2.23 成人 B 型慢性肝炎患者を対象としたエンテカビルとラミブジンの安全性及び
抗ウイルス作用を比較する第 2 相無作為化二重盲検試験(Study AI463-005)
治験実施医療 機関 39 施設(オーストラリア、ベルギー、カナダ、フランス、ドイツ、香港、イス ラエル、イタリア、マレーシア、オランダ、フィリピン、ポーランド、ロシア、 シンガポール、タイ)公表文献 Rosmaw Ati M, Lai CL, Van Vlierberghe H, Anderson F, Thomas N, DeHertogh D. A multinational trial of entecavir vs lamivudine in adults with chronic hepatitis B. The European Association for the Study of Liver 2001.
治験期間 最初の被験者を組み入れた日:19 年 月 日 最後の被験者が完了した日:20 年 月 日 開発のフェーズ 第 2 相 目的 本治験の主要目的は、PCR 法による 22 週目の平均 log10 HBV DNA 量を指標と して、エンテカビルの各用量群における抗ウイルス作用をラミブジンと比較する ことであった。また、log10HBV DNA 量に基づき、エンテカビル 3 用量(0.01, 0.1 及び 0.5 mg)間の用量反応性を検討した。 治験方法 本治験は、ヌクレオシド未治療(12 週間以下)の成人 B 型慢性肝炎患者[HBe 抗原の有無は問わない]を対象に、エンテカビルの 3 用量(0.01, 0.1 及び 0.5 mg) とラミブジン 100 mg を 1 日 1 回、24 週間投与により比較する無作為化二重盲検 試験であった。盲検下で治験薬を 24 週間投与し(二重盲検投与期間)、22 週目 の血清 HBV DNA 量、HBe 抗原及び血清 ALT に基づいて、各被験者における 24 週目以降の治療法を選択した。即ち、22 週目に治療効果[bDNA 法による HBV DNA 量が検出限界(0.7 MEq/mL)未満及び ALT が正常の両方を満たし、かつ投 与開始前に HBe 抗原が陽性であった被験者では HBe 抗原が陰性化した場合]が 認められた被験者は、24 週目に治験薬投与を終了して 36 週目まで無治療で観察 し、安全性及び効果の持続性を検討した。無治療観察期間に再燃(bDNA 法によ り 2 週間以上の間隔で 2 回測定した HBV DNA 量が検出限界以上又は HBe 抗原 が陽性化)が認められた場合は、48 週目まで非盲検下でラミブジン(治験薬) を投与するか、治験責任医師が推奨する治療法を開始した。22 週目に Partial Response(PR:bDNA 法による HBV DNA 量が検出限界未満であるが、HBe 抗 原が陽性か ALT が異常の少なくとも一方を満たす場合)であった被験者は、24 週目から治療効果が認められるまで又は 48 週目まで、非盲検下でラミブジン(治 験薬)を投与した。22 週目に No Response(NR:bDNA 法による HBV DNA 量 が検出限界以上)であった被験者は、24 週目に治験薬投与を終了して治験責任 医師が推奨する治療法を開始すると共に、36 週目まで安全性を観察した。 選択基準 (1) 16 歳(又は、国ごとに定められた臨床試験への参加に必要な年齢)以上の
男女。
(2) 以下の条件をすべて満たすことにより、B 型慢性肝炎への感染が確認された 患者。
1) 無作為化前 24 週間以上にわたって血清 HBs 抗原が陽性
2) 2 週間以上の間隔で 2 回測定した bDNA 法による HBV DNA 量が 40 MEq/mL (143 pg/mL)以上 (3) 無作為化前 12 週間以上にわたって HBe 抗原が陽性、又は無作為化前 12 週 間以上にわたって HBe 抗原陰性かつ HBe 抗体陽性の患者。 (4) 血清 ALT が正常~基準値上限の 10 倍以下の患者。 (5) 以下の条件をすべて満たす代償性肝障害の患者。 1) プロトロンビン時間の延長が基準値から 3 秒以下、又は INR が 2.23 以下 2) 血清アルブミンが 3.0 g/dL(30 g/L)以上 3) 血清総ビリルビンが 2.5 mg/dL(42.75 µmol/L)以下 (6) 妊娠の可能性がある女性では、治験薬の投与開始前 48 時間以内に実施した 血清又は尿妊娠検査(β-ヒト絨毛性ゴナドトロピンの検出感度が 25 IU/L 以 上)の結果が陰性であること。 除外基準 (1) 無作為化前 24 週間以内に免疫抑制療法(副腎皮質ステロイドの全身投与を 含む)を受けた患者。 (2) 無作為化前 24 週間以内にインターフェロン α 又は thymosin-α を投与された 患者。 (3) 12 週間を超えてラミブジン、famciclovir、アデホビル又は lobucavir を投与 された患者。これらの薬剤は無作為化の 24 週間以上前に中止されていなけ ればならない。 (4) 既往歴、理学的検査又は血清学的検査により、ヒト免疫不全ウイルス(HIV) への感染が認められる患者。 (5) 血清クレアチニンが基準値上限の 1.5 倍を超える患者。 (6) ヘモグロビン量が 10.0 g/dL(100 g/L)未満の患者。 (7) 血小板数が 70,000 /mm3未満の患者。 (8) 顆粒球数が 1,500 /mm3未満の患者。 (9) リパーゼが基準値上限の 2.1 倍以上の患者、あるいは膵炎の臨床症状又は既 往(無作為化前 24 週間以内)がある患者。 (10) 肝性脳症、出血性静脈瘤、又は利尿剤や穿刺術を要する著しい腹水の合併あ るいはそれらの既往がある患者。 (11) 既往歴、肝生検又は血清学的検査により、他の肝疾患(C 型又は D 型肝炎 への重複感染、自己免疫性肝炎など)に罹病している患者。 (12) 肝細胞癌の既往歴、あるいは理学的検査又は X 線像により肝腫瘤が認めら れる患者。
(13) 血清 α-フェトプロテインが 20 ng/mL を超える患者。血清 α-フェトプロテイ ンが 21~100 ng/mL に上昇していた場合は、2 週間後の再測定で持続的な上 昇が認められず、かつ無作為化前に実施した肝の超音波検査又はコンピュー タ断層撮影(CT)検査により癌を示唆する病巣が認められない場合のみ、 組入れを可能とする。 (14) 以前にエンテカビルを使用した患者。 (15) アルコール又は非合法薬に対する依存症で、適切な服用ができないか、肝毒 性の危険性が高いと治験責任医師が判断した患者。 (16) 不妊手術を受けておらず、治験期間を通じて適切な避妊法(既承認の経口、 注射用又は埋込み型避妊薬、子宮内装具、殺精子薬を添加したペッサリー又 はコンドーム、性交渉を行わない)を実施する意思のない妊娠可能な女性及 びそのパートナー。 (17) 妊婦及び授乳婦。 (18) 経口投与ができない患者。 (19) 末梢血管の穿刺が困難な患者。 (20) その他の重篤な病状により本治験を完了することが困難な患者。 治験薬/用量 及び投与方法 (1) 治験薬 治験薬 剤型 バッチ番号 エンテカビル 0.01 mg カプセル N99108, N99152 エンテカビル 0.1 mg カプセル N99047 エンテカビル 0.5 mg カプセル 外観上相互に識別不能な灰色 の不透明なカプセル剤 N99049 ラミブジン 100 mg 錠 (カプセルに封入) 市販の 100 mg 錠を灰色の不 透明なカプセルに封入し、エ ンテカビルカプセルと外観上 識別不能にしたもの N99014, 9A432, 9G435, 9C433 04688.01 (2) 用量及び投与方法 治験薬(エンテカビル 0.01, 0.1, 0.5 mg 又はラミブジン 100 mg)1 カプセルを 1 日 1 回、毎日ほぼ一定の時刻(食事の前後 2 時間以内を避ける、午前 10 時を推 奨)に服用した。 評価項目/評価 基準 【有効性】 (1) 主要評価項目 PCR 法による 22 週目の平均 log10HBV DNA 量(エンテカビルの各用量群とラ ミブジン群との比較) (2) 副次評価項目 • PCR 法による 4, 12 及び 22 週目の log10HBV DNA 量の推移 • PCR 法(検出限界:400 copies/mL)による 12 及び 22 週目の HBV DNA 量 及び bDNA 法(検出限界:700,000 copies/mL 又は 0.7 MEq/mL)による 22 週目の HBV DNA 量が検出限界未満になった被験者の割合
陰性化した割合及びセロコンバージョン(HBe 抗原の消失及び HBe 抗体の 出現)した割合 • 投与開始前に血清 ALT が異常(WHO 毒性グレード 1 以上 = 基準値上限の 1.25 倍以上)であった被験者のうち、22 週目に正常化(WHO 毒性グレード 0 = 基準値上限の 1.25 倍未満)した割合 • 22 週目に治療効果が認められた被験者の割合 <治療効果の定義>
投与開始前に HBe 抗原が陽性であった被験者では、bDNA 法による HBV DNA 量が検出限界未満で HBe 抗原が陰性化し、かつ ALT が正常であること。投与開 始前に HBe 抗原が陰性であった被験者では、bDNA 法による HBV DNA 量が検 出限界未満かつ ALT が正常であること。
• 無治療で 12 週間観察後(36 週目)に治療効果が持続していた被験者の割合 • HBV DNA 量が検出限界未満になった後、治験薬投与中に 1 log10以上増加し
た被験者における、HBV の遺伝子的な耐性株出現頻度
