44:174 <総 説>
黄疸の成因と病態
足立 幸彦
諸岡 留美
要旨:黄疸はビリルビンの生成から肝細胞でのグルクロン酸抱合を経て腸管内への排泄までの いずれかの代謝・輸送段階の破綻によって発症する.近年肝細胞の抱合酵素 UGT1A1,毛細胆 管膜の ATP 依存性輸送蛋白 MRP2,類洞側膜局在の MRP3 等の核内レセプターによる発現調節についての解明が進んでいる.Gilbert 症候群を起こす UGT1A1 遺伝子多型(UGT1A1*
6)と薬 物や発癌物質の代謝にかかわる UGT1A アイソザイムの遺伝子多型(UGT1A6* 2,UGT1A7* 3) とのリンクが認められ,また有機アニオン系薬物を輸送する MRP2 活性が Dubin-Johnson 症候 群で欠如,肝細胞内輸送蛋白 GSTα が Rotor 症候群で欠如していることから,各種体質性黄疸 において薬物代謝遅延が生じ得る.これらの代謝酵素,輸送蛋白は黄疸を伴う後天性の肝胆道 疾患でも低下が報告されており,薬物代謝の面からも大きな問題となるので注意が必要である. 索引用語: 黄疸 核内レセプター 遺伝子多型 薬物代謝 オーダーメイド医療 はじめに 黄疸ではビリルビンの貯留により全身の皮膚,粘膜 が黄染するが,一般に血清ビリルビンが 2.5mg!dl 前後 を超えないと顕性とはならない.1.8∼2.5mg!dl の範囲 では眼球強膜の特に周辺部がわずかに黄染するのみな ので特に亜黄疸(subicterus,scleral icterus)とも呼ん でいる.臨床でよく遭遇する胆汁うっ滞は黄疸が主症 状となるが,胆汁酸,コレステロールを始め各種胆汁 成分の体内貯留を来す点で黄疸と同義ではない. 黄疸時,血清で上昇しているビリルビンには非抱合 型と抱合型,およびデルタ(δ)ビリルビンの 3 種類が 存在する.血清ビリルビンの種類を含め黄疸の病態生 理を理解するために,まず肝細胞を中心としたビリル ビンの代謝,排泄について簡単に紹介し,各種病態時 における変化,薬物代謝との関連等を遺伝子レベルの 研究成果を含めて述べることとする1). ビリルビンの生成から胆汁中排泄まで(図 1) 成人の場合 1 日 250∼300mg のビリルビンが体内でヘ ムから生成されているが,その約 8 割は老廃赤血球の ヘモグロビン由来,残りの約 2 割が骨髄での無効造血 や肝での代謝回転の早いヘムに由来している.後者は シャントビリルビンと呼ばれている. ビリルビンは水溶性が非常に低く血中では通常アル ブミンに結合され運搬される.肝小葉において,微量 の蛋白非結合のビリルビンが肝細胞シヌソイド側膜に 局在する電位依存性(Na+非依存性)の輸送蛋白 OATP2
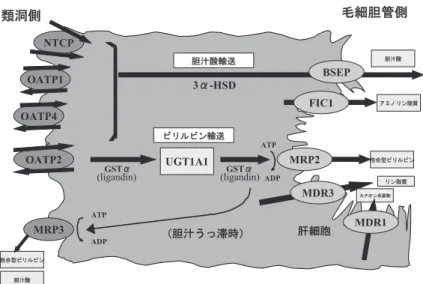
(organic anion-transporting polypeptide 2;OATP-C,OATP1B1 とも呼称される)により血中から肝細胞 内へ電位依存性に摂取される.この摂取は,肝細胞か ら血中に放出されるグルタチオンの輸送と共役してい ると考えられており,また胆汁うっ滞時には逆方向の 輸送も起こりうる.一方,ビリルビンがアルブミン結 合状態のまま肝細胞内に摂取されているとの in vitro の観察報告もあり,摂取機構が完全に解明されたわけ ではない2). ビリルビンは肝 細 胞 内 で は 結 合 蛋 白 GSTα(glu-tathione S-transferaseα;ligandin)に結合されて小胞 体 に 運 ば れ,小 胞 体 膜 上 の UGT1A1(UDP-glucuronosyltransferase 1A1)によりグルクロン酸抱合 を受けて水溶性の抱合型ビリルビン(bilirubin monoglu-curonide 及び bilirubin diglumonoglu-curonide)となる.抱合型 ビリルビンは,再び GSTα に結合され運搬されると考 えられているが,毛細胆管膜において ATP 依存性の輸 送 蛋 白 MRP2(multidrug resisitance-associated pro-tein 2;ABCC2)によって胆汁中へ能動的に排泄される. なお,毛細胆管膜には MRP2 のほかに ATP 依存性の輸 送蛋白,FIC1(ATP8B1)(アミノリン脂質の膜内輸送), BSEP(ABCB11)(胆汁酸を輸送),MDR1(ABCB1) 桑名市民病院内科 胆 道 23 巻 2 号 174∼180(2009)
