バナジウム酸化物デバイス界面に出現する新たな電
子相に関する研究
著者
志賀 大亮
学位授与機関
Tohoku University
博士論文
バナジウム酸化物デバイス界面に
出現する新たな電子相に関する研究
志賀 大亮
目次
第1章
はじめに
1
1.1. 強相関エレクトロニクス
1
1.2. 金属-絶縁体転移(MIT)
3
1.2.1. モット転移 3 1.2.2. パイエルス転移 3 1.2.3. MIT の制御 41.3. 強相関酸化物 VO
26
1.3.1. VO2のMIT 6 1.3.2. 電界効果による VO2のMIT 制御 8 1.3.3. キャリア制御による VO2の電子相変化 9 1.3.4. 次元性制御による VO2の電子相変化 101.4. 本研究の目的
13
第
2章
実験手法
19
2.1. 放射光電子分光
19
2.1.1. 光電子分光(PES) 19 2.1.2. 放射光 21 2.1.3. X 線吸収分光(XAS) 22 2.1.4. 軟 X 線分光の表面敏感性 222.2. その場放射光 PES – レーザー分子線エピタキシ(MBE)複合装置 24
2.3. 放射光電子分光装置
25
2.4. 酸化物薄膜の作製
30
2.4.1. パルスレーザー堆積法 30 2.4.2. レーザーMBE 装置 31 2.4.3. 薄膜の成長様式 332.5. アルカリ金属原子蒸着による表面キャリア注入法
35
2.6. 分析・試料評価手法
37
2.6.1. 反射高速電子線回折(RHEED) 37 2.6.2. 低速電子線回折(LEED) 38 2.6.3. 原子間力顕微鏡(AFM) 39 2.6.4. X 線回折(XRD) 40 2.6.5. 電気抵抗率測定 41 2.6.6. 走査型透過電子顕微鏡(STEM) 41第
3章
VO
2エピタキシャル極薄膜の作製
43
3.1. はじめに
43
3.2. VO
2極薄膜の作製条件
45
3.3. TiO
2(001)基板
48
3.4. VO
2極薄膜の作製条件最適化
50
3.4.1. 内殻準位測定によるヘテロ界面の急 峻しゅん性評価 53 3.4.2. AFM 観察による表面形態の評価 55 3.4.3. XRD 測定によるエピタキシャル関係の評価 56 3.4.4. 電気輸送特性の評価 58 3.4.5. 電子線回折による表面結晶性の評価 593.5. 本章のまとめ
61
第
4章
表面電子注入された
VO
2薄膜における新たな電子相の出現
65
4.1. はじめに
65
4.2. 実験条件
66
4.3. 結果と考察
68
4.3.1. VO2薄膜への放射光照射による光損傷の検討 68 4.3.2. VO2薄膜のMIT に伴う PES スペクトルの変化 71 4.3.3. K 蒸着による絶縁体相 VO2薄膜の金属化 75 4.3.4. K/VO2におけるキャリア誘起金属相 78 4.3.5. K/VO2の大気暴露に伴う電子状態変化 84 4.3.6. V-V 二量化に伴うd//*状態の形成 86 4.3.7. 偏光依存 XAS による V-V 二量化の観測 87 4.3.8. VO2薄膜の酸素K 端 XAS(O K XAS) 90 4.3.9. K/VO2におけるキャリア誘起金属相の結晶構造 91 4.3.10. K/VO2の電子相境界における相分離の検証 93
4.4. 本章のまとめ
97
第
5章
VO
2極薄膜における電子・結晶構造の膜厚依存性
100
5.1. はじめに
100
5.2. 実験条件
102
5.3. 結果と考察
103
5.3.1. VO2極薄膜の輸送特性の膜厚依存性 103 5.3.2. VO2極薄膜における価電子帯スペクトルの膜厚依存性 105 5.3.3. VO2極薄膜におけるO K XAS スペクトルの膜厚依存性 107 5.3.4. VO2の2 次元極限に発現する絶縁体相の起源 1085.4. 本章のまとめ
112
第
6章
まとめと今後の展望
115
謝辞
121
研究業績一覧
125
第
1 章
はじめに
はじめに、研究背景として、強相関酸化物の基礎物性、及び強相関酸化物が示す金属-絶 縁体転移を動作原理とする次世代の電子デバイス「モット・トランジスタ」について概説する。 本論文は、このモット・トランジスタのチャネル材として最も注目されている強相関酸化物 「二酸化バナジウム」を対象とし、ナノサイズのデバイス構造に組み込まれたときの挙動とそ の起源について、電子分光学の観点からのべたものである。本章後節では、本研究アプロー チの詳細と、その学術上・応用上の位置づけ・重要性についてのべる。1.1. 強相関エレクトロニクス
電気伝導をはじめとする固体の物性を担っているのは、その固体中の電子の挙動であり、 その電子状態をどの様に記述するかということが物性を理解する上で基本となる。通常の物 質では、電子は結晶の周期的に並んだ陽イオンと自分以外の電子からなる平均場のポテンシ ャルの中を運動するという「一電子近似」がよく成り立っており、従来の半導体デバイスの挙 動を予測したりする上での基礎理論となっている。一方で、遷移金属などのd 軌道の電子が 物性を担う場合には、その軌道の広がりによって電子分布が空間的に制限されるために電子 の局在性が強くなり、二つの電子が近接した際の電子間相互作用(電子相関)が無視できなく なる。特に、例えば遷移金属酸化物の場合では、最外殻のs 軌道が酸素とのイオン結合に使 用され、d 電子が露わに伝導を担う様になるため、この電子相関の影響が大きくなってくる。 この様な物質系は電子同士が互いに強く影響し合う「強相関電子系」と呼ばれる。べた一電子近似ではその電子状態を記述することが困難であり、その物性を理解するために は多体効果を取り入れた理論モデルが必要となる。 この強相関電子系材料が示す劇的な金属-絶縁体転移(MIT)[1]を外部電場によって制御 する「モットトランジスタ」は、後述する理由から、従来の電界効果トランジスタ(FET)[図 1.1.1(a)]にとって代わる技術要素として実現が期待されている。通常 FET では、半導体チ ャネル層に外部電圧を引加し、キャリアを注入することで、電荷蓄積層を形成する。これに よる電気抵抗の制御が基本動作である。この FET の半導体チャネル材を強相関電子材料に 置き換えると、材料が示す劇的な電子相転移を外場制御することが可能になる[図1.1.1(b)]。 ここで、(1)この電子相転移(MIT)は非常に高速であり、(2)相転移前後で電流を大きく変化 させることができるため、高速かつエネルギー損失が少ないトランジスタ動作を実現できる と考えられている。このチャネル材として、急激なMIT を示す二酸化バナジウム[1–16]が近 年注目を集めている。 図1.1.1. (a)FET の一種である薄膜トランジスタの模式図.ゲート電極に電圧を印加し材料表面に蓄 積層を形成することで電気抵抗を制御する.(b)モット・トランジスタの模式図.強相関酸化物の急激な MIT を基本的な動作原理とする.急激な On/Off スイッチ比と超高速動作の実現が期待される.