• 投与開始前及び 22 週目に肝生検を実施した被験者における肝の組織学的改 善[Knodell Histologic Activity Index(HAI)スコアが 2 ポイント以上減少] 【安全性】 以下に該当する被験者の割合 • 臨床検査値異常、自他覚症状にかかわる有害事象、原疾患の悪化又は死亡に よる治験薬投与の中止 • 重篤な有害事象 • 有害事象及び臨床検査値異常 • グレード 3(重度)~4(非常に重度)と判定された有害事象及び臨床検査 値異常 • 血清 ALT、AST 及び総ビリルビンが投与前値の 3 倍以上に上昇 統計手法 (1) 有効性の解析 有効性の解析対象集団は評価可能集団とした。評価可能集団は、22 週目に PCR 法で血清 HBV DNA 量を測定し、その時点で治験薬投与を受けていた被験者(投 与終了/中止後 1 週間までは許容)と定義した。 連続変数は平均、中央値、標準偏差、最小値及び最大値により要約した。主要 評価項目の解析においては、投与前の HBV DNA 量及び HBe 抗原の有無を共変 量、投与群を独立変数とする回帰モデルを適用し、22 週目に PCR 法で測定した 血清 HBV DNA 量について、エンテカビルの各用量群とラミブジン群との差を算 出し、t 検定による比較を行った。その際、3 組の 2 群間比較のそれぞれに対し て異なる回帰モデルを適用すると共に、多重比較を考慮し、Hochberg の方法を 用いて有意水準を調整した。エンテカビル(0.01, 0.1, 0.5 mg)の用量反応性は、 log10HBV DNA 量を log10変換した投与量に対して一次回帰し、その傾きに t 検定
を適用することにより評価した。また、主要評価項目と同様の方法で各用量の 2 群間比較を行った。 二値反応変数は各カテゴリーに分類された被験者の頻度及び割合で要約し、投 与前の HBe 抗原の有無を層別因子とする Cochran-Mantel-Haenszel 検定を用いて 比較した。 (2) 安全性の解析 安全性の解析は、治験薬の投与を開始した被験者を対象に行った。ただし、重 篤な有害事象はスクリーニングを受けた被験者を対象として報告した。安全性デ ータの集計は、二重盲検投与期間(投与終了後 5 日目まで)、投与終了後観察期 間(HBV に対する治療の有無及び種類は問わない)別に行った。なお、22 週目 に治療効果が認められた被験者及び NR であった被験者における投与終了後観 察期間は 12 週間、22 週目に PR であった被験者における投与終了後観察期間は 24 週間であった。 計画被験者数 無作為化例数として 180 例(1 群 45 例) 【設定根拠】 1 群の被験者数は、PCR 法による 22 週目の平均 log10HBV DNA 量(主要評価 項目)の差に基づいて算出した。即ち、以下が成り立つものと仮定すると、1 群 の被験者数を 45 例に設定することにより、エンテカビルのある用量群のラミブ ジン群に対する優越性について、約 90%の検出力が確保される。 • log10HBV DNA 量の平均値の差が 1.0 である。
• log10HBV DNA 量の標準偏差が 1.25 で、投与開始前と 22 週目の HBV DNA
量の間に相関性がない。 • 各群で少なくとも 40 例が治験薬投与を完了する。 • エンテカビルの 3 用量群とラミブジン群との多重比較を考慮して調整を行 う。 報告書の日付 20 年 月 日 【結 果】 (1) 被験者の内訳 1) 治験の完了状況 スクリーニングを受けた被験者は 431 例で、そのうち 185 例が無作為割付けされ、177 例(エ ンテカビル 0.01 mg 群:54 例、0.1 mg 群:36 例、0.5 mg 群:46 例、ラミブジン群:41 例)に盲 検下で治験薬が投与された。 169 例(エンテカビル 0.01 mg 群:52 例、0.1 mg 群:34 例、0.5 mg 群:43 例、ラミブジン群: 40 例)が 24 週目まで投与を完了した。24 週未満で中止した被験者は 8 例で、そのうち 4 例が有 害事象又は臨床検査値異常によるものであった。
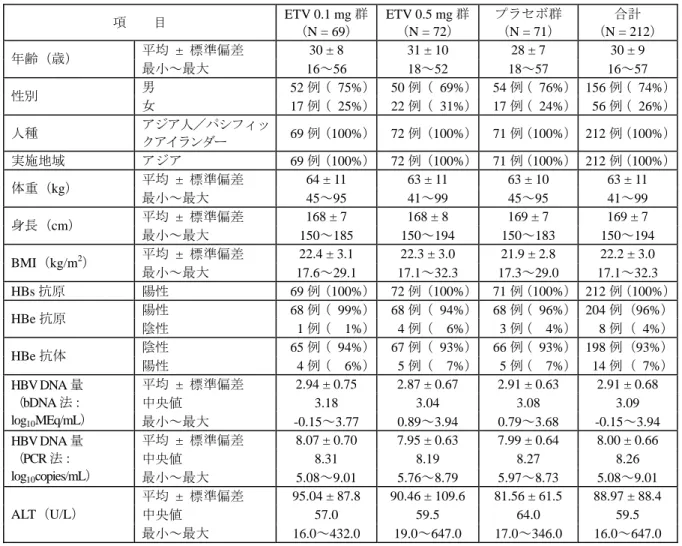
表 2.23-1: 治験の完了状況 エンテカビル 0.01 mg 群 0.1 mg 群 0.5 mg 群 ラミブジン 100 mg 群 合計 スクリーニングを受けた被験者 431 例 無作為割付けされた被験者 54 例 40 例 48 例 43 例 185 例 治験薬を投与された被験者 54 例 36 例 46 例 41 例 177 例 24 週未満で中止 2 例 2 例 3 例 1 例 8 例 有害事象又は臨床検査値異常 1 例 2 例 0 例 1 例 4 例 不適格 1 例 0 例 0 例 0 例 1 例 追跡不能 0 例 0 例 2 例 0 例 2 例 中止理由 コンプライアンス不良 0 例 0 例 1 例 0 例 1 例 24 週完了 52 例 34 例 43 例 40 例 169 例 2) 解析対象集団 22 週目に PCR 法で血清 HBV DNA 量を測定し、その時点で治験薬投与を受けていた被験者は 169 例(エンテカビル 0.01 mg 群:52 例、0.1 mg 群:34 例、0.5 mg 群:43 例、ラミブジン群:40 例)で、評価可能集団に採用され、有効性の解析対象となった。なお、中止例のうち中止時期が 22 週目以降であった 1 例は評価可能集団に採用され、完了例のうち 22 週目に来院がなかった 1 例は評価可能集団から除外された。 安全性は治験薬を投与された 177 例(エンテカビル 0.01 mg 群:54 例、0.1 mg 群:36 例、0.5 mg 群:46 例、ラミブジン群:41 例)を対象に評価した。 (2) 人口統計学的因子及び他の基準値の特性 大部分の被験者が男性で、アジア人が多く、平均年齢は 31.3~35.7 歳であった。ラミブジン群 (15%)では他の群(24~35%)と比較して、女性の割合が小さかった。 B 型慢性肝炎の病態について見ると、大部分の被験者が HBe 抗原陽性であり、bDNA 法及び PCR 法による HBV DNA 量(平均)はそれぞれ 2.7~2.8 log10MEq/mL 及び 7.9~8.1 log10copies/mL で、
群間に偏りは認められなかった。エンテカビル 0.5 mg 群では ALT が正常(基準値上限の 1.25 倍 未満)であった被験者の割合が他の群と比較して小さく、基準値上限の 2.5 倍を超えていた被験 者の割合が大きかった。
大部分の被験者は B 型肝炎の治療歴がなく、インターフェロンの使用経験がある被験者は 20 ~25%、ラミブジンの使用経験がある被験者は 0~6%であった。
表 2.23-2: 人口統計学的因子及び他の基準値の特性(治験薬を投与された被験者) エンテカビル 項 目 0.01 mg 群 (N = 54) 0.1 mg 群 (N = 36) 0.5 mg 群 (N = 46) ラミブジン 100 mg 群 (N = 41) 男 41 例(76%) 25 例(69%) 30 例(65%) 35 例(85%) 性別 女 13 例(24%) 11 例(31%) 16 例(35%) 6 例(15%) アジア人/パシフィック アイランダー 32 例(59%) 23 例(64%) 23 例(50%) 23 例(56%) 白人 19 例(35%) 11 例(31%) 16 例(35%) 16 例(39%) 黒人 1 例( 2%) 1 例( 3%) 4 例( 9%) 0 例 ヒスパニック/ラテン系 0 例 0 例 1 例( 2%) 0 例 人種 その他 2 例( 4%) 1 例( 3%) 2 例( 4%) 2 例( 5%) 平均 ± 標準偏差 35.7 ± 11.1 31.4 ± 10.1 32.5 ± 12.0 31.3 ± 11.9 年齢(歳) 最小~最大 16.0~60.0 17.0~52.0 16.0~68.0 16.0~60.0 HBe 抗原 陽性 44 例(81%) 31 例(86%) 36 例(78%) 33 例(80%) 平均 ± 標準偏差 2.8 ± 0.7 2.7 ± 0.9 2.8 ± 0.8 2.8 ± 1.1 HBV DNA 量 (bDNA 法: log10MEq/mL) 最小~最大 0.3~4.1 -0.2~3.9 0.1~4.4 -0.2~4.2 平均 ± 標準偏差 8.1 ± 0.7 7.9 ± 0.9 8.1 ± 0.9 8.0 ± 1.2 HBV DNA 量 (PCR 法: log10copies/mL) 最小~最大 5.9~9.6 6.1~9.6 5.7~9.4 3.7~9.6 < 1.25 × ULN a 22 例(41%) 16 例(44%) 14 例(30%) 18 例(44%) 1.25~2.5 × ULN 18 例(33%) 11 例(31%) 15 例(33%) 10 例(24%) > 2.5~5.0 × ULN 10 例(19%) 4 例(11%) 12 例(26%) 9 例(22%) > 5.0~10 × ULN 3 例( 6%) 5 例(14%) 4 例( 9%) 2 例( 5%) > 10 × ULN 1 例( 2%) 0 例 1 例( 2%) 2 例( 5%) 中央値 61.5 60.5 80.0 65.0 ALT(IU/L) 最小~最大 10.0~564.0 17.0~455.0 15.0~409.0 11.0~1,521.0 インターフェロン 11 例(20%) 9 例(25%) 11 例(24%) 8 例(20%) 前治療 ラミブジン 1 例( 2%) 2 例( 6%) 0 例 1 例( 2%) ULN(Upper Limit of Normal):基準値上限
(3) 有効性の結果 1) 主要評価項目
PCR 法で測定した 22 週目の血清 HBV DNA 量(投与前の HBV DNA 量及び HBe 抗原の有無で 調整)について、エンテカビルの 0.01, 0.1 及び 0.5 mg 群とラミブジン群との差(95%信頼区間) はそれぞれ 0.98(0.53~1.42)、-0.97(-1.42~-0.51)及び-1.28(-1.69~-0.86)log10copies/mL であ
った。エンテカビル 0.01 mg 群における HBV DNA 量の減少作用はラミブジン群よりも劣ってい たが、0.1 及び 0.5 mg 群についてはラミブジン群に対する優越性が示された。
