修士論文
新規無機化合物の合成と高選択的生体分子 吸着・検出への応用
平成
23年度
三重大学大学院工学研究科 博士前期課程分子素材工学専攻
山内 善博
目次 第
1節 序論
1.1 ハイドロキシアパタイト (HAp
について)
1.2 ハイドロキシアパタイトの形態とタンパク質吸着について
1.3
メソポーラスシリカ (MPS)について
1.4
バイオセンサーについて
1.5 本研究の背景と目的
第
2節 実験試薬
2.1 研究に使用した試薬
第
3節 ハイドロキシアパタイト
(HAp)のタンパク質分離材としての応用
3.1 HAp
粒子の合成
3.1.1 低pH
条件下での
HAp粒子の合成
3.1.2 高pH条件下での
HAp粒子の合成
3.2 HAp
粒子の構造解析
3.2.1
自動比表面積
/細孔分布測定装置
(BET) 3.2.2 X線回折装置 (XRD)
3.2.3 電界放射型走査電子顕微鏡 (FE-SEM) 3.2.4 誘導結合プラズマ発光分析 (ICP-OES) 3.2.5 透過型電子顕微鏡 (TEM) 3.2.6 ζ電位
3.3 HAp
のタンパク質吸着実験
3.3.1 タンパク質3.3.2 タンパク質吸着量測定用検量線の作成
1 2 3 4 5
6
7 7 7
8 8 9 9 10 10 11
11 13
3.3.3 HAp
のタンパク質吸着実験
3.4 タンパク質の選択的吸着
3.4.1 SDS-PAGE
ゲルの調製
3.4.2 サンプル溶液の調製 3.4.3 SDS-PAGE
3.5 加水分解酵素スブチリシンの活性評価
3.5.1 スブチリシンのHAp
への固定化
3.5.2 酵素活性評価
3.6 タンパク質の高次構造解析 3.6.1 二次構造の測定
3.6.2
三次構造の測定
3.7 結果及び考察
3.7.1 HAp
粒子の構造解析
3.7.2 HAp
粒子のタンパク質吸着実験の結果
3.7.3 SDS-PAGE
の結果
3.7.4 加水分解酵素スブチリシンの活性評価の結果
3.7.5
タンパク質の高次構造解析
3.7.5.1 二次構造の測定 3.7.5.2 三次構造の測定 3.7.6 総括
13
13 13 14 14
15 15 15
16 16 17
17 17 24 26 29 31 31 31 35
第
4節 メソポーラスシリカ (MPS)薄膜の高感度バイオセンサーとしての応用
4.1 MPS
薄膜の作成
4.2 MPS
薄膜の構造解析
4.2.1 XRD4.2.2
接触角
4.2.3 DFM4.2.4 エリプソメトリー
4.3 タンパク質吸着実験 4.3.1 タンパク質
4.3.2 Protein A
の吸着測定用検量線の作成
4.3.3 Protein A
の吸着
4.3.4 FITC-IgG
の吸着測定用検量線の作成
4.3.5 Protein A-IgG
結合
4.3.6 DFM
による
Protein Aの吸着の確認
4.3.7 DFM
による
Protein A-IgG結合の確認
4.3.8 BET
によるタンパク質吸着の確認
4.4
バイオセンサーへの応用としての
ELISA法
4.4.1 MPS薄膜上での
ELISA法
4.4.2 DFM
による薄膜表面でのタンパク質吸着状態の検討
4.5 薄膜上での触媒反応 4.5.1 タンパク質
4.5.2 Cytochrome C
の吸着測定用検量線の作成
4.5.3 Cytochrome C
の吸着
4.5.4 Cytochrome Cの触媒反応
4.5.5 Cytochrome C
の繰り返し反応
37
38 38 38 38 39
39 39 40 40 41 41 41 41 42
42 42 43
43 43 43 44 44 45
4.6 結果及び考察
4.6.1 MPS
薄膜の構造解析
4.6.2 タンパク質吸着実験
4.6.3 DFM
によるタンパク質の吸着の確認
4.6.4 BET
によるタンパク質吸着の確認
4.6.5 バイオセンサーへの応用としてのELISA
法
4.6.6 DFM
による薄膜表面でのタンパク質吸着状態の検討
4.6.7 薄膜上での触媒反応 4.6.8
総括
第
5節 総括
5.1 ハイドロキシアパタイト (HAp)の分離材としての応用
5.2 メソポーラスシリカ (MPS)薄膜の高感度バイオセンサーとしての応用
第
6節 謝辞
第
7節 学会発表
第
8節 参考文献
45 45 50 51 55 57 57 61 6568
68 70
71
72
第
1節 序論
1.1 ハイドロキシアパタイト(HAp)について
水酸アパタイトは正式名をハイドロキシアパタイトと言い,リン酸カルシウム化合物 の一種である.弱酸性水溶液中で析出する第一リン酸カルシウムは,水に容易に溶解す るが,第二,第三となるにつれて水に難溶となる.そして,水酸アパタイトは最も水に 難溶なリン酸カルシウム化合物である.水酸アパタイトの最大の特徴は,非化学量論性 にある.化学量論的組成は,化学式が
Ca(PO4)6(OH)2であるため,Ca/P モル比は
1.67,H2O=1.79%であるが,非化学量論性のためにCa/P
モル比が理想値である
1.67にならな
い.化学量論的なハイドロキシアパタイトを合成するには,合成温度の上昇,長時間の 熟成などの技術が必要となってくる.
Ca/P<1.67の場合,アパタイトは吸着水,格子水,
HPO4
2-
を含み,H
2O>1.79%となる.H2O含有率は,Ca/P モル比,乾燥温度によって変動 する.Ca/P モル比が
1.67以上のアパタイトは,PO
43-サイトが
CO32-
,H
4O44-
,CO
3F3-な どに置換されることが考えられる.一方で,Ca/P<1.67 のアパタイトの場合,構造柔軟 性や表面活性が増加するために,吸着および触媒作用は一般に強化されるため,材料と しての関心が高まっている.
