学位論文内容の要約
Co-Occurrence of 22q11 Deletion Syndrome and HDR Syndrome
(22q11 欠失症候群と HDR 症候群の合併を認めた一例)
Ryoko Fukai
深井 綾子
Department of Neurology and Stroke Medicine,
Yokohama City University Graduate School of Medicine
横浜市立大学大学院医学研究科 医科学専攻
神経内科学・脳卒中医学
(
Research Supervisor:Naomichi Matsumoto,Professor)
(研究指導教員:松本 直通 教授 遺伝学)
(
Doctoral Supervisor:Fumiaki Tanaka,Professor)
(指導教員:田中
章景 教授)
1
学位論文内容の学位論文内容の要約
Co-Occurrence of 22q11 Deletion Syndrome and HDR Syndrome (22q11欠失症候群とHDR 症候群の合併を認めた一例) http://www.ncbi.nlm.nih.gov/pubmed/23918631 【背景と目的】 発達障害の診断法として,欧米では 2004 年頃よりマイクロアレイによる網羅的な CNV 解析 が急速に普及し,これまでにマイクロアレイを用いて,発育遅延(developmental delay:DD), 先天性多発奇形(Multiple congenital anomalies:MCA), ASD などのゲノムコピー数異 常が多数報告されている[Miller et al., 2010].マイクロアレイの登場により, 数十 kb のサイズの微細な CNV をゲノムワイドに網羅的に検出することが可能になった[Miller et al., 2010; Cooper et al., 2011].22q11 欠失症候群は,22q11.2 領域の 1.5 Mb-3.0 Mb の微細欠失を伴う先天奇形症候群である.頻度は約 4,000 人出生に 1 人に認める比較的頻 度が高い欠失症候群である[Kitsiou-Tzeli et al., 2004; Kobrynski and Sullivan 2007; McDonald-McGinn and Sullivan 2011; Gennery 2012].特異的な症状として,特異的顔貌, 心血管奇形,胸腺低形成,副甲状腺低形成に伴う低カルシウム血症などを呈する.その他, ID,言語発達障害,精神障害(統合失調症やうつ病など),難聴,鎖肛,鼠径ヘルニア, 腎尿路奇形,低身長,血小板減少,自己免疫疾患など,さまざまな症状を認めることも知 られている[Perez and Sullivan 2002; Digilio et al., 2005; Goldmuntz 2005; Kobrynski and Sullivan 2007; McDonald-McGinn and Sullivan 2011].一方 10p13-p14 の領域は DiGeorge 症候群/verlocardiofacial 症候群複合-2 (DiGeorge syndrome/velocardiofacial syndrome complex-2: DGS2) critical region (DGCR2) と言われており,欠失により 22q11.2 欠失に 似た症状を呈する[Lichtner et al., 2000].10p14 にはGATA3が含まれており,GATA3の ハプロ不全は,副甲状腺機能低下症,感音性難聴,腎形成不全を三主徴とする常染色体優 性遺伝形式の稀な疾患である HDR disease 症候群(OMIM 146255)を引き起こす.これまで に約 50 人の報告がある[Greenberg et al., 1986; Shapira et al., 1994; Gottlieb et al., 1998; Schuffenhauer et al., 1998; Lichtner et al., 2000; Van Esch et al., 2000; Muroya et al., 2001; Thakker 2001; Skrypnyk et al., 2002; Mino et al., 2005; Benetti et al., 2009; Lindstrand et al., 2010; Fukami et al., 2011].
症例は,臨床症状から 22q11 欠失症候群が疑われ,FISH 法により同症候群の確定診断にいた った.しかし増悪する腎機能不全や感音性難聴,重症な低カルシウム血症やてんかんは, 22q11 欠失単独で説明づけることが出来ず, 22q11 欠失以外の遺伝学的要因を検索する目的 で, Cytogenetics Whole-Genome 2.7 M Array (Affymetrix) を用いた網羅的なコピー数解 析を行った.非典型症例に対してマイクロアレイ等の網羅的解析を行うことで詳細な遺伝 的原因の解明を目的とした研究を行った.