表 2.23-3: PCR 法による 22 週目の血清 HBV DNA 量の薬剤間比較(評価可能集団) 0.01 mg 群 – ラミブジンン群 0.1 mg 群 –ラミブジン群 0.5 mg 群 – ラミブジン群 N=52, N=40 N=34, N=40 N=43, N=40 差の推定値a(95%信頼区間) 単位:log 10copies/mL 0.98(0.53~1.42) -0.97(-1.42~-0.51) -1.28(-1.69~-0.86) P < 0.0001 P < 0.0001 P = 0.0001 a 投与前の HBV DNA 量及び HBe 抗原の有無で調整 2) 副次評価項目 a) 血清 HBV DNA 量 PCR 法で測定した 4, 12 及び 22 週目における log10HBV DNA 量の推移は、図 2.23-1 に示すとお りで、22 週目における投与前からの平均変化量は、エンテカビルの 0.01, 0.1 及び 0.5 mg 群でそ れぞれ-2.41, -4.31 及び-4.72 log10copies/mL、ラミブジン群で-3.36 log10copies/mL であった。
図 2.23-1: PCR 法で測定した log10HBV DNA の投与前からの平均変化量の推移(評価可能集団)
PCR 法で測定した 22 週目の血清 HBV DNA 量(投与前の HBV DNA 量及び HBe 抗原の有無で 調整)について、エンテカビル 3 用量間の比較を行った結果、明らかな用量反応性が認められた。 即ち、0.1 及び 0.5 mg 群の HBV DNA 量の減少作用は 0.01 mg 群と比較して明らかに大きく、更 に、0.5 mg 群は 0.1 mg 群よりも優っていた。
表 2.23-4: PCR 法による 22 週目の血清 HBV DNA 量の用量間比較(評価可能集団) 0.01 mg 群 – 0.1 mg 群 0.01 mg 群 – 0.5 mg 群 0.1 mg 群 – 0.5 mg 群 N=52, N=34 N=52, N=43 N=34, N=43 差の推定値a(95%信頼区間) 単位:log 10copies/mL 1.92(1.53~2.31) 2.28(1.93~2.64) 0.38(0.07~0.69) P < 0.0001 P < 0.0001 P = 0.0180 a 投与前の HBV DNA 量及び HBe 抗原の有無で調整
PCR 法による 22 週目の血清 HBV DNA 量が 2 log10以上減少するか、400 copies/mL 未満になっ
た被験者の割合は、エンテカビル 0.1, 0.5 mg 群及びラミブジン群ではいずれも高く(92.5~100%)、 群間に差は認められなかったが、エンテカビル 0.01 mg 群では 65.4%で、他の 3 群と比較して小 さかった。HBV DNA 量が 400 copies/mL 未満になった被験者の割合は、エンテカビルの 0.1 及び 0.5 mg 群でそれぞれ 26.5%及び 25.6%であり、ラミブジン群(17.5%)よりも大きかったが、有意 差は認められなかった。
bDNA 法による 22 週目の HBV DNA 量が 0.7 MEq/mL 未満になった被験者の割合は、エンテカ ビル 0.5 mg 群(83.7%)がラミブジン群(57.5%)よりも優っていた。エンテカビル 0.1 mg 群(61.8%) とラミブジン群の間には差がなく、0.01 mg 群(23.1%)はラミブジン群よりも劣っていた。 表 2.23-5: 22 週目における血清 HBV DNA 量(評価可能集団) エンテカビル 0.01 mg 群 (N = 52) 0.1 mg 群 (N = 34) 0.5 mg 群 (N = 43) ラミブジン 100 mg 群 (N = 40) PCR 法 34 例(65.4%) 34 例(100%) 43 例(100%) 37 例(92.5%) 2 log10以上減少 or < 400 copies/mL P = 0.002 – a – a – 1 例(1.9%) 9 例(26.5%) 11 例(25.6%) 7 例(17.5%) < 400 copies/mL P = 0.010 P = 0.201 P = 0.373 – bDNA 法 12 例(23.1%) 21 例(61.8%) 36 例(83.7%) 23 例(57.5%) < 0.7 MEq/mL P = 0.001 P = 0.558 P = 0.008 – a 期待セル度数が 5 未満のため、計算不能。 b) 血清学的検査 投与開始前に HBe 抗原が陽性であった被験者を対象に HBe 抗原の陰性化及びセロコンバージ ョンを検討したが、いずれの群においても 22 週目に HBe 抗原が陰性化した被験者及びセロコン バージョンした被験者はわずかであった。
表 2.23-6: 22 週目における血清学的検査結果(評価可能集団 a) エンテカビル 0.01 mg 群 (N = 43) 0.1 mg 群 (N = 30) 0.5 mg 群 (N = 34) ラミブジン 100 mg 群 (N = 32) HBe 抗原の陰性化 0 例 4 例(13%) 0 例 2 例(6%) セロコンバージョン 0 例 2 例( 7%) 0 例 1 例(3%) a 投与開始前に HBe 抗原が陽性で、22 週目に血清学的評価が可能な被験者 c) 血清 ALT の正常化 投与開始前に ALT が異常であった被験者のうち 22 週目に正常化した割合は、エンテカビルの 0.1 及び 0.5 mg 群でそれぞれ 83.3%及び 69.0%で、いずれもラミブジン群(59.1%)よりも大きか ったが、評価対象例数が少なかったため、有意差は認められなかった。 表 2.23-7: 22 週目における血清 ALT の正常化(評価可能集団 a) エンテカビル 0.01 mg 群 (N = 30) 0.1 mg 群 (N = 18) 0.5 mg 群 (N = 29) ラミブジン 100 mg 群 (N = 22) 15 例(50%) 15 例(83.3%) 20 例(69.0%) 13 例(59.1%) ALT の正常化 P = 0.543 P = 0.098 P = 0.468 – a 血清 ALT が投与開始前に異常で、22 週目に評価可能な被験者 d) 治療効果 22 週目に治療効果が認められた被験者は 24 例のみで、その割合はエンテカビルの 0.01, 0.1, 0.5 mg 群及びラミブジン群でそれぞれ 6%, 24%, 16%及び 15%であった。エンテカビル 0.01 mg 群で は、他の群と比較して NR と判定された割合が大きかった。 表 2.23-8: 22 週目における治療効果(評価可能集団) エンテカビル 0.01 mg 群 (N = 52) 0.1 mg 群 (N = 34) 0.5 mg 群 (N = 43) ラミブジン 100 mg 群 (N = 40) 治療効果ありa 3 例( 6%) 8 例(24%) 7 例(16%) 6 例(15%) PRb 9 例(17%) 13 例(38%) 29 例(67%) 17 例(43%) NRc 40 例(77%) 13 例(38%) 7 例(16%) 16 例(40%) 欠測 0 例 0 例 0 例 1 例(3%) a
治療効果あり:bDNA 法による HBV DNA 量が検出限界(0.7 MEq/mL)未満及び ALT が正常の両方を満た し、かつ投与開始前に HBe 抗原が陽性であった被験者では HBe 抗原が陰性化した場合
b
PR(Partial Response):bDNA 法による HBV DNA 量が検出限界未満であるが HBe 抗原が陽性か ALT が異常 の少なくとも一方を満たす場合
c
e) 効果の持続性 22 週目に治療効果が認められた 24 例のうち、36 週目(無治療で 12 週間観察後)にも治療効果 が持続していた被験者は、エンテカビルの 0.01, 0.1 及び 0.5 mg 群でそれぞれ 1/3 例、6/8 例及び 3/7 例、ラミブジン群で 3/6 例であった。 f) 肝の組織学的改善 評価可能集団のうち、投与開始前及び 22 週目に肝生検を実施し、Knodell HAI スコアが評価可 能な被験者は 25 例であった。大部分の被験者では投与前と比較して Knodell HAI スコアが変化し なかったが、6 例(エンテカビル 0.01 mg 群:1 例、0.5 mg 群:3 例、ラミブジン群:2 例)では 改善、3 例(エンテカビル 0.01 mg 群:1 例、0.5 mg 群:2 例)では悪化が認められた。なお、上 記に加えて、評価可能集団から除外されたエンテカビル 0.5 mg 群の 1 例において、Knodell HAI スコアの改善が見られた。 表 2.23-9: 22 週目における肝の組織学的改善(評価可能集団 a) エンテカビル 0.01 mg 群 (N = 5) 0.1 mg 群 (N = 4) 0.5 mg 群 (N = 8) ラミブジン 100 mg 群 (N = 8) 改善 b 1 例(20%) 0 例 3 例(38%) 2 例(25%) 不変 c 3 例(60%) 4 例(100%) 3 例(38%) 6 例(75%) 悪化 d 1 例(20%) 0 例 2 例(25%) 0 例 a 投与開始前及び 22 週目に肝生検を実施し、Knodell HAI スコアが評価可能な被験者 b 投与前と比較して Knodell HAI スコアが 2 ポイント以上減少 c 投与前と比較して Knodell HAI スコアの変化が 2 ポイント未満 d 投与前と比較して Knodell HAI スコアが 2 ポイント以上上昇 g) HBV 耐性遺伝子変異 HBV DNA 量が検出限界未満になった後、二重盲検投与期間に 1 log10以上増加した被験者が、 エンテカビルの 0.01, 0.1 及び 0.5 mg 群で各 1 例、ラミブジン群で 2 例認められた。エンテカビル 群の 3 例において、HBV の遺伝子変異株は検出されなかった。 (4) 安全性の結果 1) 治験薬への曝露 二重盲検投与期間に治験薬を投与された被験者は 177 例(エンテカビル 0.01 mg 群:54 例、0.1 mg 群:36 例、0.5 mg 群:46 例、ラミブジン群:41 例)で、投与期間(中央値)はいずれの群に おいても 169 日であり、エンテカビルは最長 182 日間、ラミブジンは最長 180 日間投与された。 投与終了後観察期間には、上述の 177 例中 85 例(48%、内訳はエンテカビル 0.01 mg 群:14 例、 0.1 mg 群:17 例、0.5 mg 群:30 例、ラミブジン群:24 例)に非盲検下でラミブジン(治験薬又