1.2. 金属-絶縁体転移(MIT)
1.2.1 モット転移
強相関電子系において、一電子近似が完全に破綻する最もシンプルな例の一つとして、電 子相関に起因した系の絶縁化が挙げられる[1]。通常の一電子近似におけるバンド構造にお いて、伝導バンドにサイトあたり 1 つの電子が存在している場合(ハーフフィリング)を考 える。1 つのエネルギー準位には上下スピンをもった二つの電子が存在することが許容され るので、電子が1 個存在している場合にはバンドが半分埋まった状態となっており、電子相 関がなければこの系は金属的な伝導を示すはずである。ここで強い電子相関が存在すると、 電子間に大きなクーロン反発力が生じるために、1 つのサイトに 1 個の電子が存在した場合 に、同一サイトに二つ目の電子が入れなくなる。このため、最終的には全てのサイトに1 個 ずつ電子が存在しており、電子がどのサイトにも移動できなくなるという状態が生じる(図 1.2.1)。この様にして絶縁化している物質を「モット絶縁体」と呼ぶ。モット絶縁体は、実効 的なクーロン反発力の大きさが変化したり、ハーフフィリングからのずれが生じたりするこ とにより金属となるため、温度変化や化学組成の変化、更には外場の印加によっても MIT を引き起こす。 図 1.2.1. モット絶縁体の模式図.モット転移は、強相関効果による電子の局在化によって実現する絶 縁体転移現象である。1.2.2 パイエルス転移
モット転移は電子-電子間相互作用に起因するものであるが、別機構の MIT の一つとして、 電子-格子間相互作用に起因する「パイエルス転移」が挙げられる。先ほどと同様に、伝導バ ンドに 1 サイトあたり 1 個の電子が存在しており、通常のバンド描像であれば金属状態と なっている場合を考える。ここで結晶格子がひずむことによって、元々の2 サイトが 1 サイ トとなる様な周期性の変調を起こしたとすると、再構成されたバンドには 2 個の電子が収なる。この様な現象が生じるためには、バンド構造にエネルギーギャップが生じることによ る電子系全体のエネルギー(利得)が、結晶格子をひずませるために必要なエネルギー(損失) を上回る必要がある。1 次元(1D)結晶の場合には、結晶軸方向に 2 倍周期の格子変調を生じ させることにより、元々伝導方向が1D に制約されていたバンドの伝導方向にエネルギーギ ャップが生じるため(図1.2.2)、系は必ず絶縁体となる。この様な機構による MIT はパイエ ルス転移と呼ばれる。 図1.2.2. 1D 結晶におけるパイエルス転移の模式図.パイエルス転移は、もともと等間隔に並んでいた V イオンの「二量化」によってエネルギーギャップが形成され、これによって生まれる「電子系のエネル ギー利得」が、「格子変調によるエネルギー損失」を上回ることで生じる。
1.2.3 MIT の制御
この様な多体相互作用に起因する電子相転移現象は、僅かな刺激によって劇的な集団応答 を引き起こすため、デバイス応用の観点からも注目を集めている。電子の集団的な特長を生 かし、磁場・電場・圧力・光など外部から僅かな摂動を与えることで、電子集団自体にMIT の 様な相転移を起こし、その結果生じる劇的かつ超高速の物性変化をデバイスの出力として利 用する「強相関デバイス」の創成が期待されている。例えば FET の様なスイッチング素子に ついて考えると、従来の半導体特性を利用した FET デバイスでは、少数のキャリアの電荷 としての特徴を利用するが、基本的には独立した1電子の動きを制御する。したがって、出 力の大きさは注入したキャリアの量に応じた値となる。それに対して強相関電子系の MIT を利用すると、電子相関の影響で局在化していた電子が僅かな変調により一斉に伝導に寄与 するため、原理的にはキャリア数が一挙にサイト数オーダー(>1022 cm-3)まで増大することになり、非常に高速かつ巨大なスイッチング応答が期待できる。 この様な FET 構造を用いた強相関電子系の電子相制御は、強相関デバイスとしての応用 という観点のみならず、化学的摂動による強相関電子系の物性変調といった課題として、基 礎物性の観点からも非常に興味がもたれている。これまで強相関電子系へのキャリアドープ は、価数の異なるイオンを化学的に置換することにより行われることが一般的であったが、 化学置換の場合には結晶中にキャリアのみならず格子変形や結晶の乱れなどを導入するこ とになるため、純粋なキャリアドーピングの効果を必ずしも反映しない場合があった。これ に対して、FET 構造を用いたゲート電圧印加によるキャリア注入法では、系に純粋にキャリ アのみを導入することが可能であり、物性変調に対するキャリアの効果を純粋に調べること ができる。強相関電子系において、金属相は 1022 cm-3オーダーものキャリア濃度をもつた め遮蔽長が非常に長く、従来の固体デバイスの様なゲート誘電体を用いてキャリア注入によ る MIT を起こすことは非常に困難であった。しかし近年、イオン液体をゲート誘電体とし て用いた電気二重層トランジスタ(EDLT)構造を用いて従来の FET 構造と比較して 10 倍以 上と格段に多くのキャリアを固体中に誘起(注入)することが可能となり[16]、これを利用し た様々な強相関電子系のキャリア注入による電子相制御が注目を集めている。特に実用化の 期待が高まっているのが、チャネル材料として、事項にあげる理由から最も盛んに研究が行 われている典型的な強相関酸化物の一つ、二酸化バナジウム(VO2)[1–16]である。
1.3. 強相関酸化物 VO
21.3.1. VO
2の
MIT
VO2の最大の特徴は、室温付近で構造相転移を伴ったMIT を示す点にある。この 1 次相 転移に伴うステップ様の抵抗率変化は、3 桁以上と非常に巨大で、かつ非常に急 峻しゅんである。 更に、近年、EDLT 構造を用いたゲート電圧印加により VO2のMIT が制御可能であること が報告された[18]。わずか 1 V 程度の微弱な電圧印加により、絶縁体から金属への相転移に 伴う、巨大かつ急激な抵抗変調が得られる(図1.3.1(b))。このため、超低消費電力で高速か つ急 峻しゅんなOn/Off スイッチングの実現が期待される。以上の理由から、VO2は次世代デバイ スのチャネル材料の有力候補として、そのデバイス応用研究は、国内外で精力的に行われて いる[16–22]。この VO2が示すMIT について更に詳しく説明する。図 1.3.1. (a)VO2がチャネル材料として組み込まれた EDLT(VO2-EDLT)の模式図[18].(b)VO2
-EDLT 構造における電界印加による MIT 制御.On/Off スイング(S 値)は約 100mV/decade である [18].これは,従来の MOSFET の理論下限値に匹敵する性能値である.