2 【対象・方法】 症例 (対象) 血族婚のない両親の第一子で 22 歳の日本人男性.周産期に異常はなく 37 週で出生した. 出生体重 2,412g (-1.4 SD), 身長 45 cm (-1.9 SD), 頭囲 (OFC) 31.5 cm (-1.3 SD). 本 症例が呈した顔貌の特徴として,腫れぼったい眼瞼,幅広い鼻梁,耳介低位,小さい口を認め た.また,口蓋裂,臍ヘルニア,鼠径ヘルニア,先細り指を認めたことから先天奇形症候群が 疑われた.6 か月時の身長 61.6 cm (-2.5 SD), 体重 6,000g (-2.2 SD), 頭囲 43 cm (-0.23 SD). 1 才身長 73 cm (-0.77 SD), 体重 7,850 g (-1.57 SD), 頭囲 46.5 cm (þ0.28 SD). 5 才 1 か月身長 93.1 cm (-3.2 SD), 体重 12.5 kg (-2.0 SD), 13 才 9 か月身長 130 cm (-3.8 SD), 体重 27 kg (-2.2 SD)であった.頚定は 6 か月,座位 15 か月,歩行は 2 歳 9 か月で発育 障害を認めた.5 か月時に痙攣重責を認め, West 症候群の診断となり内服加療が開始され た.6 か月時に施行された聴性脳幹反応は>90dB であり,重度の感音性難聴と診断された.3 歳時には,社会性障害,コミュニケーション障害,興味の偏り,手の常同行動が著明とな り,DSM-IV に基づき自閉症症候群と診断された他,田中ビネー式検査では,重度精神運動発 達遅延と判定された. 5 か月時の血液検査で低カルシウム血症 (Ca, 5.2 mg/dl; 正常値: 9.6–11.6 mg/dl) を指 摘され,Ca 剤とビタミン D 誘導体 (alfacalcidol)を内服加療が開始された.しかし 10 歳時 の血液検査で,再度低 Ca 血症 (Ca, 6.4 mg/dl; 正常値: 8.5–10.2 mg/dl),高感度副甲状腺 ホルモン低値(<100 mg/ml; 正常値: 160–520 mg/ml) を指摘された.また, 3 歳時に施行し た血液検査で腎機能障害 (creatinine 1.0 mg/dl (正常値:0.3–0.5 mg/dl)), 血液尿素窒 素 (BUN) 65 mg/dl (正常値: 5–18 mg/dl))を指摘されており,10 歳時に施行した排泄性尿 路造影検査にて右腎と尿管の描出が困難であった. 方法 1. ゲノム DNA の抽出
患児及びその両親の末梢血白血球から,QuickGene-610L(FUJIFILM, Tokyo, Japan)を用 いて,プロトコールに従いゲノム DNA を抽出した.
2. マイクロアレイ
患児のゲノム DNA を用いて,SNP アレイを行った (尚,マイクロアレイの実施は以前遺伝 学教室に在籍していた大学院生により,データ解析以降を著者・深井が行った).
Cytogenetics Whole-Genome 2.7 M Array (Affymetrix)
3
ロトコールに従って反応を行った.データは Chromosome Analysis Suite version1.2 (Affymetrix) NA32 (hg19) により解析を行った.構造異常の絞り込みは,欠失:連続す る異常マーカー数≧20,欠失サイズ≧10kb,信頼性≧89%;重複フィルター:連続する異常 マーカー数≧20,重複サイズ≧100kb,信頼性≧90%とした.
