Nrf2の活性状態は食道異形成を引き起こし食道の発
がんにおける細胞選択性を誘導する
著者
堀内 真
学位授与機関
Tohoku University
学位授与番号
11301甲第19135号
URL
http://hdl.handle.net/10097/00129230
博士論文
Nrf2 の活性状態は食道異形成を引き起こし
食道の発がんにおける細胞選択性を誘導する
東北大学大学院医学系研究科医科学専攻 外科病態学講座消化器外科学分野 堀内 真目次 1. 要約 --- 3 2. 研究背景 --- 5 3. 研究目的 --- 9 4. 研究方法 --- 10 5. 研究結果 --- 16 6. 考察 --- 34 7. 結論 --- 42 8. 文献 --- 43 9. 図 --- 52 10. 表 --- 77 11. 謝辞 --- 79
1. 要約
酸化ストレスや親電子性物質に応答して細胞保護に働く転写因子Nrf2 は、化学発
がんに対する抑制効果とがん細胞の悪性化の両側面に貢献する。しかしながら、生
体においてNrf2 が食道の発がんにどのような役割を担っているのかは不明な点が多
い。そこで、Nrf2 やその抑制因子である Keap1 を成獣の時点で誘導的に欠失する
マウス、Keratin5-CreERT2::Nrf2flox/flox(K5CreERT2-Nrf2F/F)マウスと
Keratin5-CreERT2::Keap1floxB/floxB(K5CreERT2-Keap1FB/FB)マウスを用いて、
Nrf2 が 4-ニトロキノリン-1-オキサイド(4-nitroquinoline-1-oxide;4NQO)によ って誘導される食道の化学発がんにどのように貢献しているのかを検証した。 タモキシフェンの投与によりKeap1 の欠失を誘導した K5CreERT2-Keap1FB/FB マウスの食道は、Nrf2 の活性化を認め、上皮の増殖能の亢進と分化障害により異形 成を示した。興味深いことに、DNA 障害のマーカーであるリン酸化ヒストン H2A.X(gH2A.X)を Keap1 の欠失を免れた上皮基底細胞で認め、これらの細胞は DNA 障害を受けやすい状況下にあることが示唆された。K5CreERT2-Keap1FB/FBマ ウスを用いた4NQO 発がん実験では、あらかじめ Keap1 を欠失させることで 4NQO による発がんが促されることを見出した。ただし、ほとんどの腫瘍は Keap1
を発現している細胞集団であり、腫瘍化したクローンはKeap1 の欠失を免れた細胞 であった。Keap1 の欠失した細胞は異型細胞となったが、その周囲で DNA 障害を 受けたKeap1 陽性細胞が 4NQO によって腫瘍化しやすくなる可能性が考えられ た。一方で、タモキシフェンの投与によりNrf2 の欠失が誘導された K5CreERT2-Nrf2F/Fマウスの食道は、特徴的な表現型を認めなかった。K5CreERT2-Nrf2F/Fマ ウスを用いた4NQO 発がん実験においては、Nrf2 欠失細胞は 4NQO によるストレ ス環境下で消失してしまうため、Nrf2 の欠失を免れた細胞のみが腫瘍化した。 本研究は当初、正常細胞においてNrf2 が発がんを抑制する側面を観察するために 発がん物質を投与する前の遺伝子改変実験を、がん細胞においてNrf2 ががん形成を 促進する側面を観察するために発がん物質を投与した後の遺伝子改変実験を行っ た。しかしながら、食道扁平上皮に部分的に遺伝子欠失が成立したことで予想と反 する結果が得られた。Keap1 欠失細胞の出現による発がん促進効果の明確なメカニ ズムは不明ではあるが、上皮細胞間の相互作用や環境適応に、細胞におけるNrf2 の 活性状態が密接に関わっていることを証明した。本研究では、様々な細胞クローン が存在する食道扁平上皮の発がん過程において、Nrf2 の活性状態が細胞の運命決定 に作用することを示した。
2. 研究背景 食道がんは最も予後の悪い悪性腫瘍の一つであり、世界でも6番目に死亡数が多い 腫瘍として知られている 1)。食道がんに対する有効な分子標的薬はなく、外科手術、 化学療法、および放射線療法を組み合わせた集学的治療の進歩があるにも関わらず、 未だに予後は悪く2)、新たな治療法や予防法の開発は急務である。食道がんの由来は、 食道管腔の大部分を覆う扁平上皮が最も多く、特に東アジアやアフリカでは食道扁平 上皮がんが大多数を占める 3)。食道の他、皮膚や頭頸部なども構成する扁平上皮は、 外界に曝露されて物理的・化学的ストレスを受けやすい。酸化ストレスや親電子性物 質は食道の発がん過程における重要なメディエーターであるが、食道扁平上皮の発が んにおける最大のリスクファクターであるタバコでも、酸化ストレスや親電子性物質 の関与があるとされている4)。例えば、タバコの煙には、活性酸素種の産生を誘導す るニコチンだけでなく、過酸化水素などの活性酸素種5)、また、親電子性物質である クロトンアルデヒドなども含まれている。がんは、これらのメディエーターが誘導す るDNA の突然変異により形成され、食道がんにおいては TP53、CDKN2A、あるい はNRF2 の遺伝子である NFE2L2 などの変異を高頻度に認める6)。それゆえに、食 道における発がんを抑えるためには、酸化ストレスや親電子性物質がどのように発が
んに寄与しているのかを理解する必要がある。 Nrf2 は酸化ストレスや親電子性物質のような環境ストレスに対して保護的な作用 を示すマスター転写因子である7, 8)。定常状態では、Nrf2 は Keap1 という抑制因子 に捕えられており、ユビキチン化を受けてプロテアソームにより分解されている9, 10)。 一方、酸化ストレスや親電子性物質に曝露されると、Keap1 のシステイン残基に修飾 が加わることで Nrf2 はユビキチン化を免れる 11)。それにより、Nrf2 は核内へ移行
し、抗酸化剤応答配列(Antioxidant Response Element; ARE)に結合することで、
解毒代謝酵素群(NAD(P)H:quinone oxidoreductase 1; Nqo1)、g-グルタミルシステ
イン合成酵素(Glutamate-cysteine ligase catalytic subunit; Gclc)などの遺伝子群
を発現し、酸化ストレスや親電子性物質に対する防御作用を発揮する(図 1)。実際 に、Nrf2 欠失マウスは細胞保護作用の欠落から、様々な化学発がん物質に対して感 受性が高く、発がんを起こしやすい12-15)。一方で、抑制因子である Keap1 の欠失マ ウスは、恒常的にNrf2 の活性化が誘導されることで、生後間もなく食道に過角化を 起こして致死となる16)。この事実により、食道におけるKeap1 の欠失、つまりは食 道におけるNrf2 の活性化を誘導したマウスを実験に利用することができず、食道発 がんにおけるKeap1-Nrf2 システムの解析は困難であった。 Nrf2 が化学発がんを抑制する一方、様々ながんにおける Nrf2 の過剰発現は、がん
細胞に対する保護的な役割と悪性化に寄与することも分かってきている 17, 18)。実際 に、KEAP1やNRF2の体細胞変異が様々ながんで確認されており、それらはNRF2 の恒常的な発現を誘導する18, 19)。特に、食道扁平上皮がんでは NRF2の体細胞変異 が高頻度に見られ、NRF2 の過剰発現が予後悪化に関与している20)。そして、これら NRF2 が高発現しているがんは Nrf2-addicted cancers と呼ばれている21)。 4NQO はマウスの口腔や食道における発がん物質として用いられ、酸化ストレスに よるDNA 酸化障害と NQO1 による代謝物であるヒドロキシアミノキノリン-1-オキ
サイド(4-hydroxyaminoquinoline-1-oxide ; 4HAQO)の DNA 付加により、遺伝子
変異を引き起こしてがんを誘導する(図 2)。過去の報告によると、4NQO を用いた 食道の発がん実験において、Nrf2 欠失マウスは 4NQO に対して感受性が高く、反対 にKeap1 低発現マウスは抵抗性であることが示されている22)。しかし、このNrf2 が 活性化したKeap1 低発現マウスにおける 4NQO 発がんに対する抵抗性の機序は、食 道における過角化による物理的な作用であるのか、もしくは解毒酵素活性の上昇によ るのかは不明なままであった。さらに、Nrf2 の活性は、がん微小環境における免疫細 胞へ関与することも明らかにされている。例えば、骨髄由来免疫抑制細胞(MDSC) や制御性T 細胞において Nrf2 の活性が低下すると、がんに対する免疫機構が減弱し、 がんの進展や転移を促す23, 24)。以上から、食道上皮細胞におけるNrf2 の活性が発が