3d1電子配置をもつVO
2のMIT は V イオンの二量体形成(V-V 二量化)による構造相転移
とともに数桁にわたる巨大かつ急激な電気抵抗率の変化を伴う[図1.3.1(b)及び図 1.3.2(a)]
[1–16]。このため、モットトランジスタのチャネル材料[16,18]として注目され、最も盛んに 研究されている。温度がMIT 温度 TMITより高い場合、キャリア密度はV 原子密度(Natom)に
匹敵し、ハーフフィリングな金属相として振る舞う。T ~ TMITでは、数桁にも及ぶ急激な電
気抵抗及びキャリア密度変化を示す。一方T < TMITでは、キャリア密度が温度に対して指数
関数的に減少する絶縁体として振る舞う。絶縁体状態の活性化エネルギーは、アレニウスプ
ロットから約0.3 eV と推定され、これは VO2の大きなエネルギーギャプ(~0.6 eV)の約半分
図 1.3.2. (a)VO2/TiO2(001)薄膜における電気抵抗率rの温度依存性(r-T)[23],及び単斜晶系絶縁 体相とルチル型金属相VO2の結晶構造の模式図[3].MIT に伴い V イオンが cR軸に沿って二量化す る.この急激な電子相転移を利用した次世代デバイスの開発が進められている.(b)VO2バルク単結 晶における温度依存MIT に伴う価電子帯スペクトルの変化[10,12].V 3d 状態が O 2p バンドと分離し てフェルミ準位近傍に存在する. ここで、高温の金属相から低温の絶縁体相への転移に伴い、V4+サイトがc R軸方向に沿っ て二量化する[図 1.3.2(a)参照]。これにより、結晶系が正方晶系ルチル型(P42/mnm)から単 斜晶系(P21/c)へと変化する[3,4]。その結果、3d 電子はスピン・シングレット状態を形成して、 二量化したV サイトに局在する[3]。 この様なVO2におけるMIT の発現機構を解明しようとする研究は古くから行われてきた が、未だ議論が続いている[1–15]。一般に、結晶構造変化を伴った MIT は、電子-格子間相 互作用を支配的起源とするパイエルス転移型[6,7]であると考えられるが、最近の光電子分光 の結果[図 1.3.2(b)]において、ルチル型金属(RM)相から単斜晶系絶縁体相(MI)相への相転 移に伴い、フェルミ準位(EF)近傍に1 eV 程度のエネルギーギャップが形成されることが報 告されている[12]。この大きさは、TMITのエネルギースケールが高々数十ミリ電子ボルトオ ーダーであるためパイエルス転移から予想されるものより一桁以上大きいこととなり[6,7]、 強相関効果も重要だと考えられている(モット-ハバード型 MIT)。そのため、現在では、こ の素子動作の基本となるVO2の特異なMIT は、主にパイエルス転移(V-V 二量化)とモット 転移(強相関効果)とが“協調的”に作用した結果であると考えられている(図 1.3.3)[4–15]。 しかしながら、二つの相転移不安定性がVO2のMIT にどの様に関わっているのかについて O 2p V 3d
Intensity
(arb.
units)
VO2 Valence band hν = 700 eV 10Binding Energy (eV)
8 6 4 2 EFResistivity
ρ (
Ω
cm)
Temperature (K)
101 100 10−1 10−2 10−3 10−4 250 300 350Rutile
Metal
Rutile
Metal
Rutile
Metal
cR V OMonoclinic
Insulator
Monoclinic
Insulator
Monoclinic
Insulator
V-V dimer 12Monoclinic
Insulator
Monoclinic
Insulator
Monoclinic
Insulator
Rutile
Metal
Rutile
Metal
Rutile
Metal
また、この動作中のチャネル層の挙動が未解明であることが、素子開発の 妨さまたげとなってい る。
バルクVO2結晶(若しくは緩和薄膜)のTMITは室温をはるかに上回るが(~340 K)[2]、エピ
タキシャルひずみによって広い範囲にわたって制御することができる[23,24]。r-T 曲線は、
MIT の 1 次性に起因する明確な熱ヒステリシスループを描き[図 1.3.2(a)]、TMIT付近では、
RM 相と MI 相はほぼ縮重して双安定である。 図1.3.3. VO2のMIT における二つの相転移不安定性の均衡を表す概念図.
1.3.2. 電界効果による VO
2の
MIT 制御
前述のとおり、およそ10 年前、中野ら[18]により、VO2薄膜への純粋なキャリアドープの 手法として、EDLT 構造における電界効果を利用したキャリア注入による電子相制御が報告 され、注目を集めた。チャネル材料であるVO2薄膜に、ゲート絶縁層として電解質の一種で あるイオン液体(IL)を乗せ、ゲート電圧(VG)を印加することで、1015 cm-2もの膨大な量の電 荷を薄膜表面に誘起する。すると、cR軸に沿って集合的な格子変形が生じ、電子が非局在化 する。その結果、図1.3.1 に示す様に、僅かな電圧で VO2のMIT が極低温まで抑制される [18]。電界制御に伴う輸送特性の変化は、化学ドーピング[図 1.3.4(a)]によるものと類似し ていることからも、その起源は静電的なキャリア注入によるものであると考えられてきた。Peierls instability
2cR π/2cR EF Peierls gap V-V dimer-izationMott instability
Bandfilling Mott gap DOS U W U/W Mott Insu-lator EF Metal Metal 1/2 LHB UHBしかしながら、近年、VO2-EDLT の動作モデルを巡って国内外で多くの議論が噴出してい る。図1.3.4(b)に、Jeong ら[Jeong]が報告した、VO2-EDLT 構造における“ゲーティング”後の IL 除去前後でのチャネル層の輸送特性変化を示す。ここでは、電界誘起金属相が、IL 除去 後もなお長時間にわたってその特性を維持することが実証されている。彼らは、チャネル層 であるVO2薄膜における電界誘起MIT の起源が、IL を用いたゲーティング時のエッチング に伴う「酸素脱離」によるものであると主張している[19]。一方で、Ji ら[20]や渋谷ら[21]は、 この系において、ゲーティングに伴うプロトン等のイオン挿入による影響を排除できないこ とを指摘している。この様に、VO2-EDLT 構造における電界誘起 MIT の起源については、中 野らが提唱している「静電電荷蓄積(キャリア注入)モデル」以外にも、電気化学反応に起源 を求める化学反応モデル[19,20,21]が提案されていおり、未だこの現象の統一的な理解には 至っていない。この起源解明し、VO2 を用いた相転移デバイスの実現へと繋げるためには、 VO2チャネル表面(界面)への純粋なキャリア注入の実現と、その時のチャネル層領域の挙動 を明らかにすることが不可欠である。 図1.3.4. (a)化学ドーピングした VO2(V1-xWxO2)薄膜におけるr-T 特性の変化[26].W6+置換量の増
加に伴いMIT は抑制され,やがて x ≥ 0.07 で金属基底状態が出現する.(b)VO2-EDLT における“ゲ
ーティング”後のIL 除去前後でのチャネル層の輸送特性変化[19].電界誘起金属状態(赤色)は,IL の 完全除去後 1 日以上にわたって変化を示さない(緑色).このことは,電界誘起 MIT の起源が静電的 なものではない(化学反応に起源をもつ)ことを示唆している.
