Pdと光を利用したラジカル多成分連結反応に関する
研究
著者
房野 暁
内容記述
学位授与大学: Osaka Prefecture University(大阪
府立大学), 学位の種類: 博士(理学), 学位記番号:
論理第99号, 学位授与年月日: 2012-03-31, 指導教
員: 柳 日馨.
Pd と光を利用したラジカル多成分連結反応に関する研究
Studies on Radical Multi-Component Coupling Reactions
under Pd/Light Combined Conditions
房野 暁
Akira Fusano
大阪府立大学大学院 理学系研究科
Graduate School of Science
Osaka Prefecture University
目次
総論... 1 参考文献... 7 本論 第1 章 多成分連結反応によるカルボン酸エステル、ラクトン誘導体合成反応... 9 第1 節 緒言... 9 第2 節 アルキルヨウ化物、アルケン、一酸化炭素、アルコールを用いた 4 成 分連結反応によるカルボン酸エステル誘導体の合成... 13 第3 節 アルキルヨウ化物、アルケニルアルコール、一酸化炭素を用いた 3 成 分連結反応によるラクトン誘導体の合成... 20 第4 節 反応機構の考察... 24 第5 節 結論... 31 第6 節 実験の部... 32 第7 節 参考文献... 55 第2 章 3 成分連結反応によるアルキルアルキニルケトン合成反応 ... 58 第1 節 緒言... 58 第2 節 アルキルヨウ化物、一酸化炭素、芳香族アセチレンを用いた 3 成分連 結反応... 61 第3 節 アルキルヨウ化物、一酸化炭素、脂肪族アセチレンを用いた 3 成分連結反応... 65 第4 節 反応機構の考察... 68 第5 節 結論... 70 第6 節 実験の部... 71 第7 節 参考文献... 79 第3 章 3 成分連結反応によるカルバモイルアセテート誘導体合成反応 ... 81 第1 節 緒言... 81 第2 節 アルキルヨウ化物、一酸化炭素、アミンを用いた 3 成分連結反応. 84 第3 節 結論... 89 第4 節 実験の部... 90 第5 節 参考文献... 96 第4 章 その他の検討... 97 第1 節 アルキルヨウ化物、アルキン、一酸化炭素、アルコールを用いた 4 成 分連結反応による,-不飽和エステル誘導体の合成... 97 第2 節 アルキルヨウ化物、アルケン、一酸化炭素、有機金属試薬を用いた 4 成分連結反応による非対称ケトン誘導体の合成... 100 第 3 節 光照射条件における[Pd2(CNMe)6][PF6]2錯体を用いた多成分連結反 応... 104
第4 節 結論... 108 第5 節 実験の部... 109 第6 節 参考文献... 116 総括... 117 論文リスト... 119 謝辞... 120
略語一覧 Ac acetyl 9-BBN 9-borabicyclo[3.3.1]nonane bmim 1-butyl-3-methylimidazolium Bn benzyl Bu normal butytl
t-Bu tertiary butyl
cat. catalyst DCC N,N'-dicyclohexylcarbodiimide DMAP 4-dimethylaminopyridine DMF dimethylformamide DMSO dimethylsulfoxide dppf 1,1'-bisdiphenylphosphinoferrocene dppp 1,3-bis(diphenylphosphino)propane e.s.r. electron spin resonance
Et ethyl
EWG electron withdrawing group
GC gas chromatography
GCMS gas chromatography mass spectrometry HOMO highest occupied molecular orbital
HPLC high performance liquid chromatography HRMS high resolution mass spectrometry
Imd. imidazole
LAH lithium aluminium hydride
LDA lithium diisopropylamide
Me methyl
MS mass spectroscopy
NHC N-heterocyclic carbene
NMR nuclear magnetic resonance NOE nuclear Overhauser effect
Ph phenyl
rt room temperature
SOMO singly occupied molecular orbital
TBS tert-butyldimethylsilyl
TEMPO 2,2,6,6-tetramethylpiperidine 1-oxyl
tert tertiary
Tf trifluoromethanesulfonyl
TFP tetrafluoropropanol
THF tetrahydrofuran
TLC thin layer chromatography
TMS trimethylsilyl
総論
現代の有機合成化学において、多様な構造および官能基を有する化合物を効率的 かつ高収率で合成することは学術的な興味に留まらず、関連する産業界においても 環境負荷の低減、コスト削減、スピードの向上といった観点から重要となっている。 そのため、効率的な炭素―炭素結合形成反応の開発ならびに官能基変換反応の開発 は有機化学における恒久的なテーマとなっている。 多成分連結反応(Multi-Component Reactions: MCRs)は 3 種類以上の出発成 分を1つの反応容器で反応させ、複数の炭素―炭素(ヘテロ原子)結合形成反応を 一挙に行うことにより全ての出発成分を生成物へ取り込む反応形式である1)。その 高い効率性と有用性はすでに医薬分野におけるリード化合物の探索研究において 注目されており、多様性に富んだ化合物ライブラリーの拡充に寄与している 2)。 MCRs 研究の歴史は古く、古典的な反応として Mannich 反応3)やRobinson 反応 4)が広く知られており、近年ではUgi 反応 5)がその代表例となっている。これらを 含む既報の MCRs の反応様式について目を向けてみると、イオン反応または有機 金属反応を利用した反応例が多く、ラジカル反応を経た報告例は決して多くない6)。 これはラジカルが非常に反応性に富んだ活性種であるため、多成分系における反応 の制御が難しいことに起因していると思われる。しかしラジカル反応は、(1)中性 条件下での反応が可能であり、高い官能基許容性がある(2)完全な水の除去を必 ずしも必要としない(3)イオン反応とは異なった官能基選択性、位置選択性が発 現するといった利点があり、反応工程の短縮や操作の簡便化が期待できる反応様式 である。かかる背景の下、著者の研究室ではこれまでに一酸化炭素をC1 シントン (合成素子)として利用したラジカル多成分連結反応を数多く報告してきた7)。そ の中でも光照射条件下におけるヨウ素原子移動カルボニル化反応を利用したアル キルヨウ化物からのカルボン酸エステル、アミド合成反応は、入手容易な出発原料 から汎用性の高いカルボン酸誘導体を与えることができるという観点から極めて 有用である(eq. 1, 2)8)。また、遷移金属触媒を用いたカルボニル化反応は数多く 報告されているが、sp3-ハロゲン結合への酸化的付加はsp2炭素―ハロゲン結合へ のそれよりも一般的に困難であるため9)、ラジカル機構を経る本反応は遷移金属触 媒反応の課題点を補うことができ、相補的な関係にあると言える。近年、原子移動 カルボニル化反応の応用が幾つかのグループにより行われており10)、特に11CO を 取り込んだ放射性化合物の合成に関してLångström らが精力的な研究を行っている 11)。I CO Et2NH 12 h Et3N hexane NEt2 O 20 atm h (Xe, Pyrex) 88% I CO EtOH 15 h Et3N hexane OEt O 20 atm h (Xe, Pyrex) 72% (eq. 1) (eq. 2)
しかしながら、上記の原子移動カルボニル化(Atom Transfer Carbonylation: ATC)反応は第 2 級および第 3 級のアルキルヨウ化物を用いた場合にはスムーズに 進行するものの、第1 級のアルキルヨウ化物では多大な反応時間を要する。これは アシルラジカルへのヨウ素の移動速度が第 1 級のアルキルヨウ化物では緩慢であ ることに起因している(Scheme 1)12)。 Scheme 1 一方、渡部らは以前に白金触媒と光照射を組み合わせた条件における第1 級アル キルヨウ化物からのカルボン酸エステル合成反応を報告している(eq. 3)13)。この 反応では、白金触媒非存在下では目的の反応は全く進行しないことが示されている。 また鈴木、宮浦らはパラジウム触媒と光照射条件におけるアルキルヨウ化物、一酸 化炭素、アルキルボランからの非対称ケトン合成反応を報告している (eq. 4) 14)。 これらいずれの反応においても遷移金属触媒からアルキルヨウ化物への 1 電子移
動(Single Electron Transfer: SET)を経たラジカル機構の介在が提案されている が、どの段階でラジカルが関与するのかについては、確認された訳ではない。
最近、著者の研究室ではこれら遷移金属触媒からのSET を利用することで、第 1 級のアルキルヨウ化物の ATC 反応が飛躍的に加速されることを見出し、従来の ATC 反応の課題点を克服することに成功している(eq. 5, 6)15)。これらの反応で は、いずれもパラジウム触媒からの SET により生成したアルキルラジカルが一酸 化炭素を捕捉することでアシルラジカルが形成され、続くパラジウム触媒とのカッ プリングによりアシルパラジウム中間体を経て反応が進行していると考えられて いる(eq. 7)。 著者はこれまでに述べてきた Pd/h系の素反応としてのポテンシャルに着目し、 これを更に応用・発展させることで新たなラジカル多成分連結反応を見出すことが できると考え、研究を行った。この方法論に基づいた新規多成分連結反応の開発が