んへどのように関与するかを解析する際は、上皮特異的な遺伝子改変マウスを利用す る必要があり、加えて食道の過角化を避けることが理想であると考えられた。 また、食道を含む臓器は様々な遺伝子変異をもつ細胞集団で構成されており、それ らのクローンは同一組織内でせめぎ合って存在している6, 25)。あるクローンが食道の 扁平上皮組織内に存続するためには、細胞の生存能力と分化せずに幹細胞性を維持す る能力が必要となる26)。Nrf2 の働きは細胞の生存能力を亢進する一方、基底層から の分化を促進することが示唆されており 27)、細胞競合には有利にも不利にも働く可 能性がある。また、曝露される環境によっても上皮の生存能力や分化能力に影響を与 えるため、それらのバランスが細胞競合に反映されると考えられる。前述のように、 Nrf2 は様々な側面からがんに関与することが分かってきたが、同一組織内の細胞集 団において、上皮細胞間の相互作用や細胞競合とNrf2 の関係性は明らかになってい ない。
3. 研究目的 扁平上皮特異的かつ誘導的な遺伝子欠失により、食道上皮の Keap1 欠失を時期特 異的に誘導することで、今までは困難であった成獣マウスの Keap1 欠失食道の表現 型を解析する。また、扁平上皮特異的にNrf2 あるいは Keap1 の欠失を誘導したマウ スを用いることで、食道発がんの過程で上皮細胞におけるNrf2 の役割を明らかにす る。
4. 研究方法
4-1. 遺伝子改変マウス
扁平上皮特異的かつ誘導的な遺伝子欠失マウスは、ヒトKeratin5をプロモーター
としてCre タンパク質を発現し、かつタモキシフェン誘導的に遺伝子欠失をするマウ
ス(
K5CreERT2)を東北大学医化学分野にて作製した。蛍光発現モニター(Rosa26-tdTomato)マウス(#026006)は Jackson Laboratory から購入した。Nrf2-flox マウ
ス28)およびKeap1-floxB マウス29)はそれぞれJingbo Pi 博士、Sham Biswal 教授よ
り譲渡いただいた。Keap1-floxA マウスは東北大学医化学分野で作製した30)。7-9 週 齢雄性の C57BL/6J および BDF1 の混合系統マウスを利用した。すべてのマウスは 東北大学大学院医学系研究科附属動物実験施設で飼育し、東北大学動物実験委員会の 承認を得て動物実験を遂行した。7 日間における 20%の体重減少を人道的エンドポイ ントとした。 4-2. K5CreERT2 マウスを用いた遺伝子欠失誘導 それぞれの遺伝子型マウスを交配することで、K5CreERT2:: Rosa26-tdTomato
(K5CreERT2-Keap1FB/FB)マウス、Keap1FA/FAマウス、およびNrf2flox/flox
(Nrf2F/F)マウス、K5CreERT2::Nrf2flox/flox(K5CreERT2-Nrf2F/F)マウスを作製
した。Cre タンパク質を発現させ遺伝子欠失を誘導させる際は、タモキシフェン
(Sigma-Aldrich, #T5648)をコーン油(富士フィルム和光純薬社, #032-17016)で
20 mg/ml の濃度に溶解し、成獣マウスに腹腔内投与した。0.1 mg/g 体重の容量で 3
日連続投与した。K5CreERT2 の発現臓器の確認は、K5CreERT2::
Rosa26-tdTomato マウスの蛍光を観察することにより行った。
7-9 週齢の K5CreERT2:: Rosa26-tdTomato マウスにタモキシフェンを投与した
後、マウスを安楽死させ、それぞれの臓器を4%パラホルムアルデヒド(PFA;ナカ
ライテスク社、#09154-85)で 12 時間固定した。20%スクロース溶液で処理した後
にOCT コンパウンド(サクラファインテック社、#4583)を用いて組織を包埋し
た。14 µm 厚の凍結切片を作製し、Fluorescence Mounting Medium(ダコ社、
#S3023)を滴下した後に、倒立蛍光顕微鏡 (KEYENCE BZ-8000)で観察した。
4-3. 4NQO を用いた食道発がんモデル
食道の発がんモデルとして、化学発がん物質である4NQO(Sigma-Aldrich,
ルスルホキシド(ナカライテスク社、#09659-85)で 10 mg/ml の濃度に溶解した後 に、水で0.1 mg/ml に希釈し、自由飲水投与した。1つ目のプロトコールは、4-2 と同様に7-9 週齢のマウスにタモキシフェンを 3 日腹腔内投与して Keap1 あるいは Nrf2 の欠失を誘導し、その1週間後より 4NQO の投与を開始した(pre-protocol)。4NQO を 12 週間投与し、その後さらに通常水に戻して 12 週間観察後に 解析した。2 つ目のプロトコールは、4NQO を 12 週投与したのちにタモキシフェン を3 日腹腔内投与して遺伝子欠失を誘導した(post-protocol)。タモキシフェン投与 と同時に通常水に切り替えて12 週間観察後に解析した。観察期間内は、1週間に一 度、体重測定と飲水の交換を行った。組織の採取は、イソフルラン麻酔下で安楽死 後に行った。マウス食道を長軸方向に割を入れ開き、粘膜面全体を撮影した。その 画像をもとに、Image J Fiji ソフトウェアを用いて腫瘍径を測定し、最大径が 1 mm 以上の腫瘍を数えた。
4-4. 定量逆転写 PCR(Quantitative reverse transcription-PCR;qRT-PCR)
採取したマウス食道は長軸方向に開き、0.5 g/l トリプシン /0.53 mmol/l-EDTA 溶
液(ナカライテスク社、#32778-05)に約 8 時間浸した後、扁平上皮層のみを分離
た。RNA はセパゾール RNAⅠ Super G(ナカライテスク社、#09379-97)を用い
て精製し、ナノフォトメーターNP80(Implen 社)で RNA 濃度を測定して調製し
た。cDNA の合成は ReverTra Ace qPCR RT Master Mix with gDNA Remover(東
洋紡ライフサイエンス社, #FSQ-301)を用いた。得られた cDNA は KAPA SYBR
FAST qPCR master mix (2x) Kit (Kapa Biosystems 社、#KK4602) を用いて調製
し、qRT-PCR 解析は QuantStudio 6 Flex リアルタイム PCR システム(サーモフィ
ッシャーサイエンティフィック社)を使用した。リボソームRNA(rRNA)を内部
標準として、得られた値を補正した。使用したプライマーの配列は表1 に示す。
4-5. DNA 組換え効率の測定
Cre タンパク質によって誘導された DNA の欠失は、DNA の組換え効率として定
量PCR により評価した。採取したマウス食道は長軸方向に開き、0.5 g/l-トリプシン /0.53 mmol/l-EDTA 溶液(ナカライテスク社、#32778-05)に約 8 時間浸した後、 扁平上皮層のみを分離した。DNA を精製し、ナノフォトメーターNP80(Implen 社)でDNA濃度を測定して調製した。qPCR は前述の 4-4 と同様に行った。b-アク チンを内部標準として、得られた値を補正した。使用したプライマーの配列は表2 に示す。
4-6. 組織学的解析 マウス食道を長軸方向に開き、あるいは横断的に輪切りとし、マイルドホルム 10N(富士フィルム和光純薬社、#131-10317)で固定した。パラフィン包埋とし、 4 µm 厚の薄切切片を作製した。 ヘマトキシリン・エオジン(HE)染色を行った組織標本は、食道上皮の細胞層と 角化層の厚さを1組織あたり3箇所測定し、平均値を評価した。発がん実験を行っ た組織においては、HE 染色で 6-7 横断面の中から浸潤がんを認める箇所を数えた。 免疫組織化学染色はそれぞれの一次抗体に対して増感法を用いて行った。使用し
た一次抗体は、抗Nrf2 (D9J1B) 抗体(Cell Signaling Technology 社、#14596;
400 倍希釈)、抗 Keap1 抗体31)(4 倍希釈)、抗 Nqo1 抗体 (アブカム社、
#ab2346;200 倍希釈)、抗 Ser139 リン酸化ヒストン H2A.X (20E3) 抗体 (Cell Signaling Technology 社、#9718;300 倍希釈)を使用した。
パラフィン包埋した薄切切片をキシレンとエタノールで脱パラフィン後、10 mM
クエン酸緩衝液(pH6)に浸し、高圧蒸気滅菌器による 121℃、10 分間の加熱によ
り抗原賦活化処理を施した。3%過酸化水素を含むメタノール溶液に 10 分間浸して
内因性ペルオキシダーゼ活性を阻害した。Protein Block Serum-Free(ダコ社、
抗体に4℃で一晩浸した。ラット抗体に対してはシンプルステインマウス MAX-PO
(Rat;ニチレイバイオサイエンス、#414311)に 30 分浸し、ポリマー複合体を形
成した。ウサギ抗体に対してはEnVision+ Dual Link System-HRP(ダコ社、
#K4063)に 30 分間浸した。ヤギ抗体に対しては Rabbit anti-Goat
