九州大学学術情報リポジトリ
Kyushu University Institutional Repository
構造規制された2つのSi-H基がもたらす特異的反応性
星, 香花
https://doi.org/10.15017/4060207
出版情報:九州大学, 2019, 博士(工学), 課程博士 バージョン:
権利関係:
構造規制された 2 つの Si-H 基がもたらす特異的反応性
星 香花
i
Ⅰ.序論
1.構造規制された近接官能基と反応 ··· 2
2.構造規制された近接シリル基を有する化合物の反応 ··· 3
3.ジシラメタラサイクル骨格を有する遷移金属錯体と触媒反応 ··· 6
4.本研究の動機と目的 ··· 9
5.引用文献 ··· 12
Ⅱ.カルボン酸の還元によるアルデヒド合成 Ⅱ‐1.序論 ··· 16
1 1.アルデヒドの有用性 ··· 16
1 2.カルボン酸誘導体のヒドリド還元 ··· 18
1 3.カルボン酸誘導体のヒドリド還元によるアルデヒドの合成 ··· 20
1 4.ヒドロシランを用いたカルボン酸誘導体の還元 ··· 27
1 5.本研究の目的··· 34
Ⅱ‐2.結果と考察 ··· 37
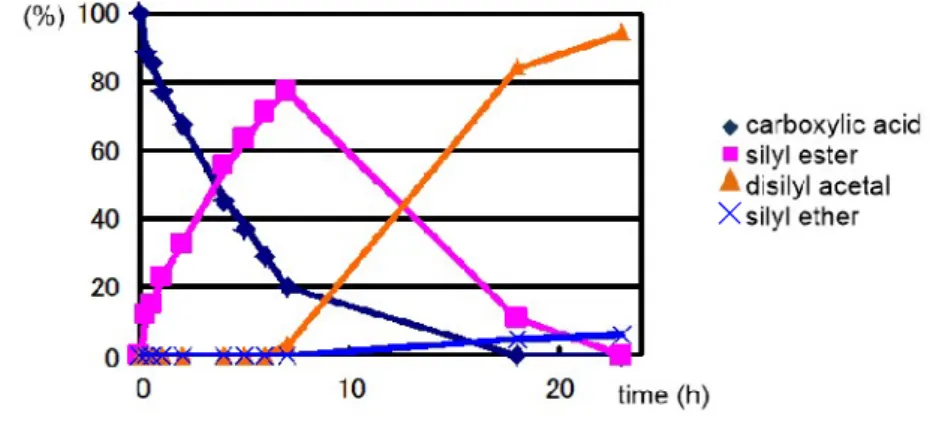
2 1.環状ジシリルアセタールの安定性 ··· 37
2 2.環状ジシリルアセタールの合成 ··· 44
2 3.カルボン酸の還元によるアルデヒド合成 ··· 52
2 4.ジシリルアセタールの単離と構造解析 ··· 57
2 5.考察 ··· 60
2 5 1.反応の特徴と観察事項に関する考察 ··· 60
2 5 2.反応の優れている点、今後応用が期待される点 ··· 62
2 5 3.反応機構の考察 ··· 63
2 5 4.本研究の位置付けと価値 ··· 65
Ⅱ‐3.結論 ··· 70
Ⅱ‐4.実験項 ··· 71
4 1.一般 ··· 71
4 2.化合物の合成とスペクトルデータ ··· 71
4 3.p-アニス酸(II-3a)の還元を用いたヒドロシランの反応性の検討 ··· 73
4 4.アルデヒドの合成・単離 ··· 74
ii
4 5.-位に不斉炭素を有するカルボン酸の光学純度を保った還元 ··· 79
4 6.環状シロキサンの同定 ··· 79
4 7.ジシリルアセタールの単離 ··· 80
Ⅱ‐5.引用文献 ··· 82
Ⅲ.DFT 計算を用いたジシラメタラサイクル骨格錯体とアルケンの水素化反応研究 Ⅲ‐1.序論 ··· 90
1 1.不飽和化合物の触媒的水素化 ··· 90
1 2.1,2-ビス(ジメチルシシル)ベンゼン (III-1)を配位子とし、ジシラメタラサイクル骨 格を有する錯体 M(CO)2(H)2(1,2-(SiMe2)2C6H4)2 (M=Fe (III-2) 4, Ru (III-3))の合成、反応、 理論計算 ··· 93
1 3.本研究の動機と目的 ··· 96
Ⅲ‐2.結果と考察 ··· 98
2 1.汎関数の設定··· 98
2 1 1.汎関数の設定法 ··· 98
2 1 2.本研究における汎関数の決定 ··· 98
2 2.錯体 M(CO)2(H)2(1,2-(SiMe2)2C6H4)2 (M=Fe, Ru, Os)の構造最適化 ··· 102
2 2 1.6 つの安定構造 ··· 102
2 2 2.鉄錯体 III- 2 の構造最適化と結晶構造解析結果との比較 ··· 103
2 2 3.ルテニウム錯体 III- 3 の構造最適化と結晶構造解析結果の比較 ··· 105
2 2 4.オスミウム錯体 III- 4 の構造最適化 ··· 109
2 2 5.鉄錯体 III- 2、ルテニウム錯体 III- 3、オスミウム錯体 III- 4 の最適化構造比較 ··· 111
2 2 6.SISHA の理論的解釈と NMR ··· 113
2 3.動的挙動 ··· 115
2 3 1.錯体 III-2、錯体 III-3、錯体 III-4 の動的挙動 ··· 115
2 3 2.トランス体の鉄錯体 III-2、ルテニウム錯体 III-3、オスミウム錯体 III-4 におけ る動的挙動 ··· 116
2 3 3.シス構造を持つ、鉄錯体 III-2、ルテニウム錯体 III-3、オスミウム錯体 III-3 における動的挙動 ··· 120
iii
2 3 4.鉄錯体 III-2、ルテニウム錯体 III-3、オスミウム錯体 III-4 における動的挙動の
比較 ··· 127
2 4.触媒サイクルのアウトラインと触媒活性種の発生 ··· 131
2 5.シス型中間体を経る水素化の反応機構 pathway-A ··· 136
2 5 1.鉄錯体 III- 2 を触媒とするエチレンの水素化計算 pathway-A ··· 136
2 5 2.ルテニウム錯体 III- 3 を触媒とするエチレンの水素化計算 pathway-A ···· 137
2 5 3.オスミウム錯体 III- 4 を触媒とするエチレンの水素化計算 pathway-A ···· 141
2 6.シス型中間体を経る水素化の反応機構 pathway-B ··· 144
2 6 1.鉄錯体 III- 2 を触媒とするエチレンの水素化計算 pathway-B ··· 144
2 6 2.ルテニウム錯体 III- 3 を触媒とするエチレンの水素化計算 pathway-B ···· 146
2 6 3.オスミウム錯体 III- 4 を触媒とするエチレンの水素化計算 pathway-B ···· 149
2 7.トランス型中間体を経る水素化の反応機構 pathway-C ··· 152
2 7 1.鉄錯体 III- 2 を触媒とするエチレンの水素化計算 pathway-C ··· 152
2 7 2.ルテニウム錯体 III- 3 を触媒とするエチレンの水素化計算 pathway-C ···· 154
2 7 3.オスミウム錯体 III- 4 を触媒とするエチレンの水素化計算 pathway-C ···· 157
2 8.水素-水素結合開裂機構に関する考察 ··· 161
2 9.計算結果のまとめと考察 ··· 164
Ⅲ‐3.結論 ··· 168
Ⅲ‐4.実験項 ··· 172
4 1.DFT 計算 ··· 172
4 2.計算精度の検証 ··· 172
Ⅲ‐5.引用文献 ··· 174
Ⅳ.結論 結論 ··· 180
謝辞 謝辞 ··· 183
1
Ⅰ.序論
2
Ⅰ.序論 1.構造規制された近接官能基と反応
多くの有機反応が開発され、世の中に必要な多数の有機化合物が合成されている。一般 に、有機反応を起こす鍵は有機化合物に含まれている官能基である。有機反応の中で最も 簡単な反応である酸塩基反応を例に挙げると、ルイス塩基であるアミンは容易にプロトン と反応してアンモニウム塩を生じる(Scheme I-1, a)。一方、ルイス酸であるボランは容易 にヒドリドと反応してボレートを生じる(Scheme I-1, b)。興味深いことに、プロトン化反 応におけるアミンの塩基性は複数のアミノ基を含む化合物で、かつアミノ基どうしがある 距離に存在するように構造規制されている場合に強くなることが発見され、現在では総説 にまとめられているI-1, I-2, I-3。特に 1,8-ジメチルアミノナフタレン(Scheme I-1, c)は、
