高周波熱プラズマによるリチウムイオン二次電池電 極材料のナノ粒子合成
曽根, 宏隆
http://hdl.handle.net/2324/4060135
出版情報:九州大学, 2019, 博士(工学), 課程博士 バージョン:
権利関係:
高周波熱プラズマによる
リチウムイオン二次電池電極材料の ナノ粒子合成
令和2年3月 博士論文
九州大学大学院 工学府 化学システム工学専攻
曽根 宏隆
題名 『高周波熱プラズマによるリチウムイオン二次電池 電極材料のナノ粒子合成』
目次
第 1 章 序論
1
1.1 プラズマ 1
1.1.1 プラズマ 1
1.1.2 熱プラズマ 2
1.1.3 熱プラズマの発生方法および特徴 3
1.2 電池 6
1.2.1 電池の分類と特徴 6
1.2.2 リチウムイオン二次電池 8
1.2.3 リチウムイオン二次電池の正極材料 10
1.2.4 リチウムイオン二次電池の負極材料 14
1.3 ナノ粒子 17
1.3.1 ナノ粒子の特性 17
1.3.2 ナノ粒子の合成方法 18
1.4 熱プラズマによるナノ粒子の合成 21
1.4.1 金属ナノ粒子の合成 21
1.4.2 合金および金属間化合物ナノ粒子 23
1.4.3 酸化物および複合酸化物ナノ粒子 26
1.4.4 窒化物ナノ粒子 34
1.4.5 ホウ化物ナノ粒子 37
1.4.6 炭化物ナノ粒子 39
1.5 リチウムイオン二次電池の正極材料ナノ粒子の合成 42
1.5.1 Li-Co 複合酸化物ナノ粒子 43
1.5.2 Li-Mn複合酸化物ナノ粒子 44
1.5.3 Li-Ni 複合酸化物ナノ粒子 46
1.5.4 Li-Fe 複合酸化物ナノ粒子 47
1.5.5 Li-Mn-Me 複合酸化物ナノ粒子 48
1.6 リチウムイオン二次電池の負極材料ナノ粒子の合成 49
1.6.1 結晶性シリコンナノ粒子 49
1.6.2 Li-Ti 複合酸化物ナノ粒子 54
1.7 研究目的および構成 54
第 2 章 高周波熱プラズマ実験装置とナノ粒子の分析方法
95
2.1 高周波熱プラズマ実験装置 95
2.1.1 高周波熱プラズマ装置構成 95
2.1.2 原料粉体 96
2.1.3 実験条件 96
2.1.4 実験手順 96
2.2 分析方法および解析方法 97
2.2.1 粉末X 線回折(XRD)による生成物の同定 97
2.2.2 透過型電子顕微鏡(TEM)による生成物の観察 98
2.2.3 粒径分布測定 98
2.2.4 エネルギー分散型 X 線分析(TEM-EDS)による生成物の元素分析 98
2.2.5 走査型電子顕微鏡(SEM)による生成物の観察 98
2.2.6 制限視野電子線回折(TEM- Diffraction)による元素分析および解析 98
2.2.7 結晶構造、電子・核密度および結晶外形の可視化 98
2.3電池評価 99
2.3.1電気化学特性および電池評価 99
第 3 章 Li-Mn 酸化物ナノ粒子の合成と電池特性 105
3.1 緒言 105
3.2 実験方法 106
3.2.1 実験装置の構成と手順 106
3.2.2 分析 107
3.3 ナノ粒子合成の実験結果 107
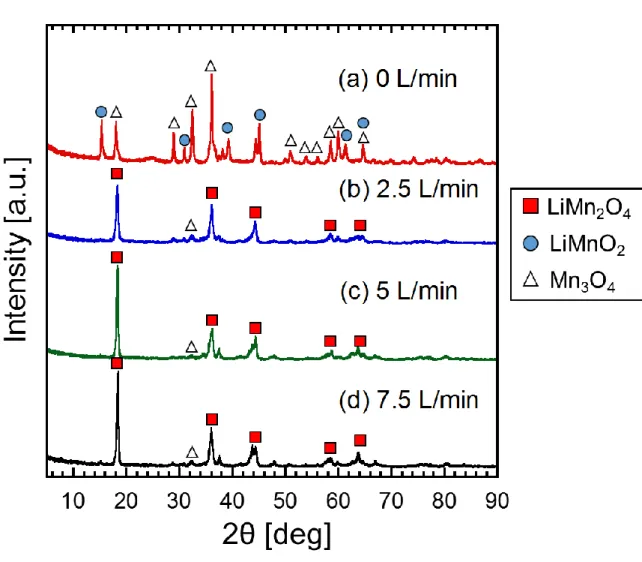
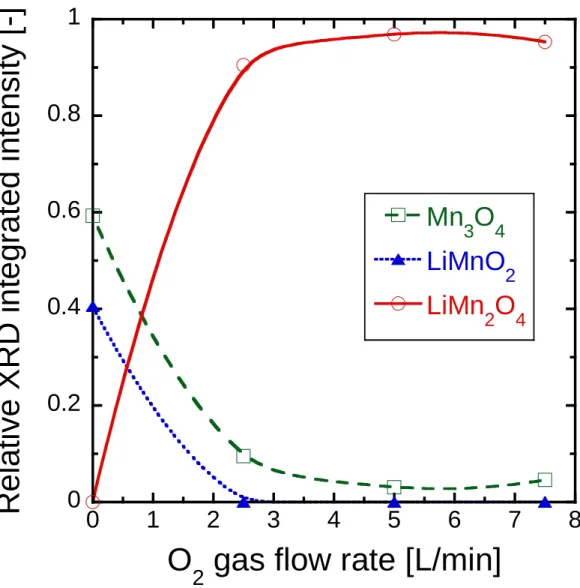
3.3.1 O2 モル分率変化による結晶構造への影響 107
3.3.2 Li-Mn モル分率による結晶構造への影響 110
3.3.3 プラズマ出力による結晶構造への影響 112
3.3.4 原料供給量によるLiMn複合酸化物の結晶構造への影響 113
3.4 ナノ粒子合成の考察 113
3.4.1 LiMnO2およびLiMn2O4におけるLiとOモル比の相安定性効果 113
3.4.2 核生成温度の推算 114
3.4.3 熱力学的検討 115
3.4.4 Gibbsの自由エネルギーの計算 115
3.4.5 Li系複合酸化物の生成反応式 117
3.4.6生成エネルギー 117
3.4.7 Li-Mn複合酸化物ナノ粒子の作製 119
3.5 電気化学特性および電池特性の実験結果 120
3.5.1 電気化学特性 120
3.5.2 電池容量特性 120
3.5.3 レート特性 121
3.6 まとめ 121
第 4 章 Li-Ni 酸化物ナノ粒子の合成と電池特性 163
4.1 緒言 163
4.2 実験方法 164
4.2.1実験装置の構成と手順 164
4.2.2 分析 164
4.2.3 電気化学測定 164
4.3 ナノ粒子合成の実験結果 165
4.3.1 Li/Ni組成比による結晶構造への影響 165
4.3.2 酸素モル分率による結晶構造への影響 167
4.3.3 プラズマ出力による結晶構造への影響 167
4.4 ナノ粒子合成の考察 168
4.4.1 非化学量論的Li1-xNi1+xO2におけるLiとOモル比の相安定性効果 168
4.4.2 核生成温度の推算 169
4.4.3 熱力学的検討 170
4.5 電気化学特性および電池特性の実験結果 171
4.5.1 電池容量特性 171
4.5.2 電気化学特性 172
4.6 まとめ 173
第 5 章 Li-Ni-Mn 酸化物ナノ粒子の合成と電池特性 203
5.1 緒言 203
5.2 実験方法 204
5.2.1 実験装置の構成と手順 204
5.2.2 分析 204
5.2.3 電気化学測定 205
5.3 ナノ粒子合成の実験結果 205
5.3.1 Ni/(Ni+Mn)組成比による結晶構造への影響 205
5.3.2 酸素モル分率による結晶構造への影響 207
5.4 ナノ粒子合成の考察 209
5.4.1 核生成温度の推算 209
5.4.2 熱力学的検討 209
5.5 電気化学的特性および電池特性の実験結果 210
5.5.1 電池容量特性 211
5.6 まとめ 211
第 6 章 Li-Nb-Mn 酸化物ナノ粒子の合成と電池特性 243
6.1 緒言 243
6.2 実験方法 243
6.2.1 実験装置の構成と手順 243
6.2.2 分析 244
6.2.3 電気化学測定 244
6.3 ナノ粒子合成の実験結果 244
6.3.1 Li/(Ni+Mn)組成比による結晶構造への影響 244
6.3.2 酸素モル分率による結晶構造への影響 246
6.4 ナノ粒子合成の考察 247
6.4.1 核生成温度の推算 247
6.4.2 熱力学的検討 247
6.5 電気化学特性および電池特性の実験結果 248
6.5.1 電池容量特性 249
6.5.2 レート特性 249
6.6 まとめ 249
第 7 章 Si 複合ナノ粒子の合成と電池特性 271
7.1緒言 271
7.2 実験方法 271
7.2.1実験装置の構成と手順 271
7.2.2 分析 272
7.2.3 電池測定 272
7.3 ナノ粒子合成の実験結果 272
7.3.1 SiおよびTiナノ粒子合成によるナノ粒子構造への影響 272
7.3.2 Si-Ti複合ナノ粒子合成によるナノ粒子構造への影響 273
7.4ナノ粒子合成の考察 275
7.4.1 核生成温度の推算 275
7.4.2 熱力学的検討 275
7.4.3 Si複合ナノ粒子の表面モデル 276
7.5 電気化学特性および電池特性の実験結果 277
7.5.1 Si/Ti比率による充放電特性への影響 277
7.5.2 Si-Ti複合ナノ粒子の充放電特性 277
7.6 まとめ 278
第 8 章 結論 305
8.1 本論文のまとめ 305
8.1.1 Li-Mn酸化物ナノ粒子の合成と電池特性 305
8.1.2 Li-Ni酸化物ナノ粒子の合成と電池特性 306
8.1.3 Li-Ni-Mn 酸化物ナノ粒子の合成と電池特性 306
8.1.4 Li-Nb-Mn酸化物ナノ粒子の合成と電池特性 306
8.1.5 Si 複合負極材料ナノ粒子の合成と電池特性 307
8.2 今後の課題と展望 307
謝辞
309※なお,本論文掲載の全頁の表および図は本論文著者が作成したものである.