図 1 肝細胞のビリルビン代謝と輸送の模式図
胆汁酸の膜輸送蛋白等も一緒に示す.NTCP:Na + /taurocholate co-transport
-ing protein,OATP:organicanion-transporting polypeptide,MRP:multidrug resisitance-associated protein,BSEP:bile saltexportpump,FIC1:progres -sive familialintrahepaticcholestasistype 1(Byler病)にて欠損している ABC ト ラ ン ス ポ ー タ ー,MDR:multidrug resistance gene product,UGT1A1: UDP-glucuronosyltransferase 1A1,GSTα:glutathione S-transferase α, 3α-HSD:3α-hydroxysteroid dehydrogenase.
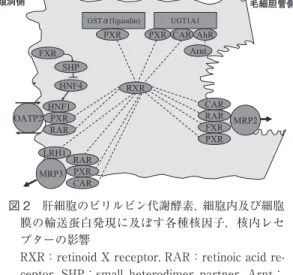
(カチオン系薬物などを輸送),MDR3(ABCB4)(リン 脂質の膜内輸送)といった輸送蛋白が存在し3),MDR1 を除いてこれらの機能異常にても胆汁うっ滞,黄疸を 発症しうる. 胆汁うっ滞時には MRP2 の表出が低下し抱合型ビリ ルビンの細胞内貯留が起こるが,シヌソイド側の肝細 胞膜上に健常時にはわずかにしか発現していない ATP 依存性輸送蛋白 MRP3(ABCC3)が誘導されて,抱合 型ビリルビンや胆汁酸の血中への逆流を来す4)5).また, OATP2 による抱合型ビリルビンの血中への逆輸送も起 こりうる. 肝細胞におけるこれら多くのビリルビン代謝に関わ る酵素,輸送蛋白の発現は,薬物代謝や胆汁酸代謝の 調節と同様に,核内受容体である PXR(pregnane X receptor;アゴニストとして dexamethasone,rifam-picin など),FXR(farnesoid X receptor;アゴニスト と し て cholic acid な ど),CAR(constitutive andro-stane receptor;ア ゴ ニ ス ト と し て phenobarbital な ど),AhR(aryl hydrocarbon receptor),Nrf2(nu-clear factor E2-related factor 2),PPARα(peroxisome proliferators activator receptorα;アゴニストとして
fi-brate など)等や,肝細胞核因子の HNF4α(hepatocyte nuclear factor 4α),HNF1α 等の直接作用またはクロス トークにより主に転写レベルで調節を受けており,胆 汁うっ滞時などの病態時における肝細胞障害の回避に 合目的的に発現の誘導や抑制が行われているようであ る(図 2)6)7).これらは肝細胞の胆汁酸代謝とも連動し ている.またこれら因子の発現に個人差のあることか ら,ビリルビン代謝に個人差の存在することも説明可 能となる.一方,UDCA による MRP2 誘導のように核 内レセプターを介さない誘導も観察されている. 更に,胆汁うっ滞時の腎においては尿細管上皮細胞 の管腔側膜上に MRP2 が誘導されて抱合型ビリルビン の尿中排泄が増加する8). 胆汁へ排泄された抱合型ビリルビンは一時胆嚢にて 貯留,濃縮され,食事摂取と共に十二指腸へと排泄さ れる.小腸において細菌性のβ グルクロニダーゼで脱 抱合されたビリルビンは更にウロビリノゲンを含むウ ロビリン体へ還元され大部分は便中に排泄される.ウ ロビリノゲンの一部は小腸粘膜上皮細胞により再吸収 され肝に至り,大部分は胆汁中へと排泄される(腸肝 循環)が,更に一部は尿中に排泄されている.新生児