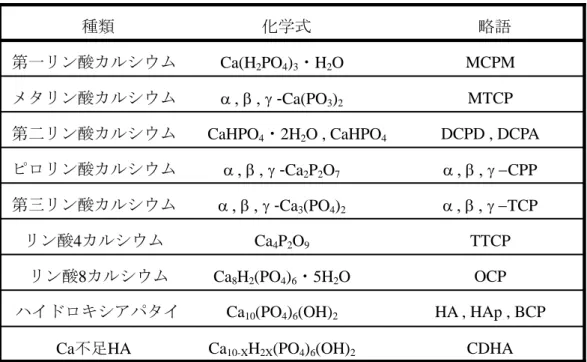
また,リン酸カルシウム化合物には様々な種類があり,多かれ少なかれアパタイトと 関係をもっている.リン酸カルシウム化合物の代表的なものを
Table 1.1にまとめる.
例えば,Ca 不足
HAは,カルシウム不足ハイドロキシアパタイト
(Ca-deficient hydroxyapatiteまたは
ACP amorphous calcium phosphate)と呼ばれ,カルシウムが不足している分,水素原子が置換していることを表している.これは,ハイドロキシアパタイ トの合成実験で,前駆物質として生成する.最近では、ACP は
Ca不足
HAとは構造が 異なるため,別の物質であるとされている.なお,
HAp,メタリン酸カルシウム,ピロリン酸カルシウムにおいて,PO
43-をオルトリン酸,PO
3-をメタリン酸,P
2O74-
をピロリ
ン酸と呼ぶ.オルトリン酸は正リン酸,メタリン酸は環状につながっている事を意味し,
ピロリン酸はオルトリン酸が二つ繋がっているもので縮合リン酸と言う.三つ以上縮合
している場合,トリポリ,テトラポリ,ペンタポリリン酸イオンと言う.メタリン酸カ
ルシウム,ピロリン酸カルシウム,そして第三リン酸カルシウムには,型の結晶
が存在する.
-は同じ組成だが,構造や性質が異なる多形のものである.多形とは,HA , HAp , BCP Ca10(PO4)6(OH)2
ハイドロキシアパタイ ト
OCP Ca8H2(PO4)6
・5H
2Oリン酸8カルシウム
TTCP Ca4P2O9
リン酸4カルシウム
TCP
-Ca3(PO4)2
第三リン酸カルシウム
CPP
-Ca2P2O7
ピロリン酸カルシウム
DCPD , DCPA CaHPO4
・2H
2O , CaHPO4第二リン酸カルシウム
MTCP
-Ca(PO3)2
メタリン酸カルシウム
MCPM Ca(H2PO4)3
・H
2O第一リン酸カルシウム
略語 化学式
種類
Ca不足HA Ca10-XH2X(PO4)6(OH)2 CDHA
Table 1.1
リン酸カルシウムの種類
(1)同一の化学組成にも関わらず,複数の異なる結晶形を取ることを言う.メタリン酸カル シウム,ピロリン酸カルシウム,そして第三リン酸カルシウムにおいて,同一の化学組 成だが,,,型と構造が異なる多形の物質であり,型が高温安定型,そして以下
と続いて熱安定性が異なる物質となる.さらに,物理化学的性質が異なっていくた
め,タンパク質吸着特性も同様に変化していくものと考えられる
(1).
1.2 ハイドロキシアパタイト (HAp)の形態とタンパク質吸着について
骨や歯あるいは天然鉱物のアパタイト結晶の形態は,基本的にはすべて六角柱状ある いは板状の形態を有する.六角柱状の結晶において,側面部分を
a面,上下の平面部分 を
c面という.アパタイトが結晶成長する場合には,通常
c軸方向から伸長する.また,
短時間で合成したアパタイトは細長い針状の結晶となることもある.針状の結晶になる のは,構造周期が短い (格子定数が小さい)ため成長する確率が高いということである.
そして,ある程度
c軸方向に伸長した後,a 軸方向に伸びて肉厚になっていく.したが
って,成長初期では,a 軸に伸長する前の状態であるため,針状結晶となる.逆に、肉
厚なアパタイト結晶を酸で溶解した場合,a 軸方向から溶解していく.つまり,細長く
なった後,c 軸方向が溶解する (Fig.1.1).酸などの多くの外部影響を受ける歯のエナメ
結晶成長 結晶成長
溶解 溶解
ル質のアパタイト結晶は,表面に向かって
c軸に配向することで,外部からの化学的影 響を極力低下させている
(2).
また結晶面は,a 面はカルシムイオンが多く露出しているために正に帯電し,一方で
c面はリン酸イオンが多く露出しているために負に帯電している.一般に,
HApと有機 物との相互作用は表面吸着型である.そのために,有機物の吸着が起きる場合,a 面で はカチオンが,c 面ではアニオンが吸着しやすくなる.このことから,アパタイトは静 電気力によりタンパク質などを吸着していることが考えられる
(3).
c
面
a
面
Fig. 1.1 HAp
の結晶成長と溶解の模式図
1.3 メソポーラスシリカ (MPS)について
メソポーラスシリカ (MPS)は,1990 年に早稲田大学の黒田らの研究グループにより 開発され,その後,1992 年に米の石油会社モービルの研究グループらによりゾルゲル 法を用いた作成方法が報告された.
MPSは直径が
2 ~ 50 nmの均一なメソ細孔を有して おり,細孔のタイプもヘキサゴナル,キュービック,ラメラ構造などを形成し,高い表 面積,高い細孔体積を有するナノ材料である.また,MPS の構造は,鋳型となる界面 活性剤やトリブロックコポリマーなどの構造指向剤を調整することで,組成及び配向性,
細孔径,細孔体積,表面積を制御することが可能である.さらに,1999 年に (株)豊田
中央研究所の稲垣らのグループは,シランカップリング剤を用いてシリカ表面に官能基
を結合させた有機・シリカハイブリッド
MPSの合成に成功し,メソポーラス材料の機
能化に成功した
(4) – (6).
ゾルゲル法を用いることでその形状は粒子だけでなく,薄膜化が可能である.MPS の薄膜化は,
(1)浸漬法,(2) ディップコート法,(3) スピンコート法の液相法により作 成が行われる.特にスピンコート法は,簡便であり,膜圧をコート溶液の粘性及びスピ ン速度により制御することが容易であり,工業化にも適している
(6).
この様なユニークな構造及び官能基の結合により機能化した
MPSは,吸着材料及び 触媒担体,浄化技術としての分離センサー,絶縁体などの応用に重要な効果を持つ材料 である.また,ドラッグデリバリーシステム及びトランスフェクションデバイス,再生 医療などのバイオ分野への利用が期待される
(7),(8).