できれば、有機合成における新たな効率的合成方法を提供できるものと期待される。 第 1 章では、Pd/h系における位に電子求引性基を持つアルキルヨウ化物、オ レフィン、一酸化炭素、アルコールを用いた多成分連結反応の検討結果について述 べる(eq. 8)。位に電子求引性基を有するアルキルラジカルは求電子性を有する ため、一酸化炭素の捕捉よりもオレフィンへの付加が速やかに進行する。続いて生 成するアルキルラジカルと一酸化炭素の付加反応は平衡反応であるが、一酸化炭素 を加圧することでアシルラジカル側に平衡を偏らせることが可能となる。著者はこ の様な条件設定によりラジカルの制御が可能となり、ラジカル多成分連結反応が達 成できると考え検討を行った。また更にオレフィンとアルコールの代わりにアルケ ニルアルコールを用いることで分子内環化反応が進行し、ラクトン環形成が可能で あると考えた(eq. 9)。 また本章では反応機構についてその詳細を明らかにするための検討も行った。上 述の渡部、鈴木・宮浦らはアルキル金属中間体への一酸化炭素の挿入によってアシ ル金属種が形成される反応機構を提唱している。一方、著者の研究室ではアルキル ラジカルと一酸化炭素のカップリングにより生成するアシルラジカルを経由して アシル金属種が形成されるメカニズムを提案している(Scheme 2)。そこで Pd/h 系におけるエステル化反応において、著者らが提唱する機構で進行していることを 検証するとともに、渡部、鈴木・宮浦らが提唱する反応機構に関する検討も併せて 行った。
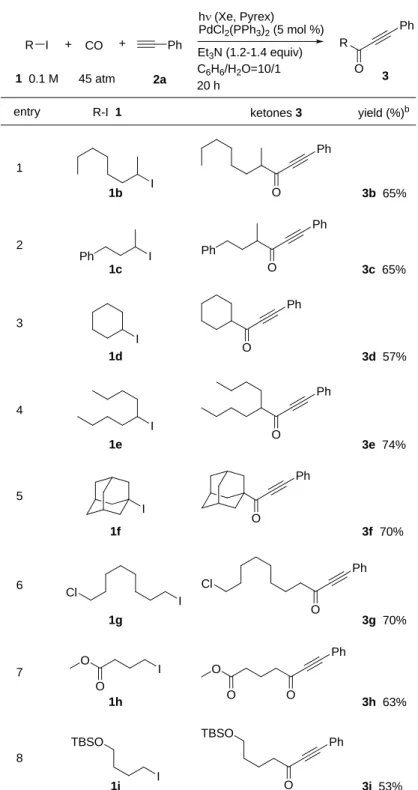
Scheme 2 第2 章では、Pd/h系におけるアルキルヨウ化物、一酸化炭素、末端アセチレン による3 成分連結を経たアルキルアルキニルケトン合成反応について述べる。これ までにカルボニル化を伴った薗頭反応に関する報告例は数多くあるが、いずれも芳 香族ハロゲン化物やビニルハロゲン化物を用いた反応であり、単純なアルキルハロ ゲン化物からの合成例はこれまでになかった(eq. 10)。著者は Pd/h系においてア ルキルヨウ化物からアシルパラジウム中間体が形成されるという仮説に則ること で、これまでに不可能であったアルキルヨウ化物からのアルキニルケトン合成が可 能となると考え検討を行った。鋭意検討の結果見出された3 成分連結反応は、アル キルハロゲン化物に対する広い構造、官能基許容性を示し、また末端アセチレンに ついても芳香族アセチレンだけではなくアルキルアセチレンの場合も良好な結果 を与えることを示した(eq. 11)。 R X + CO + R' R O R' R = aryl, vinyl R X + CO + R' Pd/h R O R' R = alkyl This work Pd Previous work (eq. 10) (eq. 11) 第 3 章では、Pd/h系における位に電子求引性基を持つアルキルヨウ化物と一 酸化炭素、アミンによるカルバモイルアセテート合成反応について述べる。一般的
に位に電子求引性基を持つアルキルヨウ化物は塩基存在下、アミンと速やかに反 応が進行してしまう。しかしながら、一酸化炭素雰囲気下、Pd/h系で反応を行う と良好に3 成分連結反応が進行し、カルバモイルアセテート誘導体が得られること を見出した。なお、本反応に類似した3 成分系によるカルバモイルアセテート合成 反応の報告例は極めて少ない。本章では条件検討から基質の一般性に関する検討結 果についてその詳細を述べる。 第4 章では Pd/h系を利用した上記以外の新規反応探索に関する検討結果につい て述べる。 なお、参考文献や化合物の合成法などについては各章でまとめており、理解を助 けるため重複している箇所があることを了承していただきたい。
参考文献
1 (a) Bienayme, H.; Hulme, C.; Oddon, G.; Schmitt, P. Chem. Eur. J. 2000, 6, 3321. (b) Multicomponent Reactions; Zhu, J., Bienayme, H., Eds.; Wiley-VCH: Weinheim, 2005. (c) Synthesis of Heterocycles via Multicomponent Reactions I; Orru, R. V. A. Ruijter, E., Eds.; Springer: Heidelberg, 2010.
2 Weber, L. CombinatorialChemistry, 2002, 7, 143. 3 Mannich, C.; Kröschl, W. Arch. Pharm. 1912, 250, 647. 4 Robinson, R. J. Chem. Soc. 1917, 111, 762.
5 (a) Ugi, I.; Steinbrückner, C. Chem. Ber. 1961, 94, 734. (b) For a review, see: (b) Dömling, A.; Ugi, I. Angew. Chem., Int. Ed. 2000, 39, 3168.
6 For radical multi-component reactions, (a) in Multicomponent Reactions; Zhu, J., Bienayme, H., Eds.; Wiley-VCH: Weinheim, 2005, pp. 169-198. Also see reviews: (b) Malacria, M. Chem. Rev. 1996, 96, 289. (c) Godineau, E.; Landais, Y.
Chem. Eur. J. 2009, 15, 3044.
7 (a) Ryu, I.; Yamazaki, H.; Kusano, K.; Ogawa, A.; Sonoda, N. J. Am. Chem. Soc. 1991, 113, 8558. (b) Ryu, I.; Kusano, K.; Yamazaki, H.; Sonoda, N. J. Org. Chem. 1991, 56, 5003. (c) Ryu, I.; Yamazaki, H.; Ogawa, A.; Kambe, N.; Sonoda, N. J. Am. Chem. Soc. 1993, 115, 1187. (d) Tunoi, S.; Ryu, I.; Fukushima, H.; Tanaka, M.; Komatsu, M.; Sonoda, N. Synlett 1995, 1249. (e) Tunoi, S.; Ryu, I.; Yamasaki, S.; Fukushima, H.; Tanaka, M.; Komatsu, M.; Sonoda, N. J. Am. Chem. Soc. 1996, 118, 10670. (f) Miura, K.; Tojino, M.; Fujisawa, N.; Hosomi, A.; Ryu, I.
Angew. Chem., Int. Ed. 2004, 43, 2423. Also see reviews, (g) Ryu, I.; Sonoda, N.
Angew. Chem., Int. Ed. 1996, 35, 1050. (h) Ryu, I.; Sonoda, N.; Curran, D. P.
Chem. Rev. 1996, 96, 177.
8 (a) Ryu, I.; Nagahara, K.; Kambe, N.; Sonoda, N.; Kreimerman, S.; Komatsu, M. Chem. Commun. 1998, 1953. (b) Nagahara, K.; Ryu, I.; Komatsu, M.; Sonoda, N. J. Am. Chem. Soc. 1997, 119, 5465. Also see reviews: (c) Chatgilialoglu, C.; Crich, D.; Komatsu, M.; Ryu, I. Chem. Rev. 1999, 99, 1991. (d) Ryu, I. Chem. Soc. Rev. 2001, 30, 16.
9 For overviews of the difficulty of metal catalyzed coupling reactions starting from alkyl halides, see: (a) Cárdenas, D. J. Angew. Chem., Int. Ed. 2003, 42, 384. (b) Luh, T. Y.; Leung, M. K.; Wong, K. T. Chem. Rev. 2000, 100, 3187. (c) Urata,
H.; Maekawa, H.; Takahashi, S.; Fuchikami, T. J. Org. Chem. 1991, 56, 4320. 10 Sugiura, M.; Hagio, H.; Kobayashi, S. Chem. Lett. 2003, 898.
11 (a) Itsenko, O.; Kihlberg, T.; Långström, B. J. Org. Chem. 2004, 69. 4356. (b) Itsenko, O.; Långström, B. J. Org. Chem. 2005, 70, 2244. (c) Itsenko, O.; Långström, B. Org. Lett. 2005, 7, 4661. (d) Itsenko, O.; Kihlberg, K.; Långström, B. Synlett 2005, 3160. (e) Itsenko, O.; Kihlberg, T.; Långström, B. Eur. J. Org. Chem. 2005, 3830.
12 Matsubara, H.; Ryu, I.; Schiesser, C. H. Org. Biomol. Chem. 2007, 5, 3320. 13 (a) Kondo, T.; Tsuji, Y.; Watanabe, Y. Tetrahedron Lett. 1988, 29, 3833. (b) Kondo, T.; Sone, Y.; Tsuji, Y.; Watanabe, Y. J. Organomet. Chem. 1994, 473, 163. 14 Ishiyama, T.; Murata, M; Suzuki, A.; Miyaura, N. J. Chem. Soc. Chem. Commun. 1995, 295.
15 (a) Ryu, I.; Kreimerman, S.; Araki, F.; Nishitani, S.; Oderaotoshi, S.; Minakata, S.; Komatsu, M. J. Am. Chem. Soc. 2002, 124, 3812. (b) Fukuyama, T.; Nishitani, S.; Inouye, T.; Morimoto, K.; Ryu, I. Org. Lett. 2006, 8, 1383.