胆 道 23 巻 2 号(2009) 46:176
図 2 肝細胞のビリルビン代謝酵素,細胞内及び細胞 膜の輸送蛋白発現に及ぼす各種核因子,核内レセ プターの影響
RXR:retinoid X receptor,RAR:retinoicacid re ceptor, SHP:small heterodimer partner, Arnt: Ahrnucleartranslocator.他は本文を参照.多く の核内受容体が RXRと heterodimerを作ってトラ ン ス ポ ー タ ー や 酵 素 の 遺 伝 子 を 調 節 し て い る. 点線は heterodimer形成を示す.SHPは HNF4に 抑制的に作用する. 黄疸のように大量のビリルビンが腸内に存在する場合 には,非抱合型ビリルビンの腸管からの再吸収も臨床 的に問題となる.一方,Crigler-Najjar 症候群 I 型では 抱合型ビリルビンが生成されないので胆汁中にはビリ ルビンがほとんど含まれないが,代償的に非抱合型ビ リルビンの腸管上皮からの排泄が起こると報告されて いる9). 黄疸の分類と病態 健常者における血中ビリルビンはほとんどが非抱合 型ビリルビンで占められているが,肝細胞の UGT1A1 による抱合段階以降の障害があれば,抱合型ビリルビ ンおよび抱合型ビリルビンが血管内でアルブミンと共 有結合して生じたδ ビリルビンが増加する10).非抱合型 ビリルビンは大部分が反応促進剤を添加しないとジア ゾ反応が進まないが,一方抱合型ビリルビンとδ ビリル ビンは大部分が反応促進剤添加なしでジアゾ反応が進 む.従って,臨床においては前者を間接ビリルビン, 後 2 者を直接ビリルビンと呼んで,それぞれ非抱合型, 抱合型ビリルビンとほぼ同義に使用している.高間接 ビリルビン血症(間接ビリルビンが総ビリルビンの 80% 以上)の場合に非抱合型ビリルビンの増加している病 態,高直接ビリルビン血症(直接ビリルビンが 50% 以 上)を抱合型ビリルビンが増加している病態と見なし て臨床的に診断,治療に用いている.語義の混乱も生 じているが,δ ビリルビンを除外して抱合型ビリルビン のみを測定できる“直接ビリルビン測定キット”も病 態を良く把握できるので,臨床使用されている. 高間接ビリルビン血症 ビリルビンの生成増加を来す溶血性貧血,新生児黄 疸,シャント高ビリルビン血症や,UGT1A1 によるビ リルビンのグルクロン酸抱合能が低下する体質性黄疸 (Gilbert 症候群,Crigler-Najjar 症候群 I,II 型)や酵素 活性阻害作用のある薬物投与時などの場合に軽度∼高 度の黄疸を認める.Crigler-Najjar 症候群 I 型や高度の 新生児黄疸では核黄疸の危険性があるが,それ以外で は一般に黄疸そのものが予後を左右することは無い. Gilbert 症候群は頻度が高く人口の 2∼7% 存在しており, UGT1A1 活性が健常者の 25∼30% に低下しているため 血清ビリルビンが 2∼6mg!dl に軽度に上昇している. 各症候群の診断はそれぞれの基準によるので詳細は略 するが,最近は診断手段として遺伝子 UGT1A1 の解析 が行われている.UGT1A1 はイリノテカン,エチニル エストラジール,ロラゼパムなどのグルクロン酸抱合 も司っている.近年 UGT1A1 のプロモーター領域の TATA box に お い て A(TA)6TAA の と こ ろ
A(TA)7TAA と変異している多型(UGT1A1*28)をも
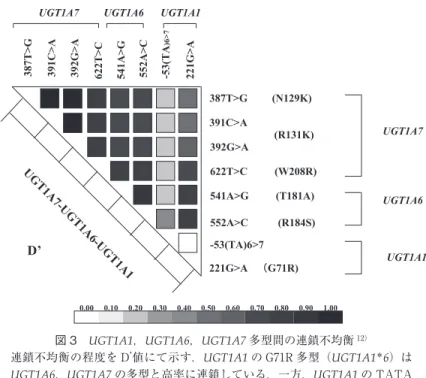
