九州大学学術情報リポジトリ
Kyushu University Institutional Repository
O3型遷移金属層状岩塩酸化物の合成とそのナトリウ ム電池特性
坪田, 隆之
https://doi.org/10.15017/4060201
出版情報:九州大学, 2019, 博士(理学), 課程博士 バージョン:
権利関係:
O3 型遷移金属層状岩塩酸化物の合成と そのナトリウム電池特性
坪田 隆之
九州大学大学院総合理工学府
量子プロセス理工学専攻
目次
第 1 章 序論
1.1. 緒言 ... 7
1.2. リチウムイオン二次電池 ... 9
正極活物質の高エネルギー密度化の検討 ... 10
負極活物質の高エネルギー密度化の検討 ... 11
1.3. ナトリウムイオン二次電池 ... 12
ナトリウムイオン二次電池用の層状岩塩構造の正極材料 ... 13
1.4 本研究の目的 ... 16
参考文献 ... 18
第 2 章 リチウムイオン二次電池のサイクル劣化機構
2.1. 緒言 ... 222.2. 実験 ... 24
Li(Ni1/3Mn1/3Co1/3)O2電極の作製 ... 24
グラファイト電極の作製,電池の作製 ... 24
Li(Ni1/3Mn1/3Co1/3)O2電極,グラファイト電極を用いた電池の作製 ... 25
Si負極電極の作製,電池の作製 ... 25
充放電試験,交流インピーダンス法,TEM観察,XPSの測定条件 ... 26
2.3. 結果と考察 ... 28
2.3.1. Li(Ni1/3Mn1/3Co1/3)O2電極の1C×100 サイクルにおける劣化機構 ... 28
1C×100サイクルにおける電池特性/充放電特性,及び内部抵抗 ... 28
1C×100サイクルにおける Li(Ni1/3Mn1/3Co1/3)O2正極活物質の劣化解析 . 31 2.3.2. Li(Ni1/3Mn1/3Co1/3)O2電極の2C×6600サイクルにおける劣化機構 ... 34
充放電サイクル特性 ... 34
内部抵抗分離解析 ... 36
単極容量の評価 ... 41
正極活物質内のリチウムイオン拡散抵抗の評価 ... 42
正極の酸化還元反応解析 ... 44
正極の劣化解析 ... 45
グラファイト負極の劣化解析 ... 50
2.3.3. Si負極電極のサイクル劣化機構 ... 53
充放電サイクル試験 ... 53
内部抵抗解析 ... 53
電極の構造変化,表面被膜の調査 ... 55
内部抵抗の解析 ... 63
2.4結論 ... 69
Li(Ni1/3Mn1/3Co1/3)O2電極の 1C×100サイクルにおける劣化機構 ... 69
Si負極電極のサイクル劣化機構 ... 69
参考文献 ... 71
第 3 章
ナトリウムイオン電池用 O3 型遷移金属層状岩塩酸化物の合成と電池特 性
3.1. 緒言 ... 74
3.2. 実験 ... 76
Na(Ni1/3Mn1/3Co1/3)O2(Na-NMC)の合成 ... 76
Na(Fe1/3Mn1/3Co1/3)O2(Na-FMC)の合成 ... 76
Na(Ni1/3Fe1/3Co1/3)O2(Na-NFC)の合成 ... 76
Na(Ni1/3Mn1/3Fe1/3)O2(Na-NMF)の合成 ... 77
合成した活物質のキャラクタリゼーション ... 77
電気化学特性... 78
Na-FMCの酸化還元反応 ... 78
3.3. 結果と考察 ... 79
合成したナトリウム層状岩塩構造酸化物のキャラクタリゼーション ... 79
電気化学的特性 ... 84
Na-NMC,Na-FMC,Na-NFC,Na-NMFの酸化還元反応解析 ... 87
Na-FMCの充電状態における酸化還元反応解析 ... 92
3.4. 結論 ... 98
参考文献 ... 99
第 4 章
O3 型遷移金属層状岩塩酸化物 Na(Fe
1/3Mn
1/3Co
1/3)O
2のサイクル劣化機 構
4.1. 緒言 ... 102
4.2. 実験 ... 102
Na(Fe1/3Mn1/3Co1/3)O2の合成 ... 102
Na(Fe1/3Mn1/3Co1/3)O2電極の作製と電池の作製 ... 103
充放電試験,交流インピーダンス法,TEM観察の条件 ... 103
4.3. 結果と考察 ... 105
出力特性 ... 105
充放電サイクル特性 ... 106
サイクル試験における内部抵抗 ... 108
サイクル試験における結晶構造変化 ... 109
第一原理計算によるカチオンミキシングにおける活性化エネルギー評価 .... 115
Li(Ni1/3Mn1/3Co1/3)O2 と Na(Fe1/3Mn1/3Co1/3)O2の比較 ... 118
4.4. 結論 ... 121
参考文献 ... 122
第 5 章
総括 ... 124謝辞 ... 127
第1章
序論
1.1. 緒言
近 年 , 蓄 電 池 の 需 要 が 高 ま っ て い る . リ チ ウ ム イ オ ン 電 池 は ,ニ ッ ケ ル 水 素
(Ni-MH)電池などの従来の水系二次電池と比較して,小型で軽量であり,かつ
電圧が高いことからエネルギー密度が高く,スマートフォンなどのモバイル機器 に加え,近年では国内自動車会社において,ハイブリッド自動車や電気自動車な ど車載用蓄電池への適用が進められている.
その背景にはアメリカ合衆国のカリフォルニア州におけるZEV(Zero Emission Vehicle)規制の強化がある.2018年からはHV(hybrid vehicle)が対象から除外 され,2019 年の導入された中国の NEV(New Energy Vehicle)規制においても HV が対象から除外された.日本は環境対応自動車として HV 技術により世界を牽引 してきたが,これからはPHV(Plug in Hybrid Vehicle),EV(Plug in Hybrid Vehicle),
あるいは FCV(Fuel Cell Vehicle)へシフトせざるを得ない規制環境の変化があり,
PHV,EV,FCVの実現のためにはいずれも大容量の蓄電池が必要となる.
NEDOプロジェクト「革新型蓄電池実用化促進基盤技術開発」では,現行のガ ソリン車並みの航続距離を目指し,2020年に 250~350kmを,2030年には 500km 超を実現す る,これ ま でのリチウ ムイオン 電 池(LIB)の性能を はる かに凌駕す るエネルギー密度を持つ革新型電池として,ナノ界面制御電池(ハロゲン化物),
亜鉛空気電池,ナノ界面制御電池(コンバージョン)及び硫化物電池などの新型 電池の研究開発が行われている.
また,2019年度からは,NEDOプロジェクト「先進・革新蓄電池材料評価開発
(第2期)」にて,自動車・蓄電池・材料メーカーおよび大学・公的研究機関が連 携・協調し,全固体リチウムイオン電池の生産プロセス,EV 搭載に向けた研究 開発がスタートするなど,基礎研究から実用化に向けた検討に進んでいる.
自動車業界では 100年に1度の変革期と言われているが,2016年のパリモータ ーショーにて,ダイムラーの CEOが中期経営計画の中で提唱した「CASE」が注 目を集めている.これは「Connected:コネクティッド」「Autonomous:自動運転」
「Shared/Service:シェアリング/サービス」「Electric:電動化」の 4 つの頭文字 をとったものである.電動化には蓄電池は必須であり,自動運転と電動化は相性 が良い.国内においても自動運転タクシーの実証実験が進められているが,自動 運転普及の観点からも電動化が進むと予想され,蓄電池の適用が進むと考えられ る.
さらに定置型蓄電池の普及も進んでいる.2011年に発生した東日本大震災によ ってもたらされた東京電力の福島第一原子力発電所の炉心融解を含む事故をきっ かけに,国内の原子力発電所はすべて停止に追い込まれた.その後,一部の原子 力発電所は再稼働しているが,政府は太陽光発電,風力発電などの再生可能エネ ルギーの導入拡大を目指している.ところが天候によって出力変動が大きい再生 可能エネルギーの割合が増加すると,逆潮流により周波数変動が大きくなり,電 力の安定供給に悪影響が出ることが顕在化している.2018 年の 10 月には九州電 力が国内初となる出力抑制を実施した.急激に増えている太陽光発電の電力が受 け入れ可能量を上回る可能性があったためである.電力の安定供給のために,ピ ークカット,ピークシフトを行う MWh 級の大型蓄電池に期待が高まってきてい る.