Aldrich 社から強いプロトン捕捉剤という意味で Proton Sponge という商標で市販されて おり、現在では一連の構造規制された 2 つのアミノ基を持ち強いプロトン捕捉効果をもつ 化合物の総称として Proton Sponge が使われることが多い。Proton Sponge の特徴は、1 つのプロトンが 2 つのアミノ基で挟まれた[N…H…N]+ 構造を持つことであり、2 つの塩 基性の官能基が 1 つのプロトンと水素結合で結合していることがプロトン捕捉効果の原因 と考えられている(Scheme I-1, c)。同様な考え方で、Hydride Sponge が 1985 年に Katz により報告された。2 つのボリル基が近接した 1,2-ボリルブタンが 1,4-ジボリルブタンよ りもルイス塩基と反応しやすいことは知られていたがI-4、Katz は 1,8-ジボリルナフタレ ン(I-2)が KH と反応して 2 つのB が1 つのヒドリドを挟み込む形で捕捉することを見出し、
X 線結晶構造解析でその構造を確認している(Scheme I-1, d)。
Scheme I-1 酸塩基反応とプロトン/ヒドリドの補足剤
3
2.構造規制された近接シリル基を有する化合物の反応
このような構造規制されている 2 つの官能基が 1 つの基質と相互作用して基質捕捉する 例は、現在では数多くの例が知られている。その中で、本論文でとりあげるベンゼン環の 1,2-位に 2 つのシリル基を持つ化合物における特異的な反応性としては、1990 年に玉尾ら が 1,2-ビス(ジフルオロフェニル)ベンゼン(I-3)が F-の捕捉剤として有効に作用することを 報告している(Scheme I-2)I-5。先に述べたプロトンやヒドリドの捕捉剤は、1,8-位に官能基 を持つナフタレン骨格を利用していた。これはプロトンやヒドリドの原子半径が小さいこ とに由来している。プロトンやヒドリドよりも原子半径が大きい F-を捕捉するには、むし ろ 1,2-位にシリル基をもつベンゼン骨格のほうが構造的に有利である。
Scheme I-2 フッ素イオン捕獲剤 1,2-ビス(ジフルオロフェニル)ベンゼン
1,2-位にシリル基をもつベンゼン骨格は、遷移金属の捕捉剤としても有効に作用する。
1973 年にイギリスの Eaborn により、1,2-ビスジメチルシリルベンゼン(以下、BDSB と略 称する)を白金錯体と反応させると水素の発生を伴い、2 つのケイ素に金属が結合した5員 環のジシラメタラサイクル化合物が生成することが報告された(Scheme I-3, a)I-6。さらに、
1975 年にはスイスの Fink により、鉄錯体及びルテニウム錯体について同様の反応が報告 された(Scheme I-3, b)I-7。それ以降、ジシラメタラサイクルは多くの有機金属化学研究者 の興味の対象となり、現在では Figure I-1 に示すような数多くの遷移金属を含む錯体が単 離され、構造解析されている。I-8, I-9
Si Si
H H
Si Si
H
H + M3(CO)
1-7a(M = Fe) 1-7b(M = Ru)
Si Si
M(CO)4 + CO + H2
+ Pt
PPh3 PPh3
Si Si
Pt(PPh3)2 + H2+ CH2=CH2
I-4 I-5 I-6
(a)
(b)
I-4
Scheme I-3 BDSB を用いたジシラメタラサイクル骨格を有する有機金属錯体の合成
4
Figure I-1 ジシラメタラサイクル骨格を有する有機金属錯体I-9
注目すべきことに、1,2-ビスジメチルシリルベンゼン骨格の特異的な機能が、さらに反 応へと展開した例が報告されている。先に述べたように、玉尾らは 1,2-ビス(フルオロジメ チルシリル)ベンゼンがフッ素アニオンの捕捉に有効であることを示したが(Scheme I-2)、
その後丸岡らは、この機能をフッ素アニオンにより誘起されるアリルシランを用いるアル デヒドへのアリルアニオン付加反応に適用すると大幅な加速効果が得られることを報告し ている(Scheme I-4) I-11。すなわち、BDSB 骨格と 2 つのアリル基を持つ化合物 1,2-ビス(ア リルジメチルシリル)ベンゼン(I-16)が普通のアリルシランよりもはるかに効率的に F-と 反応し、アルデヒドのアリル化が迅速に進行する。加速化機構として、I-16 の 2 つのシリ ル基が F-を挟み込むように捕捉することで、フルオロシリケートが安定に生成し、アリル アニオンの容易な発生とアルデヒドへの求核付加反応が達成される。
Scheme I-4 1,2-ビス(アリルジメチルシリル)ベンゼンを用いたアルデヒドのアリル化
5
もう一つの、BDSB 骨格の利用は、向山のラクトン化反応であるI-12。ロジウム触媒は、
ヒドロシランとアルコールやカルボン酸の反応により脱水素シリル化を起こすことはよく 知られている。向山らは、Table I-1 に示すように、ヒドロキシカルボン酸前駆体と BDSB から 2 つのジメチルシリル基を含む中~大環状ラクトンを合成したのちシリルジトリフレ ートで処理すると、中~大員環のラクトンが効率的に生成することを報告している。一般 に,-ヒドロキシカルボン酸からのラクトン合成は、5~6 員環ラクトンでは容易であるが、
中~大員環のラクトン合成は分子内反応(ラクトンの生成)と分子間反応(オリゴマーの生 成)が混ざるために難しい。通常の反応試薬を用いると、目的の分子内のカルボン酸とアル コールが脱水して得られるラクトン以外に、2 分子のヒドロキシカルボン酸が互いのカル ボキシ基とアルコキシ基で脱水した環状エステル化であるジオリドが副生成物として発生 する。Table I-1 で示すように反応は段階的に進み、1 段階目はカルボン酸のシリル化、2 段階目はアルコールのシリル化であるが、BDSB のジメチルシリル基がベンゼン環の 1,2- 位に存在しているために 2 つのジメチルシリル基を含む中~大環状ラクトンが生成しやす い。さらにシリルジトリフレートを作用させるとジシロキサンが発生してラクトンを生じ るが、この 5 員環ジシロキサンが BDSB 骨格を持つために極めて安定であることから、反 応は熱力学的に有利となる。このように、BDSB 骨格を用いた構造規制はラクトン合成に 有効な手段として活用されている。
Table I-1 ロジウム触媒と BDSB を用いる中員環ラクトン合成
6
3.ジシラメタラサイクル骨格を有する遷移金属錯体と触媒反応
遷移金属ジシラメタラサイクルの特異的な反応の最初の例は、Curtis による Ir ジシラメ タラサイクルである(Figure I-1, I-8)I-13。この錯体はジシロキサンの不均化(Redistribution) の中間体であると提案されている。一方、BDSB 骨格を含むジシラメタラサイクルの特異 的な反応の最初の例は、Corriu の Scheme I-5 に示す量論反応である。2 つのジメチルシリ ル基が近接しているために、鉄‐ケイ素結合にニトリルの C≡N 結合が挿入した後、脱水 素を起こして 5 員環ジシリルエナミンが生じる。向山ラクトン化のジシロキサンと同様に、
BDSB 骨格は 5 員環ジシリルエナミンの安定性に貢献しているI-14。
Scheme I-5 鉄カルボニル BDSB 錯体とニトリルの反応
田中、石川らは、白金及びニッケルのジシラメタラサイクルを中間体とした触媒反応を 開発している。Scheme I-6 にまとめるように、ホスフィンを支持配位子とする白金やニッ ケル錯体は BDSB との反応でジシラメタラサイクルを生じるが、その金属‐ケイ素結合に はアルキンやアルケン等の不飽和結合を持つ化合物が挿入し、還元的脱離反応を経て 2 つ のジメチルシリル基を含む環状ジシリル化合物を生じるI-15,I-16。
Scheme I-6 ジシラメタラサイクル中間体を経由する白金、ニッケルの触媒反応