3. Fluorescence in situ hybridization;FISH 法
患者と両親の末梢血リンパ球又はリンパ芽球細胞株をカルノア液(メタノール:酢酸 = 3: 1)固定し,染色体スライド標本を作成した. プローブは,RPC-11 細菌人口染色体(BAC)を用 いた.患者の欠失領域に RP11-91K20 (chr10:8,303,515-8,458,288 bp)と RP11-1057H19 (chr22:19,310,701-19,484,643 bp) を選択し,リファレンスとして RP11-1069N9
(chr10:90,801-288,527 bp)と RP11-81N15 (chr22:42,605,111-42,773,705 bp) を使用した. 蛍光標識は,Nick Translation Kit(Abbott Molecular, Des Plaines, IL, USA)を用い て,Cy3 又は Spectrum Green で標識した.染色体上に蛍光プローブをハイブリダイズさせ 洗浄後,DAPI で核を染色した.その後 Axioplan2 蛍光顕微鏡(Carl Zeiss, Oberkochen, Germany)を用いてシグナルの観察を行った. 【結果と考察】 CNV 解析の結果,22q11.2 (19.0-22.0Mb)の欠失(図 1a)と 10p14(7.4-8.9Mb)の欠失(図 1b)を検出した.10p14 に認めた 1.5Mb の欠失の中には HDR 症候群を引き起こすGATA3が含 まれていた.2 ヶ所の欠失領域で FISH 解析を行い,de novo (両親に欠失は認めない) 変化 であることを確認した.10p14 欠失領域の断端は chr10: 7,398,380 と chr10: 8,934,448 bp であり,Sanger 法により GTTGTT (chr10: 7,398,366 -7,398,371 bp)の欠失を伴っている 事を同定した. 図 1.患者の細胞遺伝学的解析結果 SNP アレイの結果 (ab): 22q11.2(19.0-22.0Mb)の欠失(a)と 10p14(7.4-8.9Mb)の欠失(b)
4
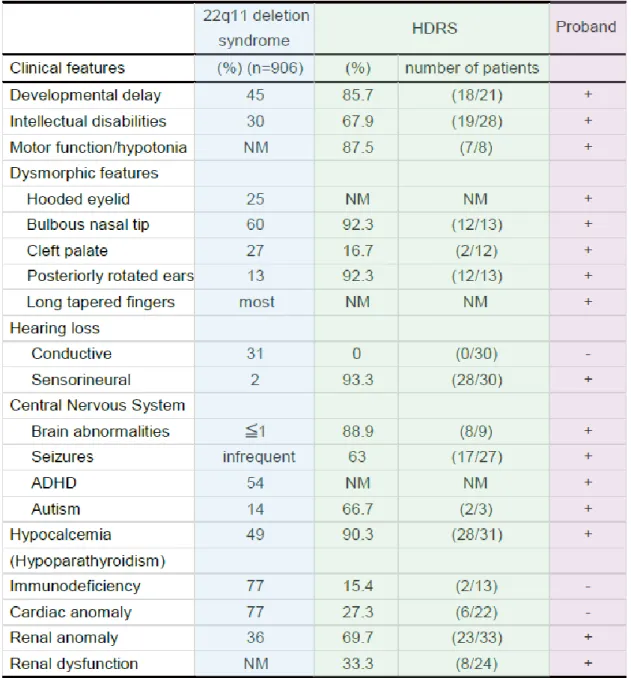
2つの症候群の臨床症状と患者のそれとを比較すると,患者の特異的顔貌は 22q11 欠失症 候群に基づくと考えられる (表 1).一方,増悪する腎機能不全や感音性難聴や,副甲状腺低 形成に伴う難治性の低カルシウム血症は HDR 症候群に起因すると考えられた.GATA3は zinc finger タイプの転写因子GATAファミリーの一員であり,血球系の細胞や腎臓,内耳,副甲 状腺,中枢神経,胸腺などでの発現が報告されている[Oosterwegel et al., 1992; Ting et al., 1996].また,GATA3 ノックアウトマウスでは,頭蓋顔面異常,造血障害および中枢神 経の形成異常,腎低形成を起こし,胎生 11-12 日で死に至る事が多いことが報告されてい る[Pandolfi et al., 1995; Lim et al., 2000].TBX1は DNA 結合ドメインを有する T-box ファミリーの一員であり,心血管系,胸腺や副甲状腺に主に発現している[Chapman et al., 1996; Garg et al., 2001; Jerome and Papaioannou 2001; Lindsay et al., 2001; Merscher et al., 2001; Yagi et al., 2003; Zahirieh et al., 2005].ヒト 22q11 欠失症候群のモ デルマウスに相当すると考えらえるTBX1 +/-マウスは心排出路に高頻度で奇形を持つ一方 で,TBX1 -/- マウスは短い頸,顎の低形成,耳介低位,大動脈弓の異常,胸腺および副甲 状腺低(無)形成を認め,呼吸器障害により生後致死的であることが報告されている[Jerome and Papaioannou 2001]. GATA3 とTBX1の発現臓器は似ているが,過去に両遺伝子の機能的関連の報告はない.GATA3 は内耳と聴神経に発現しており,GATA3+/-と-/-マウスは聴神経伝達の障害を呈する事が知 られている[Karis et al., 2001].TBX1は第 1 鰓弓と耳胞に発現しており,TBX1-/-マウス で耳の構造異常を認めた[Lawoko-Kerali et al., 2004; Arnold et al., 2006].これらの 違いは 22q11 欠失症候群において免疫不全や耳の構造異常が寄与し,慢性中耳炎が起こり 伝音声難聴を招くことが考えられる.一方で HDR 症候群は,内耳や聴神経が関与している ことから,感音性難聴がメインである.本症例における重度の感音性障害は,2遺伝子異 常の相乗効果の可能性も考えられる.本患者のように,症状が類似する2疾患に罹患してい る場合,症状がどちらの症候群由来かを判断する事が難しいことも多い.これまでに同一 患者に疾患関連 CNV や関連遺伝子変異を同時に認める報告もあり,複数の遺伝子異常が症例 の症状の重篤度と関連している可能性が示唆される[Miller et al., 2010; Cooper et al., 2011].本症例は,22q11.2 と 10p14 の合計 2 か所にde novoの欠失を合併する稀少な症例 であり,微細な染色体構造異常を網羅的に解析することにより遺伝的原因を解明する事が できた.表現型と遺伝型の詳細な解析の重要性を明瞭に示している.