つ Gilbert 症候群症例でイリノテカン代謝遅延の起こる ことが報告された11).現在癌のオーダーメイド治療が広 まりつつあるが,体質性黄疸を伴う癌患者へのイリノ テカン投与は特に慎重に行う必要がある.また,UGT 1A遺伝子群はUGT1A1からUGT1A13Pの13個(内UGT 1A2P,1A11P,1A12P,1A13P は偽遺伝子のため計 9 個)のアイソザイム遺伝子からなるが,日本人におい ては UGT1A1 の G71R 多型(UGT1A1* 6;Gilbert 症候 群を発症)が UGT1A6,UGT1A7 遺伝子の多型(UGT 1A6* 2,UGT1A7* 3;それぞれ酵素活性の著明低下をき たす)と高率にリンクしていることが判明した(図 3)12). 酵素 UGT1A6 はフェノール系薬物の,UGT1A7 はフェ ノール環をもつ高分子発癌物質の,それぞれ代謝を司っ ており,我々は UGT1A7* 3多型が Brinkman 喫煙指数と 同レベルに高い肺癌発症リスクであることを見出して おり,高率にリンクしている UGT1A1* 6多型を有する Gilbert 症候群患者での注意が必要と考えている13). 高直接ビリルビン血症 UGT1A1 によるグルクロン酸抱合を受けてから以降
表 1 高直接ビリルビン血症の分類 ①肝細胞障害性黄疸 ウイルス肝炎,急性肝不全,肝硬変,自己免疫性肝 炎,薬物性肝障害,うっ血肝など ②肝内胆汁うっ滞 胆汁うっ滞型のウイルス肝炎・薬物性肝障害,敗血 症,Weil病,原発性胆汁性肝硬変,原発性硬化性胆 管炎,進行性家族性肝内胆汁うっ滞 1,2,3型,良 性反復性肝内胆汁うっ滞 1,2型,妊娠性胆汁うっ 滞,先天性胆道閉鎖症,Alagille症候群など ③肝外胆汁うっ滞(閉塞性黄疸;肝門部から Vater
乳頭までの閉塞による黄疸)
総胆管結石,胆道系の炎症,Mirizzi症候群,総肝 管・総胆管・Vater乳頭部・膵頭部の癌など ④体質性黄疸
Dubin-Johnson症候群,Rotor症候群 図 3 UGT1A1,UGT1A6,UGT1A7多型間の連鎖不均衡12)
連鎖不均衡の程度を D’ 値にて示す.UGT1A1の G71R多型(UGT1A1 * 6)は UGT1A6,UGT1A7の多型と高率に連鎖している.一方,UGT1A1の TATA
boxの A (TA) 7TAA多型(UGT1A1 * 28)は余り連鎖していない.因みに,
UGT1A6 * 2は図中の 2つのアミノ酸置換の,UGT1A7 * 3は 3つのアミノ酸置 換の,それぞれ重複したアイソザイム遺伝子で,酵素活性の著明低下を来す. のビリルビン代謝の障害にて発症する.肝細胞障害性 黄疸,肝内胆汁うっ滞,肝外胆汁うっ滞(閉塞性黄疸; 肝門部から Vater 乳頭までの胆道閉塞による黄疸),体 質性黄疸に分類される(表 1).肝細胞障害性黄疸の場 合は,血中に ALT,AST といった肝逸脱酵素の上昇が 著しいのに比し肝内及び肝外胆汁うっ滞では一般的に 胆道系酵素(ALP,LAP,γ-GTP など)の上昇が強く, 抱合型ビリルビンのほか胆汁中へ排泄される各種成分 (胆汁酸,コレステロール,銅など)も血中で上昇する. 参考として,薬物性肝障害の診断時に用いられている 肝細胞障害性と胆汁うっ滞性との鑑別基準を表 2 に示 す.一方,体質性黄疸はビリルビン代謝障害に基づく 黄疸であるので胆汁酸などの上昇を伴っていない点で 区別される. 肝細胞障害に起因する黄疸では,トランスアミナー ゼの上昇と共に抱合型ビリルビンとδ ビリルビンとが血 中で増加する.黄疸の進行期にはδ ビリルビンよりも抱 合型ビリルビンが優位を占めるが,回復期には抱合型 ビリルビンは急速に肝へ再摂取されて消失して行く一 方で代謝の遅れるδ ビリルビンが血中に残り優位となる ことから,黄疸の病態の把握に抱合型ビリルビンとδ ビリルビンの割合の比較が行われる14).抱合型ビリルビ ンのみに着目して経過を観察することも可能である. また,劇症肝炎や非代償性肝硬変のように肝機能の著 明低下を来した場合には,肝細胞のビリルビン抱合酵 素 UGT1A1 の活性低下を伴うことから血中の非抱合型 ビリルビンの割合が増加してくるので病態診断にも用 いられる.特に急性肝不全においては総ビリルビン 10