一方、永島らは 2 つの近接したシリルヒドリドがカルボニル化合物のヒドロシリル化反 応を大きく加速することを発見し、これを広範に展開して新しい化学を創出しているI-17。 この化学の端緒となる発見は、RhCl(PPh3)3触媒を用いるケトンのヒドロシリル化反応で
7
ある。この反応は一般に 1 級、2 級、3 級のヒドロシランを用いて行われるが、1 級、2 級 のヒドロシランと比較して 3 級のヒドロシランを用いた反応は遅い。反応部位が 3 級のヒ ドロシランであるにもかかわらず、ヒドロシランとして、1,2-ビスジメチルシリルエタン (BDSE)や 1,2-ビスジメチルシリルベンゼン(BDSB)を用いると、反応がエチルジメチルシ ランを用いた場合より数倍から 50 倍速く、さらに、分子内に存在する 2 つのジメチルシ リル基のうち、1 つだけが反応した化合物が得られる(Scheme I-7)。2 つ目のジメチルシリ ル基の反応性はエチルジメチルシランと同程度であり、2 つの近接した Si-H 基が強い加速 効果をもたらしている。その後の研究により、RhCl(PPh3)3と BDSB の反応からいくつか のジシラメタラサイクル錯体(Scheme I-7, I-22、I-23、I-24)が得られることが明らかとな っており、加速反応の鍵がジシラメタラサイクル中間体の生成である可能性が示唆されて いるI-18。
Scheme I-7 ケトンのヒドロシリル化における近接 Si-H 基の反応加速効果
2 つの近接した Si-H 基によるヒドロシラン還元の加速効果は RhCl(PPh3)3触媒特有の反 応ではなく、他の金属触媒においても広範にみられることがその後の研究で明らかになっ ている。例えば、アセナフチレンが配位した 3 核ルテニウムカルボニル錯体(I-25)はケト ンやアルデヒド、エステルだけでなくアミドのヒドロシラン還元によるアミンの合成に良 好な触媒となるが、3 級アミドと比較して反応性に乏しい 2 級アミドの 2 級アミンへの変 換反応においては BDSE や 1,1,3,3-テトラメチルジシロキサン(TMDS)が優れた加速結果 を与える。また、イリジウム触媒を用いる 3 級アミドのヒドロシラン還元は近接した 2 つ
8
の Si-H を持つヒドロシランの中でも TMDS のみが高い活性を示し、かつ、生成物は 3 級 アミンではなくエナミンである。白金触媒及び鉄カルボニル触媒も 3 級アミドの 3 級アミ ンへのヒドロシラン還元によい活性を示すが、興味深いことにこれらの触媒では他の二官 能性ヒドロシランよりも BDSB のほうが良い効果を与える。二官能性ヒドロシランと有機 金属触媒により達成されたカルボニル化合物の還元反応を Scheme I-8 にまとめるI-18~I-20。
(ACE)Ru3(CO)7
Si-H
R O
R R'
O R OR'
O NR'2 R R
O[Si]
R' R NR'2
R O[Si]
Ru CO
OC
OC CO
CO OC
O Ru Ru Rh cat. RhCl(PPh3)3
(Wilkinson's complex) Ru cat.
Ru(CO)2(H)2((SiMe2)2C6H4) (I-25)
Pt cat. H2PtCl6/iPrOH Pt2[(CH2=CHSiMe2)2O]3 Pt(dba)2
(dba = dibenzylideneacetone)
Ir cat. IrCl(CO)(PPh3)2 (Vaska's complex) IrCl(CO)[P(OC6F5)3]2 Fe cat. Fe(CO)4, Fe3(CO)12
[Fe3(CO)11(h-H)]2Fe(DMF)4 Fe(CO)2(H)2((SiMe2)2C6H4)
Si-H [Si]
Rh, Ru, Pt, Fe cat.
Ar-H
O NR'2
R O[Si]
MR'2 R
NR'2 O
R NHR'
O NH2 R
R CN R NHR' O
R H
TsNH2 Rh, Ru, Pt, Fe cat.
Ru, Pt, Fe cat.
Ir cat.
Ru, Pt cat.
Ru cat.
Ru cat.
Ru cat.
Ru, Fe cat.
R Ar
R NTs R
O[Si]
H
Scheme I-8 近接 Si-H 基を有するヒドロシランとカルボニル化合物の反応
Scheme I-8 に示した反応のうち、白金触媒を用いる BDSB と 3 級アミドの反応につい ては有機合成化学に有用な触媒反応としての研究から中間体と強い関連を持つ白金ジシラ メタラサイクル錯体の単離、構造決定、反応の研究、さらには、中谷らとの共同研究によ る理論計算による反応機構解析が行われ、ジシラメタラサイクル中間体が鍵中間体である ことが明らかとなっているI-21。白金触媒を用いるアミドのヒドロシラン還元は強いヒドロ シラン依存性を持ち、ジメチルフェニルシランに代表されるモノヒドロシランは反応性が なく、Si-H が近接している二官能性ヒドロシランの中でも BDSB と TMDS、ポリメチル ヒドロシロキサン(PMHS)のみが良好な結果を与える。ジメチルスルフィドは反応を全く 阻害しないことから、ジメチルスルフィド存在下で白金前駆体と BDSB の反応を行うこと により Scheme I-9 に示す 3 つの白金錯体(I-26、I-27、I-28)が単離、構造決定された。こ れらはいずれも良好な触媒活性を示す。これらは触媒サイクル中間体の類縁体であり、こ
9
れらの構造をもとにした DFT 計算により Scheme I-9 に示すような触媒サイクルが提案さ れている。BDSB が高い反応性を示す鍵は中間体 I-26 であり、同一平面上に存在する 2 つ のシリル基の強いトランス影響によりヒドリドとしての性質が強くなっている。
Scheme I-9 ジシラプラチナサイクル中間体を鍵とする 3 級アミドの還元
4.本研究の動機と目的
以上に述べた、2 つの近接したケイ素基が反応にもたらす特殊な効果についてまとめる。
一つは、玉尾‐丸岡のフッ素アニオン捕捉とフッ素アニオンによる炭素‐ケイ素結合活性 化及び向山のマクロラクトン化における効果である。これらにおいては BDSB 骨格が 2 つ のケイ素官能基を効果的に近くし、環状の反応中間体を圧倒的に有利にしている。もう一 つは、遷移金属種が 2 つの近接したケイ素基の間に取りこまれ、安定なジシラメタラサイ クル錯体を作ることである。特に、BDSB 骨格は BDSB がベンゼン環を介して 2 つのケイ 素基を Rigid に固定することから、ジシラメタラサイクル生成に対して速度論的にも熱力 学的にも有利に働く。構造的興味で開始されたジシラメタラサイクル研究は、その後 Corriu、Tanaka の先駆的な研究を経て、当研究室が系統的に開発してきた 2 つの近接した Si-H 基によるヒドロシラン還元反応の加速化を鍵にした広範な触媒反応化学へと展開し ている。特筆すべきことは、白金触媒に代表されるように BDSB が最も良い効果を与える 例が見いだされ、これらが BDSB 骨格により速度論的にも熱力学的にもジシラメタラサイ クル生成が有利になっているという点である。言い換えれば、BDSB 骨格は「構造規制さ
10
れた 2 つのジメチルシリル基」を生み出し、また、これがもたらす特異的な反応の開発余 地はまだまだ広く存在すると考えられる。
