CTD 第2部
2.7 臨床概要
2.7.6 個々の試験のまとめ
略号·用語一覧
AI accumulation index 累積係数
ALL acute lymphblastic leukemia 急性リンパ性白血病
ALT alanine aminotransferase アラニン·アミノトランスフェラーゼ AST aspartate aminotransferase アスパラギン酸アミノトランスフェラーゼ AUC(TAU) area under the plasma concentration vs time curve 血漿中濃度曲線下面積
AUC(INF) area under the plasma concentration vs time curve from time zero to infinity 0 時間から無限時間までの血清中濃度曲線下面積 AUC(0-T)
area under the plasma concentration vs time curve from time zero to last time of measurable concentration
0 時間から最終測定可能時間までの血清中 濃度曲線下面積
B5D twice a day, 5 days on and 2 days off schedule 1 日 2 回 5 日間投与 2 日間休薬 B7D twice a day, continuous dosing schedule 1 日 2 回連日投与
BCR-ABL a protein tyrosine kinase チロシンキナーゼ蛋白の一種 BID twice a day 1 日 2 回投与
CCyR complete cytogenetic response 細胞遺伝学的完全寛解 CD4 cluster of differentiation 4 ヘルパーT 細胞表面上の抗原 CHR complete hematologic response 血液学的完全寛解
CI confidence interval 信頼区間
CK creatine kinase クレアチンキナーゼ
CL clearance クリアランス
CLo apparent oral clearance 定常状態の見かけの経口クリアランス CLpBSA systemic serum clearance based on body surface area 体表面積に基づく全身血清クリアランス Cmax maximum plasma concentration 最高血中濃度
Cmin minimum plasma concentration 最低血中濃度 CML chronic myelogenous leukemia 慢性骨髄性白血病 CRKL substitute of BCR-ABL BCR-ABL 蛋白の基質 CyR cytogenetic response 細胞遺伝学的寛解 DLT Dose-limiting toxicity 用量規制毒性
ECOG Eastern Cooperative Oncology Group 米国東海岸癌臨床試験グループ EGFR epidermal growth factor receptor 上皮細胞増殖因子受容体 FACT-G Functional Assessment of Cancer Therapy General QOL 調査票のひとつ
Hb hemoglobin ヘモグロビン
IC50 concentration at which 50% inhibition is obseved 無処置対照群の細胞の増殖に対して細胞の増殖を 50%抑制するのに必要な薬物濃度 λZ elimination rate constant 消失速度係数
INR international normalization ratio 国際標準比 MAD maximum administered dose 最大許容量
MaHR major hematological response 血液学的major 寛解
MDA MD Anderson Cancer Center MD アンダーソン癌センター MedDRA Medical Dictionary for Regulatory Activities Terminology 国際医薬用語集
MCyR major cytogenetic response 細胞遺伝学的major 寛解 MMR major molecular response 分子遺伝学的major 寛解 MiHR minor hematologic responce 血液学的minor 寛解 mRNA messenger ribonucleic acid メッセンジャーリボ核酸 MRT mean residence time 平均滞留時間
MUGA multiple gated acquisition scan 放射性核種を用いた同期心室撮影 N number of subjects 被験者数
NA not available 評価不能
NCI-CTC National Cancer Institute – Common Toxicity Criteria 米国国立癌研究所–毒性共通基準 NEL no evidence of leukemia 血液学的部分寛解
OHR overall hematological response 血液学的寛解 OS overall survival 全生存期間
PCR polymerase chain reaction ポリメラーゼ連鎖反応 PCyR partial cytogenetic response 細胞遺伝学的部分寛解 PD progressive disease 病勢の進行
PDGF platelet-derived growth factor 血小板由来増殖因子 PFS progression free survial 無増悪生存期間
Ph+ Philadelphia chromosome positive フィラデルフィア染色体陽性
PK pharmacokinetics 薬物動態
PS performance status 一般状態
PT prothrombin time プロトロンビン時間
PTT partial thromboplastin time 部分トロンボプラスチン時間 Q5D once daily 5 days on and 2 days off schedule 1 日 1 回 5 日間投与 2 日間休薬
QD once daily 1 日 1 回投与
QoL quality of life 生活の質
QT the interval between the beginning of the Q-wave and the end of the T-wave on an electrocardiogram 心電図上のでの間隔 Q 波の開始から T 波の終わりま QTc corrected Q to T wave interval on electrocardiogram 補正QT 間隔
RQ-PCR real time quantitative PCR リアルタイム定量PCR SAE serious adverse event 重篤な有害事象
SD stable disease 安定
SRC a protein tyrosine kinase チロシンキナーゼ蛋白のひとつ t1/2 terminal elimination half-life 終末消失半減期
TDD total daily dose 1 日投与量
TLast time to last measurable serum concentration 血清中濃度の最終測定可能時点までの時間 Tmax time to maximum serum concentration 最高血清中濃度到達時間
UCLA University of California, Los Angeles カリフォルニア大学 Vss volume of distribution at steady-state 定常状態分布容積 Vz/F apparent volume of distribution at elimination phase 見かけの分布容積 WHO World Health Organization 世界保健機構
1 臨床試験一覧表
第2 部 項番号 標題 第5 部 項番号 2.7.6-2.1 CA180-037: 健康成人を対象としたダサチニブ 50 mg 錠 1 錠及び 20 mg 錠 1 錠に対するダサチニブ 70 mg 錠 1 錠の生物学的同等性試験 5.3.1.2-1 2.7.6-2.2 CA180-019: 健康な男性被験者における[14C]BMS-354825 のマスバランス、薬物動態 及び代謝 5.3.3.1-1 2.7.6-2.3 CA180-016: 健康成人におけるダサチニブの製剤比較試験 5.3.3.1-4 2.7.6-2.4 CA180-034: イマチニブに抵抗性又は不耐容の慢性期慢性骨髄性白血病を対象とし たダサチニブの1 回 50 mg 又は 70 mg の 1 日 2 回投与あるいは 1 回 100 mg 又は140 mg の 1 日 1 回投与 2x2 比較試験-中間成績- 5.3.5.1a-2 2.7.6-2.5 CA180-017: 400-600 mg/日のイマチニブに抵抗性の慢性期慢性骨髄性白血病を対 象とした、ダサチニブ又はイマチニブ(800 mg)の無作為化臨床第Ⅱ相 オープンラベル試験-中間成績- 5.3.5.1a-1 2.7.6-2.6 CA180-002: メシル酸イマチニブに抵抗性又は不耐容の慢性期、移行期並びに急性期 慢性骨髄性白血病及びフィラデルフィア染色体陽性急性リンパ性白血病 を対象とし、安全性、薬物体内動態及び薬力学の検討を目的とした BMS-354825 の臨床第 I 相試験 5.3.3.2-1 5.3.5.2a-1 2.7.6-2.7 CA180-013: イマチニブに抵抗性又は不耐容の慢性期慢性骨髄性白血病を対象とし たダサチニブの臨床第II 相試験-中間成績- 5.3.5.2a-4 5.3.5.2a-4.1 2.7.6-2.8 CA180-005: イマチニブに抵抗性又は不耐容の移行期慢性骨髄性白血病を対象としたダサチニブの臨床第 II 相試験-中間成績- 5.3.3.2-4 5.3.5.2a-2 5.3.5.2a-2.1 2.7.6-2.9 CA180-006: イマチニブに抵抗性又は不耐容の骨髄芽球性急性期慢性骨髄性白血病を対象としたダサチニブの臨床第 II 相試験-中間成績- 5.3.3.2-7 5.3.5.2a-3 5.3.5.2a-3.1 2.7.6-2.10 CA180-015: イマチニブに抵抗性又は不耐容のリンパ芽球性急性期慢性骨髄性白血 病又はフィラデルフィア染色体陽性急性リンパ性白血病を対象としたダ サチニブの臨床第II 相試験-中間成績- 5.3.5.2b-1 5.3.5.2b-1.1 5.3.5.2b-1.2 2.7.6-2.11 CA180-031: BMS-354825 の Philadelphia 染色体陽性慢性骨髄性白血病及び急性リンパ性白血病に対する臨床第 I/II 相試験 5.3.5.2a-5 2.7.6-2.12 CA180-036: BMS-354825 の Philadelphia 染色体陽性慢性骨髄性白血病及び急性リン パ性白血病に対する継続投与試験 5.3.5.2a-6 2.7.6-2.13 CA180-009: 健康成人における BMS-354825 の薬物動態に対する低脂肪食及び高脂 肪食の影響 5.3.3.4-1 2.7.6-2.14 CA180-022: 健康成人におけるシンバスタチンの薬物動態に対するダサチニブの影響 5.3.3.4-3 2.7.6-2.15 CA180-032: 健康成人におけるダサチニブ経口投与時の薬物動態に対するリファン ピシンの影響 5.3.3.4-5 2.7.6-2.16 CA180-020: 健康成人におけるダサチニブ経口投与時の生物学的利用能に対する制 酸剤の影響を検討する第1 相臨床試験 5.3.3.4-8 2.7.6-2.17 CA180-021: 進行性固形癌患者におけるダサチニブの薬物動態に対するケトコナゾ ールの影響及び薬力学的マーカーに対するダサチニブの影響を検討する 第1 相臨床試験 5.3.3.4-10 5.3.3.4-10.1 2.7.6-2.18 CA180-035: イマチニブに抵抗性又は不耐容の移行期・急性期の慢性骨髄性白血病及 びフィラデルフィア染色体陽性急性リンパ性白血病を対象としたダサチ ニブの1 回 70 mg の 1 日 2 回投与又は 1 回 140 mg の 1 日 1 回投与の比 較試験-中間成績- 5.3.5.1a-3 2.7.6-2.19CA180-138: BMS-354825 の Philadelphia 染色体陽性または BCR-ABL 陽性慢性期慢性 骨髄性白血病に対する臨床第Ⅱ相試験-1 回 100mg(1 日 1 回)または 1 回50mg(1 日 2 回)投与における検討- -中間成績-
2 個々の試験の概要
2.1 Study
CA180-037
試験課題名: 健康成人を対象としたダサチニブ 50 mg 錠 1 錠及び 20 mg 錠 1 錠に対 するダサチニブ70 mg 錠 1 錠の生物学的同等性試験 治験責任医師: 治験実施医療機関: 公表論文: なし 治験期間: 最初の被験者の治験登録日:20 年 月 日 最後の被験者の治験終了日:20 年 月 日 開発のフェーズ: 第1 相 目的: 主要目的: ダサチニブ 50 mg 錠 1 錠及び 20 mg 錠 1 錠の組み合わせとダサチニブ 70 mg 錠 1 錠との生物 学的同等性を検証する。 副次目的: ダサチニブの安全性の検討 治験方法: 本治験は、健康成人を対象とした非盲検、ランダム化、2 期 2 処置のクロスオーバー試験であ った。治験組み入れの21 日前以内にスクリーニング検査を行い、被験者の適格性について評価し た。被験者は1 期の投与開始前日から治験実施医療機関に入院し、1 期の 2 日目に一時的に退院 したのち、投与後7 日間以上の休薬期間を経て、2 期の投与開始前日に再入院し、治験終了時(2 期の2 日目)まで施設内で管理下に置かれた。被験者はランダムに割り付けられた順に処置 A(ダ サチニブ50 mg 錠 1 錠及び 20 mg 錠 1 錠)又は処置 B(ダサチニブ 70 mg 錠 1 錠)の投与を受け た。薬物動態(PK)用検体の採血は、それぞれの処置でダサチニブ投与後 24 時間まで経時的に行 った。また、本治験では予定された時間に理学的検査、バイタルサイン、12 誘導心電図(ECG) 及び臨床検査をそれぞれ実施し、有害事象(AE)については治験期間を通して観察した。 被験者数: 総計229 例の被験者が治験に参加(文書による同意を取得)した。ランダム化された 64 例のう ち、61 例が治験を完了した。なお、2 例の被験者が有害事象により治験を中止し、1 例が同意を 撤回した。 主要な選択基準: 文書による同意が得られた、18~50 歳の健康な男女で、体格指数(BMI)が 18~30 kg/m2の者 とした。また、女性については閉経後又は外科的手術により不妊である者のみを対象とした。 治験薬、用量、剤型、投与経路、製造/ロット番号: 薬剤 剤型 用量 投与経路 ロット番号 製造番号 ダサチニブ 錠剤 70 mg×1 錠 経口 5F00381 5F04871 対照薬、用量、剤型、投与経路、製造/ロット番号: 薬剤 剤型 用量 投与経路 ロット番号 製造番号 ダサチニブ 錠剤 20 mg×1 錠 経口 5B02159.5A 5C06213 ダサチニブ 錠剤 50 mg×1 錠 経口 5B02174.5A 5C05064 投与期間: 7 日間以上の休薬期間を設け、単回投与を 2 回行った。 評価基準: 有効性: 処置A ダサチニブ 50 mg 錠+20 mg 錠 処置B ダサチニブ 70 mg 錠 処置A ダサチニブ 50 mg 錠+20 mg 錠 処置B ダサチニブ 70 mg 錠 1 期 スクリーニン グ・登録 2 期 ランダ ム 割 付 け 休薬期間 休薬期間 治験終了 休薬期間は7 日間以上
該当せず。 安全性及び忍容性: 安全性評価は、有害事象(AE)報告の他、バイタルサイン、ECG、理学的検査及び臨床検査の 結果に対する医学的な評価に基づいて実施した。発現した有害事象は一覧表にし、臨床上の意義 及びその重要性について検討した。 薬物動態: ダサチニブの経口投与後における血漿中濃度の経時的推移から、ダサチニブの単回投与時にお けるPK パラメータ(Cmax、Tmax、AUC(0-T)、AUC(INF)及び t1/2)を算出した。 薬力学: 該当せず。 ファーマコゲノミクス: 該当せず。 統計方法: 被験者数: ダサチニブ70 mg 錠 1 錠を投与したときと、50 mg 錠 1 錠及び 20 mg 錠 1 錠を同時に投与した ときの生物学的利用率に差がないと仮定すると、被験者60 例の場合、生物学的に同等であると結 論するための検出力はCmax では 91%、AUC(INF)では 95%であった。しかしながら、生物学的 利用率に5%の差があると仮定すると、被験者を 60 例とした場合に生物学的に同等であると結論 するための検出力はCmax で 82%、AUC(INF)で 88%であった。脱落例が出る可能性を考慮して、 本治験の被験者数を64 例とした。 統計解析: 被験者背景: 性別及び人種別の頻度分布を表に示し、年齢、体重、身長及びBMI については要約統計量を表 に示した。 安全性: 報告されたAE はすべて、器官分類、基本語及び処置別に一覧表に示した。バイタルサイン及 び臨床検査値は、処置及び時間ごとに一覧表に示した。臨床上重要と思われる診察所見及び臨床 検査値については列挙した。ECG のパラメータはチャートを基に評価し、異常が認められた場合 は表に示した。 薬物動態:
処置A に対する処置 B の Cmax 及び AUC(INF)の幾何平均値比(処置 B/処置 A)の 90%信頼区 間が80%~125%に完全に含まれるとき、処置 B は処置 A と生物学的に同等であるとした。90% 信頼区間は、Cmax 及び AUC(INF)の対数変換値における分散分析の結果から算出した。なお、多 重性に関する補正はしていない。それぞれの解析における因子は、処置群、処置群内の被験者、 時期及び薬剤であった。処置群内の被験者をランダム効果として扱っているため、処置群の効果 に関する F 値は、処置群内の被験者に対する処置群のタイプ I の平均平方比として算出された。
対数スケールでの90%信頼区間をオリジナルスケールに変換し、結果を示した。薬剤の力価につ いて補正したCmax 及び AUC(INF)の幾何平均値比の点推定値及び 90%信頼区間を分散分析によ り算出した。Cmax 及び AUC(INF)と同様に AUC(0-T)についても解析した。
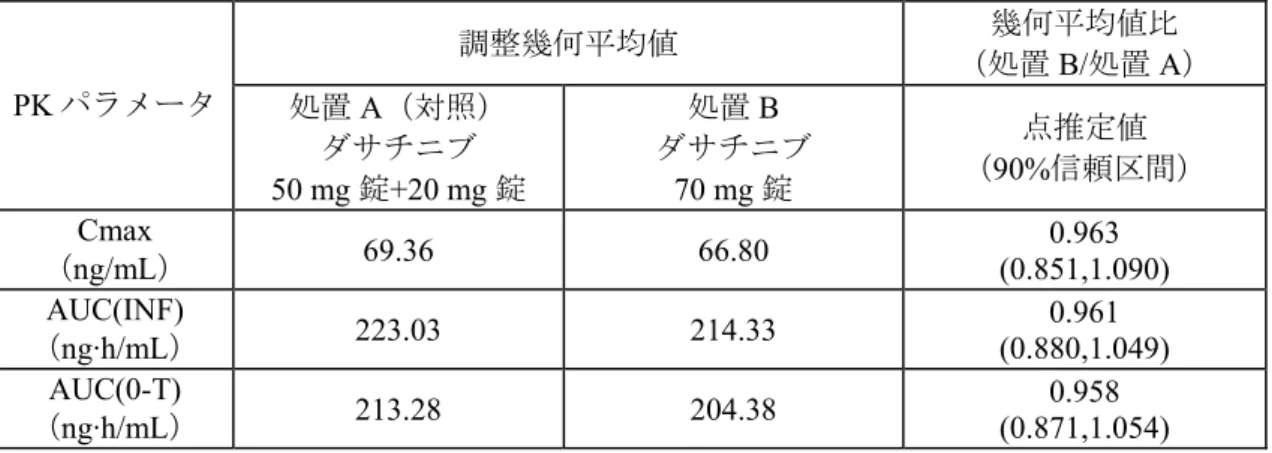
ダサチニブの PK パラメータの要約統計量を表に示した。Cmax、AUC(INF)及び AUC(0-T)は、 幾何平均値及び変動係数で示した。なお、Tmax は中央値及び範囲、t1/2は算術平均値及び標準偏 差で示した。 結果: 被験者背景: 被験者背景の要約を以下に示す。 表 2.1-1: 被験者背景の要約 年齢(歳) 平均値(範囲) 32(19~50) 男性(%) 46 (72) 性別 女性(%) 18 (28) 白人(%) 54 (84) 黒人/アフリカ系(%) 7 (11) 人種 アジア人(%) 3 (5) BMI(kg/m2) 平均値(範囲) 25.8(19.2~31.5) N = 64 薬物動態: ダサチニブのPK パラメータの要約統計量を表 2.1-2 以下に示す。 表 2.1-2: ダサチニブの薬物動態パラメータ(要約統計量) Cmax (ng/mL) AUC(INF) (ng·h/mL) AUC(0-T) (ng·h/mL) Tmax (h) t1/2 (h) 処 置 被験者数 幾何平均値 (変動係数%) 幾何平均値 (変動係数%) 幾何平均値 (変動係数%) 中央値 (最小, 最大) 算術平均値 (標準偏差) 処置A ダサチニブ 50 mg 錠+20 mg 錠 61 (39) 69.39 223.16 (37) 213.42 (38) (0.50, 5.00) 1.00 (0.88) 3.58 処置B ダサチニブ 70 mg 錠 61 (40) 66.83 214.42 (36) 204.48 (37) (0.50, 3.00) 1.00 (1.38) 3.77
Cmax , AUC(INF) 及び AUC(0-T)の対数値について分散分析を行い、これらのパラメータの幾何 平均値比の90%信頼区間を算出した。これらの結果を表 2.1-3 に示す。
表 2.1-3: ダサチニブの Cmax , AUC(INF) 及び AUC(0-T)に関する統計解析結果 調整幾何平均値 幾何平均値比 (処置B/処置 A) PK パラメータ 処置A(対照) ダサチニブ 50 mg 錠+20 mg 錠 処置B ダサチニブ 70 mg 錠 点推定値 (90%信頼区間) Cmax (ng/mL) 69.36 66.80 0.963 (0.851,1.090) AUC(INF) (ng·h/mL) 223.03 214.33 0.961 (0.880,1.049) AUC(0-T) (ng·h/mL) 213.28 204.38 0.958 (0.871,1.054) N = 61/処置群