胆 道 23 巻 2 号(2009) 48:178
表 2 薬物性肝障害診断における肝細胞障害型と胆汁うっ滞型の病型 分類
肝細胞障害型
ALT> 2N & ALP<= N または ALT比 /ALP比>= 5 胆汁うっ滞型
N>= ALT & ALP> 2N または 2>= ALT比 /ALP比 混合型
ALT> 2N & N> ALP かつ 5> ALT比 /ALP比> 2 (症例の例示)症例 1:60歳女性.結核性頸部リンパ節炎に対して投与したリ
ファンピシンと INHにより肝障害を発症.AST(単位 IU/L,以下同様)1300 (基 準 値 上 限 40,以 下 同 様),ALT(IU/L)1250(35),ALP(IU/L)900 (340),γ-GTP(IU/L)460(37),総ビリルビン(mg/dl)9.9(1.1).ALT比 /
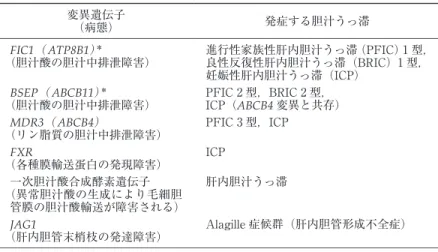
ALP比= 35.7/2.65= 13.5であるので,表から「肝細胞障害型」に分類する. 症例 2:57歳女性.バセドウ氏病に処方されたチアマゾールにて肝障害を発 症.AST(IU/L)95(40),ALT(IU/L)93(35),ALP(IU/L)1360(340), γ-GTP(IU/L)395(37),総ビリルビン(mg/dl)14.0(1.1).ALT比 /ALP 比= 2.66/4= 0.665であるので,表から「胆汁うっ滞型」に分類する. 表 3 遺伝子異常と肝内胆汁うっ滞 発症する胆汁うっ滞 変異遺伝子 (病態) 進行性家族性肝内胆汁うっ滞(PFIC)1型, 良性反復性肝内胆汁うっ滞(BRIC)1型, 妊娠性肝内胆汁うっ滞(ICP) FIC1( ATP8B1) * (胆汁酸の胆汁中排泄障害) PFIC 2型,BRIC 2型, ICP(ABCB4変異と共存) BSEP( ABCB11) * (胆汁酸の胆汁中排泄障害) PFIC 3型,ICP MDR3( ABCB4) (リン脂質の胆汁中排泄障害) ICP FXR (各種膜輸送蛋白の発現障害) 肝内胆汁うっ滞 一次胆汁酸合成酵素遺伝子 (異常胆汁酸の生成により毛細胆 管膜の胆汁酸輸送が障害される) Alagille症候群(肝内胆管形成不全症) JAG1 (肝内胆管末梢枝の発達障害)
*γ-GTPは正常範囲.PFIC1,BSEP,MDR3は毛細胆管膜上の ATP依存性輸送蛋白で,
それぞれアミノリン脂質(フォスファチジルセリン,フォスファチジルエタノールアミ ン)の細胞膜外膜から内膜への膜内輸送(flippase作用),胆汁酸の輸送,リン脂質(フォ スファチジルコリン)の細胞膜内膜から外膜への膜内輸送(flippase作用)を司る.FXR は核内オーファンレセプター.JAG1(jagged1)は Notch receptorの ligandでありさま ざまな細胞の分化・発育に関与する.PFIC1の異常(欠損)による胆汁酸の胆汁中排泄 障害発現の機序の詳細は充分解明されていない. mg!dl 以上と黄疸の増悪を来すとともに直接ビリルビ ン!総ビリルビン比 0.6 以下へ低下する症例は予後不良 と考えられる15). 肝内胆汁うっ滞はウイルス肝炎,薬物性肝障害,原 発性胆汁性肝硬変,原発性硬化性胆管炎のように臨床 的に比較的よく遭遇する疾患に伴って発症するものの 他,先天性胆道閉鎖症や毛細胆管膜の各種トランスポー ターの遺伝子変異などで起こる先天的(遺伝的)な胆 汁うっ滞(表 3)が知られている.長期にわたれば,掻 痒感,全身倦怠感も出現する.また,胆汁酸排泄低下 による脂肪便,コレステロール排泄低下による高脂血 症,黄色腫や,脂溶性ビタミン吸収障害による骨粗鬆 症(vitamin D 欠乏),プロトロンビン時間延長(vita-min K 欠乏)などを合併する.