5
表 1. 22q11 欠失症候群と HDR 症候群と症例の主な臨床症状
NM, 記載なし; +, 症状あり; - , 症状なし.
6 【参考文献】
Arnold JS, Braunstein EM, Ohyama T, Groves AK, Adams JC, Brown MC, Morrow BE. 2006. Tissue-specific roles of Tbx1 in the development of the outer, middle and inner ear, defective in 22q11DS patients. Hum Mol Genet 15:1629-1639. Benetti E, Murer L, Bordugo A, Andreetta B, Artifoni L. 2009. 10p12.1 deletion: HDR
phenotype without DGS2 features. Experimental and molecular pathology 86:74-76.
Chapman DL, Garvey N, Hancock S, Alexiou M, Agulnik SI, Gibson-Brown JJ, Cebra-Thomas J, Bollag RJ, Silver LM, Papaioannou VE. 1996. Expression of the T-box family genes, Tbx1-Tbx5, during early mouse development.
Developmental dynamics : an official publication of the American Association of Anatomists 206:379-390.
Cooper GM, Coe BP, Girirajan S, Rosenfeld JA, Vu TH, Baker C, Williams C, Stalker H, Hamid R, Hannig V, Abdel-Hamid H, Bader P, McCracken E, Niyazov D, Leppig K, Thiese H, Hummel M, Alexander N, Gorski J, Kussmann J, Shashi V,
Johnson K, Rehder C, Ballif BC, Shaffer LG, Eichler EE. 2011. A copy number variation morbidity map of developmental delay. Nature genetics 43:838-846. Digilio M, Marino B, Capolino R, Dallapiccola B. 2005. Clinical manifestations of
Deletion 22q11.2 syndrome (DiGeorge/Velo-Cardio-Facial syndrome). Images in paediatric cardiology 7:23-34.
Fukami M, Muroya K, Miyake T, Iso M, Kato F, Yokoi H, Suzuki Y, Tsubouchi K,
Nakagomi Y, Kikuchi N, Horikawa R, Ogata T. 2011. GATA3 abnormalities in six patients with HDR syndrome. Endocrine journal 58:117-121.
Garg V, Yamagishi C, Hu T, Kathiriya IS, Yamagishi H, Srivastava D. 2001. Tbx1, a DiGeorge syndrome candidate gene, is regulated by sonic hedgehog during pharyngeal arch development. Developmental biology 235:62-73.
Gennery AR. 2012. Immunological aspects of 22q11.2 deletion syndrome. Cellular and molecular life sciences : CMLS 69:17-27.
Goldmuntz E. 2005. DiGeorge syndrome: new insights. Clinics in perinatology 32:963-978, ix-x.
Gottlieb S, Driscoll DA, Punnett HH, Sellinger B, Emanuel BS, Budarf ML. 1998. Characterization of 10p deletions suggests two nonoverlapping regions contribute to the DiGeorge syndrome phenotype. American journal of human genetics 62:495-498.
7
Hypoparathyroidism and T cell immune defect in a patient with 10p deletion syndrome. The Journal of pediatrics 109:489-492.
Jerome LA, Papaioannou VE. 2001. DiGeorge syndrome phenotype in mice mutant for the T-box gene, Tbx1. Nature genetics 27:286-291.
Karis A, Pata I, van Doorninck JH, Grosveld F, de Zeeuw CI, de Caprona D, Fritzsch B. 2001. Transcription factor GATA-3 alters pathway selection of olivocochlear neurons and affects morphogenesis of the ear. The Journal of comparative neurology 429:615-630.