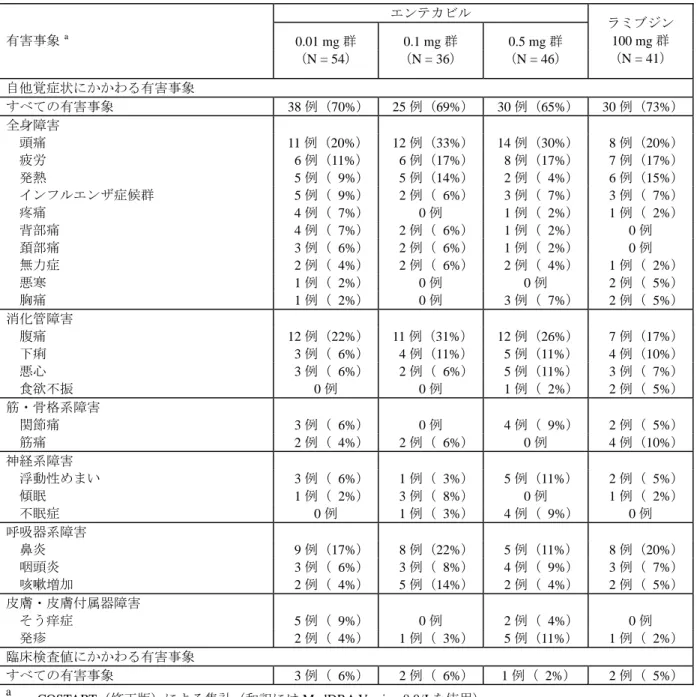
は市販品)が投与されていた。 2) 有害事象 二重盲検投与期間における自他覚症状にかかわる有害事象の発現率は、エンテカビルの 0.01, 0.1 及び 0.5 mg 群でそれぞれ 70%, 69%及び 65%、ラミブジン群で 73%で、群間に明らかな違いは 認められなかった。全体の発現率が最も高かったのは頭痛で、エンテカビル群で 20~33%、ラミ ブジン群で 20%認められた。次いで腹痛、疲労、鼻炎及び発熱が多かった。神経系障害に分類さ れる有害事象(浮動性めまい、不眠症、傾眠など)の発現率は、ラミブジン群よりもエンテカビ ル群、特に 0.5 mg 群で高い傾向が見られたが、明らかな差ではなかった。有害事象の程度は大部 分がグレード 1(軽度)又は 2(中等度)であった。 二重盲検投与期間における臨床検査値にかかわる有害事象の発現率は、エンテカビルの 0.01, 0.1 及び 0.5 mg 群でそれぞれ 6%, 6%及び 2%、ラミブジン群で 5%であり、個々の事象の発現率は いずれも 5%未満であった。
表 2.23-10: いずれかの群で 5%以上認められた有害事象(二重盲検投与期間) エンテカビル 有害事象 a 0.01 mg 群 (N = 54) 0.1 mg 群 (N = 36) 0.5 mg 群 (N = 46) ラミブジン 100 mg 群 (N = 41) 自他覚症状にかかわる有害事象 すべての有害事象 38 例(70%) 25 例(69%) 30 例(65%) 30 例(73%) 全身障害 頭痛 11 例(20%) 12 例(33%) 14 例(30%) 8 例(20%) 疲労 6 例(11%) 6 例(17%) 8 例(17%) 7 例(17%) 発熱 5 例( 9%) 5 例(14%) 2 例( 4%) 6 例(15%) インフルエンザ症候群 5 例( 9%) 2 例( 6%) 3 例( 7%) 3 例( 7%) 疼痛 4 例( 7%) 0 例 1 例( 2%) 1 例( 2%) 背部痛 4 例( 7%) 2 例( 6%) 1 例( 2%) 0 例 頚部痛 3 例( 6%) 2 例( 6%) 1 例( 2%) 0 例 無力症 2 例( 4%) 2 例( 6%) 2 例( 4%) 1 例( 2%) 悪寒 1 例( 2%) 0 例 0 例 2 例( 5%) 胸痛 1 例( 2%) 0 例 3 例( 7%) 2 例( 5%) 消化管障害 腹痛 12 例(22%) 11 例(31%) 12 例(26%) 7 例(17%) 下痢 3 例( 6%) 4 例(11%) 5 例(11%) 4 例(10%) 悪心 3 例( 6%) 2 例( 6%) 5 例(11%) 3 例( 7%) 食欲不振 0 例 0 例 1 例( 2%) 2 例( 5%) 筋・骨格系障害 関節痛 3 例( 6%) 0 例 4 例( 9%) 2 例( 5%) 筋痛 2 例( 4%) 2 例( 6%) 0 例 4 例(10%) 神経系障害 浮動性めまい 3 例( 6%) 1 例( 3%) 5 例(11%) 2 例( 5%) 傾眠 1 例( 2%) 3 例( 8%) 0 例 1 例( 2%) 不眠症 0 例 1 例( 3%) 4 例( 9%) 0 例 呼吸器系障害 鼻炎 9 例(17%) 8 例(22%) 5 例(11%) 8 例(20%) 咽頭炎 3 例( 6%) 3 例( 8%) 4 例( 9%) 3 例( 7%) 咳嗽増加 2 例( 4%) 5 例(14%) 2 例( 4%) 2 例( 5%) 皮膚・皮膚付属器障害 そう痒症 5 例( 9%) 0 例 2 例( 4%) 0 例 発疹 2 例( 4%) 1 例( 3%) 5 例(11%) 1 例( 2%) 臨床検査値にかかわる有害事象 すべての有害事象 3 例( 6%) 2 例( 6%) 1 例( 2%) 2 例( 5%) a
COSTART(修正版)による集計(和訳には MedDRA Version 8.0/J を使用)
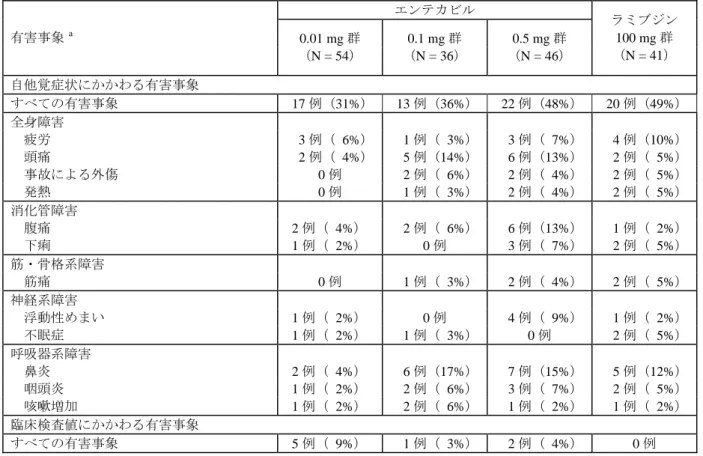
投与終了後観察期間における自他覚症状にかかわる有害事象の発現率は、エンテカビルの 0.01, 0.1 及び 0.5 mg 群でそれぞれ 31%, 36%及び 48%、ラミブジン群で 49%であり、認められた事象の 種類及び程度は二重盲検投与期間と類似していた。
投与終了後観察期間における臨床検査値にかかわる有害事象の発現率は、エンテカビルの 0.01, 0.1 及び 0.5 mg 群でそれぞれ 9%, 3%及び 4%であり、ラミブジン群では認められなかった。
表 2.23-11: いずれかの群で 5%以上認められた有害事象(投与終了後観察期間) エンテカビル 有害事象 a 0.01 mg 群 (N = 54) 0.1 mg 群 (N = 36) 0.5 mg 群 (N = 46) ラミブジン 100 mg 群 (N = 41) 自他覚症状にかかわる有害事象 すべての有害事象 17 例(31%) 13 例(36%) 22 例(48%) 20 例(49%) 全身障害 疲労 3 例( 6%) 1 例( 3%) 3 例( 7%) 4 例(10%) 頭痛 2 例( 4%) 5 例(14%) 6 例(13%) 2 例( 5%) 事故による外傷 0 例 2 例( 6%) 2 例( 4%) 2 例( 5%) 発熱 0 例 1 例( 3%) 2 例( 4%) 2 例( 5%) 消化管障害 腹痛 2 例( 4%) 2 例( 6%) 6 例(13%) 1 例( 2%) 下痢 1 例( 2%) 0 例 3 例( 7%) 2 例( 5%) 筋・骨格系障害 筋痛 0 例 1 例( 3%) 2 例( 4%) 2 例( 5%) 神経系障害 浮動性めまい 1 例( 2%) 0 例 4 例( 9%) 1 例( 2%) 不眠症 1 例( 2%) 1 例( 3%) 0 例 2 例( 5%) 呼吸器系障害 鼻炎 2 例( 4%) 6 例(17%) 7 例(15%) 5 例(12%) 咽頭炎 1 例( 2%) 2 例( 6%) 3 例( 7%) 2 例( 5%) 咳嗽増加 1 例( 2%) 2 例( 6%) 1 例( 2%) 1 例( 2%) 臨床検査値にかかわる有害事象 すべての有害事象 5 例( 9%) 1 例( 3%) 2 例( 4%) 0 例 a
COSTART(修正版)による集計(和訳には MedDRA Version 8.0/J を使用)
3) 死亡 本治験において死亡は報告されなかった。 4) その他の重篤な有害事象 自他覚症状にかかわる重篤な有害事象が 6 例[エンテカビル 0.01 mg 群:3 例(6%)、0.5 mg 群:1 例(2%)、ラミブジン群:2 例(5%)]で報告された。これらのうち 0.5 mg 群の 1 例は投与 開始前の肝生検時に肺出血を生じたもので、治験薬投与を開始しなかった。また、ラミブジン群 の 1 例では投与開始前より肝炎のフレアが認められ、1 日目の肝機能検査結果が得られた時点で 入院、投与中止となった。上記以外の 4 例における重篤な有害事象の発現時期はいずれも二重盲 検投与期間で、有害事象の内訳は外傷による背部痛及び浮腫が 1 例(0.01 mg 群)、甲状腺の新生 物(腺腫様甲状腺腫)が 1 例(0.01 mg 群)、胆道痛(胆石による胆道仙痛)が 1 例(0.01 mg 群)、 咽頭炎(扁桃炎)が 1 例(ラミブジン群)であった。 臨床検査値にかかわる重篤な有害事象は 3 例[エンテカビル 0.01 mg 群:2 例(4%)、0.1 mg 群:1 例(3%)]で報告された。内訳は高ビリルビン血症が 1 例(0.01 mg 群)、ALT 増加が 1 例 (0.01 mg 群)、ALT 増加及び AST 増加が 1 例(0.1 mg 群)で、高ビリルビン血症は二重盲検投 与期間、その他の事象は投与終了後観察期間に認められた。高ビリルビン血症を発現した被験者 では治験薬の投与が中止された。
表 2.23-12: 重篤な有害事象の発現状況(治験薬を投与された被験者) エンテカビル 有害事象 a 0.01 mg 群 (N = 54) 0.1 mg 群 (N = 36) 0.5 mg 群 (N = 46) ラミブジン 100 mg 群 (N = 41) 自他覚症状にかかわる重篤な有害事象 すべての有害事象 3 例(6%) 0 例 0 例 2 例(5%) 全身障害 1 例(2%) 0 例 0 例 0 例 浮腫 1 例(2%) 0 例 0 例 0 例 背部痛 1 例(2%) 0 例 0 例 0 例 消化管障害 1 例(2%) 0 例 0 例 1 例(2%) 胆道痛 1 例(2%) 0 例 0 例 0 例 肝炎 0 例 0 例 0 例 1 例(2%) 内分泌障害 1 例(2%) 0 例 0 例 0 例 甲状腺の新生物 1 例(2%) 0 例 0 例 0 例 呼吸器系障害 0 例 0 例 0 例 1 例(2%) 咽頭炎 0 例 0 例 0 例 1 例(2%) 臨床検査値にかかわる重篤な有害事象 すべての有害事象 2 例(4%) 1 例(3%) 0 例 0 例 ALT 増加 1 例(2%) 1 例(3%) 0 例 0 例 高ビリルビン血症 1 例(2%) 0 例 0 例 0 例 AST 増加 0 例 1 例(3%) 0 例 0 例 a
COSTART(修正版)による集計(和訳には MedDRA Version 8.0/J を使用)
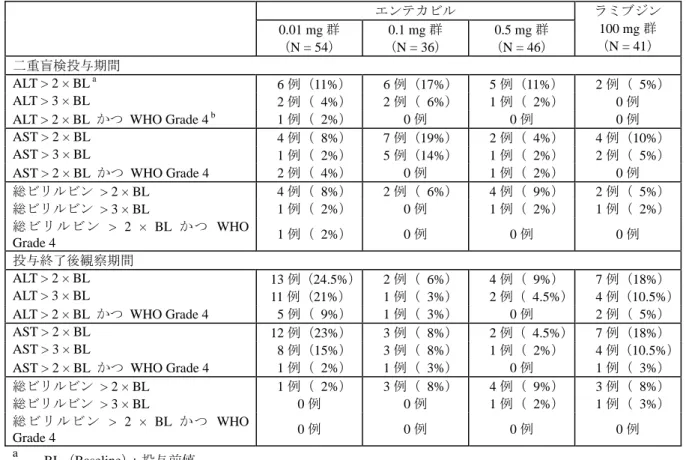
5) 有害事象による中止 自他覚症状又は臨床検査値にかかわる有害事象のために治験薬の投与を中止した被験者が 4 例 (エンテカビル 0.01 mg 群:1 例、0.1 mg 群:2 例、ラミブジン群:1 例)認められた。0.01 mg 群の 1 例(グレード 4 の高ビリルビン血症)及びラミブジン群の 1 例(肝炎のフレア)は上述の 重篤な有害事象発現例であり、0.1 mg 群の 2 例の内訳は、嗜眠及び光線過敏が 1 例、急性 HIV 感 染が 1 例であった。 6) 臨床検査値の異常 二重盲検投与期間に ALT が投与前値の 2 倍又は 3 倍を超えて上昇した被験者は、ラミブジン群 よりもエンテカビルの各用量群で多い傾向が見られた。投与前値の 2 倍を超えかつ WHO 毒性グ レード 4(基準値上限の 10 倍超)に該当する ALT 上昇を示したのは、0.01 mg 群の 1 例のみであ った。 エンテカビルの 0.01 mg 群では、投与終了後観察期間に ALT 又は AST が投与前値の 2 倍又は 3 倍を超えて上昇した被験者が、他の用量群及びラミブジン群と比較して多かった。