以上のように車載用途,定置用途のそれぞれに,リチウムイオン二次電池の適 用範囲は拡大していくと考えられるが,問題となるのは電池内で電荷を運ぶリチ ウムと,正極で酸化還元反応を担う遷移金属の埋蔵量,生産量の制約である.リ
チウムと比較して資源量比が1000 倍多いナトリウムを,正極では Ni,Coに代わ り鉄やチタンなどの安価な遷移金属が使用できれば,コストが大幅に低減できる.
小型蓄電池,あるいは車載用蓄電池ではエネルギー密度が重視されるが,材料 費のウエイトが大きくなる大型蓄電池の場合には,コストパフォーマンスが最優 先される.
本 研 究 に て , リ チ ウ ム イ オ ン 電 池 の 正 極 活 物 質 と し て 使 用 さ れ て い る O3 型 Li(Ni1/3Co1/3Mn1/3)O2 を ベースに,ナトリウ ム をインターカレーシ ョ ンゲストに,
遷移金属を鉄に置き換えた,ナトリウムイオン電池用遷移金属層状岩塩酸化物に 着目した背景について順次概説する.
1.2. リチウムイオン二次電池
リチウムイオン電池は,電気自動車(EV)などの車載用電池や,定置型大型蓄 電池,モバイル機器用小型電池として用いられている.正極はリチウムを含有す る遷移金属酸化物,負極にグラファイトなどの炭素材料が用いられている.2019 年のノーベ ル化学 賞を 受賞した吉 野彰氏 によ り,コバル ト酸リ チウ ム(LiCoO2) を正極に,炭素材料を負極としたリチウムイオン電池の構成,正極のアルミ箔集 電体,オレフィン製微多孔膜による発熱時のシャットダウン機能などの基本概念 が 1985 年に確立されている.その後,ソニー・エナジー・テック株式会社より 1991年に実用化された.
リチウムイオン電池の構成概要図をFig. 1-1に,反応式を以下に示す.
正極: LiCoO2 ⇄ LiCoO2 + 0.5Li+ + 0.5e- 負極: C6 + Li+ +e-⇄ LiC6
全反応式:2LiCoO2 + C6 ⇄ 2LiCoO2 + LiC6
Fig. 1-1 リチウムイオン電池の構成概要図.
正極活物質の高エネルギー密度化の検討
リチウムイオン電池の正極材料には層状酸化物 LiCoO2 はモバイル機器にて広 く使用されてきた.しかしながら希少金属である Co は,世界の生産量の約 60%
がコンゴ共和国であるが,政情が不安定なこともあり,カントリーリスクと価格 上 昇 が 課 題 と な っ て い る . そ こ で ,Co の 一 部 を Ni,Mn で 置 き 換 え た Li(Ni1/3Co1/3Mn1/3)O2 ( NCM111 )が開発された[1, 2].車載用としての検討が進めら
れていた2010 年当時は,NCM111の他,スピネル構造を持つ LiMn2O4や,作動電 圧が低いものの安価で優れた熱安定性を有している LiFePO4が使用されていた.
し か し な が ら 航 続 距 離 の 観 点 か ら エ ネ ル ギ ー 密 度 の 高 い 正 極 材 料 が 志 向 さ れ , 2019 年現在では,車載用電池の正極材料は NCM 系に収斂し,EV 用のリチウム イオン電池への適用が進められている.
NCM111における充放電において優先的に起こる酸化還元反応はNi2+ / Ni4+ で
あることから,さらなる高エネルギー密度化のためにNiの比率を高めた Li(Ni0.5Co0.2Mn0.3)O2 (NCM523),Li(Ni0.6Co0.2Mn0.2)O2 (NCM622),
Li(Ni0.8Co0.1Mn0.1)O2 (NCM811) の適用検討が進められている[3, 4].
負極活物質の高エネルギー密度化の検討
リチウムイオン電池には,負極は主に炭素材料が使用されているが,グラファ イトの理論容量は372 mAh/g であり,実効容量としてすでに理論容量の90%以上 を使用している.負極の高エネルギー密度化のために,グラファイトと比較して 大きな理論容量を持つSi,Sn といった合金系負極の研究開発が行われている.ま た高容量の合金系負極を用いることで,電極の合材層を薄くできることから使用 材料の低減によるコストダウンも期待できる.しかしながら,優れたLiの吸蔵能 力が故に,約4.4倍の体積膨張となり,充放電サイクルでのLi 吸蔵・放出に伴う,
膨張・収縮が大きく,微粉化して急激に容量が低下する課題がある.サイクル寿 命を向上させるために,SiO2マトリックスに Siのナノクラスタを分散させたSiO が研究されている[5, 6].また,SiCやSiO-Cの検討も進められている[7, 8].粒 子の形態制御についても検討が進められており,Siのナノ粒子やナノワイヤー化 の検討も進められている[9, 10].
1.3. ナトリウムイオン二次電池
前述のようにリチウムイオン電池は実用化され,電気自動車(EV)や大型の定 置型電源へ適用が進んでいる.しかしなら,材料費のウエイトが大きくなる大型 電源では,環境負荷低減やコストパフォーマンスが優先されることから,クラー ク数が小さく希少元素であるリチウムから,埋蔵量にして約 1000倍のナトリウム に電荷のキャリアを置き換えたナトリウムイオン電池の開発が期待されている.
一方で,Table 1-1に示すようにナトリウムはリチウムに対し,標準電極電位が
0.3 V以上高くなる上,イオン体積にして 2倍以上,原子量にして3倍以上かさば
るなどの違いがあり,リチウムイオン電池用正極活物質の探索指針をそのまま適 用することはできない.ナトリウムイオン電池用正極材料としての新たな設計指 針が必要となる.
Table 1-1. リチウムとナトリウムの比較.
特性 リチウム ナトリウム
資源量比 1 1,000
コスト( 炭酸塩 ) $ 5,000/t $ 150/t
原子量 6.9 g/mol 23 g/mol
イオン体積 1.84 Å3 4.44 Å3
理論容量 3,829 mAh/g 1,165 mAh/g
標準電極電位 vs. SHE - 3.045 V - 2.714 V
ナトリウムイオン二次電池用の層状岩塩構造の正極材料
ナトリウムイオン二次電池の正極材料の検討の歴史は古く,酸化物系正極材料,
リン酸塩系正極材料,硫化物系正極材料,有機材料系正極材料など,多くの研究 が行われている.
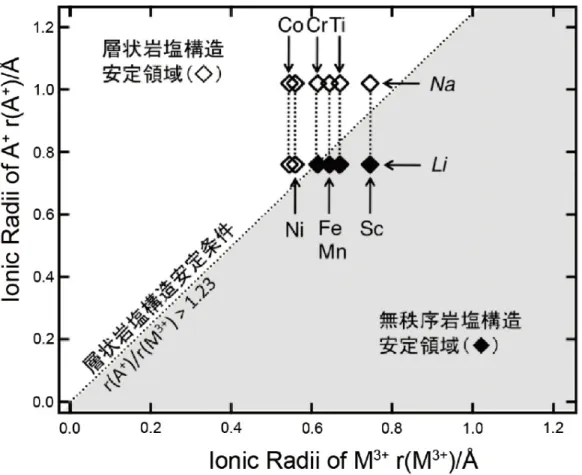
酸化物系の正極材料では,NaFeO2の研究に始まり,層状岩塩構造を持つ酸化物 はナトリウムイオン二次電池の正極材料の有力候補として検討されている.Table 1-2に代表的なナトリウムイオン電池の酸化物系の正極材料の報告例を示す.