1
第 1 章 序論
1.1 プラズマ 1.1.1 プラズマ
プラズマとは正の電荷を帯びた粒子と負の電荷を帯びた電子とがほぼ同じ密度で混在し,
系全体として電気的中性を保って分布している粒子集団のことである.プラズマは荷電粒 子と中性粒子とにより構成され、集団的ふるまいをする.プラズマの発現は主に気体などの 流体に電流または電圧などのエネルギーを印加することで気体分子を原子に解離させ,原 子がイオンと電子に電離しプラズマとなる.プラズマは気体からの単なる物質の状態変化 ではなく,外的エネルギーによって発現される.したがって,プラズマと気体とでは持って いるエネルギーに大きな差があり,固体,液体,気体とは異なる性質を有するところから物 質の第四の状態とも表現される.下記にプラズマの身近な現象と工業的役割,プラズマの歴 史について示す.
(a) プラズマの身近な現象と工業的役割
宇宙の物質の 99%以上はプラズマであり,身近な自然界のプラズマとしては,太陽,稲 妻,オーロラなどがある.また,人工的に生み出されたプラズマのうち,身近なものは蛍光 灯,ネオンランプ,燃焼炎などが挙げられる.人工的にプラズマを発生させるためには,大 きなエネルギーを必要とするため,加熱してエネルギーを得るよりも,気体放電による電気 的なエネルギー供給の方が効率よく発生させることができる.
プラズマは熱の平衡状態によって低温プラズマと熱プラズマに大別される.低温プラズ マは電子温度が高く10,000-100,000 K程であるがイオンや中性子の温度が低く300-1,000 K 程の非平衡プラズマである.非平衡プラズマは粒子密度が小さく,さらにイオンと電子の質 量が極端に違っているため衝突によるエネルギー交換が起こり難い.したがって,イオンと 電子とが別々の温度を持ちイオンや中性子の温度は室温に近く,電子温度は高温となる.非 平衡プラズマの多くは半導体の分野で使用されている.一方,熱プラズマは粒子密度が高く 電子温度とイオン,中性子の温度がほぼ等しく組成も平衡状態に近いプラズマである.工業 界では熱プラズマと低温プラズマを用いて物質を処理している.
(b) プラズマの歴史
プラズマは1879年にイギリスの化学者・物理学者であるサー・ウィリアム・クルックス がクルックス管の中で初めて認識し,著書の中でradiant matterと記述したのが起源とさ れている.さらに,1897年にイギリスの物理学者ジョセフ・ジョン・トムソンがクルック ス管で発生した陰極線の正体を電子の流れと特定した.プラズマが物理学に登場したのは 1928年であり,アーヴィング・ラングミュアが希薄気体放電管の陽光柱の部分をプラズマ
2
と呼び,新しい研究分野として注目を浴びるようになった.ラングミュアは,デバイ遮断や プラズマ振動などのプラズマの基本的性質を明らかにした.その後,この電離気体が第四の 物質状態であると表現し,1928年にラングミュアがこの電離気体をプラズマと名付けた.
1835年にはマイケル・ファラデーがプラズマを実験室で安定に発現させた.それ以降,プ ラズマに対する研究は半導体,プラズマ物理,プラズマ数値解析,プラズマ材料工学,廃棄 物処理,宇宙物理学および原子核融合などの工学的応用において進展している.
1.1.2 熱プラズマ
熱プラズマは大気圧および大気圧に近い圧力で局所熱平衡状態になり,温度は数千から 数万Kとなる.局所熱平衡状態(LTE, Local Thermodynamic Equilibrium)はプラズマ中にて,
電子とイオン,中性原子などの重粒子の温度がほぼ等しく,組成が熱平衡に近い状態をいう.
熱プラズマの性質としては以下の4つ特徴が挙げられる[1, 2].
(a) 高温度および高熱容量領域の創生
熱プラズマはプラズマ全体が高温であり,プラズマの滞留時間が長く熱容量が大きい.そ のため,電子・イオンおよび原子などの重粒子も約100,000 Kの高い温度領域とすることが できる.熱プラズマは0.5気圧(50.65 kPa)以上の圧力状態であるため,プラズマのエネル ギー密度が大きく,被加熱物を短時間で高温にすることができる.また化学反応速度は温度 に対して指数関数的に増大するため,熱プラズマ中では反応速度は著しく大きくなる.
(b) 荷電粒子および熱プラズマ流の制御
熱プラズマはプラズマ中に存在する電子やイオンなどの荷電粒子を容易に制御できる.
プラズマ中の電子やイオンは電磁場の影響を受けるため,外部からの電磁場を調節するこ
とで,RadialおよびTangentialなどの各種プロセスに応じたプラズマ流を発生させること
ができる.
(c) ラジカルおよび化学種を利用した高化学反応性
熱プラズマはプラズマ中の電子の衝突によってラジカルなどの様々な化学種を生成する.
熱プラズマ中はプラズマがイオンやラジカル源となるため,ラジカル反応を利用した高化 学反応性の材料プロセスとして利用することができる.
(d) 熱プラズマ処理雰囲気の選択性
熱プラズマはプラズマ中の処理雰囲気を自由に選択できる.Ar を用いた不活性雰囲気,
O2を用いた酸化雰囲気,H2を用いた還元雰囲気,炭化水素を用いた炭素雰囲気など各種プ ロセスにおける化学反応場として広く利用できる.燃焼反応を用いた高温プロセスでは,燃
3
焼ガス中に生じる多くの化学種によって目的反応が阻害されてしまう.しかし,熱プラズマ ではそのような副反応を回避することができる.
1.1.3 熱プラズマの発生方法および特徴
熱プラズマは直流アーク,交流アークおよび高周波熱プラズマに分類できる.Fig. 1-1は,
直流アーク,交流アークおよび高周波熱プラズマを示している.さらに,熱プラズマの発生 方法は電極を使用する方式および電極を使用しない方式に分類できる.熱プラズマ発生方 法の違いから各熱プラズマの特徴,利点およびナノ粒子合成時の問題点を下記に示す.