また、薬剤の力価で補正したCmax, AUC(INF)及び AUC(0-T)の幾何平均値及び幾何平均値比の 点推定値及び90%信頼区間を表 2.1-4 に示す。
表 2.1-4: 力価で補正したダサチニブの Cmax, AUC(INF)及び AUC(0-T)の統計解析結果
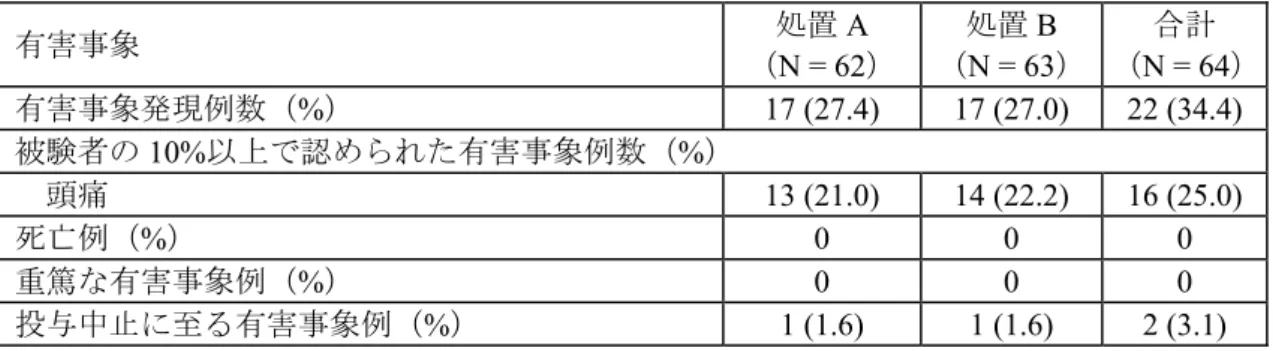
パラメータ 処置A(対照) ダサチニブ 50 mg 錠+20 mg 錠 処置B ダサチニブ 70 mg 錠 幾何平均値比(B/A) 点推定値 (90%信頼区間) ロット番号 05C05064 (50 mg) 05C06213 (20 mg) 05F04871 - 含量(%表示量) 99.1% 99.6% - 補正因子 1.0086 1.0043 - Cmax 幾何平均値 (ng/mL) 69.96 67.09 0.959 (0.847,1.085) AUC(INF)幾何平均値 (ng·h/mL) 224.96 215.25 0.957 (0.876, 1.045) AUC(0-T)幾何平均値 (ng·h/mL) 215.13 205.26 0.954 (0.867, 1.050) N = 61/処置群 Cmax 及び AUC(INF)の幾何平均値比の 90%信頼区間は、薬剤の力価の補正の有無の関わらず予 め設定した範囲(0.80~1.25)に完全に含まれた。したがって、ダサチニブ 50 mg 錠及び 20 mg 錠の組み合わせとダサチニブの70 mg 錠は生物学的に同等であることが裏付けられた。 安全性及び忍容性: 有害事象の要約を表 2.1-5 に示す。
表 2.1-5: 有害事象発現状況 有害事象 処置A (N = 62) 処置B (N = 63) 合計 (N = 64) 有害事象発現例数(%) 17 (27.4) 17 (27.0) 22 (34.4) 被験者の10%以上で認められた有害事象例数(%) 頭痛 13 (21.0) 14 (22.2) 16 (25.0) 死亡例(%) 0 0 0 重篤な有害事象例(%) 0 0 0 投与中止に至る有害事象例(%) 1 (1.6) 1 (1.6) 2 (3.1) 処置A:ダサチニブ 50 mg 錠 + 20 mg 錠同時投与(対照群) 処置B:ダサチニブ 70 mg 錠投与 本治験において、死亡及び重篤な有害事象の発現例は認められなかった。報告された有害事象 はすべて軽度及び中等度であった。ほとんどの有害事象で治験薬との因果関係は「関連あるかも しれない」であった。治験薬投与により発現した有害事象のうち、両処置群で最も高頻度に認め られたのは頭痛であった。16 例(25.0%)の被験者から合計 27 件の頭痛が報告され、これらはす べて軽度(grade 1)であった。頭痛はほとんどが 1 日以内に消失し、持続期間は 5 時間~6 日間 であった。この他に、被験者の10%以上に発現した有害事象は認められなかった。被験者 2 例が、 臨床検査値異常により治験を中止した。このうち1 例は grade 1 の血清ブドウ糖上昇(131 mg/dL; 基準範囲68~112 mg/dL)を示し、14 時間以内に消失した。他の 1 例は grade 1 の血中クレアチン ホスホキナーゼ(CPK)上昇(454 U/L;基準範囲 49~397 U/L)で 3 日以内に消失した。いずれ の被験者も特に治療は必要とせず、治験責任医師により治験薬投与との因果関係はないものと判 断された。
CTCAE(Common Terminology Criteria for Adverse Events)grade において、投与前値と比較して grade 1 と判定された臨床検査値異常が 1 項目以上に認められた被験者数は 22 例(34.4%)であっ たが、数例の被験者にのみ検査値異常が観察された臨床検査項目がほとんどであった。10%以上 の被験者に発現した臨床検査値異常は認められなかった。
ECG 検査では、1 例から 450 msec を超える QTcB(451 msec)が 2 期の処置 A の薬剤投与 2 時間 後に認められた。この被験者の投与前(2 期、1 日目)の QTcB は、445 msec であった。さらに他 の1 例において、投与前(2 期、1 日目)の QTcB が 455 msec を示した。また、1 例から 450 msec を超えるQTcF(454 msec)が 2 期の処置 B の薬剤投与 2 時間後に認められた。この被験者の投与 前(2 期、1 日目)の QTcF は 449 msec であった。
結論: • Cmax 及び AUC(INF)においてダサチニブ 70 mg 錠 1 錠とダサチニブ 50 mg 錠 1 錠及び 20 mg 錠1 錠は生物学的に同等であった。 • 70 mg 錠 1 錠又は 50 mg 錠 1 錠及び 20 mg 錠 1 錠を投与したときの安全性及び忍容性が認め られた。 報告書作成日:20 年 月 日
2.2 Study
CA180-019
試験課題名: 健康な男性被験者における[14C]BMS-354825 のマスバランス、薬物動 態及び代謝
治験責任医師:
治験実施医療機関: Bristol Myers Squibb Clinical Research Center Hamilton, NJ
公表文献 なし 治験期間: 最初の被験者の治験登録日:20 年 月 日 最後の被験者の治験終了日:20 年 月 日 開発のフェーズ: 第1 相 主要目的: 健康成人男子に100 mg(120 μCi)の[14C]BMS-354825を単回経口投与 した時のマスバランス、薬物動態、代謝及び排泄を検討する。 副次目的: 健康成人男子に100 mg(120 μCi)の[14C]BMS-354825を単回経口投与 した時の安全性を検討する。 治験方法: 本試験は非盲検、非ランダム化、単回投与試験であった。合計8 例の健康成人男子を本試験に 登録した。被験者は、本試験に適格であることを判断するためのスクリーニング検査を登録前21 日以内に受診した。被験者は投与開始2 日前の夕方に治験実施医療機関へ入院した。選択基準を 満たし、除外基準に該当しない被験者を登録し、試験前日に被験者番号を割り当てた。試験1 日 目に、全被験者は、総放射能120 μCi に相当する 100 mg の[14C]BMS-354825 溶液の投与を受けた。 試験期間を通じて有害事象を観察した。バイタルサイン、理学的検査、心電図及び臨床検査の 結果を、試験期間を通じて定期的に観察し、安全性を評価した。ダサチニブとその代謝物 BMS-606181 及び総放射能(TRA)の薬物動態及び代謝を検討するため、あらかじめ設定したサ ンプリングポイントで7 日間採血した。各被験者の尿及び糞便を 7 日間又は退院するまで採取し た。1 日目は 12 時間間隔の蓄尿を行い、その後、24 時間間隔の蓄尿を行った。尿中のダサチニブ、 BMS-606181 及び TRA を分析し、また、糞便中の TRA を測定した。尿及び糞便中のその他の代 謝物についても分析した。その結果は、別途作成する報告書に記載する。 治験実施医療機関を退院する前の排便を容易にするため、6 日目の午前中に、下剤であるマグ ネシアミルク(マグネシア®)30 mL を被験者に単回経口投与した。24 時間間隔の蓄尿(1 日目は 12 時間ごとの蓄尿)及び糞便中 TRA を測定したが、測定結果を得るのに尿及び糞便サンプルの 採取から3 日間必要であった。7 日目の午前中に得られた蓄尿及び糞便中 TRA が投与した放射能 の1%以下という条件の下、被験者は試験 10 日目の午前中に蓄尿及び糞便採取終了後、治験実施 医療機関を退院することとした。 被験者数: 登録被験者数 8 薬剤を投与した被験者数 8 試験を中止した被験者数 0
主要な選択基準: 既往歴、理学的検査、12 誘導心電図及び臨床検査により、適格と判断された健康成人男子を本 試験に組み入れた。本試験は放射性同位体で標識した薬剤を使用し、標識体を投与したときの情 報は雄ラットのみを用いた試験から得られていたので、女性は対象外とした。 治験薬、用量、バッチ番号: ラベルバッチ番号 外 観 含 量 製品バッチ番号 5A10945 透明なガラス製バイアルに封入 された白色~微黄白色の粉末 110 mg/バイアル 5A10853 治験薬、用法及び用量、バッチ番号:該当せず。 投与期間:100 mg (120 μCi) の[14C] BMS-354825 を溶液として単回経口投与した。試験期間は 8 ~14 日間であった。 評価基準: 有効性: 該当せず。 安全性及び忍容性: 安全性の評価は、有害事象報告の他、バイタルサイン、心電図、理学的検査及び臨床検査の 結果に対する医学的な評価に基づいて実施した。有害事象は一覧表にし、臨床上の意義並びに その重要性を評価した。 薬物動態: 試験期間中に血液、血漿、尿及び糞便を採取し、これら生体試料中のTRA を測定した。また、 血漿中及び尿中のダサチニブ及びBMS-606181 濃度を測定した。ダサチニブ単回投与後の血漿 中ダサチニブ、BMS-606181 及び TRA 濃度推移から、薬物動態パラメータ(Cmax, Tmax, AUC(INF), t1/2及びCLR(腎クリアランス))を算出した。プロトコールには記載されていなか
ったものの、ダサチニブ、BMS-606181 及び TRA の血漿中濃度データから、AUC(0-T)を算出し た。また、血液中TRA 濃度については測定感度が不十分であったため、プロトコールに記載さ れていたものの薬物動態パラメータは算出できなかった。尿中排泄データから、ダサチニブ、 BMS-606181 及び TRA の尿中排泄率(%UR)を算出した。糞便中排泄データから、TRA の糞 便中排泄率(%FE)を算出した。尿及び糞便中の TRA を合わせた排泄率(%TOTAL)を投与 した放射能量に対する割合として算出した。
血漿中TRA の AUC(INF)に対する血漿中ダサチニブ及び BMS-606181 の AUC(INF)の割合を それぞれ算出した。更に、プロトコールには記載されていなかったものの、Cmax についても 同様に割合を算出した。また、測定感度が不十分であったため血液中TRA の AUC(INF)に対す る血漿中TRA の AUC(INF)の割合は算出できなかった。
薬力学: 該当せず。