AMO2型岩塩構造の Structure Field マップを Fig. 1-2に示す.A+とM3+のイオン 半径が近いとお互いが混じり合って不規則岩塩構造が安定相となり,正極として の活性度を示さなくなる.そのため,リチウム含有系では安価なLiFeO2や LiTiO2
の層状岩塩相を安定相として得ることは困難である.ところが,ナトリウム含有
系では NaFeO2を含めすべての 3d 遷移金属が層状岩塩型 NaMO2を安定相にもつ
ため,これらの電気化学活性な層状岩塩相を得ることができる.さらに,ナトリ ウム含有系では A+と M3+のイオン半径の違いが大きいため,結晶内でこれらの原 子が入れ替わるカチオンミキシングがリチウム含有系と比較して起こりにくいと 考えられる.これは充放電サイクルにおけるカチオンミキシングを伴う結晶構造 変化による電池特性の劣化を抑制できると予想され,ナトリウム含有系層状岩塩 の設計指針における重要な観点となる.
なお,リチウムイオン二次電池用正極材料として使用されているスピネル構造 をもつ酸化物であるLiMn2O4については,リチウムをナトリウムで置換した材料 が存在しない.リチウムイオンは酸素の八面体サイト,四面体サイトに存在する が,ナトリウムの場合にはイオン体積がリチウムよりも2倍以上大きいため,四 面体サイトに存在することができないためである.
Table 1-2. 代表的なナトリウムイオン電池の酸化物系の正極材料の報告例.
*容量の一部は,初回放電にてナトリウム対極により補償されている
Formula Voltage
(V vs. Na+/Na)
Capacity (mAh / g)
Redox Couples
Coulombic
Efficiency (%) Cycling Performance
(number)
Structural
stability References Initial
cycle After cycling
O3-NaFeO2 3.3V ~80 Fe4+/Fe3+ 75% (30) poor 11, 12
O3-NaMnO2
2.63 V,
2.7-3.8 V 185 Mn4+ / Mn3+ ~86% ~90% ~71% (20) moderate 13 O3-NaNiO2 2.5-3.5 V 120 Ni4+ / Ni3+ 83.8% 99.2% 94% (20) moderate 14
O3-NaCrO2 ~3V 120 Cr4+ / Cr3+ ~83% (50) good 15, 16
O3-NaxVO2 ~1.7 V,
1.8-2.4 V 120 V4+ / V3+ ~100% (14) moderate 17, 18
O3-NaxFe0.5Mn0.5O2 1.5-4.2 V 100-110 Fe4+ / Fe3+
Mn4+ / Mn3+ ~65% (30) good 19
O3-NaNi0.5Mn0.5O2 2.2-3.8 V 100-125 Ni4+ / Ni2+ 75% (50) good 20 O3-NaNi1/3Mn1/3Co1/3O2 2.5-3.75 V 120 Ni4+ / Ni2+
Co3+ / Co4+ ~96% (50) moderate 21
O3-NaNi1/3Mn1/3Fe1/3O2 2.0-4 V 130 Ni4+ / Ni3+ ~70% >99% 77% (150) good 22
P2-NaxCoO2 2.0-3.9 V ~120 Co4+ / Co3+ good 23-25
*P2-NaxMnO2 2.0-3.8 V ~140 Mn4+/Mn3+ ~30% (10) poor 26
P2-NaxVO2
1.6 V,
1.6-2.4 V ~100 V4+ / V3+ 110% (10) moderate 27
*P2-Na2/3Fe0.5Mn0.5O2 1.5-4.3 V 190 (1.5-4.3 V)
Me4+ / Me3+
(Me=Fe0.5Mn0.5) ~80% (30) good 29
P2-Na2/3Co2/3Mn1/3O2 1.5-4.0 V ~90 Co4+ / Co3+ good 27
P2-Na2/3Nil/3Mn2/3O2 3.0-4.5 V 160 Ni4+ / Ni2+ good 28
*P2-Na0.45
Ni0.22Co0.2Mn0.66O2
2.1-4.3 V 135 Mn4+ / Mn3+, Co4+ / Co3+, Ni4+ / Ni2+
99.7% 72% (275)
good
29
*P2-Na0.67Ni0.15Co0.2Mn0 .65Al0.05O2
2.3V,3.65V,
4.25V 141 130% >99% 88% (50) 30
P2-NaLi0.2Ni0.5Mn0.75Oy 2.0-4.2 V ~100 Ni4+ / Ni2+ 85% >99% ~98% (50) good 31 Na0.44MnO2 2.0-4.0 V ~110 Mn4+ / Mn3+ ~100% ~77% (1000) good 32, 33
Fig. 1-2 層状岩塩構造AMO2の Structure Fieldマップ.
1.4 本研究の目的
Ni,Mn,Co を遷移金属として使用する O3 型層状岩塩酸化物である正極材料 Li(Ni1/3Mn1/3Co1/3)O2を使用したリチウムイオン電池は,電気自動車の電池や定置 型蓄電池として使用されており,市場での実績もある.ただし,今後の EV 化や 再生可能エネルギーの増加に伴うピークカット,ピークシフトの需要に応えるた めに,リチウムイオン電池の生産量が拡大すると,リチウムの供給に問題が出る 可能性がある.さらに市場からの低コスト化の要請がある.
著者らは,リチウムイオン二次電池にて市場で実績のある O3 型層状岩塩構造 をベースに,ナトリウムイオンをインターカレーションゲスト,遷移金属の一部 に鉄を使用した O3 型遷移金属層状岩塩酸化物に着目した.ナトリウム含有系で はA+とM3+のイオン半径の違いが大きいため,結晶内でこれらの原子が入れ替わ るカチオンミキシングがリチウム含有系と比較して起こりにくいと考えた.充放 電サイクルにおけるカチオンミキシングを伴う結晶構造変化による電池特性の劣 化を抑制できると予想され,この検証を行った.
第2章では,リチウムイオン二次電池について,交流インピーダンス法による 内部抵抗の分離解析と,結晶構造変化,活物質表面に形成されるSEI被膜の解析 との複合解析による劣化機構について議論した.
第 3 章 で は ナ ト リ ウ ム を イ ン タ ー カ レ ー シ ョ ン ゲ ス ト と し た Na(Ni1/3Mn1/3Co1/3)O2 ( Na-NMC ), 遷 移 金 属 の 一 部 を 鉄 に 置 き 換 え た Na(Ni1/3Fe1/3Co1/3)O2(Na-NFC),Na(Ni1/3Mn1/3Fe1/3)O2(Na-NMF), 新 規材 料 と な るNa(Fe1/3Mn1/3Co1/3)O2(Na-FMC)を合成し,放射光硬 X 線XAFS により遷移金 属の価数を議論した.
第4章では,ナトリウムイオン電池正極材料として新規合成したNa-FMC につ いて,充放電時における遷移金属の価数変化を放射光硬X 線 XAFS,57Fe メスバ
ウア測定により酸化還元反応機構を議論した.第5章ではナトリウムイオン電池 正極材料として新規合成したNa-FMC について,充放電サイクル試験後のカチオ ンミキシングを Cs-STEM観察,EELS分析により評価し,第一原理計算によるカ チオンミキシングにおける活性化エネルギー計算を行い,Li(Ni1/3Mn1/3Co1/3)O2と 対比しながらカチオンミキシングを伴うサイクル劣化機構について議論した.
参考文献
[1] N. Yabuuchi, Y. Koyama, N. Nakayama, T. Ozuku, J. Electrochem. Soc., 152 (7) (2005) A1434.
[2] N. Yabuuchi and T. Ozuku, J. Power Sources, 146 (2005) 636.
[3] H.-J. Noh, S. Youn, C.S. Yoon, Y-K. Sun, J. Power Sources, 233 (2013) 121.
[4] A. Verma, K. smith, S. Santhanagopalan, D. Abraham, K. P. Yao, P. P. Mukherjee, J.
Electrochem. Soc., 164 (13) (2017) A3380.
[5] Y. Hwa, C.-M. Park, H.-J. Sohn, J.Power Sources, 222 (2013) 129.
[6] K. Pan, F. Zou, M. Canova, Y. Zhu, J.-H. Kim, J.Power Sources, 413 (2019) 20.
[7] J. Saint, M. Morcrette, D. Larcher, L. Laffont, S. Beattie, J.‐P. Pérès, D. Talaga, M.