(a) 直流アーク
直流アークは,大気圧下で利用でき,発生が比較的容易なことから,代表的な熱プラズマ の一つである.アークは陰極からの熱電子放出により持続しており,陰極中に存在する自由 電子が仕事関数以上のエネルギーを持つことで,陰極から熱電子として放出される.直流ア ーク放電を用いたプラズマ発生方法には,非移送式アークと移送式アークがある.非移送式 アークは,陽極部と陰極部の間でアークを発生させ,ノズル部分の熱的ピンチ効果を利用し て高温の熱プラズマ流を得ている.電極間でプラズマガスが加速され,ノズル状の陽極から 熱プラズマが高速で噴出することから,この機構をプラズマジェットという.プラズマジェ ットは熱効率が 30%程度であるが,対象物が導電性でなくても良いため自由度が高い.ま た移送式アークでは,導電性ノズルに僅かに電位をかけてアークが発生し,ノズルから離れ た導電性の物質に主たる電位をかけ,プラズマアークを発生させる.プラズマアークでは熱
効率が 70%以上と高いので溶接,ナノ粒子合成等に用いられている.欠点として非伝導性
の材料には適用できないことが挙げられる[3-5].
(b) 交流アーク
交流アークは本質的には直流アークと変わらないが電流値が時間に関して変化するので プラズマの物性値も時間変動を持つ.そのため,アーク電圧と電流との瞬間的な関係は直 流アークの時のように一義的には定まらない. また,交流アークは半周期ごとに電流の 瞬時値がゼロになる瞬間があり,この瞬間はアークが消弧する.そのため,半周期ごとに アークの再点弧が必要となり,直流電源よりもアークの維持が難しいが,交流アークの不 安定性を改善するために多相交流電源を用いた方式が考案されている.
多相交流アークは交流電源により複数の電極間でアークを発生させたものである.特徴 として高温領域が広く,流速の遅いプラズマ領域を発生できるため,ナノ粒子合成のプロ セスとして期待されている.一方で,直流アークに比べて電極の消耗が大きく,長時間の 安定な放電が困難であるという問題点もある.
4 (c) 多相交流アーク
多相交流アークは位相の異なる交流電圧を複数の電極に印加させ,電極間に熱プラズマ を発生させる.最近の研究から電極数を増やすことで,大口径のプラズマを得ることに成 功している.3相までの交流アークに関する研究は古くからなされているが,6本または 12本の電極を放射状に配置した多相交流アークは,渡辺研究室および松浦らにより開発さ れた[6-9].多相交流アークは電極を6本または12本用い放射状に配置し,それぞれの電 極に異なる位相の交流電圧を印加することで電極間に熱プラズマを発生させる.交流電源 は,工業用の交流アーク溶接機を12台使用し結線することで交流用の電源(200 V, 50 Hzま
たは60 Hz)としている.
多相交流アークの特徴としては,熱プラズマの直径が約100 mmと大きく,ガス流速が 遅いので,粉体を効率良く処理することができる.また,複数の電極トーチ間にアークを 発生させるため,トーチにプラズマ領域が制限されず,プロセスに応じた反応器を設計で きる.しかし,多相交流アークは電極を有するのでプラズマ中に電極由来の不純物が混入 する.また,アーク放電の安定性や長時間の連続運転の実績がないなどの問題点がある.
(d) 高周波熱プラズマ
高周波熱プラズマは,高周波電磁場により誘導的に熱プラズマを発生させる手法であり,
無電極放電であるため高純度な材料合成が可能である.
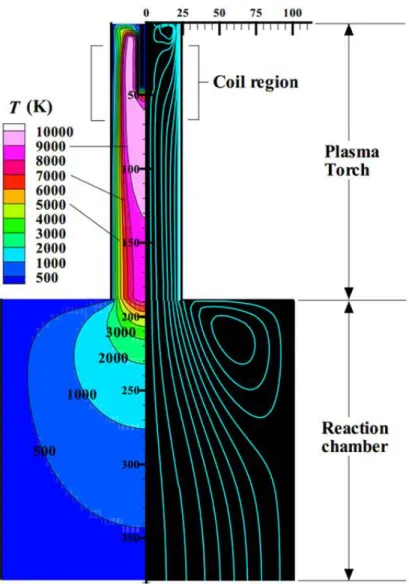
高周波熱プラズマの発生原理は,水冷された石英管内または窒化珪素管内にアルゴンガ スを流し,その管周囲の銅製の誘電コイルに数MHzの高周波電流を流すことで,トーチ内 中心軸方向に周波数に応じた磁界が発生する.高周波電流から発生する電磁場によりトー チ内のアルゴンガスが誘導的にプラズマ化する.さらに,磁界を打ち消す方向にトーチ内で 渦電流が形成される.そのため,原子やイオンは抵抗となり,発生したジュール熱により熱 プラズマが維持される[10, 11].Fig. 1-2は,プラズマを発生させた状態の装置内の温度分布 と流線を示している.トーチ中心部は約10,000 Kに達している.
高周波熱プラズマの利点として,高純度材料の生成,雰囲気制御の選択,長い滞留時間(10
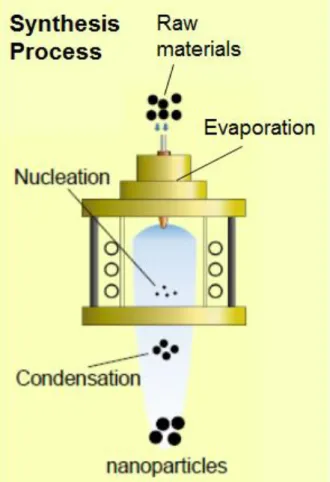
〜100 ms),高化学反応性そして急速冷却(104-106 K/s)が挙げられる.高純度材料の生成で は無電極放電のため,プラズマ中に電極材料が不純物として混入しない.雰囲気制御の選択 では,各種の反応性ガスを利用した酸化雰囲気,還元雰囲気そしてその分圧制御など自由に 選択可能である.長い滞留時間では,直流アークと比べてガス流速が一桁程度遅く,反応物 質への滞留時間が長いため,十分に化学反応を進行させることができる.Fig. 1-3は,ナノ 粒子の合成プロセスを示している.発生した熱プラズマはプラズマの直径が大きい(約 50~60 mm)という特徴を有しているため,ナノ粒子材料合成に広く用いられている.高化学 反応性では,金属材料,酸化物材料,セラミックス材料および高融点材料など様々な粉末材 料を容易に蒸発し目的の状態に変化させることができるため,高融点金属やセラミックの 球状化,成膜,表面改質といった高品質,高選択性そして高性能材料の製造へと適応されて
5
いる.さらに,高周波熱プラズマはプラズマ火炎テールで迅速な急冷が可能であるため,通 常では得ることが困難な化学組成,形態,結晶構造およびナノ材料の生成に有効である[12].
高周波熱プラズマはナノ粒子合成プロセスとして多くの利点をもつ一方で,外的要因お よび大量の粉体導入によるプラズマの揺動に不安要素がある.近年の研究では外的要因や 大量の粉体導入による熱プラズマの不安定性を克服するため,吉田らが開発したハイブリ ッド型トーチ[13]や,上杉・吉田らが開発したタンデム型トーチ[14]がある.ハイブリッド型 トーチは,高周波熱プラズマの上部にDCアークプラズマを設置したトーチである.DCア ークプラズマにより,予め粉体が約10,000 Kで溶解可能となり,その溶解した材料が高周 波熱プラズマ中に導入されることで,大量の粉体導入,粉体による急激な温度変化および分 圧変化によってもプラズマ安定性の確保が可能となる.さらに,DCアークプラズマの影響 によりプラズマガス流速が速くなり冷却効果が向上ため,より均一なナノ粒子合成が可能 となる.タンデム型トーチは高周波熱プラズマを発生する大小二つのコイルを縦に二段設 置したトーチである.一段目のコイルで原料を十分予熱できるため,効率のよい加熱処理が 可能となり,熱プラズマの安定性も向上する.