Kitsiou-Tzeli S, Kolialexi A, Fryssira H, Galla-Voumvouraki A, Salavoura K, Kanariou M, Tsangaris GT, Kanavakis E, Mavrou A. 2004. Detection of 22q11.2 deletion among 139 patients with Di George/Velocardiofacial syndrome features. In Vivo 18:603-608.
Kobrynski LJ, Sullivan KE. 2007. Velocardiofacial syndrome, DiGeorge syndrome: the chromosome 22q11.2 deletion syndromes. Lancet 370:1443-1452.
Lawoko-Kerali G, Rivolta MN, Lawlor P, Cacciabue-Rivolta DI, Langton-Hewer C, van Doorninck JH, Holley MC. 2004. GATA3 and NeuroD distinguish auditory and vestibular neurons during development of the mammalian inner ear. Mech Dev 121:287-299.
Lichtner P, Konig R, Hasegawa T, Van Esch H, Meitinger T, Schuffenhauer S. 2000. An HDR (hypoparathyroidism, deafness, renal dysplasia) syndrome locus maps distal to the DiGeorge syndrome region on 10p13/14. Journal of medical genetics 37:33-37.
Lim KC, Lakshmanan G, Crawford SE, Gu Y, Grosveld F, Engel JD. 2000. Gata3 loss leads to embryonic lethality due to noradrenaline deficiency of the sympathetic nervous system. Nature genetics 25:209-212.
Lindsay EA, Vitelli F, Su H, Morishima M, Huynh T, Pramparo T, Jurecic V, Ogunrinu G, Sutherland HF, Scambler PJ, Bradley A, Baldini A. 2001. Tbx1
haploinsufficiency in the DiGeorge syndrome region causes aortic arch defects in mice. Nature 410:97-101.
Lindstrand A, Malmgren H, Verri A, Benetti E, Eriksson M, Nordgren A, Anderlid BM, Golovleva I, Schoumans J, Blennow E. 2010. Molecular and clinical
characterization of patients with overlapping 10p deletions. American journal of medical genetics Part A 152A:1233-1243.
McDonald-McGinn DM, Sullivan KE. 2011. Chromosome 22q11.2 deletion syndrome (DiGeorge syndrome/velocardiofacial syndrome). Medicine 90:1-18.
8
Russell RG, Factor S, Tokooya K, Jore BS, Lopez M, Pandita RK, Lia M, Carrion D, Xu H, Schorle H, Kobler JB, Scambler P, Wynshaw-Boris A, Skoultchi AI, Morrow BE, Kucherlapati R. 2001. TBX1 Is Responsible for Cardiovascular Defects in Velo-Cardio-Facial/DiGeorge Syndrome. Cell 104:619-629.
Miller DT, Adam MP, Aradhya S, Biesecker LG, Brothman AR, Carter NP, Church DM, Crolla JA, Eichler EE, Epstein CJ, Faucett WA, Feuk L, Friedman JM, Hamosh A, Jackson L, Kaminsky EB, Kok K, Krantz ID, Kuhn RM, Lee C, Ostell JM, Rosenberg C, Scherer SW, Spinner NB, Stavropoulos DJ, Tepperberg JH, Thorland EC, Vermeesch JR, Waggoner DJ, Watson MS, Martin CL, Ledbetter DH. 2010. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. American journal of human genetics 86:749-764.
Mino Y, Kuwahara T, Mannami T, Shioji K, Ono K, Iwai N. 2005. Identification of a novel insertion mutation in GATA3 with HDR syndrome. Clinical and experimental nephrology 9:58-61.
Muroya K, Hasegawa T, Ito Y, Nagai T, Isotani H, Iwata Y, Yamamoto K, Fujimoto S, Seishu S, Fukushima Y, Hasegawa Y, Ogata T. 2001. GATA3 abnormalities and the phenotypic spectrum of HDR syndrome. Journal of medical genetics
38:374-380.
Oosterwegel M, Timmerman J, Leiden J, Clevers H. 1992. Expression of GATA-3 during lymphocyte differentiation and mouse embryogenesis. Developmental
immunology 3:1-11.
Pandolfi PP, Roth ME, Karis A, Leonard MW, Dzierzak E, Grosveld FG, Engel JD, Lindenbaum MH. 1995. Targeted disruption of the GATA3 gene causes severe abnormalities in the nervous system and in fetal liver haematopoiesis. Nature genetics 11:40-44.
