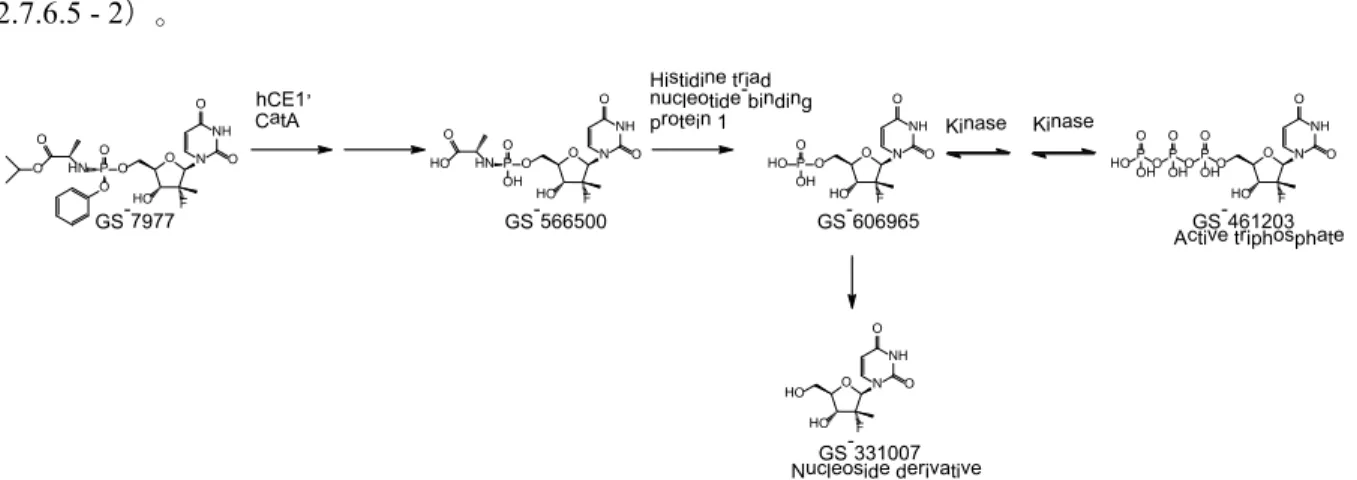
ソバルディ
®
錠
400 mg
第
2 部(モジュール 2):CTD の概要(サマリー)
2.7.6 個々の試験のまとめ
2.7.6.1 - 1
1. 臨床試験一覧
Type of Study Number Study Objective(s) Study Design Study and Control Drug Regimens Duration of Treatment Number of Subjects
Study Population/ Entry Criteria Study Status; Type of Report Comparative
BA/BE P7977-0111 Compare the rates and extent of absorption of GS-9851 and Sofosbuvir and estimate the effect of a high-fat meal on the PK of Sofosbuvir and its metabolites Phase 1 randomized, single-dose, 3-way crossover study Treatment A: GS-9851 200 mg (2 × 100 mg capsule) while fasting
Treatment B: Sofosbuvir
200 mg (2 × 100-mg tablet) while fasting
Treatment C:
Sofosbuvir 200 mg (2 × 100-mg tablet) with a high-fat meal
Subjects received a single dose of each treatment according to 1 of 6 treatment sequences (ABC, BCA, CAB, CBA, ACB, or BAC) Single dose of each of the 3 treatments administered over three 4-day treatment periods, with a 1-week washout period between doses. Overall: 24 treated 24 completed treatment Treatment A: 24 treated 24 completed Treatment B: 24 treated 24 completed Treatment C: 24 treated 24 completed Healthy adult
subjects Study Completed;
Type of Study Number Study Objective(s) Study Design Study and Control Drug Regimens Duration of Treatment Number of Subjects Study Population/ Entry Criteria Study Status; Type of Report Comparative
BA/BE P7977-1318 Compare the rates and extent of absorption of 2 formulations of Sofosbuvir administered following an overnight fast and estimate the effect of a high-fat meal on the PK of Sofosbuvir and its metabolites
Phase 1 randomized, single-dose, 3-way crossover study Treatment A: Sofosbuvir (2 × 200-mg tablet) while fasting Treatment B: Sofosbuvir (1 × 400-mg tablet) while fasting Treatment C: Sofosbuvir (1 × 400-mg tablet) with a high-fat meal
Subjects received a single dose of each treatment according to 1 of 6 treatment sequences (ABC, BCA, CAB, CBA, ACB, or BAC) Single dose of each of the 3 treatments administered over three 4-day treatment periods, with a 1-week washout period between doses. Overall: 40 treated 38 completed treatment Treatment A: 40 treated 39 completed Treatment B: 40 treated 40 completed Treatment C: 40 treated 38 completed Healthy adult
subjects Study Completed;
2.7.6.1 - 3 Type of
Study Number Study Study Objective(s) Design Study and Control Drug Regimens Duration of Treatment Number of Subjects
Study Population/ Entry Criteria Study Status; Type of Report Healthy Subject PK Bridging Study GS-US-334-0
111 Investigate the PK of SOF and metabolites (GS 566500 and GS 331007) following administration of SOF in healthy Japanese and Caucasian subjects Investigate the PK of SOF, metabolites (GS 566500 and GS 331007), and LDV following administration of SOF/LDV FDC tablet in healthy Japanese and Caucasian subjects
Assess the safety and tolerability of SOF and SOF/LDV in healthy Japanese and Caucasian subjects
Phase 1, open label,
single dose study Group 1: Single dose of SOF 200 mg
Group 2: Single dose of SOF
400 mg
Group 3: Single dose of SOF
800 mg
Group 4: Single dose of
SOF/LDV FDC 400 mg/90 mg 1 day Overall: 64 treated 63 completed Group 1: 8 treated 7 completed Group 2: 8 treated 8 completed Group 3: 8 treated 8 completed Group 4: 16 treated 16 completed Healthy adult subjects (Japanses and Caucasian) Study completed; m5.3.3.1.1, Final CSR Healthy Subject PK and Initial Tolerability
P7977-0312 Explore the routes and rates of elimination of [14C]SOF Phase 1, nonrandomized, open-label, single-dose, mass balance study
Single PO dose of SOF
(1 × 400-mg capsule) containing a mixture of [12C]SOF and
[14C]SOF with approximately
100 μCi of radioactivity.
Single dose Overall:
7 treated 7 completed treatment
Healthy adult
subjects Study completed;
m5.3.3.1.2, Final CSR
Type of
Study Number Study Study Objective(s) Design Study and Control Drug Regimens Duration of Treatment Number of Subjects
Study Population/ Entry Criteria Study Status; Type of Report Intrinsic Factor PK P2938-0515 (SOF-specific data) Characterize the PK of SOF and metabolites over 7 days of dosing in HCV-infected subjects with varying degrees of hepatic impairment compared to historical PK data
Phase 1, open-label
PK/PD study Child-Pugh Class B group (moderate hepatic impairment): SOF 400 mg QD PO on Days 1−7
Child-Pugh Class C group (severe hepatic impairment): SOF 400 mg QD PO on Days 1−7 7 days Overall: 17 treated 16 completed treatment Class B group: 9 treated 8 completed Class C group: 8 treated 8 completed Adult subjects with chronic HCV infection and mild, moderate, or severe hepatic impairment according to Child-Pugh classification Subjects were naive to DAA treatment and had cirrhosis documented by historical biopsy Study completed; m5.3.3.3.1, Final CSR
2.7.6.1 - 5 Type of
Study Number Study Study Objective(s) Design Study and Control Drug Regimens Duration of Treatment Number of Subjects
Study Population/ Entry Criteria Study Status; Type of Report Intrinsic
Factor PK P7977-0915 Characterize the PK of SOF and metabolites following single doses of SOF in subjects with varying degrees of renal impairment compared to matched healthy subjects Phase 1, open-label,
single-dose study Group A (normal renal function): Single dose of SOF 400 mg PO Group B (mild chronic renal impairment): Single dose of SOF 400 mg PO
Group C (moderate chronic renal impairment): Single dose of SOF 400 mg PO
Group D (severe chronic renal impairment and not on dialysis): Single dose of SOF 400 mg PO Group E (end-stage renal impairment disease requiring dialysis): SOF 400 mg PO (a single dose prior to dialysis during Period 1 and a single dose following dialysis during Period 2)
Normal and mild, moderate, and several renal function groups: Single dose End-stage renal impairment group: 2 single doses separated by a washout period of at least 2 weeks Overall: 30 treated 30 completed treatment Groups A–E: 6 treated 6 completed Adult subjects with normal renal function and varying degrees of renal impairment Study completed; m5.3.3.3.2, Final CSR
Type of
Study Number Study Study Objective(s) Design Study and Control Drug Regimens Duration of Treatment Number of Subjects
Study Population/ Entry Criteria Study Status; Type of Report Comparative BA/BE (Cohort 5) Extrinsic Factor PK (Cohorts 1−4) GS-US-334-0
131 Evaluate the PK of SOF on coadministration with EFV/FTC/
TDF, DRV/r, RAL, and RPV relative to SOF alone; the PK of TFV, FTC, EFV, DRV, RTV, RAL, RPV on coadministration with SOF relative to administration of these agents alone; the safety and tolerability of coadministration of SOF and HIV medications; and the single-dose PK of a tablet containing SOF Form Phase 1, open-label, multiple-dose, fixed-sequence, single-center, PK, DDI study
Cohort 1: Fasted, single dose of
SOF 400 mg PO on Day 1, EFV 600 mg/ FTC 200 mg/ TDF 300 mg QD PO on Days 5−18, and single doses of SOF 400 mg + EFV 600 mg/ FTC 200 mg/TDF 300 mg PO on Day 19
Cohort 2: Fed, single dose of
SOF 400 mg PO on Day 1, DRV/r 800/100 mg QD PO on Days 5–14, and single doses of SOF 400 mg PO
+ DRV/r 800/100 mg PO on Day 15
Cohort 3: Fasted, single dose of
SOF 400 mg PO on Day 1, RAL 400 mg BID PO on Days 5−14, and single doses of SOF 400 mg + RAL 400 mg PO on Day 15
Cohort 4: Fed, single dose of
SOF 400 mg PO on Day 1, RPV 25 mg QD PO on Days 5−14, and single doses of SOF 400 mg PO + RPV 25 mg PO on Day 15
Cohort 5: Fasted, single dose of
SOF Form 400 mg PO on Day 1 Cohort 1: 19 days Cohorts 2−4: 15 days Cohort 5: 1 day Overall: 88 treated 86 completed treatment Cohort 1: 17 treated 16 completed Cohort 2: 19 treated 18 completed Cohort 3: 19 treated 19 completed Cohort 4: 17 treated 17 completed Cohort 5: 16 treated 16 completed Healthy adult
subjects Study completed;
m5.3.3.4.1, Final CSR
2.7.6.1 - 7
Type of
Study Number Study Study Objective(s) Design Study and Control Drug Regimens Duration of Treatment Number of Subjects
Study Population/ Entry Criteria Study Status; Type of Report Extrinsic
Factor PK P7977-0814 Evaluate the effect of steady-state SOF 400 mg on the steady-state PK of (R)- and (S)-methadone Phase 1, open-label, single-sequence, DDI study
Subjects on stable methadone therapy received methadone 30- to 130-mg QD PO on Days −1 through 11 and SOF 400 mg QD PO on Days 1−7
Methadone alone for 5 days and SOF+methadone for 7 days during the in-clinic portion of the study Overall: 15 treated 15 completed treatment Healthy adult subjects on daily oral methadone prescribed for opiate addiction Study completed; m5.3.3.4.2, Final CSR Extrinsic
Factor PK P7977-1819 Evaluate the effect of SOF coadministration on single-dose PK of CsA and tacrolimus and the effect of CsA and tacrolimus coadministration on single-dose PK of SOF and its metabolites Phase 1, randomized, open-label, 3-period crossover, DDI study
Group 1 (SOF+CsA): Single
dose of SOF 400 mg PO, single dose of CsA 600 mg PO, and single dose of SOF 400 mg PO + CsA 600 mg PO, according to randomized treatment sequence, with doses separated by a 2-week washout period