IgGs/Biotinylated(ダコ社、#E0466)に 30 分間、Streptavidin/HRP (ダコ社、 #P0397)に 30 分間浸した。その後、SignalStain DAB Substrate Kit(Cell Signaling Technology 社, #8059S)を用いて一次抗体に応じた時間で発色し、ヘマ トキシリンによる核染、エタノール脱水処理、キシレン透徹、および封入を行っ た。gH2A.X 陽性細胞は 1 切片における基底細胞数に対する割合を定量した。 4-7. 統計学的解析 結果は平均と標準偏差を示した。統計学的な有意差は、二群間の解析はStudent’s t検定で算出した。二因子の関連多群について、全体の傾向の解析はtwo-way ANOVA検定で算出した。P値は0.05以下を統計学的に有意差があるとした。
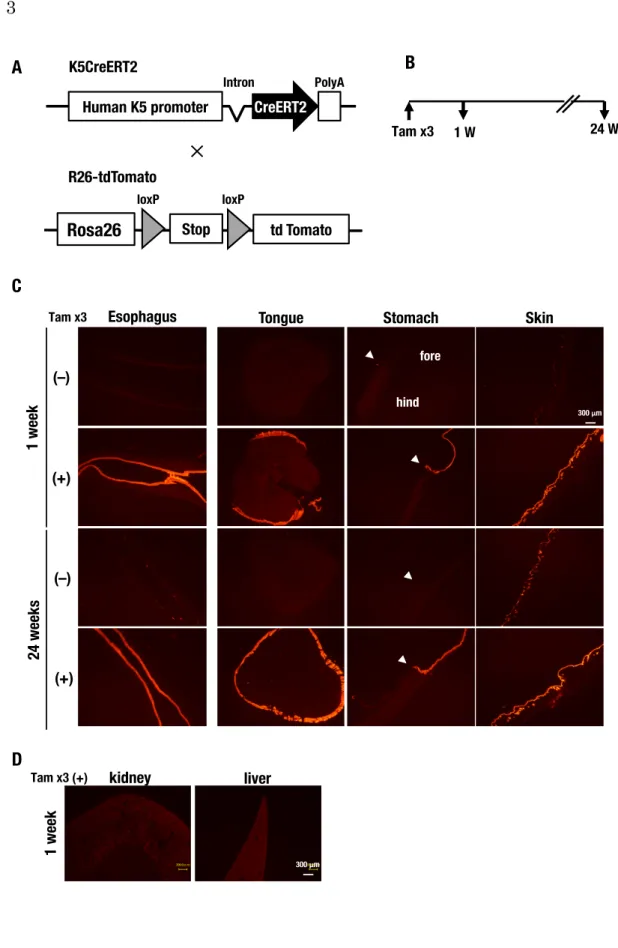
5. 研究結果 5-1. 扁平上皮特異的かつ誘導的遺伝子改変マウスの発現解析 重層扁平上皮は基底層、有棘層、顆粒層、角質層からなるが、自己複性能を持つ 基底細胞が分裂し、一定の割合で管腔側の有棘層細胞に分化することで恒常性が保 たれている25)。本研究では、基底細胞で発現するKeratin 5 をプロモーターとした Cre タンパク質を利用することで、基底細胞とその細胞を基に分化・構成される基 底層上部の細胞に欠失を誘導し、扁平上皮全層の遺伝子改変を試みた。扁平上皮特 異的かつ誘導的遺伝子欠失マウスであるK5CreERT2 マウスにおいて、Cre タンパ ク質の発現臓器を確認した。Cre タンパク質存在下で停止コドンが欠失して蛍光を
発現するK5CreERT2:: Rosa26-tdTomato マウスの蛍光を解析した(図 3A)。タモ
キシフェンあるいは溶媒を投与し、Cre タンパク質の発現を誘導して tdTomato を 発現させた後、1週後と24 週後に解析した(図 3B)。24 週は 4NQO 発がん実験を 完遂するのに要する期間である。1 週後の解析では、タモキシフェンの誘導によ り、食道、舌、前胃、および皮膚の扁平上皮部にtdTomato の蛍光を確認した(図 3C)。扁平上皮組織ではない腎臓や肝臓には発現を認めず、扁平上皮特異的に Cre タンパク質が発現していることが確認された(図3D)。24 週後においても、1週後
と同様に、食道、舌、前胃、および皮膚の扁平上皮部にtdTomato の蛍光を確認し た(図3C)。ただし、タモキシフェン非投与群においても、食道と皮膚で蛍光が観 察され、タモキシフェンによる誘導がなくともRosa26-tdTomato アリルではわずか に欠失が誘導されることが分かった(図3C)。この意図せぬ誘導は本研究に影響し ないと考えられた。以上より、K5CreERT2 はタモキシフェン誘導的に扁平上皮特 異的に発現し、少なくとも24 週間は欠失の誘導された細胞が維持されることを確認 した。このことは、全ての基底細胞において遺伝子改変が成立した場合、上皮のタ ーンオーバーや寿命に関わらず、複製を繰り返すことで遺伝子改変の成立した基底 細胞が半永久的に維持されることが示唆された。 5-2. 食道上皮における誘導的な Keap1 欠失は上皮の異形成を生じる タモキシフェン投与により扁平上皮のKeap1 を欠失するマウス、K5CreERT2-Keap1FB/FBマウスを用いて、成獣期に誘導的にKeap1 を欠失した食道の解析を行っ た。対照群はCre タンパク質を発現しない Keap1FB/FBマウスとし、タモキシフェン 投与後1 週間で解析した(図 4A)。食道断面の HE 染色像を図 4B と図 4C に示す。 通常、基底細胞は規則正しく基底層に並んでおり、管腔側へと分化が進み、最終的 には脱核して角化層に至る(図4B;黄色い点線は基底膜を示す)。一方、
K5CreERT2-Keap1FB/FBマウスの食道は、基底層に核の腫大を伴う異型細胞の集団 が認められ、それらが粘膜下層側に突出する形態をとった(図4C)。その異型細胞 の深部への突出に伴い、異型を認めない基底細胞集団が管腔側へ押し上げられ、基 底膜が蛇行した。食道横断面における基底細胞の総数は、対照マウスと比較して K5CreERT2-Keap1FB/FBマウスで増加した(図 4D)。また、K5CreERT2-Keap1FB/FBマウスの食道で細胞層と角層の厚さはどちらも肥厚しており、特に細胞 層の肥厚が顕著であった(図4B、4C;黄色い矢印は細胞層の厚さ、黒い矢印は角 層の厚さを示す、4E、4F)。以上から、K5CreERT2-Keap1FB/FBマウスの食道は異 型細胞を有し、かつ上皮の肥厚を伴う異形成を生じており、上皮組織が垂直および 水平方向に拡大していることが分かった。 続いて、Keap1 の欠失とそれに伴う Nrf2 の活性化を確認するため、K5CreERT2-Keap1FB/FBマウスの食道において免疫組織化学染色を行った。Keap1 は、対照マウ スでは基底層を中心に発現が高く、その発現は基底層で均一に広がることが確認さ れた(図4G 上段)。一方、K5CreERT2-Keap1FB/FBマウスでは、Keap1 の欠失は飛 び石状に起きていることが確認された(図4H 上段)。そして、その Keap1 が欠失 した細胞は、腫大した核を伴う異型細胞に一致した(図4H;白い点線は異型細胞の 境界を示す)。Nrf2 の染色においては、対照マウスでは Nrf2 の核蓄積を認めなかっ
た(図4G 中段)。これは、定常状態では Nrf2 が速やかに Keap1 により分解される ことを示している32)。同様に、Nrf2 の標的遺伝子である Nqo1 の発現も対照マウス では弱い傾向にあるが、主に基底層を中心に発現した(図4G 下段)。一方で K5CreERT2-Keap1FB/FBマウスでは、Keap1 が欠失した細胞に一致して Nrf2 の核 濃染を認め(図4H 中段)、同様に Nqo1 の発現も同部位で有意に増加した(図 4H 下段)。 興味深い結果として、DNA 二重鎖切断のマーカーであるリン酸化ヒストン H2A.X(gH2A.X)を免疫組織化学染色で確認したところ、K5CreERT2-Keap1FB/FB マウスではgH2A.X 陽性細胞が増加しており、その陽性細胞は Keap1 の欠失を免れ た細胞で顕著に増えていた(図4J)。gH2A.X 陽性細胞の割合を定量すると、 K5CreERT2-Keap1FB/FBマウスで有意に増加した(図4K)。以上をまとめると、 Keap1 欠失細胞は Nrf2 の活性化を示し、組織学的には異型細胞となり食道の異形 成を形成した。ただし、これらKeap1 欠失細胞の出現は、周囲の Keap1 陽性細胞 のDNA 障害を何らかの機序で誘導したと考えられた。 K5CreERT2-Keap1FB/FBマウスは皮膚や舌においても遺伝子欠失による影響が観 察可能であるが、皮膚や舌においては特筆すべき表現型を認めなかった(データ未 掲載)。また、前胃においては全身性Keap1 欠失マウスと同様に、過角化が再現さ
れた(データ未掲載)。 5-3. Keap1 欠失により誘導された食道異形成は経時的変化を伴う 全身性Keap1 欠失マウスあるいは Keap1 低発現マウスは、表現型として食道や 前胃において過角化を示す16, 33)。それゆえに、誘導的にKeap1 を欠失させた K5CreERT2-Keap1FB/FBマウスの表現型が、時間経過とともに過角化へ移行するの ではないかと考え、タモキシフェン投与2 週、4 週、および 12 週後の表現型を同様 に解析した(図5A)。HE 染色像を確認すると、2 週、4 週、および 12 週後におい ては特に過角化は認めなかった(図5B)。角層の厚さを定量すると、同週齢の対照 マウスと比較しても差を認めなかった(図5E)。また、対照マウスと比較すると、 K5CreERT2-Keap1FB/FBマウスでは深層への上皮細胞の突出などが認められ、組織 異形成は保たれていた(図5B)。基底層の細胞数は、K5CreERT2-Keap1FB/FBマウ スで多い傾向にあり、12 週後のみで有意差を認めた(図 5F)。細胞層の厚さは K5CreERT2-Keap1FB/FBマウスで有意に肥厚する傾向にあったが、時間経過ととも に徐々に差異が軽減する現象が見られた(図 5G)。以上から、K5CreERT2-Keap1FB/FBマウスの表現型は、過角化よりむしろ細胞層の肥厚を中心とする異形成 であると結論づけた。