統計手法: 被験者数: 当該試験の被験者数は、統計的な検出力を考慮して設定したものではない。被験者6 例では ダサチニブのCmax 及び AUC(INF)の幾何平均値が真の母集団値の±20%以内にある信頼率が、 それぞれ53 及び 60%以上となり、脱落例が出る可能性を考慮して、本治験の被験者数を 8 例 とした。 統計解析: 被験者背景: 人種の度数分布を一覧表にした。年齢、体重、身長及び体格指数(BMI)を一覧表にした。 安全性: 記録された有害事象はすべて、器官分類、基本語及び処置群別に一覧表にした。重篤な有害 事象はいずれも一覧表に記載した。バイタルサイン及び臨床検査値は一覧表にして要約した。 臨床上重要と思われる診察所見、心電図及び臨床検査結果を一覧表にした。心電図の記録は治 験責任医師により評価され、もし異常があれば一覧表に記載した。また、心電図のパラメータ 及びベースラインからの変化の要約統計量を測定時点ごとに一覧表にした。臨床検査値異常は、 処置及び被験者別に一覧表にした。 薬物動態: ダサチニブ、BMS-606181 及び TRA の薬物動態パラメータの要約統計量を一覧表にした。TRA の%UR、%FE 及び%TOTAL を採取間隔及び累積採取期間別に要約した。ダサチニブ及び BMS-606181 の累積尿中排泄率についても要約統計量を示した。ダサチニブ及び BMS-606181 は、一般的に投与後24 時間以内に尿中排泄されるので、採取間隔ごとに要約しなかった。 結果: 被験者背景: 被験者背景の要約を表 2.2-1 に示す。 表 2.2-1 被験者背景の要約 年齢(歳) 平均値(範囲) 30 (21~41) 男性 (%) 8 (100) 性別 女性 (%) 0 (0) 白人 (%) 4 (50) 人種 黒人/アフリカ系(%) 4 (50) BMI(kg/m2) 平均値(範囲) 25.2 (20.0~30.1) N = 8 薬物動態: 薬物動態パラメータは、ノンコンパートメント法により算出した。健康成人男子に 100 mg の[14C] BMS-354825 を単回経口投与した時、血漿中では未変化体が主化合物であったのに対し、
循環代謝物であるBMS-606181 はわずかであった。
血漿中TRA の Cmax に対するダサチニブ及び BMS-606181 の Cmax の割合は、それぞれ 48.8 及び1.38%であり、AUC(INF)ではそれぞれ 29.06 及び 1.34%であった(表 2.2-2)。 薬剤由来の放射能の主たる排泄経路は糞便中であった。[14C]ダサチニブ 100 mg(120 μCi) を単回投与した時、投与後9 日までの糞便及び尿中放射能排泄率の総和の平均値は約 89%であ った(表2.2-3)。糞便中及び尿中へ排泄された放射能は、それぞれ投与量の 85%及び 4%であっ た。ダサチニブ及び BMS-606181 の尿中排泄率は約 1%であった。このことから、ダサチニブ を腎機能障害者に投与した時、用量調整は必要ないものと考えられた。 表 2.2-2: Cmax 及び AUC(INF)の割合の要約統計量 被験者数 比 較 Cmax の割合(%) AUC(INF)の割合(%) BMS-606181/ダサチニブ 2.84 (27) 4.87 (30) ダサチニブ/TRA 48.8 (15) 29.06 (40) N = 8 BMS-606181/TRA 1.38 (41) 1.34 (44) 表 2.2-3: TRA、ダサチニブ及び BMS-606181 の尿及び糞便中排泄率の要約統計量 項 目 排泄率(%) 尿中TRA 3.58 (1.17) 糞便中TRA 85.32 (17.28) 尿中ダサチニブ 0.12 (0.05) 尿中BMS-606181 1.2 (0.49) 平均値(標準偏差)、N = 8 安全性及び忍容性: 有害事象発現状況の要約を表 2.2-4 に示す。
表 2.2-4: 有害事象発現状況の要約 有害事象 ダサチニブ100 mg 投与(N = 8) 有害事象発現例数 6 (75.0) 主たる有害事象例数a 頭痛 倦怠感 5 (62.5) 2 (25.0) 死亡例(%) 0 重篤な有害事象発現例(%) 0 有害事象による試験中止例(%) 0 a 2 つ以上の器官で 2 件以上の有害事象を報告した被験者、又は 1 例の被験者が同一の器官で 2 件以上の有害事 象を報告したケースがあるので、個々の有害事象の発現件数の総数は有害事象を発現した被験者の総数を上回 る。 ( )内の数値はダサチニブ 100mg 投与群 8 例に対する割合(%) 100 mg の[14C] BMS-354825 投与は安全で、忍容性は良好であった。被験者 8 例のうち 6 例で 有害事象が報告されたものの、その頻度は高くなかった。被験者2 例以上に発現した有害事象 は、頭痛と倦怠感のみであった。すべての有害事象は、治験責任医師により軽度から中等度と 評価され、治験薬との因果関係が「関連あるかもしれない」であった。重篤な有害事象は認め られず、試験中止例はなかった。 当該試験において、CTC grade 2 以上の臨床検査値異常は認められなかった。血液学的検査を 含めた試験開始前の検査値と比較して、CTC grade 1 の臨床検査値異常が 3 例の被験者でみられ た。治験責任医師により、1 例の臨床検査値異常(血小板減少)が有害事象であると判断され た。当該試験において、QTc 延長はみられなかった。 結論: • 薬物由来の放射能は主に糞便中へ排泄された。投与後9 日にわたる糞便中及び尿中 TRA 排泄 率はそれぞれ約85%及び 4%であり、合計で約 89%であった。 • ダサチニブ及びBMS-606181 は尿中へわずかに排泄された(投与量の約 1%)。 • 100 mg の[14C] BMS-354825 を単回経口投与した時の血漿中の主化合物は、未変化体であった。 血漿中では、BMS-606181 は微量の代謝物であった。 • 100 mg の[14C] BMS-354825 を単回投与したときの安全性及び忍容性は良好であった。 報告書作成日:20 年 月 日
2.3 Study
CA180-016
治験課題名: 健康成人におけるダサチニブの製剤比較試験 治験責任医師: (1) , (2) 治験実施医療機関: (1) , (2) 公表論文: なし 治験期間: 最初の被験者の治験登録日:20 年 月 日 最後の被験者の治験終了日:20 年 月 日 開発のフェーズ: 第1 相 治験目的: 主要目的: BMS-354825(ダサチニブ)の臨床試験用製剤 5 mg 錠(投与群 B)、20 mg 市販用製剤(投与群 C)、及び 50 mg 市販用製剤(投与群 D)を用いたときの生物学的利用率を、50 mg 臨床試験用製 剤(投与群 A)と比較検討する。 副次目的: ・健康成人にダサチニブ100 mg を単回投与したときの薬物動態(PK)を検討する。 ・健康成人にダサチニブ100 mg を単回投与したときの安全性及び忍容性を検討する。 ・健康成人にダサチニブ100 mg を単回投与したときの QTc 時間に及ぼす影響を検討する。 治験方法: 本試験は、健康成人における非盲検、ランダム化、4 投与群による単回投与試験であり、臨床 試験用と市販用ダサチニブ製剤を比較する試験であった。被験者は以下の4 投与群のいずれかに 割り付けられ、1 日目にダサチニブ 100mg が投与された。 投与群 A: 50 mg × 2 ダサチニブ 臨床試験用製剤 投与群 B: 5 mg × 20 ダサチニブ 臨床試験用製剤 投与群 C: 20 mg × 5 ダサチニブ 市販用製剤 投与群 D: 50 mg × 2 ダサチニブ 市販用製剤 被験者は、投与開始2 日前に実施医療機関に入院し、予定された内容を終了するまで拘束され、 2 日目に退院した。 PK 解析のため、ダサチニブ投与後のあらかじめ設定された時間に血液と尿 サンプルを採取した。安全性は、有害事象、臨床検査、バイタルサイン及び心電図(ECGs)所見に より評価した。被験者数: 被験者合計75 例(投与群 A、C 及び D はそれぞれ 19 例、投与群 B は 18 例)が登録され、脱 落・中止例はなかった。 主要な選択基準: 選択/除外基準を満たした健康成人 試験製剤、投与量、投与方法及び製造番号: ダサチニブ 投与量 投与方法 製造番号 臨床試験用製剤(錠剤) 単回, 5 mg 錠× 20 経口 4C88975 市販用製剤(錠剤) 単回, 20 mg 錠× 5 経口 4L77202 市販用製剤(錠剤) 単回, 50 mg 錠× 2 経口 4L77205 標準製剤、投与量、投与方法及び製造番号: ダサチニブ 投与量 投与方法 製造番号 臨床試験用製剤(錠剤) 単回, 50 mg 錠× 2 経口 4C91969 投与期間: 単回投与 評価基準: 有効性: 該当せず。 薬物動態: ダサチニブ経口投与後の血漿中濃度推移から、ダサチニブ単回投与時のPK パラメータ(Cmax, Tmax, AUC(INF), AUC(0-T), t1/2及び尿中排泄率)を算出した。
主要な安全性: 安全性評価は、有害事象、バイタルサイン、ECG、理学的検査及び臨床検査の結果の医学的な 評価に基づいて行った。有害事象は一覧表にし、臨床上の意義及びその重要性について検討した。 副次的安全性: QTc 時間、PR 間隔及びそれらのベースライン(投与開始前日の値)からの変化を評価した。 薬力学: 該当せず。 ファーマコゲノミクス: 該当せず。
統計手法: 被験者数: 被験者数は統計学的検出力に基づいて設定されていないが、被験者数が20 例の場合、Cmax 及 びAUC(INF)の、投与群 A に対する投与群 B、投与群 C あるいは投与群 D の幾何平均値比の推定 値が真の母集団値の±20%以内である信頼率はそれぞれ 75%及び 80%であった。 解析: 被験者背景: 性別及び人種別被験者数を製剤ごと(投与群 A、B、C 又は D)に表に示し、年齢、体重、身長及 び体格指数(BMI)の要約統計量を表に示した。 安全性: すべての有害事象について、投与群ごとに、器官分類、基本語及び製剤ごとに要約し、一覧表 にした。バイタルサイン及び臨床検査値は、製剤ごとに一覧表にし、要約した。臨床上重要と思 われる診察所見、ECG 所見及び臨床検査値を表にして示した。ダサチニブの QTc 間隔への影響に ついては、QTc 最大値及びベースラインからの QTc 変化量の最大値の頻度分布、並びにこれらの 要約統計量により評価した。さらに、ベースラインからのQTc 平均変化量の経時的推移をプロッ トし、血漿中ダサチニブ濃度に対するQTc 及びベースラインからの QTc 変化量の散布図を作成し た。また、ダサチニブのQTc 間隔に対する影響を、ダサチニブの曝露量(Cmax 及び平均濃度) に対するQTc 測定値(ダサチニブ濃度が Tmax 時の QTc 変化量及び 24 時間の QTc 平均変化量) の線形回帰分析により評価した。上記QTc 解析に加えて、最大心拍数及びベースラインからの最 大心拍数変化量並びに最大PR 及びベースラインからの最大 PR 変化量について頻度分布及び要約 統計量を示し、散布図を作成した。 薬物動態: 投与群A に対して投与群 B、C 及び D を比較するため、log(Cmax)、log(AUC[0-T])及び log(AUC [INF])による分散分析を行った。製剤(A、B、C、又は D)を分散分析の要因とした。製剤ごと の幾何平均値比の点推定値及び 90%信頼区間を分散分析結果から算出した。 なお、多重性に関 する補正は実施しなかった。製剤ごとにダサチニブのPK パラメータの要約統計量を表にした。