表 2.23-13: 二重盲検投与期間及び投与終了後観察期間における肝機能検査値の異常 エンテカビル 0.01 mg 群 (N = 54) 0.1 mg 群 (N = 36) 0.5 mg 群 (N = 46) ラミブジン 100 mg 群 (N = 41) 二重盲検投与期間 ALT > 2 × BL a 6 例(11%) 6 例(17%) 5 例(11%) 2 例( 5%) ALT > 3 × BL 2 例( 4%) 2 例( 6%) 1 例( 2%) 0 例 ALT > 2 × BL かつ WHO Grade 4 b 1 例( 2%) 0 例 0 例 0 例 AST > 2 × BL 4 例( 8%) 7 例(19%) 2 例( 4%) 4 例(10%) AST > 3 × BL 1 例( 2%) 5 例(14%) 1 例( 2%) 2 例( 5%) AST > 2 × BL かつ WHO Grade 4 2 例( 4%) 0 例 1 例( 2%) 0 例 総ビリルビン > 2 × BL 4 例( 8%) 2 例( 6%) 4 例( 9%) 2 例( 5%) 総ビリルビン > 3 × BL 1 例( 2%) 0 例 1 例( 2%) 1 例( 2%) 総ビリルビン > 2 × BL かつ WHO Grade 4 1 例( 2%) 0 例 0 例 0 例 投与終了後観察期間 ALT > 2 × BL 13 例(24.5%) 2 例( 6%) 4 例( 9%) 7 例(18%) ALT > 3 × BL 11 例(21%) 1 例( 3%) 2 例( 4.5%) 4 例(10.5%) ALT > 2 × BL かつ WHO Grade 4 5 例( 9%) 1 例( 3%) 0 例 2 例( 5%) AST > 2 × BL 12 例(23%) 3 例( 8%) 2 例( 4.5%) 7 例(18%) AST > 3 × BL 8 例(15%) 3 例( 8%) 1 例( 2%) 4 例(10.5%) AST > 2 × BL かつ WHO Grade 4 1 例( 2%) 1 例( 3%) 0 例 1 例( 3%) 総ビリルビン > 2 × BL 1 例( 2%) 3 例( 8%) 4 例( 9%) 3 例( 8%) 総ビリルビン > 3 × BL 0 例 0 例 1 例( 2%) 1 例( 3%) 総ビリルビン > 2 × BL かつ WHO Grade 4 0 例 0 例 0 例 0 例 a BL(Baseline):投与前値 b
WHO Grade 4:ALT/AST;> 10 × 基準値上限、総ビリルビン;> 5 × 基準値上限
二重盲検投与期間及び投与終了後観察期間における肝機能以外の臨床検査値異常の発現状況は、 エンテカビルの各用量群及びラミブジン群でほぼ同様であった。 (5) 結論 有効性の主要評価項目である PCR 法で測定した 22 週目の血清 HBV DNA 量について、エンテ カビルの 0.01, 0.1 及び 0.5 mg 群とラミブジン群との変化量の差(95%信頼区間)はそれぞれ 0.98 (0.53~1.42)、-0.97(-1.42~-0.51)及び-1.28(-1.69~-0.86)log10copies/mL で、エンテカビル 0.1 及び 0.5 mg 群のラミブジン群に対する優越性が示された。また、エンテカビルによる血清 HBV DNA 量の減少作用には、明らかな用量反応性が認められた。安全性について見ると、自他覚症状 及び臨床検査値にかかわる有害事象の発現状況は、エンテカビルの各用量群とラミブジン群とで ほぼ同様であった。 以上より、ヌクレオシド未治療(12 週間以下)の成人 B 型慢性肝炎患者において、エンテカビ ル 0.1 及び 0.5 mg の 1 日 1 回 22 週間投与は、ラミブジン 100 mg と比較して優れた血清 HBV DNA 量の減少作用を示し、耐容性も良好であった。
2.24 成人 B 型慢性肝炎患者を対象に BMS-200475(エンテカビル)経口投与時の
安全性と抗ウイルス作用の検討を目的とした用量漸増による無作為化二重盲
検プラセボ対照パイロット試験(Study AI463-004)
治験実施医療 機関 9 施設(アルゼンチン、ベルギー、カナダ、香港、イタリア、オランダ、アメリ カ) 公表文献 抄録 2 件:ICAAC(2000 年 9 月)、AASLD(2000 年 10 月)論文1件:De Man RA, Wolters L, Nevens F, Chua D, Sherman M, Lai CL, et al. Safety and Efficacy of Oral Entecavir Given for 28 Days in Patients with Chronic Hepatitis B Virus Infection. Hepatology 2001; 34: 578-582.
(2001 年 1 月現在) 治験期間 最初の被験者の組入れ日:19 年 月 日 最後の被験者の完了日:20 年 月 日 開発の フェーズ 第 2 相 目的 (1) 主要目的
1) 投与終了時に血清 HBV DNA 量が bDNA 法により 2 log10以上減少したか、
HBV DNA 量が bDNA 法により検出限界(2.5 pg/mL 又は 0.7 MEq/mL)未満 で、かつ PCR 法により 2 log10以上減少した被験者の割合をエンテカビル群 とプラセボとで比較する。 2) B 型慢性肝炎患者においてエンテカビル経口投与時の安全性を評価する。 治験方法 この治験は、成人 B 型慢性肝炎患者を対象としてエンテカビルの安全性と抗 ウイルス活性を検討した、用量漸増による無作為化二重盲検プラセボ対照試験 であった。エンテカビルは 4 用量(0.05, 0.1, 0.5 及び 1 mg)を 1 日 1 回、28 日 間投与した(当初は 0.1, 0.5, 1 及び 2.5 mg の投与が予定されていたが、0.1 mg 投 与時に認められた抗ウイルス活性が高かったため、2.5 mg に代えて 0.05 mg を投 与する治験実施計画書に変更した)。各用量群には 10 例(エンテカビル 8 例、 プラセボ 2 例)を無作為に割り付けた。投与終了前に薬剤関連毒性以外の理由 で被験者が脱落した場合には、代わりの被験者を組み入れることとした。 投与終了後には遅延性毒性を観察するために、最低 12 週間の無治療期間を含 む 24 週間の投与終了後観察期間を設けた(当初は 24 週間の投与終了後観察期 間を通じて被験者に対する抗 HBV 療法を禁止したが、その後禁止期間を最初の 12 週間に変更した)。 投与期間中は、毎週 bDNA 法により HBV DNA 量を測定して抗ウイルス効果 を評価した。また、1 日目の投与開始前と 28 日目の最終投与時には、更に高感 度の PCR 法(検出限界:400 copies/mL)により HBV DNA 量を測定した。投与
期間終了後は、42 及び 56 日目に HBV DNA 量を測定した。HBV の血清学的検 査も 1, 28 及び 196 日目に実施した。 エンテカビルの薬物動態を解析するため、投与期間中に全例からトラフ時の 血液試料を採取した(1, 7, 14, 21 及び 28 日目)。実施医療機関 013(ロッテルダ ム)で組み入れられた被験者は、薬物動態解析のため 1 及び 14 日目に投与後 24 時間にわたって(投与 1.5, 3, 6 及び 10 時間後)血液を採取した。また、HBV DNA 量に関する薬力学的検討のため血液試料を頻回に採取し、完全閉環二本鎖(ccc) DNA 量の減少を評価するため肝生検を 1, 14 及び 28 日目に実施した。 安全性評価(有害事象、血液生化学的検査、血液学的検査)は、投与期間中 には毎週の来院日、24 週間の投与終了後観察期間中には 4 週間ごとに 1 回実施 した。 選択基準 (1) 16 歳以上の男女。ただし法的基準がある国では 18 歳以上。 (2) 以下に該当する B 型慢性肝炎の患者。 1) 血清 HBs 抗原が 24 週間以上にわたって検出されている。 2) bDNA 法を使用して最低 2 週間の間隔で 2 回測定した HBV DNA 量が 70 pg/mL(20 MEq/mL)以上で、2 回の測定値の差が 1 log10を超えない。 (3) 以下の条件をすべて満たす代償性肝障害の患者。 1) プロトロンビン時間の延長が基準値から 3 秒以下、又は INR が 1.95 以下。 2) 血清アルブミン値が 3 g/dL(30 g/L)を超える。 3) ビリルビン値が 3 mg/dL(51.3 µmol/L)未満。 4) ALT 及び AST が基準値上限の 5 倍以下。 (4) 妊娠可能な女性は、治験薬投与開始前 72 時間以内に血清又は尿妊娠検査(β-ヒト絨毛性ゴナドトロピンの最小検出感度が 25 IU/L)が陰性であること。 除外基準 (1) 治験参加の 6 ヵ月以内に免疫抑制療法(ステロイドを含む)、又は B 型肝炎 に対して効果のある薬物(例えばインターフェロン α、ラミブジン、 famciclovir、lobucavir、アデホビル)の投与を受けた患者。 (2) 既往歴、身体所見又は血清学的検査所見から、ヒト免疫不全ウイルス(HIV) への感染が確認されている患者。 (3) 血清クレアチニン値が基準値上限の 1.3 倍を超える患者。 (4) ヘモグロビン量が 11.0 g/dL(110 g/L)未満の患者。 (5) 血小板数が 75,000 /mm3(75 × 109 /L)未満の患者。 (6) 顆粒球数が 1,500 /mm3未満の患者。 (7) リパーゼが基準値上限の 1.9 倍以上、又は膵炎の臨床症候がある患者。 (8) 6 ヵ月以内に膵炎に罹患した患者。 (9) 明らかな腹水(利尿薬や穿刺術が要求される)、肝性脳症又は出血性静脈瘤 の合併又は既往のある患者。 (10) 既往歴、血清学的検査又は肝生検で他の肝疾患(例えば C 型肝炎や D 型肝