Group 2 (SOF+tacrolimus):
Single dose of SOF 400 mg PO, single dose of tacrolimus 5 mg PO, and single dose of SOF 400 mg PO + tacrolimus 5 mg PO, according to randomized treatment sequence, with doses separated by a 2-week washout period Single doses of SOF, SOF+CsA/ tacrolimus, and CsA/tacrolimus, each separated by a 2-week washout period Overall: 40 treated 34 completed treatment Group 1: 20 treated 19 completed Group 2: 20 treated 15 completed Healthy adult
subjects Study completed;
m5.3.3.4.3, Final CSR
Type of
Study Number Study Study Objective(s) Design Study and Control Drug Regimens Duration of Treatment Number of Subjects
Study Population/ Entry Criteria Study Status; Type of Report Extrinsic
Factor PK P7977-1910 (As of the 12 September 2012 interim data cut) Evaluate whether SOF significantly influences the PK parameters of ATV/r, DRV/r, EFV, TFV, FTC, RAL, ZDV, and 3TC Phase 1, open-label, single-sequence, DDI study Cohort 1: SOF 400 mg QD PO + EFV 600 mg/FTC 200 mg/ TDF 300 mg QD PO Cohort 2: SOF 400 mg QD PO + EFV 600 mg QD PO + ZDV 300 mg/3TC 150 mg BID PO Cohort 3: SOF 400 mg QD PO + ATV/r 400/100 mg + FTC 200 mg/TDF 300 mg QD PO Cohort 4: SOF 400 mg QD PO + DRV/r 800/100 mg + FTC 200 mg/TDF 300 mg QD PO Cohort 5: SOF 400 mg QD PO + RAL 400 mg BID PO + FTC 200 mg/ TDF 300 mg QD PO 7 days Overall: 34 treated 33 completed treatment Cohort 1 12 treated 11 completed Cohort 2 4 treated 4 completed Cohort 3 8 treated 8 completed Cohort 4 5 treated 5 completed Cohort 5 5 treated 5 completed HCV/HIV-coi nfected adult subjects Study ongoing; m5.3.3.4.4, Interim CSR
2.7.6.1 - 9
Type of
Study Number Study Study Objective(s) Design Study and Control Drug Regimens Duration of Treatment Number of Subjects
Study Population/ Entry Criteria Study Status; Type of Report Healthy Subject PD and PK/PD P7977-0613 Demonstrate lack of effect of SOF administration on cardiac repolarization as determined by the baseline-adjusted QTcF effect of each active regimen relative to placebo following a single oral dose targeting therapeutic and supratherapeutic exposures Phase 1, single-dose, randomized, blinded, placebo- and positive-controlled, 4-period crossover study
Subjects were randomized to 1 of 4 treatment sequences and received the following 4 treatments:
Treatment A: SOF 400 mg PO,
SOF placebo PO, moxifloaxcin placebo PO
Treatment B: SOF 1200 mg PO,
moxifloaxcin placebo PO
Treatment C: SOF placebo PO,
moxifloxacin placebo PO
Treatment D: moxifloxacin 400
mg PO, SOF placebo
5 days (single dose of placebo on Day −1 and study treatment on Day 1 of each of 4 treatment periods) Overall: 60 treated 54 completed treatment Treatment A: 60 treated 59 completed Treatment B: 59 treated 57 completed Treatment C: 57 treated 57 completed Treatment D: 59 treated 59 completed Healthy adult
subjects Study completed;
m5.3.4.1.1, Final CSR
Type of Study Number Study Objective(s) Study Design Study and Control Drug Regimens Duration of Treatment Number of Subjects Study Population/ Entry Criteria Type of Report Study Status;
Patient
PD and PK/PD P7977-0221 Assess the PK, PD, safety, and tolerability of SOF administered in combination with Peg-IFN+RBV for 28 days Phase 2a, randomized, double-blind, placebo-controlled, parallel-group, dose-ranging, multicenter study SOF 100 mg + Peg-IFN+RBV group: SOF 100 mg QD PO on
Days 0–27 and Peg-IFN+RBV Weeks 1–48
SOF 200 mg + Peg-IFN+RBV group: SOF 200 mg QD PO on
Days 0–27 and Peg-IFN+RBV Weeks 1–48
SOF 400 mg + Peg-IFN+RBV group: SOF 400 mg QD PO on
Days 0–27 and Peg-IFN+RBV Weeks 1–48
Placebo+Peg-IFN+RBV group: SOF-matching placebo
QD PO on Days 0–27 and Peg-IFN+RBV Weeks 1−48 For all groups, Peg-IFN dose was 180 µg/week SC and RBV dose was 1000 or 1200 mg/day (divided daily dose)a PO.
If a subject had detectable HCV RNA at Week 12,
Peg-IFN+RBV treatment was continued to Week 24. If a subject had detectable HCV RNA at Week 24,
Peg-IFN+RBV treatment was discontinued. 48 weeks (SOF or matching placebo + Peg-IFN+RBV for 4 weeks followed by an additional 44 weeks of Peg-IFN+RBV alone) Overall: 63 treated 48 completed SOF treatment period (Days 0−27) SOF 100 mg +Peg-IFN+RBV group: 16 treated 16 completed SOF 200 mg +Peg-IFN+RBV group: 18 treated 17 completed SOF 400 mg +Peg-IFN+RBV group: 15 treated 15 completed Placebo +Peg-IFN+RBV group: 14 treated 14 completed Noncirrhotic, treatment-naive adult subjects with chronic genotype 1 HCV infection
Study completed;
m5.3.4.2.1, Final CSR
2.7.6.1 - 11
Type of Study Number Study Objective(s) Study Design Study and Control Drug Regimens Duration of Treatment Number of Subjects Study Population/ Entry Criteria Type of Report Study Status;
Controlled Clinical Studies Pertinent to the Claimed Indication P7977-1231
(FISSION) Assess the efficacy (proportion of subjects with SVR12) and safety of SOF+RBV administered for 12 weeks compared with Peg-IFN+RBV administered for 24 weeks in treatment-naive subjects with genotype 2 or 3 HCV infection Phase 3, randomized, active-controlled, open-label, multicenter study SOF+RBV group: SOF 400 mg QD PO + RBV 1000 or 1200 mg/day (divided daily dose)a PO
Peg-IFN+RBV group: Peg-IFN 180 µg/week SC + RBV 800 mg/day (divided daily dose)PO SOF+RBV group: 12 weeks Peg-IFN+RBV group: 24 weeks Overall: 499 treated 464 completed through SVR12 assessment SOF+RBV group: 256 treated 239 completed Peg-IFN+RBV group: 243 treated 225 completed Treatment-naive adult subjects with chronic genotype 2 or 3 HCV infection; up to 20% of subjects may have the presence of cirrhosis. Study completed; m5.3.5.1.1, Interim CSR and synoptic CSR Controlled Clinical Studies Pertinent to the Claimed Indication GS-US-334-0107
(POSITRON) Assess the efficacy (proportion of subjects with SVR12) and safety of SOF+RBV compared with placebo administered for 12 weeks in subjects with genotype 2 or 3 HCV infection who are IFN intolerant, IFN ineligible, or unwilling to take IFN Phase 3, randomized, double-blind, placebo-controlled, multicenter study SOF+RBV group: SOF 400 mg QD PO + RBV 1000 or 1200 mg/day (divided daily dose)a PO
Placebo group:
SOF placebo QD PO + RBV placebo (divided daily dose)PO 12 weeks Overall: 278 treated 171 completed through SVR12 assessment SOF+RBV group: 207 treated 171 completed Placebo group: 71 treated 0 completed
Adult subjects with chronic genotype 2 or 3 HCV infection who were IFN intolerant, IFN ineligible, or unwilling to take IFN; up to 20% of subjects may have the presence of cirrhosis
Study completed;
m5.3.5.1.2, Interim CSR and synoptic CSR
Type of Study Number Study Objective(s) Study Design Study and Control Drug Regimens Duration of Treatment Number of Subjects Study Population/ Entry Criteria Type of Report Study Status; Controlled Clinical Studies Pertinent to the Claimed Indication GS-US-334-0108
(FUSION) Assess the efficacy (proportion of subjects with SVR12) and safety of SOF+RBV administered for 12 weeks compared with 16 weeks in subjects with genotype 2 or 3 HCV infection who failed prior treatment with IFN
Phase 3, randomized, double-blind, multicenter study
SOF+RBV 12-week group:
SOF 400 mg
+ RBV 1000 or 1200 mg (divided daily dose)a QD PO
for 12 weeks followed by SOF placebo + RBV placebo (divided daily dose) QD PO for 4 weeks
SOF+RBV 16-week group:
SOF 400 mg
+ RBV 1000 or 1200 mg (divided daily dose)a QD PO
for 16 weeks SOF+RBV 12-week group: 12 weeks SOF+RBV 16-week group: 16 weeks Overall: 201 treated 127 completed through SVR12 assessment SOF+RBV 12-week group: 103 treated 54 completed SOF+RBV 16-week group: 98 treated 73 completed Treatment-experienced adult subjects with chronic
genotype 2 or 3 HCV infection; up to 30% of subjects may have the presence of cirrhosis
Study completed;
m 5.3.5.1.3, Interim CSR and synoptic CSR
2.7.6.1 - 13
Type of Study Number Study Objective(s) Study Design Study and Control Drug Regimens Duration of Treatment Number of Subjects Study Population/ Entry Criteria Type of Report Study Status;
Controlled Clinical Studies Pertinent to the Claimed Indication GS-US-334-0133
(VALENCE) Assess the efficacy (proportion of subjects with SVR12) and safety of SOF+RBV administered for 12 weeks or 24 weeks in subjects with genotype 2 or 3 HCV infection Phase 3, randomized, double-blind, multicenter study
SOF+RBV 12 Week genotype 2 group: Subjects with
genotype 2 HCV infection received SOF 400 mg administered once daily+RBV total daily dose of 1000 or 1200 mg administered in a divided daily dose for 12 weeks
SOF+RBV 12 Weeks genotype 3 group: Subjects
with genotype 3 HCV infection received SOF 400 mg
administrated once daily + RBV total daily dose of 1000 or 1200 mg administered in a divided daily dose for 12 weeks
SOF + RBV 24 Weeks genotype 3 group: Subjects
with genotype 3 HCV infection received SOF 400 mg
administrated once daily + RBV total daily dose of 1000 or 1200 mg administered in a divided daily dose for 24 weeks
Placebo group: Subjects with
genotype 2 or 3 HCV infection received SOF placebo
administered once daily + RBV placebo BID for 12 weeks.