1.4 バイオセンサーについて
バイオセンサーとは,生体起源の分子認識機構を利用した化学センサーの総称であり,
バイオセンサーの原理は
1962年に Leland C. Clark により提唱され、最初の論文は
1967年に
UPDIKEと
HICKSにより報告された.その報告は,酵素 (グルコースオキシダー
ゼ)をゲルを使って担持した電極により基質 (グルコース)の有無を検出する系について
であった
(9).現在までに,酵素などの生体材料を感応部に用いるセンサー,生体反応を
電気信号に変換し検出するセンサーなどが開発されている.例えば,固定化酵素 (グル
コースオキシダーゼ,GOx)を用いたグルコースセンサーが糖尿病患者の自己血糖モニ
タリング用センサーである.これはグルコース (Glc)が
GOxによって酸化される際の酸
素の減少,または反応産物である過酸化水素を電極でとらえ,電気信号に変換する.セ
ンサー酵素としては酸素を消費または生成する反応や,発光反応を触媒するものを利用
する.環境分野では,固定化微生物を利用した
BOD (Biological Oxygen Demand,生物化学的酸素要求量)センサーが実用化され,排水モニタリングや水質汚濁検査に利用さ
れている.近年,バイオセンサーを利用した測定技術は,選択的・簡便な測定を可能に
することから広範囲の分野における利用が期待されている.
1.5 本研究の背景と目的
バイオセンシング技術とは,バイオセンサーを利用した測定技術であり,選択的・簡 便な測定を可能にすることから広範囲の分野における利用が期待される.バイオセンサ ーとは,生体起源の分子認識機構を利用した化学センサーの総称であり,以下の三つに 分類することができる.(1)酵素を使用し,酵素反応生成物を検出するバイオを利用す るセンサー (2)細胞~組織~生体中の化学物質濃度を簡便かつ選択的に測定するバイ オを計る・知るセンサー (3)ヒト・動物などの能力を加味したバイオに学ぶセンサーで ある.現在,市販されている腫瘍マーカーや環境ホルモン測定における測定範囲は,
1 –100 ng/mL
であるため,早期に発見するためにもより高選択性及び高感度を持つバイオ
センサーの開発が求められている.
本研究の目的は,
(1) a面と
c面の二つの結晶面が存在し,それぞれの結晶面で吸着す るタンパク質が異なることが報告されている
HApの形態を制御し,HAp が持つ吸着特 性及び生体適合性を評価することにより,高選択的分離材として応用することを目的と
した
(2).
(2)ゾルゲル法を用いてMPS薄膜作製し,
MPS材料が持つユニークな構造を利
用する高感度バイオセンサーへの応用及びメソ細孔の影響について評価し、高感度バイ
オセンサーとしての応用することを目的とした.各種無機材料は,
XRD,BET,FE-SEMなどを用いて測定し,細胞培養試験には,マウス骨芽細胞様細胞株を用いて,タンパク
質吸着特性試験には三種類のタンパク質などを用いて評価を行った.
第
2節
2.1
研究に使用した試薬
本研究で使用した試薬を
Table 2.1に示す.その他の試薬は,市販品・特級を使用した.
試薬名 製造元 製品番号
硝酸カルシウム四水和物 (Ca(NO
3)2・4H
2O)和光純薬工業(株)
039-00735リン酸水素二アンモニウム ((NH
4)2HPO4)和光純薬工業(株)
016-03325 25%アンモニア水 (NH
4OH)和光純薬工業(株)
010-03166ウシ血清アルブミン (BSA) 和光純薬工業(株)
309-60563ミオグロビン(Myoglobin)
SIGMA-ALDRICH M0630リゾチーム (Lysozyme)
SIGMA-ALDRICH L6876Protein Assay Bio-Rad 500-0006
スブチリシン (Subtilisin)
SIGMA-ALDRICH P5380酢酸エチル 和光純薬工業(株)
051-00356硫酸マグネシウム (MgSO
4)和光純薬工業(株)
138-00415アセトン (CH
3COCH3)和光純薬工業(株)
012-00343(R/S)-フェニルエチルアセテート
研究室合成品
ト リ ス ヒ ド ロ キ シ メ チ ル ア ミ ノ メ タ ン
(Tri-HCl)和光純薬工業(株)
514-82371テトラメチレンジアミン (TEMED) 和光純薬工業(株)
17919Coomassie brilliant blue Bio-Rad 161-0435
ドデシル硫酸ナトリウム (SDS) 和光純薬工業(株)
196-08675P123 SIGMA-ALDRICH 14611DB
F127 BASF
テトラエトキシシラン (TEOS) 信越化学工業(株)
LS-2430エタノール (EtOH) 和光純薬工業(株)
057-00456塩酸 (HCl) 関東化学(株)
18078-00Table 2.1
使用した実験試薬
第
3節 ハイドロキシアパタイト (HAp)の分離材としての応用
3.1 HAp
粒子の合成
3.1.1 低pH
条件下での
HAp粒子の合成
0.05 M
硝酸カルシウム水溶液 200 mL あるいは
0.05 M酢酸カルシウム水溶液 200
mL及び,
0.03 Mリン酸水素二アンモニウム水溶液 200 mL を混合し,
20℃で
5分攪 拌した.その後,得られた懸濁液を
1℃/分の昇温速度で合成温度 (25 ℃あるいは、
60
℃)まで上昇した後,各温度で
3時間攪拌した.また,合成温度が
120℃あるいは,
180
℃の場合は、水熱処理を行った.水熱処理とは,高温・高圧の熱水存在下でおこな う加熱処理のことである.方法としては,オートクレーブ内に出発物質と蒸留水を混合 して,容器を密閉した状態で加熱することで生成物を得た.今回の処理としては,
1℃
/分の昇温速度で合成温度 (120
℃あるいは,180 ℃)まで上昇させた後,各温度で
3時
間攪拌した.その後,懸濁液を冷却し,吸引濾過し,合成された試料を分離,蒸留水で 洗浄した後,60 ℃で一晩乾燥させた.合成した粒子は,合成温度の低温から順に
CP-1~ CP-4
と記載する.