炎)がある患者。 (11) ヌクレオシド類縁体によるアレルギーの既往歴がある患者。 (12) 以前にエンテカビルを使用した患者。 (13) アルコール又はドラッグの濫用者。 (14) 不妊手術を受けておらず、治験期間中にわたって適切な避妊法(既承認の 経口、注射用又は埋込み型避妊薬、子宮内装具、殺精子薬を添加したペッ サリーやコンドーム、性交渉を行わない)を実施する意思のない妊娠可能 な女性及びそのパートナー。 (15) 妊婦又は授乳婦。 (16) 経口投与ができない患者。 (17) 末梢静脈の穿刺が困難な患者。 (18) その他の重篤な病状により治験を完了することが困難な患者。 (19) 片頭痛、群発頭痛又は頻繁な緊張性頭痛の既往がある患者。 治験薬/用量 及び投与方法 (1) 治験薬 治験薬 剤型 バッチ番号 エンテカビル 0.1 mg カプセル N98071 エンテカビル 0.5 mg カプセル N98156, N97138 エンテカビル 0.05 mg カプセル N99110 プラセボカプセル 0 号の灰色の不 透明な硬カプセ ル N96158 (2) 用量及び投与方法 エンテカビル 0.1, 0.5 又は 0.05 mg 群に無作為割付けされた被験者は、エンテ カビル 1 カプセル(0.1, 0.5 又は 0.05 mg)を 1 日 1 回服用した。また、1 mg 群 に割り付けられた被験者は、0.5 mg を 2 カプセル 1 日 1 回服用した。プラセボ 群に割り付けられた被験者は、エンテカビルカプセルと外観上識別不能なプラ セボ 1 カプセル(1 mg 投与時は 2 カプセル)を 1 日 1 回服用した。治験薬の服 用は毎日ほぼ一定の時刻(食事の前後 2 時間以内を避ける、午前 10 時を推奨) に行った。 評 価 項 目 / 評 価基準 【有効性】 (1) 主要評価項目
投与終了時に血清 HBV DNA 量が bDNA 法により 2 log10以上減少したか、又
は HBV DNA 量が bDNA 法により検出限界未満で、かつ PCR 法により 2 log10以
上減少した被験者の割合。 (2) 副次評価項目 1) 投与終了時における PCR 法による HBV DNA 量のベースライン時と比較し た減少度。 2) 投与終了時及び無治療観察期間に bDNA 法で HBV DNA 量が検出限界未満 となった被験者の割合。 3) 無治療観察期間に HBV DNA 量がベースライン時の値 ± 10%に戻った被験
者の割合及びベースライン時の値に戻るまでの期間の中央値。 4) 投与終了時及び無治療観察期間終了時に HBe 抗原が陰性となった被験者の 割合。 【安全性】 治験薬の投与を 1 回でも受けたすべての被験者について安全性の評価を実施 した。初回投与時から試験終了時又は中止時までに収集された臨床所見と臨床 検査値データを投与前に測定したベースライン値と比較した。毒性が認められ た場合には、治験薬投与との関連性(「関連あり」「関連あるかもしれない」「関 連なし」)の判定を実施した。 【薬物動態・薬力学】 エンテカビル経口投与後のトラフ時の薬物濃度。 統計手法 (1) 解析手法 離散変数はそれぞれのカテゴリーに分類された頻度及び割合で要約した。連 続変数は平均値、中央値、標準偏差、最小値及び最大値で要約した。信頼区間 はプールしない t 分布を用いて算出した。 連続変数である HBV DNA 量の平均値の比較は、HBV DNA のベースライン値 からの変化量に基づいて Welch の t 検定を行った。HBV DNA 量の経時的グラフ はベースライン値からの変化に基づいて作成した。二値反応変数の比較は Fisher 直接確率計算法を使用した。主要な比較に対する有意水準は、Bonferroni 型の調 整を行った。プラセボ群は各用量群でプラセボを投与した被験者を合計した。 (2) 主要評価項目の解析 薬剤の毒性のため投与が中断された被験者は、主要評価項目を無効として扱 った。 (3) 副次評価項目 ベースライン時に HBe 抗原陽性の被験者において、HBe 抗原の陰性化を治療 群間で比較した。また、ベースライン時に ALT が異常値(グレード 0 を超える) の被験者では ALT の正常化(グレード 0)の割合を治療群間で比較した。 (4) 安全性 投与中止の頻度及び中止理由を治療群ごとに集計した。臨床検査値異常につ いては、ベースライン時に該当項目が正常値であった被験者と異常値であった 被験者を分け、治験中にグレード 1 又は 2 の毒性を示した被験者とグレード 3 又は 4 の毒性を示した被験者の百分率を算出した。 計画被験者数 組入れ予定数は、各用量共 10 例(計 40 例)で、10 例中 8 例はエンテカビル、2 例はプラセボを投与。 【設定根拠】 この治験の主要評価項目に対して、ヌクレオシド類縁体である lobucavir のパ イロット試験では 90%を超える反応率を示し、一方プラセボ群 5 例では反応し
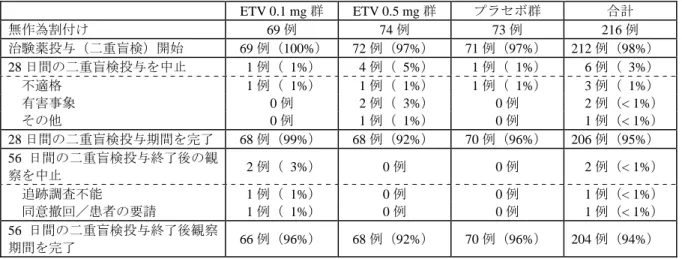
なかった。 被験者数を 8 例として二項分布を仮定すると: (1) HBV DNA 量に対する真の反応率が 90%であれば、95%の確率で 6 例以上の 反応例が観察される。 (2) 真の用量制限毒性発現率が 50%であれば、85%の確率で毒性による中止例が 3 例以上観察される。 報告書の日付 20 年 月 日 【結 果】 (1) 被験者の内訳 計画被験者数は各用量群 10 例(エンテカビル投与 8 例、プラセボ投与 2 例)の計 40 例であっ たが、投与前の HBV DNA 量が選択基準に満たなかった 0.1 mg 群の 1 例及び血清リパーゼ増加の ために治験薬投与が中断された 0.5 mg 群の 1 例に代わって、投与開始後に予備被験者を組み入れ たため、治験薬を投与された被験者は計 42 例(0.05 mg 及び 1 mg:各 8 例、0.1 mg 及び 0.5 mg: 各 9 例、プラセボ:8 例)となった。これら被験者のうち、0.1 mg 群及び 1 mg 群の各 1 例では有 害事象のため治験薬の投与が中止されたため、28 日間の投与を完了した被験者は 40 例であった。 その他、スクリーニング時に重篤な有害事象を発現した 1 例が無作為化の対象から除外された。 したがって、有効性の評価対象は無作為割付けされた 42 例から 1 例(0.1 mg の投与前 HBV DNA 量が選択基準に満たなかった被験者)を除いた 41 例が採用され、安全性の評価対象は治験薬が投 与された 42 例となった(表 2.24-1)。 表 2.24-1: 被験者の内訳 0.05 mg 群 0.1 mg 群 0.5 mg 群 1 mg 群 プラセボ群 合計 スクリーニングを受けた被験者 43 例 無作為割付けされた被験者 8 例 9 例 9 例 8 例 8 例 42 例 治験薬を投与された被験者 8 例 9 例 9 例 8 例 8 例 42 例 中止例 0 例 1 例 0 例 1 例 0 例 2 例 投与を完了した被験者 8 例 8 例 9 例 7 例 8 例 40 例 有効性の評価対象 8 例 8 例 9 例 8 例 8 例 41 例 安全性の評価対象 8 例 9 例 9 例 8 例 8 例 42 例 大部分の被験者が男性で、年齢はおよそ 40 歳、大部分がアジア人で、約 1/3 が白人であった。 0.05 mg 群では年齢がやや低く(平均 34 歳)、0.5 mg 群では体重がやや低かった(平均 64.2 kg)。 被験者はすべて B 型慢性肝炎であった。HBe 抗原陰性の被験者は 4 例であった。ベースライン 時の bDNA 法による HBV DNA 量は、0.05 mg 群で他の用量群よりやや低かった。また、ALT は、 中央値で比較すると 0.1 mg 群で低く、1 mg 群で高い値を示した。
を受けた被験者の割合に偏りが認められた[0.5 mg 群:0 例、0.05 mg 群:4 例(50%)]。 表 2.24-2: 被験者の背景 1(年齢、性別、体重、人種) 項 目 0.05 mg 群 (8 例) 0.1 mg 群 (9 例) 0.5 mg 群 (9 例) 1 mg 群 (8 例) プラセボ群 (8 例) 平均 ± 標準偏差 33.75 ± 17.86 45.11 ± 12.72 35.22 ± 14.02 41.13 ± 17.3 42.13 ± 15.12 中央値 24.0 40.0 33.0 36.0 43.5 年齢(歳) 最小~最大 17.0~63.0 29.0~63.0 18.0~58.0 19.0~75.0 24.0~73.0 男性 6 例(75%) 8 例(89%) 7 例(78%) 8 例(100%) 7 例(88%) 性別 女性 2 例(25%) 1 例(11%) 2 例(22%) 0 例 1 例(13%) 平均 ± 標準偏差 66.84 ± 10.56 73.12 ± 16.35 64.2 ± 9.29 78.6 ± 7.73 69.57 ± 9.71 中央値 70.25 73.0 60.8 77.1 71.4 体重(kg) 最小~最大 52.4~81.0 46.4~106.6 55.3~83.7 71.0~90.0 54.0~84.0 ア ジ ア / パ シ フ ィ ッ クアイランダー 5 例(63%) 6 例(67%) 3 例(33%) 2 例(25%) 6 例(75%) 白人 2 例(25%) 3 例(33%) 4 例(44%) 3 例(38%) 2 例(25%) そ の 他 ( ア ラ ス カ な ど) 1 例(13%) 0 例 1 例(11%) 0 例 0 例 ラテン系 0 例 0 例 0 例 3 例(38%) 0 例 人種 黒人 0 例 0 例 1 例(11%) 0 例 0 例 表 2.24-3: 被験者の背景 2(有効性評価項目など) 項 目 0.05 mg 群 (8 例) 0.1 mg 群 (9 例) 0.5 mg 群 (9 例) 1 mg 群 (8 例) プラセボ群 (8 例) HBs 抗原 陽性 8 例(100%) 9 例(100%) 9 例(100%) 8 例(100%) 8 