SOF+RBV 12-week group: 12 weeks SOF+RBV 24-week group: 24 weeks Overall: 419 treated 331 completed GT2 SOF+RBV 12 wks: 73 treated 73 completed GT3 SOF+RBV 12 wks: 11 treated 8 completed GT3 SOF+RBV 24 wks: 250 treated 246 completed GT 2/3 Placebo: 85 treated 4 completed Treatment-naïve and treatment experienced adult subjects with chronic genotype 2 or 3 HCV infection; up to 40% of subjects may have the presence of cirrhosis.
Study ongoing;
m5.3.5.1.4, Interim CSR
Type of Study Number Study Objective(s) Study Design Study and Control Drug Regimens Duration of Treatment Number of Subjects Study Population/ Entry Criteria Type of Report Study Status; Controlled Clinical Studies Pertinent to the Claimed Indication P7977-0422
(PROTON) Assess the safety and tolerability of SOF administered with Peg-IFN+RBV for 12 weeks Phase 2b, placebo-controlled, dose-ranging, multicenter study in treatment-naive subjects with chronic genotype 1 HCV infection and an open-label assessment in patients with genotype 2 or 3 HCV infection Randomized, Double-Blind Groups (Genotype 1): SOF 200 mg + Peg-IFN+RBV group: SOF 200 mg QD PO + Peg-IFN+RBV SOF 400 mg + Peg-IFN+RBV group: SOF 400 mg QD PO + Peg-IFN+RBV Placebo+Peg-IFN+RBV group: SOF-matching placebo
QD PO + Peg-IFN+RBV Open-Label Group (Genotype 2/3): SOF 400 mg + Peg-IFN+ RBV group: SOF 400 mg QD PO + Peg-IFN+RBV For genotype 1 subjects, Peg-IFN dose was 180 µg/week SC, and RBV dose was 1000 or 1200 mg/day (divided daily dose)a PO.
For genotype 2/3 subjects, Peg-IFN dose was 180 µg/week SC and RBV dose was 800 mg/day (divided daily dose). Randomized, Double-Blind Groups: SOF or matching placebo+ Peg-IFN+RBV for 12 weeks, followed by Peg-IFN+RBV for an additional 0−36 weeks (depending on treatment group and response)b Open-Label Group: 12 weeks Overall (genotype 1): 121 treated 102 completed study SOF 200 mg + Peg-IFN+RBV group: 48 treated 45 completed SOF 400 mg + Peg-IFN+RBV group: 47 treated 42 completed Placebo +Peg-IFN+RBV group: 26 treated 15 completed Overall (genotype 2/3) SOF 400 mg + Peg-IFN+RBV Group: 25 treated 24 completed Noncirrhotic, treatment-naive adult subjects with chronic genotype 1, 2, or 3 HCV infection
Study completed;
m5.3.5.1.5, Final CSR
2.7.6.1 - 15
Type of Study Number Study Objective(s) Study Design Study and Control Drug Regimens Duration of Treatment Number of Subjects Study Population/ Entry Criteria Type of Report Study Status;
Controlled Clinical Studies Pertinent to the Claimed Indication P7977-0523 (ELECTRON) (Parts 1−3)
Assess the safety and tolerability of SOF 400 mg for 8 or 12 weeks administered with and without RBV and/or Peg-IFN in subjects with genotype 1, 2, or 3 HCV infection Phase 2a, open-label, multicenter study Part 1 (treatment-naive subjects with genotype 2/3 HCV):
Group 1: SOF+RBV 12 weeks Group 2: SOF+Peg-IFN+RBV
for 4 weeks then SOF+RBV for 8 weeks
Group 3: SOF+Peg-IFN+RBV
for 8 weeks then SOF+RBV for 4 weeks
Group 4: SOF+Peg-IFN+RBV
for 12 weeks
Part 2 (treatment-naive subjects with genotype 2/3 HCV in Groups 5 and 6 and null responders with
genotype 1 HCV in Group 7): Group 5: SOF for 12 weeks Group 6: SOF+Peg-IFN+RBV
for 8 weeks
Group 7: SOF+RBV for
12 weeks
Part 3 (treatment-naive subjects with genotype 1 HCV in Group 8 and treatment-experienced subjects with genotype 2/3 in Group 9)
Groups 8 and 9: SOF+RBV
for 12 weeks
For all groups, SOF dose was 400 mg QD PO, Peg-IFN dose was 180 µg/week SC, and RBV dose was 1000 or 1200 mg/day (divided daily dose)a PO.
Groups 1−5 and 7–9: 12 weeks Group 6: 8 weeks Overall: 120 treated 118 completed through SVR12 assessment Group 1: 10 treated 10 completed Group 2: 9 treated 9 completed Group 3: 10 treated 10 completed Group 4: 11 treated 11 completed Group 5: 10 treated 10 completed Group 6: 10 treated 10 completed Group 7: 10 treated 10 completed Group 8: 25 treated 25 completed Group 9: 25 treated 23 completed Noncirrhotic, treatment-naive and treatment-experienced adult subjects with chronic genotype 1, 2, or 3 HCV infection
Study ongoing;
m5.3.5.1.6, Interim CSR
Type of Study Number Study Objective(s) Study Design Study and Control Drug Regimens Duration of Treatment Number of Subjects Study Population/ Entry Criteria Type of Report Study Status; Controlled Clinical Studies Pertinent to the Claimed Indication P2938-0721 (QUANTUM) (SOF-specific data)
Assess the efficacy (proportion of subjects with SVR12), PK, PD, safety, and tolerability of regimens containing SOF and RBV in subjects with chronic HCV infection Phase 2, randomized, double-blind, multicenter study Group C: SOF 400 mg QD PO + RBV PO for 12 weeks Group G: SOF 400 mg QD PO + RBV PO for 24 weeks For all subjects, RBV dose was 1000 or 1200 mg/day (divided daily dose)a. Group C: 12 weeks Group G: 24 weeks Overall: 50 treated 48 completed through the SVR12 assessment Group C: 25 treated 23 completed Group G: 25 treated 25 completed Treatment-naive adult subjects with chronic genotype 1−6 HCV infection; up to 10% of subjects may have the presence of cirrhosis Study completed; m5.3.5.1.7, Interim CSR Controlled Clinical Studies Pertinent to the Claimed Indication GS-US-334-0123
(PHOTON-1) Assess the efficacy (proportion of subjects with SVR4) and safety of SOF+RBV administered for 12 weeks in treatment-naive genotype 2 or 3 HCV subjects who are coinfected with HIV Phase 3, open-label, multicenter study SOF 400 mg QD PO + RBV 1000 or 1200 mg/day (divided daily dose)a PO
12 weeks 31 treated
23 completed through SVR4 assessment
Treatment-naive adult subjects with chronic genotype 2 or 3 HCV and coinfected with HIV; up to 20% of subjects may have the presence of cirrhosis Study ongoing; m5.3.5.1.8, Interim synoptic CSR Controlled Clinical Studies Pertinent to the Claimed Indication P7977-2025 (As of the 24 January 2013 interim data cut)
Assess if the administration of SOF+RBV (for up to 24 weeks) to HCV-infected subjects with HCC meeting the Milan criteria prior to undergoing liver transplant could prevent reinfection 12 weeks posttransplant Phase 2, open-label, multicenter study SOF 400 mg QD PO + RBV 1000 or 1200 mg/day (divided daily dose)a PO
Up to 24 weeks depending on time to liver transplant 61 treated 28 received study treatment and underwent liver transplant
Adult subjects with chronic HCV infection (any genotype) who met the Milan criteria and were undergoing liver transplant for HCC secondary to HCV-related cirrhosis with a MELD score of < 22 and a HCC- weighted MELD score of ≥ 22 and a
Child-Pugh score ≤ 7
Study ongoing;
m5.3.5.1.9, Interim synoptic CSR
2.7.6.1 - 17
Type of Study Number Study Objective(s) Study Design Study and Control Drug Regimens Duration of Treatment Number of Subjects Study Population/ Entry Criteria Type of Report Study Status;
Open-Label Study Pertinent to the Claimed Indication
GS-US-334-0118 Assess the efficacy (proportion of subjects with SVR12) and safety of SOF+RBV administered for 12 weeks in subjects with genotype 2 HCV infection. Phase 3, open-label, historical controlled, multicenter study SOF 400 mg QD PO + RBV 600, 800 or 1000 mg/day (divided daily dose)a PO SOF+RBV group: 12 weeks Overall: 153 treated 153 completed through SVR12 assessment Treatment-naïve and treatment experienced adult subjects with chronic genotype 2 or 3 HCV infection; up to 40% of subjects may have the presence of cirrhosis.