Protein A SIGMA-ALDRICH P6031
FITC-IgG SIGMA-ALDRICH F9636
Human IgG ELISA Quantitation Kit BETHYL E80-104
Cytochrome C SIGMA C2506
ABTS
東京化成工業(株)
A5200過酸化水素 (H
2O2)和光純薬工業(株)
086-07445
P V C PC V C P
P V
P
m m
1 1
0 0
3.1.2 高pH
条件下での
HAp粒子の合成
0.05 M
硝酸カルシウム水溶液 200 mL あるいは,
0.05 M酢酸カルシウム水溶液 200
mL及び,0.03 M リン酸水素二アンモニウム水溶液 200mL それぞれの
pHをアンモニ ア水を滴下し,
pHを
10に調整した.その後,溶液を混合し,得られた混合液に対して
3.1.1
と同様の手順で合成を行った.合成した粒子は,合成温度の低温から順に
pHCP-1~ pHCP-4
と記載する.
3.2 HAp
粒子の構造解析
3.2.1 自動比表面積/細孔分布測定装置 (BET)
BET (Brunauer Emmett Teller)法は,粉体の比表面積を測定する方法のひとつで,単分
子吸着層理論である
Langmuir理論を
Brunauer,Emmettおよび
Tellerらによって
1938年に多分子吸着に拡張した,比表面積の計算法として有名な理論である.液体窒素温度 において窒素の吸着等温線を測定し,窒素の断面積を
16.2Å
2として算出する方法が最 も一般的である.BET 式は次のようになる.
P :
吸着平衡圧
P0 :飽和蒸気圧
V :吸着平衡圧
Pにおける吸着量
Vm :単分子層吸着量
C : BET定数
この関係式は
P/ P0が
0.05~0.35の範囲でよく成立する.測定した
Vと
P/ P0のプロット から
Vmを算出し,得られた
V mを吸着分子の個数に換算し,それに同分子の断面積を 掛けた値が試料の比表面積となる.
今回,生成物の比表面積を測定するために自動比表面積/細孔分布測定装置を用いて
比表面積の測定を行った.測定には,島津製作所製,TriStar 3000 を使用した.窒素吸着
法を用い,測定温度
77 Kで行い,比表面積は
BET法によって算出した.
3.2.2 X
線回折装置 (XRD)
粉末
X線回折 (X-Ray Diffraction ; XRD )法は, 提案者の名をとって,
Debye-Scherrer (デバイ・シェラー)法とも呼ばれる.この方法の利点としては以下のものが挙げられる.
①原理的に
Braggの式を直接利用するのみの簡単なもの.Bragg の式は以下に示す.
d ;
格子面距離
n = 1 , 2 ,… (整数) 波長②試料がいかなる物質から成立しているかを半定量的に決定できる.
③粉末を使用するために,平均的な結果が得られる.
X
線回折の装置の原理としては,高電圧 (30~50 kV)で発生した
X線は,単波長にす るために適当なフィルターを透過させる.すなわち,純金属に加速電子流を当てて得ら れる特性
X線のうち最も強い
K線のみとする.例えば,Feに対して
Mnフィルター,
Cu
に対して
Niフィルターなどである.そして,回折された
X線はカウンターで受け,
増幅され,各角度でのカウンター数がコンピューターに取り込まれる.
今回は,合成されたリン酸カルシウム種及びその結晶性を確認するために,Rigaku 製,RINT2000/PC を使用して
X線回折測定を行った.モノクロメータにより単色化し
た
CuK線にて,印可電流30 mA,印可電圧40 kV,スキャン速度2.000 deg/min,走査範囲 2θ= 3.0 – 60.0 で測定を行った.
3.2.3
電界放射型走査電子顕微鏡
(FE-SEM)電界放射型走査電子顕微鏡 (Field Emission Scanning Electron Microscope : FE-SEM)は,
真空中に置いた試料を電子線で
X-Yの二次元方向に走査を行い,試料表層から発生す る二次電子などの信号を検出し,ブラウン管上に二次電子像を映し出して,試料表面の 形態,微細構造を観察できる.電界放射型で,電子線を極細くしぼれることができ,電 流を多く流せることから,通常の
SEMに比べて高輝度,高分解能な像観察が可能であ る.さらに,付属のエネルギー分散型
X線分析装置を用いることで組成元素の分布,
定性,定量の分析もできる.
今回,生成物の表面組織を加速電圧 10 kV の条件で観察した.測定には,日立製作 所製,電界放射型走査電子顕微鏡
S-4300を使用した.サンプルは,エタノール中に分 散させた後,アルミニウム製試料台に滴下し乾燥させた.乾燥後,観察試料は予め白金 をスパッタ (日立製作所製,E-1020,ION SPUTTER )によりコーティングした.
3.2.4 誘導結合プラズマ発光分析 (ICP-OES)
誘 導 結 合 プ ラ ズ マ
(Inductively Coupled Plasma)は , 発 光 分 析 (Optical Emission
Spectrometry)法の一つの手法である.分析試料にプラズマのエネルギーを外部から与えると,含有されている成分元素 (原子)が励起される.その励起された原子が低いエネ ルギー準位に戻るときに発光線 (スペクトル線)が放出され,光子の波長に相当する発 光線を測定する方法である.発光線の位置から成分元素の種類を判定し,その強度から 各元素の含有量を求める.プラズマの生成には,アルゴンガスを流しトーチ管の先端部 においたワークコイルには高周波電流を流す.高周波電流によりトーチ管内に生成され る電磁場によりアルゴンガスを電離しプラズマにより試料を励起発光させる.溶液試料 は霧化された状態でトーチ管の中央の細管よりプラズマ内に導入される.
今回,ICP-OES により生成物の
Ca/Pモル比を測定した.測定には,サーモ・エレク トロン社製、IRIS Advantage を用いた.0.01 M の塩酸
10 mLで試料
5 mgを溶解させ,
溶液を蒸留水により
5倍希釈し,フィルター濾過後,測定を行った.
3.2.5
透過型電子顕微鏡
(TEM)透過型電子顕微鏡(TEM)は,観察対象に電子を当てて,透かして観察することになるた め,対象をできるだけ薄く (厚さ
100nm以下)切ったり,電子を透過するフィルムの上 に薄く塗りつけたりして観察する.対象の構造や構成成分の違いにより,どのぐらい電 子線を透過させるかが異なるため,場所により透過してきた電子の密度が変わり,これ が顕微鏡像となる.
今回,TEM によって生成物の形態を観察した.観察には
JOEL社製,JEM-3010 を使 用した.試料はエタノールで試料台 (Cu マイクログリッド)に滴下し固定した.乾燥後,
余分な試料を除去し,加速電圧
300 kV,ビーム電流の条件で観察を行った.