1.3.3. キャリア制御による VO
2の電子相変化
VO2 へのキャリア注入としては、W などの高原子価元素を用いた化学ドーピングが古く からよく行われている。一般的には、パイエルス不安定性は電子ドーピングにより減少し、 またモット絶縁体においてもハーフフィリングからのずれは絶縁体相を不安定化させるた め(モット不安定性の抑制)、金属相が安定となり、TMITは急激に低下することが予想される。 実際、W6+の化学置換による電子ドーピングにより化学ポテンシャルがシフトし、MIT が劇 的に抑制されることが報告されている(図1.3.5)。V1−xWxO2エピタキシャル薄膜(膜厚t = 30– 40 nm)の低濃度置換領域(x ≤ 0.07)においては、MIT に伴う酸素 K 端の X 線吸収分光スペク トルの変化がVO2薄膜(x = 0)と同じ傾向を示し、MI 相への転移に伴う V-V 二量化によりパ イエルスギャップが開くことが発見された[25]。しかしながら、強相関酸化物において、イ オン半径の異なる電子供与体による化学置換は、不可避的に結晶格子乱れを誘発し、その効 果は物性に大きな影響を与えてしまう可能性がある。 図1.3.5. (左)V1-xWxO2薄膜の MIT に伴う価電子帯,及び EF近傍おける硬X 線光電子スペクトル の変化[25].(右)V1-xWxO2薄膜における電子相図[26].W6+置換量の増加に伴いTMITは単調減少す る.
1.3.4. 次元性制御による VO
2の電子相変化
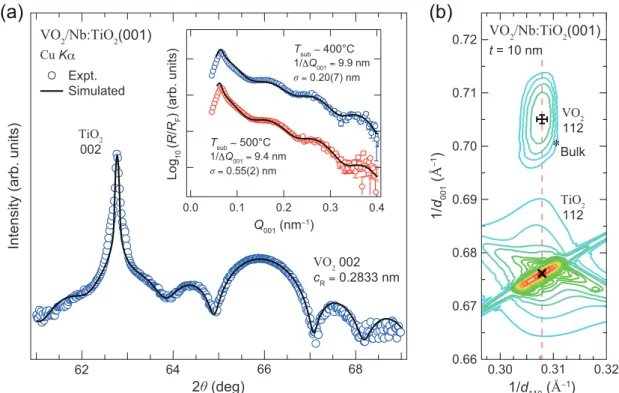
一般に、酸化物薄膜を用いたデバイスにおいては、その特性がサイズやヘテロ界面の構造 に非常に敏感であることが知られている[36–39]。強相関 FET の場合、チャネル層の設計厚 さは数ナノメートルとなるため、精密なデバイス設計に向けて、VO2ナノ構造が示す特性の 400 300 200 100 0 TMI (K) 0.10 0.08 0.06 0.04 0.02 0.00 x in V1−xWxO2 M/I Monoclinic Insulator Rutile Metalサイズ(膜厚)依存性に関する知見(VO2 はどの厚さまでその特性を保つのか)が不可欠であ る。この様な背景の下、近年、VO2ナノ構造のサイズ制御により電子相変化を誘起し、その 起源を特定しようとする研究が行われている[27–35]。 膜厚を制御した VO2極薄膜の作製とその電子特性評価は、2007 年の長嶋ら[27]の報告を 皮切りに、既に様々な研究グループで行われている[28–35]。いずれの研究においても、VO2 の極膜化によって MIT が抑制されることが報告されているが[図 1.3.5(a)]、膜厚に依存し てVO2が示す挙動については、報告毎にばらつきがある。VO2が示す電子構造変化の膜厚依 存性について、本質的な情報を得るためには、膜厚以外の実験パラメータを可能な限り固定 する必要がある。更に、VO2の場合には、MIT に密接に絡む結晶構造(V-V 二量化特性)変化 についても同時に追跡する必要がある。しかしながら、いずれの研究においても、下記に挙 げる問題点が報告されており、この系においての体系的な理解が進んでいない主な要因とな っている。 • 基板からの不均一なひずみ(膜厚に依存したエピタキシャル関係の変化)[27–29] • 大気下での表面過酸化層の形成[27–33][図 1.3.4(b,c)] • 構成カチオン種の相互拡散によるヘテロ界面の乱れ[27–35][図 1.3.4(c)]
図1.3.5. (a)t = 2.5–10 nm の VO2/TiO2(001)極薄膜におけるr-T 特性の膜厚依存性[23,29].膜厚 の減少に伴い MIT が抑制される様子が観測されている.(b)大気下で測定された t = 1 及び 7.5 nm のVO2/TiO2(001)極薄膜における V 2p 内殻準位の硬 X 線光電子スペクトル.表面過酸化層(~0.5 nm) の形成を示唆するV5+成分に由来するピーク構造が明瞭に観測されている[33].(c)VO 2/TiO2(001)薄 膜表面の走査透過電子顕微鏡(STEM)像,及び電子エネルギー損失分光(EELS)測定から見積もら れたヘテロ界面におけるカチオンの分布プロファイル[30].大気下 STEM-EELS 解析においても 1 nm 程度の表面過酸化層の形成が示唆されている.