本論文では、BDSB を研究の出発点とした 2 つの研究成果をまとめる。1つは、有機合 成化学で広く知られているカルボン酸のアルデヒドへの選択的還元反応である。有機化学 の教科書にあるように、カルボン酸を LiAlH4で還元するとアルコールが得られるが、その 素反応過程としては、カルボン酸の C=O 結合へのヒドリドの付加反応が第一段階、生成 したアセタールアニオンの C-O 結合が還元的に切断される反応が第 2 段階である。この 2 つの素反応は、それぞれ反応速度が異なる。中間体であるアセタールアニオンはアルデヒ ド等価体であり、もし、第一段階の反応速度と比較して第二段階の反応速度が圧倒的に遅 い場合、カルボン酸からアセタールアニオンを経てアルデヒドを選択的に生成する反応が 可能である。カルボン酸をヒドロシランで還元する場合、触媒の作用で Si-H 基が開裂し、
カルボニル基に付加する反応が第一段階である。得られた中間体は酸素上がシリル基とな っているジシリルアセタールであり、アセタールの中では安定なため、加水分解に気を付 ければ単離可能な化合物である。先に、向山のラクトン化反応において、BDSB がベンゼ ン環の 1,2-の位置にジメチルシリル基を持つことが、,-ヒドロキシカルボン酸と BDSB の 1:1 反応による 2 つのジメチルシリル基を含むラクトン合成に有利であること、ならび に、ラクトン化終了後にケイ素を含む化合物として得られる環状ジシロキサンの熱力学的 安定性に対して BDSB 骨格が大きな効果を持つことを示した。本論文では、このような中 間体、生成物への BDSB 骨格を活用した構造規制の導入が、カルボン酸からジシリルアセ タールへの選択的転換と得られた 7 員環ジシリルアセタールの安定化に極めて有効である ことを明らかにした。その結果、カルボン酸からアルデヒドへの変換反応が可能となった (Scheme I-10)。
Scheme I-10 BDSB を用いたカルボン酸を原料とするアルデヒド合成
一方、先に述べてきたように、当研究室では触媒反応の中間体として一連のジシラメタ ラサイクル錯体の合成、構造、反応に関する研究を蓄積してきた。最近の特筆すべき成果 として、鉄とルテニウムを含む新しいジシラメタラサイクル錯体
Fe (CO)2(H)2{(SiMe2)2C6H4} (I-29)I-22と Ru (CO)2(H)2{(SiMe2)2C6H4} (I-30)が砂田によ り合成され、その特異的な構造が明らかとなった。この反応性の検討から、鉄錯体 I-29 が アルケンの水素化、アルケンのヒドロシリル化、ケトンのヒドロシリル化の触媒となるこ と、活性は I-29 より低いがルテニウム錯体 I-30 もアルケンの水素化活性を持つことを明 らかにしている(Scheme I-11)。田原らは、特に鉄錯体 I-29 の構造、動的挙動、水素化機
11
構について、DFT 計算を用いて、H2Fe(VI)Si4コアに Si…H…Si の 2 次的な相互作用をも つこれまでにないユニークな構造及び水素の Fluctuation を含む低エネルギーでの異性体 間相互変換さらに鉄中心ではなく鉄‐水素結合上での水素の H-H 結合の活性化、という これまでにない新しい成果を報告したI-23。これらは従来の水素化触媒にない数々の新しい 事実を含んでおり、本研究開始時には、ルテニウム錯体 I-30 でも同様な現象がみられるか、
についての検討が急がれた。DFT 計算は得られた実験結果の合理的説明だけでなく、まだ 実現していない実験の予測も可能である。本研究では、鉄錯体 I-29、ルテニウム錯体 I-30 に加えオスミウム錯体 I-31 の計算を、CO 配位子がcis-の位置関係にある錯体、trans-の 位置関係にある錯体の合計 6 つの化合物に対して行い、田原らの H2M(VI)Si4コアに Si…
H…Si の 2 次的な相互作用をもつこれまでにないユニークな構造、水素の Fluctuation を含 む低エネルギーでの異性体間相互変換、鉄中心ではなく鉄‐水素結合上での水素の H-H 結 合の活性化、が一般的な現象であること、並びに錯体の幾何構造の違い、中心金属の違い が何を生み出すかについて体系的な検討をおこなった。
Scheme I-11 ジシラメタラサイクル骨格を有する有機金属錯体とエチレンの水素化反応 機構
以下本論では、カルボン酸の還元によるアルデヒド合成について第二章に、DFT 計算を 用いたジシラメタラサイクル骨格錯体とアルケンの水素化反応研究について第三章にまと め、最後に第四章に本研究の結論をまとめる。
12 5.引用文献
I-1 総説:Staab, H. A.; Saupe, T. Angew. Chem., Int. Ed. Engl., 1988, 27, 865. and references cited therein.
I-2 総説:Alder, R. W. Chem. Rev. 1989, 89, 1215. and references cited therein.
I-3 総説: Llamas-Saiz, A. L.; Faces-Focesa, C.; Elguerob, J.; Struct, J. M. 1994, 328, 297.
I-4 Shriver, D. F.; Biallas, M. J. J. Am. Chem. Soc. 1967, 89. 1078.
I-5 Tamao, K.; Hayashi, T.; Ito, Y.; Shiro, M. J. Am. Chem. Soc. 1990, 112, 2422.
I-6 Eaborn, C.; Metham, T. N.; Pidcock, A. J. Organomet. Chem., 1973, 54, C3 I-7 a) Fink, W. Helv. Chim. Acta, 1975, 58, 1464.
b) Fink, W. Helv. Chim. Acta, 1976, 59, 606.
I-8 総説:Shimada, S.; Tanaka, M. Coord.Chem. Rev. 2006, 250, 991.
I-9 総説:Sunada, Y.; Nagashima, H. Dalton Trans. 2017, 46, 7644.
I-10 a)Greene, J.; Curtis, M. D. J. Am. Chem. Soc. 1977, 99, 5176.
b) Curtis, M. D.; Greene, J. J. Am. Chem. Soc. 1978, 100, 6362.
c) Curtis, M. D.; Greene, J.; Butler, W. M. J. Organomet. Chem. 1979, 164, 371.
d) Bell, L. G.; Gustavson, W. A.; Thanedar, S.; Curtis, M. D. Organometallics, 1983, 2, 740.
e) Osakada, K.; Hataya, K.; Nakamura, Y.; Tanaka, M.; Yamamoto, T. J. Chem. Soc. Chem.
Commun. 1993, 576.
f) Mitchell, G. P.; Tilley, T. D. Organometallics, 1996, 15, 3477.
g)Shimada, S.; Tanaka, M. Coord. Chem. Rev. 2006, 250, 991.
h) Losa, M.; Faller, J. W.; Crabtree, R. H. Inorg. Chem. 1995, 34, 2937.
i)Dionaev, V. K.; Yoo, B. R.; Procopio, L.; Carroll, P. J.; Berry, D. H. J. Am. Chem. Soc.