Couzi, J.‐M. Tarascon, Adv. Funct. Mater., 17 (2017) 1765.
[8] M. Yamada, A. Ueda, K. Matsumoto, T. Ozuku, J. Electrochem. Soc., 158 (4) (2011) A417.
[9] P. Hovington, M. Dontigny, A. Guerfi, J. Trottier, M. Lagace, A. Mauger, C. M.
Julien, J.Power Sources, 248 (2014) 457.
[10] C. K. Chan, R. Ruffo, S. S. Hong, Y. Cui, J.Power Sources, 189 (2009) 1132.
[11] N. Yabuuchi, H. Yoshida, S. Komaba, Electrochem., 80 (2012) 716.
[12] J. Zhao, L. Zhao, N. Dimov, S. Okada, T. Nishida, J. Electrochem. Soc, 160 (2013) A3077.
[13] X. M- Ma, H. L. Chen, G. Ceder, Electrochem. Soc., 158(2011) A1307.
[14] P. Vassilaras, X. H. Ma, X. Li, G. Ceder, J. Electrochem. Soc., 160 (2013) A207.
[15] S. Komaba, C. Takei, T. Nakayama, A. Ogata, N. Yabuuchi, Electrochem. Commun., 12(2010) 355.
[16] J. J. Ding, Y. N. Zhou, Q. Sun, Z. W. Fu, Electrochem. Commun., 22(2012) 85.
[17] D. Hamani, M. Ati, J.M. Tarascon, P, Rozier, Electrochem. Commun., 13 (2011) 938.
[18] C. Didier, M. Guignard, C. Denage, 0. Szajwaj, S. Ito, I. Saadoune, J. Darriet, C.
Delmas, Electrochem. Solid State Lett., 14 (2011) A75.
[19] N. Yabuuchi, M. Kajiyama, J. lwatate, H. Nishikawa, S. Hi omi, R. Okuyama, R.
Usui, Y. Yamada, S. Komaba, Nat. Mater., 11 (2012) 512.
[20] S. Komaba, N. Yabuuchi, T. Nakayama, A. Ogata, T. Ishikawa, I. Nakai, lnorg.
Chem., 51 (2012) 6211.
[21] M. Sathiya, K. Hemalatha, K. Ramesha, J.M. Tarascon, A. S. Prakash, Chem. Mater., 24 (2012) 1846.
[22] D. Kim, E. Lee, M. Slater, W. Q. Lu, S. Rood, C. S. Johnson, Electrochem.
Commun., 18 (2012) 66.
[23] R. Berthelot, D. earlier, C. Delmas, Nat. Mater., 10 (2011) 74.
[24] J. J. Ding, Y. N. Zhou, Q. Sun, X. Q. Yu, X. Q. Yang, Z. W. Fu, Electrochim. Acta, 87 (2013) 388.
[25] M. D'Arienzo, R. Ruffo, R. Scotti, F. Morazzoni, C. M. Maria, S. Polizzi, Phys.
Chem. Chem. Phys., 14 (2012) 5945.
[26] A. Caballero, L. Hernan, J. Morales, L. Sanchez, J. S. Pena, M.A. G. Aranda, J.
Mater. Chem., 12 (2002) 1142.
[27] D. Carlier, J. H. Cheng, R. Berthelot, M. Guignard, M. Yoncheva, R. Stoyanova, B.
J. Hwang, C. Delmas, Dalton Trans., 40 (2011), 9306.
[28] Z. H. Lu and R. Dahn, J. Electrochem. Soc, 148 (2001) A1225.
[29] D. Buchholz, A. Moretti, R. Kloepsch, S. Nowak, V. Siozios, M. Winter, S.
Passerini, Chem. Mater., 25 (2013) 142.
[30] D. D. Yuan, W. He, F. Pei, F. Y. Wu, Y. Wu, J. F. Qian, Y. L. Cao, X. P. Ai , H. X.
Yang, J. Mater. Chem. A, 1 (2013) 3895.
[31] D. Kim, S. H. Kang, M. Slater, S. Rood, J. T. Vaughey, N. Karan, M.
Balasubramanian, C. S. Johnson, Adv. Energy Mater., 1 (2011) 333.
[32] F. Sauvage, L. Laffont, J.M. Tarascon, E. Baudrin, lnorg. Chem., 46 (2007) 3289.
[33] Y. L. Cao, L F. Xiao, W. Wang, D. W. Choi, Z. M. Nie, J. G. Yu, L.V. Saraf, Z. G.
Yang, J. Liu, Adv. Mater., 23 (2011) 3155.
第 2 章
リチウムイオン二次電池のサイクル劣化機構
2.1. 緒言
リチウムイオン二次電池は,Ni-MH電池などの従来の二次電池と比較して小型 で軽量であり,かつセル電圧が高いことからエネルギー密度が高く,スマートフ ォンなどのモバイル機器に加え,近年ではハイブリッド自動車や電気自動車,定 置型蓄電池への適用が進められている.しかしながら,高容量化,高出力化,長 寿命化,安全性の向上,低コスト化の課題があり,電極材料や電解液,セパレー タの開発は活発に行われている.特に,長期使用が想定される車載用や定置型の リチウムイオン電池では,耐久性向上の検討や寿命予測の観点で,正極・負極の いずれが支配的に劣化しているかを明確にすることは,さらなる長寿命,高入出 力特性の電池を開発する上で重要である.リチウムイオン電池の正極材料には層
状酸化物 LiCoO2 がモバイル機器にて広く使用され,劣化機構の研究が行われて
きた[1]. しかしながら希少金属である Coは価格上昇が課題がり,Coの一部を Ni
で置き換えた Li(NixCo1-x)O2や[2,3],Co と Mn で置き換えた Li(Ni1/3Co1/3Mn1/3)O2
(NCM111)が開発された[4 - 6].車載用としての検討が進められていた 2010年当時
は,NCM111の他,スピネル構造を持つ LiMn2O4や,作動電圧が低いものの安価
で優れた熱安定性を有している LiFePO4が使用されていた.しかしながら航続距 離の観点からエネルギー密度の高い正極材料が志向され,2019年現在では,車載 用電池の正極材料はNCM系に収斂し,EV用のリチウムイオン電池への適用が進 められており,市場での長期耐久性についても.NCM111 における充放電におい て,優先的に起こる酸化還元反応はNi2+ / Ni4+ であることから,さらなる高エネ ル ギ ー 密 度 化 の た め に Ni の 比 率 を 高 め た Li(Ni0.5Co0.2Mn0.3)O2 (NCM523), Li(Ni0.6Co0.2Mn0.2)O2 (NCM622),Li(Ni0.8Co0.1Mn0.1)O2 (NCM811) の適用検討が進め られている[7 - 10].
一方,負極については活物質に炭素材料,特にグラファイトを用いることが主
流であり,その理論容量は372 mAh/g である.一方,高容量を発現する負極活物 質としてSiが知られており,その理論容量は 4200 mAh/gとグラファイトを大き く上回る.しかしならが,高容量が故にリチウムイオンを吸蔵した際に膨張が大 きく,リチウムイオンの挿入・脱離に伴う膨張・収縮によりクラックが発生・伸 展することで,微粉化するという課題があり,合金化やナノ粒子化,ナノファイ バー化などの検討が進められている[17 – 27].
Siや SiOなどの合金負極は,高容量化を達成するポテンシャルを持ちながらも 上記の課題により使いこなすことが難しく,グラファイト電極に少量を添加する コンポジット電極として使用されることが多い.合金負極を用いることで,電極 合材層に使用する活物質量を減らすことができ,コストダウン効果が期待できる.
リチウムイオン電池の劣化機構については,交流インピーダンス法による抵抗 の解析やGITT法(Galvanostatic Intermittent Titration Technique)が行われている
[15, 16].通常の電池構成であるフルセル(正極-負極)にて交流インピーダンス
測定を行った事例を,Fig. 2-1に示す.正極の抵抗,負極の抵抗が混在し,反応時 定数が近い場合には波形分離も困難となり,正極,負極の反応抵抗を分離した解 析手法が必要となる.