1.4. 本研究の目的
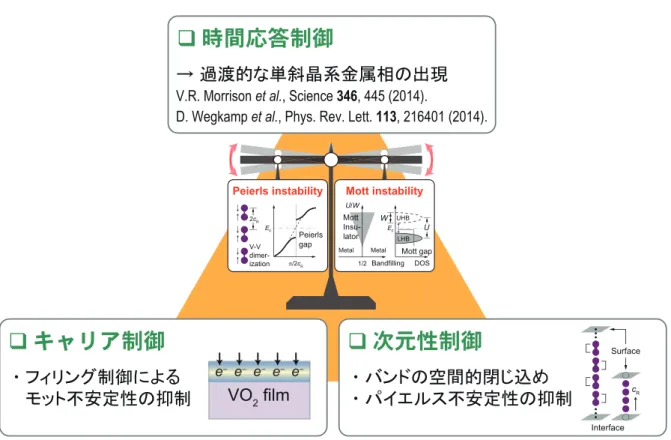
前述のとおり、VO2の急激なMIT は、主に V-V 二量化による「パイエルス転移」と強相関 効果である「モット転移」とが協調的に作用した結果であると考えられている(図1.3.32 の概 念図参照)。VO2が示す劇的なMIT のデバイス応用研究は、国内外で精力的に行われている が、デバイスを作製して輸送特性を評価するといった、従来通りの研究が主であり、デバイ ス界面における電子・結晶構造を直接観測してその動作機構を特定し、その知見に基づいた デバイス設計を行うといった研究は現在のところ、ほとんど行われていない。本研究は、そ の場放射光解析を駆使して、摂動印加時の VO2チャネル層領域における電子相変化を明ら かにするものであり、これにより、動作機構の解明を行う。このVO2のMIT の起源解明の ためには、それぞれの相転移不安定性が持つ役割を明らかにする必要がある。そこで近年、 VO2のMIT に物理的・化学的摂動を与えることによって、二つの相転移不安定性の均衡を崩 し、その時の応答を実験的に観測するといったアプローチが有効であると考えられている。 ここで、VO2のMIT に与える摂動と対応する制御パラメータを三つ挙げる(図 1.4.1)。 ■ 時間(制御パラメータ: 時間応答) 電子系の物理であるモット転移は、格子系のパイエルス転移よりも速く進行する。こ の時間スケールの差異を利用して、光照射などで誘起したMIT に伴う電子・格子構造の 「時間」変化を観測することで二つの相転移不安定性の寄与を分離する[40,41]。 ■ キャリア(制御パラメータ: バンドフィリング) 純粋なキャリアの導入によるフィリング制御によりモット不安定性及びパイエルス 不安定性を抑制する。二つの不安定性のうち、フィリング制御に敏感な方が相対的に抑 制されるため、不安定性の均衡が崩れ、協調的なMIT が抑制されると予想される。 ■ 次元性(制御パラメータ: 膜厚) VO2のサイズを制御することで次元性を変化させ、バンド幅制御(有効配位数変調)と V-V 二量化の閉じ込め、といった二つの効果を引き起こす。これにより、モット不安定 性は強化され、パイエルス不安定性は抑制されるため、相転移不安定性の均衡は崩れ、 協調的MIT は抑制されると予想される。図1.4.1. 本研究の概念図.VO2のMIT に摂動を与えることによってパイエルス不安定性とモット不安 定性の均衡を崩し、協調的モット-パイエルス転移を抑制する.本研究では、キャリアと次元性を制御す る. 本研究では、VO2の MIT に上記の摂動を与えることによって相転移不安定性の均衡を崩 し、そのときの電子・結晶構造変化をその場放射光電子分光によって調べることで、MIT に おける各相転移不安定性の役割を解明することを目的とした。図1.4.1 に示す三つの研究ア プローチのうち、「時間」を摂動パラメータとするアプローチは既に様々な研究[40,41]で行わ れている。そこで本研究では、キャリア、次元性といったパラメータを制御し、そのときの VO2における電子構造の変化を調べることにした。 また、本研究では、その場で「試料表面修飾によるキャリア注入」と「放射光電子分光解析 (光電子分光・X 線吸収分光)」とを組み合わせた研究アプローチを採用することで、摂動印 加時の VO2チャネル層で発現する MIT に伴う電子・結晶構造変化を同時に特定する。これ により、これまで未知であったチャネル層領域(界面数ナノメートル)における電子相変化を 直接観測することで、その全容を明らかにする。 Peierls instability 2cR π/2cR EF Peierls gap V-V dimer-ization Mott instability Bandfilling Mott gap DOS U W U/W Mott Insu-lator EF Metal Metal 1/2 LHB UHB
次元性制御
Surface Interface cRキャリア制御
VO2 film e− e− e− e− e−時間応答制御
V.R. Morrison et al., Science 346, 445 (2014).
本章では、研究背景として、強相関酸化物が示すMIT を外場印加により制御するモット・ トランジスタ、及びそのチャネル層物質としての実用化が期待されている VO2の基礎物性 について概説した。本研究では、キャリア・次元性といった摂動を印加した時のVO2ナノ構 造における特性変化を電子分光学的に明らかにし、特異な MIT が内包する二つの相転移不 安定性の役割を解明することを目的とする。次章では、本研究アプローチの鍵である、表面 汚染などの外的要因を排除し本質的な情報を得るための実験手法に関して、レーザー分子線 エピタキシ法によるよく定義された表面・界面をもつ良質な酸化物エピタキシャル極薄膜試 料の合成法と薄膜評価手法の原理、及び試料合成から試料の表面修飾や放射光電子分光解析 に至るまでを大気非暴露系で行う「その場放射光実験」の特徴、についてのべる。第 3 章で は、種々の評価結果に基づき、本放射光電子分光解析を行う上で必須となる高い表面清浄 性・結晶性・平坦たん性、並びにヘテロ界面急 峻しゅん性をもつ良質なVO2エピタキシャル極薄膜試料 の作製条件最適化について示す。第 4 章では、静電キャリア注入時の VO2のチャネル層領 域(表面数ナノメートル)における電子相変化に関する知見を得るために、VO2薄膜表面への アルカリ金属原子蒸着による電子注入を試み、VO2-EDLT 構造と同様のキャリア誘起 MIT を 実現した内容についてのべる。さらに、この表面電荷蓄積時の VO2 における電子構造変化 及び結晶構造変化(V-V 二量化)を放射光電子分光により一括観測した結果について示す。第 5 章では、次元性制御による VO2のチャネル層の電子相変化に関する知見を得るために、原 子レベルでサイズを制御したVO2極薄膜における電子・結晶構造の膜厚依存性をその場放射 光電子分光により評価した結果について示す。最終章では、本研究の総括として、キャリア (第4 章)、次元性(第 5 章)といった摂動印加時に VO2チャネル層領域において発現するこ とが明らかとなった2 種の「新たな電子相」について、VO2のMIT が内包する二つの相転移 不安定性の観点から、それらの発現機構についてのべる。更に、本研究で得られた知見に基 づいた最適な相転移デバイス構造の設計を提案し、次世代のモット・トランジスタの実現の 可能性を示す。
参考文献
[1] M. Imada, A. Fujimori, and Y. Tokura, Rev. Mod. Phys. 70, 1039 (1998). [2] F. J. Morin, Phys. Rev. Lett. 3, 34 (1959).
[3] J. B. Goodenough, J. Solid State Chem. 3, 490 (1971). [4] N. F. Mott, Rev. Mod. Phys. 40, 677 (1968).
[7] R. M. Wentzcovitch, W. W. Schulz, and P. B. Allen, Phys. Rev. Lett. 72, 3389 (1994). [8] A. Zylbersztejn and N. F. Mott, Phys. Rev. B 11, 4383 (1975).
[9] T. M. Rice, H. Launois, and J. P. Pouget, Phys. Rev. Lett. 73, 3042 (1994).
[10] S. Biermann, A. Poteryaev, A. I. Lichtenstein, and A. Georges, Phys. Rev. Lett. 94, 026404 (2005). [11] M. W. Haverkort, Z. Hu, A. Tanaka, W. Reichelt, S. V. Streltsov, M. A. Korotin, V. I. Anisimov, H.
H. Hsieh, H.-J. Lin, C. T. Chen, D. I. Khomskii, and L. H. Tjeng, Phys. Rev. Lett. 95, 196404 (2005). [12] T. C. Koethe, Z. Hu, M. W. Haverkort, C. Schüßler-Langeheine, F. Venturini, N. B. Brookes, O. Tjernberg, W. Reichelt, H. H. Hsieh, H.-J. Lin, C. T. Chen, and L. H. Tjeng, Phys. Rev. Lett. 97, 116402 (2006).