例(100%) HBe 抗原 陽性 7 例(88%) 8 例(89%) 7 例(78%) 8 例(100%) 8 例(100%) 平均 ± 標準偏差 2.73 ± 0.65 3.0 ± 0.99 3.22 ± 0.72 3.11 ± 0.79 3.0 ± 0.68 中央値 2.65 3.33 3.48 3.51 3.15 HBV DNA 量 (bDNA 法: log10 MEq/mL) 最小~最大 2.1~3.7 0.7~4.1 2.3~4.0 1.8~4.0 1.6~3.9 平均 ± 標準偏差 8.08 ± 0.53 8.09 ± 1.03 8.72 ± 0.95 8.22 ± 0.64 8.16 ± 0.73 中央値 8.10 8.13 8.82 8.20 8.46 HBV DNA 量 (PCR 法: log10 copies/mL) 最小~最大 7.4~8.9 6.1~9.3 7.0~9.6 7.3~9.2 7.0~9.0 平均 ± 標準偏差 77.4 ± 51.99 78.0 ±53.96 105.9 ± 73.61 124.0 ± 78.72 85.8 ± 65.43 中央値 73.0 58.0 96.0 117.5 68.5 ALT(U/L) 最小~最大 14~163 25~173 30~286 26~297 35~235 平均 ± 標準偏差 4.23 ± 0.27 4.47 ± 0.30 4.44 ± 0.32 4.19 ± 0.24 4.18 ± 0.29 中央値 4.20 4.50 4.0 4.20 4.30 アルブミン (g/dL) 最小~最大 3.9~4.7 3.8~4.9 4.0~5.0 3.9~4.5 3.8~4.5 治療薬あり 4 例(50%) 5 例(56%) 4 例(44%) 3 例(38%) 2 例(25%) インターフェロン 4 例(50%) 3 例(33%) 4 例(44%) 3 例(38%) 2 例(25%) ラミブジン 4 例(50%) 4 例(44%) 0 例 2 例(25%) 1 例(13%) 治 験 開 始 前 の HBV 治療薬 その他 0 例 1 例(11%) 0 例 1 例(13%) 1 例(13%) ベースライン時の血液学的検査値は、大多数の被験者で正常であった。また、肝機能検査値は 正常又はやや上昇しており、2 例の ALT 値が基準値上限の 5 倍を超えていた(0.5 mg 群及び 1 mg 群の各 1 例)。
(2) 有効性の結果 1) HBV DNA 量
有効性の主要評価項目は、投与終了時に血清 HBV DNA 量が bDNA 法により 2 log10以上減少し
たか、又は HBV DNA 量が bDNA 法により検出限界未満で、かつ PCR 法により 2 log10以上減少
した被験者の割合であった。このエンドポイントに到達した被験者は、エンテカビル投与例では、 0.05 mg 群の 7 例(88%)、0.1 mg 群の 4 例(50%)、0.5 mg 群の 8 例(89%)及び 1 mg 群の 6 例 (75%)で、プラセボ群では到達例はなかった。0.1 mg 群を除いた各用量群とプラセボ群との間 に有意差が認められた(Bonferroni の多重比較、P < 0.0125)。 bDNA 法で HBV DNA 量が検出限界未満であった被験者は、0.05 mg 群で 2 例、0.1 mg 群で 2 例、0.5 mg 群で 3 例及び 1 mg 群で 1 例であったが、プラセボ群では 0 例だった。PCR 法により HBV DNA 量が検出限界未満であった被験者はなかった。 エンテカビルの 4 用量はいずれも強力な抗ウイルス活性を示し、投与前と比較した投与 28 日目 の bDNA 法による HBV DNA 量の平均減少量をプラセボ群と比較すると、減少量の差は 0.05 mg 群、0.1 mg 群、0.5 mg 群及び 1 mg 群でそれぞれ-2.20, -2.27, -2.80 及び-2.54 log10MEq/mL であり、 いずれもプラセボ群と比較して有意であった(0.1 mg 群は P = 0.0002、他の用量群は P < 0.0001)。 また、PCR 法で測定した投与前と投与 28 日目の HBV DNA 量の平均減少量もプラセボ群と比較 して、各用量群でそれぞれ-2.29, -2.29, -3.08 及び-2.65 log10copies/mL と 4 用量群すべてが有意に減 少した(P < 0.0001)。 表 2.24-4: 28 日目におけるウイルス学的反応 項 目 0.05 mg 群 (8 例) 0.1 mg 群 (8 例)a 0.5 mg 群 (9 例) 1 mg 群 (8 例)a プラセボ群 (8 例) 主要評価項目到達例 7(88%) 4(50%) 8(89%) 6(75%)b 0 P 値 0.0014 0.077 0.0004 0.007 – bDNA 法による HBV DNA 量検出限界未満 2(25%) 2(25%) 3(33%) 1(13%) 0 P 値 0.467 0.233 0.206 1.0 – a 28 日目以前に治験薬投与を中止した被験者は、無効例として取り扱った b 1 例が 1 日目に HBV DNA 量が自然減少したため、有効性の評価から除外した
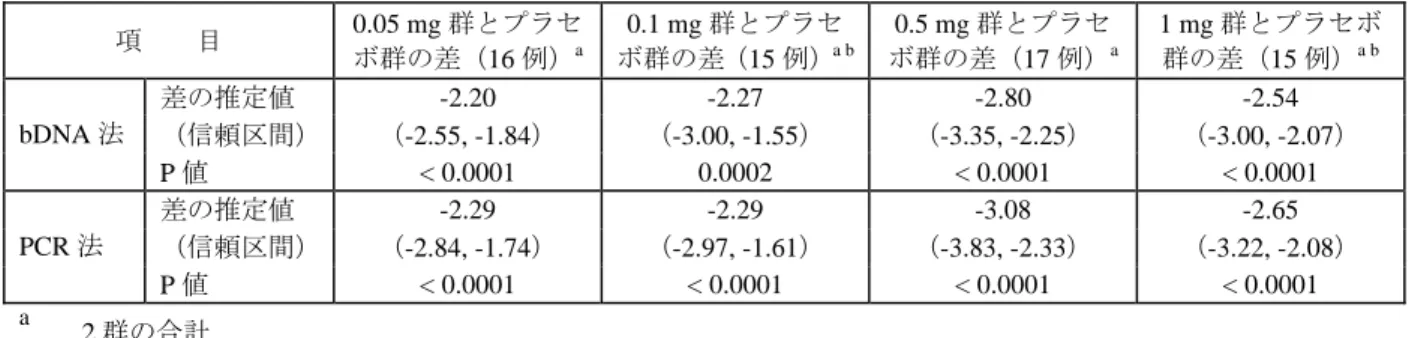
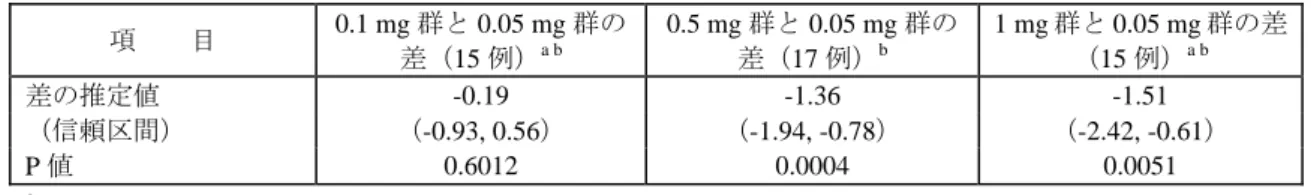
表 2.24-5: 28 日目における平均 HBV DNA 量の減少に関する各用量群とプラセボ群との比較 項 目 0.05 mg 群とプラセ ボ群の差(16 例)a 0.1 mg 群とプラセ ボ群の差(15 例)a b 0.5 mg 群とプラセ ボ群の差(17 例)a 1 mg 群とプラセボ 群の差(15 例)a b 差の推定値 -2.20 -2.27 -2.80 -2.54 (信頼区間) (-2.55, -1.84) (-3.00, -1.55) (-3.35, -2.25) (-3.00, -2.07) bDNA 法 P 値 < 0.0001 0.0002 < 0.0001 < 0.0001 差の推定値 -2.29 -2.29 -3.08 -2.65 (信頼区間) (-2.84, -1.74) (-2.97, -1.61) (-3.83, -2.33) (-3.22, -2.08) PCR 法 P 値 < 0.0001 < 0.0001 < 0.0001 < 0.0001 a 2 群の合計 b 28 日目以前に治験薬投与を中止した被験者は除外した
bDNA 法による HBV DNA 量はエンテカビルの 4 用量群とも急速に減少した。HBV DNA 量の 減少は、ほとんどが投与開始後 14 日目までに認められた。28 日間の投与終了時におけるウイル ス量減少度は、エンテカビルの各用量群でほぼ同程度であった。投与終了後 1 ヵ月以内に、ほと んどの被験者の HBV DNA 量は再び上昇したが、ベースライン値を上回ることはなかった。投与 終了後の HBV DNA 量は、投与終了時とは異なり明らかな用量反応性を示した。投与終了後のウ イルス抑制作用は、0.5 mg 群及び 1 mg 群のエンテカビル高用量群では 0.05 mg 群又は 0.1 mg 群 より強かった。
表 2.24-6: 56 日目における各用量群の平均 HBV DNA 減少量の差 項 目 0.1 mg 群と 0.05 mg 群の 差(15 例)a b 0.5 mg 群と 0.05 mg 群の 差(17 例)b 1 mg 群と 0.05 mg 群の差 (15 例)a b 差の推定値 -0.19 -1.36 -1.51 (信頼区間) (-0.93, 0.56) (-1.94, -0.78) (-2.42, -0.61) P 値 0.6012 0.0004 0.0051 a 28 日目以前に治験薬投与を中止した被験者は除外した b 2 群の合計 2) HBe 抗原の陰性化 HBe 抗原の陰性化は、わずかな被験者にしか見られなかった。0.05 mg 群では、1 例が投与終了 時に陰性化したが、24 週間の投与終了後観察期間の終了時には陽性に戻っていた。0.05 mg 群の 別の 1 例では、投与終了時まで HBe 抗原は陽性だったが、投与終了後観察期間終了時には陰性化 していた。0.5 mg 群では 1 例が投与終了時に陰性化したが、投与終了後観察期間終了時には陽性 化した。0.1 mg 群と 1 mg 群では、28 日間の投与期間中に HBe 抗原が陰性化した例はなかった。 これは投与期間が短いためと考えられた。 3) 血清 ALT の正常化 血清 ALT の正常化は 2 例の被験者にしか見られなかった。これは投与期間が短いためと考えら れた。 (3) 薬物動態/薬力学の結果
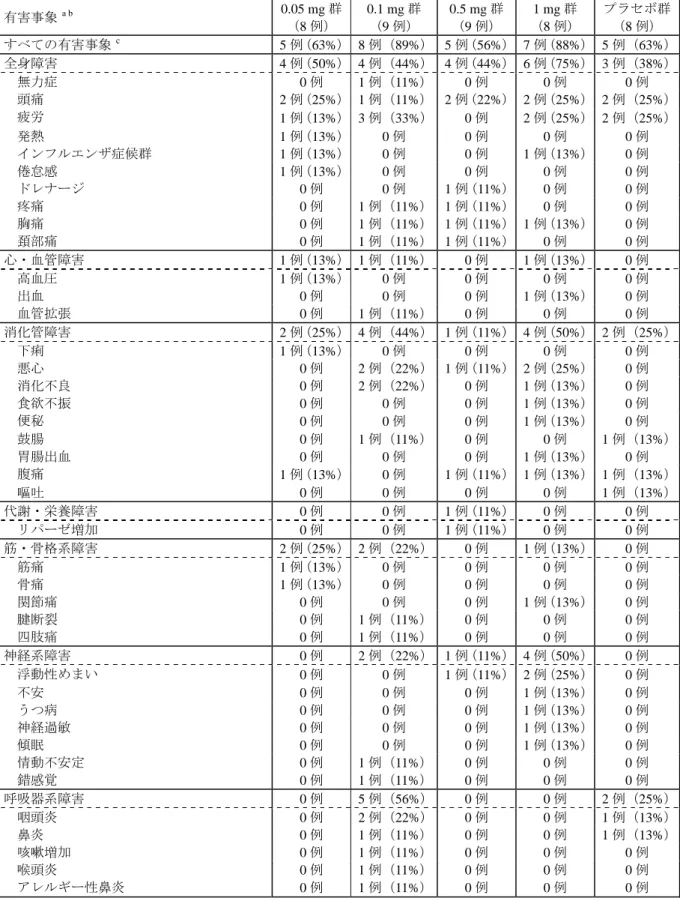
薬物動態/薬力学についての結果は、Study AI463-004, AI463-005, AI463-014 をまとめて、別の 報告書とする。 (4) 安全性の結果 1) 治験薬への曝露 治験薬の投与例数と平均投与期間は、0.05 mg 群が 8 例に 28.50(28~29)日間、0.1 mg 群が 9 例に 27.11(20~28)日間、0.5 mg 群が 9 例に 28.22(28~29)日間、1 mg 群が 8 例に 25.38(8 ~28)日間及びプラセボ群が 8 例に 28.13(28~29)日間であり、各用量間は類似していた。また、 治験薬へのコンプライアンスは良好でほぼ治験計画書のとおり投与がなされた。 2) 治療期間中の有害事象 ほとんどの被験者で治療期間中に軽度から中等度の有害事象が報告されたが、各有害事象の発 現率は、エンテカビル投与群とプラセボ群に明らかな差は認められなかった。最も頻度が高い有 害事象は、頭痛と疲労であった。頭痛は 9 例(0.05 mg:2 例、0.1 mg:1 例、0.5 mg:2 例、1 mg: 2 例、プラセボ:2 例)で、すべて軽度から中等度であった。1 mg 群の 1 例で 7 日目に 12 時間を 超えて軽度から中等度の頭痛が持続したため、治験実施計画書の規定に従って投与が中止された。
