東北医科薬科大学 審査学位論文(博士)
氏名(本籍) ヤン ジェ
楊 (中国)
学位の種類 博士(薬科学)
学位記番号 博薬科第 22 号
学位授与の日付 令和2年3月10日
学位授与の要件 学位規則第4条1項該当
学位論文題名 EpCAM regulates cell adhesion and migration in cancer cells
論文審査委員
主査 教 授 細 野 雅 祐
副査 教 授 関 政 幸
副査 教 授 顧 建 国
EpCAM regulates cell adhesion and migration in cancer cells
令和 2 年 3 月
東北医科薬科大学大学院薬学研究科 楊 婕
Contents
1. Introduction ... 1
2. Materials and methods ... 3
3. Results ... 7
4. Discussion... 14
5. Reference ... 16
6. Abbreviation ... 19
7. Acknowledgement ... 20
1
1. Introduction
Cell adhesion is essential for various biological and pathological functions of multicellular organisms. There are two major types of cell adhesions, cell-cell and cell- extracellular matrix (ECM) adhesion [1].These adhesions have a different function and recognize different ligands. Cadherins and immunoglobulins are homophilic cell adhesion molecules (CAMs) for cell-cell adhesion, while integrins and selectins are heterophilic CAMs for cell-ECM adhesion. Cooperation or cross-regulation between cell–cell and cell–ECM adhesion has been implicated in multiple tissues, developmental processes and cancer metastasis. CAMs are cell surface receptors that not only promote cell adhesion in the ECM but also allow cells to interact and communicate with each other. In CAMs, epithelial cell adhesion molecule (EpCAM) is a unique type I membrane protein that is not one of the four major CAM families:
cadherin, integrin, selectin, and immunoglobulin-like-CAMs [2]. In the majority of cancer cells, EpCAM overexpression strongly correlates with the worse survival rates and poor prognoses for cancer patients [3-5]. For example, the expression levels of EpCAM are significantly associated with tumor recurrence in colon [6] and liver [7]
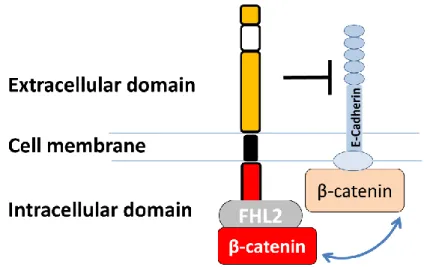
cancers, and have been correlated with a decrease in the overall survival rate of patients with gallbladder cancer [8]. Furthermore, the gene silencing of EpCAM has been shown to lead to a decrease in cell proliferation[9], migration, and lymph-node metastasis in primary and metastatic breast cancer [10]. In cell proliferation, EpCAM is sequentially cleaved by ADAM17 and presenilin-2, and then a fragment of the intracellular domain is translocated into the nuclear with FHL2, β-catenin, and Lef-1 to drive growth-signal genes such as c-Myc [9]. On the other hand, EpCAM promotes homophilic cell-cell interactions and provides functional antagonism for E-cadherin-mediated adhesions [11]
(Fig1). In addition, due to a common sequence with nidogen, a component of ECM, EpCAM is also thought to be involved in cell-ECM adhesion [12]. However, the connection between EpCAM expression and cell migration remains largely unknown.
2
Integrins are heterodimeric cell-surface receptors that mediate cell adhesion in the ECM typically by promoting epithelial cell adhesion to the basement membrane, and these receptors also contribute to the migration, proliferation and survival of tumor cells [13]. Integrins and EpCAM both span the cell membrane and promote cellular signaling and cancer migration, which is why elucidating the precise mechanisms involved in EpCAM-mediated adhesion is fundamental to understanding coordinated cell migration.
Previous studies have associated EpCAM with a tetraspanin-enriched microdomain in highly metastatic cells, where integrin α3 has also been observed, which suggests a functional association of EpCAM and ECM adhesion [14].
In the present study, we hypothesized the possibility of an interplay between EpCAM and integrin in cell adhesion, migration and proliferation. To elucidate this hypothesis, we established EpCAM-null cell lines, and found that the association between EpCAM and integrin β1 regulated cell adhesion and proliferation as well as cellular signaling. This association may shed light on the roles of EpCAM expression in cancer progression and metastasis, which could be useful in the development of new cancer drugs.