続いて、同組織において免疫組織化学染色を行い、Keap1 および Nrf2 の発現を 確認した。Keap1 の発現は、対照マウスでは基底層を中心に均一な発現を認めるが (図5C 上段左)、K5CreERT2-Keap1FB/FBマウスでは飛び石状にKeap1 の欠失が 保たれていた(図5C 上段右 3 パネル)。Nrf2 の発現は、Keap1 の欠失が誘導され たと考えられる細胞領域でNrf2 の核濃染を確認することができたが、時間経過とと もに発現が減弱する傾向があった(図5C 中段右 3 パネル)。Nrf2 の標的因子である Nqo1 の発現は、Keap1 欠失細胞において亢進されたまま保たれていたが、細胞質 の発現は時間とともに減弱する傾向にあった(図5C 下段右 3 パネル)。 K5CreERT2-Keap1FB/FBマウスではgH2A.X 陽性細胞が増加する傾向にあった(図 5D)。gH2A.X 陽性細胞の定量においても、対照群で時間経過ごとにばらつきがある ものの、K5CreERT2-Keap1FB/FBマウスで一貫して陽性率が高い傾向にあった(図 5H)。 5-4. 誘導的 Keap1 欠失による Nrf2 の活性化と経時的な Nrf2 活性の減弱 K5CreERT2-Keap1FB/FBマウスにおける誘導的なKeap1 の欠失は部分的に起こる ことが組織像から示唆された。また、Keap1 欠失による Nrf2 の活性は経時的に変 化することも組織像から示唆された。続いて、時間経過において、食道上皮全体を
標本として、DNA や RNA レベルで詳細な定量解析を行った(図 6A)。DNA の組 換え効率は、残存したKeap1のアリルを定量しており、タモキシフェン投与1 週後 で約40%、12 週後で約 30%欠失した(図 6B)。この結果は、組織像の解析と矛盾 しないと考えられた。また、K5CreERT2-Keap1FB/FBマウスでは一貫してKeap1 mRNA の発現が抑制されており(図 6C)、Keap1 の欠失細胞が維持されていること を確認した。Nrf2 の標的因子であるNqo1とGclcの遺伝子発現は、タモキシフェ ン投与1 週後における K5CreERT2-Keap1FB/FBマウスで顕著に増加しており、Nrf2 の活性化が確認された(図6D)。ただし、その発現増加率は 2 週後になると軽減さ れ、4 週後以降では対照群レベルに戻る現象が見られた(図 6D)。Nqo1 の免疫組織 化学染色では、Keap1 欠失細胞における発現の亢進は時間とともに減弱傾向も、 Keap1 欠失細胞の境界が判別可能な程度に維持されていた。一方で Nqo1 が発現し ていない細胞もK5CreERT2-Keap1FB/FBマウスで見受けられたので、食道全体とし てはNqo1 の発現は対照群と比較して差がなくなったと考えられた。Nrf2 mRNA の 発現は、タモキシフェン投与後全ての時点で有意差を認めなかった。Keap1 欠失に よるNrf2 の活性化は、遺伝子レベルでなくタンパク質レベルで分解が抑制されるた め9)、矛盾しない結果であると考えられた。また、時間経過とともにNrf2 の活性化 が軽減される現象は、Keap1 以外の Nrf2 タンパク質の制御が関連していることが
示唆された。 5-5. 誘導的 Keap1 欠失は分化障害と増殖促進を引き起こす 食道における誘導的なKeap1 の欠失は、過角化ではなく細胞層の肥厚を中心とし た異形成を示すことが明らかとなったので、続いて増殖や分化マーカーの遺伝子発 現を同様の時間経過で確認した。基底層のマーカーであるKeratin 5の発現は、タ モキシフェン投与1 週後における K5CreERT2-Keap1FB/FBマウスで顕著に増加した (図7A)。ただし、その発現の増加率は 2 週後になると軽減され、4 週後以降では 対照群レベルに戻る現象が見られた(図7A)。Nrf2 の標的因子の遺伝子発現と同一 の挙動であると考えられた。一方、扁平上皮分化の最終段階で発現するLoricrin は、タモキシフェン投与1 週後における K5CreERT2-Keap1FB/FBマウスで顕著に抑 制された(図7B)。こちらも、その発現の抑制率は 2 週後になると軽減され、4 週 後以降では対照群レベルまで戻った。以上から、Keap1 の誘導的な欠失は、食道扁 平上皮における分化障害をきたし、その発現変化はNrf2 の活性化度合に相関するこ とが示唆された。 扁平上皮の増殖状態を反映するマーカーであるKeratin 6は、Keap1 欠失誘導後 に発現が顕著に亢進するものの、時間経過により対照群レベルまで抑制された(図
7C)。増殖マーカーであるKi67の発現は、タモキシフェン投与1 週後では発現が亢
進するものの、2 週後以降は抑制された(図 7D)。K5CreERT2-Keap1FB/FBマウス
の表現型である細胞層の肥厚は、食道上皮の分化障害に加え、増殖能の亢進も大き
く寄与していると考えられた。
5-6. 誘導的 Keap1 欠失マウスと Keap1 低発現マウスは食道の表現型が異なる
Keap1 低発現マウスである Keap1FA/FAマウスはKeap1 欠失マウスの致死的な過
角化を緩和することが報告されており、成獣マウスの解析が可能である33)。
K5CreERT2-Keap1FB/FBマウスとの比較のため、Keap1FA/FAマウスの食道を詳細に
解析した。対照マウスとして、Cre タンパク質の非存在下で Keap1 が正常に発現す
るKeap1FB/FBマウスを用いた。HE 染色像を確認すると、角化層は Keap1FA/FAマウ
スで有意に厚くなった(図8A、8B)。一方、細胞層の厚さは有意差を認めなかった
(図8A、8C)。分化マーカーの発現は、Keap1FA/FAマウスでKeratin 5は有意に抑
制される一方、Loricrinは有意差を認めず、Keratin 6は発現が亢進した(図8D)。
これらの結果から、Keap1 低発現マウスは食道上皮における基底層からの分化を促
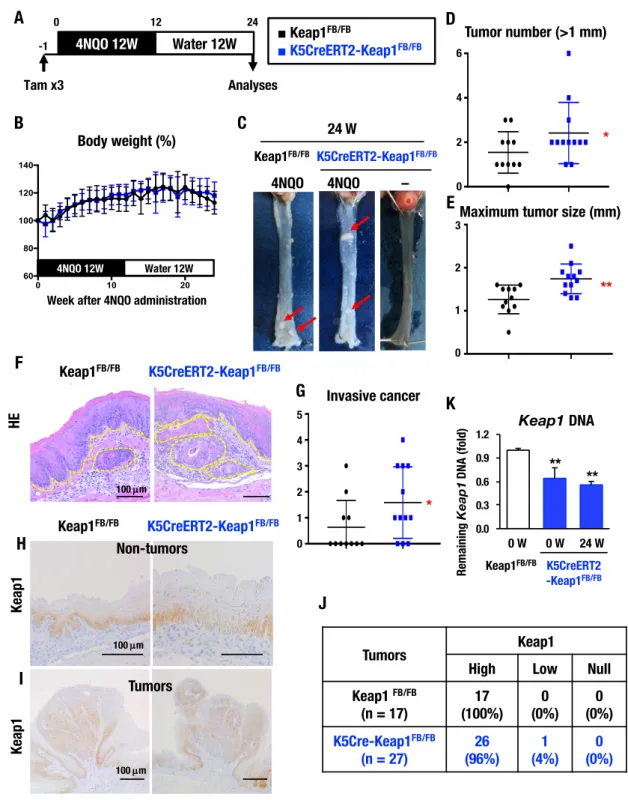
5-7. 4NQO 発がん pre-protocol における Keap1 の欠失はがん形成を促す Keap1 の欠失、つまりは Nrf2 の活性化が食道の発がんにどのように貢献するか を検証した。口腔がんや食道がんのマウスモデルに利用される4NQO を用いた発が ん実験で、対照マウスをKeap1FB/FBマウスとし、K5CreERT2-Keap1FB/FBマウスを 用いた。pre-protocol として、まず成獣マウスにタモキシフェンを投与することで Keap1 を欠失させ、1 週後に 4NQO の投与を開始し 12 週間投与した。その後、通 常水に変えてさらに12 週間観察し、解析した(図 9A)。発がん実験の過程では、体 重の変化に両群で差を認めなかった(図9B)。タモキシフェン投与 24 週後におい て、4NQO を投与した対照マウスおよび K5CreERT2-Keap1FB/FBマウスの食道に、 肉眼的に腫瘤が形成されていることを確認した(図9C)。一方、食道における誘導 的なKeap1 欠失は異形成を示すが、4NQO の投与を行わなければ、タモキシフェン 投与24 週後の K5CreERT2-Keap1FB/FBマウスの食道に腫瘤の形成は確認できなか った(図9C)。形成された腫瘤を詳細に解析したところ、長径 1 mm 以上の腫瘤の 数はK5CreERT2-Keap1FB/FBマウス群で有意に増加し(図9D)、腫瘤の最大径も同 様にK5CreERT2-Keap1FB/FBマウス群で有意に大きくなった(図9E)。組織学的な 解析において、粘膜下層以深へ浸潤したがん部の組織像を図9F に示す。組織学的に 浸潤を認めるがん部の個数を定量すると、肉眼初見と同様に
K5CreERT2-Keap1FB/FBマウス群で増加した(図9G)。 Nrf2 は酸化ストレスや親電子性物質に対する細胞保護的な働きをする重要な因子 であり、化学発がん物質の遺伝子変異に対しても抵抗性を示すと考えられている 12)。それゆえ、Keap1 の欠失により Nrf2 の活性化を誘導した群でなぜ腫瘍の形成 が促されたのか、その理由を探るべく、腫瘍に対するKeap1 の発現を免疫組織化学 染色で確かめた。対照群では基底層を中心に均一にKeap1 の発現が見られたが、 K5CreERT2-Keap1FB/FBマウス群ではまだらに発現した(図9H)。ただし、完全に 染色が陰性となる上皮領域は両群ともに認めなかった。腫瘤におけるKeap1 の発現 を確認すると、両群ともにKeap1 強陽性の腫瘤であり(図 9I)、対照群は 100%、 K5CreERT2-Keap1FB/FBマウス群では96%の腫瘍が強陽性であった(図 9J)。これ らの結果は、腫瘤を形成した細胞がKeap1 の欠失が誘導された異型細胞ではなく、 Keap1 の欠失を免れた細胞であることを示している。食道上皮におけるKeap1の欠 失の組換え効率を、発がん実験を完遂した時点で確認すると、約40%のアリルで Keap1が欠失していた(図9K)。 以上の結果から、4NQO 発がん実験前に Keap1 を欠失させることで食道がんの形 成が有意に促されたが、それら腫瘤の由来細胞はKeap1 を欠失させた異型細胞では なく、周囲にあるKeap1 を保持した細胞群であることが示された。Keap1 欠失によ
って起こったDNA 障害の誘導が影響し、がん形成を促した可能性が考えられた。