3.2.6 ζ電位
ζ電位 (ゼータ電位)は,溶液に別の相 (電極やコロイド粒子など)が接触したとき,
その界面では電荷分離が起き,電気二重層が形成され電位差が生じる.溶液に対して接 触した相が相対的に運動しているとき,接触相の表面からある厚さの層にある溶液は粘 性のために接触相とともに運動する.この相の表面 (滑り面)と界面から充分に離れた 溶液の部分との電位差を言う.
測定の原理,滑り面が接触相と同じ速度で運動するため,溶液と接触相の運動の早退速 度と等しくなる.滑り面が一定速度で運動している時,そこに働く力は釣り合っている.
溶液に対して一定の電圧
Vをかけた場合,この電圧による静電気力と溶液の粘性によ る摩擦力が釣り合う.その結果,ζ電位と溶液の接触相の運動の相対速度
vとの間には 以下の関係が成り立つ.
溶液の誘電率
溶液の粘度
この関係から,電気浸透や電気泳動によって測定する.
今回,生成物を
pH 7.4のリン酸緩衝液にサンプルを
0.4 mg/mLの濃度溶液で分散さ せた後,大塚電子製,ゼータ電位・粒径測定システム (ELSZ-2)を使用して測定を行っ た.
3.3 HAp
粒子のタンパク質吸着実験
3.3.1 タンパク質
酸性,中性,塩基性の性質における
HApのタンパク質吸着特性を検討するために.
今回の実験では,以下 (a)~(c)に示した
3種類のタンパク質を用いた.
(a) BSA
酸性タンパク質として
BSA (ウシ血清アルブミン, Albumin, from Bovine Serum,分子量
67,200, pI 4.7)を使用した.血清アルブミンは血漿中に多く存在するタンパク質で組織
や体からの分泌物に存在する.これは,いくつかの異なる種類の不溶性分子に結合する ことができる輸送タンパク質で,血液を通して体中にそれらを運ぶ.血清アルブミンに よって輸送される最も重要な分子のいくつかは脂肪酸である.脂肪細胞によって放出さ れた脂肪酸は,体内における多くの反応で必要とされる
ATPを放出するために代謝が 行われる体内のほかの細胞に運ばれる必要がある.脂肪酸は,その疎水性炭素鎖が周囲 の水から遮断される深い凹部で血清アルブミンと結合する.血清アルブミンは,他の不 溶性分子,とりわけ,体中に運ぶためにイブプロフェン,アスピリジン,ジアゼパム,
そして他のものとも結合する
(10).
(b) Myoglobin
中性タンパク質として
Myoglobin (From Hourse Skeletal Muscle, pI 7))を使用した.Myoglobin
は筋肉中に存在し,ヘモグロビンと同様に酸素分子と結合する.また,ヘモ
グロビンよりも酸素に対する親和性が高いために,血中のヘモグロビンから酸素を受け 取り貯蔵することができる.中央に酸素を結合する鉄原子を持つプロトポルフィン(ヘ ム)グループが分子についており,ヘモグロビン内のヘムのように,ミオグロミンのヘ ムも一酸化炭素,二酸化炭素,シアン化物のような酸素以外の分子を結合することもで きる.一酸化炭素やシアン化物への親和性は酸素よりも大きいため,酸素結合の阻害に より死に至ることもある.このように,ミオグロビンの構造と機能は,ヘモグロビンと 類似性が高いが,ヘモグロビンが四量体であるのに対してミオグロビンは単量体である 点が大きく異なる
(10).
(c) Lysozyme
塩基性タンパク質として
Lysozyme (分子量14,000, pI 11)を使用した.Lysozymeは,真
正細菌の細胞壁を構成する多糖類を加水分解する酵素である.この作用があたかも細菌
を溶かしているように見えることから溶菌酵素とも呼ばれる.ヒトの場合涙や鼻汁,母
乳などに含まれている.工業的には卵白から抽出したリゾチームが食品や医薬品に応用
されている
(10).
3.3.2 タンパク質吸着量測定用検量線の作成
1.5 mL
エッペンドルフチューブ中に,
BSA,Myoglobinまたは
Lysozymeを
1 mgを
11 mMリン酸緩衝液 pH 7.4) 1 mL 中に加え溶解させた.この
1 mg/mL濃度のタンパク質 溶液に,同様のリン酸緩衝液を用いて希釈し,1, 0.5, 0.25, 0.125, 0.0625, 0.03125 mg/mL の各濃度のタンパク質溶液を調整した.各濃度の溶液を
Bradford法に基づいて,光学密 度 Optical Density : O.D.)を測定し,タンパク質を定量した.
96 wellマイクロプレートに 調整した溶液を
10 Lとタンパク質定量試薬 BIO-RAD)200 L 加え,アルミホイルで遮 光し約
2分間振盪,20 分間室温で静置後,分光光度計 Perkin Elmer 社製, Wallac 1420
ARVOSX)を用いて波長595 nm
で,O.D.値を測定した.2 回測定後,平均値を算出し,
各タンパク質濃度の検量線を作成した.
3.3.3 HAp
粒子のタンパク質吸着
1.5mL
エッペンドルフチューブ中に合成した各リ
HAp粒子を
5 mg及び,
0.5 mgタン
パク質/11 mM リン酸緩衝液 pH 7.4)1 mL 加え,4 ℃で一晩撹拌し,粒子にタンパク質 を吸着させた.撹拌後、遠心分離 4 ℃、12,000 r.p.m、10 分)をし,上澄みの
10 Lを 分取し,3.3.2 と同様の
Bradford法により
O.D.値を測定した.さらに,上記で作成した検量線を利用して,上澄み中の含有タンパク質量を定量した.以上の操作より,上澄み 中のタンパク質量の減少量を,粒子に吸着したタンパク質量に換算し,さらに粒子の形 状によるタンパク質吸着特性の違いを比較するため,
BET法により得られた比表面積か ら単位表面積当たりのタンパク質吸着量を算出した.
3.4 タンパク質の選択的吸着
3.4.1 SDS-PAGE
ゲルの調製
12 %分離用ゲルは,12 %アクリルアミド,0.41 %
ビスアクリルアミド, 0.1 % SDS,
375 mM Tris-HCl (pH 6.8),0.01 %
過硫酸アンモニウム及び
0.001 % TEMEDを混合する
ことで調整した.濃縮用ゲルは,3.89 %アクリルアミド,0.11 % ビスアクリルアミド,
0.1 % SDS,125 mM Tris-HCl (pH 8.3),0.01 %
過硫酸アンモニウム及び
0.001 % TEMEDを混合することで調製した.