Study ongoing;
m5.3.5.2.1, Interim CSR
[12C]- = radiolabeled carbon 12; [14C]- = radiolabeled carbon 14; 3TC = lamivudine; API = active pharmaceutical ingredient; ART = antiretroviral therapy; ATV = atazanavir; BA =
bioavailability; BE = bioequivalence; BID = twice daily; CPT = Child-Pugh-Turcotte; CsA = cyclosporine (cyclosporin A); CSR = clinical study report; DAA = direct activing antivirals; DDI = drug-drug interaction; DRV = darunavir; EFV = efavirenz; FDC = fixed-dose combination; FTC = emtricitabine; HCV = hepatitis C virus; HIV = human immunodeficiency virus; Peg-IFN = pegylated interferon or Peg-IFN-alfa-2a; PD = pharmacodynamic(s); PK = pharmacokinetic(s); QD = once daily; QTcF = QT interval corrected for heart rate using the Fridericia formula; /r = boosted with ritonavir; RAL = raltegravir; RBV = ribavirin; RNA = ribonucleic acid; RPV = rilpivirine; RTV = ritonavir ; SOF = sofosbuvir, TDF = tenofovir disoproxil fumarate; ZDV = zidovudine.
MELD = model for end-stage liver disease
a For subjects who weighed < 75 kg, the RBV dose was 1000 mg/day divided into 2 doses, and for subjects who weighed ≥ 75 kg, the RBV dose was 1200 mg/day divided into 2 doses. b For Study P7977-0422, genotype 1 HCV-infected subjects with HCV RNA below the limit of detection on Day 28 through Week 12 (ie, extended rapid virologic response) received an
additional 12 weeks of Peg-IFN+RBV after the SOF/placebo treatment period. Genotype 1 HCV-infected subjects with HCV RNA not below the LOD on Day 28 or with HCV RNA above the LOD at any time from Day 28 through Week 12 received an additional 36 weeks of Peg-IFN+RBV after treatment with SOF and Peg-IFN+RBV. Genotype 1 HCV-infected subjects who received placebo and achieved an eRVR received an additional 36 weeks of Peg-IFN+RBV.
2. P7977-0111 試験
(参考資料:第5.3.1.2.1 項) 本試験は、GS-9851 100 mg カプセルとソホスブビル(SOF、GS-7977) 100mg 錠のバイオアベ イラビリティを比較する、第1 相、ランダム化、3 期(3-way)クロスオーバー、単回投与試験で あった。適格な被験者を William’s ラテン方格に従い 6 種類の投与順の 1 つに割り付けた。各被 験者はTreatment A~C の全ての投与を受けた。 表 2.7.6.2 - 1 P7977-0111 試験:試験の概要 項目 内容 試験番号 P7977-0111 試験の標題 PSI-7851 (GS-9851)カプセルの PSI-7977(SOF)錠に対する相対的バイオア ベイラビリティ及びPSI-7977 錠の食事の影響を評価する単回投与、ランダム 化、3 期、クロスオーバー試験 開発相 第1 相 目的 本試験の主要目的は次のとおりであった。 • 健康被験者において一晩絶食後にGS-9851 100 mg カプセル又は SOF 100 mg 錠を 200 mg 単回投与したときの吸収を比較する。 本試験の副次的目的は次のとおりであった。 • プロドラッグのGS-9851 と SOF の薬物動態を検討する。 • 健康被験者において、GS-9851 カプセルと SOF 錠を 200mg 単回経口投与 したときの安全性及び忍容性を検討する。 • 健康被験者において、SOF 100mg 錠を 200mg 単回投与したときの SOF とその代謝物の薬物動態に及ぼす高脂肪食の影響を検討する。 試験デザイン 24 例の被験者を William’s のラテン方格に従い 6 種類の投与順の 1 つに割り付 けた。各被験者は、以下のいずれの投与期 の単回投与を受け、各投与期の間 には7 日以上のウォッシュアウト期間を設けた。 • GS-9851(カプセル)投与期:GS-9851 200 mg(GS-9851 100mg カプセル ×2)を投与(投与期 A) • SOF(錠)投与期:SOF 200mg(SOF 100mg 錠×2)を投与(投与期 B) • SOF(錠)+高脂肪食投与期:SOF 200mg(SOF 100 mg 錠 × 2)を高脂 肪食摂取下で投与(投与期C) 被験者は、各投与期のDay 1 から 24 時間の薬物動態測定用検体採取と他の全 ての評価を完了するDay 2 の朝まで治験実施医療機関に入院した。48 時間と 72 時間の評価は外来で行った。各被験者は、各投与期の Day 1 に GS-9851 又 はSOF(絶食下及び高脂肪食摂取下)のいずれかの単回投与を受け、72 時間 後までの安全性及び薬物動態を評価した。 診断及び主な選 択基準 本試験には、18~55 歳、BMI が 19~32 kg/m2の男性並びに妊娠/授乳中では ない女性を組入れた。問題となる既往歴がなく、スクリーニング評価時に治 験責任医師が健康状態良好と判断した者とした。 被験者数(計画 時及び解析時) 計画時:24 例 解析時:24 例項目 内容 治験薬、用量、 用法及びロット 番号: • GS-9851(カプセル)投与期:GS-9851 200 mg(GS-9851 100mg カプセル ×2)を経口投与(投与期 A) • SOF(錠)投与期:SOF 200mg(SOF 100mg 錠×2)を経口投与(投与期 B) • SOF(錠)+高脂肪食投与期:SOF 200mg(SOF 100 mg 錠 × 2)を高脂 肪食摂取下で経口投与(投与期 C) ロット番号: (GS-9851)、 (SOF) 投与期間 GS-9851 200 mg を絶食下で単回投与 SOF 200 mg を絶食下で単回投与 SOF 200 mg を高脂肪食摂取下で単回投与 評価基準 薬物動態:各投与期にGS-9851、SOF、GS-566500(旧 PSI-352707)及び GS-331007(旧 PSI-6206)の血漿中濃度測定のため、投与前及び投与後 0.5、 1、1.5、2、3、4、6、8、10、12、16、24、48 及び 72 時間に採血した。 GS-9851、SOF、GS-566500 及び GS-331007 の尿中濃度測定のため、投与前の ベースライン(投与前排尿後の残尿)及び0-6、6-12、12-24、24~48 及び 48 ~72 時間の間隔での採尿を実施した。 次のような薬物動態パラメータをGS-9851、SOF、GS-566500 及び GS-331007 (可能な場合)について算出した:Cmax、Tmax、Clast、AUC0-last、AUCinf、 AUCexp、λz、t½、CL/F、Vz/F、尿中回収率、CLrenal
安全性:安全性は、有害事象、血液学的検査、血液生化学検査、尿検査、バ イタルサイン測定及び心電図の間隔を評価した。
解析方法 薬物動態:各被験者のGS-9851、SOF、GS-566500 及び GS-331007 の血漿中 及び尿中濃度-時間データはノンコンパートメントモデルにて解析した。 AUCinf、AUC0-las及びCmaxは対数変換して解析した。SOF 錠と GS-9851 カプ セルとの比較は、GS-331007 の AUCinf、AUC0-last及びCmaxについて自然対数 変換して行った。最小二乗幾何平均(GLSM)比と 90%信頼区間(CI)は、 GS-9851 カプセルに対する SOF 錠について算出した。
SOF 錠の高脂肪食摂取下と絶食下との比較は、GS-331007 の AUCinf、AUC0-last 及びCmax.について自然対数変換して行った。絶食投与時に対する高脂肪摂取 時の最小二乗幾何平均比と90%信頼区間を算出した。 安全性:安全性は投与を受けた薬剤毎に要約した。記述統計量又は被験者の 一覧のみで示した。 実施医療機関 1 施設(米国) 試験実施期間 2009 年 11 月 30 日(第 1 被験者のスクリーニング日)~ 2010 年 1 月 27 日(最後被験者の最終観察日) 2.7.6.2 - 2
2.1 被験者の内訳と人口統計学的特性
本試験には合計 24 例の被験者が組入れられ、ランダム化された。全被験者が 3 種の投与を全 て受け、試験を完了した。 被験者の平均年齢は 27 歳、年齢範囲は 18~51 歳であった。被験者の大半が白人で[24 例中 18 例(75%)]、男性が多かった[24 例中 14 例(58%)](表 2.7.6.1 - 2). 表 2.7.6.2 - 2 P7977-0111 試験:人口統計学的特性(安全性解析対象集団) Characteristic Overall (N = 24) Sex, n (%) Male 14 (58.3) Female 10 (41.7) Age, years Mean (SD) 27 (9.3) Median 24 Min, Max 18, 51 Race, n (%)American Indian or Alaska Native 1 (4.2) American Indian or Alaska Native, White 1 (4.2)
Asian 3 (12.5)
Black or African American, White 1 (4.2)