5-8. 4NQO 発がん post-protocol における Keap1 欠失はがん形成に影響を与えない
がんにおけるNrf2 の活性化は細胞保護作用や増殖能を獲得し、がんの悪性化に寄
与する17, 18)。続いて、4NQO を投与したのちに Keap1 の欠失、つまりは Nrf2 の活
性化を誘導するpost-protocol で、がん化した細胞における Nrf2 の貢献の解析を試
みた。マウスはpre-protocol と同様に Keap1FB/FBマウスとK5CreERT2-Keap1FB/FB
マウスを用いた。4NQO を 12 週間飲水投与した後、タモキシフェンを投与するこ とでKeap1 を欠失させ、通常水に変えてさらに 12 週間観察し、解析した(図 10A)。この実験が成立するためには、腫瘍において Cre タンパク質による欠失が誘 導されることが必要である。免疫組織化学染色により、4NQO で形成した食道腫瘤 はKeratin 5 が確実に発現していた(図 10B)。体重変化を見ると、タモキシフェン 投与後からK5CreERT2-Keap1FB/FBマウスの体重は劇的に減少したが、タモキシフ ェン投与3 週後にはタモキシフェン投与時の体重まで回復した(図 10C)。ただし、 対照マウスの水準までの回復は認めなかった。また、K5CreERT2-Keap1FB/FBマウ ス群において、7 日間で体重が 80%以下に減少した 4 匹は 21- 22 週目に人道的エン ドポイントを迎えた。K5CreERT2-Keap1FB/FBマウスにおいて体重減少をきたした
タモキシフェン投与後1 週間の時点で食道を観察すると、K5CreERT2-Keap1FB/FB マウスの食道は細胞層の肥厚に加えて過角化の所見を認めた(図 10D)。post-protocol における一過性の体重減少は、Nrf2 活性化による食道の閉塞による食餌困 難が原因であると予想され、時間経過とともに表現型が緩和して体重が回復したと 考えられた。タモキシフェン投与後1 週間の時点では肉眼的に明らかな腫瘤は認め なかったが、Keap1 の組換え効率を確認すると、食道上皮のおよそ 50%のアリルで 欠失していた(図10E)。肉眼的に腫瘍を解析したところ、腫瘤の形成に差はなく (図10F)、1 mm 径以上の腫瘤の数(図 10G)、あるいは最大径に差を認めなかっ た(図10H)。組織学的な解析においても、K5CreERT2-Keap1FB/FBマウスと対照 群に、食道粘膜の全面的な異形成を生じており差は認めず、腫瘍の形態にも両者に 差を認めなかった(図10I)。がんの浸潤部の数にも差を認めなかった(図 10J)。 以上より、発がん誘導後のKeap1 欠失による Nrf2 の活性化は、がんの形成に影響 を与えないという結果を得た。DNA 障害により将来的にがん化すると予想される細 胞にもKeap1 の欠失は確かに誘導されていると考えられるが、Keap1 を欠失した細 胞におけるNrf2 の活性が早期に減弱した可能性、Keap1 欠失細胞が消えてしまっ た可能性などが考えられた。
5-9. 食道上皮における誘導的な Nrf2 の欠失は特異的な表現型を示さない 食道におけるNrf2 の活性化による様々な影響が示されたので、続いては誘導的な Nrf2 の欠失による影響を解析した。対照群は Nrf2F/Fマウスとし、タモキシフェン 投与により扁平上皮のNrf2 が欠失する K5CreERT2-Nrf2F/Fマウスを用いた。タモ キシフェン投与後、1 週後と 12 週後の表現型を解析した(図 11A)。欠失誘導後に 残存するNrf2のアリルのDNA 組換え効率は1週後と 12 週後で同程度に約 50%で 維持されていた(図11B)。また、DNA の欠失と一致して、Nrf2とその標的因子で あるNqo1の発現抑制が認められ、1週後と12 週後で同程度に維持されることが分 かった(図11C)。Nrf2 の欠失は部分的な細胞に起こっていたが、それらの細胞と 遺伝子発現は時間経過に関わらず維持されていることが確認された。組織レベルで は、K5CreERT2-Nrf2F/Fマウスで特記すべき変化は認められなかった(図11D)。 角層と細胞層の厚さをそれぞれ評価したが、対照群と有意差を認めなかった(図 11G、11H)。Nrf2 の免疫組織化学染色では、両群ともに検出されず(図 11E)、 Nrf2 は速やかに分解され、定常状態では活性化していない結果であると考えられた 32)。DNA 二重鎖切断マーカーであるgH2A.X も確認したところ、基底細胞にわずか に陽性細胞を認めるのみで(図11F)、K5CreERT2-Nrf2F/Fマウスと対照マウスに 差を認めなかった(図11I)。以上から、誘導的な Nrf2 の欠失は特異的な表現型を
示さなかったが、定常状態ではそもそもNrf2 の活性化は認めないために表現型は現 れないと考えられた。 5-10. 発がん pre-protocol における Nrf2 の欠失はがん形成に影響を与えない 全身性Nrf2 欠失マウスは 4NQO による発がんに脆弱であることが示されている 22)。続いては、食道における誘導的なNrf2 欠失が発がんにどのように影響を与える かを解析した。まず、4NQO の投与に先んじて Nrf2 の欠失を誘導する pre-protocol を行った(図12A)。対照マウスは Nrf2F/Fマウスとし、K5CreERT2-Nrf2F/Fマウス を用いた。24 週間で体重の変化に差を認めなかった(図 12B)。肉眼的に腫瘍を解 析したところ、腫瘤の形成に差はなく(図12C)、1 mm 径以上の腫瘤の数(図 12D)あるいは最大径(図 12E)に差を認めなかった。組織学的な解析においても 腫瘍の形態(図12F)や、がんの浸潤部の数(図 12G)にも差を認めなかった。以 上から、発がんpre-protocol における Nrf2 の欠失はがん形成に影響を与えないとい う結果を得た。 5-11. Nrf2 欠失細胞は 4NQO 投与環境下で消失する 続いて、Nrf2 欠失 pre-protocol で得られた検体において(図 13A)、Nrf2 の発現
を免疫組織化学染色で確認した。定常状態ではNrf2 の発現を認めなかったが(図 13E)、24 週後の食道粘膜においては、腫瘍化の有無にかかわらず Nrf2 の核濃染つ まりはNrf2 が活性化した細胞が散見された(図 13B)。そして、Nrf2 を欠失したは ずのK5CreERT2-Nrf2F/Fマウス群においても、同様にNrf2 活性化細胞を認めた (図13B)。Nrf2 の標的因子である Nqo1 の発現を免疫組織化学染色で確認する と、両群ともにNqo1 が発現している腫瘤が多く(図 13C)、Nqo1 陰性の腫瘤は対 照群で0%、K5CreERT2-Nrf2F/Fマウス群で12%であった(図 13D)。これらの結 果から、4NQO は Nrf2 の欠失を免れた細胞において Nrf2 を活性化させ、これらの 細胞が選択的に腫瘍化したのではないかという可能性が考えられた。 上記の可能性を検証すべく、タモキシフェンを投与後、4NQO を 12 週投与した 直後の組織を解析した(図13E)。まず、食道の粘膜を観察すると、長径 1 mm に満 たない結節を認めたが、本研究で腫瘍と定義する病変は認めなかった(図13F)。続 いて、食道におけるNrf2 欠失細胞の割合が、4NQO 投与下で維持されているか否 かを確認するため、食道上皮におけるDNA の組換え効率を調べた。タモキシフェ ン投与1 週後ではNrf2の欠失はおよそ50%であったのに対し(図 13G:0 週)、 4NQO を 12 週投与した食道においてはNrf2のアリルの割合は対照群と同程度とな り、Nrf2 が欠失したアリルは消失した(図 13G:12 週)。この結果から、Nrf2 の欠
失が誘導された細胞は4NQO のストレス環境下では消失してしまい、代わりに Nrf2 の欠失を免れた細胞が食道上皮を占拠したということが示された。 以上をまとめると、全身性Nrf2 欠失マウスは 4NQO による発がんに脆弱である ことが報告されているが、本研究の発がん実験pre-protocol における Nrf2 の欠失は がん形成に影響を与えなかった。その理由として、4NQO 投与後に Nrf2 欠失細胞 は食道上皮から消失するためと考えられた。 5-12. 発がん post-protocol における Nrf2 の欠失はがん形成に影響を与えない Nrf2 の活性を獲得したがん(Nrf2 addicted cancer)は予後が悪いことが示され ており、そのようながんに対してはNrf2 を抑制することが治療として期待されてい る20, 34)。続いては、食道がんにおけるNrf2 の欠失ががん形成にどのような影響を 与えるかを検証すべく、4NQO を投与した後に Nrf2 の欠失を誘導する実験を行な った。つまり、対照マウスはNrf2F/Fマウスとし、K5CreERT2-Nrf2F/Fマウスを用 い、post-protocol を行なった(図 14A)。発がん実験の経過中、体重変化は両群に差 を認めなかった(図14B)。食道に腫瘍の形成は確認でき(図 14C)、肉眼的な腫瘍 の解析においては、最大径1 mm 以上の腫瘤の数(図 14D)、腫瘤の最大径(図 14E)共に差を認めなかった。また、組織学的な解析においても、がんの浸潤する