Fig. 1. Schematic structure of EpCAM.
EpCAM consists of extracellular domain, transmembrane domain and intracellular domain, which can bind with FHL2 and β-catenin, resulting in an inhibition of E-cadherin-mediated cell-cell adhesion.
3
2. Materials and methods
2.1 Antibodies and reagents
Antibodies for Western blot included the following: monoclonal antibodies(mAb) against integrin β1 (610468), integrin α5 (610634), integrin αV (611013), p-focal adhesion kinase (FAK) (611807), and FAK (610088) were purchased from BD Biosciences; mAbs for EpCAM (sc-25308) and integrin α3 (sc-6592) were acquired from Santa Cruz Biotechnology; mAbs against p-AKT (4060s), AKT (9272s), p-ERK 1/2 (4370s), and ERK 1/2 (4695s) were acquired from CST; and, mAbs against α-tubulin (T6199) were acquired from Millipore Sigma. For flow cytometry analysis and immunostaining, the following antibodies were used: EpCAM (Billerica, CBL251), integrin β1 (Development Studies Hybridoma Bank, University of Iowa, clone P5D2), integrin α3 (Santa Cruz Biotechnology, sc-32237), integrin α5 (Millipore Sigma, MAB1999), and integrin αV (BioLegend, 327902). Fibronectin (F0895) and laminin-511 (NO.892011) were purchased from Millipore Sigma and Matrixome Japan, respectively.
2.2. Cell culture
The CW-2, 293T and HeLa cell lines were purchased from RIKEN, Japan. A431 and MIA PaCa-2 cells were obtained from ATCC and Japanese Cancer Research Resources Bank, respectively. All cell lines were maintained at 37 °C in Dulbecco's modified Eagle's medium (DMEM), supplemented with 10% fetal bovine serum (FBS), under a humidified atmosphere containing 5% CO2.
2.3. Genomic deletion of EpCAM-KO in CW-2 and A431 cells
CRISPR/Cas9-based EpCAM-KO cells were established as described previously [15, 16]. Briefly, the target sequence of human EpCAM (TAATGTTATCACTATTGATC) was chosen from the CRISPR-Cas9 KO library and cloned into PX458 (plasmid 48138; Addgene). EpCAM-KO cell lines were created by electroporating cells according to the manufacturer's recommendations (Amaxa cell line
4
Nucleofectorkit R). Two days after transfection, GFP-positive cells were sorted using FACSAria II (BD Biosciences). Approximately 10 days later, the EpCAM-positive cells were removed by sorting. The negative selections were repeated 3 times to obtain a stable EpCAM-KO cell line.
2.4. Western blot and immunoprecipitation analysis
Cell lysates were prepared with lysis buffer containing 20 mM Tris-HCl at pH 7.4, 150 mM NaCl, 1% Triton X-100, protease inhibitors, and phosphatase inhibitors (Nacalai Tesque, Kyoto, Japan) on ice, which was centrifuged at 15,000 g for 10 min at 4 °C. After removing the nuclei by centrifugation, proteins were normalized by a bicinchonini cacid protein assay kit (Thermo Fisher Scientific). An equal amount of protein from each sample was subjected to SDS-PAGE and transferred to a PVDF membrane (Millipore Sigma). After blocking with 5% non-fat milk in TBS/0.05%
Tween-20 (TBST) and washing, the proteins were probed with indicated antibodies overnight. The membrane was then incubated with secondary antibodies for 2 h.
Detection was performed via chemiluminescence (Millipore Sigma). For immunoprecipitation, cells were lysed in 0.1% Triton in TBS buffer (20 mM Tris-HCl pH7.4, 150 mM NaCl) with a protease inhibitor and then centrifuged. The supernatants were immunoprecipitated and incubated with integrin β1 (P5D2) and a high-capacity protein-A mutant, Ab-Capcher Mag (ProteNova, Japan). After washing twice, the immunocomplex was eluted in2.5x loading buffer and subjected to SDS-PAGE.