本論
第1 章 多成分連結反応によるカルボン酸エステル、ラクトン誘導体合成反応 第1 節 緒言 原子移動カルボニル化(ATC)反応は、脂肪族ハロゲン化物からカルボン酸エス テル誘導体を与える有用な手法として広く認識されている。しかしながら、第 2,3 級の脂肪族ハロゲン化物の反応は容易に進行するものの、第1 級の場合では多大な 反応時間を要することが課題となっていた。これは、第1 級のアルキルラジカルの 安定性が低いため、アシルラジカルへのハロゲン原子移動が起こりにくいことに起 因していることが考えられている1)。近年、著者の研究室では渡部2)、鈴木・宮浦 3)らの先駆的な研究を基にして見出した Pd/h条件において第 1 級のアルキルヨウ 化物を出発原料としたカルボニル化反応が良好に進行することを報告し、ATC 反 応の課題点を克服することに成功した(Scheme 1-1)4)。そこで著者はこのPd/h 系を利用した ATC 反応に着目し、このシステムを多成分連結反応へ応用すること を考えた。 Scheme 1-1 ラジカル多成分連結反応は、イオン反応や有機金属反応を利用した多成分反応と比較すると報告例が少ない5)。これはラジカルが非常に反応性に富んだ活性種であ ることに起因していると考えられるため、多成分系への応用にあたっては生成する ラジカルを如何にコントロールするかが鍵となる。そこで著者はScheme 1-2 に示 した官能基移動反応に着目した。―(フェニルセレニル)カルボニル化合物に対 し、光照射条件下、一酸化炭素とアルケンを作用させるとフェニルセレニル基の官 能基移動を伴った3 成分連結反応が進行し、アシルセレニド基を有する化合物が得 られる6)。この反応は、各段階で生成するラジカルの特徴を把握することで複数の 炭素―炭素結合形成を高度に制御している。 Scheme 1-2 著者はこの反応をScheme 1-1 で述べた Pd/h系に適応させることでカルボン酸 エステル誘導体の合成が可能と考えた。すなわち、位に電子求引性基を有するア ルキルヨウ化物とアルケン、一酸化炭素、アルコール用いることで、所望の4 成分 連結反応の進行を期待した(Scheme 1-3)。上述のアシルセレニド基はその後アル デヒドへと高収率で変換可能でるものの、直接的にカルボン酸エステル基を導入す る手法を確立することができれば素反応としての有用性は更に高まるものと思わ れる。
Scheme 1-3 EWG I R CO Pd/h R EWG OR' O R'OH EWG OR' R O EWG R CO R'OH C2 synthon C1 synthon EWG I Pd / h またオレフィンの2 重結合炭素の官能基化に関し、両炭素へのカルボニル基導入 反応は数多く報告されているが7)、炭素ユニットとカルボニル基をそれぞれの炭素 へ一挙に導入した反応例は決して多くなく、適応可能な基質も限定的である (Scheme 1-4)8)。このような背景の下、著者はPd/h系を利用した新規ラジカル 多成分連結反応の開発に着手した。 Scheme 1-4 CCl4 CO EtOH Pd(OAc)2, PPh3 K2CO3 50oC, 24 h solvent 40 atm Cl3C CO2Et Cl3C Cl 50% 33% CO 50 atm PdCl2(PPh3)2 KF H2O, 80oC, 48 h I F2 C C F2 F2 C C F2 I F2 C C F2 F2 C C F2 93% CO2H HO2C 以下、第2 節では位に電子求引性基を有するアルキルヨウ化物とアルケン、一 酸化炭素、アルコールを用いた4 成分連結反応について述べる。第 3 節ではアルケ ンに換えてアルケニルアルコールまたはアルケニルアミンを用いることで分子内 環化反応が進行し、ラクトンやラクタム環を合成できると考え検討を行ったのでそ の詳細を述べる(Scheme 1-5)。第 4 節では反応機構の解明に向けた検討ならびに 渡部、鈴木・宮浦らが提唱する反応機構の検証について述べる。
Scheme 1-5 YH EWG I CO Pd/h O Y EWG n n Y = O or NR
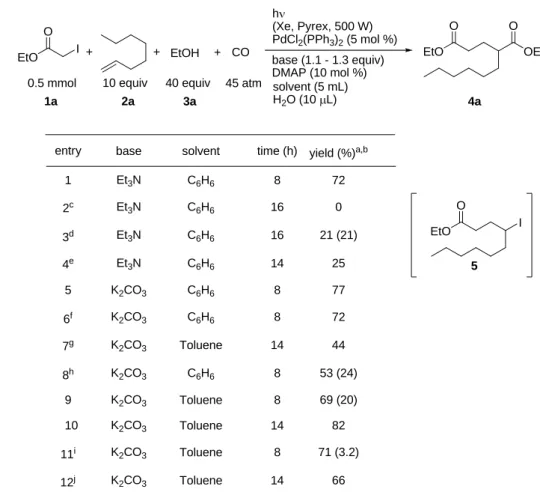
第2 節 アルキルヨウ化物、アルケン、一酸化炭素、アルコールを用いた4 成分連結反応に よるカルボン酸エステル誘導体の合成 基質としてヨード酢酸エチル(1a)、1-オクテン(2a)、エタノール(3a)及び 一酸化炭素を用いて検討を行った(Table 1-1)。触媒として PdCl2(PPh3)(2 5 mol %) を用い、トリエチルアミンおよびDMAP 触媒存在下、ベンゼンを溶媒として水を 添加し、Xe ランプ(500 W)照射下 8 時間反応を行ったところ、目的としたジエ ステル4a が単離収率 72%で得られた(entry 1)。一方、水を加えずに反応を行っ た場合、目的物は全く得られなかった(entry 2)。本反応における水の詳細な役割 は不明であるが、近年、2 価パラジウムの 0 価への還元過程において水の存在が還 元反応を加速させることが報告されていることから9)、この反応においても0 価パ ラジウム発生に寄与しているものと考えられる。次に本反応における光照射及びパ ラジウム触媒の必要性を明確にするため、光非照射及び触媒の非添加条件下におい て反応を行った。光照射を行わず、パラジウム触媒のみを用い、80℃にて反応を行 ったところ目的物4a の収率は 21%まで低下し、同時にヨウ素原子移動生成物 5 が 21%で得られた(entry 3)。またパラジウム触媒を添加せず光照射のみにおいて反 応を行った場合においても、ジエステル4a の収率は 25%にまで低下した(entry 4)。 以上の結果から光照射とパラジウム触媒の両方が本反応において必須であること が明らかとなった。次に塩基をトリエチルアミンから炭酸カリウムへ変更して反応 を行ったところ、ジエステル4a の収率は僅かに向上した(entry 5)。同条件にお いてオクテンの使用量を10 当量から 5 当量へ減らして反応をおこなったところ、 僅かな収率低下がみられた(entry 6)。また 1.3 当量まで減らした場合、4a の収率 は大きく減少した(entry 7)。また DMAP を加えない場合、アルキルヨウ化物 5 の生成が顕著であった(entry 8)。次に溶媒をトルエンへ変更し、反応時間をベン ゼンの時と同様に8 時間で行ったところ、4a および 5 の収率はそれぞれ 69%,20% であった(entry 9)。そこで反応時間を 14 時間へ延長して同様の反応を検討した ところ82%まで単離収率が向上した(entry 10)。これまで一酸化炭素は 45 気圧で 検討してきたが、75 気圧および 15 気圧で検討を行った。その結果、いずれに場合 においても収率の低下が観測された(entry 11, 12)。
Table 1-1 CO + + h (Xe, Pyrex, 500 W) PdCl2(PPh3)2(5 mol %) base (1.1 - 1.3 equiv) DMAP (10 mol %) 1a EtO O I EtO O OEt O + 3a 4a EtOH 2a
entry yield (%)a,b
1 2c 8h 72 53 (24) 9 69 (20) 10 82 11i 71 (3.2) 6f 77 base solvent time (h) Et3N C6H6 8 72 K2CO3 C6H6 8 K2CO3 C6H6 8 K2CO3 C6H6 8 K2CO3 Toluene 8 14 K2CO3 Toluene K2CO3 Toluene 8 5 Et3N C6H6 16 0 44 K2CO3 Toluene 14 7g 12j K 66 2CO3 Toluene 14 0.5 mmol solvent (5 mL) H2O (10 L) 40 equiv EtO O I 5 Et3N C6H6 16 21 (21) 3d Et3N C6H6 14 25 4e
a: Isolated yield after silicagel choromatography. b :Parentheses showed isolated yield of 5. c: Without additon of H2O.
d: The reaction was performed without photo-irradiation conditon at 80℃. e: The reaction was carried out without Pd catalyst.
f: 1-Octene ( 5.0 equiv.) was used. g: 1-Octene ( 1.3 equiv.) was used. h: Without DAMP.
i: The reaction was carried out under CO(75 atm). j: The reaction was carried out under CO(15 atm).
45 atm 10 equiv
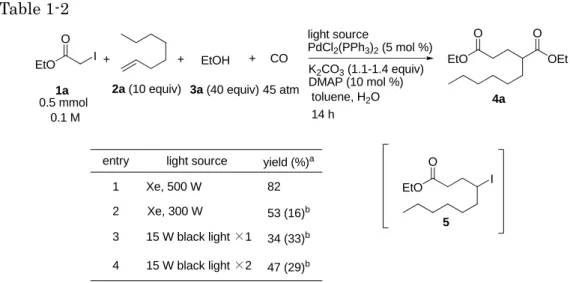
続いて光源の検討を行った(Table 1-2)。Xe ランプ(300 W)を用いて反応を行 ったところ、ジエステル4a が 53%、アルキルヨウ化物 5 が 16%で得られた(entry 2)。また 15 W の black light(ピーク波長:352 nm)1 本を用いて検討したとこ ろ、4a が 34%、5 が 33%で得られた(entry 3)。また black light の本数を 2 本用
いて反応を行った場合、若干の収率向上が見られた(entry 4)。以上の結果から、 光源の出力低下により目的物4a の収率は減少し、アルキルヨウ化物 5 の生成量が 増加することが明らかとなった。すべての場合において原料であるアルキルヨウ化 物1a は消失していることから、1a からのラジカルの発生および 1-オクテンへの付 加はいずれの光源を用いても効率的に進行していること考えられ、光源の出力の差 は中間体5 からのラジカルの発生に効いていると考えられる。
Table 1-2 CO + + light source PdCl2(PPh3)2(5 mol %) K2CO3(1.1-1.4 equiv) DMAP (10 mol %) 45 atm 1a 0.5 mmol 2a (10 equiv) EtO O I EtO O OEt O + 3a (40 equiv) toluene, H2O 4a EtOH 0.1 M
entry light source yield (%)a
15 W black light ×1 15 W black light ×2 1 2 47 (29)b 34 (33)b 3 4 Xe, 500 W Xe, 300 W 82 14 h 53 (16)b EtO O I 5
a: Isolated yield after silicagel choromatography. b :Parentheses showed isolated yield of 5.