また,石垣・作田らが開発したパルス変調 高周波熱プラズマ(PMITP, pulse-modulated induction thermal plasma)がある[15, 16].プラズマ発生に時間制御の考えを新たに導入し,パ ルス的に高周波電力の供給をオン,オフすることにより,プラズマの超高温場と低温場をミ リ秒の時間オーダーで交互に実現する特異な反応場を実現している.この新発生法の開発 は,プラズマ温度の時間制御とともに,活性化学種を高濃度に発生できる可能性がある.
近年では,田中らにより,原料の間歇的供給システム(TCFF, time controlled feeding of feed
stocks)とPMITPを組み合わせたナノ粒子合成プロセスが開発された[17].このプロセスは,
PMITP の周期に合わせ,プラズマ場が高温になったときに,原料の供給を行う.この方法
は効率的に原料を蒸発させることができるため,ナノ粒子の大量合成が可能である.実際に
PMITPと TCFFを組み合わせたプロセスにより,熱プラズマの電力変調に同期させ大量の
原料を間歇的に供給し,クエンチングガスを導入することでナノ粒子を大量生成する手法 を考案している.これにより粒径が制御されたAl-doped TiO2 ナノ粒子を約400 g/h という 高効率での生成が可能であることを見出している[18-20].このときのナノ粒子生成速度は,
通常の高周波熱プラズマに比べて,約10〜20倍の生成速度であると報告している.さらに,
高周波熱プラズマを維持する数百アンペアにも達するコイル電流の振幅を意図的に AM 変 調させて,熱プラズマに擾乱を与えて平均的な温度を制御する方法も開発されている.任意 波形変調熱プラズマ(AMITP,arbitrary-waveform modulated induction thermal plasma)である.
TiO2ナノ粒子以外では,Siナノ粒子材料の合成も行われている.20 kWのAr-H2パルス変 調高周波熱プラズマ(PMITP)を金属Si粉末供給原料(TCFF)の時間制御供給と共に用 いて,1 g/hで大量のSiナノワイヤーを迅速に合成している[21].
6 1.2 電池
1.2.1 電池の分類と特徴
電池はエネルギーによって直流の電力を生み出す電力機器であり,主に化学電池,物理電 池に分類できる.化学電池とは化学物質のもつ化学エネルギーを直接電気エネルギーに変 換する電池である.物理電池とは熱や光といった物理エネルギーから電気を作る電池であ る.さらに,化学電池は一次電池,二次電池,燃料電池,生物電池に分類でき,物理電池は 太陽電池,熱電池,原子力電池に分類できる.
(a) 一次電池
一次電池は放電と呼ばれる化学エネルギーを電気エネルギーに一方向に変換することの みが一度だけ可能な電池である.すなわち,活物質の電気化学反応が終了すると電気を取り 出すことができなくなる電池である.一次電池の種類は次の電池が挙げられる.主な電池は マンガン電池,アルカリマンガン電池,ニッケル系一次電池,リチウム電池,水銀電池,空 気亜鉛電池,酸化銀電池である.
(b) 二次電池
二次電池は放電時と逆方向に電流を流すことにより放電後に電池内に存在する反応生成 物を逆反応させて元に戻し,活物質を再生させることで繰り返し使用できる電池である.す なわち,放電過程で内部の化学エネルギーが電気エネルギーに変換し、充電過程で電気エネ ルギーを化学エネルギーに変換して蓄積が可能な電池である.二次電池の種類は次の電池 が挙げられる.一般的な二次電池として鉛蓄電池,ニッケル・カドミウム蓄電池,ニッケル・
水素蓄電池,リチウムイオン二次電池,ナトリウム・硫黄(NaS)電池,ニッケル・亜鉛蓄電 池が挙げられる.ニッケル水素電池の体積エネルギー密度は 150〜350 Wh/L で電池容量は 少ないが高出力電池であるため,ハイブリッド車載用途に使用されている.特にリチウムイ オン二次電池はニッケル水素電池に比べ高容量型電池であるため,近年急速に発展を遂げ てきた比較的新規な二次電池である.
(c) 燃料電池
燃料電池は燃料である水素,メタノールおよび天然ガスを金属触媒にて改質し,化学反応 させ発電を行う電池である.触媒反応は高温を必要とするものが多く,使用する電解質や燃 料の種類により以下の電池に分類できる.最近注目されている,固体酸化物形燃料電池 (SOFC)は電池性能としては高い性能を発揮するが,電解質に酸素イオン伝導性のセラミッ クスを用いるため,1,000℃付近の高温領域での使用が課題となっている.リン酸形燃料電 池(PAFC)は SOFC より性能は劣るが電解質にリン酸を用いることで,100℃-1,000℃の中温 領域の使用を可能としている.固体高分子形燃料電池は電解質に水を含む高分子を用いる ため,最も低温の100℃付近の低温領域で使用できるが,課題として寿命に問題がある.溶
7
融炭酸塩形燃料電池 (MCFC)は電解質に溶融したアルカリ金属の炭酸塩を用が,課題として 高温領域で炭酸が揮発し,性能が劣化する.使用温度領域は 100℃-1,000℃の中温域で使用 可能である.直接メタノール型燃料電池 (DMFC) は燃料にメタノールを使用しているため 安全性およびコストが高いのが課題である.
(d) 生物電池
生物電池は生物自身の持つ化学反応、つまり化学エネルギーを電気エネルギー変換利用 した電池である.バイオ電池とも言われ注目を浴びている.主に土壌細菌を利用し,マグネ タイトなどを媒体とした燃料電池が注目されている.さらに,地下水中で微生物が合成する チューブ形状の酸化鉄が現行のリチウムイオン二次電池の炭素負極材料よりも優れた電池 特性を示すことも報告されている[22].
(e) 太陽電池
太陽電池は光起電力効果を利用し,光エネルギーを電気エネルギーに変換する電池であ る.一次電池や二次電池のような電力を蓄える蓄電池とは異なり,光起電力効果によって光 を瞬時に電力に変換し出力する発電機である.太陽電池は2種類に分類でき,シリコンを用 いた化合物半導体太陽電池と有機材料を薄膜化した色素増感型太陽電池に分けられる.
(f) 熱電池
熱電池は熱エネルギーを電気エネルギーに変換する電池である.ゼーベック効果,ペルチ ェ効果,トムソン効果など熱と電気の関係を利用した電池である.熱電池の仕組みとしては,
2種類の異種金属(または半導体)の両端を接続し,その両端に温度差を設け起電力を発生 させる.
(g) 原子力電池
原子力電池は放射性元素が原子核崩壊を起こす際に発生する原子力エネルギーを電気エ ネルギーに変換する電池である.主に人工衛星や宇宙探査機に使用される.原子力電池の仕 組みとしては,放射性元素の原子核崩壊の際に発生するエネルギーを利用して電力を発生 させる.α崩壊を起こすプルトニウム 238やポロニウム210 などが主に用いられる.この タイプは放射されたアルファ線が物質に吸収されて生じた熱を利用している.ストロンチ ウム90のように長い半減期をもつ同位体を用いる場合は,寿命の長い電源が得られる.
(h) 電池の歴史
電池は1791年ルイージ・ガルヴァーニ(イタリア)がガルバニ電池を発見したことに始 まり,今日まで様々な電池が誕生している.1800 年にはアレッサンドロ・ボルタ(イタリ ア)が,ボルタ電池を発明している.1802年には物理学者ヨハン・ウィルヘルム・リッター
8
(ドイツ)が小型一次電池を発明した.1866年ジョルジュ・ルクランシェ(フランス)はル クランシェ電池(マンガン乾電池の原型)を発明した.さらに,今までの電池で使われてい た電解液をゲル状にすることで現行使われる乾電池の原型を作製した.1899 年のユングナ ー(スウェーデン)は,ニッケル・カドミウム蓄電池を発明した.1900年にはトーマス・エ ジソン(米国)がニッケル・鉄蓄電池を発明した.最近では,1959年にエバレディ (Eveready)
(米国)がアルカリ乾電池を開発した.その後,1985 年にジョン・グッドイナフ(米国), ラシド・ヤザミ(フランス),吉野彰(日本)らはリチウムイオン電池を発明した.1991年 に西美緒らはソニーから世界で初めてリチウムイオン電池を商品化した.小型で軽量なモ バイル電子機器(携帯電話やスマートフォンなど)の実現に大きく貢献し,今後は自動車用 としても普及が見込まれている.