White 18 (75.0) Ethnicity, n (%) Non-Hispanic/Non-Latino 22 (91.7) Hispanic/Latino 2 (8.3) BMI, kg/m2 Mean (SD) 25.6 (3.28) Median 25.5 Min, Max 19.3, 30.7
Source: Section 15.1, Table 14.1.2; Appendix 16.2, Listing 16.2.4
2.2 有効性の結果
本試験では有効性の評価は行わなかった。
2.3 薬物動態の結果
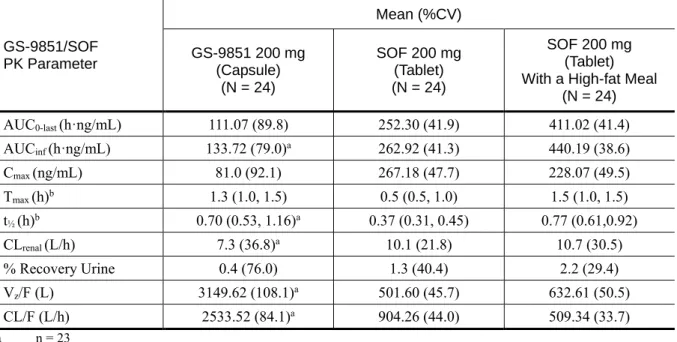
最小二乗幾何平均(GLSM)比と 90%信頼区間(CI)は 70%~143%の同等性基準の範囲内では なかった。SOF 錠による主要代謝物 GS-331007 の Cmax及びAUCinfの最小二乗幾何平均比は、
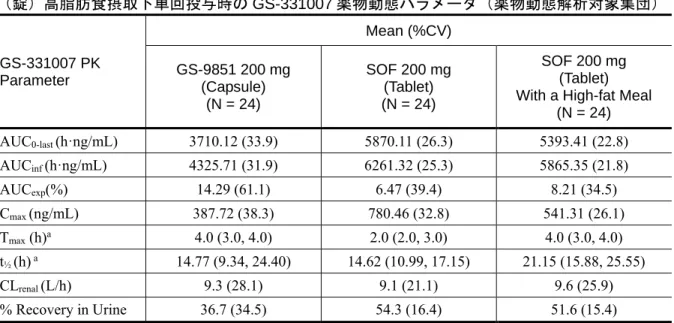
GS-9851 カプセルと比較して 207%及び 148%と高値であった(表 2.7.6.2 - 3)。SOF 錠の投与では GS-9851 カプセルの投与時よりも SOF 及び GS-566500 の平均曝露量は増加した(Cmaxはそれぞれ 3.3 倍、4.3 倍、AUCinfはそれぞれ2.0 倍、2.5 倍)(表 2.7.6.2 - 4、表 2.7.6.2 - 5)。これらのデー タは、GS-9851 カプセルを投与時と比較して比べて SOF 錠を投与時には吸収率及び吸収量が増加 することを示している。 高脂肪食摂取下投与では、絶食下投与と比較して SOF 錠の吸収速度は遅延するが、AUC を含 むPK パラメータからは、実質的な吸収量には大きな変化はなかった(表 2.7.6.2 - 6)。この結果 はSOF の代謝物 GS-566500 と GS-331007 の PK プロファイルにも影響を及ぼした。GS-331007 に ついて、Tmaxの延長(2 時間が 4 時間)、Cmaxの軽度低下(30%低下)が見られた。しかしなが
ら、GS-331007 の AUC0-last及び AUCinfは高脂肪食摂取下でも変化しなかった(表 2.7.6.1 - 3、表
2.7.6.1 - 6)。食事の影響についての同等性基準は満たさないが、Cmaxの減少は臨床的に問題とな るものではないと考えられた。 表 2.7.6.2 - 3 P7977-0111 試験:GS-9851 カプセル(絶食下)、SOF 錠(絶食下)又は SOF 錠(高脂肪食摂取下)単回投与時の GS-331007 の統計解析結果(薬物動態解析対象集団) GS-331007 PK Parameter %GLSM Ratio SOF (Tablet)/ GS-9851 (Capsule) 90% CI %GLSM Ratio SOF (Tablet) With a
High-fat Meal/ SOF (Tablet) Fasted
90% CI Cmax 207.38 (185.05, 232.40) 70.40 (62.8, 78.90) AUC0-last (h·ng/mL) 162.25 (148.13, 177.72) 92.69 (84.62, 101.53) AUCinf (h·ng/mL) 147.88 (136.50, 160.21) 94.46 (87.19, 102.33)
Source: Section 15.1, Table 14.2.19.1
表 2.7.6.2 - 4 P7977-0111 試験:GS-9851(カプセル)絶食下、SOF(錠)絶食下又は SOF (錠)高脂肪食摂取下単回投与時の GS-9851/SOF 薬物動態パラメータ (薬物動態解析対象集団) GS-9851/SOF PK Parameter Mean (%CV) GS-9851 200 mg (Capsule) (N = 24) SOF 200 mg (Tablet) (N = 24) SOF 200 mg (Tablet) With a High-fat Meal
(N = 24) AUC0-last (h·ng/mL) 111.07 (89.8) 252.30 (41.9) 411.02 (41.4) AUCinf (h·ng/mL) 133.72 (79.0)a 262.92 (41.3) 440.19 (38.6) Cmax (ng/mL) 81.0 (92.1) 267.18 (47.7) 228.07 (49.5) Tmax (h)b 1.3 (1.0, 1.5) 0.5 (0.5, 1.0) 1.5 (1.0, 1.5) t½ (h)b 0.70 (0.53, 1.16)a 0.37 (0.31, 0.45) 0.77 (0.61,0.92) CLrenal (L/h) 7.3 (36.8)a 10.1 (21.8) 10.7 (30.5) % Recovery Urine 0.4 (76.0) 1.3 (40.4) 2.2 (29.4) Vz/F (L) 3149.62 (108.1)a 501.60 (45.7) 632.61 (50.5) CL/F (L/h) 2533.52 (84.1)a 904.26 (44.0) 509.34 (33.7) a n = 23 b median (Q1, Q3)
Source: Section 15.1, Tables 14.2.10, 14.2.11, 14.2.12, 14.2.41, 14.2.42, and 14.2.43
表 2.7.6.2 - 5 P7977-0111 試験:GS-9851(カプセル)絶食下、SOF(錠)絶食下又は SOF (錠)高脂肪食摂取下単回投与時の GS-566500 薬物動態パラメータ(薬物動態解析対象集団) GS-566500 PK Parameter Mean (%CV) GS-9851 200 mg (Capsule) (N = 24) SOF 200 mg (Tablet) (N = 24) SOF 200 mg (Tablet) With a High-fat Meal
(N = 24) n = 23 n = 24 n = 24 AUC0-last (h·ng/mL) 97.93 (77.6) 437.66 (35.6) 615.67 (23.4) Cmax (ng/mL) 33.72 (54.1) 146.22 (37.2) 135.24 (22.0) Tmax (h)a 2.0 (1.5, 3.0) 1.0 (1.0, 1.5) 2.0 (1.8, 3.0) n = 19 n = 24 n = 24 AUCinf (h·ng/mL) 193.26 (50.0) 480.29 (32.3) 658.93 (22.2) AUCexp(%) 39.2 (60.9) 9.90 (56.2) 6.88 (34.8) t½ (h) 2.30 (1.83, 4.25) 1.80 (1.63, 1.90) 2.14 (1.98, 2.31) CLrenal (L/h) 6.7 (38.5) 6.8 (24.4) 8.3 (31.0) % Recovery in Urine 0.7 (56.4)b 2.1 (37.9) 3.4 (24.5) a median (Q1, Q3) b n = 24
表 2.7.6.2 - 6 P7977-0111 試験:GS-9851(カプセル)絶食下、SOF(錠)絶食下又は SOF (錠)高脂肪食摂取下単回投与時の GS-331007 薬物動態パラメータ(薬物動態解析対象集団) GS-331007 PK Parameter Mean (%CV) GS-9851 200 mg (Capsule) (N = 24) SOF 200 mg (Tablet) (N = 24) SOF 200 mg (Tablet) With a High-fat Meal
(N = 24) AUC0-last (h·ng/mL) 3710.12 (33.9) 5870.11 (26.3) 5393.41 (22.8) AUCinf (h·ng/mL) 4325.71 (31.9) 6261.32 (25.3) 5865.35 (21.8) AUCexp(%) 14.29 (61.1) 6.47 (39.4) 8.21 (34.5) Cmax (ng/mL) 387.72 (38.3) 780.46 (32.8) 541.31 (26.1) Tmax (h)a 4.0 (3.0, 4.0) 2.0 (2.0, 3.0) 4.0 (3.0, 4.0) t½ (h) a 14.77 (9.34, 24.40) 14.62 (10.99, 17.15) 21.15 (15.88, 25.55) CLrenal (L/h) 9.3 (28.1) 9.1 (21.1) 9.6 (25.9) % Recovery in Urine 36.7 (34.5) 54.3 (16.4) 51.6 (15.4) a median (Q1, Q3)
Source: Section 15.1, Tables 14.2.16, 14.2.17, 14.2.18, 14.2.47, 14.2.48, and 14.2.49
2.4 安全性の結果
本試験において、死亡、重篤な有害事象、妊娠、及び治験薬投与中止に至った有害事象は報告 されなかった。GS-9851 カプセル投与により、24 例中 3 例(12.5%)で有害事象が認められた。 その内訳は、頭痛、上腹部痛及び瀉血による頭部ふらふら感であった[治験総括報告書(第 5.3.1.2.1 項、Table 14.3.1)]。SOF 錠投与時には有害事象は認められなかった。発現した有害事 象の重症度は、瀉血による頭部ふらふら感の 1 件が中等度であった以外は全て軽度であり、一過 性であった。頭痛と上腹部痛の 2 件は、治験薬投与と関連ありと判定された[治験総括報告書 (第 5.3.1.2.1 項、Listing 16.2.7)]。臨床検査値異常は全てグレード 1 又は 2 であった。臨床的 徴候を伴う臨床検査値異常はなかった。2.5 結論
本試験の結論は次のとおりであった。 • SOF 錠と GS-9851 カプセルを健康被験者に単回投与したときの忍容性は概して良好であ った。 • GS-9851 カプセルと SOF 錠との同等性は認められなかった。SOF 錠投与後では、体内の 主要代謝物であるGS-331007 の Cmax及びAUCinfは、GS-9851 カプセルと比べてそれぞれ107%及び 48%高値であった。
• これらの曝露量の結果は、個体間変動は小さく、安全性プロファイルはGS-9851 カプセ ル及びSOF 錠で変わらなかったことから、今後の SOF の臨床開発で SOF 錠を使用する ことを支持するものであった。
• SOF 錠投与について、高脂肪食摂取下では絶食下と比較して、GS-331007 の Cmaxは約
30%低値であった。しかしながら、AUCinfは高脂肪食摂取下と絶食下では同程度であった
2.7.6.3 - 1
3. P7977-1318 試験