また、0.1 mg 群の 1 例に眼鏡の新調が原因と考えられる軽度の頭痛を伴って視覚障害が認められ た。1 mg 群では、他の用量群より頻繁に、神経系障害に分類される軽度から中等度の浮動性めま い、不安、うつ病、神経過敏及び傾眠が報告された。
各用量群の治験薬投与に関連すると考えられる有害事象の頻度と重症度は同程度であった。治 療期間中に治験薬に関連したグレード 3 又は 4 の有害事象は見られなかった。
表 2.24-7: 治療期間中の有害事象(グレード 1 からグレード 4) 有害事象 a b 0.05 mg 群 (8 例) 0.1 mg 群 (9 例) 0.5 mg 群 (9 例) 1 mg 群 (8 例) プラセボ群 (8 例) すべての有害事象 c 5 例(63%) 8 例(89%) 5 例(56%) 7 例(88%) 5 例(63%) 全身障害 4 例(50%) 4 例(44%) 4 例(44%) 6 例(75%) 3 例(38%) 無力症 0 例 1 例(11%) 0 例 0 例 0 例 頭痛 2 例(25%) 1 例(11%) 2 例(22%) 2 例(25%) 2 例(25%) 疲労 1 例(13%) 3 例(33%) 0 例 2 例(25%) 2 例(25%) 発熱 1 例(13%) 0 例 0 例 0 例 0 例 インフルエンザ症候群 1 例(13%) 0 例 0 例 1 例(13%) 0 例 倦怠感 1 例(13%) 0 例 0 例 0 例 0 例 ドレナージ 0 例 0 例 1 例(11%) 0 例 0 例 疼痛 0 例 1 例(11%) 1 例(11%) 0 例 0 例 胸痛 0 例 1 例(11%) 1 例(11%) 1 例(13%) 0 例 頚部痛 0 例 1 例(11%) 1 例(11%) 0 例 0 例 心・血管障害 1 例(13%) 1 例(11%) 0 例 1 例(13%) 0 例 高血圧 1 例(13%) 0 例 0 例 0 例 0 例 出血 0 例 0 例 0 例 1 例(13%) 0 例 血管拡張 0 例 1 例(11%) 0 例 0 例 0 例 消化管障害 2 例(25%) 4 例(44%) 1 例(11%) 4 例(50%) 2 例(25%) 下痢 1 例(13%) 0 例 0 例 0 例 0 例 悪心 0 例 2 例(22%) 1 例(11%) 2 例(25%) 0 例 消化不良 0 例 2 例(22%) 0 例 1 例(13%) 0 例 食欲不振 0 例 0 例 0 例 1 例(13%) 0 例 便秘 0 例 0 例 0 例 1 例(13%) 0 例 鼓腸 0 例 1 例(11%) 0 例 0 例 1 例(13%) 胃腸出血 0 例 0 例 0 例 1 例(13%) 0 例 腹痛 1 例(13%) 0 例 1 例(11%) 1 例(13%) 1 例(13%) 嘔吐 0 例 0 例 0 例 0 例 1 例(13%) 代謝・栄養障害 0 例 0 例 1 例(11%) 0 例 0 例 リパーゼ増加 0 例 0 例 1 例(11%) 0 例 0 例 筋・骨格系障害 2 例(25%) 2 例(22%) 0 例 1 例(13%) 0 例 筋痛 1 例(13%) 0 例 0 例 0 例 0 例 骨痛 1 例(13%) 0 例 0 例 0 例 0 例 関節痛 0 例 0 例 0 例 1 例(13%) 0 例 腱断裂 0 例 1 例(11%) 0 例 0 例 0 例 四肢痛 0 例 1 例(11%) 0 例 0 例 0 例 神経系障害 0 例 2 例(22%) 1 例(11%) 4 例(50%) 0 例 浮動性めまい 0 例 0 例 1 例(11%) 2 例(25%) 0 例 不安 0 例 0 例 0 例 1 例(13%) 0 例 うつ病 0 例 0 例 0 例 1 例(13%) 0 例 神経過敏 0 例 0 例 0 例 1 例(13%) 0 例 傾眠 0 例 0 例 0 例 1 例(13%) 0 例 情動不安定 0 例 1 例(11%) 0 例 0 例 0 例 錯感覚 0 例 1 例(11%) 0 例 0 例 0 例 呼吸器系障害 0 例 5 例(56%) 0 例 0 例 2 例(25%) 咽頭炎 0 例 2 例(22%) 0 例 0 例 1 例(13%) 鼻炎 0 例 1 例(11%) 0 例 0 例 1 例(13%) 咳嗽増加 0 例 1 例(11%) 0 例 0 例 0 例 喉頭炎 0 例 1 例(11%) 0 例 0 例 0 例 アレルギー性鼻炎 0 例 1 例(11%) 0 例 0 例 0 例
有害事象 a b 0.05 mg 群 (8 例) 0.1 mg 群 (9 例) 0.5 mg 群 (9 例) 1 mg 群 (8 例) プラセボ群 (8 例) 皮膚・皮膚付属器障害 1 例(13%) 2 例(22%) 2 例(22%) 1 例(13%) 0 例 そう痒症 1 例(13%) 0 例 1 例(11%) 0 例 0 例 発疹 1 例(13%) 1 例(11%) 0 例 1 例(13%) 0 例 脱毛症 0 例 0 例 1 例(11%) 0 例 0 例 皮膚変色 0 例 1 例(11%) 0 例 0 例 0 例 単純ヘルペス 0 例 0 例 1 例(11%) 0 例 0 例 特殊感覚障害 0 例 1 例(11%) 0 例 0 例 0 例 視覚異常 0 例 1 例(11%) 0 例 0 例 0 例 a
COSTART(修正版)による集計(和訳には MedDRA Version 8.0/J を使用) b 各群共、グレードが 1 又は 2 で 1、2 例にのみ発現した有害事象は記載していない(グレード 3 以上のもの、 発現例数の多い事象を記載した)。重症度が特定されなかったものは記載した。 c 少数例の有害事象もすべてを含む発現例数。 3) 投与終了後観察期間の有害事象 24 週間の投与終了後観察期間中では、ほとんどの被験者に治療期間中と同様の有害事象が報告 された。 4) 死亡 治療期間中に死亡例はなかった。0.1 mg 群の 1 例がエンテカビル投与終了 6 ヵ月後に肝細胞癌 及び肺線維症と診断され、投与終了約 7 ヵ月後に肝癌で死亡した。 5) その他の重篤な有害事象 スクリーニング期間中、1 例が肝性脳症、吐血及びプロトロンビン時間延長を示し、治験薬投 与が開始されなかった。1 mg 群の 1 例では、軽度から中等度の頭痛が投与 6 日目から報告され、 治験実施計画書の規定に従って、頭痛が 12 時間を超えて持続した 7 日目に投与が中止された。こ の被験者はこれに関連した神経系の愁訴はなく、神経学的検査は正常範囲内であった。この被験 者は過去にもストレスによる頭痛が頻回に報告されており、ラミブジン投与時も同様であった。 また、エンテカビル投与中止後も間欠性頭痛を訴えたが、それ以外の状態は良好であった。 投与終了後観察期間中、上述の死亡例以外に、3 例で重篤な有害事象が報告された。0.5 mg 群 では、2 例で重篤な有害事象が報告された(1 例は 119 日目に胆石による胆嚢切除のために入院し、 他の 1 例は 57 日目にヘロイン過量投与と敗血症性ショックのために入院した)。また、1 mg 群の 1 例は、89 日目に自動車事故による頭部外傷のため入院した。0.05 mg 群とプラセボ群では、重 篤な有害事象の報告はなかった。 6) 有害事象による中止 有害事象のため投与が中止された被験者は 2 例であった。0.1 mg 群の 1 例は、血清 ALT/AST が 2 倍を超えて上昇した(無症候性)ため 19 日目に投与が中止された。この被験者のトランスア ミナーゼ値は、35 日目までにベースライン値に回復した。1 mg 群の 1 例では、「5) その他の重篤 な有害事象」に示したように軽度から中等度の頭痛が発現し、治験実施計画書に従い 7 日目に投
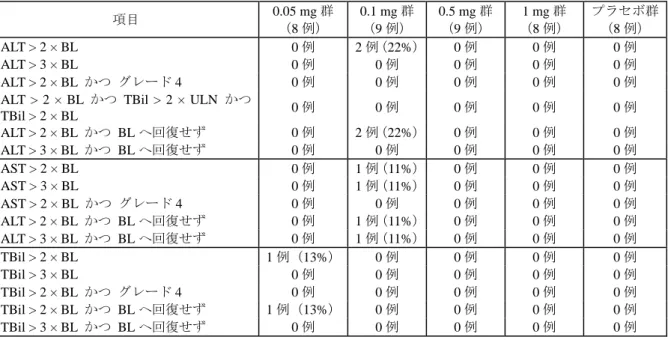
与が中止された。 7) 有害事象とされた臨床検査値の異常 治験期間中、有害事象とされた臨床検査値の異常は、4 例報告された。 治療期間中では、0.5 mg 群 1 例に無症候性の血清リパーゼ増加(グレード 3)が認められ、有 害事象として報告された。この被験者は、無作為化の 2 ヵ月前にグレード 3 の血清リパーゼ増加 を示していた。治験薬を中断して再測定をしたところアミラーゼ及びリパーゼは正常化した。こ の被験者は治験を再開し、その後膵酵素は増加せずに治験を完了した。 投与終了後観察期間には、3 例に臨床検査値異常が認められ有害事象として報告された。0.1 mg 群の 1 例に高コレステロール血症と低血糖症が報告された。0.5 mg 群の 1 例には、高脂血症が報 告された。プラセボ群の 1 例は、治験に組み入れられたときには ALT がグレード 2 であったが、 投与終了後観察期間にはグレード 3 に増加した。 表 2.24-8: 有害事象とされた臨床検査値の異常 項目 0.05 mg 群 (8 例) 0.1 mg 群 (9 例) 0.5 mg 群 (9 例) 1 mg 群 (8 例) プラセボ群 (8 例) グレード 1-4 3-4 1-4 3-4 1-4 3-4 1-4 3-4 1-4 3-4 臨床検査による有害事象 0 0 1(11) 0 2(22) 1(11) 0 0 1(13) 1(13) 代謝・栄養障害 0 0 1(11) 0 2(22) 1(11) 0 0 1(13) 1(13) ALT 増加 0 0 0 0 0 0 0 0 1(13) 1(13) 高コレステロール血症 0 0 1(11) 0 0 0 0 0 0 0 高脂血症 0 0 0 0 1(11) 0 0 0 0 0 低血糖症 0 0 1(11) 0 0 0 0 0 0 0 リパーゼ増加 0 0 0 0 1(11) 1(11) 0 0 0 0 8) 肝機能検査値の異常(グレード 1 からグレード 4) 治験開始前に ALT が異常であった被験者のうち、エンテカビル投与の 7 例とプラセボ投与の 1 例が悪化を示した。しかし、異常がグレード 3 と判定されたのはエンテカビル投与及びプラセボ 投与の各 1 例で、他はグレード 2 であった。また、投与終了後観察期間には、エンテカビル投与 の 12 例、プラセボ投与の 3 例が ALT の悪化を示した。 また、0.1 mg 群の 2 例は、治療期間中に ALT がベースライン値の 2 倍を超えたが、血清ビリル ビンの増加を伴っていなかった。ALT の増加はエンテカビルの各用量群では同様であった。