2.5. Flow cytometry analysis
Cells were detached from the culture dishes using trypsin containing1 mM EDTA, and were then stained with the indicated primary antibodies for 1 h on ice, followed by washing, and then incubation with Alexa Fluor 647 goat anti-mouse IgG (Thermo Fisher Scientific) for 30 min on ice. Finally, the cells were washed three times with PBS and analyzed via FACS Calibur (BD Biosciences).
5 2.6. Cell-migration assay
The cell-migration assay was described previously [15]. The cell migration test was accomplished using a 24-well transwell chamber system (8.0-μm pore control inserts;
BD Biosciences). Only the bottom side of the chamber was coated with 10 μg/ml fibronectin and let stand at 4 °C overnight, and was then blocked with 1% BSA. Cells were trypsinized and re-suspended in culture media. The cell suspensions were then seeded in the upper portion of the fibronectin-pretreated chamber at 8 × 104 cells in 0.4 ml 3% FBS DMEM. Then, 0.5 ml of DMEM with 3%FBS was placed in the bottom well. After incubation for 16 h, the migrated cells on the lower surface were fixed, stained with crystal violet overnight, and counted under a light microscope.
2.7. Immunofluorescence staining
Cells were seeded on a glass-bottom dish washed with PBS and fixed with 4%
paraformaldehyde. The cells were permeabilized with 0.1% TritonX-100 in PBS. The cover glasses were blocked with 0.1% Tween-20and 3% BSA in PBS and then stained with EpCAM antibody diluted 1 to100 and let stand overnight at 4 °C. After rinsing three times with PBS, cells were incubated with secondary Alexa 488-labeled goat anti- mouse IgG and then with TO-PRO-3 (Thermo Fisher Scientific) under darkness. Finally, the cells were mounted and confocal microscope images were acquired by sequential excitation using an Olympus FV1000 laser scanning confocal microscope with an UPlanSApo 60X/1.35 Oil objective, which was operated by F10-ASW ver. 4.02 software.
2.8. Cell adhesion assay
Cell adhesion assay was performed as previously described with minor modifications [16, 17]. Six-well plates were coated with laminin-511 (0.5 μg/ml) or fibronectin (10 μg/ml) in PBS overnight at 4 °C and then blocked with 1% BSA in DMEM for 1 h at 37 °C. The indicated cells were detached and suspended in DMEM without FBS. After replating either on laminin-511 (0.5 μg/ml) for 2 h or on fibronectin (1 μg/ml) for 20
6
min, the non-adherent cells were gently removed by washing with PBS 3 times. The attached cells were then fixed with 4% paraformaldehyde in PBS, and photos were then taken by phase-contrast microscopy (Olympus, Japan). Cell spreading was determined in at least 20 cells per view, and only cells that contained lamellipodia regions were counted as spreading cells.
2.9. Cell growth assay
Cell growth assays were performed as previously described [16]. Briefly, cells were seeded into 6-well culture dishes and let stand overnight. Cells were detached and counted. The fold change in the number of cells was normalized to the number on day 0.
2.10. Statistical analysis
Results are presented as the mean ± S.E.M. Statistical analyses were performed using a two-tail unpaired Welch's correction t-test using GraphPad Prism 6. (*, ** and ***
represent P-values of <0.05, 0.01, and 0.001, respectively).
7
3. Results
3.1. Comparison of the EpCAM expression levels in various cancer cell lines EpCAM overexpression is known to be essential for the proliferation, migration and invasiveness of tumor cells [10]. However, the molecular mechanisms of EpCAM that induce those phenotypes remain unclear. To gain better insight into the expressions and functions of EpCAM in cancer cells, we first examined the expression levels of EpCAM in six human cell lines: MiaPaCa-2, A431, CW-2, 293T, HeLa, MDA-MB-231, and HepG2.As shown in Fig. 2A, CW-2 and A431 cells showed relatively higher expression levels among these cells. Other cell lines only expressed a marginal level of EpCAM.
Therefore, we selected both CW-2 and A431 cells as models for examining the functions of EpCAM expression.