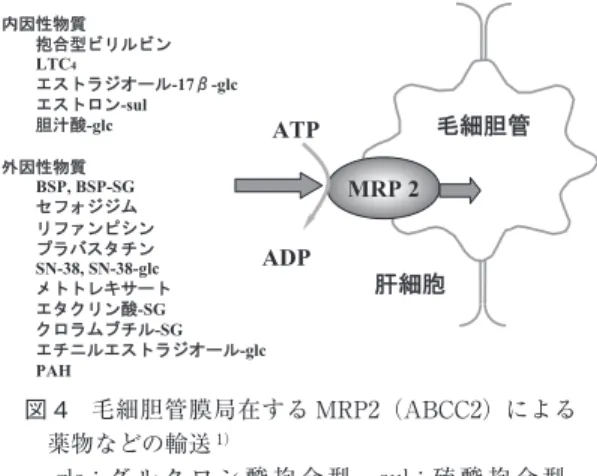
図 4 毛細胆管膜局在する MRP2(ABCC2)による 薬物などの輸送1) -glc:グ ル ク ロ ン 酸 抱 合 型, -sul:硫 酸 抱 合 型, -SG:グルタチオン抱合型 体質性黄疸としては Dubin-Johnson 症候群と Rotor 症候群とがある.胆汁酸の代謝はほぼ保たれているの で血中胆汁酸は正常である.Dubin-Johnson 症候群は MRP2遺伝子変異による毛細胆管膜の MRP2(ABCC2) の欠損により発症し,軽度の高直接ビリルビン血症, BSP テスト再上昇現象,経口胆嚢造影不良,尿中コプ ロポルフィリン I イソマー分画著増(80% 以上)など にて診断するが,MRP2 にて胆汁中へ排泄される多く の有機アニオン系薬物(グルクロン酸抱合物,硫酸抱 合物,グルタチオン抱合物を中心に)(グルタチオン抱 合型 BSP,ICG,イリノテカン代謝物の SN-38,SN-38 グルクロナイド,セフォジジム,リファンピシン,プ ラバスタチン,メトトレキサート,グルタチオン抱合 型エタクリン酸,テモカプリラートなど)や内因性物 質(抱合型ビリルビンのほか,ロイコトリエン C4,エ ストラジオール-17β-グルクロナイド,硫酸抱合型エス トロンなど)の代謝遅延を来し臨床的に問題となるの で注意を要する(図 4)16).一方 Rotor 症候群は中等度 の高直接ビリルビン血症を来し,ICG や BSP の血中消 失の著明遅延,尿中コプロポルフィリン量著増とイソ マー I 分画増加(80% 以内)から診断される.肝細胞 内のビリルビン輸送蛋白の GSTα(リガンディン)の欠 損が報告されている17)18)ので,これにより肝細胞内を輸 送される有機ア ニ オ ン 系 薬 物 の 代 謝 遅 延 が Dubin-Johnson 症候群と同様に臨床的に問題となり得る. おわりに 以上のように,肝細胞におけるビリルビン代謝の主 要代謝酵素,輸送蛋白は薬物や発癌物質の代謝とも密 接な関連を有している.従って,先天性の黄疸に限ら ず後天性の黄疸においても薬物治療などにおいて注意 を充分に払う必要がある.これらの遺伝子解析はオー ダーメイド医療として今後癌の化学療法などに広く応 用されて行くものと期待している. 文 献 1)足立幸彦.黄疸研究の進歩―薬物代謝との関連を含 めて.日本内科学会雑誌 2007;96:1980―1986 2)Adachi Y. Hepatic uptake of bilirubin re-visited.