1.2.2 リチウムイオン二次電池
リチウムイオン二次電池は電気エネルギーと化学エネルギーを交互に変換して蓄積が可 能な電池であり,主に高容量型電池,高出力型電池に分類できる.高容量型電池では 800 Wh/Lを超える電池の開発が進められており,主にバッテリーの小型化や電気自動車(EV)
の航続距離向上に適している.高出力型電池では出力密度 4,500W/kg を超える電池の開発 が進められており,主にハイブリッド自動車(HV)の出力向上および急速充電の向上に適 している.さらにリチウムイオン二次電池は平均作動電圧が3.6~4.0Vと高いため,電気自 動車やハイブリッド車の車載用途への展開が盛んに進められている.また,ノートパソコン や携帯電話等の小型化,軽量化に伴い,ポータブル電子機器の電源として急速に普及してい る.今後も電気自動車の普及や電力貯蔵量の増大のため,二次電池の高容量化が求められて いる.
(a) 特徴
リチウムイオン二次電池は単位体積・単位重量あたりのエネルギー密度が他の二次電池 と比較して大きい,自己放電が小さい,鉛やカドミウムなど有害物質を含まない,メモリー 効果が小さいという特徴を有する.
(b) 電池の構成
リチウムイオン二次電池は,正極と負極をリチウムイオンが挿入・脱離することで充放電 を行う二次電池である.Fig. 1-4は電池の全体像を示します.電池全体の構成としては,正 極にリチウム金属酸化物,負極にカーボン,リチウム金属酸化物(LTO),一酸化珪素(SiO)
またはシリコン(Si),正極バインダーにポリフッ化ビニリデン(PVDF),負極バインダー にポリアミドイミド(PAI),ポリイミド(PI),電解液にLi塩(LiPF6)を溶解した有機溶媒 エチレンカーボネート(EC),ジエチルカーボネート(DEC)が用いられる.充電では正極
9
から負極に電子が移動し,リチウムイオンが電解液を介して負極に移動する.これにより電 極間で電位差が生じ充電が完了した状態になる.一方,放電は負極から正極に電子が移動し,
負極に存在していたリチウムイオンが正極に移動することで正極材料がリチウム金属酸化 物に還元される.
(c) リチウムイオン電池の歴史
携帯電話やノートパソコンなどの携帯機器の普及により,1980 年代に高容量で小型軽量 の二次電池ニーズが高まった.しかしながら,従来のニッケル水素電池では容量に限界があ り高容量の二次電池が切望された.
1960~1970 年代は,様々なリチウムイオン電池の提案や実験が行われたが,実用化まで
には至らなかった.1976 年にエクソンのマイケル・スタンリー・ウィッティンガムは正極 に硫化チタン,負極に金属リチウムを用いた金属リチウム二次電池[23]を提案した.1974- 1976 年にミュンヘン工科大学のベーゼンハルトは黒鉛内リチウムイオンのインターカレー ション[24, 25]と陰極の酸化物へのインターカレーションを発見した[26, 27].その後,1978- 1979年にペンシルベニア大学のSamar Basuが黒鉛内でのリチウムイオンの電気化学的イン ターカレーションを実証している[28, 29].しかし当時は,負極に黒鉛を用いると電解液の プロピレンカーボネートは負極側で分解してしまう[30]ため,炭素系材料へのリチウムイオ ンのインターカレーションは困難であると考えられていた.1977年に旭化成の吉野彰らは,
白川英樹が発見したポリアセチレンに注目し,1981 年に非水溶媒系二次電池の負極材料に 適していることを見出した.
1980年代に入りリチウムイオン電池の実用化に繋がる材料が次々と発見された.1980年 にはオックスフォード大学のジョン・グッドイナフと水島公一らがリチウムと酸化コバル トを用いてコバルト酸リチウム (LiCoO2) の合成に成功し正極材料として提案した[31].こ れがリチウムイオン二次電池の正極の起源となった.1983 年にはリチウムイオン二次電池 の原型を創出した.1981 年には三洋電機から黒鉛を負極材料とする二次電池の特許が初め て出願された.1982 年にラシド・ヤザミらは固体電解質を用いて黒鉛内にリチウムイオン をインターカレーションさせることを実証した[32].京都大学の山邊時雄らは量子化学的設 計に基づいてポリアセン系高分子型炭素材料(一次元グラファイト)を提唱した[33].1981 年にカネボウの矢田静邦は安定な難黒鉛化炭素の一種であるポリアセン系有機半導体(PAS)
を作成し,2種類のバッテリーを開発,実用化した。これを機に安定なドープ,脱ドープが 可能な黒鉛炭素材料の開発が加速した. 1983年にマイケル・メイクピース・サッカレーと ジョン・グッドイナフらは,スピネル構造を有するマンガン酸リチウム(LiMn2O4)を正極 材料として提案した[34].コバルト酸リチウムと比較して安価で安全な材料を見出した.マ ンガン酸リチウムは1996年に正極材料として実用化された.1985年に吉野彰らは炭素材料 を負極とし,リチウムを含有するコバルト酸リチウムを正極としたリチウムイオン二次電 池 (LIB)の基本概念を確立した.1990年にジェフ・ダーンらは,負極に黒鉛を用いた場合の
10
電解液として,エチレンカーボネートを用い初期の充電で分解させ黒鉛表面に保護被膜を 形成することを見出し,有機電解液の分解反応を停止できることを発見した[35].1994年に 松下電池工業の電解液として採用され,現在に至っている.1991年にはソニー・エナジー・
テックは世界で初めてリチウムイオン電池を商品化した.その後1993年にエイ・ティーバ ッテリー(旭化成と東芝との合弁会社)により商品化され,1994 年に三洋電機により黒鉛 炭素質を負極材料とするリチウムイオン電池が商品化された.1997年にはAkshaya Padhiと ジョン・グッドイナフらはオリビン構造を有するリン酸鉄リチウム(LiFePO4)を正極材料 として提案した[36].リン酸鉄リチウムはコバルト酸リチウムと比較して安全で長寿命であ る.2009年にソニーからリン酸鉄リチウムイオン電池が商品化された.2008年には東芝が 負極にチタン酸リチウム (Li4Ti5O12) を用いたリチウムイオン電池を商品化した.炭素材料 に比べ安全性,長寿命化,高出力,低温動作可能といった特性をもっている.黒鉛よりも電 位が約1.5V高いため単セル電圧が低くなることやエネルギー密度が黒鉛に比べ低いといっ た背反がある.
1.2.3 リチウムイオン二次電池の正極材料
リチウムイオン二次電池の正極材料として要求される特性は,高エネルギー密度,サイク ル特性の向上,低コスト,安全性である.高容量にはリチウムイオンの納まるべきサイト数,
高電位には結晶の電子構造,サイクル特性の向上には結晶の安定性が関係する.Fig. 1-5に は,これまで研究開発されてきた電極材料の電位と放電容量密度の関係を表している.Table.
1-1に理論容量を示している.Fig. 1-5およびTable. 1-1からわかるように,現在検討される 正極材料の結晶構造は層状岩塩型,オリビン型,スピネル型の3つに大別される.
リチウムイオン二次電池の開発当時から層状岩塩型LiCoO2は正極材料として用いられて おり,現在も主流となっている.この結晶構造はリチウムイオンの脱着がスムースに生じる ため良好なサイクル特性を示し,作動電位も4 V前後と高い.しかしコバルトは希少金属で ありコストが高く,将来の量産化にむけてCoに代わる新規材料の開発が求められる.そこ
でLiCoO2に代わる材料として同じ結晶構造のLiFeO2,LiNiO2の研究も進められている.
LiMPO4(M = Mn, Ni, Fe, Co等)の組成式で表せられるオリビン型正極材料はリンと酸素
が強く結合していることから熱安定に優れ,理論容量も大きい.また鉄やマンガンを利用す ることで電池の低コスト化に期待ができる.しかし,オリビン型の全般に言える問題点とし て電気伝導度が他の正極材料と比較して極めて低いことが挙げられる.これにより電池反 応の速度が制限されてしまうため,実用化の妨げとなっている.