数に差を認めなかった(図14F, 14G)。 以上より、発がん誘導後のNf2 欠失は、がんの形成に影響を与えないという結果 を得た。しかしながら、post-protocol で Keap1 を欠失した実験系と同様に、そもそ もNrf2 の欠失が誘導できなかった可能性、Nrf2 欠失細胞が消えてしまった可能性 など、様々な理由が考えられる。今回の結果がどのような現象を観察しているか結 論づけるには、さらなる検証が必要である。
6. 考察 がんと生体の関係性においてNrf2 の活性は多面的に貢献している。Nrf2 は、正常 細胞において細胞保護的な作用により発がんを抑制する一方、一旦がん化してしまっ た細胞の進展や悪性化にも寄与する35, 36)。また、がん微小環境においては、骨髄由来 免疫抑制細胞(MDSC)や制御性 T 細胞における Nrf2 の活性が、がんの抑制効果を もたらす。さらに、がん腫によっても貢献は異なり、肝細胞がんや膵がんの発がんお いてNrf2 の活性を要することが分かっている37, 38)。このようにNrf2 は様々な側面 をはらむ中、食道上皮におけるNrf2 の活性は、発がんする前と後で食道がんへの貢 献が逆転するのではないかという仮説を検証すべく、食道上皮特異的な遺伝子欠失を 発がんの前後で行う実験を行なった。しかしながら、上皮の一部の細胞に欠失が誘導 されたことで、予想とは反する結果が得られた点で実に興味深い。本研究におけるモ デルを図15 に示す。成獣マウスの食道における Keap1 の欠失は増殖能の亢進と上皮 の分化障害をきたし、組織全体としては異形成を示した。Keap1 の欠失した細胞は Nrf2 の活性化を認め、それらが核の腫大を伴う異型細胞となった。ただし、その Keap1 欠失細胞の出現は、周囲にある Keap1 陽性細胞の DNA 障害を誘導したと考
えられた。4NQO によって Keap1 陽性細胞の発がんが促進されたが、原因の一つと
えられた。一方、成獣マウスの食道におけるNrf2 の欠失は定常状態では特記すべき 表現型を認めなかった。予想と反して、Nrf2 欠失細胞は 4NQO によりがん化しなか った。これは、Nrf2 欠失細胞は 4NQO のようなストレス存在下で残存することがで きず、Nrf2 陽性細胞が食道上皮を占拠したと考えられた。全身性 Nrf2 欠失マウスは 4NQO に対して脆弱で発がんしやすいことが示されており22)、今回のNrf2 が消失す る現象は、4NQO に対する上皮全体の生体防御機構であると言える。 全身性 Keap1 欠失マウスは生後間もなく食道の過角化により致死となることが報 告されており16)、成獣マウスにおけるKeap1 欠失食道の解析と実験応用は不可能で あったが、本研究で成獣マウスの誘導的な Keap1 の欠失は、過角化とは全く異なる 異形成を示すことを見出した。全身性 Keap1 欠失マウスの表現型である過角化は、 Nrf2 を同時に欠失することにより消失するため16)、Nrf2 の活性化に依存すると考え られる。また、Keratin5-Cre マウスを利用した上皮特異的な Keap1 欠失は、同様の 過角化を示して致死に至るため、食道の過角化はまさに食道上皮におけるNrf2 の活
性化が引き起こす表現型である33, 39)。本研究でも、Keap1 低発現の Keap1FA/FAマウ
スは過角化を示すことを確認している。これらの知見から、胎仔期からの永続的な Nrf2 の活性化が食道の過角化を形成し、成獣における誘導的な Keap1 欠失マウスで はこの致死的な過角化を回避できることが示された。
Nrf2 の活性化はペントースリン酸回路への誘導や p38 プロテインキナーゼ経路の 活性化など、代謝プロファイルを変化させることが知られており 20, 40, 41)、これらの 代謝変化は上皮細胞の増殖能を促す。本研究でもKeap1 欠失により mRNA レベルで Keratin 6やKi67の発現が亢進し、基底細胞の数が増えたので、Nrf2 の活性化は確 かに増殖能を促すことが確認された。一方、Nrf2 は基底細胞から有棘細胞層への分 化に必要であり25, 27)、Nrf2 の基底層における活性化は扁平上皮細胞の分化を促すこ とが示唆される。実際に、Nrf2 が持続的に活性化している Keap1 低発現マウスは、 Keratin 5を発現する基底細胞からの分化を促した。それとは対照的に、
K5CreERT2-Keap1FB/FB マウスにおける Keap1 を欠失誘導した食道では、mRNA レベルで
Keratin 5の発現が亢進し、Loricrinの発現が抑制されており、上皮の分化障害が示 唆された。ともにNrf2 の活性は亢進しているが、分化においては全く異なる変化を 示した。この違いはNrf2 の活性化・蓄積の程度により生み出されたと考えられ、高 発現のNrf2 が、通常は制御していない分化に関与する遺伝子の転写活性化を誘導し た可能性が考えられる。この分化様式の差異が、細胞異型や細胞層の肥厚の有無の違 いとして現れたと考えられる。Keap1 欠失誘導により、代謝プロファイルの変化によ る増殖能の亢進と、Nrf2 の転写制御の変化がもたらした分化障害によって異型細胞 が出現したと考えられた。
DNA 二重鎖切断は、電離放射線の被曝、生理的にプログラムされた DNA の切断、 酸化ストレスなどで誘導される42)。Nrf2 は抗酸化酵素を誘導して活性酸素種を消去 する作用があるため、Nrf2 の活性化は DNA 障害を抑制することが知られている43, 44)。しかしながら、本研究で Keap1 欠失細胞の周囲に存在する Keap1 陽性細胞に DNA 障害が蓄積した理由は明らかでない。Nrf2 は抗酸化剤であるグルタチオンの合 成に関与するGcl酵素群の発現を誘導するが8)、シスチン・グルタミン酸トランスポ ーターである Slc7a11 の発現誘導にも関与してグルタチオン合成に必要なシスチン の細胞内への取り込みも促している 45)。Keap1 欠失細胞によりシスチンが過度に取 り込まれた結果、周囲の Keap1 陽性細胞がシスチンを取り込むことができずに、酸 化ストレス応答ができなくなった可能性が考えられた。あるいは、Keap1 欠失細胞の 貫入により管腔側へ押し出されたKeap1 陽性細胞は 4NQO により曝露されやすくな り、がん化したとも考えられた。いずれにせよ、gH2A.X 陽性細胞の出現は、Keap1 欠失細胞が出現したことによるストレスが原因と考えられ、gH2A.X 陽性細胞自体の 内因的な原因であるp53 が関与する DNA 損傷修復異常などは関与が薄いと考えられ た。 今回、Nrf2 の食道がんへの貢献を明らかにするため、4NQO を用いた発がん実験 を行なった。4NQO の発がん機序は二つ報告されており、一つは活性酸素種の発生に
よる酸化的DNA 障害46)、もう一つは代謝物の4HAQO による直接的な DNA 付加で
ある47)。Nrf2 の活性は、抗酸化酵素の発現を亢進することで活性酸素種を消去する
作用を持つ。一方で、Nrf2 の代表的な標的因子である Nqo1 は、4NQO から 4HAQO
への代謝を促す。つまり、Nrf2 の活性は抗酸化酵素の発現上昇により 4NQO による 酸化障害を抑制する可能性と、4HAQO の産生を促進して発がんを誘導する可能性の 双方が考えられた。過去の報告では、Nrf2 欠失マウスでは 4NQO による発がんを促 進し、Nrf2 の活性化する Keap1 低発現マウスではその発がんを抑制すると結論づけ られている 22)。本研究においては、Keap1 欠失細胞は 4NQO により腫瘍化せず、 Keap1 の欠失を免れた細胞の発がんを促進したため、Nrf2 の活性化により増加した 4HAQO が遺伝子変異を誘導した可能性がある。また、Nrf2 欠失細胞は消失すること により腫瘍化しなかったのは、酸化ストレスに加えてNrf2 陽性細胞によって代謝さ れた4HAQO の蓄積が Nrf2 欠失細胞に影響することで、発がんではなく細胞死に至 るストレスが加わった可能性が考えられた。Nrf2 欠失細胞は 4NQO により選択的に 上皮組織から排除され、Keap1 欠失細胞は周囲の正常上皮細胞に DNA 障害を引き起 こした。これらの結果は、ヒト食道上皮には様々な変異を持った細胞クローンが存在 し6)、食道上皮ががん化する過程において細胞クローン同士が相互に作用することを 意味している。