第2章では,リチウムイオン電池のサイクル試験における劣化機構を明らかに するため,Li(Ni1/3Co1/3Mn1/3)O2正極材料,グラファイト負極,Si負極を用いた電 池を作製し,充放電試験を行った.交流インピーダンス法を用いた内部抵抗分離 解析技術による抵抗解析と,活物質の結晶構造変化,化学結合状態の変化の観点 から劣化メカニズムについて議論した.
Fig. 2-1. フルセルのインピーダンス測定例.
2.2. 実験
Li(Ni1/3Mn1/3Co1/3)O2電極の作製
正極は,平均粒径10μmのLi(Ni1/3Mn1/3Co1/3)O2を活物質とし,導電助剤として アセチレンブラック, 結着剤として PVDF( ポリフッ化ビニリデン ),NMP(N- メチルピロリドン,溶媒)を加えて薄膜旋回型高速ミキサーにて混練し,粘度を 適切に調整したのち,連続塗工機にて集電体 Al箔に両面塗工した.乾燥,ロール プレスにより密度調整を行い,合材層の膜厚42μm,密度 2.8g/cm3の正極とした.
グラファイト電極の作製,電池の作製
負 極 は 平 均 粒 径 35μm の グ ラ フ ァ イ ト を 活 物 質 と し , 正 極 同 様 の 工 程 に て 混 練・スラリー化し,集電体Cu箔に合材層を両面塗工し,膜厚 55μm,密度1.3g/cm3 の負極とした.
Li(Ni1/3Mn1/3Co1/3)O2電極,グラファイト電極を用いた電池の作製
露点-70℃以下に調整された Ar ガス雰囲気のグローブボックス内にて,作製し た正極と負極を,セパレータ(ポリエチレン製微多孔膜)を挟んで対向し,電解 液(1M LiPF6 / EC:DEC=1:1vol.%)を加えて,設計容量 2.8mAhのバルクセルを作 製し,0.2C(0.27mA/cm2)×3 サイクルの初期充放電を行った後に,1C×100 サイ クルの充放電試験に供試した.
同様に,露点-70℃以下に調整されたArガス雰囲気のグローブボックス内にて,
作製した正極とグラファイト負極を,セパレータ(ポリエチレン製微多孔膜)さ せ た 電 極 積 層 体 を ラ ミ ネ ー ト 外 装 材 に 格 納 し , 電 解 液 (1M LiPF6 /
EC:DEC=1:1vol.%)を加えて,設計容量 550mAh の積層型ラミネート型電池を作
製し,0.2C(0.26mA/cm2)×3 サイクルの初期充放電を行った後に,6600 サイク ルの充放電試験に供試した.
Si負極電極の作製,電池の作製
平均粒径 5μm の Si を活物質とし,導電助剤としてアセチレンブラック,結着 剤としてPVDF(ポリフッ化ビニリデン),NMP(N-メチルピロリドン,溶媒)を 加えて混練し,粘度を適切に調整したのち,連続塗工機にて集電体銅箔に塗工し
た.Fig. 2-1に塗工後の Si負極の断面観察像を示す.ロールプレスにより密度調
整を行い,合材層の膜厚42μm,密度 1.2g/cm3の負極とした.
露点-70℃以下に調整された Ar 雰囲気のグローブボックス内にて,作製した負 極と対極の金属 Liを,セパレータ(ポリプロピレン製微多孔膜)を挟んで対向さ せ た 電 極 積 層 体 を ラ ミ ネ ー ト 外 装 材 に 格 納 し , 電 解 液 (1M LiPF6 / EC:DEC=1:1vol.%)を加えて設計容量 220mAh(電極面積 21cm2)のラミネート型 電池とした.
Fig. 2-1. 塗工後の Si負極の断面 SEM観察像.
充放電試験,交流インピーダンス法,TEM観察,XPSの測定条件
充放電試験は恒温槽内に電池を設置し,菊水電子工業製のPFX2011を用いて充 放電を行った.交流インピーダンス法による抵抗測定はSolartron Analytica 社製の 電 気 化 学 測 定 装 置 Celltest System 1470E, イ ン ピ ー ダ ン ス ア ナ ラ イ ザ ーModel
1255Bを用いた.OCV(開回路電圧)に対し,振幅 10mV を重畳させた交流電圧
を,100サイクルの解析では300kHzから0.01Hz,6600サイクルでの解析では1MHz
から0.001Hzまで掃引し,応答電流から内部抵抗を求めた.
ナノ領域の結晶構造観察はTEMにより行った.露点-76℃以下,酸素 10ppm以 下に管理された Ar グローブボックス内で電極をサンプリングし,雰囲気遮断試
料台に固定後,大気非暴露状態でFIB装置内に導入した.試料の最表面保護のた め,FIB にてカーボ ン 膜,および タング ステ ン膜をコー ティン グし たのち,FIB マイクロサンプリング法にて試料小片を真空遮断ホルダの Cu 製メッシュへ摘出 した.摘出した小片は,FIB加工によりTEM観察可能な厚さまで薄片化を行った.
TEM試料は,グローブボックス内で雰囲気遮断 2軸傾斜ホルダにセッティング し,大気非暴露状態でTEM装置内に導入した.
FIB加工装置 : 日立ハイテクノロジーズ製
収束イオン/電子ビーム加工観察装置 nanoDUET NB5000 加工条件 : Gaイオン,加速電圧 30kV,クライオ仕上げ5kV(-100℃)
TEM観察装置 : 日本電子製球面収差補正機能付き走査型透過型電子顕微鏡 JEM-ARM200F
観察条件 : 加速電圧:200kV,観察時ビーム径約0.1nmφ
XPSについても同様に,大気非暴露にてサンプリングし,測定を行った.
装置 : アルバック・ファイ社製全自動走査型 X線光電子分光装置 Quantera SXM
分析条件 : X線源:単色化 Al Kα線(1486.7 eV) 分析領域:100μmφ
2.3. 結果と考察
2.3.1. Li(Ni1/3Mn1/3Co1/3)O2電極の 1C×100 サイクルにおける劣化機構
1C×100サイクルにおける電池特性/充放電特性,及び内部抵抗
試作した電池を室温にて電圧 3.0Vから 4.2Vの間で,CC充電-CC放電の 充放電サ イクル試験(1C×100サイクル)を実施した.放電極線を Fig 2-2に示す.充放電サイ クル数に伴って放電容量の低下が見られ,100サイクルにて放電容量は初期の87%に 低下した.内部抵抗測定結果をFig.2-3に示す. 約数kHz の高周波数域に 応答する反 応と,約数Hzの低周波数域に応答する反応に伴う2つの円弧と,Liの拡 散抵抗(ワ ールブルグインピーダンス)と考えられる右上がりの直線部が見られた.実数軸切片 は電子および電解質イオンの関与した抵抗成分と考えられる.サイクルに伴って高周 波数域,低周波数側の円弧が増大した. 特に,低周波数域 の 円 弧 は 顕 著 に 増 大 し た .
4.4 4.2 4.0 3.8 3.6 3.4 3.2 3.0 2.8
c e ll vo lt ag e ( V )
2.5 2.0
1.5 1.0
0.5 0.0
Capacity (mAh)
1cycle
50cycles
100cycles
Fig 2-2. サイクル試験における放電曲線.
Fig 2-3. フルセルの交流インピーダンス波形.
内部抵抗に対する正極・負極の寄与を分離するため,電池を解体し,正極・負極そ れぞれについて対極を Li とし,ハーフセルを作製,内部抵抗を測定した. 抵抗分離 の概念図を Fig. 2-4に示す.正極・負極それぞれ の内部抵抗測定結果を Fig. 2-5 に示 す.正極は高周波数域と低周波域で円弧が確認されるのに対して,負極は高周波数域 のみで円弧が確認された.電池(フルセル)では,充放電サイクルに伴って顕著に低 周波数域の反応が増加していたことから,内部抵抗増加の主要因は正極であることが 明らかとなった.