LiMn2O4に代表されるスピネル型正極材料は資源が豊富なマンガンを利用することで大 きくコストを削減できる点から研究がさかんに行われてきた.しかし他の正極材料に比べ て容量が低いこと,3 V 領域での充放電においてサイクル特性が低下してしまうことから,
Mn の一部を他の金属で置換することにより充放電特性を向上させる試みが勢力的に行わ
11
れている.置換スピネル酸化物LiMn2-xMxO4(M = Fe, Cr, Ni, Cu等)は5 V級の正極材料とし て注目されている.特にLiMn1.5Ni0.5O4はサイクル特性が良好であることから最も有望視さ れている[37].
また実用されている電池の正極材料はマイクロ粒子であるが,軽量化や高出力化が求め られている近年では,正極材料ナノ粒子の合成や電池特性の評価に関する研究が多数報告 されている.正極材料のナノ粒子化は,比表面積の増大により粒子表面のリチウムの反応抵 抗および粒子内部のリチウム拡散抵抗を小さくすることが可能であり電池の反応速度の向 上が見込まれる.
(a) コバルト系材料
現在商品化されているリチウムイオン二次電池の正極材料は,その多くがLiCoO2である.
LiCoO2の正極活物質への実用化は,1980年にMizushima らによってLiMeO2型(Me:遷移
金属)がリチウム金属に対し4V 電位であるのを見い出したことから始まった[31].LiCoO2
の結晶構造はFig. 1-6に示すように,六方晶の黒色酸化物である.立方最密充填している酸 素(111)面の片側の八面体位置にLi+イオンが,反対側の八面体位置にCo3+が交互に配列した,
α-NaFeO2 型の層状岩塩型構造である.合成方法は,リチウム化合物(炭酸リチウム,水酸 化リチウムなど)とコバルト化合物(水酸化コバルト,炭酸コバルト,酸化コバルトなど)
を混合し,空気中で800℃に加熱すると容易に得られる[38].また,LiCoO2 は充放電特性に 優れ,出発原料を変えてもLiCoO2の特性に大きな差は認められない[39, 40].LiCoO2の理論
容量は 274mAh/g であるが,充電時の引き抜くリチウムの量によって結晶構造が保持でき
なくなるため,理論容量のすべてを電池として利用できるわけではない.一般にLixCoO2は,
X が 0.5 から1 の範囲,すなわち120~140mAh/g を限度として充放電を繰り返すようにす ると,構造が崩壊せずに安定したサイクル特性が得られる[41, 42].また,LiCoO2のCo を 他の遷移金属元素で置換して低コスト化を図り,高エネルギー密度,優れたサイクル特性,
安全性などの特性を向上しようとする試みが盛んである.中でもAl 置換については,放電 特性を犠牲にしてコストを低減するという目的で研究が行われている.また,アルミニウム の価数が変化しないために,正極材料中では電気化学的に不活性であると考えられていた
が, Ceder らはAl 置換したLiCoO2の充放電特性が向上することを見いだし,その原因と
して金属原子と酸素原子の間を電子が容易に移動するためであることを明らかにした[43].
一方Jangらも同様のLiCoO2を合成し,容量特性が向上することに加えて結晶構造が安定化 することを見いだし,Co はその半分をAl が置換できることを見いだしている[44].さらに
Tukamoto と West によって,Mg 置換の場合にも同様の効果があることが報告されている
[45].また過剰のLi によってLiCoO2の電極としての電気化学的特性が向上し,充放電特性
も安定化することが明らかになった.以上のように,Co 位置を異種元素で置換することに よってその特性は大幅に向上することが明らかになったため,現在でも遷移金属の導入、Li 過剰系材料など様々な観点から検討されている.
12 (b) ニッケル系材料
LiNiO2は理論容量が275mAh/g と大きく,LiCoO2と同じ層状岩塩型の結晶構造である.
さらにNi材料は Co 材料に比べ 安価であるため,LiCoO2と同様に古くから正極材料とし て研究が行われてきた.しかし,LiNiO2は LiCoO2のように容易に作製できるわけでなく,
合成時にNi3+はNi2+に還元され易く,その結果 Li が不足した Li1-XNi1+XO2が生成しやすく なる[46].また,LiNiO2からLi がデインターカレートする際にはNiがLi サイトへ移動し てしまうこと[47],Li が多い場合にはNi位置をLi が占有してしまうことも明らかになっ ている[48].このような化学両論組成からのずれは充放電特性に大きく影響する.したがっ て,NiやLi出発原料の選択,酸素雰囲気制御[49]および焼成条件の工夫により,化学量論
組成のLiNiO2合成が試みられている.Dutta らは,酸素中800℃以上の温度で長時間加熱す
ることによって化学量論組成の LiNiO2が合成できることを明らかにした[50].一方,Li2O と NiO を湿潤空気中で加熱することによってほぼ化学量論組成に近い LiNiO2を合成でき るが,真の化学量論組成物は得られなかった[51].また,三元系材料のLiaNibCocMndO2(NCM),
LiaNibCocAldO2(NCA)のように Ni を Co,Mn,Al などの遷移元素で置換することによって 電池特性が向上する.中でもCo 置換は最も効果的であり,電池特性は大きく向上すること が明らかになっている[52].例えば,Niの20mol%をCo で置換した試料は充放電特性に加 えてサイクル特性も向上した.リチウムイオン二次電池の重要な問題は,充電する際に発熱 する正極の安定性である.DSC の測定から,LiNiO2 からの酸素の発生による発熱量は
LiCoO2やLiMn2O4に比べて低く,安全性は有利である[53].また,LiXNiO2 は電池として利
用できるLi の組成領域がX=0.3 程度までとLiCoO2に比べて広いため,有望な正極材料と して今後も検討されると思われる.
(C) マンガン系材料
LiMn2O4は,LiCoO2やLiNiO2に比べて理論容量は小さいが,中心金属のマンガンが低コ ストで埋蔵量が豊富である,環境負荷が比較的低いこと,さらに充電時の安全性が高いなど の特徴から,現在最も研究開発が盛んな材料のひとつと言える.LiMn2O4 はFig. 1-7に示す ようにスピネル構造であり,空間群はFd3m である.Liと Mn はそれぞれ,立方最密充填 している酸素の四面体位置(8a サイト)と八面体位置を占める[54].Li が構造内を拡散す るときには,8a サイトのすぐ隣にある空の八面体位置である16c サイトを経由して,8a→
16c→8a→のようにジグザグに進むことが明かになっている.通常は,炭酸リチウムとマン ガン酸化物を空気中で焼成することによって作製されるが,作製方法によって電池特性は 異なる[55, 56].またLiMn2O4 は過剰の Li を取り込むことが可能であり,その場合には正 方晶Li2Mn2O4が生成される.この原因としては,スピネル中の Mn3+が有機電解液中に溶出 すること,デインターカレートによって生成する Li 不足の LiMn2O4 構造は不安定である こと,さらに MnO6 八面体はヤーン・テラー効果のために歪みやすいことなどが考えられ ている.このため,Mn の一部を他の金属元素M で置換したLiMn2-XMXO4を合成し,サイ
13
クル特性を向上させようという試みも盛んである.この時,単位格子のc/a は1より大きく なるが,放電電圧はほぼ3Vに保たれる[57].しかし,この相転移は大きな体積膨張(6%)
を伴うため,3V を挟んで充放電を繰り返すと徐々にサイクル特性は低下する.一方4V領 域でLiMn2O4 からリチウムを脱離させた場合では,スピネル構造を保ったまま,充電が進 行する.つまり,4V 領域において充放電を繰り返せば,サイクル特性の劣化を抑えること ができる.このため LiMn2O4は,特に 4V 級のリチウムイオン電池用正極活物質として有 望視されている[58].しかし充放電を繰り返すと容量は大幅に減少する.数々の特徴を有す る LiMn2O4ではあるが,その理論容量はLiCoO2やLiNiO2の約半分であるためにLiCoO2や
LiNiO2と同程度の理論容量を持つLiMnO2に注目が集まっている[59].しかし,LiMnO2はそ
の構造が不安定なために原料粉末を混合して空気中にて焼成するだけでは生成せず[60],そ の合成方法が模索されている状況である[61].また最近,LiMnO2 には斜方晶(o-LiMnO2) と単斜晶(m-LiMnO2)で表される2種類の結晶構造が存在することが明らかになった[62].