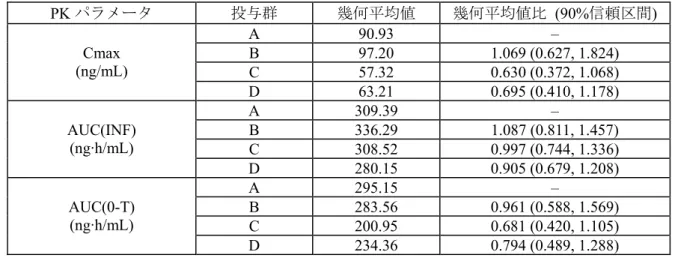
結果: 被験者背景: 被験者背景の要約を表 2.3-1 に示す。 表 2.3-1: 被験者背景の要約 投与群A 投与群B 投与群C 投与群D 50 mg 錠 x 2 5 mg 錠 x 20 20 mg 錠 x 5 50 mg 錠 x 2 臨床試験用製剤 臨床試験用製剤 市販用製剤 市販用製剤 N = 19 N = 18 N = 19 N = 19 年齢 平均値 35 40 37 41 (範囲) (19~47) (24~48) (18~49) (20~50) 性別 男性(%) 13 (68) 13 (72) 12 (63) 14 (74) 女性(%) 6 (32) 5 (28) 7 (37) 5 (26) 人種 白人(%) 15 (79) 8 (44) 13 (68) 11 (58) 黒人/アフリカ系(%) 4 (21) 8 (44) 4 (21) 8 (42) アジア人(%) 0 0 1 (5) 0 その他 0 2 (11) 1 (5) 0 BMI (kg/m2) 平均値 25.6 26.2 25.2 25.8 (範囲) (20.7~31.2) (19.9~30.8) (20.7~31.8) (20.9~31.7) 薬物動態学的評価: PK パラメータはノンコンパートメント解析により算出した。 表 2.3-2: ダサチニブの薬物動態パラメータ(要約統計量) Cmax (ng/mL) AUC(INF) (ng·h/mL) AUC(0-T) (ng·h/mL) Tmax (h) t1/2 (h) 尿中排泄率 (%) 投 与 群 被験者数 幾何平均値 (変動係数%) 幾何平均値 (変動係数%) 幾何平均値 (変動係数%) 中央値 (最小, 最大) 算術平均値 (標準偏差) 算術平均値 (標準偏差) A 18 90.93 (47) 309.39 (48) 295.15 (49) (0.50, 2.00)1.00 (1.14) 3.79 (0.08) 0.14 B 18 97.20 (56) 336.29(47) a 283.56 (55) (0.50, 5.00)1.25 (0.97) 4.06 (0.07) 0.16 b C 19 57.32 (60) 308.52 (45) a 200.95 (59) (0.50, 4.00)1.00 (3.00) 4.83 (0.06) 0.12 b D 19 63.21 (63) 280.15 (49) c 234.36 (56) (0.50, 4.00)1.00 (1.98) 4.45 (0.08) 0.13 a a N = 17, b N = 16, c N = 18 投与群A:臨床試験用製剤 50 mg 錠 x 2 投与群B:臨床試験用製剤 5 mg 錠 x 20 投与群C:市販用製剤 20 mg 錠 x 5 投与群D:市販用製剤 50 mg 錠 x 2
投与群A 及び B の Cmax 及び AUC(0-T)の幾何平均値比は 100±10%以内であり、これら 2 群の Cmax 及び AUC(0-T)は類似していた。投与群 C 及び D の Cmax 及び AUC(0-T)の幾何平均値は投 与群A に比較して低値を示したが、Cmax と AUC(0-T)の個々の値の分布範囲は標準治療群(投与 群A)のそれと同程度であった。
表 2.3-3: ダサチニブの Cmax, AUC(INF)及び AUC(0-T)に関する統計解析結果
PK パラメータ 投与群 幾何平均値 幾何平均値比 (90%信頼区間) A 90.93 – B 97.20 1.069 (0.627, 1.824) C 57.32 0.630 (0.372, 1.068) Cmax (ng/mL) D 63.21 0.695 (0.410, 1.178) A 309.39 – B 336.29 1.087 (0.811, 1.457) C 308.52 0.997 (0.744, 1.336) AUC(INF) (ng·h/mL) D 280.15 0.905 (0.679, 1.208) A 295.15 – B 283.56 0.961 (0.588, 1.569) C 200.95 0.681 (0.420, 1.105) AUC(0-T) (ng·h/mL) D 234.36 0.794 (0.489, 1.288)
Cmax 及び AUC(0-T):N = 74、 AUC(INF):N = 70 投与群A:臨床試験用製剤 50 mg 錠 x 2
投与群B:臨床試験用製剤 5 mg 錠 x 20 投与群C:市販用製剤 20 mg 錠 x 5 投与群D:市販用製剤 50 mg 錠 x 2
安全性: 安全性成績の要約を表 2.3-4 に示す。 表 2.3-4: 有害事象発現状況の要約 有害事象 投与群 A 投与群 B 投与群 C 投与群 D 合計 (N = 19) (N = 18) (N = 19) (N = 19) (N = 75) 有害事象発現例数 (%) 12 (63.2) 13 (72.2) 13 (68.4) 10 (52.6) 48 (64.0) 高頻度に認められた有害事象例数 (%) 頭痛 7 (36.8) 12 (66.7) 10 (52.6) 10 (52.6) 39 (52) 筋痛 5 (26.3) 4 (22.2) 7 (36.8) 3 (15.8) 19 (25.3) 悪心 2 (10.5) 4 (22.2) 4 (21.1) 4 (21.1) 14 (18.7) 嘔吐 1 (5.3) 2 (11.1) 2 (10.5) 3 (15.8) 8 (10.7) 死亡例 (%) 0 0 0 0 0 重篤な有害事象例 (%) 0 0 0 0 0 投与中止例 (%) 0 0 0 0 0 投与群A:臨床試験用製剤 50 mg 錠 x 2 投与群B:臨床試験用製剤 5 mg 錠 x 20 投与群C:市販用製剤 20 mg 錠 x 5 投与群D:市販用製剤 50 mg 錠 x 2 有害事象はいずれも軽度から中等度であり、ほとんどの有害事象で治験薬との因果関係は「関 連あるかもしれない」であった。頭痛が認められた39 例中 32 例(82.1%)は軽度(grade 1)で あり、7 例(17.9%)は中等度(grade 2)であった。軽度の頭痛はいずれも治療は必要なかったが、 中等度の頭痛では全例がアセトアミノフェンによる治療を受けた。頭痛発現例は、回復までに 3 日から5 日を要した 3 例を除き、すべて 1 日以内に回復した。投与前値と比較した臨床検査値異 常が1 件以上報告されたのは 18 例(24.3%)であり、そのうち 2 例以上に報告された臨床検査値 異常はリンパ球減少のみであり、6 例(8.1%)であった。臨床検査値異常はすべて軽度(grade 1) であり、有害事象あるいは臨床的に重要な所見とは判断されていない。 投与前日に、投与群 C の 1 例が QTcLP(対数線形性回帰に基づく補正 QT 値、log-linear population correction for QT)が最大値 451msec を示したが、投与後の QTcLP の最大値が 450msec を超える 被験者は認められなかった。また、4 例で QTcLP のベースラインからの最大変化量が 30~60msec を示した。ダサチニブTmax における QTcLP のベースラインからの変化量に、Cmax の増加に伴 う増加は認められなかった。また、ダサチニブ平均血漿中濃度(Cavg(0-T))の増加に伴い、QTcLP (0-24 時間平均値)のベースラインからの変化量の減少が認められた。
結論: • 投与群B(20x5mg 臨床試験用製剤)の投与群 A(2x50mg 臨床試験用で標準製剤)に対する Cmax 及び AUC(0-T)の差は±10%以内であり、曝露量は投与群 A 及び B で類似していた。 • 投与群C(5x20mg の市販用製剤)及び D(2x50mg 市販用製剤)の Cmax と AUC(0-T)の幾何 平均値は、投与群 A に比較して低値を示したが、Cmax と AUC(0-T)の分布範囲は投与群 A と同程度であった。 • 健康成人にダサチニブ100mg を単回投与したとき、安全性及び忍容性は良好であった。 • ダサチニブのTmax における QTcLP のベースラインからの変化量に、Cmax の増加に伴う増 加は認められなかった。 • ダサチニブ血漿中濃度(Cavg(0-T))の増加に伴い、QTcLP(0-24 時間平均値)のベースライ ンからの変化量に有意な減少が認められた。 報告書作成日:20 年 月 日
2.4 Study
CA180-034