3.4.2 サンプル溶液の調製
1.5mL
エッペンドルフチューブ中に
0.25 mg BSA及び
0.25 mg Myoglobinを
11 mMリ ン酸緩衝液 (pH 7.4) 1 mL に溶解させ混合タンパク質溶液を作製し,合成した
pHCP-4粒子を
5 mgを加え,
4℃で一晩撹拌し,粒子にタンパク質を吸着させた.遠心分離後,
上澄みを別容器に移し,66 mM リン酸緩衝液 (pH 7.4) 1 mL で粒子を再懸濁させ,4 ℃ で一晩撹拌することで,粒子に吸着したタンパク質を脱離させた.
SDS-PAGE用のサン プル溶液として,タンパク質吸着前,タンパク質吸着後,タンパク質脱離後の溶液を使 用した.
3.4.3 SDS-PAGE
タンパク質吸着前,タンパク質吸着後,タンパク質脱離後の溶液から
10 L分取し、
sample buffer (114 mM Tris-HCl (pH 6.4)
,3.64 % SDS ,25.4 % グ リセ ロ ー ル ,
9 %-mercaptoennol blue) 10 Lと混和し,
5分間煮沸した.調製した
12 %分離用ゲル及び濃縮用ゲルから成るミニスラブゲルと
Running buffer (0.1 % SDS,25 mM Tris, 52 mMglycine (pH 8.3))を泳動槽にセットし,混和したサンプル10 L
をゲルに加え,500 V,
30 mA
で約
1時間電気泳動した.泳動後,ゲルを蒸留水で
3回振盪洗浄し,Coomassie
Brilliant Blue
を加え
30分間振盪することでタンパク質を染色した.染色後,ゲルを蒸
留水で
3回振盪洗浄し,さらに一晩蒸留水で振盪洗浄した.洗浄後,ゲルをろ紙の上で
乾燥させた.
3.5 加水分解酵素スブチリシンの活性評価
3.5.1 スブチリシンのHAp
への固定化
HAp
吸着状態におけるタンパク質の機能を評価するために,モデルタンパク質とし て加水分解酵素スブチリシンを使用した.スブチリシンの
HApへの固定化は,
1.5mLエ ッペンドルフチューブ中に合成した各
HAp粒子を
5 mg及び,1.5 mg スブチリシン/1.1
mMリン酸緩衝液 (pH 7.4) 900 L を加え,4 ℃で一晩撹拌し,粒子にタンパク質を吸 着させた.スブチリシンの吸着量は, 3.3.3 と同様の手順で算出した.
3.5.2 酵素活性評価
1.5mL
エッペンドルフチューブ中に合成した
HAp粒子を
5 mg及び,
1.5 mgスブチリ
シン/11 mM リン酸緩衝液 (pH 7.4) 900 L を加え,4 ℃で一晩撹拌し,粒子にタンパク 質を吸着させた.攪拌後,遠心分離し,上澄みを別容器に移した.その後,900
Lの リン酸緩衝液 (pH 7.4)を加え、5L/100L の基質 (R, S)-1-phenylethyl acetate/acetone を 加え,室温で
2日間混合することで反応させた.反応後,遠心分離し,上澄みを別の容 器へ移し,
1mLの酢酸エチルを加え,十分に攪拌し,上澄みを別の容器に移し硫酸マグ ネシウムを加え脱水した後に,ろ過しガスクロマトグラフィーで評価した.また,
66 mMのリン酸緩衝液を用いて,吸着させた
HApからスブチリシンを脱離させ,同様の手順 で酵素活性を評価した.脱離したスブチリシンの量は,
3.3.3と同様の手順で算出した.
ガスクロマトグラフィーの測定条件は,
Table 3.1にまとめる.加水分解反応の反応式
を
scheme 1に示す.
カラム
VARIAN CP-Chrirasil-DexCB column (25 mm x 0.25 mm x 0.25 mm)カラム温度
135℃
注入温度
180℃
検出器温度
180℃
流速
50 mL/minSchme 1 R, S-1-phenylethyl acetate/acetone
の加水分解反応
3.6
タンパク質の高次構造解析
3.6.1 二次構造の測定
タンパク質の二次構造の測定は,
CDスペクトルを用いて行った.
CDスペクトルは,
直線偏光が円偏光二色性を持つ物質中を通過すると,その直線偏光を構成していた左円 偏光と右円偏光に振幅の差が生じるため楕円偏光に変化する.円偏光の波長に対して,
円偏光二色性の大きさ (通常はモル楕円率)をプロットしたものである.これによって,
タンパク質が持つ二次構造 (-helix,
-sheet,random)の割合を得ることができる.測定は,
BSA及び
lysozymeを使用し,測定波長が
190 ~ 250 nm,濃度が10-4 ~ 10-5 Mで行い,
吸着前のタンパク質及び脱離後のタンパク質の二次構造について検討した
(11).
Table 3.1
ガスクロマトグラフィー測定条件
CH3
OAc
+H2O
CH3
OH
S-1-Phenylethyl alcohol (R, S)-1-Phenylethyl acetate
3.6.2 三次構造の測定
タンパク質の三次構造は,タンパク質中に含まれるトリプトファン (Trp)の蛍光を測 定することで検討した.測定条件は,励起波長
280 nm,蛍光波長 300 ~ 400 nmで測定 を行い,吸着前のタンパク質及び
HAp吸着状態のタンパク質について検討した.吸着 前のタンパク質及び
HAp吸着状態のタンパク質の三次構造について検討した
(11).
3.7 結果及び考察
3.7.1 HAp
粒子の構造解析
各リン酸カルシウム粒子の構造解析結果を以下の
1) - 5)に示す.1) X
線回折装置 (XRD)
(ⅰ)
低
pH条件下での
HAp粒子の合成
各生成物の
XRDパターンを
Fig.3.1に示す.25 ℃におけるパターンは、26
o,32
o,
46 o付近に見られる
HApの特徴的なピークが観察されなかったが,第二リン酸カルシ ウム(DCPD)に見られる特徴的なピークが観察されたため,DCPD が得られたことが分 かった.一方で,60 ℃,120 ℃,180 ℃では,HAp に見られる特徴的なピークが観察 された.また,合成温度の上昇に伴いピークはシャープになり,結晶性が向上すること が分かった.