最近の研究ではMn サイトの一部をCr およびFe で置換することでm-LiMnO2が生成する ことを見出し,さらにm-LiMnO2は構造安定性に優れおり,サイクル特性も良いことを確認 している[63, 64].また, m-LiMnO2は充放電にともなって生じる構造変化を吸収できるこ とが明らかになり,これがm-LiMnO2の優れたサイクル特性要因であることが報告されてい る[65].しかし,単斜晶が高温相、斜方晶が低温相で安定であると理解されているなど,結 晶学的には理解し難いことに加えてその構造は不安定であると考えられているため,多く の問題が山積していることも事実である.現在でもm-LiMnO2は様々な角度から検討されて いる物質のひとつである.
(d) 鉄系材料
Fe は Co,Ni,Mn の遷移金属元素に比べコストや毒性などのリスクが少ない.しかし
LiFeO2はLiMnO2と同様に構造が不安定であると考えられ,通常の焼結による固相反応法で
は合成することが困難であった.LiFeO2 に代わり,近年注目を集めているのがオリビン型
構造の LiFePO4 である.Fig. 1-8 に LiFePO4 のオリビン型構造を示す.1997 年には
Fe(CH3COO)2,(NH4)3PO4,Li2CO3から合成されたLiFePO4 は,リチウムの挿入・脱離が可 能であることが明かになった[66].また,LiFePO4 の理論容量は170mAh/g と大きくはない
が,その95%以上の充放電に利用でき,サイクル特性も優れている[67].したがって,LiFePO4
は最も有望な正極材料の一つと考えられるようになった.定比組成のLiFePO4中でFeは2 価であるが,空気中で焼成する場合には酸化され易いためにFe3+となってしまう.このよう な酸化を防止するため,焼成をアルゴンや窒素などの不活性ガス雰囲気中で行う必要があ る[68].また,2価のFeを含む原料は一般的ではないために高価であり,取り扱いが難しい ことなどの問題もある.Fe 原料は将来的にも安価で取り扱い易いため,原料コストの低減 策はFe原料の材料開発に掛かっている.
14 1.2.4 リチウムイオン二次電池の負極材料
リチウム一次電池の負極材料として用いられていた金属リチウムは,二次電池の負極材 料としても使用可能とするため,多くの研究が進められていた.しかしながら,充電時に活 性な金属リチウムが樹枝(デンドライト)状に成長・析出するため[69],金属リチウムの脱 落またはデンドライトが正極まで到達して微小な短絡を引き起こし,充放電サイクル寿命 が短くなり実用化が困難であった.上記の課題は,電気化学的にリチウムイオンをグラフェ ン面間にインターカレート可能な炭素材料[70-72]を負極材料に用い解決した.ただし,充放 電サイクル寿命は大きく伸ばすことができたが,エネルギー密度の低下が生じる結果とな った.現在は,黒鉛材料が実用化され,現状のリチウムイオン二次電池に至っている.
最近では,負極活物質は現状の黒鉛系炭素材料の理論容量の372 mAh/gから,合金系負極 活物質を用いることで,Li金属の理論容量である3862 mAh/gまで約10 倍近く向上させる 可能性を有している.Table 1-2 に各種提案されている負極活物質の理論容量と作動電位(vs.
Li / Li+)を示す[73].その中で,Si は理論容量が4200 mAh/g、9786 mAh/cmとリチウム金属
の理論容量と同等で作動電圧も低いため,資源量の観点でも次世代のリチウムイオン二次 電池の負極活物質として注目されている.このように,次世代負極活物質としてはSi 系負 極が最有望であり,リチウムイオン二次電池の更なる高容量化と高エネルギー密度化には 炭素負極に代わるSi 系負極の実用化を見据えた研究が重要である.
(a) 炭素材料
負極材料として用いられる炭素材料は,結晶構造は配向が比較的均一な黒鉛(グラファイ ト)と,結晶の配向がランダムで長距離秩序性のない難黒鉛化炭素(ハードカーボン)に大 きく分別される.黒鉛にはフッ化水素酸や高温の熱処理で不純物を除去した天然黒鉛と,コ ールタールピッチ等の原料から 3200℃の高温下で黒鉛化して製造される人造黒鉛が用いら れている.理論容量は372 mA h /gを有する.
天然黒鉛は安価ではあるが,かさ密度が低く電極密度を上げにくいといった欠点がある.
一方,人造黒鉛は天然黒鉛に比べて,かさ密度が高く電極密度を上げやすいが高価である.
ハードカーボンは樹脂等を800~1400℃の熱処理を施して炭化したもので,黒鉛に比べて 安価で容量が大きいという利点がある.しかしながら,かさ密度が大きく初期の充放電のク ーロン効率が低く放電電位が平坦でないという欠点も有している.
現在,リチウムイオン電池の主な負極材料として使用されている黒鉛は既に理論値 372
mA h/gに近づいており,黒鉛の理論容量を超える炭素材料や無機化合物材料などの新たな
材料の開発が望まれている.
(b) シリコン材料
シリコンはグラファイト(372 mAh/g)の10倍の理論容量(4200 mAh/g)を有する.また,比 較的低い作動電位を有し,Si 元素は天然に豊富に存在するため環境に優しいことから,グ
15
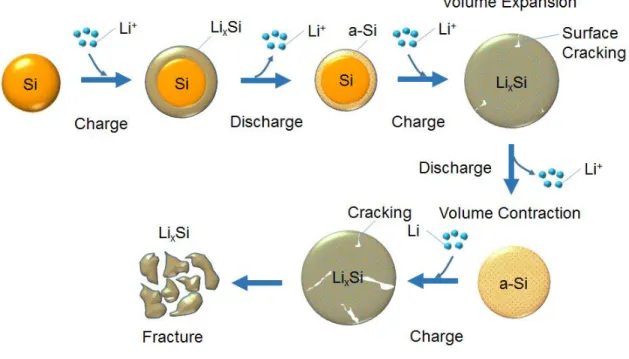
ラファイトに代わる負極材料として期待されている.しかし,Si の使用に関連する課題と して充放電時の体積変化が挙げられる.Si粒子はLiイオンの挿入・脱離により最大400 % まで体積変化し最終的に崩壊する.Fig.1-9はSi粒子へのLiイオンの挿入・脱離を示してい る.そのため,サイクル寿命が短く,急激かつ不可逆的な容量減少および低いクーロン効率 が引き起こされる.また,Si粒子の表面のSEI(Solid Electrolyte Interphase)膜が過剰に形成さ れることによる内部抵抗の増加も課題のひとつである.リチウムの脱離によるナノ構造の 収縮に伴ってSEIが破損すると,シリコンの新しい表面が電解液に露出して SEIが形成さ れ,充放電を繰り返すごとに厚くなることは電気容量の低下につながる.これらの課題を解 決するアプローチとして,ナノ粒子化,非晶質化および複合材料の利用など多くの研究が行 われている.
一方、SiOxはSiと比較すると体積変化が250 %と小さく,その構造はSiO2マトリックス 中にナノSi が分散した構造をとっている.しかしながら,SiOxは初回充放電に伴う不可逆 容量が大きいため,SiOx単独での負極材料の実現を困難にしている.その要因として,SiO2
マトリックス中の反応生成物であるリチウムシリケート化合物Li4SiO4の生成が挙げられる.
Table1-3 にリチウムシリケート化合物とクーロン効率を示した.リチウムシリケート化合
物の生成はクーロン効率の低下を引き起こし不可逆容量の一因となっている,これらの課 題を解決するため,SiOx 改良および粒子形状の制御など副反応抑制の研究が盛んに行われ ている.