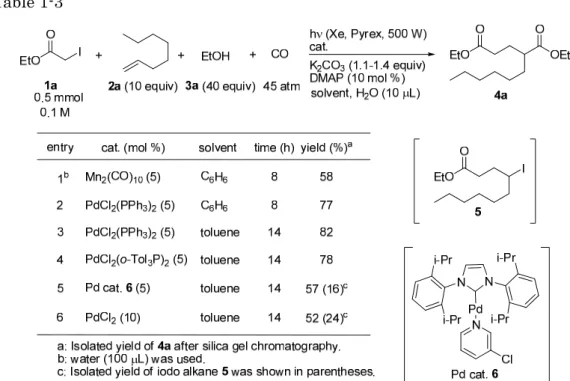
次に触媒の検討を行った(Table 3)。Mn2(CO)1010)を用いて反応を検討したとこ
ろ、同条件におけるPdCl2(PPh3)2の収率よりも低い結果となったものの、Mn 触媒
も本反応に適応可能であることが明らかとなった(enry 1 vs entry 2)。Pd 触媒の 配位子をより電子豊富にすることで収率向上が期待できると考え、PdCl2(o-Tol3P)2
を用いて反応を行ったところ、PdCl2(PPh3)2と同程度の収率でジエステル4a が得
られた(entry 3 vs entry 4)。またより電子的に豊富な NHC(N-Heterocyclic Carbene)配位子を持つ錯体 6 を用いて反応を検討したが、4a の単離収率は低下 し5 の残存が確認された(entry 5)。次に PPh3配位子の効果を確認するため、配 位子を持たないPdCl2を用いて反応を検討したところ、ジエステル4a を 52%およ びアルキルヨウ化物5 を 24%で与えた(entry 6)。この結果は光照射単独条件にお ける 4a の収率(25%)よりも高く、配位子非存在下でも Pd 触媒による反応の加 速効果が観測された。一方でPdCl2(PPh3)2の結果とすると。4a の収率は低下し 5 の残存が認められることから、配位子の存在は反応の加速効果を高めることが示さ れた。
Table 1-3 これまでの検討から最適条件を見出すことができたため、本反応におけるアルキ ルヨウ化物、オレフィンおよびアルコールの適応範囲に関する検討を行った(Table 1-4)。ヨード酢酸エチル(1a)をその臭化物 1b へ変更して反応を行ったところ、 反応系が複雑化するとともにジエステル4a の収率は大きく減少した(entry 2)。 ベンジルアルコールまたはメタノールを用いた場合でも反応は良好に進行し、対応 するジエステルを高収率で与えた(entry 3, 4)。またメタノールの場合、40 当量 を用いていることから原料1a 由来のエチルエステル部分のトランスエステル化が 進行するため、メチルエステルとなっている。オレフィンの官能基に関しては Cl 基やPh 基も許容された(entry 5, 6)。また末端オレフィンだけではなく、内部オ レフィンであるシクロヘプテンを用いた場合でも反応は良好に進行し、目的とする ジエステル4f を単一の異性体として与えた(entry 7)。次にアルキルヨウ化物の検 討を行った。パーフルオロヘキシルヨージド(1c)を用い、オレフィン 2a, 2c と反 応させたところ、それぞれ対応するエステル4g, 4h を良好な収率で与えた(entry 8, 9)。ヨードアセトニトリル(1d)およびヨードメチルフェニルスルホン(1e) を用いてもそれぞれ対応するエステル4i, 4j を与えた(entry 10, 11)。
続いてラジカル反応の特徴であるカスケード反応を利用した多成分連結反応に ついて検討を行った。アリル基を有するアルキルヨウ化物1f と末端オレフィン 2e、 エタノールを一酸化炭素 45 気圧下、反応させたところシクロペンタン環を有する ジエステル4k が 55%で得られた。また末端に Cl 基を有するオレフィン 2b を用い ても良好に反応が進行し、56%でジエステル 4l を与えた(Scheme 1-6)。次にメチ レンシクロヘキサン 2f を用いて同様に反応を行ったところ、スピロ骨格を有する ジエステル4m が 62%の収率で得られた(Scheme 1-7)。この様に、本反応を応用 することで合成に多段階を要する炭化水素骨格の構築を効率的に達成できること が示された。 Scheme 1-6 cheme 1-7 S 次にヨード酢酸エチル(1a)と 1,5-ヘキサジエン(2g)を用いた、2 分子の一酸 化炭素取り込みを経る5 成分連結反応の検討を行った(Table 1-5)。溶媒にベンゼ ン、塩基にトリエチルアミンを用いて一酸化炭素65 気圧下反応を行ったところ、 21%の収率でシクロペンタノン骨格を有するジエステル 4n が得られた(entry 1)。 次に溶媒にトルエンを用い、塩基を炭酸カリウムへ変更したところ、収率が 36%
へ向上した。また5-exo 環化を起こさずエステル基が形成された化合物 4o も 25% で得られた(entry 2)。エタノールの当量の削減や一酸化炭素の加圧を試みたが収 率への大きな影響は観測されなかった(entry 3, 4)。Pd 触媒を 20 mol %へ増量し たところ収率の向上が認められ、この条件において一酸化炭素を95 気圧まで加圧 することで47%の収率でジエステル 4n を得ることができた(entry 5, 6)。しかし ながら、いずれの条件検討においても未環化体 4o の生成量を抑えることはできな かった。 Table 1-5 以上、第2 節では電子求引性官能基をα位に有するアルキルヨウ化物とオレフィ ン、一酸化炭素、アルコールによる多成分連結反応が遷移金属触媒存在下、光照射 により良好に進行することを明らかにした。
第3 節
アルキルヨウ化物、アルケニルアルコ 酸化炭素を用いた3 成分連結反応に
ラクトン骨格は、様々な天然物や医農薬化合物を始めとする生理活性化合物の基
酢酸エチル(1a, 0.25 mmol)と 4-ペンテン-1-オール(7a)を基質として ール、一 よるラクトン誘導体の合成 本骨格として広く存在している11)。その一般的な合成方法は、分子内にあるカルボ ン酸とアルコールによる酸性条件下での脱水縮合または DCC/DMAP や向山 -Corey’s 試薬、山口試薬といった縮合剤によるカルボン酸の活性化を経た縮合反応 により合成されている12)。一方、著者の研究室では水酸基を分子内に有するアルキ ルヨウ化物に対してラジカル条件下、一酸化炭素と反応させることでラクトン環が 構築できることを報告している13)。本節では、第2 節で述べた 4 成分連結反応にお いてオレフィンとアルコールの代わりにアルケニルアルコールを用いることで分 子内環化反応が進行し、ラクトン環合成が可能になると考え検討を行った。これま でにアルキルヨウ化物とアルケニルアルコール、一酸化炭素の3 成分によるアルケ ンの両炭素の官能基化を経たラクトン環合成反応の報告例はなく、この合成反応が 実現できればラクトン誘導体の高効率的な合成反応を新たに提供できると考えら れる。 ヨード 最適条件の探索を行った(Table 1-6)。まず第 1 節で述べた 4 成分連結反応におい て当初見出した条件において反応を行ったところ、42%の収率で目的とする-ラク トン8a を得ることができた(entry 1)。また一酸化炭素を捕捉せずに環化するこ とで形成されるテトラヒドロフラン環を持つ副生成物も25%で得られた。所望のラ クトン環が形成されることが確認できたため、まず水の添加量を50, 100 L と増 量して検討を行ったところ、50 μL において最も高い収率が得られた(entry 2, 3)。 次に反応濃度について検討を行った。その結果、0.05 M において顕著な収率向上 が観測された(entry 4, 5)。一方、いずれの条件においても主な副生成物として上 述のテトラヒドロフラン誘導体が得られたため、一酸化炭素の圧力を45 気圧から 60 気圧へ加圧して反応を行ったが、収率の向上は見られなかった(entry 6)。次に 塩基を炭酸カリウムへ変更して行ったが、収率に大きな変化はなかった(entry 7)。 また溶媒としてトルエンを用いたところ、収率に変化はなかったがGC および TLC 分析において反応系の単純化が観測された(entry 8)。光源の出力を 500 W から 300 W へ変更して検討を行ったところ、大幅な収率向上を達成することができた (entry 9)。これは 500 W の光源装置による発熱によって 8a の開環やその他の副
反応が誘起されていたことに起因していると考えられる。 Table 1-6 3 成分連結反応によるラクトン環形成反応の最適条件を見出すことができたため、 基質一般性について検討を行った(Table 1-7)。アルケニルアルコールのメチレン 鎖長について検討したところ、3-ブテン-1-オール(7b)を用いた場合でも良好に 反応が進行し、所望の-ラクトン誘導体 8b が 72%で得られた(entry 2)。また-ラクトン環の形成においては、テトラヒドロピラン環形成との競合がありながらも 中程度の収率で所望のラクトン誘導体8c を与えた(entry 3)。一方、-ラクトンの 形成はその立体的な歪みによりまったく進行せず、また反応系が複雑化する結果と なった(entry 4)。 Table 1-7
C6F13 I OH 1c 7e 1a 2 3b 4 5 7 8 O EtO O O 8e 63% 6 S O O 40% 7a EtO O 8b O O OH 7b 1a 72% O C6F13 O 8g 77% C6F13 8h 69% O O EtO O 8c 50% O O S O O O O 8j 49% 1c 7a 7b 1a S I O O 9 OH 7c O EtO O 9 O C6F13 O 63% 1e O EtO O O OH 7f 1a OH 7d 8f 8i 1a c 7e EtO O O O OH 10 18% 21% 16% 10 11 EtO O 8d O 0% O a O O 8l 50% NC 1d
a: Isolated yield after silica gel chromatography. b: DMAP was 20 mol % used and CO was 85 atm. NC I 8k 1e 7b S O O 32% S O O O O 60% 11 12 11