イマチニブに抵抗性又は不耐容の慢性期慢性骨髄性白血病を対象としたダサチニブの 1 回 50 mg 又は 70 mg の 1 日 2 回投与あるいは 1 回 100 mg 又は 140 mg の 1 日 1 回投与 2 x 2 比較試験 -中間成績- 試験方法の概略を表 2.4-1 に示す。 表 2.4-1 試験方法の概略 項目 内容 治験の相 第Ⅲ相 治験の目的 主要目的: イマチニブ抵抗性の慢性期CML 患者に、ダサチニブを 1 日 1 回投与(QD)及び 1 日 2 回投与(BID) のスケジュールで少なくとも6 ヵ月間投与したときの MCyR 率を比較する。6 ヵ月後の MCyR 率 の差(MCyRRQD-MCyRRBID)の95%信頼区間の下限が-15%以上の場合、1 日 1 回投与の有効性は、 1 日 2 回投与に劣らないと判断する。 副次目的: 1.イマチニブ抵抗性患者における MCyR 率を 1 日用量 100mg と 140mg で比較する。 2.イマチニブ抵抗性患者において、投与スケジュール別、1 日投与量別及び各投与群別に以下を 検討する。 ・MCyR 率及び CHR 率を推定する。 ・MCyR あるいは CHR に到達するまでの期間及び持続期間を推定する。 ・PFS 及び OS を評価する。 ・イマチニブ不耐容患者におけるMCyR 及び CHR を評価する。 3.全例を対象に以下を評価する。 ・投与スケジュール、1 日投与量及び各投与群別にダサチニブの安全性を評価する。 ・各投与スケジュール、各1 日投与量、各投与群別に、体液貯留、胸水/心嚢液貯留、骨髄抑制、 有害事象による減量の発現率を投与スケジュール間及び1 日投与量間で比較する。4.薬物体内動態を検討し、投与群別に QoL(Health Utility measurements)を評価する。 5.BCR-ABL 遺伝子の突然変異及び発現量を検討する。 治験 デザイン 無作為化、オープンラベル試験 本治験の投与群は下記のように、2 要因、計 4 群からなる。 投与スケジュール 1 日 1 回投与 1 日 2 回投与 100 mg グループ1 (100 mg) グループ2 (50 mg) 1 日投与量 140 mg グループ3 (140 mg) グループ4 (70 mg) イマチニブ抵抗性と不耐容は各群に均一に割付け、1:1:1:1 のブロック無作為化を用い、1 群あた りイマチニブ抵抗性87 例を登録する。 対象疾患 対象疾患:イマチニブに初期又は獲得抵抗性、あるいは不耐容の18 歳以上の慢性期 CML 患者 選択/ 除外基準 選択基準 1. 以下の基準のいずれかに該当する慢性期 CML。 ①末梢血及び骨髄中の芽球が15%未満。 ②末梢血及び骨髄中の芽球と前骨髄球の和が30%未満。 ③末梢血中の好塩基球が20%未満。 ④血小板数が100,000/mm3以上(前治療の副作用の影響による場合を除く)。 ⑤髄外浸潤がない(肝・脾を除く)。 2. 以下の基準のいずれかに該当するイマチニブ抵抗性。 a.初期抵抗性 ・イマチニブ1 日 400mg~800mg の投与を 4 週間以上行っても白血球の減少がみられない。 ・イマチニブ1 日 400mg 以上の投与を 3 ヵ月間以上行っても CHR が得られない。 ・イマチニブ1 日 400mg 以上の投与を 6 ヵ月間以上行っても MCyR が得られない。 ・イマチニブ1 日 400mg 以上の投与を 12 ヵ月間以上行っても CCyR が得られない。
表 2.4-1 試験方法の概略 (つづき) 項目 内容 選択/ 除外基準 b.獲得抵抗性 ・イマチニブ治療によりMCyR が得られた後、Ph+の分裂中期細胞が 30%以上増加して MCyR の基 準を満たさなくなった。 ・10%以上の Ph+の分裂中期細胞の増加を伴う分子生物学的効果の消失。 ・イマチニブ治療によりMCyR が得られたが、BCR-ABL 遺伝子に新たな変異がみつかった。 ・イマチニブ治療によりCHR が得られた後、2 週間以上の間隔をおいた一連の血液検査のすべてで 白血球数が10,000/mm3を超えて増加し、CHR の基準を満たさなくなった。 c.以下に定義するイマチニブ不耐容 ・イマチニブ400 mg/日以下の投与中に因果関係が否定できない Grade 3 以上の毒性が発現し、イマ チニブの投与を中止した。 イマチニブ400 mg/日では忍容性が認められたものの、CCyR が得られず、600 mg/日以上の増量 が忍容できない場合はイマチニブ抵抗性とする。 3. 肝機能が十分保持されていること。 ①総ビリルビン値が施設正常値上限の2 倍以内であること。 ②ALT 及び AST が施設正常値上限の 2.5 倍以内であること。 4. 十分な腎機能を有し、血清クレアチニン値が正常値上限の 1.5 倍以下であること。 5. 血清カリウム及びマグネシウムが正常範囲内で、血清カルシウムが正常値下限以上であること。 6. 12 ヵ月以上の生存可能であり、ECOG の PS が 0 又は 2 であること。 除外基準 1. 観察期間において、移植が可能かつ移植を希望している患者。 2. 治験開始 1 ヵ月前から終了 3 ヵ月後までの期間、適切な避妊法を実施する意思がないか、あるい は実施することが不可能な妊娠の可能性がある女性。 3. 妊婦及び授乳婦。 4. 治験薬投与開始前に行った妊娠検査の結果が陽性である女性。 5. パートナーが妊娠可能な女性であって、治験期間中及び治験終了後少なくとも 3 ヵ月間、適切な 避妊法を用いる意思がないか、あるいは用いることができない男性。 6. ダサチニブの投与に障害となる重度の合併症や感染症がある患者。 7. コントロール不良又は重度の心疾患。 8. CML に関連しない出血性疾患の既往。 9. CML 以外の治療困難な悪性腫瘍。 10.これまでに移行期又は急性期 CML と診断されたことがある患者。 被験者数 724 例が登録され、670 例が無作為化され、各投与群に割り付られた。うち、662 例がダサチニブの 投与を受けた。 投与方法 1. 使用薬剤 ダサチニブ錠:20 mg 錠及び 50 mg 錠 2. 用法、用量及び投与期間 1 回 50 mg 又は 70 mg の 1 日 2 回投与、あるいは 1 回 100 mg 又は 140 mg の 1 日 1 回投与を開始 用量とし、疾患の増悪がみられた場合、あるいは副作用が発現した場合、100 mg QD では 1 回 80 ~140 mg、140 mg QD では 1 回 80~180 mg まで、50 mg BID では 1 回 40~70 mg まで、70 mg BID では1 回 40~90 mg までの範囲で増減量を可とした。但し投与回数の変更は行わなかった。投与は、 用量調節にも関わらず、疾患の増悪あるいは許容できない副作用の発現がみられるまで、又は投与 中止基準に該当するまで継続する。 有効性・ 安全性の 評価項目 有効性 主要評価項目:6 ヵ月間以上の観察期間における MCyR 率 細胞遺伝学的効果は、可能な限り20 個以上の骨髄試料中の分裂中期細胞により評価する。細 胞遺伝学的効果に関する定義を以下に示す。 細胞遺伝学的寛解の分類 骨髄中分裂中期細胞におけるPh+染色体の割合 CCyR 0% PCyR > 0%、< 35% Minor CyR > 35%、< 65% Minimal CyR > 65%、< 95% No Response > 95%、< 100% 上記のCCyR と PCyR を合わせて MCyR とする。
表 2.4-1 試験方法の概略 (つづき)
項目 内容
有効性・安 全性の評価 項目
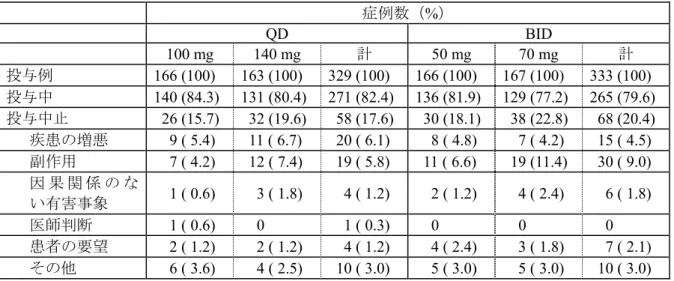
副次評価項目:CHR 率、MCyR が得られるまでの期間、MCyR 及び CHR の持続期間、PFS、OS 血液学的効果:以下の基準のすべてに該当し、当該状態を4 週間以上持続した場合に、CHR を 得たと判断する。 ・白血球数が施設上限以下 ・血小板数が450,000/mm3未満 ・末梢血中に芽球又は前骨髄球を認めない ・末梢血中の骨髄球及び後骨髄球の和が5%未満 ・末梢血中の好塩基球が20%未満 ・肝腫大及び脾腫を含む髄外白血病を認めない 安全性:有害事象、臨床検査値の異常変動、有害事象による投与中止・減量・中断 解析方法 解析対象 有効性評価対象例:全割付症例 安全性評価対象例:全投与症例 1 日 1 回投与と 1 日 2 回投与、及び 1 日 100 mg と 140 mg における MCyR 率の差を 95%信頼区 間とともに算出する。1 日 1 回投与と 1 日 2 回投与の差の信頼区間の下限値が-15%以上の場合、 1 日 1 回投与は 1 日 2 回投与に比較して非劣性であるとした。 CHR 率は投与スケジュール別、1 日投与量別及び各投与群別に算出する。MCyR 及び CHR が 得られるまでの期間、持続期間、PFS、OS については、Kaplan-Meier 法を用いて中央値及び 95% 信頼区間を求める。 安全性:有害事象はNCI CTC Version 3.0 に基づいて重症度を判定する。有害事象のコーディン グにはMedDRA Version 9.1 を用いる。 治験期間 2005 年 7 月 13 日から 2.4.1 症例の内訳 症例の内訳を図 2.4-1 及び表 2.4-2 に示す。 図 2.4-1 症例の内訳 組み入れ症例数 N = 724 無作為化せず N = 54 無作為化 N = 670 1 日 1 回投与 N = 334 1 日 2 回投与 N = 336 100 mg 投与 N = 167 140 mg 投与 N = 167 50 mg 2 回投与 N = 168 70 mg 2 回投与 N = 168 投与せず N = 1 不適格 投与せず N = 4 同意撤回2 不適格2 投与せず N = 2 不適格 同意撤回 投与せず N = 1 不適格 100 mg QD 解析対象 N=166 140 mg QD 解析対象 N=163 100 mg BID 解析対象 N=166 140 mg BID 解析対象 N=167
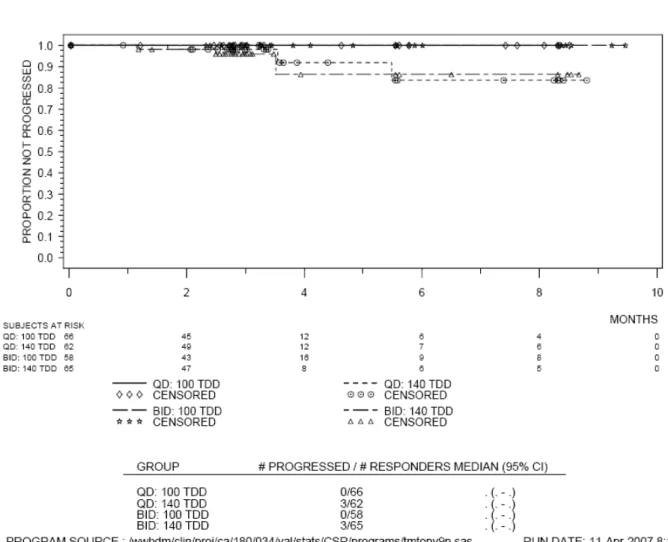
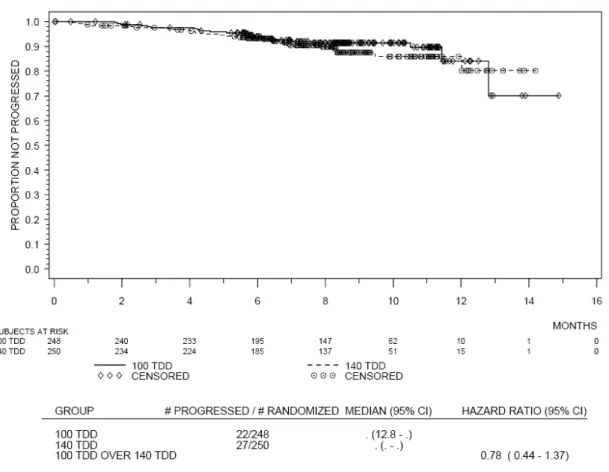
表 2.4-2: 症例の内訳 症例数(%) QD BID 100 mg 140 mg 計 50 mg 70 mg 計 投与例 166 (100) 163 (100) 329 (100) 166 (100) 167 (100) 333 (100) 投与中 140 (84.3) 131 (80.4) 271 (82.4) 136 (81.9) 129 (77.2) 265 (79.6) 投与中止 26 (15.7) 32 (19.6) 58 (17.6) 30 (18.1) 38 (22.8) 68 (20.4) 疾患の増悪 9 ( 5.4) 11 ( 6.7) 20 ( 6.1) 8 ( 4.8) 7 ( 4.2) 15 ( 4.5) 副作用 7 ( 4.2) 12 ( 7.4) 19 ( 5.8) 11 ( 6.6) 19 (11.4) 30 ( 9.0) 因 果 関 係 の な い有害事象 1 ( 0.6) 3 ( 1.8) 4 ( 1.2) 2 ( 1.2) 4 ( 2.4) 6 ( 1.8) 医師判断 1 ( 0.6) 0 1 ( 0.3) 0 0 0 患者の要望 2 ( 1.2) 2 ( 1.2) 4 ( 1.2) 4 ( 2.4) 3 ( 1.8) 7 ( 2.1) その他 6 ( 3.6) 4 ( 2.5) 10 ( 3.0) 5 ( 3.0) 5 ( 3.0) 10 ( 3.0) 本治験では2005 年 7 月 13 日から 20 年 月 日までの間に計724 例が登録され、うち 670 例 が4 つの治療群のいずれかに無作為に割り付けられた。