(ⅱ)
高
pH条件下での
HAp粒子の合成
各生成物の
XRDパターンを
Fig.3.2に示す.上述の
pHを調整した場合とは異なり
25℃においても,26
o,32
o,46 °付近に見られる
HApの特徴的なピークが観察され た.また,60 ℃,120 ℃,180 ℃と合成温度が上昇するに伴い,ピークはシャープに なり,結晶性が向上することが分かった.
以上のことから,合成時の
pHを塩基性に調整することによって,低温においても
HApが生成することが確認できた.これは,懸濁液中にヒドロキシルイオンが多量に
存在することになるために,HAp の核形成が起きやすくなったためであると考えられ
る.
2)
自動比表面積/細孔分布測定装置 (BET)
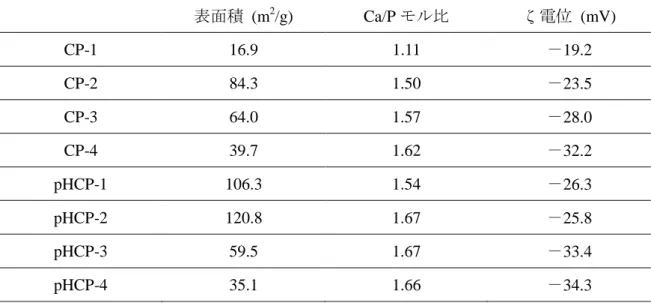
各リン酸カルシウム粒子の比表面積を Table3.2 に示す.低
pH条件下で合成した粒子 は,16.9 ~ 64.0 m
2/gと変化し,高
pH条件下で合成した粒子は,37.2 ~ 109.3 m
2/gと変化した.
3)
誘導結合プラズマ発光分析 (ICP-OES)
各リン酸カルシウム粒子の
Ca/Pモル比を Table3.2 に示す.低
pH条件及び高
pH条件 の両方において,合成温度の上昇に伴い
Ca/Pモル比が,
HApの理論値である
1.67に接 近した.また,高
pH条件においては,低
pH条件と比較して,より
1.69の
Ca/Pモル比 に接近した結果となった.XRD の結果と合わせて,合成温度の上昇に伴い,結晶性が 高く,Ca 欠損の少ない
HAp結晶が得られたことが分かった.
4)
ζ電位
各リン酸カルシウム粒子のζ電位を Table3.2 に示す.pH を調整した場合,あるいは 無調整,どちらにおいても合成温度の上昇に伴い,つまり結晶性の向上とともに,電位 は負の方向へ増加した.
表面積 (m
2/g) Ca/Pモル比 ζ電位 (mV)
CP-1 16.9 1.11
-19.2
CP-2 84.3 1.50
-23.5
CP-3 64.0 1.57
-28.0
CP-4 39.7 1.62
-32.2
pHCP-1 106.3 1.54
-26.3
pHCP-2 120.8 1.67
-25.8
pHCP-3 59.5 1.67
-33.4
pHCP-4 35.1 1.66
-34.3
Table 3.2 BET
、
ICP-OES、ζ電位の結果
5) 電界放射型走査電子顕微鏡 (SEM) (ⅰ) pH
無調整
Fig.3.2
に
FE-SEM画像を示す.25 ℃の場合,約
16 mの板上の大きな結晶が得られ
た.60 ℃の場合,長さは約
210 nm,幅は約50 nmの柱状結晶,120 ℃の場合,長さは
約
230 nm,幅は約45 nmの柱状結晶,180 ℃の場合,長さは約
250 nm,幅は約40 nmの柱状結晶であることが分かった.結晶の長さは,FE-SEM 画像から算出した.
(ⅱ) pH
を
10に調整
Fig.3.2
に
FE-SEM画像を示す.25 ℃の場合,約
11 mの板上の大きな結晶が得られ
た.
60℃の場合,約
32 nmの粒状結晶,120 ℃の場合,長さは約
54 nm,幅は約32 nmの柱状結晶,180 ℃の場合,長さは約
108 nm,幅は約48 nmの柱状結晶であることが 分かった.結晶の長さは,FE-SEM 画像から算出した.
6) 透過型電子顕微鏡 (TEM)
Fig.3.3
に
TEM画像を示す.使用したサンプルは,合成時の
pHが粒子形態に与える
影響を検討するために,SEM による観察で最も違うが観察された
CP-4及び
pHCP-4を 用いた.
CP-4
の場合,SEM で観察された形態と同様に柱状の粒子形態であることが観察され
た.一方,pHCP-4 の場合,CP-4 とは異なり
c軸方向への結晶成長が抑制され,柱状で
はなく粒状に変化していた.
Fig.3.1
異なる合成温度で合成したリン酸カルシウムの
XRDを示す.
(a) pH
無調整で合成した粒子(b) pH 10 に調整し合成した粒子.
0 10 20 30 40 50 60
2q
a.u
(a)
CP-1
CP-2 CP-3 CP-4
0 10 20 30 40 50 60
2q
a.u
(b) pHCP-1
pHCP-2
pHCP-3
pHCP-4
(a)
(b)
Fig.3.2 FE-SEM
による粒子形態の観察
(a) pH
無調整で合成した粒子 (b) pH 10 に調整し合成した粒子.
10 m 1 m
pHCP-1 pHCP-2
1 m 1 m
pHCP-3 pHCP-4
CP-1 CP-2
10 m 1 m
1 m 1 m
CP-3 CP-4
Fig.3.3 TEM
による粒子形態の観察 (a) CP-4 (pH 無調整), (b) pHCP-4 (pH 10 を調整).