次にアルケニルアルコールへの置換基導入を検討したところ、7e を用いた場合 は良好に反応が進行したが、7f の場合では目的のラクトン誘導体は低収率となった。 これは立体障害により分子内環化が阻害されたことに起因すると考えられる (entry 5, 6)。続いてアルキルヨウ化物の検討を行った。パーフルオロヘキシルヨ ージド(1c)を用いて各種のアルケニルアルコールとの反応を検討した結果、いず れの場合においても高収率で対応するラクトン誘導体を与えた(entry 7, 8, 9)。ま たヨードメチルフェニルスルホン(1e)やヨードアセトニトリル(1d)も本反応に 適応可能であることが示された(entry 10, 11, 12)。なお、ヨードメチルフェニル スルホンの場合、その還元体であるメチルフェニルスルホン 11 が副生成物として 得られた。この結果からその他のアルキルヨウ化物の場合においても同様の還元反 応が副反応として進行している可能性が示唆される。 以上、第 3 節では Pd/h系を利用することでα-置換アルキルヨウ化物とアルケ ニルアルコール、一酸化炭素による3 成分連結反応が良好に進行し、5-7 員環ラク トンが中~高収率で得られることを見出した。
第4 節 反応機構の考察 これまで述べてきた Pd/h系を利用した原子移動型カルボニル化反応の反応機 構を考察するにあたり、まず反応の開始段階におけるパラジウム触媒の役割につい て検討を行った。すなわち、炭素―ヨウ素結合への酸化的付加によるアルキルパラ ジウム種の形成を経ているのか、またはパラジウムからの1 電子移動を経たアルキ ルラジカルの発生を経ているかについて明らかにすることとした。そこで6-ヨード オクテン(12)を用いたラジカル条件下と Pd/h条件でのジアステレオ選択性比較 実験を行った(Scheme 1-8)。一般的なラジカル開始条件である Et3B を用い反応 を行ったところ、目的とした環化生成物13 が NMR 収率 47%、cis/trans 比 73/27 で得られた。一方、Pd/h条件による環化反応も同様の生成物を単離収率 43%、 cis/trans 比 73/27 で与えた。また、Pd 触媒非添加条件(光照射のみ)では全く環 化生成物13 は生成しなかった。一酸化炭素、エタノール共存におけるカルボニル 化反応も良好に進行し、シクロペンタン骨格を有するエチルエステル14 を 62%で 与え、cis/trans 比も同様の結果(74/26)を与えた。以上の結果から、Pd/h条件 における開始段階は、炭素―ヨウ素への酸化的付加を経由した反応ではなくラジカ ル機構で進行しており、更にPd 触媒はラジカル開始段階に寄与していることが示 唆された。また、本実験により得られたcis/trans 比は、既知の 1-メチル-5-ヘキセ ニルラジカルの5-exo 環化速度と一致していることも本反応のラジカル機構介在を 支持している14)。
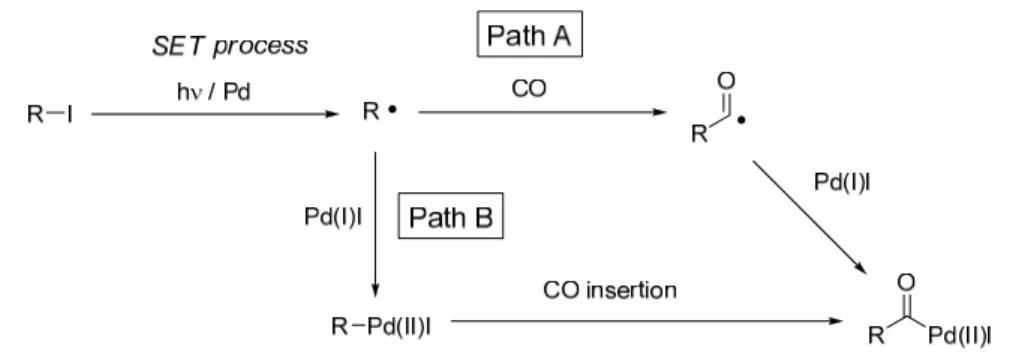
Scheme1-8 次に一酸化炭素の捕捉段階について考察を行った。一酸化炭素を捕捉する経路と しては、①パラジウム触媒からアルキルヨウ化物への1 電子移動により生じたアル キルラジカルが一酸化炭素を捕捉することでアシルラジカルが形成される機構 (Path A)、②アルキルラジカルがパラジウムを捕捉することで形成されるアルキ ルパラジウム種が一酸化炭素を捕捉する機構(Path B)の 2 種類が考えられる (Figure 1-1)。
Figure 1-1 そこでカルボニル化段階の機構解明のため、2-ヨード-3-メチルヘプタン(15, ジ アステレオ比 88/12)を用いた光照射カルボニル化反応におけるパラジウム触媒の 存在/非存在下での生成物の立体化学を比較した(Scheme 1-9)。Pd 触媒非存在下、 光照射のみでカルボニル化反応を行ったところ、対応するメチルエステル 16 が 29%で得られ、その anti/syn 比は 42/58 であった15)。一方、同様の条件にPd 触媒 を添加した結果、反応の加速効果が認められ単離収率61%でメチルエステル 16 が 得られ、anti/syn 比は 43/57 であった。これらの結果は、Pd 触媒の存在、非存在 に関わらずカルボニル化段階における炭素―炭素結合形成はラジカル機構(Path A)で進行していることを示唆している。 Scheme 1-9 次に上記の立体化学比較実験を応用することで、渡部らにより報告されている白 金触媒と光照射 2)または白金触媒と熱条件 16)によるカルボニル化反応における反 応機構を明らかにすることが可能であると考え、その検討を行った(Scheme 1-10,11)。
Scheme 1-10 Scheme 1-11 PtCl2(PPh3)2 触媒と光照射下におけるカルボニル化反応では高圧水銀灯を用い ており、その収率は中程度(>40%)となっている。またこの報告では、non-chain ラジカル機構が提案されており、アルキルプラチナ種の形成とその後のCO 挿入に よりアシルプラチナ中間体が得られていると考えられている(Figure 1-1, Path B)。 そこで同様の白金触媒存在下、アルキルヨウ化物 15 を出発原料として用いて Xe ランプ照射による反応を行った結果、上述のPd/h系と同じ anti/syn 比を有する生 成物16 が得られた(Scheme 1-12)。また PtCl2(PPh3)2触媒と熱条件においても同 様のカルボニル化反応が進行することを報告しており、この反応においても Pt(0) 触媒の酸化的付加によるアルキルプラチナ種形成を経て反応が進行していると考 えられている。そこでこの Pt/熱条件におけるカルボニル化反応についても同様の 方法による検討を行った結果、この条件においても同じ anti/syn 比を有する生成 物を得ることができた。以上の結果より、白金触媒を用いた光条件および熱条件に おけるカルボニル化反応は、アルキルラジカルの発生とカルボニル化段階までのラ ジカル機構の介在を経て進行していることが示唆された。
Scheme 1-12 一方、著者の研究室ではPd/h条件下、アルキルヨウ化物、一酸化炭素、アミン の 3 成分反応においてケトアミド誘導体が主生成物として得られることを報告し ている(Scheme 1-13)4)。またこの反応においてMn 触媒を用いた場合、アミド 誘導体が主生成物として得られ、ケトアミドは全く生成しない。 Scheme 1-13 類似のPd 触媒によるダブルカルボニル化を経るケトアミド形成反応はすでに多 数の報告例があり17)、その生成機構については山本らによって下記の経路が提唱さ れている(Figure 1-2)18)。
Figure 1-2 一般的にラジカル反応においてアシルラジカルへの更なるカルボニル化は困難 であるため19)、Pd/h条件下におけるアルキルヨウ化物を用いたケトアミド形成反 応もこの生成機構に従ってアシルパラジウム中間体を経て進行していることが考 えられる。すなわち、Pd/h条件における Pd 触媒の機能として、アルキルヨウ化 物からのアルキルラジカル発生とアシルラジカルからのアシルパラジウム形成に 寄与していることが既述の実験結果より考えられる。 これまでの考察を基にした Pd/h系におけるカルボニル化反応の反応機構につ いてヨード酢酸エチルと1-オクテン、一酸化炭素、エタノールの 4 成分連結反応を 例に示した(Scheme 1-14)。反応の開始段階として Pd(0)からの 1 電子移動により アルキルヨウ化物のアニオンラジカルを経由してアルキルラジカルが発生する。こ のアルキルラジカルは求電子性を有するため、1-オクテンへの付加がスムーズに進 行する。この付加により生成したアルキルラジカルが一酸化炭素を捕捉することで アシルラジカルとなり、Pd(I)とのカップリングによりアシルパラジウム中間体を 与え、続くアルコリシスによりジエステルが生成し、同時にPd(0)が再生される。 Pd(0)を始めとする低原子価遷移金属触媒からアルキルヨウ化物への 1 電子移動を 経たアルキルラジカルの発生はすでに提唱されており20)、パーフルオロアルキルヨ ウ化物とオレフィンの反応においてカスケード反応の生起や e.s.r.スペクトルによ りラジカルの生成が実証されている21)。またこの電子移動により生成するPd(I)は そのダイマーとの平衡反応が存在し、Pd ダイマーへの光照射によって Pd(I)が発生 することがすでに報告されている 22)。更に本反応において Pd(I)は persistent
radical としての役割を果たしていると考えられる23)。 Scheme 1-14 EtO O O Pd(II)I Pd(I)I Pd(0) EtOH (Pd(I)I)2 h h 1a EtO O SET EtO O O CO EtO O 2a EtO O OEt O 4a EtO O I + + CO+ EtOH Pd(0) / h 1a 2a 3a 4a 以上、第4 節では Pd/h系における反応機構について検討を行った。本反応では 光照射条件下、Pd 触媒からの電子移動を経たアルキルラジカルの存在が考えられ、 一酸化炭素によるカルボニル段階までラジカル機構で進行していることが示唆さ れた。またPd 触媒は開始段階だけではなくアシルパラジウム中間体を形成する段 階においても重要な役割を果たしていると考えられる。
第5 節 結論 Pd/h系を利用した ATC の多成分連結反応への応用を検討した結果、下記に示す 結果を得ることができた。 1. 位に電子求引性官能基を有するアルキルヨウ化物とオレフィン、一酸化炭素、 アルコールによる4 成分連結反応が Pd/h条件において良好に進行し、対応す るカルボン酸エステル誘導体が高収率で得られることを見出した。本反応は広 い官能基許容性を有しているとともに、様々な電子求引性官能基を有するアル キルヨウ化物を用いることができ、また末端オレフィンだけではなく内部オレ フィンも適応可能であることが示された。 2. 上記 4 成分連結反応においてオレフィンとアルコールに換えてアルケニルアル コールを用いた場合、分子内環化反応が良好に進行し、ラクトン誘導体が高収 率で得られることを見出した。この反応は5,6 員環ラクトンだけではなく 7 員 環ラクトンの構築にも有効であることが示された。 3. Pd/h系を利用したカルボニル化反応における機構解明について検討を行った 結果、本反応はアルキルヨウ化物の Pd(0)への酸化付加経由ではなく Pd(0)か らの1 電子移動によるラジカル機構を経て進行していることが示された。また カルボニル化段階はアルキルラジカルの一酸化炭素捕捉により生成するアシ ルラジカルを経て進行しており、さらにその後アシルパラジウム中間体を形成 していることが示唆された。 4. 過去に報告された白金触媒/光照射条件および白金触媒/熱条件におけるカルボ ニル化反応についてその反応機構解明のための検討を行った結果、これらの反 応もアルキルラジカルからのアシルラジカル形成を経て進行していることが 示された。