(参考資料:第5.3.1.2.2 項) 本試験は、健康被験者を対象に一晩絶食後にSOF 400 mg を単回投与したときの SOF 200 mg 錠 の吸収(率及び量)を SOF 400 mg 錠と比較して検討する第 1 相、単回投与、ランダム化、3 期 クロスオーバー試験であった。 表2.7.6.3 - 1 P7977-1318 試験:試験の概要 項目 内容 試験番号 P7977-1318 試験の標題 PSI-7977(SOF) 200 mg 錠と 400 mg 錠との生物学的同等性の評価及び 400 mg 錠のBA に対する食事の影響を評価する単回投与、ランダム化、3 期、クロス オーバー試験 開発相 第1 相 目的 本試験の主要評価項目は次のとおりである。 • 健康被験者に対し一晩絶食後に SOF 200 mg 錠又は 400 mg 錠を 400 mg の 用量で単回投与したときの吸収をGS-331007 の曝露量から比較する。 本試験の副次評価項目は次のとおりである。 • 健康被験者に対し一晩絶食後に SOF を投与したときの SOF 及び代謝物の 薬物動態特性を評価する。 • 健康被験者に SOF 400 mg 錠を単回投与したときの安全性及び忍容性を評 価する。 • 健康被験者に SOF 400 mg 錠を単回投与したときの SOF 及び代謝物の薬物 動態に及ぼす高脂肪食の影響を評価する。 試験デザイン 本第1 相、単回投与、ランダム化、3 期、クロスオーバー試験では、健康被験 者において一晩絶食後にSOF 200 mg 錠又は 400 mg 錠を 400 mg の用量で単回 投与したときの吸収を比較した。被験者をランダム化し、以下の投与法で単回 投与した。各投与間に最短7 日のウオッシュアウト期間を設けた。 • SOF 400 mg(200 mg 錠×2)を絶食下で投与(投与法 A) • SOF 400 mg(400 mg 錠×1)を絶食下で投与(投与法 B) • SOF 400 mg(400 mg 錠×1)を高脂肪食摂取下で投与(投与法 C) 診断名及び主な 組入れ基準 年齢18~55 歳、体格指数(BMI)19~36 kg/m2で、問題となる既往歴を有さ ず、治験薬の初回投与前30 日以内に実施されたスクリーニング評価で治験責 任医師により全身状態が良好であると判定された男性又は女性(女性は、妊娠 していない又は授乳婦でないこととした)を本試験に組入れた 被験者数 計画時:40 例 解析時:安全性解析対象集団40 例 薬物動態解析対象集団39 例項目 内容 治験薬、用法・ 用量及びロット 番号 被験薬 SOF 400 mg(400 mg 錠×1)を絶食下で単回投与(投与法 B) SOF 400 mg(400 mg 錠×1)を高脂肪食摂取下で単回投与(投与法 C) ロット番号: 対照薬 SOF 400 mg(200 mg 錠×2)を絶食下で単回投与(投与法 A) ロット番号: 投与期間 本試験には、4 日間の SOF 単回投与期が 3 期、各投与期の間での 1 週間のウオ ッシュアウト期間を含めた。 評価項目 薬物動態:各投与期のDay 1 に以下の時点の血液検体を採取した;投与前及び 投与後0.25、0.5、1、1.5、2、3、4、6、9、12、16、24、48 及び 72 時間。 次の薬物動態パラメータをSOF、GS-566500 及び GS-331007 について算出し た:Cmax、Clast、Tmax、AUC0−t、λz、t½、AUC0-inf、%AUCexp、CL/F 及び Vz/F. 安全性:安全性評価は有害事象及び併用薬のモニタリング、臨床検査値、バイ タルサイン測定、12 誘導心電図(ECG)及び理化学検査について試験期間中、 事前に取り決めた間隔で実施した。
解析方法 薬物動態:薬物動態パラメータは記述統計を用いて要約した。SOF 200 mg 錠 (対照製剤、投与法A)及び SOF 400 mg 錠(被験製剤、投与法 B)の生物学 的同等性はGS-331007 の AUC0-inf、AUC0−t及びCmaxの薬物動態パラメータ用い て検討した。これらの検討は、投与順序、投与期及び投与群を固定効果、投与 順序を加味した被験者をランダム効果とした混合効果モデルから導いた最小二 乗幾何平均比及びその90%信頼区間に基づいた。AUC0-inf、AUC0−t及びCmaxの 最小二乗幾何平均比及びその90%信頼区間は比較ごとに算出した。2 つの製剤 間の生物学的同等性は比の90%信頼区間が同等性の基準範囲 80%~125%内で あった場合に成立することとした。
SOF 400 mg 錠のバイオアベイラビリティに及ぼす高脂肪食の影響についても GS-331007 の AUC0-inf、AUC0−t及びCmaxの薬物動態パラメータ用いて検討し た。当該検討には投与順序、投与期及び投与群を固定効果、投与順序を加味し た被験者をランダム効果とした混合効果モデルから導いた最小二乗幾何平均比 及びその90%信頼区間を用いた。高脂肪食摂取下で SOF 400 mg を投与時(投 与法C)の曝露に関する薬物動態パラメータについて、絶食下で SOF 400 mg を投与時(投与法B)と比較したときの最小二乗幾何平均比の 90%信頼区間が 80%~143%の範囲内であった場合は、高脂肪食摂取下の投与は SOF 400 mg 錠 を投与したときのSOF の薬物動態を変化させないと結論付けた。 安全性:安全性解析対象集団は治験薬を1 回以上投与された全ての被験者とし た。安全性評価は有害事象及び併用薬のモニタリング、臨床検査値、バイタル サイン測定及び理化学検査を含めた。安全性データは初回投与日から最終投与 後30 日間までに得られたデータを含め、投与期ごとに解析した。有害事象名 はMedDRA(Medical Dictionary for Regulatory Activities)Version 15.0 にて読み 替えた。
実施医療機関 1 施設(米国)
試験期間 2011 年 8 月 2 日(最初の被験者のスクリーニング日)~ 2011 年 9 月 6 日(最後の被験者の最終観察日)
2.7.6.3 - 3
3.1 被験者の内訳と被験者背景
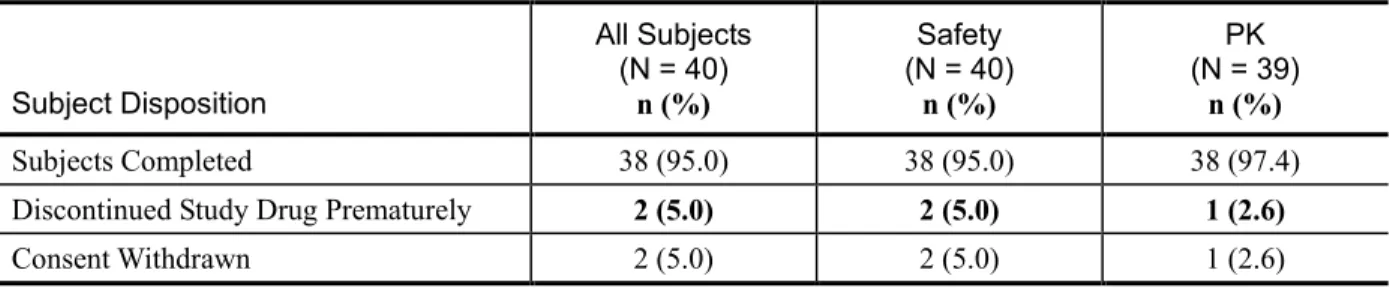
被験者計 40 例が本試験に組入れられ、38 例が試験を完了した。2 例は同意を撤回し、治験薬 の投与を完了することなく試験を中止した(表2.7.6.3 - 2)。 表2.7.6.3 - 2 P7977-1318 試験:被験者の内訳 Subject Disposition All Subjects (N = 40) n (%) Safety (N = 40) n (%) PK (N = 39) n (%) Subjects Completed 38 (95.0) 38 (95.0) 38 (97.4) Discontinued Study Drug Prematurely 2 (5.0) 2 (5.0) 1 (2.6)Consent Withdrawn 2 (5.0) 2 (5.0) 1 (2.6)
Source: Section 15.1, Table 9.1, Appendix 16.2, Listings 1 and 2
被験者の人口統計学的特性及びベースライン時の特性を表 2.7.6.3 - 3 に示す。被験者の平均年 齢は 34.4 歳、範囲は 18~55 歳であった。被験者の大半は女性[40 例中 23 例(57.5%)]、白人 [40 例中 39 例(97.5%)]であった。
表2.7.6.3 - 3 P7977-1318 試験:人口統計学的特性及びベースライン時の特性 (安全性解析対象集団)
Characteristic All Subjects (N = 40) Sex, n (%) Male 17 (42.5) Female 23 (57.5) Age, years Mean (SD) 34.4 (10.17) Median 32.5 Min, Max 18, 55 Race, n (%) White 39 (97.5)
Black or African American 1 (2.5)
Ethnicity, n (%)
Hispanic or Latino 2 (5.0)
Not Hispanic or Latino 38 (95.0)
Body Mass Index, kg/m2
Mean (SD) 28.3 (4.87)
Median 28.2
Min, Max 19.5, 36.0
3.2 薬物動態の結果
SOF 錠(400 mg 錠×1)と SOF 錠(200 mg 錠×2)との生物学的同等性及び SOF 錠 400 mg 錠 を投与したときの食事(高脂肪食摂取下)の影響を評価するために、GS-331007 の薬物動態パラ メータを算出した。絶食下でSOF 錠(200 mg 錠×2、対照製剤)又は SOF 錠(400 mg 錠×1、被 験製剤)並びに高脂肪食摂取下で SOF 400 mg 錠単回投与後の GS-331007 の薬物動態パラメータ の最小二乗幾何平均比及びその 90%信頼区間を用いた統計学的比較の要約を表 2.7.6.3 - 4 に示す。 表2.7.6.3 - 4 P7977-1318 試験:SOF 錠(2 × 200 mg 錠又は 1 × 400 mg 錠)投与後の GS-331007 の薬物動態パラメーターの統計学的比較(薬物動態解析対象集団) GS-331007 PK Parameter GLSM GLSM Ratio (90% CIs) Sofosbuvir 400 mg (2 × 200-mg tablet) Fasted (N = 39)a Sofosbuvir 400 mg (1 × 400-mg tablet) Fasted (N = 39)a Sofosbuvir 400 mg (1 × 400-mg tablet) Fasted / Sofosbuvir 400 mg (2 × 200-mg tablets) Fastedc Cmax (ng/mL) 1175.29 1168.89 99.5 (91.9, 107.7) AUC0-inf (ng·h/mL) 13,685.20 13,002.51 95.0 (91.0, 99.2) AUC0-t (ng·h/mL) 12,475.19 11,954.32 95.8 (91.3, 100.6) Sofosbuvir 400 mg (1 × 400-mg tablet) Fasted (N = 39)a Sofosbuvir 400 mg (1 × 400-mg tablet) High-Fat Meal (N = 38)a, b Sofosbuvir 400 mg (1 × 400-mg tablet) High-Fat Meal / Sofosbuvir 400 mg (1 × 400-mg tablet) Fastedc Cmax (ng/mL) 1168.89 886.35 75.8 ( 70.0, 82.2) AUC0-inf (ng·h/mL) 13,002.51 13,420.26 103.2 (98.8, 107.8) AUC0-t (ng·h/mL) 11,954.32 11,892.35 99.5 (94.7, 104.5)
a Subject 020 was not eligible for the PK analysis set and was excluded from the PK summary statistics.
