 2.7.6.2.2-7 に示す。 治験薬との関連性が否定できない眼局所以外の有害事象は多くはみられなかった。概して消失 に至った軽度のものであった。 表 2.7.6.2.2-7 眼局所以外の治験薬との関連性を否定できない有害事象 TRA/TIM (N=15) TRA (N=15) TIM (N=14) 有害事象名 (MedDRA Term) N % N % N % 神経系障害 頭痛 - - - - 2 14.3 注意力障害 - - - - 1 7.1 呼吸器、胸郭及び縦隔障害 後鼻漏 1 6.7 1 6.7 1 7.1 全身障害及び投与局所様態 口渇 1 6.7 - - - - 臨床検査 眼圧低下 1 6.7 - - - - ②PG 製剤に特徴的な所見(眼の充血) 薬物動態を主目的としたクロスオーバー試験であったため、標準写真を用いた充血判定は行な われなかった。 有害事象の判定基準は特別には設けなかったが、好ましくない眼の充血、すなわち眼の充血に より試験を中止した場合、又は患者から訴えがあった場合については有害事象として取り上げる こととした。 有害事象として取り上げられた眼の充血(眼充血及び結膜充血)は、前述の通り、TRA/TIM 群 13 例(眼充血 1 例 6.7%、結膜充血 12 例 80.0%)、TRA 群 9 例(眼充血 2 例 13.3%、結膜充血 7 例 46.7%及び TIM 群 4 例(結膜充血 4 例 28.6%)であった(いずれも軽度)。 ③脈拍、血圧 脈拍、血圧の測定は実施しなかった。
2.7.6 個々の試験のまとめ(5.3.3.1.2(C-10*)) 29 *:新薬承認審査情報提供時に置き換えた ④眼科検査所見 視力、角膜、虹彩、前房、水晶体、眼底(網膜/黄斑/脈絡膜、視神経、硝子体)所見、C/D 比に ついて、治験薬との関連性を否定できない悪化はいずれの投与群でもみられなかった。 ⑤臨床検査 臨床検査は実施しなかった。 2.7.6.2.3 結論 血漿中の AL-5848(トラボプロスト遊離酸代謝物)濃度は、トラボプロスト 0.004%/チモロール 0.5%配合点眼液(1 日 1 回点眼)及びトラボプロスト 0.004%点眼液(1 日 1 回点眼)ともに極め て低かった。 血漿中のチモロール濃度は、トラボプロスト 0.004%/チモロール 0.5%配合点眼液(1 日 1 回点 眼)とチモロール 0.5%点眼液(1 日 1 回点眼)で大きな差はみられなかった。 トラボプロスト 0.004%/チモロール 0.5%配合点眼液(1 日 1 回点眼)の外国人健康被験者に対 する安全性が確認された。
30 2.7 臨床概要 2.7.6 個々の試験のまとめ( 5.3.5.1.2 ( C-1 0 *) ) 表 2.7.6.2.2-8 程度別有害事象一覧表(全ての有害事象・眼局所) 全ての有害事象
安全性評価対象例:15 例 TRA/TIM (15) TRA (15) TIM (14)
有害事象 軽度 中等度 重度 計 軽度 中等度 重度 計 軽度 中等度 重度 計 有害事象発現例数 14 14 9 9 6 6 有害事象発現率 93.3 93.3 60.0 60.0 42.9 42.9 有害事象発現件数 24 24 17 17 13 13 発現例数(眼局所) 13 13 9 9 4 4 発現率(眼局所) 86.7 86.7 60.0 60.0 28.6 28.6 発現件数(眼局所) 20 20 14 14 6 6 有害事象項目 N % N % N % N % N % N % N % N % N % N % N % N % 眼局所 結膜充血 12 80.0 12 80.0 7 46.7 7 46.7 4 28.6 4 28.6 眼刺激 2 13.3 2 13.3 1 6.7 1 6.7 眼乾燥 2 13.3 2 13.3 1 6.7 1 6.7 眼充血 1 6.7 1 6.7 2 13.3 2 13.3 霧視 1 6.7 1 6.7 1 7.1 1 7.1 眼瞼そう痒症 1 6.7 1 6.7 眼そう痒症 1 6.7 1 6.7 眼部不快感 1 6.7 1 6.7 1 7.1 1 7.1 眼痛 1 6.7 1 6.7 結膜出血 1 6.7 1 6.7
31 2.7 臨床概要 2.7.6 個々の試験のまとめ( 5.3.5.1.2 ( C-1 0 *) ) *:新薬承認審査情報提供時に置き換えた 表 2.7.6.2.2-9 程度別有害事象一覧表(全ての有害事象・眼局所以外) 全ての有害事象