Nrf2 が同組織内の相互作用や細胞競合にどのように関与するかは、はっきりとし た共通認識は得られていない。Nrf2 欠失細胞は 4NQO によるストレス負荷のない環 境下においては食道上皮内に定着することができたが、4NQO 投与により消失した。 この結果は、生存能力が低下した細胞の排除が働いたと考えられる。Nrf2 と細胞競 合に関しては、ショウジョウバエの翅を用いた研究で、Nrf2 が高発現する細胞は隣 り合う細胞に排除されるとの報告がある48)。一方で、マウスを用いた研究で、Nrf2 の 活性が高く、抗酸化酵素を高発現している細胞は酸化ストレスに強く、食道上皮内に 広がることが示されている 25)。扁平上皮における細胞競合に勝つ細胞クローンの特 徴は、増殖能力が強く、分化が抑制され、生存能力の高い細胞である26, 49)。Nrf2 の 基底細胞における活性化は、細胞の生存能力をあげて分化を促進するので、細胞競合 の観点からは有利にも不利にも働くと考えられる。Nrf2 が適切なバランスで発現す る こ と が 、 細 胞 競 合 に お い て は 重 要 で あ る こ と が う か が え る 。 K5CreERT2-Keap1FB/FBマウスの Keap1 欠失細胞は、適切に分化することができなかったために 異型細胞となり、組織内にとどまった。反対に、K5CreERT2-Nrf2F/FマウスのNrf2 欠失細胞は、その生存能力や組織内の細胞選択により 4NQO のストレス化で排除さ れた。これらの結果は、様々なNrf2 活性程度にある細胞を包含している食道扁平上 皮において、Nrf2 欠失細胞が選択的に排除され、Nrf2 の活性化している細胞は異型
細胞となることを示している。扁平上皮の集団において、Nrf2 の活性化具合が隣り 合う細胞との相互作用や細胞の適応能と運命を決定している。 誘導的 Keap1 欠失に伴う Nrf2 活性化の経時的な減弱は、大変興味深い結果であ る。先天的な遺伝子欠失を認めるKeap1 低発現マウスや条件付 Keap1 欠失マウスで はNrf2 の活性化を認めるものの、経時的な Nrf2 の活性変化を観察した報告はない。 Keap1 欠失マウスにおいて、まず出現する生体において有害な表現型が食道の過角 化・組織異型であり、食道上皮においては異常なNrf2 活性化に対して Keap1 とは独 立した制御機構が働く可能性が示唆される。本研究では、食道上皮におけるKeap1 の 欠失誘導によりNrf2 の活性化を確認できたが、Keap1 欠失細胞が存在するにも関わ らず、時間経過とともにNrf2 活性化が低下した。Nrf2 の転写に差は認めなかったた め、タンパク質レベルでの Keap1 以外の制御が働いた可能性がある。考えられる仮 説としては、Keap1 とは異なる Nrf2 タンパク質分解系であるbTrCP の活性化である 50)。食道扁平上皮がんにおいて、Keap1-Nrf2 経路とbTrCP 系の上流にあたる Akt 経 路の遺伝子変異が共存しやすいということが報告されており 51)、食道上皮における 異常なNrf2 活性化維持には Akt -bTrCP 経路の異常が必要条件となる可能性が示唆 される。さらに、ヒト食道扁平上皮がんの特徴的な遺伝子プロファイルの一つとして、 幹細胞性維持に寄与するSox2 の異常増幅が挙げられるが52)、このSox2 の下流には
Akt 経路が存在している53)。Sox2 増幅に始まる Sox2-Akt-bTrCP 経路の異常とそれ
による Nrf2 分解能の低下が、食道扁平上皮がんにおける Nrf2 異常活性化を成立さ
せる可能性がある。本研究のKeap1 post-protocol では、Keap1 欠失によるがん形成
に影響を与えなかったが、やはりこの実験系においてもNrf2 の活性化は一過性で、
この発がん実験ではbTrCP 系の異常を来さなかったために Nrf2 の活性化が抑制され
た可能性がある。Nrf2 の変異は正常の食道上皮には存在せず、食道扁平上皮がんに
特異的で、この乖離は他の遺伝子と比較しても特筆されることから6)、食道扁平上皮
7. 結論 食道扁平上皮における成獣期の Keap1 の欠失誘導は、食道異形成を引き起こした。 この成獣マウスにおける誘導的な Keap1 欠失が 4NQO により腫瘍化した細胞は Keap1 欠失を免れた Keap1 陽性細胞であった。一方、Nrf2 の欠失誘導では、Nrf2 欠 失細胞は4NQO により上皮から排除され、発がんしなかった。細胞の Nrf2 活性程度 は、上皮組織内での適応能と相互作用に密接に関わっており、発がんする細胞の選択 性にも影響する。
8. 文献
1. Bray F, Ferlay J, Soerjomataram I, et al: Global Cancer Statistics 2018: Globocan Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin 2018;68:394-424.
2. Mayanagi S, Irino T, Kawakubo H, et al: Neoadjuvant Treatment Strategy for Locally Advanced Thoracic Esophageal Cancer. Ann Gastroenterol Surg 2019;3:269-275.
3. Arnold M, Soerjomataram I, Ferlay, J, et al: Global incidence of esophageal cancer by histological subtype in 2012. Gut 2015;64:381-387.
4. Mena S, Ortega A, Estrela JM: Oxidative Stress in Environmental-Induced Carcinogenesis. Mutat Res 2009;674:36-44.
5. Bavarva JH, Tae H, Mclver L, et al: Nicotine and oxidative stress induced exomic variations are concordant and overrepresented in cancer-associated genes. Oncotarget 2014;5:4788-4798.
6. Yokoyama A, Kakiuchi N, Yoshizato T, et al: Age-Related Remodelling of Oesophageal Epithelia by Mutated Cancer Drivers. Nature 2019;565:312-317. 7. Yamamoto M, Kensler TW, Motohashi H: The Keap1-Nrf2 System: A
Thiol-Based Sensor-Effector Apparatus for Maintaining Redox Homeostasis. Physiol Rev 2018;98:1169-1203.
8. Itoh K, Chiba T, Takahashi S, et al: An Nrf2/Small Maf Heterodimer Mediates the Induction of Phase Ii Detoxifying Enzyme Genes through Antioxidant Response Elements. Biochem Biophys Res Commun 1997;236:313-322.
9. Kobayashi A, Kang MI, Okawa H, et al: Oxidative Stress Sensor Keap1 Functions as an Adaptor for Cul3-Based E3 Ligase to Regulate Proteasomal Degradation of Nrf2. Mol Cell Biol 2004;24:7130-7139.
10. Itoh K, Wakabayashi N, Katoh Y, et al: Keap1 Represses Nuclear Activation of Antioxidant Responsive Elements by Nrf2 through Binding to the Amino-Terminal Neh2 Domain. Genes Dev 1999;13:76-86.
11. Dinkova-Kostova AT, Holtzclaw WD, Cole RN, et al: Direct Evidence That Sulfhydryl Groups of Keap1 Are the Sensors Regulating Induction of Phase 2 Enzymes That Protect against Carcinogens and Oxidants. Proc Natl Acad Sci U S A 2002;99:11908-11913.