200 nm 200 nm
(a) (b)
以上の構造解析から,HAp の合成において合成温度が上昇することで,高い結晶性 の
HApが得られた.これは,XRD による結晶相の分析から,26
o,32
o,46
o付近に見 られる
HApの特徴的なピークが観察され,さらにピークがシャープになったことから 判断される.しかし,合成温度
25℃,pH 無調整で合成した
CP-1の
XRDでは
HApの 特徴的なピークは観察されず,10
o,28
o付近にシャープなピークが見られた.このピ ークから,得られた粒子は
DCPDであることが分かった.BET の結果では,合成温度 が
25℃と
60℃の間で大きく比表面積が増加した.これは,FE-SEM の画像とあわせ て,
25℃の場合に生成した板状の結晶が溶解し,溶解後
HApの柱状の結晶に再結晶し ているものと考えられる.さらに,合成温度
60℃の
FE-SEM画像で,板状の結晶が裂 けて,柱状の結晶が生成している様子が観察されなったことから,溶解,再結晶の結晶 生成過程を示唆している.また,FE-SEM の画像から, pH を調整することで微細な粒 子を得られたことが分かった.この結果から,懸濁液中に過剰の水酸化物イオンが存在 することで,懸濁液中の多くの所で,同時に結晶生成が起きたためと考えられる.また,
pH
を
10に調整し合成した粒子の
XRDは,合成温度が
25℃であっても
HApの特徴的 なピークが観察された.生成途中の結晶が水酸化物イオンを取り込みやすいために,低 い合成温度においても
HApが生成したものと思われる.
ICP-OES
による
Ca/Pモル比の測定では,同合成温度の場合においても
pHを調整し
た方が,HAp の
Ca/Pモル比の理論値である
1.67に接近していた.また,合成温度の上
昇に伴い,モル比は
1.67に接近していった.このことから,高い合成温度で合成する
ことで,モル比は理想値に接近し,高い結晶性を持ち,c 軸方向に結晶成長した
HAp粒子が得られることが分かった.
3.7.2 HAp
粒子のタンパク質吸着実験の結果
各タンパク質の単位面積当たりの吸着量の結果を
Fig.3.4に示す.
pH無調整で合成し た
CP-1については,
HApではなく
DCPDであったことから,以降の実験では使用しな いこととした.
1) BSA
(ⅰ) pH
無調整で合成した
HAp合成温度
25℃の生成物
CP-1では,BSA の吸着量が
2.49 mg/m2と最大になった.合 成温度
60℃の生成物
CP-2において,吸着量は一端減少したが,合成温度の上昇に伴 い吸着量は増加した.
(ⅱ) pH10
に調整し合成した
HAppH
無調整の場合と比較して,全体的な吸着量は低下した.しかし,吸着の傾向とし ては,
pH無調整で合成した生成物と同様に,合成温度の上昇に伴い吸着量が増加した.
2) Myoglobin
(ⅰ) pH
無調整で合成した
HAp合成温度
25℃の生成物
CP-1においては,全く吸着しなかった.合成温度
60℃の生
成物
CP-2では,
0.729 mg/m2の吸着を示し,最大となった.その後,合成温度の上昇に
伴い吸着量は減少した.
(ⅱ) pH10
に調整し合成した
HAppH
無調整の粒子と比較すると,全体的に吸着量は減少した.しかし,吸着の傾向と しては,
pH無調整の場合と同様に,合成温度
60℃の生成物
pHCP-2の吸着量が最大と なり,その後,減少した.
3) Lysozyme
(ⅰ) pH
無調整で合成した
HAp合成温度の上昇に伴い吸着量は
0.17から
0.69 mg/m2へと増加した.合成温度
180℃ の生成物
CP-4において,0.69 mg/m
2と最も高い吸着量を示した.
(ⅱ) pH10
に調整し合成した
HAp無調整の場合と同様に合成温度の上昇に伴い から
2へと増加した.
BSA
の単位面積当たりの吸着量は,HAp の合成温度が上昇するに伴い増加した.タ ンパク質の
HApへの吸着が,HAp とタンパク質間の静電気力によってのみ起きている と仮定すると,合成温度が上昇し,結晶性が向上するに伴い,BSA の吸着量は減少す ると考えられる.ζ電位の結果から,HAp 粒子は負に帯電し,合成温度の上昇つまり 結晶性の向上に伴い負の方向へ増加していった.さらに,
BSAは
pI = 4.7の酸性タンパ ク質であるために,BSA を
pH 7.4のリン酸緩衝液に溶解させた場合,タンパク質は負 に帯電していると考えられる.つまり,HAp 粒子とタンパク質間で静電反発が起こる と考えられるためである.しかし,今回の検証では,減少とは逆の結果になった.
Myoglobin
の吸着では,HAp の結晶性が向上するにつれて吸着量の減少が見られた.
これは,Myoglobin が
pI = 7.4のため,リン酸緩衝液に溶解させると,タンパク質の電 荷は±0 付近になると考えられる.そして,
HAp結晶の表面電位は負に帯電しているた めに,タンパク質と
HAp間での静電相互作用が弱くなり,BSA と比較して吸着量が減 少したと考えられる.pH を調整して得られた結晶では,低い合成温度であっても
HAp粒子が得られ,
pH無調整で得られた粒子よりも負に帯電しているために,
pH無調整の 粒子よりも低い吸着量となったと考えられる.
Lysozyme
の吸着では,合成温度の上昇つまり結晶性の向上に伴い吸着量が増加した.
Lysozyme
は,pI = 11 であるために,pH 7.4 のリン酸緩衝液に溶解させると正に帯電し
ていると考えられる.また,HAp 粒子の表面は負に帯電している.これにより,HAp 粒子とタンパク質間で静電相互作用が起き,互いに引き合うことにより吸着するために 吸着量が増加したと考えられる.
以上の
BSA及び
Lysozymeの吸着量増加傾向という結果から,タンパク質の
HApへ
の吸着は静電気力だけではないことが示唆された.タンパク質の
HApへの吸着機構は,
HAp
の形態及び結晶性,水素結合,イオン結合などの結合などにより複雑であると考
えられるため,今後も様々なタンパク質において検討していく必要があると考えられる.
3.7.3 SDS-PAGE
の結果
SDS-PAGE
によるタンパク質の選択的吸着の結果を
Fig.3.5に示す.
使用した粒子は,
3.7.2の結果より,
BSA及び
Lysozymeの吸着において高い最も選択 性を示した
pHCP-4を使用した.
(a)は,BSA及び
Myoglobinを含む混合タンパク質溶液,
(b)は,pHCP-4
へのタンパク質吸着後の上澄み中に含まれるタンパク質,
(c)は,HApか
らタンパク質を脱離させた後の上澄みに含まれるタンパク質,(d)は,タンパク質分子 量マーカーに対する
SDS-PAGEの結果である.
(a)から,混合タンパク質溶液中にBSA
および
Myoglobinが存在することが観察され
た.(b)では,BSA のバンドがわずかに薄く,狭くなったことが観察された. さらに,
(c)において,Myoglobin