-50
-40
-30
-20
-10
0
Z " (Ωcm
2)
70 60
50 40
30 20
10 0
Z' (Ωcm
2)
1Hz
10kHz
0.1Hz 10Hz
0.01Hz
100Hz
1サイクル (OCV=4.179V)
50サイクル (OCV=4.167V)
100サイクル (OCV=4.130V)
Fig. 2-4. 内部抵抗分離の概念図.
Fig. 2-5. サイクル劣化後の正極・負極の内部抵抗.
セパレーター Li
正極 正極
負極 セパレーター
負極 Li
セパレーター
正極-Liセル
負極-Liセル セパレーター
Li 正極 正極
負極 セパレーター
負極 Li
セパレーター
正極-Liセル
負極-Liセル
-30 -25 -20 -15 -10 -5 0
Z " (Ωcm
2)
80 70
60 50
40 30
20 10
0
Z' (Ωcm
2)
-30 -25 -20 -15 -10 -5 0
Z " (Ωcm
2)
10kHz 100Hz
10Hz
1Hz
0.1kHz
0.01kHz
0.01kHz 0.1kHz
1Hz 10Hz 100Hz 10kHz
正極(100サイクル後)-Li (OCV=4.122V)
負極(100サイクル後)-Li (OCV=0.093V)
1C×100サイクルにおける Li(Ni1/3Mn1/3Co1/3)O2正極活物質の劣化解析
充電時には,Liイオンは正極活物質から引き抜かれ,負極活物質に挿入される.放 電時には,負極活物質から Li イオンが引き抜か れ, 正極活物質に挿入される. よっ て,充放電サイクル試験においては,Liイオンが正極-負極間でやりとりされ,正 極 ・ 負極の活物質で Li イオ ンの挿入・脱離による構造変化が繰り返される. 正極の劣化 機構解明のため,正極のナノ構造をTEMにより調査した.
1サイクル後,100 サイクル後の放電状態の正極活物質の断面 TEM 観察結果と制 限視野電子回折像(SAED)をそれぞれFig.2-6,Fig.2-7に示す.正極活物質は,粒径 約1μmの一次粒子が凝 集し,平均粒径 10μmの 二次粒子を構成している.SAEDから 1サイクル後ではLi(Ni1/3Mn1/3Co1/3)O2の層状岩塩構造を確認した.一方,100サイク ル後では,層状岩塩構造のスポットの間に弱い回折スポットが確認された.結晶構造 の変化が示唆されたため, ナノ電子線回折像(NBED)によりナノ領域の構造を調査 した.
1サイクル後,100 サイクル後の放電状態の正極活物質の断面 TEM 写真と NBED
を Fig.2-8,Fig.2-9 に示す.1サイクル後では,最表面は内部と比較して若干の構造
変化が確認された.一方,100 サイクル後では,表面の 10nm程度の厚さ まで立方晶 に構造転移していることを確認した.
Fig.2-6. 1サイクル後のTEM観察像,SAED像.
50nm SAED pattern
Li(Ni
1/3Mn
1/3Co
1/3)O
2アセチレンブラック
+バインダー
シミュレーションパターン
R3m構造 100入射
50nm SAED pattern
Li(Ni
1/3Mn
1/3Co
1/3)O
2アセチレンブラック
+バインダー
シミュレーションパターン
R3m構造 100入射
Fig.2-7. 100サイクル後の TEM観察像,SAED像.
Li(Ni
1/3Mn
1/3Co
1/3)O
2アセチレンブラック
+バインダー
50nm SAED pattern
シミュレーションパターン
R3m構造 100入射
Li(Ni
1/3Mn
1/3Co
1/3)O
2アセチレンブラック
+バインダー
50nm SAED pattern
シミュレーションパターン
R3m構造 100入射
2.3.2. Li(Ni1/3Mn1/3Co1/3)O2電極の 2C×6600サイクルにおける劣化機構
充放電サイクル特性
試作したラミネート型電池について,室温にて電圧2.7Vから 4.2Vの間で,2C
(2.6mA/cm2)にて6100回のCC充電-CC放電の充放電サイクル試験を実施した.
2ItAでのサイクル劣化挙動を Fig. 2-8に,充放電曲線を Fig.2-9に示す.充放電 サイクル数の増加に伴って,充電曲線が高電圧側に,放電曲線が低電圧側にシフ トした.この過電圧の増加は,抵抗が増加していることを示している.また,充 放電容量の低下が見られ,6100 サイクルにて 2Cでの放電容量は初期の 46%に低 下した.
充放電容量の調査のため,2.7Vから 4.2Vの間で 0.2ItA にてCCCV 充電(定電 流定電圧充電,定電圧保持1時間),0.2C にてCC放電(定電流放電)を実施した.
充放電曲線を Fig. 2-10に示す.低レートでの充放電は過電圧の影響が少なく,電 池の充放電可能な容量を評価することができる.充放電サイクル数の増加に伴っ て充放電容量の低下が見られ,6100サイクルにて放電容量は初期の74%に低下し た.よって充放電サイクルにより,内部抵抗の増加と,容量の低下が起こってい る.
Fig. 2-8. 2Cレートのサイクル試験における放電容量の推移.
Fig. 2-9. 2Cレートのサイクル試験における放電曲線.
Fig. 2-10. 容量確認試験における充放電曲線(0.2C).
内部抵抗分離解析
内部抵抗増加因子を特定することは,充放電サイクル耐久性や入出力特性向上 のための研究開発に重要な知見を与える.しかしながら,内部抵抗の構成因子は 複雑であり,活物質/電解液界面の電荷移動抵抗,活物質中のリチウムイオン拡散 抵抗,活物質/活物質界面・活物質/導電助剤界面・活物質/集電体界面などの電子 抵抗,電解液中のイオン伝導抵抗など,リチウムイオンが関わる反応,電子が関 わる反応が存在する.内部抵抗の解析においては,時定数の異なる反応を分離す ることが出来る交流インピーダンス法が有用であるが,反応時定数が近接してい る場合には,電池(フルセル)のまま測定した交流インピーダンス波形の解析の
みで,抵抗因子を分離することは困難である.
そこで,内部抵抗に対する正極・負極・セパレータ,電解液の寄与を分離する ための,内部抵抗の分離を行った.Fig. 2-11に正極,負極の抵抗分離解析の概念 図を示す.正極・負極それぞれについて対極をリチウムとした抵抗解析用のハー フセルを作製し,内部抵抗を測定することで,正極,負極の抵抗増加の寄与を分 離することが可能となる.
Fig. 2-11. 内部抵抗分離の概念図.
Fig. 2-12,Fig. 2-13に劣化電池の正極,負極を用いて構成した解析用電池(フ
ルセル)の,満充電状態での内部抵抗測定結果を示す.内部抵抗測定は交流イン ピーダンス法により行った.周波数変調した微弱電圧(電流)を電池に印加し,
応答電流(電圧)の振幅,位相差から時定数の異なる反応素過程を分離する手法 である.OCV(開回路電圧)に対し,振幅 10mV を重畳させた交流電圧を 1MHz
から 1mHz まで印加し応答電流から内部抵抗を求めた.10kHz 付近の高周波数域 に応答する反応に対応する円弧と,10Hzから 0.1Hzの低周波数域に応答する反応 に伴う円弧と,リチウムイオンの拡散抵抗(ワールブルグインピーダンス)を示 す右上がりの直線部が見られ,サイクルの増加に伴って低周波数側の円弧が顕著 に増大していることが分かる.
Fig. 2-12. 解析用フルセルの内部抵抗(ナイキスト線図).
Fig. 2-13. 解析用フルセルの内部抵抗(ボード線図).