2003, 125, 8936.
j) Sunada, Y.; Imaoka, T.; Nagashima, H. Organometallics, 2010, 29, 6157.
k) Sunada, Y.; Imaoka, T.; Nagashima, H. Organometallics, 2013, 32, 2112.
I-11 Asao, N.; Shibato, A.; Itagaki, Y.; Jourdan, F.; Maruoka, K. Tetrahedron Lett. 1998, 39, 3177.
I-12 Mukaiyama, T.; Izumi, J.; Shiina, I. Chem. Lett. 1997, 187.
I-13Curtis, M. D.; Greene, J. J. Am. Chem. Soc., 1978, 100, 6362.
I-14 Corriu, R. J. P.; Moreau, J. J. E.; Pataud-Sat, M. J. Org.Chem., 1981, 46, 3372.
I-15 a) Hayashi, T.; Kobayashi, T.; Kawamoto, A. M.; Yamashita, H.; Tanaka, M.
Organometallics, 1991, 10, 16.
b) Tanaka, M.; Uchimaru, Y.; Lautenschlager, H. J. J. Organomet. Chem., 1992, 428, 1.
c) Shimada, S.; Tanaka, M.; Coord. Chem. Rev., 2006, 250, 991.
I-16 a) Ishikawa, M.; Naka, A.; Ohshita, J. Organometallics, 1993, 12, 4987.
13
b) Naka, A.; Lee, K. K., Yoshizawa, K.; Yamabe, T.; Ishikawa, M. Organometallics, 1999, 18, 4524.
I-17 総説:Nagashima, H. Synlett, 2015, 866.
I-18 a) Nagashima,H.; Tatebe, K.; Ishibashi, T.; Sakakibara, J.; Itoh, K. Organometallics, 1989, 8, 2495.
b) Nagashima, H.; Tatebe, K.; Itoh, K. J. Chem. Soc. Perkin Trans. 1989, 1707.
c) Nagashima, H.; Tatebe, K.; Ishibashi, T.; Nakaoka, A. Sakakibara, J.; Itoh K.
Organometallics, 1995, 14, 2868.
d) Sunada, Y.; Fujimura, Y.; Nagashima, H. Organometallics, 2008, 27, 3502.
I-19 a) Nagashima, H.; Suzuki, A.; Iura, T.; Ryu, K.; Matsubara, K. Organometallics, 1989, 8 2495.
b) Hanada, S.; Ishida, T.; Motoyama, Y.; Nagashima, H. J. Org. Chem. 2007, 72, 7551.
c) Hanada, S.; Motoyama, Y.; Nagashima, H. Eur. J. Org. Chem. 2008, 4097.
d)Sunada, Y.; Kawakami, H.; Imaoka, T.; Motoyama, Y.; Nagashima, H. Angew. Chem. Int.
E. 2009, 48, 9511.
e) Zhou, S.; Junge, K. D.; Addis, D.; Das, S.; Beller, M. Angew. Chem. Int. Ed. 2009, 48, 9507.
f) Tsutsumi, H.; Sunada, Y.; Nagashima, H. Chem. Commun. 2011, 47, 6581.
I-20 a)Motoyama, Y.; Aoki, M.; Takaoka, N.; Aoto, R.; Nagashima, H. Chem. Commun.
2009,1574.
b) Tahara, A.; Miyamoto, Y.; Aoto, R.; Shigeta, K.; Une, Y.; Sunada, Y.; Motoyama, Y.;
Nagashima, H. Organometallics, 2015, 34, 4985
c) Une, Y.; Tahara, A.; Miyamoto, Y.; Sunada, Y.; Nagashima, H. Organometallics, 2019, 38, 852.
I-21 a) Hanada, S.; Motoyama, Y.; Nagashima, H. Tetrahedron Lett. 2006, 47, 6173.
b) Hanada, S.; Tsutsumi, E.; Motoyama, Y.; Nagashima, H. J. Am. Chem. Soc. 2009, 131, 15032
c)Tsutsumi, T.; Sunada, Y.; Nagashima, H. Organometallics, 2011, 30, 68
d) Nakatani, N.; Hasegawa, J.; Sunada, Y.; Nagashima, H. Dalton Trans. 2015, 44, 19344.
I-22 Sunada, Y.; Tsutsumi, H., Shigeta, K.; Yoshida, R.; Hashimoto, T.; Nagashima, H.
Dalton Trans., 2013, 42, 16687
I-23 Tahara, T.; Tanaka, H.; Sunada, Y.; Shiota, Y.; Yoshizawa, K. Nagashima, H. J. Org.
Chem. 2016, 81, 10900.
14
15
Ⅱ.カルボン酸の還元によるアルデヒド合成
16
Ⅱ‐1.序論 1 1.アルデヒドの有用性II-1
カルボニル化合物のうちアルデヒドとケトンは、それぞれ RHC=O、R2C=O (R=アルキ ル基、アリール基等の炭化水素基)という一般式で表され、天然に存在する化合物に多く含 まれている官能基である。特にアルデヒドは、シトロネラールやバニリン等、香料の芳香 を与える官能基として知られている(Figure II-1)。
Figure II-1 代表的なアルデヒド
アルデヒド、ケトンの炭素‐酸素二重結合は炭素(+)と酸素(-)に分極しており、反応性 に富む。特に、求核剤との反応はこれらカルボニル化合物に特徴的なものである。アルデ ヒドは一般的にケトンよりも求核反応を受けやすく、さらに、カルボニル化合物に分類さ れるカルボン酸誘導体の多くと比較しても反応性が高い。この性質を利用して、アルデヒ ドは多くの有機合成反応における反応中間体として広く利用されている。Scheme II-1 に、
その代表的な例を示す。アルデヒドは、Grignard 試薬やアルキルリチウム(Scheme II-1, a) やエノレート(Scheme II-1, b)と反応して 2 級アルコールを与えるII-2。硫黄やリンを含む イリドのような求核剤との反応では、エポキシド(Scheme II-1, c)、アルキン(Scheme II-1, d)、アルケン(Scheme II-1, e)を合成することも可能であるII-3。また、Scheme II-1 の(f) はアルカロイド骨格を形成する有名な反応であるが、この反応では、まずアルデヒドがア ミンと反応してイミンが生成し、酸性条件下でイミニウム塩が求電子置換反応を起こして 複素環骨格を生成する。電子供与基を有するアリールエチルアミンにおいて、高収率で イソキノリン骨格が形成されるII-4。