8
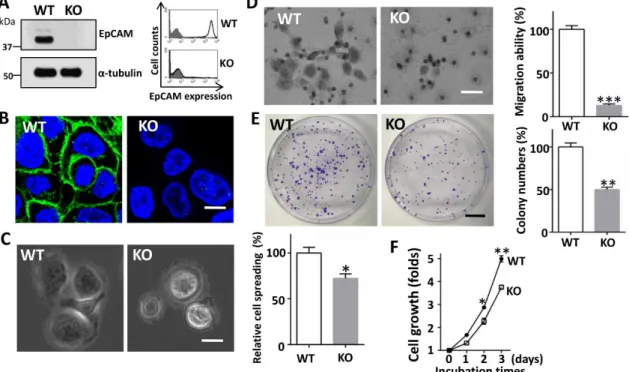
Fig.2. Establishment of EpCAM knockout (KO) CW-2 cells.
Expression levels of EpCAM in different cancer lines were examined by Western blot. Cell lysates of indicated cell lines were probed with EpCAM, and reprobed with anti-α-tubulin antibody. B) The efficiency of an EpCAM knock- out gene, produced using the CRISP/Cas9 system, was assessed by Western blot in CW-2 cell lysates. C) Expression levels of EpCAM on the cell surface (black line, no fill) were verified by flow cytometry analysis. The grey shadow represents the negative control. D) Phase contrast for both WT and EpCAM KO CW-2 cells developed by normal plastic culture. Representative images were taken via phase-contrast microscope. Scale bar is 50 mm. E) After incubation, representative images were taken via a phase-contrast microscope. Insert: a magnified view of the area, as indicated by the box. Scale bar, 130 mm. The percentages of spread cells were statistically analyzed. Data represent the mean
± S.E.M; ***p < 0.01 vs. WT CW-2 cells (n = 5). WT set as 100%.
3.2. EpCAM is important for cell adhesion on the ECM
To further address the contribution of EpCAM to the adhesion, migration and proliferation of carcinoma cells, we began by generating CRISPR/Cas9-mediated KO cells using the colorectal cancer CW-2 cell line, which was confirmed by Western blotting of total cell lysates and flow cytometry analysis using anti-EpCAM antibody (Fig. 2B and C).The wild type (WT) of CW-2 cells exhibited a slightly elongated morphology, while the KO cells exhibited a round cell morphology on normal culture dishes (Fig. 2D), which suggested that EpCAM may affect cell adhesion in the ECM. We then compared the cell adhesion of KO cells with that of the WT cells on cultured dishes coated with laminin- 511, a major component of the ECM of epithelia cells. As shown in Fig. 2E, the KO cells showed significantly decreased cell spreading compared with that in the WT cells. These results clearly suggested that EpCAM is involved in cell adhesion on the ECM.
9
3.3. Cellular signaling was decreased in the EpCAM-KO cells
FAK activation is well known to involve integrin receptor clustering with the binding of cells in the ECM via cell migration [18]. The activation of AKT and ERK is also involved in the integrin signaling pathway[19]. To examine whether these intracellular signals were affected by EpCAM deficiency, we compared the phosphorylation levels of FAK, AKT and ERK between the WT and KO cells. Interestingly, the phosphorylated FAK (p-FAK) and AKT (p-AKT) were significantly decreased in the KO cells, compared with those in the WT cells (Fig. 3A upper and middle panel). In agreement with a previous report [20], p-ERK was also decreased in the KO cells (Fig. 3A lower panel). These results suggested that EpCAM affects integrin-mediated signaling.
10
Fig. 3. Effects of EpCAM on the activation of FAK, AKT and ERK signaling.
A) Equal amounts of cell lysates from WT and KO cells were analyzed by immunoblot with indicated antibodies. B) Quantitative analysis of p-FAK, p-AKT and p-ERK expression levels normalized to the total levels of FAK, AKT and ERK in WT and EpCAM-KO CW-2 cells. Error bars: S.E.M. of three independent samples for each cell line; *p < 0.05, **p < 0.01 vs. WT CW-2 cells.
3.4. Involvement of EpCAM in cell spreading and migration in A431 cells
To understand whether the phenotypes described in CW-2 cells were general observations, we further inactivated EpCAM genes using CRISPR/Cas9 technology in A431 cells, because these cells express a relatively high level of EpCAM (Fig. 2). As Fig.