b Subject 034 did not receive Sofosbuvir 400 mg (1 × 400-mg tablet) with high-fat meal and was excluded from the PK summary statistics.
c All available data were used in the mixed-effect model with treatment sequence, period, and treatment as fixed effect, not just paired data.
Source: Section 15.1, Tables 11.9; and 11.10 Appendix 16.2, Listing 23
主代謝物 GS-331007 の評価では、SOF 錠(1×400 mg 錠)は SOF 錠(2×200 mg 錠)と絶食下 投与で生物学的に同等であった。最小二乗幾何平均比及びその 90 信頼区間はあらかじめ規定し た80%~125%の範囲内であった。絶食下投与では、SOF 及び GS-566500 の薬物動態パラメータ の平均値もSOF 錠(1×400 mg 錠)と SOF 錠(2×200 mg 錠)で、ほぼ同じであった。 高脂肪食摂取下の投与では、絶食下投与と比較して SOF の Tmaxが0.5 時間から 1.5 時間へ延長 したことから吸収速度は低下したが、AUC などの薬物動態パラメータの推定値で示されたよう に、吸収の程度には実質的な変化はなかった。この結果は GS-566500 及び GS-331007 の両代謝物 の薬物動態プロファイルに反映された。GS-331007 では、高脂肪食摂取下での投与によって Tmax が3 時間から 4 時間へ延長し、Cmaxは軽度低下(24%)したが、AUC に変化はなかった。食事の
2.7.6.3 - 5 影響を否定する判定基準である 80%~143%には合致しなかったが、Cmaxの低下は臨床的に重要 とはみなさなかった。
3.3 安全性の結果
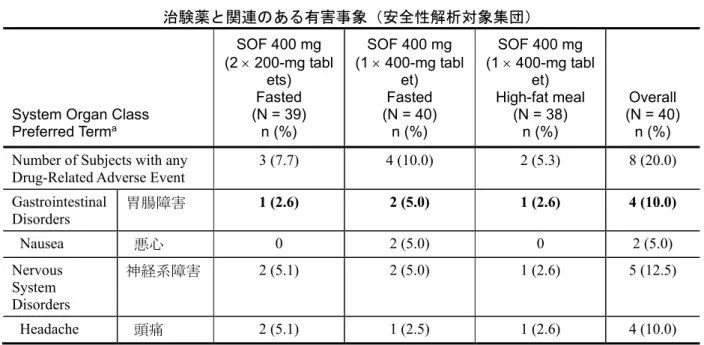
本試験では、死亡、重篤な有害事象、妊娠、治験薬の投与中止に至った有害事象の報告はなか った。最も多く報告された有害事象は頭痛(6 例)及び悪心(2 例)であった(表 2.7.6.3 - 5)。 その他の有害事象で 2 例以上の被験者で報告されたものはなかった。中等度の頭痛 1 件を除き、 有害事象は全て一過性であり、軽度であった。治験薬と関連ありと判定された有害事象は、頭痛 (4 例)、悪心(2 例)、腹痛(1 例)、消化不良(1 例)、筋肉痛(1 例)及び副鼻腔炎性頭痛 (1 例)であった(表 2.7.6.3 - 6)。Grade 3 の臨床検査値異常としては血清中リパーゼ増加の報 告が 1 件あった。その他の臨床検査値異常は Grade 1 又は 2 であった。臨床検査値異常は、いず れも臨床的徴候や症状を伴うものではなかった。 表2.7.6.3 - 5 P7977-1318 試験:2 例以上の被験者で報告された有害事象 (安全性解析対象集団)System Organ Class Preferred Terma SOF 400 mg (2 × 200-mg tablets) Fasted (N = 39) n (%) SOF 400 mg (1 × 400-mg tablet) Fasted (N = 40) n (%) SOF 400 mg (1 × 400-mg tablet) High-fat meal (N = 38) n (%) Overall (N = 40 ) n (%) Gastrointestinal Disorders 胃腸障害 1 (2.6) 2 (5.0) 1 (2.6) 4 (10.0) Nausea 悪心 0 2 (5.0) 0 2 (5.0) Nervous System Disorders 神経系 障害 3 (7.7) 4 (10.0) 1 (2.6) 7 (17.5) Headache 頭痛 3 (7.7) 3 (7.5) 1 (2.6) 6 (15.0)
a Adverse events were coded using MedDRA version 13.0
Note: Subject 020 did not receive SOF 400 mg (2 × 200-mg tablets) fasted or SOF 400 mg (1 × 400-mg tablet) with high-fat meal and Subject 034 did not receive SOF 400 mg (1 × 400-mg tablet) with high-fat meal, thus were not included in the tabulation of adverse events in the individual treatments.
表2.7.6.3 - 6 P7977-1318 試験:いずれかの投与期に 5%以上の被験者で報告された 治験薬と関連のある有害事象(安全性解析対象集団)
System Organ Class Preferred Terma SOF 400 mg (2 × 200-mg tabl ets) Fasted (N = 39) n (%) SOF 400 mg (1 × 400-mg tabl et) Fasted (N = 40) n (%) SOF 400 mg (1 × 400-mg tabl et) High-fat meal (N = 38) n (%) Overall (N = 40) n (%) Number of Subjects with any
Drug-Related Adverse Event 3 (7.7) 4 (10.0) 2 (5.3) 8 (20.0) Gastrointestinal Disorders 胃腸障害 1 (2.6) 2 (5.0) 1 (2.6) 4 (10.0) Nausea 悪心 0 2 (5.0) 0 2 (5.0) Nervous System Disorders 神経系障害 2 (5.1) 2 (5.0) 1 (2.6) 5 (12.5) Headache 頭痛 2 (5.1) 1 (2.5) 1 (2.6) 4 (10.0)
a Adverse events were coded using MedDRA version 13.0
Note: Subject 020 did not receive SOF 400 mg (2 × 200-mg tablets) fasted or SOF 400 mg (1 × 400-mg tablet) with high-fat meal and Subject 034 did not receive SOF 400 mg (1 × 400-mg tablet) with high-fat meal, thus were not included in the tabulation of adverse events in the individual treatments.