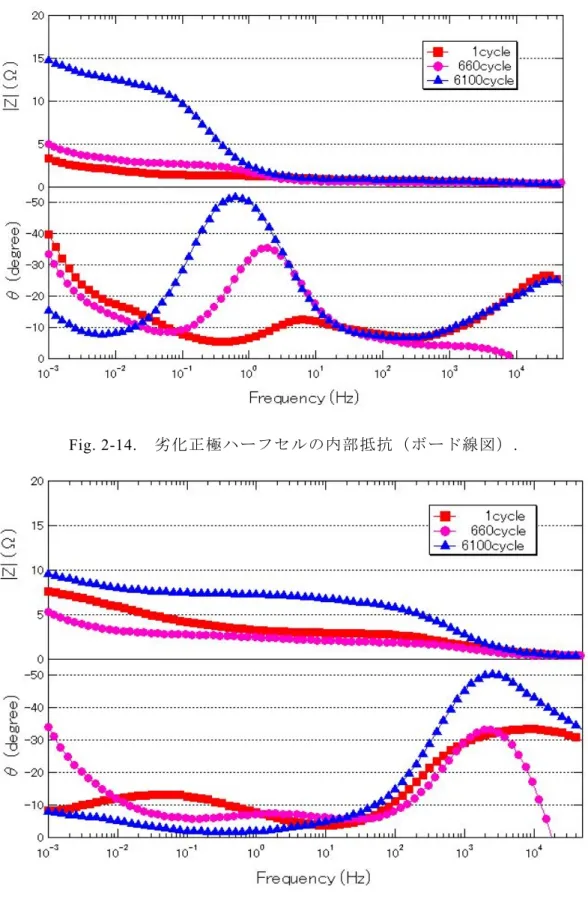
Fig. 2-14に正極ハーフセル,Fig. 2-15に負極ハーフセルの満充電状態での内部
抵抗測定結果を示す.正極は低周波数域に電荷移動抵抗が確認され,フルセルと 同様に,抵抗の増大が確認された.一方で,負極は主に高周波数域にて電荷移動 抵抗の増大が確認された.今回の充放電サイクル試験では,フルセルにおいて顕 著に低周波数域の反応が増加していたが,抵抗分離解析により,この低周波数域 の反応は正極の電荷移動抵抗であり,内部抵抗増加の主要因は正極であることが 明らかとなった.
Fig. 2-14. 劣化正極ハーフセルの内部抵抗(ボード線図).
Fig. 2-15. 劣化負極ハーフセルの内部抵抗(ボード線図).
単極容量の評価
作製したハーフセルを用いて単極容量測定を実施した結果を,Fig. 2-16に示す.
解析用フルセルでは充放電サイクル試験により容量低下が確認された.一方で,
正極ハーフセル,負極ハーフセルでは,フルセルに見られる容量低下が確認され なかった.ハーフセルは対極が金属Li であるため充放電に必要なリチウムイオン は十分に存在する.つまり,今回の,充放電サイクル試験では,正極,負極の活 物質は,リチウムイオンを挿入脱離する能力を失ってはいないが,充放電可能な リチウムイオンが減少しているために,容量低下が起こっていると考えられる.
なお,負極での容量増加は,副反応が起こっていると考えられ,電解液の分解に よる皮膜生成反応が活性化していることが示唆される.
Fig. 2-16. ハーフセルでの単極容量測定結果.
正極活物質内のリチウムイオン拡散抵抗の評価
作製した正極ハーフセルについて,リチウムイオンの拡散抵抗を,定電流間欠 滴定法(GITT:Galvanostatic Intermittent Titration Technique)により評価した[16]. GITTのパルス波形の例を Fig. 2-17に示す.IR領域は主に電解液中のイオン伝導 抵抗,電子抵抗,電解液/活物質界面の電荷移動抵抗が含まれる.ΔEτ 領域にはリ チウムイオンの拡散が含まれるが,特に,反応時定数の遅い電解液/活物質界面の 電荷移動抵抗と,リチウムイオンの拡散抵抗を正確に分離するためには,反応時 定数を評価できる交流インピーダンス法との複合解析が必要となる.
50%充電状態での GITT 波形を解析し,式(1)を用いてリチウムイオンの拡散係 数を算出した.Lは電極合材層の厚み,ΔEs はGITT測定前後の電位差である.
2 2
4
∆
= ∆
+
πτ
Eτ
Es
DLi L (
τ
<< L2/ DLi+)・・・(1)放電状態での正極活物質内のリチウムイオン拡散係数を,Fig. 2-18に示す.充 放電サイクル数の増加に伴って,拡散係数が低下していることが明らかとなった.
Fig. 2-17. 正極ハーフセルにおける GITT波形.
Fig. 2-18. 正極活物質内のリチウムイオンの拡散係数.
正極の酸化還元反応解析
NCM111活物質における Niは,リチウムイオンの挿入脱離に伴う電荷補償を担
うとされ,活物質の劣化を調べる上で重要な元素である.それ故に,天然存在比
で約1.1%含まれる 61Ni核はMössbauer核種である事を考えると,超微細相互作用
を通じてNCM111の劣化挙動を調べる事は大変興味深い.しかしながら,線源で
ある 61Co の半減期は 1.65 時間と短く報告事例は少ない.一方,放射光を用いた 無反跳共鳴吸収効果の利用実験の進歩により,様々な核種についてSPring-8で測 定できるようになっている[11, 12].著者らは放射光を用いた61Ni Mössbauer分光 測定を試み NCM111 のスペクトルを初めて得ることに成功し[13],第一原理計算 を用いてNiのカチオンミキシングを評価している[14].
測定はSPring-8 BL11XUで実施した.入射X線は Si(111)を用いて単色化し,61Ni を86%富化した Ni84V16合金箔を基準物質として用い,速度範囲+/- 7.838 mm / sec で駆動させた.検出器は8素子 Si avalanche photodiode detectorを用いた.サンプ
ルはSOC0%,及び SOC50%に調整された電極を用い,液体ヘリウムで 6Kに冷却
し,測定を実施した.
放射光による 61Ni Mössbauerスペクトルを Fig. 2-19 に示す.NMC のNéel点は
150 K程度とされるが,6 Kにおいて磁気分裂成分と非磁性成分の重畳と解釈でき
るスペクトルが得られた.解析の結果,NCM111は半数のNiだけが充放電に関わ っており,充電状態のNCM111はNi2+とNi3+の混合原子価状態である事が判明し た.
Fig. 2-19 放射光による61Ni Mössbauerスペクトル(6K).
正極の劣化解析
充電時には,リチウムイオンは正極活物質から引き抜かれ,負極活物質に挿入 される.放電時には逆の反応が起こり,充放電サイクル試験においては,正極・
負極の活物質にてリチウムイオンの挿入・脱離による構造変化が繰り返される.
正極の劣化機構解明のため,正極の結晶構造変化をCs-STEMにより調査した.
リチウムイオンは水分と反応し容易に状態が変化するため,劣化後の試作電池を 露点-70℃以下に調整された Ar 雰囲気下にて解体し,取り出した正極を,不活性 ガス雰囲気を保持したまま FIB(収束イオンビーム)装置に挿入した.その後,
断面マイクロサンプリング法によりTEMサンプルを摘出し,FIB加工により薄片 化した.
660 サ イ ク ル 後 の 正 極 活 物 質 の 断 面 TEM 観 察 結 果 を Fig. 2-20 に 示 す . Li(Ni1/3Mn1/3Co1/3)O2(NMC111) は 六 方 晶 系 の 層 状 岩 塩 構 造 で あ る が , 表 層 の 構 造はリチウムが脱離した立方晶岩塩構造に結晶構造転移していることをナノ電子 線回折像,およびシミュレーションパターンとの比較により同定した.
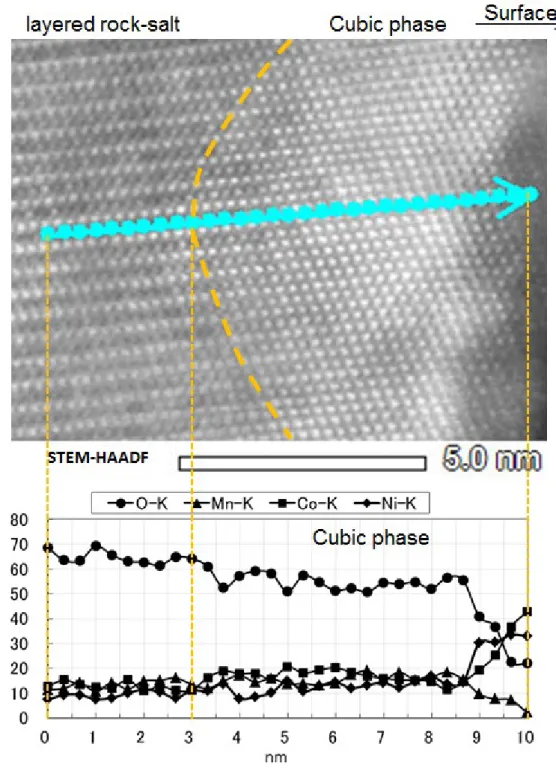
Fig. 2-21 に原子分解能で観察した正極活物質表層の断面 Cs-STEM-HADDF像
を示す.遷移金属元素の並ぶ層の間にリチウムイオンが挿入・脱離されるが,本 来リチウムイオンが存在すべきサイトに遷移金属のNi,Co,Mnが移行するカチ オンミキシングが起こっていることが確認された.また,STEM-EDXライン分析 により,立方晶岩塩構造では酸素欠損が,さらに最表層では Mn 溶出に伴う Ni, Coの濃化が確認された.