4A shows, the EpCAM expressions were completely diminished in the total cell lysate and on the cell surface. Furthermore, immunostaining analysis revealed that EpCAM were localized mainly at the cell-cell contacts in WT cells, while the immunostaining for EpCAM was completely negative in KO cells (Fig. 4B). Consistent with the data obtained from CW-2 cells, cell spread on a fibronectin-coated dish was also suppressed in EpCAM-KO A431 cells (Fig. 4C). Moreover, a transwell assay showed that cell migration was also significantly attenuated by the depletion of EpCAM (Fig. 4D), suggesting that the expression of EpCAM plays an important role in integrin-mediated cell migration. Furthermore, we found a similar tendency for the down-regulation of p- FAK and p-ERK in the KO of CW-2 (Fig. 3) and A431 cells compared with WT cells (data not shown). Several in vitro and in vivo studies have demonstrated that EpCAM predominately act on the induction of cell proliferation [21]. Therefore, we performed colony-formation and cell-proliferation assays (Fig. 4E and F). As expected, the EpCAM KO cells significantly inhibited colony formation and cell proliferation in A431 cells.
3.5. Co-Immunoprecipitation experiment demonstrated that EpCAM associates with integrin β1
Integrin β1 subunits involve 12 integrin constituents, the largest integrin subfamily, that play important roles in cell adhesion [13]. To understand EpCAM regulation and
11
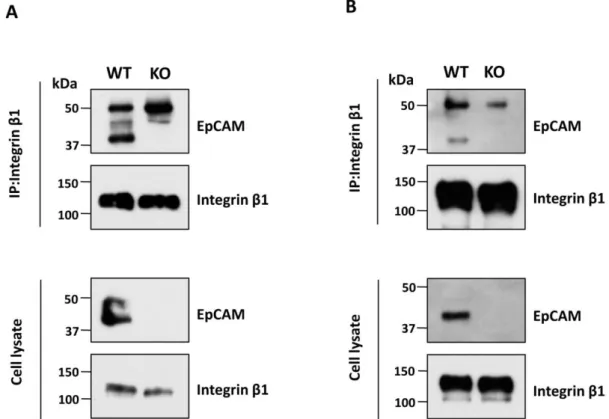
integrin-mediated cellular function and signaling, we examined integrin-mediated complex formation. Interestingly, co-immunoprecipitation using anti-integrin β1 antibody showed EpCAM proteins existing in the immunoprecipitates of CW-2 cells (Fig.5A) and A431 cells (Fig. 5B).
Fig. 4. Comparison of cell adhesion, migration, proliferation and colony formation in WT and EpCAM-KO A431 cells.
A) EpCAM-KO cells were confirmed by immunoblot with anti-EpCAM antibody, α-tubulin was used as a loading control (left). Expression levels of EpCAM on the cell surface were tested using flow cytometry analysis in WT and KO cells (Black line, no fill). The grey shadow represents negative controls without the first antibody (right). B) WT and EpCAM-KOA431 cells were cultured on a coverslip, immunostained with anti-EpCAM antibody (green) and TO-PRO-3 for nuclei (blue). Scale bar is 10 mm. C) Cell spreading was observed on a fibronectin- coated dish, and representative images were taken via a phase-contrast microscope after replating for 20 min. Insert: a magnified view of the area indicated by the box.
Scale bar, 30 mm (left). The percentages of spread cells were statistically analyzed.
WT was set as 100% (right). D) Cell migration toward fibronectin was determined by a transwell assay, as described in “Materials and Methods.” Migrated cells were stained with crystal violet solution. Representative images were shown using phase- contrast microscopy (left). Scale bar is 35 mm. The cell numbers were obtained from five random fields. WT was set as 100% (right). E) WT and KO cells (1000/per well) were grown for 14 days, then fixed and stained with crystal violet
12
and acquired. Scale bar is 100 mm (left). The numbers of foci were counted in each well. WT was set as 100% (right). F) Cells were cultured in DMEM containing 10%
FBS, and cell numbers were counted on days 1, 2, and 3 after seeding. All graphs represent the mean ± S.E.M; *p < 0.05, **p < 0.01, ***p < 0.001 vs. WT A431 cells (n = 5).