Source: Section 15.1, Table 10.3; Appendix 16.2, Listing 10
3.4 結論
本試験の結論は次のとおりであった。 • GS-331007 の薬物動態パラメータで評価すると、絶食下投与での SOF 錠(400 mg 錠×1) は、SOF 錠(200 mg 錠×2)と生物学的に同等であった。 • SOF 錠(200 mg 錠×2)及び SOF 錠(400 mg 錠×1)は、食事の有無にかかわらず、概し て良好な忍容性を示した。 • 高脂肪食によって SOF の吸収速度は低下したが、吸収の程度に実質的な変化はなかった。 GS-331007 の Cmaxの低下は臨床的に重要とはみなされず、SOF は食事の影響を受けない と考えられた。2.7.6.4 - 1
4. GS-US-334-0111 試験
(評価資料:第5.3.3.1.1 項) 本試験は、日本人及び白人健康被験者での GS-7977(ソホスブビル、SOF)(Group 1~3)及 び SOF/レジパスビル(LDV)の固定用量配合剤(FDC)(Group 4)の薬物動態、安全性及び忍 容性を評価する第1 相、非盲検、単回投与臨床試験であった。 試験には計64 例を 4 群のうちの 1 群に各 16 例(日本人 8 例、白人 8 例)組み入れた。 表2.7.6.4 - 1 試験 GS-US-334-0111 の要約 項目 内容 試験番号 GS-US-334-0111 試験の標題 日本人及び白人健康被験者でのGS-7977(SOF)並びに GS-7977 及び GS-5885 のFDC の薬物動態、安全性及び忍容性を評価する第 1 相、単回投与臨床試験 開発相 第1 相 目的 本試験の目的は以下のとおりであった。 • 日本人及び白人健康被験者にソホスブビル(SOF、GS-7977)を投与した ときのSOF 及びその代謝物(GS-566500 及び GS-331007)の薬物動態を検 討する • 日本人及び白人健康被験者にSOF 及びレジパスビル(LDV、GS-5885)の FDC を投与したときの SOF 及びその代謝物(566500 及び GS-331007)並びに LDV の薬物動態を検討する • 日本人及び白人健康被験者でのSOF 及び SOF/LDV の FDC の安全性及び 忍容性を検討する 試験デザイン 日本人及び白人健康被験者でのGS-7977SOF(Group 1~3)及び SOF/LDV の FDC(Group 4)の薬物動態、安全性及び忍容性を評価する第 1 相、非盲検、 単回投与臨床試験。計64 例を 4 群のうちの 1 群に各 16 例(日本人 8 例、白人 8 例)組み入れた。 治験薬投与開始28 日以内にスクリーニングを実施した。スクリーニング及び ベースライン(Day 0)の手順を完了後、Day 1 に適格被験者に対し、割り付け られた以下の投与群に対応した治験薬の経口単回投与を絶食下で実施した。 • Group 1;SOF 200 mg(200 mg 錠×1 錠) • Group 2:SOF 400 mg(400 mg 錠×1 錠) • Group 3:SOF 800 mg(400 mg 錠×2 錠) • Group 4:SOF/LDV FDC 400 mg/90 mg[FDC 錠(400 mg/90 mg)×1 錠] 投与群の投与順序は治験依頼者が決定した。適格となった被験者はベースライ ン(Day 0)に来院し、Group 1~3 では 96 時間、Group 4 では 144 時間までの 試験評価完了及び安全性上の懸念が消失するまで治験実施施設に入院した。被 験者は、安全性追跡調査のためにGroup 1~3 では投与後約7~10 日、Group 4 では投与後約8~10 日に来院した。項目 内容 診断及び主な選択 基準 年齢が18~45 歳(組入れ時)、体格指数(BMI)が 18~30 kg/m2(組入れ 時)で、腎機能正常であり、スクリーニング時に治験担当医師により良好な健 康状態と判断された被験者を選択した。 日本人被験者は一世とした。日本で出生した被験者で、10 年を超えて日本以 外に居住しておらず、両親及び祖父母が日本人であることを父母の家系で確認 できることとした。食事を含めた生活習慣が、日本を離れてから大きく変わら ないこととした。 白人被験者は日本人又はアジア人の子孫ではなく、被験者の両親及び祖父母の 出生地が日本又はアジア以外であることとした。 被験者数(計画時 及び解析時) 計画時: 約 64 例の被験者を次の 4 群に割り付けることとした。 • Group 1(SOF 200 mg):16 例(日本人 8 例及び白人 8 例) • Group 2(SOF 400 mg):16 例(日本人 8 例及び白人 8 例) • Group 3(SOF 800 mg):16 例(日本人 8 例及び白人 8 例) • Group 4(SOF/LDV FDC 400 mg/90 mg):16 例(日本人 8 例及び白人 8 例) 解析時: 64 例の被験者が次の 4 群に割り付けられた。 • Group 1(SOF 200 mg):16 例(日本人 8 例及び白人 8 例) • Group 2(SOF 400 mg):16 例(日本人 8 例及び白人 8 例) • Group 3(SOF 800 mg):16 例(日本人 8 例及び白人 8 例) • Group 4(SOF/LDV FDC 400 mg/90 mg):16 例(日本人 8 例及び白人 8 例) 治験薬、用量、用 法及びロット番 号: 治験薬(SOF 又は SOF/LDV FDC)は、下記の割り付けられた投与群に従い、 Day 1 に単回投与された。 • Group 1;SOF 200 mg(200 mg 錠×1 錠) • Group 2:SOF 400 mg(400 mg 錠×1 錠) • Group 3:SOF 800 mg(400 mg 錠×2 錠) • Group 4:SOF/LDV FDC 400 mg/90 mg[FDC 錠(400 mg/90 mg)×1 錠] 治験薬(SOF 又は SOF/LDV)は、一夜絶食後の Day 1 の朝に水(非炭酸水) 240 mL で投与した。被験者は絶食を継続し、投与 4 時間後の薬物動態検討の ための採血後まで食事摂取を制限した。治験薬服用に用いた240 mL の水以外 は、飲水も投与1 時間前から投与 2 時間後まで制限した。各群の治験薬投与順 序は治験依頼者により決定した。 SOF 200 mg 錠のロット番号は 、SOF 400 mg 錠のロット番号は 、SOF/LDV 400 mg/90 mg FDC のロット番号は であっ た。 投与期間 1 日間[Group 1、2 及び 3 では、それぞれ SOF 200 mg、400 mg 又は 800 mg の 単回傾向投与、Group 4 では SOF/LDV FDC(400 mg/90 mg)の単回傾向投与]
2.7.6.4 - 3
項目 内容
評価基準 有効性:本試験では有効性は評価しなかった。
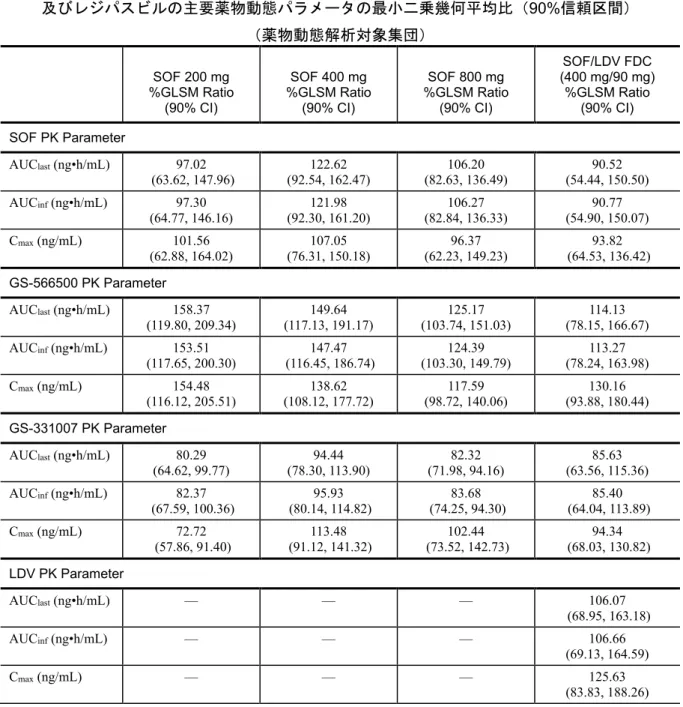
薬物動態:本試験の主要 薬物動態評価項目は、測定した SOF、GS-566500、 GS-331007(全群)、及び LDV(Group 4 のみ)の血漿中薬物動態パラメータ AUCinf、AUClast及びCmaxとした。腎クリアランス及び尿中排泄率%も求めた (Group 1~3)。 Group 1、2 及び 3 では、血漿 SOF 及び代謝物(GS-566500 及び GS-331007)濃 度を解析するため、次の時点で連続的に採血した。投与前(5 分以内)、投与 0.5、1、1.5、2、3、4、4.5、5、6、8、10、12、24、36、48、72 及び 96 時間 後。尿中SOF 及び代謝物(GS-566500 及び GS-331007)濃度を解析するため、 次の区間で採尿した。投与前の放尿、投与後0−6、6−12、12−24、24−48、 48−72、及び 72−96 時間。 Group 4 では、血漿 SOF 及び代謝物(GS-566500 及び GS-331007)及び LDV 濃 度を解析するため、次の時点で連続的に採血した。投与前(5 分以内)、投与 0.25、0.5、1、1.5、2、3、4、4.5、5、6、8、10、12、24、36、48、72、96、 120 及び 144 時間後。 安全性:本試験の主要安全性評価項目は、有害事象の発現頻度、臨床検査値異 常の評価、及び12-誘導心電図異常の評価であった。 解析方法 有効性:本試験では有効性の評価は行わなかった。 薬物動態:測定した化合物(SOF、GS-566500、GS-331007 及び LDV)の薬物 動態パラメータを被験者毎に記載し、記述統計量を用いて投与群別、日本人、 白人被験者別に要約した。評価化合物(SOF、GS-566500、GS-331007 及び LDV)の利用可能データから算出した薬物動態パラメータ(AUCinf、AUClast及 びCmax)を自然対数変換し、線型混合効果モデルを用いた分散分析(正規化) を実施した。混合効果モデルでは日本人及び白人を固定効果とした。薬物動態 パラメータの最小二乗幾何平均値では、各測定化合物での日本人と白人の比に ついて90%信頼区間を算出した。
用量比例性の情報は、AUClast、AUCinf及びCmaxを基にGroup 1~3 での全用量 の血漿中薬物濃度を比較することにより得られた。用量比例性の評価はパワー モデルを用いた。用量比例性のもう一つの評価では、用量で標準格化した薬物 動態パラメータAUCinf、AUClast及びCmaxの分散分析を用いて行った。評価値 は治療用量(すなわち、SOF 400 mg)に標準化し、用量を固定効果とした分 散分析モデルを用いて用量の違いが有意となるかを検定した。用量を標準化し たAUC 及び Cmaxの最小二乗幾何平均比の90%信頼区間が 0.7~1.43 の範囲に 収まれば、用量比例性があると結論した。 安全性:安全性データは被験者毎に記載し、投与群別及び日本人及び白人被験 者別に有害事象又は異常値の発現頻度又は適切な場合は記述統計量を要約し た。治験薬投与中、投与30 日後までの全ての安全性データを収集し要約し た。本報告で議論する有害事象及び臨床検査値異常は全て治験薬投与下で発現 したもので、本報告では有害事象及び臨床検査値異常と記載した。 実施医療機関 1 施設(米国) 試験実施期間 2012 年 4 月 13 日(最初の被験者のスクリーニング日)~ 2012 年 11 月 2 日(最後の被験者の最終観察日)