第6 節 実験の部
共通項
核磁気共鳴(1H NMR)スペクトルは特に記載のない限り CDCl3を溶媒とし、JEOL
JNM ECP-500 (500 MHz)型または JEOL AN-400 (400 MHz)型を用いて測定した。 ケミカルシフト()は parts per million(ppm)で表記し、TMS を 0.00 ppm とし て測定した。核磁気共鳴(13C NMR)スペクトルは JEOL JNM ECP-500 (125 MHz) 型またはJEOL AL-400 (100 MHz)型を用い、77.00 ppm を基準として測定した。 核磁気共鳴(19F NMR)スペクトルは JEOL JNM ECP-500 (470 MHz)型を用い、 CF3COOH を-76.55 ppm として測定した。赤外線吸収(IR)スペクトルは、JASCO FT/IR 4100 型を用いた(吸収は cm-1単位で表記した)。質量分析スペクトルおよ び高分解能質量分析スペクトル(HRMS)は JEOL MS700 型で測定した。溶媒は
すべて適切に蒸留したものを使用した。生成物はNacalai Tesque, INC., Silica Gel 60, 70-230 mesh を使用してフラッシュカラムクロマトグラフィーにより精製し た。必要に応じて分取HPLC (Japan Analytical Industry Co., Ltd., LC-908)型を 用い、GPC カラム(JAIGEL-1H + JAIGEL-2H columns)によりクロロホルムを溶 媒として精製を行った。一酸化炭素加圧下における光照射は、クオーツガラスを有
するステンレスオートクレーブを用い、特にことわりのない限り Pyrex 製内挿管
(30 mm)を使用して行った。光源としては、500 W Xe ランプ(Ushio Co. Ltd., lamp house: SX-UI500XQ, Xenon short arc lamp: UXL-500SX, power supply: BA-X500)または 300 W Xe ランプ(Asahi spectra Co. Ltd., Xenon short arc lamp type: UV, mirror module type: UV, power supply: MAX-301)を使用した。
Iodomethyl phenyl sulfone(1e)の合成24)
ベンゼンスルフィン酸ナトリウム(2.02 g, 12.3 mmol)とジヨードメタン(3.28 g, 12.2 mmol)を DMF(25 mL)へ加え、窒素雰囲気下、80-90℃で終夜反応を行っ
た。反応終了後、水を加えた後ジエチルエーテルで2 回抽出した。合わせた有機層
をシリカゲルカラムクロマトグラフィー(ヘキサン/ジエチルエーテル=2/1)によ り精製した。得られた油状生成物へエタノールを少量加え、3 時間氷冷することで 結晶化させた後、ヘキサン中でトリチュレートすることで表題の化合物を白色結晶 (1.63 g, 43%)として得た。 1H NMR (CDCl3, 400 MHz) 7.98 (d, J = 7.6 Hz, 2H), 7.71 (t, J = 8.4 Hz, 1H), 7.61 (dd, J = 8.4, 7.6 Hz, 2H), 4.47 (s, 2H). Ethyl 2-iodopent-4-enoate(1f) アルゴン雰囲気下、Rh2(OAc)4(50.0 mg, 0.113 mmol)とアリルヨージド(6.93 g, 41.3 mmol)の混合物へジアゾ酢酸エチル(1.63 g, 14.3 mmol)のシクロヘキサン (15 mL)溶液を 20 時間かけて滴下した後、さらに室温で 2 日間撹拌した。反応 終了後、ジエチルエーテルを加えて希釈し、セライトろ過により不溶物を除去した。 ろ液を水、brine で洗浄後、硫酸マグネシウムで乾燥した。ろ過、減圧濃縮により 得た粗製をフラッシュシリカゲルクロマトグラフィー(ヘキサン→ヘキサン/ジエ チルエーテル=40/1)により精製することで表題の化合物を無色油状(1.59 g, 66%) で得た。 1H NMR (CDCl3, 500 MHz) 5.75-5.67 (m, 1H), 5.18-5.14 (m, 2H), 4.30 (dd, J = 8.2, 7.3 Hz, 1H), 4.24-4.18 (m, 2H), 2.84-2.81 (m, 1H), 2.74-2.70 (m, 1H), 1.28 (t, J = 6.9 Hz, 3H). Methyl-2,2-dimethyl-4-pentenoate O O Br O O + LDA THF -78 °C to rt アルゴン雰囲気下、ジイソプロピルアミン(5.071 g, 50.11 mmol)の THF(37 mL) 溶液を0℃へ冷却後、n-ブチルリチウムの 1.6 M ヘキサン溶液(35 mL, 56 mmol) を滴下し、そのまま30 分撹拌した。調製した LDA 溶液を-78℃へ冷却し、メチル
イソブチレート(5.626 g, 55.09 mmol)を 20 分かけてゆっくり滴下した後、-78℃ で1.5 時間、-40℃で 1 時間撹拌した。臭化アリル(7.786 g, 64.35 mmol)を滴下 後、室温で終夜撹拌した。塩化アンモニウム水溶液でクエンチ後、ジエチルエーテ ルで3 回抽出した。合わせた有機層を brine 洗浄後、硫酸マグネシウムで乾燥させ、 ろ過、減圧濃縮により粗製を得た。フラッシュシリカゲルクロマトグラフィーによ り精製することで表題の目的物(5.739 g, 81%)を得た。 1H NMR (CDCl3, 400 MHz) 5.71 (m, 1H), 5.03 (m, 2H), 3.65 (s, 3H), 2.27 (d, J = 7.2 Hz, 2H), 1.16 (s, 6H). 2,2-Dimethylpent-4-en-1-ol(7e) Methyl-2,2-dimethyl-4-pentenoate(726.0 mg, 5.11 mmol)の THF(5 mL)溶液 を0℃へ冷却し、LAH(427.2 mg, 11.26 mmol)をゆっくりと加え、30 分撹拌し た。その後、室温までで昇温し、一晩攪拌した。反応溶液を0℃へ冷却後、水 0.4 mL、 15% NaOH 水溶液 0.4 mL、水 1.2 mL を順に加え、30 分攪拌した。その後生成し た白色沈殿をセライト濾過にて除き、溶媒を留去した。残渣をジエチルエーテルで 抽出し、水で洗浄後、水層の再抽出、brine で洗浄した。MgSO4で乾燥、濾過後、 溶媒の減圧留去により得られた粗製をフラッシュシリカゲルクロマトグラフィー により精製することで表題の化合物(253.4 mg, 43%)を得た。 1H NMR (CDCl3, 400 MHz) 5.84 (m, 1H), 5.05 (dd, J = 14.8, 2.8 Hz, 2H), 3.32 (s, 2H), 2.02 (d, J = 7.6 Hz, 2H), 1.39 (br, 1H), 0.88 (s, 6H). 2-Methylhex-5-en-2-ol(7f) Hex-5-en-2-one(460.2 mg, 4.69 mmol)の THF(20 mL)溶液を 0℃へ冷却し、 MeMgBr の 3M 溶液(2.0 mL, 6.00 mmol)をゆっくり滴下後、室温で 2 時間撹拌 した。反応溶液を0℃へ冷却し、飽和塩化アンモニウム水溶液でクエンチした。ジ
エチルエーテルで3 回抽出した後、brine で洗浄し、硫酸マグネシウムで乾燥した。 ろ過、減圧濃縮により得た粗製をフラッシュシリカゲルクロマトグラフィーにより 精製することで表題の化合物(340.4 mg, 60%)を得た。 1H NMR (CDCl3, 400 MHz) 5.85 (m, 1H), 5.04 (dq, J = 18.8, 1.6 Hz, 1H), 4.95 (dq, J = 10.0, 1.6 Hz, 1H), 2.18-2.12 (m, 2H), 1.57 (m, 2H), 1.26 (br, 1H), 1.23 (s, 6H). 6-Iodooct-1-ene(12)の合成 (COCl)2(2.9 mL, 30.3 mmol)の CH2Cl2(50 mL)溶液をアルゴン雰囲気下、-78℃ へ冷却し、DMSO(4.7 mL, 66.6 mmol)をゆっくりと滴下した。そのまま 10 分 撹拌後、5-Hexene-1-ol(2.5 mL, 20.8 mmol)の CH2Cl2(7.5 mL)溶液を滴下し、 -78℃で時間撹拌した。Et3N(9.2 mL, 66.2 mmol)を一気に加え、そのまま室温 になるまで撹拌した。飽和塩化アンモニウム水溶液を加えた後、水で希釈し、ジク ロロメタンで3 回抽出した。合わせた有機層を brine 洗浄した後、硫酸マグネシウ ムで乾燥し、ろ過、減圧濃縮により粗製を得、これをシリカゲルクロマトグラフィ ーによりにより精製することでアルデヒド(1.85 g, 91%)を得た。 アルデヒド(0.8356 g, 8.53 mmol)の THF(40 mL)溶液をアルゴン雰囲気下、0℃ へ冷却し、EtMgBr の 1M THF 溶液(10.2 mL, 10.2 mmol)を滴下した。0℃で 1 時間撹拌後、再び0℃へ冷却し、飽和塩化アンモニウム水溶液でクエンチした。ジ エチルエーテルで3 回抽出し、合わせた有機層を brine で洗浄し、ろ過、減圧濃縮 により得た粗製をシリカゲルクロマトグラフィーにより精製することでアルコー ル(0.5858 g, 71%)を得た。 PPh3(1.5188 g, 5.8 mmol)とイミダゾール(0.3893 g, 5.7 mmol)の CH2Cl2(30 mL)溶液をアルゴン雰囲気下、0℃へ冷却し、I2(1.4635 g, 5.8 mmol)を加えた 後、そのまま10 分撹拌した。アルコール(0.4662 g, 3.6 mmol)の CH2Cl2(6 mL) 溶液を加え、遮光下、室温で 1 時間撹拌した。飽和 Na2S2O3水溶液を加えた後、 ジクロロメタンで3 回抽出した。合わせた有機層を brine で洗浄し、硫酸マグネシ ウムで乾燥後、ろ過、減圧濃縮により粗製を得た。これをシリカゲルクロマトグラ