Fig. 5. Complex formation between EpCAM and integrin β1 in both cells.
Cell lysates obtained from the WT and EpCAM-KO of CW-2 (A) and A431 (B) were immunoprecipitated using anti-β1 antibody and then immunoblotted with EpCAM (upper panels) and β1 (low panels) antibody for detection.
3.6 EpCAM-KO A431 cells reduced the expression levels of integrin α5
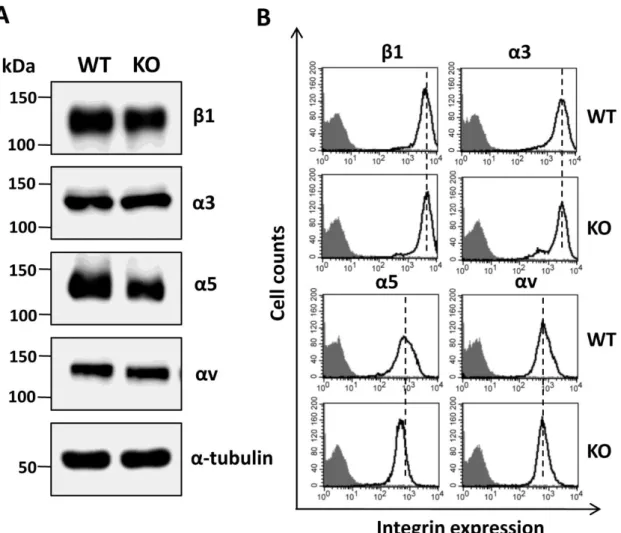
Furthermore, analysis by Western blot showed that the expression levels of a5 (not α3, αV and β1) were decreased in total cell lysates (Fig. 6A), which was also confirmed by flow cytometry analysis of EpCAM-KO A431 cells (Fig. 6B). These results strongly suggest that EpCAM expression and function may at least partially be dependent on integrins.
13
Fig 6. Comparison of integrin β1, α3, α5, and αV in WT and KO A431 cells.
A) Cell lysates from WT and KO cells were immunoblotted with indicated integrin antibodies. a-Tubulin was used as a loading control. B) Cells were stained with integrin antibodies (bold line, no fill) or without (grey shadow) and subjected to flow cytometry analysis to examine the expression levels on the cell surface.
14 4. Discussion
In the present study, we used CW-2- and A431 EpCAM-deficient cancer cell lines to examined the involvement of EpCAM in cell adhesion, migration and proliferation, as well as in the activation of FAK, AKT and ERK. Mechanistically, it is plausible that the physical interaction with integrin β1 could be one of the main connections between EpCAM and ECM adhesion that includes cell spreading and migration. In fact, this hypothesis was greatly supported by the evidence that EpCAM shares a common sequence with nidogen, which is an important component of ECM [12]. It is well known that the interdependence between cell-cell and cell-ECM adhesion plays an important role in cell biological functions [22]. For example, the loss of cell-ECM adhesion resulted in an aberrant distribution of the actin cytoskeleton, a reduction in cell migration [23], and an increase in cell-cell adhesion [24]. Conversely, the disruption of cell-cell adhesion induced by TGF-β, increased the cell-ECM adhesion [25]. EpCAM is known to exist in tetraspanin-enriched micro-domains [14]. FHL2 might be involved in interaction between EpCAM and integrins (Fig. 7). Although the underlying molecular mechanism for the interaction between EpCAM and integrin β1 remains unclear, it is clear that EpCAM regulates cell-ECM adhesion.
Curiously, in previous reports EpCAM negatively regulated cell migration and ERK phosphorylation in a polarized MDCK cell line and in Xenopus laevis development [26, 27]. Thus far, the differences in the potential roles of EpCAM in various cells remain unclear, but it is reasonable to postulate that EpCAM plays different roles in normal cells than in cancerous cells. Actually, cancer tissues differentially express integrins, which are associated with disease progression and diminished patient survival [13]. In cancers, EpCAM could associate with β1 to help integrins drive cancer cell migration and proliferation.