一方で,6100サイクル後の正極活物質の表層では,立方晶岩塩構造への結晶構 造転移層の厚みは30nm程度で顕著な増加はみられなかったが,Fig. 2-22に示す
STEM-EELS 結合状態マッピング像により,活物質表層で形成された立方晶岩塩
構造層上に,新たにMn,Coの酸化物,フッ化物由来の無機系界面層の形成も確 認された.
Fig. 2-20. 正極表面の結晶構造転移層のCs-STEM像.
Fig. 2-21. 正極表面の STEM-HAADF像とEDXライン分析.
Fig. 2-22. 正極表面の STEM-EELS結合状態マッピング像(6,100サイクル後).
グラファイト負極の劣化解析
660サイクル後の負極活物質の表面皮膜の結合状態をXPS により調査した.主 要元素の狭域スペクトルを Fig. 2-23 に示す.有機系の皮膜や LiFや Li2CO3など の 無 機 系 の 皮 膜 に 加 え , 正 極 活 物 質 か ら 溶 出 し 負 極 上 で 析 出 し た と 考 え ら れ る Mnの存在を確認した.皮膜厚さは,SiO2換算で約 30nmであった.
6100サイクル後の負極活物質の主要元素の狭域スペクトルを Fig. 2-24に示す.
660サイクル後にみられた有機系の皮膜やLiFや Li2CO3などの無機系の皮膜,Mn の析出に加え,新たにLi酸化物が確認された.皮膜厚さは,SiO2換算で約 140nm であり,660 サイクルから 4倍以上増加した.LiF や Li2CO3など加え,長期サイ クルにおける LiOx の生成が,充放電可能なリチウムイオンを失活させ,充放電 容量の低下を引き起こしていると考えられる.
Fig. 2-23. 負極表面の XPS深さ方向分析(660サイクル).
Fig. 2-24. 負極表面の XPS深さ方向分析(6100サイクル).
2.3.3. Si負極電極のサイクル劣化機構 充放電サイクル試験
試作したラミネート型電池について,室温にて電圧0Vから1.6Vの間で充放電 サイクル試験を実施した.充放電曲線をFig. 3-2 に示す.約 3500mAh/g相当のリ チウムイオンを動かす深い充放電と,約1250mAh / g 相当の浅い充放電をそれぞ れ行った.深い充電を行ったセルの放電容量は,浅い充放電を行ったセルの放電 容量と同等であり,Siに挿入した Liが脱離できていない状態であった.
内部抵抗解析
内部抵抗測定は交流インピーダンス法により行った.周波数変調した微弱電圧
(電流)を電池に印加し,応答電流(電圧)の振幅,位相差から時定数の異なる 反応素過程を分離する手法である.OCV(開回路電圧)に対し,振幅10mV を重 畳させた交流電圧を 300kHz から 1mHz まで印加し応答電流から内部抵抗を求め た.抵抗測定結果を,Fig. 2-25, Fig. 2-26に示す.3サイクル目の放電では,深 い充放電を行ったセルにて,10mHz 付近の低周波域での顕著な反応抵抗が確認さ れた.
Fig. 2-25. 初回の充放電容量.
Fig. 2-26. 3サイクル目の内部抵抗(ボード線図).
1.5
1.0
0.5
0.0
V o lt age ( V )
3500 3000
2500 2000
1500 1000
500 0
Capacity (mAh/g)
1250mAh/g charge-discharge 3500mAh/g charge-discharge
200 150 100 50 0
|Z | (Ω )
10-3 10-2 10-1 100 101 102 103 104 105
Frequency (Hz)
-80 -60 -40 -20 0
θ (d e gr e e )
1250mAh/g charge-discharge
3500mAh/g charge-discharge
電極の構造変化,表面被膜の調査
理論容量の 30%である 1260 mAh/g まで充電した Si負極の断面 SEM 観察像を Fig. 2-27に,満充電状態の Si負極の断面 SEM観察結果を Fig. 2-28に示す.リチ ウムイオンが挿入された箇所は暗いコントラストになっている.充電により合金 化した箇所が微粉化している.微粉化により電子伝導パスが切れることで,結果 としてリチウムイオンの脱離が阻害されていると推察される.
XRDの結果を Fig. 2.29に示す.1250mAh/gまで充電した場合には,結晶Siに 加え,非晶質に変化している.3500mAh/gまで充電した場合には,Li15Si4の結晶 が形成されている.この Li15Si4は不可逆との報告があり[27],電解液界面の電荷 移動抵抗が,交流インピーダンスにおける低周波数域に出現した反応抵抗である と考えられる.
Fig. 2-27. 理論容量の 30%である1260mAh / gまで充電した Si負極の 断面SEM観察像.
Fig. 2-28. 満充電状態の Si負極の断面SEM観察像.
Fig. 2-29. Liを挿入した Si負極の X線回折.
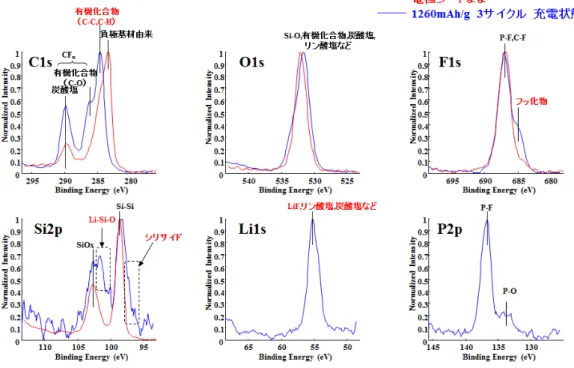
XPSによるSi負極表面の結合状態解析結果を Fig. 2-30,Fig. 2-31に示す.シリ サイドの形成を確認した.また,表層では Li-O-Si が存在していた.有機系被膜 の他,無機系被膜としてフッ化リチウム,炭酸リチウムの存在が確認された.
劣化機構解明のため,Si の結晶構造変化を Cs-STEM により調査した.リチウ ム イ オ ン は 水 分 と 反 応 し 容 易 に 状 態 が 変 化 す る た め , 劣 化 後 の 試 作 電 池 を 露 点 -70℃以下に調整された Ar 雰囲気下にて解体し,取り出した負極を,不活性ガス 雰囲気を保持したまま FIB(収束イオンビーム)装置に挿入した.その後,断面 マイクロサンプリング法により TEMサンプルを摘出し,FIB加工により薄片化し た.
理論容量の 30%である 1260mAh/g まで充電した Si 負極の断面 TEM 観察像を Fig. 2-32に,EELSによる結合状態マッピング結果を Fig. 2-33に示す.リチウム イオンの挿入によりアモルファス化している領域が確認された.Fig. 2-34にEELS スペクトルを示す.また,アモルファス領域の表層では EELS のスペクトル形状
より,Li-Siの化合物の存在が確認された.
Fig. 2-35,Fig. 2-36に高分解能 STEM観察結果を示す.リチウムイオンの挿入 による結晶欠陥が確認された.また,Si結晶のTd サイトに存在するLi を確認し た.
Fig. 2-30. 負極表面の狭域 XPS スペクトル.
Fig. 2-31. 負極の深さ方向 XPSモンタージュスペクトル.