[13] M. M. Qazilbash, M. Brehm, B.-G. Chea, P.-C. Ho, G. O. Andreev, B.-J. Kim, S. J. Yun, A. V. Balatsky, M. B. Maple, F. Keilmann, H.-T. Kim, and D. N. Basov, Science 318, 1750 (2007). [14] J. M. Tomczak, F. Aryasetiawan, and S. Biermann, Phys. Rev. B 78, 115103 (2008).
[15] M. M. Qazilbash, A. Tripathi, A. A. Schafgans, B.-J. Kim, H.-T. Kim, Z. Cai, M. V. Holt, J. M. Maser, F. Keilmann, O. G. Shpyrko, and D. N. Basov, Phys. Rev. B 83, 165108 (2011).
[16] C. H. Ahn, A. Bhattacharya, M. Di Ventra, J. N. Eckstein, C. D. Frisbie, M. E. Gershenson, A. M. Goldman, I. H. Inoue, J. Mannhart, A. J. Millis, A. F. Morpurgo, D. Natelson, and J.-M. Triscone, Rev. Mod. Phys. 78, 1185 (2006).
[17] H. Takagi and H. Y. Hwang, Science 327, 1601 (2010).
[18] M. Nakano, K. Shibuya, D. Okuyama, T. Hatano, S. Ono, M. Kawasaki, Y. Iwassa, and Y. Tokura, Nature (London) 487, 459 (2012).
[19] J. Jeong, N. Aetukuri, T. Graf, T. D. Schladt, M. G. Samant, and S. S. P. Parkin, Science 339, 1402 (2013).
[20] H. Ji, J. Wei, and D. Natelson, Nano Lett. 12, 2988 (2012). [21] K. Shibuya and A. Sawa, Adv. Electron. Mater. 2, 1500131 (2016). [22] T. Yajima, T. Nishimura, and A. Toriumi, Nat. Commun. 6, 10104 (2015). [23] Y. Muraoka and Z. Hiroi, Appl. Phys. Lett. 80, 583 (2002).
[24] N. B. Aetukuri, A. X. Gray, M. Drouard, M. Cossale, L. Gao, A. H. Reid, R. Kukreja, H. Ohldag, C. A. Jenkins, E. Arenholz, K. P. Roche, H. A. Dürr, M. G. Samant, and S. S. P. Parkin, Nat. Phys. 9, 661 (2013).
[25] E. Sakai, K. Yoshimatsu, K. Shibuya, H. Kumigashira, E. Ikenaga, M. Kawasaki, Y. Tokura, and M. Oshima, Phys. Rev. B 84, 195132 (2011).
[26] K. Shibuya, M. Kawasaki, and Y. Tokura, Appl. Phys. Lett. 96, 022102 (2010).
[27] K. Nagashima, T. Yanagida, H. Tanaka, and T. Kawai, J. Appl. Phys. 101, 026103 (2007).
[28] K. Martens, N. Aetukuri, J. Jeong, M. G. Samant, and S. S. P. Parkin, Appl. Phys. Lett. 104, 081918 (2014).
[29] B. Zhi, G. Gao, X. Tan, P. Chen, L. Wang, S. Jin, and W. Wu, Mater. Res. Express 1, 046402 (2014). [30] N. F. Quackenbush, J. W. Tashman, J. A. Mundy, S. Sallis, H. Paik, R. Misra, J. A. Moyer, J.-H. Guo, D. A. Fischer, J. C. Woicik, D. A. Muller, D. G. Schlom, and L. F. J. Piper, Nano. Lett. 13, 4857 (2013). [31] J. W. Tashman, J. H. Lee, H. Paik, J. A. Moyer, R. Misra, J. A. Mundy, T. Spila, T. A. Merz, J. Schubert,
D. A. Muller, P. Schiffer, and D. G. Schlom, Appl. Phys. Lett. 104, 063104 (2014).
[32] H. Paik, J. A. Moyer, T. Spila, J. W. Tashman, J. A. Mundy, E. Freeman, N. Shukla, J. M. Lapano, R. Engel-Herbert, W. Zander, J. Schubert, D. A. Muller, S. Datta, P. Schiffer, and D. G. Schlom, Appl. Phys. Lett. 107, 163101 (2015).
[33] N. F. Quackenbush, H. Paik, J. C. Woicik, D. A. Arena, D. G. Schlom, and L. F. J. Piper, Materials 8, 5452 (2015).
[34] Y. Muraoka, K. Saeki, R. Eguchi, T. Wakita, M. Hirai, T. Yokoya, and S. Shin, J. Appl. Phys. 109, 043702 (2011).
[35] K. Shibuya, M. Kawasaki, and Y. Tokura, Phys. Rev. B 82, 205118 (2010).
[36] K. Yoshimatsu, K. Horiba, H. Kumigashira, T. Yoshida, A. Fujimori, and M. Oshima, Science 333, 319 (2011).
[37] M. Kobayashi, K. Yoshimatsu, E. Sakai, M. Kitamura, K. Horiba, A. Fujimori, and H. Kumigashira, Phys. Rev. Lett. 115, 076801 (2015).
[38] M. Kobayashi, K. Yoshimatsu, T. Mitsuhashi, M. Kitamura, E. Sakai, R. Yukawa, M. Minohara, A. Fujimori, K. Horiba, and H. Kumigashira, Sci. Rep. 7, 16621 (2017).
[39] J. K. Kawasaki, C. H. Kim, J. N. Nelson, S. Crisp, C. J. Zollner, E. Biegenwald, J. T. Heron, C. J. Fennie, D. G. Schlom, and K. M. Shen, Phys. Rev. Lett. 121, 176802 (2018).
[40] V. R. Morrison, R. P. Chatelain, K. L. Tiwari, A. Hendaoui, A. Bruhács, M. Chaker, and B. J. Siwick, Science 346, 445 (2014).
[41] D. Wegkamp, M. Herzog, L. Xian, M. Gatti, P. Cudazzo, C. L. McGahan, R. E. Marvel, R. F. Haglund, Jr., A. Rubio, M. Wolf, and J. Stähler, Phys. Rev. Lett. 113, 216401 (2014).