イマチニブ抵抗性例及び不耐容例が 1 日 1 回投与群及び 1 日 2 回投与群間、さらに 1 日 100 mg 投与群及び 1 日 140 mg 投与群間で均一に なるよう割付を行った。 観察期間が8 ヵ月以上になった*データ締切日の時点で投与継続中の症例は QD 群及び BID 群 で、それぞれ82%及び 80%であった。1 日用量別では、100 mg 群及び 140 mg 群で、それぞれ 83% 及び79%であり、投与群別では 100 mg QD 群が最も治療継続中の症例が多かった。 投与中止例及び中止理由に群間で大きな差は見られなかったが、ダサチニブと因果関係の否定 できない有害事象による中止は70 mg BID 群で高かった。 2.4.2 人口統計学的特性 解析対象症例の人口統計学的特性を表 2.4-3 に示す。 表 2.4-3 人口統計学的特性 QD BID 100 mg 140 mg 計 50 mg 70 mg 計 N = 167 N = 167 N = 334 N = 168 N = 168 N = 336 年齢 平均 54.6 53.7 54.1 53.3 53.7 53.5 中央値 56 54 55 55 54.5 55 最小-最大 20 - 78 20 - 84 20 - 84 21 - 84 18 - 83 18 - 84 SD 13.6 15.0 14.3 14.6 15.0 14.8 < 21 1 ( 0.6) 3 ( 1.8) 4 ( 1.2) 0 3 ( 1.8) 3 ( 0.9) 21 - 45 40 (24.0) 50 (29.9) 90 (26.9) 47 (28.0) 45 (26.8) 92 (27.4) 46 - 65 80 (47.9) 75 (44.9) 155 (46.4) 83 (49.4) 77 (45.8) 160 (47.6) 66 - 75 43 (25.7) 26 (15.6) 69 (20.7) 30 (17.9) 34 (20.2) 64 (19.0) > 75 3 ( 1.8) 13 ( 7.8) 16 ( 4.8) 8 ( 4.8) 9 ( 5.4) 17 ( 5.1) 不明 0 0 0 0 0 0 *新薬承認情報提供時に置き換え
表 2.4-3 人口統計学的特性 (つづき) QD BID 100 mg 140 mg 計 50 mg 70 mg 計 性別 男性 84 (50.3) 70 (41.9) 154 (46.1) 85 (50.6) 77 (45.8) 162 (48.2) 女性 83 (49.7) 97 (58.1) 180 (53.9) 83 (49.4) 91 (54.2) 174 (51.8) 人種 白人 141 (84.4) 125 (74.9) 266 (79.6) 135 (80.4) 142 (84.5) 277 (82.4) 黒人 8 ( 4.8) 9 ( 5.4) 17 ( 5.1) 10 ( 6.0) 10 ( 6.0) 20 ( 6.0) アメリカ先 住民 0 0 0 1 ( 0.6) 0 1 ( 0.3) アジア人 12 ( 7.2) 25 (15.0) 37 (11.1) 17 (10.1) 14 ( 8.3) 31 ( 9.2) ハワイ、太 平洋諸島 0 0 0 0 0 0 その他 5 ( 3.0) 7 ( 4.2) 12 ( 3.6) 4 ( 2.4) 2 ( 1.2) 6 ( 1.8) 不明 1 ( 0.6) 1 ( 0.6) 2 ( 0.6) 1 ( 0.6) 0 1 ( 0.3) 民族 ラテン 5 ( 3.0) 12 ( 7.2) 17 ( 5.1) 5 ( 3.0) 4 ( 2.4) 9 ( 2.7) 非ラテン 53 (31.7) 48 (28.7) 101 (30.2) 45 (26.8) 40 (23.8) 85 (25.3) 不明 109 (65.3) 107 (64.1) 216 (64.7) 118 (70.2) 124 (73.8) 242 (72.0) PS 0 119 (71.3) 115 (68.9) 234 (70.1) 132 (78.6) 111 (66.1) 243 (72.3) 1 44 (26.3) 51 (30.5) 95 (28.4) 33 (19.6) 53 (31.5) 86 (25.6) 2 4 ( 2.4) 1 ( 0.6) 5 ( 1.5) 3 ( 1.8) 4 ( 2.4) 7 ( 2.1) 3 0 0 0 0 0 0 不明 0 0 0 0 0 0 イマチニブに対する感受性 初期抵抗性 75 (44.9) 78 (46.7) 153 (45.8) 88 (52.4) 82 (48.8) 170 (50.6) 獲得抵抗性 49 (29.3) 45 (26.9) 94 (28.1) 36 (21.4) 45 (26.8) 81 (24.1) 不耐容 43 (25.7) 44 (26.3) 87 (26.0) 44 (26.2) 41 (24.4) 85 (25.3) 年齢の中央値はQD 群及び BID 群とも 55 歳であり、年齢構成及び男女比も各投与群でほぼ均 一に分布していた。症例のほとんど(81%)は白人であり、その他の人種もほぼ群間で均一であ った。ECOG の PS は QD 群では 99%、BID 群では 98%が 0 又は 1 であった。 イマチニブに対する感受性では、初期抵抗性がQD 群及び BID 群でそれぞれ 46%及び 51%、獲 得抵抗性が28%及び 24%、イマチニブ不耐容がそれぞれ 26%及び 25%であった。1 日投与量ごと の比較では、100 mg 投与群及び 140 mg 投与群で、初期抵抗性がそれぞれ 49%及び 48%、獲得抵 抗性が25%及び 27%、イマチニブ不耐容がそれぞれ 26%及び 25%であった。 病歴及び前治療について、表 2.4-4 に示す。
表 2.4-4 病歴及び前治療 症例数(%) QD BID 100 mg 140 mg 計 50 mg 70 mg 計 N = 167 N = 167 N = 334 N = 168 N = 168 N = 336 診断からの期間(月) 中央値 55.0 56.0 55.5 50.9 53.0 51.5 最小-最大 1.61 - 250.81 0.89 - 227.06 0.89 - 250.81 4.37 - 212.21 1.22 - 245.52 1.22 - 245.52 前治療 骨髄移植 10 ( 6.0) 5 ( 3.0) 15 ( 4.5) 13 ( 7.7) 7 ( 4.2) 20 ( 6.0) 放射線療法 0 2 ( 1.2) 2 ( 0.6) 4 ( 2.4) 0 4 ( 1.2) 薬剤 141 (84.4) 147 (88.0) 288 (86.2) 150 (89.3) 147 (87.5) 297 (88.4) 化学療法 39 (23.4) 41 (24.6) 80 (24.0) 52 (31.0) 43 (25.6) 95 (28.3) インターフェロン 87 (52.1) 93 (55.7) 180 (53.9) 87 (51.8) 82 (48.8) 169 (50.3) ヒドロキシカルバ ミド/anagrelide 125 (74.9) 131 (78.4) 256 (76.6) 130 (77.4) 130 (77.4) 260 (77.4) 本CA180-034 試験に登録された慢性期 CML 患者は長期の前治療を受けていたが、各前治療を 受けた症例の割合は群間でほぼ均一であった。 いずれの投与群でも、ほとんどの症例が化学療法、インターフェロン、ヒドロキシカルバミド /anagrelide を含むイマチニブ以外の薬剤による治療を受けていたが、前治療歴においても群間で 偏りはなかった。 イマチニブ抵抗性又は不耐容の理由を表 2.4-5 に示す。 表 2.4-5 イマチニブ抵抗性又は不耐容の理由 症例数(%) QD BID 100 mg 140 mg 計 50 mg 70 mg 計 N = 167 N = 167 N = 334 N = 168 N = 168 N = 336 イマチニブ初期抵抗性 75 (44.9) 78 (46.7) 153 (45.8) 88 (52.4) 82 (48.8) 170 (50.6) 12 ヵ月後 CCyR 得られず 60 (35.9) 54 (32.3) 114 (34.1) 67 (39.9) 63 (37.5) 130 (38.7) 3 ヵ月後 CHR 得られず 5 ( 3.0) 14 ( 8.4) 19 ( 5.7) 4 ( 2.4) 7 ( 4.2) 11 ( 3.3) 4 週後白血球減少せず 1 ( 0.6) 2 ( 1.2) 3 ( 0.9) 1 ( 0.6) 3 ( 1.8) 4 ( 1.2) 6 ヵ月後 MCyR 得られず 36 (21.6) 38 (22.8) 74 (22.2) 45 (26.8) 44 (26.2) 89 (26.5) イマチニブ獲得抵抗性 49 (29.3) 45 (26.9) 94 (28.1) 36 (21.4) 45 (26.8) 81 (24.1) CHR 消失 15 ( 9.0) 21 (12.6) 36 (10.8) 14 ( 8.3) 18 (10.7) 32 ( 9.5) MCyR 消失(ABL 変異出現) 2 ( 1.2) 4 ( 2.4) 6 ( 1.8) 4 ( 2.4) 7 ( 4.2) 11 ( 3.3) MCyR 消失 (Ph+ 30%以上の増加) 27 (16.2) 21 (12.6) 48 (14.4) 17 (10.1) 24 (14.3) 41 (12.2) 分子遺伝学的効果消失 (Ph+ 10%以上の増加) 7 ( 4.2) 12 ( 7.2) 19 ( 5.7) 6 ( 3.6) 6 ( 3.6) 12 ( 3.6)