アルデヒドの一般的合成法としては、アルケンのオゾン酸化とオゾニドの還元分解 II-1、 末端アルキンのヒドロホウ素化・酸化反応II-1、1 級アルコールの酸化反応II-1、カルボン酸 誘導体の還元反応等が知られているII-1。このうち、1 級アルコールの酸化反応とカルボン 酸誘導体の還元反応はアルデヒドの合成にしばしば用いられる反応であるが、ともに反応 選択性に問題がある。すなわち、1 級アルコールを酸化すると、第一段階ではアルコール
17
Scheme II-1 代表的なアルデヒドの反応
がアルデヒドに酸化(脱水素)されるが、アルデヒドが多くの酸化剤に活性であることから 第二段階のアルデヒドからカルボン酸への酸化反応がしばしば併発する。そのため、第二 段階目の酸化反応を抑制する酸化剤の研究が古くからおこなわれている。
比較的収率のよいアルコールの部分酸化の 3 つの代表例を、Scheme II-2 に示すII-5。(a) は Swern 酸化と呼ばれる、ジメチルスルホキシド(DMSO)を酸化剤とするアルデヒド合成 であるが、水に対して不安定な酸塩化物を使用することや厳密な温度コントロールが必要 であるという問題点があるII-5a,b。(b)は Oppenauer 酸化と呼ばれる、アルミニウム触媒を 用いる水素移動反応であるが、基質によっては副反応としてアルドール反応やアリルアル コールの二重結合転移反応が起こるII-5c。(c)は、Dess-Martin ペルヨージナン(DMP)と呼 ばれる高原子価ヨウ素を含む酸化剤を用いる Dess-Martin 酸化であるが、水気を含んだ DMP には爆発性があること等の問題点が残されているII-5d。
一方、カルボン酸誘導体である、カルボン酸、エステル、アミドの還元も二段階で進行 する。第一段階はこれらからアルデヒド、あるいはアセタールのようなアルデヒド等価体
18
Scheme II-2 アルコールの酸化反応によるアルデヒドの合成
に還元する反応であるが、生成したアルデヒドが還元剤に対する反応性が高いことから反 応は第一段階で止まらず、さらに 1 級アルコールまで還元されることが多い。本博士論文 研究ではこのカルボン酸の部分還元によるアルデヒドの合成手法の開発を行ったが、その 鍵となるのは、いかにして第一段階の反応を速やかに行い、かつ第二段階の還元を抑制す るかである。次節ではこのようなカルボン酸誘導体の選択的還元によるアルデヒド合成の 例を挙げ、これまでの手法ではどのようにして高選択性を達成してきたのかについてまと める。
1 2.カルボン酸誘導体のヒドリド還元II-6
1 1で述べた通り、一般に酸ハロゲン化物及び酸無水物を除くカルボン酸誘導体はケ トンやアルデヒドに比べてヒドリド反応剤による還元を受けにくい。これらカルボン酸誘 導体の還元には、LiAlH4のような還元力の強いヒドリド還元剤を用いる例、NaBH4のよう な弱いヒドリド還元剤に Lewis 酸等を加えて活性化したものを用いる例、BH3や AlH3等 それ自体が Lewis 酸性を有する金属中心を含む還元剤を用いた例、等が知られている。
強いヒドリド還元剤の代表例である LiAlH4はエステル、カルボン酸、アミドを還元する ことができる。LiAlH4は Li+と[AlH4]-からなるアート錯体であり、Al 上のヒドリドがカル ボニル炭素に対して求核攻撃することで還元反応が進行する。Scheme II-3 にまとめるよ うにこの反応は二段階で進行するが、過剰の LiAlH4との還元反応ではカルボン酸、エステ ルの還元における生成物は、第一段階で止めることができず 1 級アルコールになる。同様 に、アミドの還元では対応する 1 級アミンが生成物となる。
19
Scheme II-3 カルボン酸のヒドリド還元
NaBH4は典型的な弱い還元剤であり、単独ではカルボン酸誘導体を還元する力はないが、
Lewis 酸を添加することで反応が進行する。代表例として、NaBH4に AlCl3を添加した系 が挙げられる。Scheme II-4 に示すように、Lewis 酸がカルボニル酸素に配位することで炭 素‐酸素二重結合が活性化され、弱い還元剤である NaBH4がカルボン酸誘導体と反応す ることができるようになる。この反応では、Lewis 酸のカルボニル基活性化が鍵となるが、
カルボン酸だけでなく中間体に生成するアセタール類も Lewis 酸で活性化されるため、こ の場合も第一段階と第二段階の反応速度を制御することは難しい。
Scheme II-4 ルイス酸により活性化されたカルボニル化合物のヒドリド還元
20
Lewis 酸添加によるカルボニル基の活性化は、ボランやアラン還元剤でも重要な役割を 果たす。Scheme II-5 に示すように、これらのホウ素あるいはアルミニウム中心はそれ自 体が空軌道を有するため、カルボニル酸素がこれに配位することで基質が活性化されたの ちにカルボニル炭素にヒドリドが攻撃することで還元反応が進行する。この場合も、一般 的には還元反応の第一段階と第二段階を制御することは難しい。
R OR'
O
R OR'
OBH2 H
R OH
H H
R OR'
O BH3 BH3
+HOR' lone pair
vacant orbital
activation reduction
reductive C-O bond cleavage hydrolysis
Scheme II-5 ボランによるエステルの還元
第一段階と第二段階の選択性を、ヒドリド還元剤のヒドリド性を弱める、あるいは立体 障害でカルボニル基の活性化を阻害する等の試みは古くから行われている。例えば、
LiAlH4の誘導体である LiAl(OtBu)3H はtBuO 基の電子供与性と立体障害による効果に特 徴があり、また、ホウ素上に置換基を導入した 9-ボラビシクロ(3.3.1)ノナン(9-BBN) 等 は立体障害が BH3よりも大きいため反応選択性が異なる。これらを利用すると、場合によ りアルデヒドやケトンとカルボン酸誘導体との反応性を逆転させるような例も存在する。
しかしながらこれらの還元剤が普遍的にカルボン酸誘導体からアルデヒドを選択合成する 例は多くない。
カルボン酸誘導体の還元とその還元に用いられる主な反応剤、及び反応生成物を Table II-1 にまとめた。A はアルコールが生成することを示す。B はアミンが、C はアルデヒド が生成することを示す。なお、A’は RCH2OH と R’OH の二種類のアルコールが生成する ことを示している。空欄は反応が進行しないことを示す。後述するが、アミドの還元では 同じ還元剤を用いた場合でも適当な反応条件を選択することでアミンとアルデヒドを作り 分けることができる場合がある。B and/or C の表記はそれが可能な還元剤との組み合わせ であることを示す。
1 3.カルボン酸誘導体のヒドリド還元によるアルデヒドの合成
前述の通りアルデヒドはカルボン酸誘導体よりも還元を受けやすい。しかしながら基質 によっては反応条件を制御することで、これらの還元によりアルデヒドを得ることも可能
21
Table II-1 種々のヒドリド還元剤によるカルボニル基の選択的還元反応
である。エステルの還元でアルデヒドを得るには、-70 oC でジイソブチルアルミニウムハ イドライド(DIBAL-H)を用いる反応が一般的である(Scheme II-6)II-7。先に述べたように、
エステルの還元は 2 段階で進行する。DIBAL-H とエステルの反応では、第一段階でアル ミノヘミアセタールが生成したのち、第二段階でもう 1 分子の DIBAL-H による炭素‐酸 素結合の還元的切断が起こる。アルミニウム‐酸素結合が強くアルミノヘミアセタールの 安定性が高いことと、2 つの立体障害が大きいイソブチル基が立体的にもう一分子の DIBAL-H との第二段階を起こしにくくすると考えることができる。低温で反応を行うこ とにより第二段階の反応の速度を下げることで選択的にアルミノヘミアセタールが生成し、
加水分解することで対応するアルデヒドが得られる(Scheme II-6)。
22
Scheme II-6 DIBAL-H を用いるエステルからアルデヒドへの還元