第
2 章
実験手法
本章では、本研究において試料の電子・結晶構造の観測に用いたその場放射光電子分光、 試料作製法、及び試料評価法について説明する。まず電子分光法の原理について簡単に説明 する。次に、その場放射光電子分光の実際の装置ついて、装置全体の概略をのべた後、個々 の実験装置及び酸化物薄膜の作製法、試料評価法についてのべる。2.1. 放射光電子分光
2.1.1. 光電子分光(PES)
光電子分光(PES)は、物質に単色光を入射し、外部光電効果により放出される光電子の運 動エネルギー分布(光電子スペクトル)を測定することにより、物質の電子状態を研究する方 法である。プローブに X 線が用いられるとき、この分光法は X 線光電子分光(XPS)と呼ば れる。試料は気体、液体、固体の別を問わない。PES には様々な種類があるが、以下では本 研究で用いた角度積分PES の原理及び測定装置を中心にのべる。 図 2.1.1 に PES の基本原理を示す。励起光のエネルギーを hn、放出された光電子の運動 エネルギーを EK、その結合(束縛)エネルギーを EB、そして仕事関数をf としたときに、そ れらの間にはエネルギー保存則、 (2.1)とができる。 各軌道原子の結合エネルギーは元素ごとに異なるため、EBを測定することにより元素の 同定が可能である。また、同一元素の同一軌道のEB値は、注目している原子の周りの状態・ 環境により微妙に変化する。この変化量(化学シフトと呼ばれる)を測定することにより、元 素の電子状態分析が可能となる。 PES では、励起光によって放出された光電子をどの様に検出するかによって二つのモード に分かれる。放出された光電子を全立体角で検出するのが角度積分型、微少立体角中に放出 された光電子を検出するのが角度分解型の PES である。前者がブリュアンゾーンの全ての 点にわたって積分された電子状態、つまり状態密度を与えるのに対して、後者はブリュアン ゾーン中の特定点における電子状態、すなわち波数とエネルギーの関係(分散関係)を与え る。 図 2.1.1. PES の概略図.入射エネルギーhn により束縛エネルギーEBの電子が励起され,運動エネ ルギーEKの光電子が放出される様子を示している.また,光電子アナライザにおいて観測される光電 子スペクトルは運動エネルギーが低いほど二次電子の影響が大きくなることも示されている.
2.1.2. 放射光
電子分光測定を行う際の光源としては、軟X 線(SX)発生装置や真空紫外光線(VUV)等が 用いられてきた。しかしながら、それらの単色光源では特定のエネルギーの光しか利用する ことができない。また、光の強度が弱いため、測定に非常に時間がかかる。更に、光の強度 が強い光源を用いても分解能が低い場合がある。これらの問題を解決する一つの方法として 放射光の利用がある。光速近くまで加速された高エネルギーの荷電粒子が磁場によってその 軌道を曲げられると接線方向に電磁波が放出される。この現象をシンクロトロン放射光、又 は単に放射光という。放射光は、次の様な優れた特長をもつ[1]。 (1) 赤外線からg線まで全てを含む白色の連続光であり、分光器を用いて任意の波長の光 を取り出すことができる(光エネルギー可変)。 (2) 実験室光源と比較して、格段に高い強度・指向性をもつ。 (3) 高い偏光度をもち、また、荷電粒子の曲げ方によりその偏光特性を制御することが できる。 以上の特徴をもつ優れた光源である放射光を利用することによって、(1)「高分解能」かつ「高 効率」といった相矛盾する要求を同時に満たした測定、(2)X 線吸収分光などの他の手法と組 み合わせることで一度に電子・化学状態及び結晶構造に関する包括的な情報を得る測定、(3) 内殻励起を利用して元素選択的な情報を得る測定、(4)光励起断面積の光エネルギー依存性・ 偏光依存性を利用することで各元素の磁気や軌道状態の情報を分離して得る測定、などが可 能となる。 特に、放射光電子分光のもつ「元素選択性」は、異種物質から構成されるデバイス構造の 「埋もれた(ヘテロ)界面」の電子・化学状態及び結晶構造の分析・評価に有益である。加えて、 (1)非破壊分析である、(2)定量分析が可能、といった特徴をもつ光電子分光法は、もはや機 能性薄膜・ヘテロ構造の表面・界面のために不可欠な分析・評価手法であるといえる。 放射光を発生させる光源としては、リング型加速器において電子を周回させるために電子 軌道を曲げる偏向電磁石光源と、加速器直線部において放射光を発生させるために追加の磁 石列を配置する挿入光源が挙げられる。挿入光源には、電子軌道を大きく蛇行させることに より短波長で高輝度(高い光の強度、サブナノメールの微小な分析径)の放射光を発生させる ウィグラーと、電子軌道を周期的に複数回蛇行させることにより、蛇行部で発生する放射光 を干渉させ、特定の波長で極めて高輝度の放射光を発生させるアンジュレータの 2 種類が磁場の強さを制御することにより、放射光のピーク波長を変化させることも可能である。
本研究では、実験は一括して、つくば市にある高エネルギー加速器研究機構(KEK)の放射
光実験施設 Photon Factory(PF)の表面・界面解析アンジュレータビームライン BL-2A
MUSASHI[2–4]を使用した。このビームラインでは、ピーク波長制御域の異なる 2 台のアン ジュレータを併用することにより、非常に広いエネルギー範囲で極めて高輝度の放射光を利 用することが可能となっている。
2.1.3. X 線吸収分光(XAS)
物質に X 線を照射し徐々に光のエネルギーを変化させたとき、内殻電子が非占有状態へ 励起できる様なエネルギーで光吸収量が増加する。この吸収量を光子のエネルギーの関数と して測定することで非占有状態の情報を得る手法を X 線吸収分光法(XAS)という。エネル ギー可変で高輝度の光が必要となるため、前にのべたシンクロトロン放射光がよく使用され る。酸素K 端の XAS(O K XAS)スペクトルは O 1s – O 2p 遷移に伴う吸収を反映しており、 O 2p 軌道の非占有部分状態密度の情報が得られる。V 酸化物の場合では V と O が強く混成 しているため、V 3d(強相関電子)由来の情報も得られる[5]。 XAS の測定には、透過した X 線の吸収量を測定する透過法が直接スペクトルを得ること ができるという意味では理想的であるが、硬 X 線と異なり SX ではエネルギーが低いため 透過率が非常に小さく測定が困難である。したがって、今回の実験では全電子収量(TEY)法 を用いた。TEY 法はオージェ電子の緩和によるカスケード過程でつくられた二次電子を含 む全ての脱出電子を補償電流によって観測する手法である。この全ての脱出電子の数は光電 子の数に比例することが分かっている[5]。TEY 法を用いると、得られるシグナルが透過法 より格段に大きくなり、容易に測定できるのでSX を光源とする XAS でよく用いられる手 法である。2.1.4. 軟 X 線分光の表面敏感性
PES 測定において留意するべき点は、試料から出てくる光電子は試料内部の深い位置から は散乱を受けずに脱出することができないため、表面敏感な実験手法であるということであ る。図 2.1.2 に光電子の運動エネルギーに対する非弾性平均自由行程lを示す。また、図に は様々な実験室光源を用いた励起光を示してある。薄膜1 分子層あたりの厚さを d、膜厚を n ML としたとき、nd の深さから出てくる光電子 を検出することにより得られるスペクトルの強度 I は e-nd/l に比例する。l の値は入射する 光のエネルギーによって変化する。本研究で用いる放射光のエネルギー範囲(400–1200 eV 程 度)においては、光のエネルギーを上げることでl の値が大きくなる(よりバルク敏感にな る)。すなわち、光のエネルギーを変えてPES 測定を行うことにより、表面敏感性を変えた 測定を行うことができる。エネルギーの小さな光で測定を行えば、試料表面の電子状態の寄 与が大きいスペクトルが得られ、光のエネルギーを上げて測定すれば表面の寄与は小さくな る。そのため、二つ以上の異なる光のエネルギーで測定すれば表面とバルクのスペクトルを 分離することも可能である。実際には、表面敏感性のみからではなく、輝度の違いによる分 解能やチャージアップの度合いなどとの兼ね合いで測定エネルギーを決めることになる。 図 2.1.2. 光電子の EKに対する平均自由行程.縦軸が光電子の平均自由行程,横軸が電子の運動 エネルギーに対応する.