Hepatol Res 2005; 31: 193―194
3)設楽悦久,佐藤 均,杉山雄一.肝細胞の物質輸送 とトランスポーター.小俣政男 監修:肝疾患 Re-view 2004.東京:日本メディカルセンター,2004: 215―220
4)Zollner G, Fickert P, Zenz R, et al. Hepatobiliary transporter expression in percutaneous liver biop-sies of patients with cholestatic liver diseases. He-patology 2001; 33: 633―646
5)Soroka CJ, Lee LM, Azzaroli F, et al. Cellular local-ization and up-regulation of multidrug resistance-associated protein 3 in hepatocytes and cholangio-cytes during obstructive cholestasis in rat liver. Hepatology 2001; 33: 783―791
6)Geier A, Wagner M, Dietrich CG, et al. Principles of hepatic organic anion transporter regulation during cholestasis, inflammation and liver regen-eration. Biochim Biophys Acta 2007; 1773: 283―308 7)Urquhart BL, Tirona RG, Kim RB. Nuclear recep-tors and the regulation of drug-metabolizing en-zymes and drug transporters: Implications for in-terindividual variability in response to drugs. J Clin Pharmacol 2007; 47: 566―578
8)Tanaka Y, Kobayashi Y, Gabazza EC, et al. In-creased renal expression of bilirubin glucuronide transporters in a rat model of obstructive jaundice. Am J Physiol Gastrointest Liver Physiol 2002; 282: G656―G662
9)Kotal P, Van der Veere CN, Sinaasappel M, et al. In-testinal excretion of unconjugated bilirubin in man and rats with inherited unconjugated hyperbiliru-binemia. Pediatr Res 1997; 42: 195―200
10)井谷俊夫,足立幸彦.ビリルビン.Medical Practice 編集委員会 編:臨床検査ガイド 2007∼2008.東 京:文光堂,2007:259―261
11)Ando Y, Saka H, Ando M, et al. Polymorphism of UDP-glucuronosyltransferase gene and irinotecan toxity : a pharmacogenetic analysis. Cancer Res
胆 道 23 巻 2 号(2009) 50:180
2000; 60: 6921―6926
12)Urawa N, Kobayashi Y, Araki J, et al. Linkage dise-quilibrium of UGT1A1*
6and UGT1A1*
28in rela-tion to UGT1A6 and UGT1A7 polymorphisms. On-col Rep 2006; 16: 801―806
13)Araki J, Kobayashi Y, Iwasa M, et al. Polymor-phism of UDP-glucuronosyltransferase 1A7 gene: A possible new risk factor for lung cancer. Eur J Cancer 2005; 41: 2360―2365
14)Adachi Y, Inufusa H, Yamashita M, et al. Human se-rum bilirubin fractionation in various hepatobiliary diseases by the newly developed high perform-ance liquid chromatography. Gastroenterol Japon 1988; 23: 268―272
15)高橋 均,足立幸彦.黄疸.救急医学 1997;21: 678―679
16)König J, Nies AT, Cui Y, et al. Conjugate export pumps of the multidrug resistance protein (MRP) family: localization, substrate specificity, and MRP-mediated drug resistance. Biochim Biopys Acta 1999; 1461: 377―394
17)Adachi Y, Yamamoto T. Partial defect in hepatic glutathione S-transferase activity in a case of Ro-tor s syndrome. Gastroenterol Japon 1987; 22: 34― 38
18)Abei M, Matsuzaki Y, Tanaka N, et al. Defective he-patic glutathione S-transferase in Rotor s syn-drome. Am J Gastroenterol 1997; 90: 682―683
Molecular and genetic mechanism in disturbance of bilirubin metabolism
and clinical relevance of jaundice
Yukihiko Adachi, Rumi Morooka
This is a brief review of the regulatory mechanisms and abnormalities of bilirubin transport and metabolism in hepatocytes. The induction and suppression of hepatocyte transporters by nuclear receptors are outlined. A linkage has been recognized between UGT1A1 polymorphism (UGT1A1*6) that causes Gilbert s syndrome
and UGT1A polymorphisms (UGT1A6*2and UGT1A7*3) that cause disturbance of drug glucuronide
conjuga-tion and high incidence of lung cancer. Defects of MRP2 in the Dubin-Johnson syndrome and of GST-alpha in the Rotor s syndrome also indicate disturbance in the metabolism of anionic drugs. Hepatic enzymes and transport-ers are also reduced in acquired hepatobiliary disordtransport-ers that cause jaundice; thus, occurrence of abnormalities in the metabolism of anionic drugs should be also taken into consideration in acquired hepatobiliary diseases with jaundice.
JJBA2009; 23: 174―180
Department of Internal Medicine, Kuwana City Hospital (Kuwana)
Key Words: jaundice, nuclear receptor, genetic polymorphism, drug metabolism, tailor-made therapy