12. Ramos-Gomez M, Kwak MK, Dolan PM, et al: Sensitivity to Carcinogenesis Is Increased and Chemoprotective Efficacy of Enzyme Inducers Is Lost in Nrf2
Transcription Factor-Deficient Mice. Proc Natl Acad Sci U S A 2001;98:3410-3415.
13. Iida K, Itoh K, Kumagai Y, et al: Nrf2 Is Essential for the Chemopreventive Efficacy of Oltipraz against Urinary Bladder Carcinogenesis. Cancer Res 2004;64:6424-6431.
14. Khor TO, Huang MT, Prawan A, et al: Increased Susceptibility of Nrf2 Knockout Mice to Colitis-Associated Colorectal Cancer. Cancer Prev Res (Phila) 2008;1:187-191.
15. Satoh H, Moriguchi T, Takai J, et al: Nrf2 Prevents Initiation but Accelerates Progression through the Kras Signaling Pathway During Lung Carcinogenesis. Cancer Res 2013;73:4158-4168.
16. Wakabayashi N, Itoh K, Wakabayashi J, et al: Keap1-Null Mutation Leads to Postnatal Lethality Due to Constitutive Nrf2 Activation. Nat Genet 2003;35:238-245.
17. Ohta T, Iijima K, Miyamoto M, et al: Loss of Keap1 Function Activates Nrf2 and Provides Advantages for Lung Cancer Cell Growth. Cancer Res 2008;68:1303-1309.
18. Shibata T, Ohta T, Tong KI, et al: Cancer Related Mutations in Nrf2 Impair Its Recognition by Keap1-Cul3 E3 Ligase and Promote Malignancy. Proc Natl Acad Sci U S A 2008;105:13568-13573.
19. Kim YR, Oh JE, Kim MS, et al: Oncogenic Nrf2 Mutations in Squamous Cell Carcinomas of Oesophagus and Skin. J Pathol 2010;220:446-451.
20. Kitano Y, Baba Y, Nakagawa S, et al: Nrf2 Promotes Oesophageal Cancer Cell Proliferation Via Metabolic Reprogramming and Detoxification of Reactive Oxygen Species. J Pathol 2018;244:346-357.
21. Taguchi K, Yamamoto M: The Keap1-Nrf2 System in Cancer. Front Oncol 2017;7:85.
22. Ohkoshi A, Suzuki T, Ono M, et al: Roles of Keap1-Nrf2 System in Upper Aerodigestive Tract Carcinogenesis. Cancer Prev Res (Phila) 2013;6:149-159. 23. Hiramoto K, Satoh H, Suzuki T, et al: Myeloid Lineage-Specific Deletion of
Antioxidant System Enhances Tumor Metastasis. Cancer Prev Res (Phila) 2014;7:835-844.
24. Maj T, Wang W, Crespo J, et al: Oxidative Stress Controls Regulatory T Cell Apoptosis and Suppressor Activity and Pd-L1-Blockade Resistance in Tumor.
Nat Immunol 2017;18:1332-1341.
25. Fernandez-Antoran D, Piedrafita G, Murai K, et al: Outcompeting P53-Mutant Cells in the Normal Esophagus by Redox Manipulation. Cell Stem Cell 2019;25:329-341.
26. Doupe DP, Alcolea MP, Roshan A, et al:A single progenitor population switches behavior to maintain and repair esophageal epithelium. Science 2012;337:1091-1093.
27. Jiang M, Ku WY, Zhou Z, et al: Bmp-Driven Nrf2 Activation in Esophageal Basal Cell Differentiation and Eosinophilic Esophagitis. J Clin Invest 2015;125:1557-1568.
28. Xue P, Hou Y, Chen Y, et al: Adipose Deficiency of Nrf2 in Ob/Ob Mice Results in Severe Metabolic Syndrome. Diabetes 2013;62:845-854.
29. Blake DJ, Singh A, Kombairaju P, et al: Deletion of Keap1 in the Lung Attenuates Acute Cigarette Smoke-Induced Oxidative Stress and Inflammation. Am J Respir Cell Mol Biol 2010;42:524-536.
30. Okawa H, Motohashi H, Kobayashi A, et al: Hepatocyte-Specific Deletion of the Keap1 Gene Activates Nrf2 and Confers Potent Resistance against Acute
Drug Toxicity. Biochem Biophys Res Commun 2006;339:79-88.
31. Watai Y, Kobayashi A, Nagase H, et al: Subcellular Localization and Cytoplasmic Complex Status of Endogenous Keap1. Genes Cells 2007;12:1163-1178.
32. Katoh Y, Iida K, Kang MI, et al: Evolutionary Conserved N-Terminal Domain of Nrf2 Is Essential for the Keap1-Mediated Degradation of the Protein by Proteasome. Arch Biochem Biophys 2005;433:342-350.
33. Taguchi K, Maher JM, Suzuki T, et al: Genetic Analysis of Cytoprotective Functions Supported by Graded Expression of Keap1. Mol Cell Biol 2010;30:3016-3026.
34. Yamamoto S, Inoue J, Kawano T, et al: The Impact of Mirna-Based Molecular Diagnostics and Treatment of Nrf2-Stabilized Tumors. Mol Cancer Res 2014;12:58-68.
35. Hayes JD, McMahon M: The Double-Edged Sword of Nrf2: Subversion of Redox Homeostasis During the Evolution of Cancer. Mol Cell 2006;21:732-734. 36. Sporn MB, Liby KT: Nrf2 and Cancer: The Good, the Bad and the Importance
37. Hamada S, Taguchi K, Masamune A, et al: Nrf2 Promotes Mutant K-Ras/P53-Driven Pancreatic Carcinogenesis. Carcinogenesis 2017;38:661-670.
38. Orru C, Szydlowska M, Taguchi K, et al: Genetic Inactivation of Nrf2 Prevents Clonal Expansion of Initiated Cells in a Nutritional Model of Rat Hepatocarcinogenesis. J Hepatol 2018;69:635-643.
39. Suzuki T, Seki S, Hiramoto K, et al: Hyperactivation of Nrf2 in Early Tubular Development Induces Nephrogenic Diabetes Insipidus. Nat Commun 2017;8:14577.
40. Mitsuishi Y, Taguchi K, Kawatani Y, et al: Nrf2 Redirects Glucose and Glutamine into Anabolic Pathways in Metabolic Reprogramming. Cancer Cell 2012;22:66-79.
41. Fu J, Xiong Z, Huang C, et al: Hyperactivity of the Transcription Factor Nrf2 Causes Metabolic Reprogramming in Mouse Esophagus. J Biol Chem 2019;294:327-340.
42. Scully R, Xie A: Double Strand Break Repair Functions of Histone H2ax. Mutat Res 2013;750:5-14.
Response Mitigates Cytoplasmic Radiation-Induced DNA Double-Strand Breaks. Cancer Sci 2019;110:686-696.
44. Lan A, Li W, Liu Y, et al: Chemoprevention of Oxidative Stress-Associated Oral Carcinogenesis by Sulforaphane Depends on Nrf2 and the Isothiocyanate Moiety. Oncotarget 2016;7:53502-53514.
45. Sasaki H, Sato H, Kuriyama-Matsumura K, et al: Electrophile response element-mediated induction of the cystine/glutamate exchange transporter gene expression. J Bio Chem 2002;277:44765-44771.
46. Nunoshiba T, Demple B: Potent Intracellular Oxidative Stress Exerted by the Carcinogen 4-Nitroquinoline-N-Oxide. Cancer Res 1993;53:3250-3252.
47. Tanuma J, Hirano M, Hirayama Y, et al: Genetic Predisposition to 4nqo-Induced Tongue Carcinogenesis in the Rat. Med Princ Pract 2005;14:297-305. 48. Kucinski I, Dinan M, Kolahgar G, et al: Chronic Activation of Jnk Jak/Stat and Oxidative Stress Signalling Causes the Loser Cell Status. Nat Commun 2017;8:136.
49. Frede J, Greulich P, Nagy T, et al: A Single Dividing Cell Population with Imbalanced Fate Drives Oesophageal Tumour Growth. Nat Cell Biol
2016;18:967-978.
50. Cuadrado A: Structural and functional characterization of Nrf2 degradation by glycogen synthase kinase 3/beta-TrCP. Free Radic Biol Med 2015;88:147-157. 51. Sanchez-Vega F, Mina M, Armenia J, et al: Oncogenic Signaling Pathways in
The Cancer Genome Atlas. Cell 2018;173:321-337.
52. Cancer Genome Atlas Research N, et al: Integrated genomic characterization of oesophageal carcinoma. Nature 2017;541:169-175.
53. Gen Y, Yasui K, Nishikawa T, et al: SOX2 promotes tumor growth of esophageal squamous cell carcinoma through the AKT/mammalian target of rapamycin complex1 signaling pathway. Cancer Sci 2013;104:810-816.
9. 図 図1 図1 Keap1-Nrf2 システムの概要 Nrf2 は定常状態では Keap1 という抑制因子からユビキチン化を受け、プロテアソー ムにより分解されている。活性酸素や親電子性物質などのストレスを感知すると、 Keap1 のシステイン残基が修飾を受け、Nrf2 が Keap1 の抑制制御を免れ、核内移行 することで転写制御をする。抗酸化ストレス・解毒作用を持つ酵素群などを発現