表 2.4-5 イマチニブ抵抗性又は不耐容の理由 (つづき) 症例数(%) QD BID 100 mg 140 mg 計 50 mg 70 mg 計 N = 167 N = 167 N = 334 N = 168 N = 168 N = 336 イマチニブ不耐容 43 (25.7) 44 (26.3) 87 (26.0) 44 (26.2) 41 (24.4) 85 (25.3) 胞隔炎 0 0 0 1 ( 0.6) 0 1 ( 0.3) 貧血 0 0 0 1 ( 0.6) 0 1 ( 0.3) 関節痛/筋痛 3 ( 1.8) 3 ( 1.8) 6 ( 1.8) 2 ( 1.2) 4 ( 2.4) 6 ( 1.8) 骨痛 0 1 ( 0.6) 1 ( 0.3) 0 1 ( 0.6) 1 ( 0.3) 細気管支炎 0 0 0 0 1 ( 0.6) 1 ( 0.3) うつ病 0 0 0 1 ( 0.6) 0 1 ( 0.3) 好酸球性肺炎 0 0 0 0 1 ( 0.6) 1 ( 0.3) 疲労 1 ( 0.6) 0 1 ( 0.3) 0 0 0 疲労/片頭痛 0 0 0 0 1 ( 0.6) 1 ( 0.3) 体液貯留 1 ( 0.6) 4 ( 2.4) 5 ( 1.5) 1 ( 0.6) 1 ( 0.6) 2 ( 0.6) 消化管症状 2 ( 1.2) 1 ( 0.6) 3 ( 0.9) 6 ( 3.6) 4 ( 2.4) 10 ( 3.0) 頭痛 0 1 ( 0.6) 1 ( 0.3) 0 0 0 肝毒性 6 ( 3.6) 10 ( 6.0) 16 ( 4.8) 8 ( 4.8) 7 ( 4.2) 15 ( 4.5) 肺浸潤 1 ( 0.3) 0 1 ( 0.3) 0 0 0 ニューロパシー 1 ( 0.6) 1 ( 0.6) 2 ( 0.6) 0 0 0 好中球減少症 3 ( 1.8) 1 ( 0.6) 4 ( 1.2) 3 ( 1.8) 0 3 ( 0.9) 好中球/血小板減少症 1 ( 0.6) 2 ( 1.2) 3 ( 0.9) 1 ( 0.6) 3 ( 1.8) 4 ( 1.2) 肺臓炎 1 ( 0.6) 1 ( 0.6) 2 ( 0.6) 0 1 ( 0.6) 1 ( 0.3) 発疹 17 (10.2) 15 ( 9.0) 32 ( 9.6) 13 ( 7.7) 13 ( 7.7) 26 ( 7.7) 血小板減少症 6 ( 3.6) 3 ( 1.8) 9 ( 2.7) 6 ( 3.6) 4 ( 2.4) 10 ( 3.0) 体重増加 0 1 ( 0.6) 1 ( 0.3) 0 0 0 事象名不明 0 0 0 1 ( 0.6) 0 1 ( 0.3) 670 例のうち、3 例がイマチニブ抵抗性、不耐容のいずれにも該当していなかった。1 例は実施 計画書中の基準には該当しなかったが、治験責任医師の判断によりイマチニブ抵抗性として本治 験に組み入れられた。他の2 例はイマチニブ抵抗性として組み入れられたが、イマチニブの投与 期間が43 週であった 1 例と、イマチニブ不耐容として組み入れられたが、イマチニブの投与量が 400 mg/日未満であった 1 例である。 イマチニブ抵抗性の理由に関しても群間で偏りはなく、初期抵抗性の理由として最も多かった ものは、12 ヵ月間の投与にもかかわらず CCyR が得られなかった症例であり、次いで 6 ヵ月間の 投与にもかかわらずMCyR が得られなかった症例が多かった。獲得抵抗性の理由として最も多か ったものは、すべての投与群で30%以上の Ph 染色体陽性細胞の増加を伴う MCyR の消失であっ た。 さらに、イマチニブ不耐容例においても、不耐容の理由に群間で偏りはなかった。 イマチニブの投与量、投与期間及び最良効果について表 2.4-6 に示す。
表 2.4-6 イマチニブの投与量、投与期間及び最良効果 QD BID 100 mg 140 mg 計 50 mg 70 mg 計 N = 167 N = 167 N = 334 N = 168 N = 168 N = 336 < 400 0 0 0 0 0 0 400 - 600 106 (63.5) 111 (66.5) 217 (65.0) 113 (67.3) 111 (66.1) 224 (66.7) > 600 61 (36.5) 55 (32.9) 116 (34.7) 55 (32.7) 56 (33.3) 111 (33.0) 最高投与量 (mg/day) Unknown 0 1 ( 0.6) 1 ( 0.3) 0 1 ( 0.6) 1 ( 0.3) < 1 36 (21.6) 39 (23.4) 75 (22.5) 40 (23.8) 37 (22.0) 77 (22.9) 1 - 3 55 (32.9) 58 (34.7) 113 (33.8) 68 (40.5) 60 (35.7) 128 (38.1) > 3 76 (45.5) 68 (40.7) 144 (43.1) 60 (35.7) 71 (42.3) 131 (39.0) 投与期間 (年) Unknown 0 1 ( 0.6) 1 ( 0.3) 0 0 0 CHR 136 (81.4) 138 (82.6) 274 (82.0) 146 (86.9) 141 (83.9) 287 (85.4) No Response 28 (16.8) 28 (16.8) 56 (16.8) 17 (10.1) 25 (14.9) 42 (12.5) 最良血液学 的効果 N (%) Unknown 3 ( 1.8) 1 ( 0.6) 4 ( 1.2) 5 ( 3.0) 2 ( 1.2) 7 ( 2.1) CCyR 40 (24.0) 41 (24.6) 81 (24.3) 27 (16.1) 36 (21.4) 63 (18.8) PCyR 36 (21.6) 30 (18.0) 66 (19.8) 38 (22.6) 30 (17.9) 68 (20.2) Minimal CyR 21 (12.6) 17 (10.2) 38 (11.4) 22 (13.1) 15 ( 8.9) 37 (11.0) Minor CyR 15 ( 9.0) 18 (10.8) 33 ( 9.9) 13 ( 7.7) 12 ( 7.1) 25 ( 7.4) No Response 49 (29.3) 49 (29.3) 98 (29.3) 59 (35.1) 64 (38.1) 123 (36.6) 最良細胞遺 伝学的効果 N (%) Unknown 6 ( 3.6) 12 ( 7.2) 18 ( 5.4) 9 ( 5.4) 11 ( 6.5) 20 ( 6.0) イマチニブの投与量、投与期間、最良血液学的及び細胞遺伝学的効果に投与群間で偏りは見ら れなかった。多くの症例でイマチニブの投与量は400 mg/日~600 mg/日であり、いずれの投与群 でも約1/3 の症例が 600 mg/日を超えるイマチニブの投与を受けていた。イマチニブの投与期間が 3 年を超えた症例の割合は、QD 群で 43%、BID 群で 39%であった。イマチニブの最良細胞遺伝 学的、及び血液学的効果に関しても投与群間で差はなく、QD 群及び BID 群における MCyR 率は、 それぞれ44%及び 39%、CHR 率は 82%及び 85%であった。MCyR 率は、イマチニブの 5 年投与 時の成績として報告されている値(69%)より低かった。 投与前の血液学的状態(血球数等)及び臨床検査値についても投与群間に差はなかった。 2.4.3 薬剤の曝露 ダサチニブの投与量及び投与期間を表 2.4-7 に示す。
表 2.4-7 ダサチニブの投与量及び投与期間 QD BID 100 mg 140 mg 計 50 mg 70 mg 計 N=166 N=163 N=329 N=166 N=167 N=333 中央値 99.5 126.0 100.0 92.5 108.0 98.0 平均1 日投 与 量 (mg/ 日) 最小 - 最大 18 - 150 42 - 166 18 - 166 21 - 158 13 - 167 13 - 167 0 - 90% 59 (35.5) 84 (51.5) 143 (43.5) 79 (47.6) 96 (57.5) 175 (52.6) >90-100% 84 (50.6) 68 (41.7) 152 (46.2) 70 (42.2) 63 (37.7) 133 (39.9) Dose Intensity > 100% 23 (13.9) 11 (6.7) 34 (10.3) 17 (10.2) 8 (4.8) 25 (7.5) 中央値 8.28 8.18 8.28 8.28 7.85 8.18 投 与 期 間 (月) 最小 - 最大 1.02-12.91 0.16-13.80 0.16-13.80 0.16-14.52 0.07-14.00 0.07-14.52 中央値 7.82 7.16 7.56 7.48 6.60 7.06 曝 露 期 間 (月) 最小 - 最大 1.02-12.25 0.16-13.17 0.16-13.17 0.16-13.57 0.07-13.50 0.07-13.57 100 mg QD 群の平均 1 日投与量の中央値は 99.5 mg と、目標用量に近かったが、他の投与量群 では目標投与量よりも低い用量となった。Dose Intensity が 90%以下であった症例は、QD 群で 143 例(44%)であったのに対し、BID 群では 175 例(53%)であった。QD 群及び BID 群とも、100 mg 投与の方が 140 mg 投与に比べ Dose Intensity が高かった。ダサチニブの投与期間の中央値は QD 群及び BID 群で同程度であり、それぞれ 8.28 ヵ月及び 8.18 ヵ月であった。投与中断期間を除 いた曝露期間の中央値もQD 群及び BID 群で同程度であり、それぞれ 7.56 ヵ月及び 7.06 ヵ月で あったが、投与群別では1 回 70 mg 1 日 2 回投与群で 6.6 ヵ月と、他の投与群に比べ短かった。 ダサチニブの減量、中断及び増量について表 2.4-8 に示す。 表 2.4-8 ダサチニブの用量調節 症例数(%) QD BID 100 mg 140 mg 計 50 mg 70 mg 計 N = 166 N = 163 N = 329 N = 166 N = 167 N = 333 減量した症例 49 (29.5) 77 (47.2) 126 (38.3) 68 (41.0) 92 (55.1) 160 (48.0) 1 回 11 ( 6.6) 13 ( 8.0) 24 ( 7.3) 6 ( 3.6) 6 ( 3.6) 12 ( 3.6) 2 回 10 ( 6.0) 9 ( 5.5) 19 ( 5.8) 7 ( 4.2) 12 ( 7.2) 19 ( 5.7) 3 回 12 ( 7.2) 12 ( 7.4) 24 ( 7.3) 7 ( 4.2) 9 ( 5.4) 16 ( 4.8) 4 回以上 16 ( 9.6) 43 (26.4) 59 (17.9) 48 (28.9) 65 (38.9) 113 (33.9) 投与ミス 1 ( 0.6) 0 1 ( 0.3) 3 ( 1.8) 4 ( 2.4) 7 ( 2.1) 血液毒性 36 (21.7) 55 (33.7) 91 (27.7) 44 (26.5) 53 (31.7) 97 (29.1) 非血液毒性 11 ( 6.6) 22 (13.5) 33 (10.0) 20 (12.0) 31 (18.6) 51 (15.3) 不明 0 0 0 1 ( 0.6) 1 ( 0.6) 2 ( 0.6) 減 量 理 由 その他 1 ( 0.6) 0 1 ( 0.3) 0 3 ( 1.8) 3 ( 0.9)