In the majority of cancers, EpCAM overexpression correlates with an unfavorable prognosis, and can be utilized as a biomarker to distinguish a higher risk for recurrence in patients. Thus, EpCAM is an attractive target for the treatment of various cancers.
15
Although the use of an anti-EpCAM antibody alone failed in clinical trials, receptor binding antibodies that recognize both EpCAM and CD3 have been effective and approved for the treatment of colon cancer [28]. In the present study, we found that EpCAM associated with integrin β1 and regulated cell adhesion,and, therefore, we are motivated to speculate that a bi-specific antagonist targeting both β1 and EpCAM, or both specific functional blocking antibodies, could be useful for cancer treatment.
Fig.7 Proposed molecular for complex formation between EpCAM and integrin.
It has been reported that FHL2 can associated with either EpCAM [2] or integrin [29] via cytoplasmic domain. Therefore, FHL2 may directly or indirectly involved in both physical interaction and functional connection.
16 5. Reference
1. Kawauchi, T., Cell adhesion and its endocytic regulation in cell migration during neural development and cancer metastasis. Int J Mol Sci, 2012. 13(4): p. 4564-90.
2. Munz, M., P.A. Baeuerle, and O. Gires, The emerging role of EpCAM in cancer and stem cell signaling. Cancer Res, 2009. 69(14): p. 5627-9.
3. Spizzo, G., et al., High Ep-CAM expression is associated with poor prognosis in node-positive breast cancer. Breast Cancer Res Treat, 2004. 86(3): p. 207-13.
4. Spizzo, G., et al., Overexpression of epithelial cell adhesion molecule (Ep-CAM) is an independent prognostic marker for reduced survival of patients with epithelial ovarian cancer. Gynecol Oncol, 2006. 103(2): p. 483-8.
5. Nagrath, S., et al., Isolation of rare circulating tumour cells in cancer patients by microchip technology. Nature, 2007. 450(7173): p. 1235-9.
6. Goossens-Beumer, I.J., et al., Clinical prognostic value of combined analysis of Aldh1, Survivin, and EpCAM expression in colorectal cancer. Br J Cancer, 2014.
110(12): p. 2935-44.
7. Sun, Y.F., et al., Circulating stem cell-like epithelial cell adhesion molecule- positive tumor cells indicate poor prognosis of hepatocellular carcinoma after curative resection. Hepatology, 2013. 57(4): p. 1458-68.
8. Varga, M., et al., Overexpression of epithelial cell adhesion molecule antigen in gallbladder carcinoma is an independent marker for poor survival. Clin Cancer Res, 2004. 10(9): p. 3131-6.
9. Schnell, U., V. Cirulli, and B.N. Giepmans, EpCAM: structure and function in health and disease. Biochim Biophys Acta, 2013. 1828(8): p. 1989-2001.
10. Osta, W.A., et al., EpCAM is overexpressed in breast cancer and is a potential target for breast cancer gene therapy. Cancer Res, 2004. 64(16): p. 5818-24.
11. Gaiser, M.R., et al., Cancer-associated epithelial cell adhesion molecule (EpCAM; CD326) enables epidermal Langerhans cell motility and migration in vivo. Proc Natl Acad Sci U S A, 2012. 109(15): p. E889-97.
17
12. Simon, B., et al., Epithelial glycoprotein is a member of a family of epithelial cell surface antigens homologous to nidogen, a matrix adhesion protein. Proc Natl Acad Sci U S A, 1990. 87(7): p. 2755-9.
13. Desgrosellier, J.S. and D.A. Cheresh, Integrins in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer, 2010. 10(1): p. 9-22.
14. Schmidt, M., et al., Phase IB study of the EpCAM antibody adecatumumab combined with docetaxel in patients with EpCAM-positive relapsed or refractory advanced-stage breast cancer. Ann Oncol, 2012. 23(9): p. 2306-13.
15. Isaji, T., et al., A complex between phosphatidylinositol 4-kinase IIalpha and integrin alpha3beta1 is required for N-glycan sialylation in cancer cells. J Biol Chem, 2019. 294(12): p. 4425-4436.