フィーにより精製後、更に HPLC による精製を行うことで無色油状の表題の化合 物(0.7401 g, 85%)を得た。 1H NMR (CDCl3, 500 MHz) 5.80 (ddt, J = 17.4, 10.2, 6.4 Hz, 1H), 5.05-4.95 (m, 2H), 4.12-4.05 (tt, J = 8.7, 4.6 Hz, 1H), 2.13-2.09 (m, 2H), 1.91-1.60 (m, 5H), 1.52-1.42 (m, 1H), 1.02 (t, J = 7.4 Hz, 3H); 13C NMR (CDCl3, 100 MHz) 14.1, 28.7, 32.8, 33.7, 39.6, 42.1, 114.9, 138.2; IR (neat) 3076 (C-H), 1640 (C=C), 989 (C-H), 912 (C-H), 573 (C-I) cm-1; MS (relative intensity) m/z 238 (M+, 1), 111 (41),
69 (100), 55 (42), 41 (30); HRMS (EI) calct for C8H15I [M]+ m/z 238.0218, found:
238.0229. 2-Iodo-3-methylheptane(15)の合成 CuCN(0.9997 g, 11.2 mmol)の THF(30 mL)懸濁液をアルゴン雰囲気下、-78℃ へ冷却し、n-BuLi の 1.6M ヘキサン溶液(14 mL, 22.4 mmol)をゆっくり滴下し、 -40℃で 30 分撹拌した。再び-78℃へ冷却後、2,3-Butyleneoxide(0.9 mL, 10 mmol) をゆっくり滴下し、-20℃で 1 時間撹拌した。飽和塩化アンモニウム水溶液を加え クエンチした後、ジエチルエーテルで3 回抽出した。合わせた有機層を brine で洗 浄後、硫酸マグネシウムで乾燥し、ろ過、減圧濃縮により得た粗製をシリカゲルク ロマトグラフィーにより精製することでアルコール(0.9049 g, 70%)を得た。 PPh3(2.84 g, 10.8 mmol)とイミダゾール(0.83 g, 12.2 mmol)の CH2Cl2(50 mL) 溶液をアルゴン雰囲気下、0℃へ冷却し、I2(2.73 g, 10.7 mmol)を加えた後、そ のまま10 分撹拌した。アルコール(1.71 g, 7.13 mmol)の CH2Cl2(10 mL)溶 液を加え、遮光下、0℃で 30 分、室温で 1 時間撹拌した。飽和 Na2S2O3水溶液を 加えた後、ジクロロメタンで3 回抽出した。合わせた有機層を brine で洗浄し、硫 酸マグネシウムで乾燥後、ろ過、減圧濃縮により粗製を得た。これをシリカゲルク ロマトグラフィーにより精製後、更に HPLC による精製を行うことで無色油状の 表題の化合物(1.28 g, 75%)を得た。
Obtained as an inseparable diastereomer mixture in 12/88 ratio, by 1H NMR
chromatography; major isomer: 1H NMR (CDCl3, 500 MHz) 4.35-4.31 (m, 1H), 1.84 (d, J = 7.3 Hz, 3H), 1.48-1.43 (m, 1H), 1.37-1.12 (m, 6H), 0.98 (d, J = 6.4 Hz, 3H), 0.90 (t, J = 7.3 Hz, 3H); 13C NMR (CDCl3, 125 MHz) 14.1, 18.3, 22.9, 24.7, 29.4, 34.1, 40.0, 41.7; minor isomer: 1H NMR (CDCl3, 500 MHz) 4.37-4.32 (m, 1H), 1.89 (d, J = 7.3 Hz, 3H), 1.40-1.12 (m, 7H), 0.98 (d, J = 6.4 Hz, 3H), 0.90 (t, J = 7.3 Hz, 3H); 13C NMR (CDCl3, 125 MHz) 14.1, 17.3, 22.7, 26.6, 29.1, 37.0,
40.9, 41.6; IR (neat) 584 (C-I) cm-1; MS (relative intensity) m/z 240 (M+, 0.1), 113
(87), 71 (92), 57 (100), 43 (73), 41 (32); HRMS (EI) calct for C8H17I [M]+ m/z
240.0375, found: 240.0380.
アルキルヨウ化物とオレフィン、一酸化炭素、アルコールを用いた4 成分連結反応
Pyrex 内挿管へ撹拌子、ethyl iodoacetate (1a, 0.13 g, 0.61 mmol), 1-octene (2a, 0.69 g, 6.2 mmol), EtOH (3a, 1.2 g, 26 mmol), K2CO3 (0.10 g, 0.72 mmol),
PdCl2(PPh3)2 (0.025 g, 0.036 mmol), DMAP (0.0084g, 0.069 mmol), toluene
(5.0 mL)と H2O (10 L)を加え、光反応用オートクレーブに挿入した。一酸
化炭素(10 atm)で 3 回置換した後、45 atm まで加圧し、Xe ランプ(500 W)
にて光照射を行いながら 14 時間撹拌した。反応終了後、室温まで冷却して一酸化
炭素を抜き、反応混合物を水で希釈し、ジエチルエーテルで3 回抽出した。合わせ
た有機層をbrine 洗浄後、硫酸マグネシウムで乾燥し、ろ過、減圧濃縮により粗製
を得た。これをシリカゲルクロマトグラフィーにより精製することでジエステル 4a(0.14 g, 82%)を得た。
Diethyl 2-hexylpentanedioate (4a)
1H NMR (CDCl3, 500 MHz) 4.14-4.09 (m, 4H), 2.41-2.23 (m, 3H), 1.92-1.78 (m,
2H), 1.68-1.56 (m, 1H), 1.50-1.42 (m, 1H), 1.36-1.19 (m, 14H), 0.87 (t, J = 7.9 Hz, 3H); 13C NMR (CDCl3, 125 MHz) 14.0, 14.1, 14.2, 22.5, 27.1, 27.2, 29.2, 31.6,
32.0, 32.3, 44.8, 60.1, 60.3, 173.0, 175.6; IR (neat) 1735 (C=O) cm-1; MS (relative
(29), 69 (35), 55 (34); HRMS (EI) calct for C15H28O4 [M-C2H5O]+ m/z 227.1647,
found: 227.1650.
Ethyl 4-iododecanoate (5)
This compound was known. See; Ollivier, C.; Bark, T.; Renaud, P.; Synthesis
2000, 1598. 1H NMR (CDCl3, 500 MHz) 4.14 (q, J = 7.3 Hz, 2H), 4.16-4.11 (m, 1H), 2.58 (ddd, J = 16.5, 8.2, 6.0 Hz, 1H), 2.47 (dt, J = 16.5, 7.8 Hz, 1H), 2.10-2.03 (m, 2H), 1.93-1.85 (m, 1H), 1.74-1.67 (m, 1H), 1.56-1.48 (m, 1H), 1.44-1.37 (m, 1H), 1.33-1.24 (m, 6H), 1.26 (t, J = 7.3 Hz, 3H), 0.89 (t, J = 7.3 Hz, 3H); 13C NMR (CDCl3, 125 MHz) 14.02, 14.19, 22.55, 28.42, 29.40, 31.60, 34.40, 35.44, 38.39, 40.71, 60.50, 172.67.
1-Benzyl 5-ethyl 2-hexylpentanedioate (4b)
1H NMR (CDCl3, 500 MHz) 7.39-7.29 (m, 4H), 7.29-7.24 (m, 1H), 5.16-5.09 (m,
2H), 4.13-4.09 (m, 2H), 2.48-2.40 (m, 1H), 2.35-2.21 (m, 2H), 1.97-1.79 (m, 2H), 1.68-1.59 (m, 1H), 1.53-1.44 (m, 1H), 1.31-1.17 (m, 11H), 0.91-0.84 (m, 3H); 13C
NMR (CDCl3, 125 MHz) 13.94, 14.10, 22.44, 27.03, 27.10, 29.01, 31.51, 31.93,
32.22, 44.71, 60.26, 66.00, 128.07, 128.41, 136.00, 172.88, 175.39; IR (neat) 1737 (C=O) cm-1; MS (relative intensity) m/z 334 (M+, 2), 289 (4), 227 (76), 199 (22),
135 (9), 91 (100), 88 (22), 69 (20), 55 (10); HRMS (EI) calct for C20H30O4 m/z
334.2144, found: 334.2145.