安全性評価対象例:15 例 TRA/TIM (15) TRA (15) TIM (14)
有害事象 軽度 中等度 重度 計 軽度 中等度 重度 計 軽度 中等度 重度 計 有害事象発現例数 14 14 9 9 6 6 有害事象発現率 93.3 93.3 60.0 60.0 42.9 42.9 有害事象発現件数 24 24 17 17 13 13 発現例数(眼局所以外) 3 3 3 3 5 5 発現率(眼局所以外) 20.0 20.0 20.0 20.0 35.7 35.7 発現件数(眼局所以外) 4 4 3 3 7 7 有害事象項目 N % N % N % N % N % N % N % N % N % N % N % N % 眼局所以外 感染症及び寄生虫症 鼻咽頭炎 2 13.3 2 13.3 1 7.1 1 7.1 神経系障害 頭痛 1 6.7 1 6.7 3 21.4 3 21.4 注意力障害 1 7.1 1 7.1 呼吸器、胸郭及び縦隔障害 後鼻漏 1 6.7 1 6.7 1 6.7 1 6.7 1 7.1 1 7.1 全身障害及び投与局所様態 口渇 1 6.7 1 6.7 血管穿刺部位血腫 1 7.1 1 7.1 臨床検査 眼圧低下 1 6.7 1 6.7
32 2.7 臨床概要 2.7.6 個々の試験のまとめ( 5.3.5.1.2 ( C-1 0 *) ) 表 2.7.6.2.2-10 程度別有害事象一覧表(関連性の否定出来ない有害事象・眼局所) 関連性の否定出来ない有害事象
安全性評価対象例:15 例 TRA/TIM (15) TRA (15) TIM (14)
有害事象 軽度 中等度 重度 計 軽度 中等度 重度 計 軽度 中等度 重度 計 有害事象発現例数 13 13 7 7 6 6 有害事象発現率 86.7 86.7 46.7 46.7 42.9 42.9 有害事象発現件数 21 21 13 13 10 10 発現例数(眼局所) 13 13 7 7 4 4 発現率(眼局所) 86.7 86.7 46.7 46.7 28.6 28.6 発現件数(眼局所) 18 18 12 12 6 6 有害事象項目 N % N % N % N % N % N % N % N % N % N % N % N % 眼局所 結膜充血 12 80.0 12 80.0 7 46.7 7 46.7 4 28.6 4 28.6 眼刺激 2 13.3 2 13.3 1 6.7 1 6.7 眼乾燥 眼充血 1 6.7 1 6.7 2 13.3 2 13.3 霧視 1 6.7 1 6.7 1 7.1 1 7.1 眼瞼そう痒症 1 6.7 1 6.7 眼そう痒症 1 6.7 1 6.7 眼部不快感 1 6.7 1 6.7 1 7.1 1 7.1 眼痛 1 6.7 1 6.7 結膜出血
33 2.7 臨床概要 2.7.6 個々の試験のまとめ( 5.3.5.1.2 ( C-1 0 *) ) *:新薬承認審査情報提供時に置き換えた 表 2.7.6.2.2-11 程度別有害事象一覧表(関連性の否定出来ない有害事象・眼局所以外) 関連性の否定出来ない有害事象
安全性評価対象例:15 例 TRA/TIM (15) TRA (15) TIM (14)
有害事象 軽度 中等度 重度 計 軽度 中等度 重度 計 軽度 中等度 重度 計 有害事象発現例数 13 13 7 7 6 6 有害事象発現率 86.7 86.7 46.7 46.7 42.9 42.9 有害事象発現件数 21 21 13 13 10 10 発現例数(眼局所以外) 2 2 1 1 4 4 発現率(眼局所以外) 13.3 13.3 6.7 6.7 28.6 28.6 発現件数(眼局所以外) 3 3 1 1 4 4 有害事象項目 N % N % N % N % N % N % N % N % N % N % N % N % 眼局所以外 感染症及び寄生虫症 鼻咽頭炎 神経系障害 頭痛 2 14.3 2 14.3 注意力障害 1 7.1 1 7.1 呼吸器、胸郭及び縦隔障害 後鼻漏 1 6.7 1 6.7 1 6.7 1 6.7 1 7.1 1 7.1 全身障害及び投与局所様態 口渇 1 6.7 1 6.7 血管穿刺部位血腫 臨床検査 眼圧低下 1 6.7 1 6.7