また、エステルに対しジアミノアルミニウムハイドライドを用いることで対応するアル デヒドを選択的に得ることができるII-8。アルミニウム上に導入された窒素上の不対電子が アルミニウムの Lewis 酸を弱めているため反応全体も遅くなるが、特に第二段階が相対的 により遅くなるために選択的なアルミノヘミアセタールの生成が可能である (Scheme II-7)。
Scheme II-7 ジアミノアルニウムヒドリドを用いるエステルからアルデヒドの生成
過剰のジエチルアミン存在下にペンタン中で LiAlH4を用いた還元も可能である
(Scheme II-8)II-9。これは、一段階目の還元後に生じるアランがジエチルアミンと反応して ペンタンに不溶のアラン-アミン化合物が生じるためであると述べられている。
Scheme II-8 ジエチルアミン存在下でのエステルからアミドへの LiAlH4還元
一方、アミドのヒドリド還元においては、1 2で触れているように、一段階目の還元 によりヘミアミナールが生じ、これを加水分解することでアルデヒドが得られる。この反 応では、中間体である金属ヘミアミナールの窒素原子が還元剤に由来する金属と分子内相
23
互作用することで安定化され、二段階目の還元が遅くなることによりアルデヒドを選択的 に得ることができる。この反応では主に反応系中のアミドと還元剤の当量を制御すること が重要となる(Scheme II-9, a)。還元剤としては、LiAlH4、DIBAL-H、LiAlH(OtBu)3等が 用いられるII-10。さらに、Weinreb アミドと呼ばれる N-メトキシ-N-メチルアミドを用い たヒドリド還元では、一段階目の還元で生じたヘミアミナール上の還元剤に由来する金属 と窒素上にあるメトキシ基の酸素が相互作用することで、安定な中間体を生じるため、そ れ以上の還元は起こらない (Scheme II-9, b) II-11。
Scheme II-9 アミドからのアルデヒドへの還元
一方カルボン酸の還元では、一般に生成物は 1 級アルコールとなりアルデヒドを得るこ とは難しい。ただし、カルボン酸を基質とし添加剤によって系中でカルボン酸をアミド等 に変換し二段階目の還元を抑制する方法II-12、あるいは酸無水物等カルボン酸よりも反応 性の高い化合物に変換することで一段階目の還元を加速しアルデヒドを選択的に得る手法
II-13はある程度知られている。中間体を安定化する方法の例としては、先に述べた Weinreb
アミドを経由し DIBAL-H を用いる還元がある(Scheme II-10) II-12a。Deoxo-Fluor®は市販 されている脱酸素フッ素化試薬である。この手法では、系内でカルボン酸からカルボン酸 フッ化物を発生させ、そこにアミンを反応させることで Weinreb アミドに変換したのち還 元を行うことで、二段階目の還元を抑えることに成功している。
Scheme II-10 カルボン酸からアルデヒドへの 2 段階還元
24
カルボン酸を酸無水物に変換する例としては、山本らによるパラジウム触媒による水素 化(Scheme II-11) II-13a、同じく辻らによるパラジウム触媒によるヒドロシリル化 (Scheme II-12) II-13b等がある。これらの反応では、系中にピバル酸無水物を添加することで平衡に より生じた基質のカルボン酸無水物が還元されることで目的のアルデヒドが得られる。水 素化反応では、パラジウムに対して酸化的付加した基質の酸無水物が立体障害の大きいピ バル酸無水物よりも優先して水素化でアルデヒドが生成し、触媒と基質のカルボン酸が再 生する。一方ヒドロシリル化反応では、パラジウムに対して酸化的付加した基質のカルボ ン酸とピバル酸の酸無水物にヒドロシランが反応することでシリルエステルとアルデヒド が生成する反応サイクル、及びパラジウムに対して酸化的付加したヒドロシラン酸無水物 が反応してシリルエステルとアルデヒドが生成する反応サイクルが提唱されている。
Scheme II-11 パラジウム触媒を用いるカルボン酸からアルデヒドへの水素化還元
また、カルボキシメチレンイミニウム塩化物を経由する LiAlH(OtBu)3 による還元 (Scheme II-13) II-14aやカルボキシスルホニルメタンイミニウムの LiBH(Et)3による還元 (Scheme II-14) II-14bは、カルボン酸を混合酸無水物に変換することで一段階目の還元を加 速し、かつ基質上に窒素を含む官能基を導入することで中間体を安定するという手法を用 いている。
直接カルボン酸を還元してアルデヒドを得る手法として、ヒドロボランに嵩高い置換基 を導入してジボリルアセタールの還元を抑制することで選択的にアルデヒドを得る手法も 報告されている(Scheme II-15, a, b) II-15。エステルの還元と同様にジアミノアルミニウムハ イドライドを用いてアルミニウムの Lewis 酸性を下げることによるアルデヒドの選択的合 成も報告されている(Scheme II-15, c) II-16。
25
Scheme II-12 パラジウム触媒と Ph2SiH2を用いるカルボン酸無水物からアルデヒドの合 成
Scheme II-13 カルボン酸の活性化/ヒドリド還元によるアルデヒドの合成(1)
Scheme II-14 カルボン酸の活性化/ヒドリド還元によるアルデヒドの合成(2)
その他にカルボン酸を活性化せずに直接還元する方法としては、Wagenknecht による電 気化学を用いた還元 (Scheme II-16, a) II-17、固体触媒による水素化 (Scheme II-16, b) II-18 等が存在する。しかしながらそれぞれ反応に高温を要する、電気化学という特殊な手法を 用いる必要がありまた脂肪族のカルボン酸には適用できない、等問題も多く一般的に有用 な反応とは言い難い。
以上述べてきたように、カルボン酸誘導体の還元は本質的にヘミアセタールやヘミアミ ナール誘導体への第一段還元と炭素‐酸素結合の還元的切断の第二段還元の二段階からな る。言い換えれば、第一段還元の反応速度k1と第二段還元の反応速度k2の制御が選択的な
26
Scheme II-15 ジボリルあるいはジアルミノアセタールを経由するカルボン酸からアルデ ヒドの選択合成
Scheme II-16 その他のカルボン酸からアルデヒドへの直接還元
部分還元を達成する鍵である。これには、大別して 2 つの手法がある。1 つは中間体を安 定化してk2を抑制する方法で、エステルやカルボン酸の還元では中間体が安定になるよう 設計された置換基を有するホウ素やアルミニウムを用いることで二段階目の還元を抑える ことが可能である。アミドの還元における中間体であるヘミアミナールは中間体の窒素と 還元剤に由来する金属との分子内相互作用で安定化されている場合が多く、還元剤を過剰 量用いなければ、比較的高選択性的に部分還元が達成できる例がある(Scheme II-17)。こ のため、カルボン酸の還元では一旦系中でアミドを発生し、部分還元を達成する手法も開 発されている。もう 1 つは、カルボン酸基質をさらに反応性の高い誘導体に変換すること
27
により、k2よりもk1を圧倒的に増大させる手法であり、特にカルボン酸では系中でアルデ ヒドよりも反応性の高い酸無水物やその等価体に変換することが一般的に行われている。
Scheme II-17 アミドからアミンへの還元に必要な 2 段階反応間の速度調整
1 4.ヒドロシランを用いたカルボン酸誘導体の還元II-19
1 2及び1 3で述べたヒドリド還元剤の多くは水に対して不安定である。例えば、
LiAlH4や BH3は水と激しく反応して水素を発生する。NaBH4はこれらと比較して安定性 が高いが、水と穏やかに反応し水素を発生する。一方、ヒドリド還元剤の一種であるヒド ロシラン(R3SiH)は水や酸素に対して極めて安定であり、⾧期間湿気のある空気中に暴露 しても分解しない。一般にヒドロシランはそのままでは還元剤として作用しないが、カル ボニル基が強い Brønsted 酸や強い Lewis 酸で活性化されている場合、あるいはフッ素試 薬や遷移金属触媒を用いてヒドロシランが活性化されている場合には、カルボニル基の還 元に優れた効果を発揮する。ヒドロシランとしては、PhSiH3のような 1 級ヒドロシラン、
Ph2SiH2のような 2 級ヒドロシラン、R3SiH のような 3 級のヒドロシランが用いられる。