Perez E, Sullivan KE. 2002. Chromosome 22q11.2 deletion syndrome (DiGeorge and velocardiofacial syndromes). Current opinion in pediatrics 14:678-683.
Schuffenhauer S, Lichtner P, Peykar-Derakhshandeh P, Murken J, Haas OA, Back E, Wolff G, Zabel B, Barisic I, Rauch A, Borochowitz Z, Dallapiccola B, Ross M, Meitinger T. 1998. Deletion mapping on chromosome 10p and definition of a critical region for the second DiGeorge syndrome locus (DGS2). European journal of human genetics : EJHG 6:213-225.
Shapira M, Borochowitz Z, Bar-El H, Dar H, Etzioni A, Lorber A. 1994. Deletion of the short arm of chromosome 10 (10p13): report of a patient and review. American journal of medical genetics 52:34-38.
9
Skrypnyk C, Goecke TO, Majewski F, Bartsch O. 2002. Molecular cytogenetic
characterization of a 10p14 deletion that includes the DGS2 region in a patient with multiple anomalies. American journal of medical genetics 113:207-212. Thakker RV. 2001. Genetic developments in hypoparathyroidism. Lancet 357:974-976. Ting C-N, Olson MC, Barton KP, Leiden JM. 1996. Transcription factor GATA-3 is
required for development of the T-cell lineage. Nature 384:474-478.
Van Esch H, Groenen P, Nesbit MA, Schuffenhauer S, Lichtner P, Vanderlinden G, Harding B, Beetz R, Bilous RW, Holdaway I, Shaw NJ, Fryns JP, Van de Ven W, Thakker RV, Devriendt K. 2000. GATA3 haplo-insufficiency causes human HDR syndrome. Nature 406:419-422.
Yagi H, Furutani Y, Hamada H, Sasaki T, Asakawa S, Minoshima S, Ichida F, Joo K, Kimura M, Imamura S, Kamatani N, Momma K, Takao A, Nakazawa M, Shimizu N, Matsuoka R. 2003. Role of TBX1 in human del22q11.2 syndrome. Lancet 362:1366-1373.
Zahirieh A, Nesbit MA, Ali A, Wang K, He N, Stangou M, Bamichas G, Sombolos K, Thakker RV, Pei Y. 2005. Functional analysis of a novel GATA3 mutation in a family with the hypoparathyroidism, deafness, and renal dysplasia syndrome. The Journal of clinical endocrinology and metabolism 90:2445-2450.
10 【論文目録】
I 主論文
Co-occurrence of 22q11 deletion syndrome and HDR syndrome. Fukai R, Ochi N, Murakami A, Nakashima M, Tsurusaki Y, Saitsu H, Matsumoto N, Miyake N. Am J Med Genet A. 2013 Oct;161A(10):2576-81. doi: 10.1002/ajmg.a.36083. Epub 2013 Aug 5.
II 副論文
A de novo 1.4-Mb deletion at 21q22.11 in a boy with developmental delay.
Fukai R, Hiraki Y, Nishimura G, Nakashima M, Tsurusaki Y, Saitsu H, Matsumoto N, Miyake N. Am J Med Genet A. 2014 Apr;164A(4):1021-8. doi: 10.1002/ajmg.a.36377. Epub 2014 Jan 23.
III 参考論文
Duplication of the NPHP1 gene in patients with autism spectrum disorder and normal intellectual ability: a case series. Yasuda Y, Hashimoto R, Fukai R, Okamoto N, Hiraki Y, Yamamori H, Fujimoto M, Ohi K, Taniike M, Mohri I, Nakashima M, Tsurusaki Y, Saitsu H, Matsumoto N, Miyake N, Takeda M. Ann Gen Psychiatry. 2014 Aug 6;13:22. doi: 10.1186/s12991-014-0022-2. eCollection 2014. コピー数変化. 深井綾子, 三宅紀子, 松本直通. 脳科学辞典 2012 http://bsd.neuroinf.jp/wiki/%E3%82%B3%E3%83%94%E3%83%BC%E6%95%B0%E5%A4%89%E5%8C% 96 エキソームシーケンス 深井綾子, 松本直通. 医学のあゆみ,第 249 巻 5(号) 382 頁 平 成 26 年 5 月 3 日発行