16. Zhang, G., et al., N-acetylglucosaminyltransferase-I as a novel regulator of epithelial-mesenchymal transition. Faseb j, 2019. 33(2): p. 2823-2835.
17. Isaji, T., et al., N-glycosylation of the beta-propeller domain of the integrin alpha5 subunit is essential for alpha5beta1 heterodimerization, expression on the cell surface, and its biological function. J Biol Chem, 2006. 281(44): p. 33258-67.
18. Sulzmaier, F.J., C. Jean, and D.D. Schlaepfer, FAK in cancer: mechanistic findings and clinical applications. Nat Rev Cancer, 2014. 14(9): p. 598-610.
19. Steelman, L.S., et al., JAK/STAT, Raf/MEK/ERK, PI3K/Akt and BCR-ABL in cell cycle progression and leukemogenesis. Leukemia, 2004. 18(2): p. 189-218.
20. Gao, J., et al., By inhibiting Ras/Raf/ERK and MMP-9, knockdown of EpCAM inhibits breast cancer cell growth and metastasis. Oncotarget, 2015. 6(29): p.
27187-98.
21. Wenqi, D., et al., EpCAM is overexpressed in gastric cancer and its downregulation suppresses proliferation of gastric cancer. J Cancer Res Clin Oncol, 2009. 135(9): p. 1277-85.
18
22. Burute, M. and M. Thery, Spatial segregation between cell-cell and cell-matrix adhesions. Curr Opin Cell Biol, 2012. 24(5): p. 628-36.
23. Goodwin, K., et al., Cell-cell and cell-extracellular matrix adhesions cooperate to organize actomyosin networks and maintain force transmission during dorsal closure. Mol Biol Cell, 2017. 28(10): p. 1301-1310.
24. Hou, S., et al., Distinct effects of beta1 integrin on cell proliferation and cellular signaling in MDA-MB-231 breast cancer cells. Sci Rep, 2016. 6: p. 18430.
25. Xu, Q., et al., Roles of N-acetylglucosaminyltransferase III in epithelial-to- mesenchymal transition induced by transforming growth factor beta1 (TGF- beta1) in epithelial cell lines. J Biol Chem, 2012. 287(20): p. 16563-74.
26. Barth, A.I.M., H. Kim, and I.H. Riedel-Kruse, Regulation of epithelial migration by epithelial cell adhesion molecule requires its Claudin-7 interaction domain.
PLoS One, 2018. 13(10): p. e0204957.
27. Maghzal, N., et al., EpCAM controls actomyosin contractility and cell adhesion by direct inhibition of PKC. Dev Cell, 2013. 27(3): p. 263-77.
28. Linke, R., A. Klein, and D. Seimetz, Catumaxomab: clinical development and future directions. MAbs, 2010. 2(2): p. 129-36.
29. Wixler, V., et al., The LIM-only protein DRAL/FHL2 binds to the cytoplasmic domain of several alpha and beta integrin chains and is recruited to adhesion complexes. J Biol Chem, 2000. 275(43): p. 33669-78.
19 6. Abbreviation
CAMs: cell adhesion molecules;
ECM: extracellular matrix;
EpCAM: epithelial cell adhesion molecule;
ERK: extracellular regulated protein kinases;
FAK: focal adhesion kinase;
FBS: fetal bovine serum;
KO: knockout;
WT: wild type.
20 7. Acknowledgement
First of all, I owe my sincerely gratitude to President Motoaki Takayanagi to provide the opportunity of studying in Tohoku Medical and Pharmaceutical University.
I would like to show my gratitude to my supervisor, Prof. Jianguo Gu. This thesis would not have been possible unless Prof. Gu designed the project, analyzed the data and revised the manuscript.
I would like to thank Tomoya Isaji for the guidance on the experiments. Many thanks for Dr. Tomohiko Fukuda provided technique helps. Thanks for the helps from Mr. Guowei Zhang, Mr. Feng Qi, Mr. Chengwei Duan, and Ms. Yan Hao.
Finally, I would like to thank my family and all those people who have, directly or indirectly, made efforts and contributions to completion of this dissertation.