(c) リチウム複合酸化物
負極材料としてのリチウム複合酸化物の特徴として,Li イオン挿入時の体積膨張率の低 減,Li 挿入脱離による発熱および熱暴走の抑制,寿命および低温特性の優位性が挙げられ る.また,電池の効率を支配するSEIの形成が少ないことも特徴の一つである.中でもチタ ン酸リチウム[74, 75]は,約1.55Vに電位平坦部があり,Li[Li1/3Ti5/3]O4という化学構造式の 化合物で,スピネル型の結晶構造を取り,175 mA h/gの理論容量を有する.安定な結晶構造 であるために,充放電サイクル寿命が長く急速充放電に適しており電位曲線も平坦である が,黒鉛を負極とする電池より平均電圧が 1.3 V 程低い.電位が低いことで,SEI(Solid Electrolyte Interphase)を形成しにくく,集電体にアルミニウム箔の仕様が可能である.㈱東 芝によって2011年に商品化されている.
最近では,さらに高容量(約250 mAh/g)のH2Ti12O25[76]も開発されておりこの分野も研 究が盛んである.
(d) 合金系材料
金属リチウム極のデンドライト成長の抑制のために,Li-Al等のリチウム合金が検討され,
最近では,Liと電気化学的に合金を形成するAl, Zn, Sb, Sn, Siなどの金属および,それらの 合金が検討されている.ソニー㈱は2005年に非晶質のスズ合金を負極に採用したリチウム
16
イオン二次電池を初めて製品化しデジタルビデオカメラ用バッテリーパックに搭載してい る.低温特性と急速充電に優れた性能を有している[77, 78].
SnやSiをはじめとする合金材料はTable 1-2に示すように,Liの吸蔵量が非常に大きく,
Li 挿入時の大きな体積膨張や微粉化のため充放電サイクル寿命が短命であるという問題点 がある.しかしながら,Siは資源的にも豊富でコストの低減が見込める魅力もあり,多くの 研究が行われ実用化への期待が大きい.
(e) 酸化物材料
富士フィルム㈱によって 1996 年に高容量負極材料として非晶質Sn 酸化物が報告されたが [79, 80],電池電圧が黒鉛負極に比べて低く,初期の充放電効率が低く不可逆容量が極めて 大きいため実用化されていない.ガラスマトリックス中に分散したSnOの酸素原子と充電 時に析出するLiが反応して Snのナノ粒子が形成され,その後の充電でSnとLi が合金化 し,放電でLiが放出される.ガラス中に分散したSnのナノ粒子であるためにLiとの合金 化反応での体積膨張が緩和される.初期のSnOとLiが反応して不可逆で安定な Li2Oが形 成するため,1サイクル目のLi挿入量は1200 mAh/gと大きいがLi放出量としてはその半 分の600 mA h/gである.
(f) 窒化物材料
NTTによって黒鉛の約2倍の容量密度を示すLi2.6Co0.4Nが開発され研究されているが[81- 84],空気中で不安定であること,電池電圧が低いことから実用化されていない.
17 1.3 ナノ粒子
1.3.1 ナノ粒子の特性
固体粒子はその大きさが小さくなるにつれ,呼称が変化する.100~1000 nmでは微粒子,
1~100 nmではナノ粒子(超微粒子),1 nm以下では数個から数百程度の原子の集合体とし
てクラスターと呼ばれている.一般的にナノ粒子のサイズの定義は1~100 nmとされている.
Fig.1-10 にクラスター,ナノ粒子および微粒子の関係を示す.さらに,ナノ粒子はその特性
がバルク材料と大きく異なることが知られている.ナノ粒子の特性としては反応性,量子サ イズ効果,透明性の3つに分類できる.さらには,電磁気的,光学的,機械的特性が発現す ることも大きな魅力である.これらの特徴を活かし,薄膜やナノ複合材料などさまざまな機 能材料の応用が行われている.
(a) 反応性
ナノ粒子の反応性はバルク材料と大きく異なる.これは,粒子が小さくなると比表面積が 体積より増大する影響である.微粒子の表面は内部に比べて配位数が小さく,結合が不飽和 の状態でありエネルギーが高い.しかしながら,粒子が小さくなると結合エネルギーに比べ 表面エネルギーが無視できなくなる.そのため,粒子表面原子の部位で生じる化学反応は,
表面原子の割合が大きいナノ粒子が微粒子に比べて高くなる.ナノ粒子化に伴う表面エネ ルギーの効果としては,蒸気圧や溶解度の増大,結晶変態の安定性,融点の減少,焼結温度 の低下などが挙げられる.この性質を利用した例を下記に示す.
・電池性能の向上:軽量化、大容量化
・触媒性能の向上:反応率向上
・ナノ粒子金属ペーストの低融点化
(b) 量子サイズ効果
ナノ粒子の中でも10nm以下の粒子は、ナノ粒子中の電子の振る舞いが変化する.ナノ粒 子の小空間内に電子が閉じ込められるため,電子の動きが制限されエネルギー準位の離散 化,バンドギャップの増大が生じる.そのため,バルクや微粒子とは異なり,吸収端および 発光波長の短波長化が生じる.ナノ粒子が光を吸収する原理は,光の「電場振動」とナノ粒 子の「自由電子の振動」が共鳴するためで,この現象を「プラズモン共鳴」と呼ぶ.また,
この原理を酸化チタンなどの半導体と組み合わせると,共鳴ナノ粒子から半導体へ電子が 移動し,正と負の電荷が分離可能となる[85, 86].この現象を「プラズモン誘起電荷分離」と 呼ぶ.この他に,粒子の比表面積が体積に比べ著しく増大することから,粒子径が光の波長 や磁性体の磁区より小さくなり,反応性と活性度が増大する.2~20 nmサイズの磁性ナノ 粒子は磁場がない状態で超常磁性,すなわち磁化がゼロを示し,外部の磁気源により磁化さ れる.粒子サイズをナノ粒子化することにより,物質中の電子の振る舞いを制御し性質を変 えることが可能となる.この性質を利用した例を下記に示す.
18
・光電変換およびセンサーの性能向上:光による正・負の電荷分離精度の向上
・光触媒性能の向上:反応効率向上
・核磁気共鳴映像法(MRI:Magnetic Resonance Imaging)の性能向上:造影剤の向上
・標的特異的ドラッグデリバリー性能の向上:検出制度の向上
(c) 透明性
ナノ粒子は個々の粒子が小さく見えないため,ナノ粒子を含有した液体や固体の透明性 を高くできる.透明な液体や固体中にナノ粒子を分散させ,ナノ粒子の種類やサイズによっ て,透明性を維持しながら特定の光波長域をカット可能となる.ガラス中に金ナノ粒子を分 散させることで,鮮やかな赤色を呈するため,ステンドグラスなどに利用されている.また,
酸化チタンや酸化亜鉛ナノ粒子は紫外線をカットする機能がある.その他,紫外線カット以 外にも,特定の波長をカットする機能性光学フィルムや機能性コーティング材料にナノ粒 子が使われている.さらに,顔料をナノ粒子化すると透明性が発現され彩度も向上する.こ れは,ナノ粒子化により顔料表面の光散乱が抑制されるためである.ミクロンサイズで透明 性が乏しい顔料でも,ナノ粒子にすることで透明性と彩度が得られるため,光学フィルター などに応用されている.
1.3.2 ナノ粒子の合成方法
ナノ粒子の合成方法は,固相法,液相法と気相法に大別できる.さらに,ナノ粒子の合成 法は大きく2つに分類することができる.1つは物質を原子論的にみた集団的変化の方法論 を利用し,バルク状の原料を微細に加工・粉砕・再編成する技術をトップダウン方式という.
もう 1 つは原子や分子を正確に組み合わせることで新しい機能を持った材料を合成してい く方法で,これをボトムアップ方式という[87].トップダウン方式は主に機械・電子系の分 野で,ボトムアップ方式は化学系の分野で研究が行われている.トップダウン方式は大規模 生産が可能であることが多いが,微小なサイズの制御には限界がある.また,複雑な組成を もつナノ粒子を合成することが困難である.これに対し,ボトムアップ方式はトップダウン 方式と比較し,より微小に構造やサイズを制御しやすいという点から,様々なナノ粒子合成 に応用されている.
(a) トップダウン方式
トップダウン方式は,ボールミル,ジェットミル,ビーズミルおよびメカノケミカルなど の固相法が存在する.下記に代表的な固相法のナノ粒子方法について示す.
・ボールミル
ボールミルは,ジルコニア・アルミナなどのセラミクスや鉄などの硬質のボールと,材料