これらの活性化剤を必要としない特殊な例としては、後に触れる 5 配位ヒドロシランを用 いた還元反応がある。ヒドロシランはこれらの触媒や活性化剤の存在下でそのヒドロシリ ル基が炭素‐酸素二重結合に付加し H-C-O-Si 結合を作る、いわゆるヒドロシリル化反応 として、古くからケトンやアルデヒドのアルコールへの還元に利用されてきたII-19。カル ボン酸誘導体をヒドロシランでアルコールやアミンに還元するには、ヒドロシリル化に加 えて炭素‐酸素結合や炭素‐窒素結合の水素化分解が必要である。このため、ヒドロシラ ンを用いたカルボン酸誘導体の還元反応に関する研究は最近まであまり例がなかった。
しかしながらそれまでケトンやアルデヒドの還元に限られていた遷移金属触媒を用いる ヒドロシリル化研究は、1990 年代以降になると、アミドやエステル等のカルボン酸誘導体 のヒドロシラン還元に展開されるようになった。1991 年には Buchwald らによって Cp2TiCl2を用いたエステルのヒドロシラン還元が報告されている(Scheme II-18)II-20a。こ の反応は、Cp2TiCl2から-78 oC でnBuLi を用いて系中で活性種を発生させるものであり、
比較的温和な条件で内部及び末端オレフィン、アミン、チオフェン、エチレンオキシド等 の多様な官能基の存在下にエステルのみを還元できるのが特徴である。ただし、この報告
28
の条件下でケトンは還元される。この報告以降、チタン触媒に安価な Ti(OiPr)4を用いた 反応や、ヒドロシランに毒性の低い PMHS を用いた系も報告されているII-20b,c,d,e。
Scheme II-18 チタノセン触媒によるエステルのシリルエーテルへのシラン還元反応
1994 年には、1 当量の Ti(OiPr)4と 10 当量の PMHS を用いたカルボン酸のアルコール への還元も報告されているII-20d。また、1995 年には 5 員環、6 員環ラクトンの還元による ラクトールの選択的合成も報告されているII-20f。これらの報告では、ヒドロシランとアル コキシシランのbond metathesis によって生じるチタニウムヒドリドが鍵中間体となっ ていると述べられている。1995 年には Cutler らによってマンガンカルボニル触媒 (L)(CO)4MnC(O)CH3 (L=PPh3 or CO)とトリエトキシシランを用いて脂肪族エステルを 対応するエーテルへ比較的良好な収率で還元する反応が報告されている(Scheme II-19)
II-21。ただし、ラクトンの還元では収率が低下している。
Scheme II-19 マンガン触媒によるエステルのシリルエーテルへのシラン還元反応
1998 年には桑野、伊藤らによってロジウムホスフィン触媒とジフェニルシランを用いた 温和な条件における 3 級アミドのアミンへの還元が報告されている(Scheme II-20) II-22。カ ルボニル炭素やアミドの窒素上の置換基が嵩高い基質においては反応速度が落ちるものの、
反応終了後には高い単離収率を得ることに成功している。5 員環ラクタムにも適用可能で あり、エステルやエチレンオキシドの存在下にアミドのみを選択的にアミンへと還元する ことが可能である。ただし、アルケン及びアルキンを有する基質には適用することができ ない。
1999 年には古川らによって、同じくロジウムホスフィン触媒とジフェニルシランを用い るエステルのアルコールへの還元が報告されている(Scheme II-21) II-23。この報告では、
29
Scheme II-20 マンガン触媒によるアミドのアミンへのシラン還元反応
Wilkinson 錯体 RhCl(PPh3)3がエステルのヒドロシラン還元に対し高い活性を示すことが 明らかにされた。反応は室温で進行する。反応終了後に水酸化ナトリウムを用いた加水分 解を行うことによって、1H NMR による内部標準法で良好な収率で目的生成物が確認され ている。
Scheme II-21 ロジウム触媒を用いるエステルのアルコールへのシラン還元
さらに 2001 年には、渕上らによって Ru3(CO)12または[RuCl2(CO)3]2 / EtI, Et2NH のル テニウムカルボニル触媒とトリエチルシランを用いた系で単純なエステルを中から高程度 の転化率及び変換収率で対応するアルキルシリルアセタールに変換可能であるとの報告が なされている(Scheme II-22) II-24。生成したアルキルシリルアセタールは、塩酸を用いて対 応するアルデヒドへと加水分解が可能である。
Scheme II-22 ルテニウム触媒を用いるエステルのアルデヒドへのシラン還元
本研究発表後に報告された研究であるが、2016 年には、モリブデンカルボニル触媒 Mo(CO)6と TMDS を用い、反応温度を変えることで 3 級アミドからアルデヒドとアミン をそれぞれ選択的に合成する反応も報告されている(Scheme II-23) II-25。この反応はケトン や末端オレフィンやエステル、イミン、ニトリル、カルボン酸等を還元することなくアミ ドのみをアルデヒドへと還元することができるという大きな特徴を有する。フランやチオ フェンの含ヘテロ芳香族アミドにも適用が可能である。さらに、非常に嵩高いアダマンタ
30
ン酸アミドのアルデヒドへの還元も可能である。ただし、直鎖脂肪族アミドの還元では 対応するエナミンとアミンの混合物が生成するため、これらの基質には適さない。アミン への還元では、アルデヒド存在下にアミドのみを選択的にアミンへと還元することも可能 である。アルデヒドへの還元と同様、末端オレフィンやエステル、イミンの存在下にアミ ドを選択的にアミンへと還元が可能である。ニトロ基や共役内部オレフィンを有する基質 にも適用可能である。また、塩酸存在下にカルボキシル基を有する基質の還元も可能であ る。以上の反応をまとめたものを、Table II-2 に示す。
Scheme II-23 モリブデン触媒とヒドロシランによるアミドのアルデヒドあるいはアミ ンへの選択還元
Table II-2 カルボン酸誘導体のヒドロシラン還元のまとめ
31
これらと比較して活性や基質の汎用性の上で優れた触媒に、当研究室で開発されたルテ ニウム錯体(-Acenaphthylene)Ru3(CO)7、以下(ACE)Ru3(CO)7 (II-1)があるII-26a。 この錯体は空気に対し極めて安定であり、⾧期間大気に曝露しても分解しないという大き な特⾧を有する。錯体合成及びこの錯体を用いた触媒反応時の取り扱いにおいても、グロ ーブボックス等の設備を必要としない。(ACE)Ru3(CO)7は原料の Ru3(CO)12に対し 90%
以上の収率で合成することが可能である。また、大スケールの合成も可能である。さらに、
ケトンやアルデヒドだけでなく通常遷移金属錯体ではなく Lewis 酸を触媒とするアセター ルの還元が可能であるという特徴がある。ケトンやアルデヒドのヒドロシリル化に用いら れる Wilkinson 触媒と同様に反応性は用いるヒドロシランに大きく依存するが、Wilkinson 触媒と異なりジヒドロシランよりもビスヒドロシリル化合物を用いた場合のほうが 反応性が高くなる。また、不飽和ケトンでは還元に用いるヒドロシランを変えること で、1,4-付加と 1,2-付加を高い選択性で制御することができる。また、2002 年にはこの触 媒とエチルジメチルシランを用いてエステルのアルキルエーテルまたはシリルエーテルへ
の還元II-19x、カルボン酸のシリルエーテルへの還元II-19x、アミドのアミンへの還元II-19xも
達成している(Scheme II-24)。
Scheme II-24 ルテニウムクラスター触媒を用いるカルボン酸誘導体のヒドロシラン還元
エステルの還元では、脂肪族を中心に二重結合を含むオレイン酸メチルや 5 員環、6 員 環ラクトンについても検討を行っており、加水分解後は中から高程度の収率で目的とする アルコールまたはエーテルを得ることに成功している。また、反応性は下がるものの芳香 族カルボン酸のエステルである安息香酸メチルの還元においても中程度の収率で目的生成 物を得ることに成功している。カルボン酸の還元においても同様に、安息香酸では反応性 が下がる傾向にあるが、脂肪族カルボン酸では良好な収率で目的のアルコールを得ること