1H NMR (CDCl3, 500 MHz) 3.67 (s, 3H), 3.66 (s, 3H), 2.42-2.24 (m, 3H),
1.94-1.78 (m, 2H), 1.66-1.58 (m, 1H), 1.50-1.42 (m, 1H), 1.32-1.22 (m, 8H), 0.87 (d, J = 6.9 Hz, 3H); 13C NMR (CDCl3, 125 MHz) 13.93, 22.47, 27.09, 29.04,
31.54, 31.72, 32.21, 44.66, 51.37, 51.48, 173.36, 176.01; IR (neat) 1739 (C=O) cm-1; MS (relative intensity) m/z 244 (M+, 2), 213 (67), 185 (14), 160 (100), 152
(21), 69 (35), 55 (45); HRMS (EI) calct for C13H24O4 [M-C2H5O]+ m/z 213.1491,
found: 213.1482. Diethyl 2-(4-chlorobutyl)pentanedioate (4d) 1H NMR (CDCl3, 500 MHz) 4.17-4.10 (m, 4H), 3.52 (t, J = 6.9 Hz, 2H), 2.42-2.35 (m, 3H), 1.94-1.73 (m, 4H), 1.69-1.62 (m, 1H), 1.52-1.41 (m, 3H), 1.29-1.23 (m, 6H); 13C NMR (CDCl3, 125 MHz) 14.05, 14.14, 24.38, 27.02, 31.32, 31.80, 32.21, 44.44, 44.50, 60.23, 60.44, 172.77, 175.16; IR (neat) 1732 (C=O) cm-1; MS (relative intensity) m/z 278 (M+, 1), 243 (11), 233 (100), 188 (49),
142 (50), 114 (49), 55 (34); HRMS (EI) calct for C13H23ClO4 [M-C2H5O]+ m/z
233.0944, found: 233.0940.
Diethyl 2-phenethylpentanedioate (4e)
EtO O OEt O Ph 1H NMR (CDCl3, 500 MHz) 7.30-7.26 ( m, 2H), 7.21-7.15 (m, 3H), 4.15 (q, J = 7.4 Hz, 2H), 4.11 (q, J = 6.9 Hz, 2H), 2.65-2.55 (m, 2H), 2.46-2.38 (m, 1H), 2.37-2.24 (m, 2H), 2.03-1.81 (m, 3H), 1.81-1.72 (m, 1H), 1.28 (t, J = 6.9 Hz, 3H), 1.24 (t, J = 7.4 Hz, 3H); 13C NMR (CDCl3, 125 MHz) 14.1, 14.2, 27.1, 31.8, 33.4,
33.9, 44.2, 60.3, 125.8, 128.3, 141.3, 172.8, 175.2; IR (neat) 1731 (C=O) cm-1; MS
(relative intensity) m/z 292 (M+, 5), 247 (5), 201 (19), 188 (31), 142 (27), 114 (55),
91 (100); HRMS (EI) calct for C17H24O4m/z 292.1675, found: 292.1666.
Ethyl 2-(2-ethoxy-2-oxoethyl)cycloheptanecarboxylate (4f)
1H NMR (CDCl3, 500 MHz) 4.17-4.11 (m, 4H), 2.47-2.39 (m, 1H), 2.36-2.30 (m,
2H), 2.21 (dd, J = 15.1, 9.2 Hz, 1H), 1.83-1.77 (m, 2H), 1.76-1.69 (m, 2H), 1.68-1.64 (m, 1H), 1.62-1.56 (m, 1H), 1.55-1.41 (m, 4H), 1.29-1.23 (m, 6H); 13C
NMR (CDCl3, 100 MHz) 14.16, 25.92, 26.07, 29.08, 29.97, 31.67, 37.61, 40.65,
50.50 (2 lines superimposed), 60.16, 60.21, 172.49, 176.25; IR (neat) 1730 (C=O) cm-1; MS (relative intensity) m/z 256 (M+, 12), 211 (93), 182 (100), 169 (30), 95
(77); HRMS (EI) calct for C14H24O4 m/z 256.1675, found: 256.1676.
Ethyl 4,4,5,5,6,6,7,7,8,8,9,9,9-tridecafluoro-2-hexylnonanoate (4g) 1H NMR (CDCl3, 500 MHz) 4.18 (q, J = 7.3 Hz, 2H), 2.84-2.74 (m, 1H), 2.74-2.61 (m, 1H), 2.19-2.05 (m, 1H), 1.74-1.63 (m, 1H), 1.61-1.53 (m, 1H), 1.35-1.26 (m, 11H), 0.88 (t, J = 7.3 Hz, 3H); 13C NMR (CDCl3, 125 MHz) 13.83, 13.94, 22.51, 26.72, 28.90, 31.57, 32.91 (t, J = 22.1 Hz), 33.17, 38.03, 60.85, 108.19-118.42 (m, CF2, CF3), 174.29; 19F NMR (CDCl3, 470 MHz) -81.67 (s, 3F), -114.36 (s, 2F), -122.76 (s, 2F), -123.79 (s, 2F), -124.54 (s, 2F), -127.06 (s, 2F); IR (neat) 1740 (C=O), 1240, 1197, 1145 (C-F) cm-1; MS (relative intensity) m/z 459
(M+-OEt, 2), 420 (11), 101 (100), 73 (79); HRMS (EI) calct for C17H21F13O2 m/z
Ethyl 4,4,5,5,6,6,7,7,8,8,9,9,9-tridecafluoro-2-(2-phenylethyl)nonanoate (4h) 1H NMR (CDCl3, 500 MHz) 7.29 (dd, J = 7.3, 6.9 Hz, 2H), 7.21 (t, J = 7.3 Hz, 1H), 7.17 (d, J = 6.9 Hz, 2H), 4.20 (q, J = 7.3 Hz, 2H) 2.85-2.81 (m, 1H), 2.74-2.68 (m, 1H), 2.64 (t, J = 7.3 Hz, 2H), 2.22-2.09 (m, 1H), 2.08-2.00 (m, 1H), 1.91-1.86 (m, 1H), 1.29 (t, J = 7.3 Hz, 3H); 13C NMR (CDCl3, 125 MHz) 14.01, 32.86 (t, J = 21.1 Hz), 33.05, 34.74, 37.75, 61.05, 108.63-118.35 (m, CF2, CF3), 126.25, 128.33, 128.51, 140.57, 173.92; 19F NMR (CDCl3, 470 MHz) -81.67 (s, 3F), -114.44 (s, 2F), -122.76 (s, 2F), -123.81 (s, 2F), -124.54 (s, 2F), -127.06 (s, 2F); IR (neat) 1736 (C=O), 1240, 1145, 1030 (C-F) cm-1; MS (relative intensity) m/z 479
(M+-OEt, 17), 420 (40), 105 (52), 101 (78), 91 (100), 73 (88); HRMS (EI) calct for
C19H17F13O2m/z 524.1021, found: 524.1025.
Ethyl 2-(2-cyanoethyl)octanoate (4i)
1H NMR (CDCl3, 500 MHz) 4.17 (qd, J = 6.9, 2.3 Hz, 2H), 2.52-2.46 (m, 1H),
2.44-2.29 (m, 2H), 2.03-1.94 (m, 1H), 1.85-1.77 (m, 1H), 1.68-1.60 (m, 1H), 1.53-1.44 (m, 1H), 1.33-1.22 (m, 11H), 0.88 (t, J = 6.9 Hz, 3H); 13C NMR (CDCl3,
125 MHz) 13.91, 14.14, 15.23, 22.42, 26.81, 27.43, 28.93, 31.47, 31.94, 44.09, 60.53, 119.05, 174.60; IR (neat) 2247 (C≡N), 1731 (C=O) cm-1; MS (relative
intensity) m/z 225 (M+, 2), 180 (18), 152 (25), 141 (71), 101 (100), 69 (26), 55 (17);
HRMS (EI) calct for C13H23NO2 [M-C2H5O]+m/z 180.1388, found: 180.1395.
1H NMR (CDCl3, 500 MHz) 7.92 (dd, J = 7.3, 1.3 Hz, 2H), 7.69 (tt, J = 7.3, 1.3
Hz, 1H), 7.60 (t, J = 7.3 Hz, 2H), 4.16-4.08 (m, 2H), 3.17-3.07 (m, 2H), 2.46-2.40 (m, 1H), 2.02-1.92 (m, 2H), 1.65-1.54 (m, 1H), 1.47-1.38 (m, 1H), 1.31-1.21 (m, 11H), 0.89 (t, J = 7.3 Hz, 3H); 13C NMR (CDCl3, 100 MHz) 13.95, 14.16, 22.45,
24.62, 26.82, 28.94, 31.48, 31.92, 43.80, 53.96, 60.49, 127.97, 129.27, 133.71, 138.86, 174.59; IR (neat) 1730 (C=O), 1308 (SO2), 1152 (SO2) cm-1; MS (relative
intensity) m/z 340 (M+, 1), 295 (19), 266 (58), 256 (16), 199 (100), 171 (56), 91
(55), 77 (95), 69 (72), 55 (66); HRMS (EI) calct for C18H28O4S [M-C2H5O]+ m/z
295.1368, found: 295.1370.
Ethyl 3-butyl-4-(2-ethoxy-2-oxoethyl)cyclopentanecarboxylate(4k)
EtO2C
CO2Et
Obtained as an inseparable diastereomer mixture. 1H NMR (CDCl3, 500 MHz)
4.16-4.11 (m, 4H), 2.93-2.86 (m, 0.16H), 2.83-2.76 (m, 0.84H), 2.53-2.31 (m, 1.8 H), 2.25-1.93 (m, 3.9H), 1.78-1.43 (m, 2.3H), 1.39-1.10 (m, 12H), 0.93-0.86 (m, 3H).
Ethyl 3-(4-chlorobutyl)-4-(2-ethoxy-2-oxoethyl)cyclopentanecarboxylate(4l)
Obtained as an inseparable diastereomer mixture. 1H NMR (CDCl3, 500 MHz)
4.22-4.10 (m, 4H), 3.58-3.53 (m, 2H), 2.94-2.72 (m, 1H), 2.57-2.28 (m, 1.8H), 2.25-1.85 (m, 3H), 1.83-1.71 (m, 2.2H), 1.68-1.11 (m, 13H).