2.2. その場放射光 PES – レーザー分子線エピタキシ(MBE)複合装置
PES 測定は表面敏感な手法であるため、試料の表面に酸化膜あるいは炭化水素等の付着に よる汚染などが存在すると、物質本来の電子状態を反映したスペクトルが得られない。特に 本研究の様に、価電子帯の詳細な電子状態を測定するにあたっては、試料の清浄な表面状態 が必要不可欠である。したがってバルク物質においては、超高真空(UHV)下でのへき開やヤ スリがけを用いて清浄な表面を出し、その後に PES 測定を行うといった手法がとられてき た。しかしながら、超薄膜試料の測定を行う場合には、へき開ややすりがけといった方法を 用いることはまず不可能である。そこで本研究では、実験は一括して、作製した薄膜試料の 清浄表面を保ったままで放射光電子分光測定を行う手法として、UHV の下で作製された試 料を大気に曝さらすことなく電子分光測定装置に運び測定する「その場放射光電子分光」を行うため、PF BL-2A MUSASHI において建設・改良を行った「その場放射光 PES – レーザー分子 線エピタキシ(MBE)複合装置」(図 2.2.1)を用いて実験を行った。 その場放射光PES 装置は主に、放射光ビームラインと接続され、アナライザを備えた「PES 測定チャンバ」、真空搬送の中継をするとともに低速電子線回折測定やアルカリ金属の蒸着 等を行う「試料準備チャンバ」、測定試料や製膜用基板を大気から導入する「試料導入チャン バ」、及び酸化物薄膜試料の作製を行う「レーザーMBE 装置」から構成され、全てのチャンバ がUHV の下で接続されている。これにより、VO2が示す電子特性に関して本質的な情報を 得るために、レーザーMBE 法を用いた酸化物薄膜試料の作製から PES 測定チャンバにおけ る放射光電子分光解析に至までの一連の実験は、UHV チャンバ間で試料を搬送することで、 試料表面を一度も大気に曝さらすことなく、清浄表面を保ったまま電子分光測定を行うことが可 能である。まず、電子分光装置の各部について詳しく説明する。 図2.2.1. その場 PES – レーザーMBE 複合装置の模式図[2–4]. M U S A S H I SR
光電子分光
X線吸収分光
エピタキシャル薄膜作製
: MBEアルカリ金属原子蒸着
: RHEED LEED アナライザ2.3. 放射光電子分光装置
電子分光測定チャンバは、高分解能化、極低温化、UHV 化のために、様々な工夫がなさ れている。まず、PES 測定の高分解能化のためには測定チャンバ内に入り込む地磁気を軽減 する必要があり、真空チャンバ内部を1 層のミューメタルシールドで覆っている。また、こ のシールドは光電子分析器のミューメタルシールドとも磁気的にしっかり接合させること で、漏えい磁場の影響を排除している。茨城県では470 mG 程度の地磁気があるが、このミ ューメタルシールドによりチャンバ内の残留磁場は数ミリガウス程度に抑えられる。 本研究では、試料の温度・角度・偏光依存測定を行うため、測定試料の温度及び角度を制御 する必要がある。本装置では、アールデック社製の5 軸制御型マニピュレータ i-GONIO を 採用し、液体He フロー冷却クライオスタットを用いて試料温度を 11 K まで冷却しながら、 試料の角度を 2 軸方向に制御することが可能である。試料の角度を鉛直方向及び水平方向 に任意に制御し、ポインティングベクトル周りで回転させることで、偏光ベクトルを連続的 に変化させたり、プローブ長を制御したりすることが可能となる。 UHV を実現する真空排気は、機械的排気を行うターボ分子ポンプ(TMP)を主に用いてい る。主な真空チャンバである真空排気チャンバ、試料準備チャンバ、電子分析アナライザに おいては、全てのTMP を 2 段の直列排気にしている。更に、真空排気チャンバには、大型 のイオンポンプとクライオポンプ、チタンサブリメーションポンプ、非蒸発型ゲッターポン プを併せて用いることによって、到達圧力として1 × 10-8 Pa(~8 × 10-11 Torr)を実現している。 イオンポンプは真空排気チャンバに最下部に取付けて測定位置からの距離を十分にとって ある。静電半球アナライザにも同様の直列に接続したTMP を取付け、電子エネルギー分析 機器内でも、10-8 Pa 台の UHV を実現している。マニピュレータのローターリーフィードス ルーは差動排気型のものを用いており、小型のTMP で排気している。これらの工夫により、 測定時においても常に10-8 Pa 台の圧力が実現されている。 次に試料準備チャンバ及び試料導入チャンバについてのべる。試料を測定チャンバに搬入 する度に、電子分析チャンバなどの真空チャンバを大気に曝さらしては、到達圧力が下がらない ばかりか、ゴミなどの混入でマイクロチャンネルプレート(MCP)などの繊細な部分が傷つ く可能性がある。更に、過度なベーキングは各電極の静電場にとっても良いことは無く、火 災事故の危険性も上がる。したがって、PES 装置内部は常に UHV に保っておくべきである。 そのため、測定チャンバ内の真空を損なうことなく、大気から試料を移送するための試料導 入チャンバ及び移送機構が設けられている。導入チャンバと準備チャンバの間はバルブで仕バのみを大気リークして試料ストックに入れる。本装置では、1 度に 5 個の試料まで真空中 に入れることができる。この試料台の後端にはM4 ネジがついており、搬送の際は、ロッド の先端のネジ穴に試料台をねじ込んで行う。導入チャンバの真空排気を始めてから 2 時間 程度で試料準備チャンバに搬送可能となる。 試料準備チャンバは10-8 Pa 台の UHV を保っており、試料導入チャンバから搬送された 試料は速やかにレーザーMBE 装置や PES 測定チャンバへと更に搬送することが可能であ る。また、試料準備チャンバには低速電子線回折装置のほかに、試料加熱機構、Ar スパッ タ、アルカリ金属蒸着源、水素クラッカーセル、膜厚計等が取付けられており、試料の表面 清浄化や表面蒸着等の様々な測定前準備を行うことも可能である。
2.3.1. 光電子分析器
光電子放出された電子の運動エネルギーを測定する装置が静電半球型電子分析アナライ ザと電子レンズであり、本装置の心臓部でもある。本装置では、VG Scienta 社の高分解能 PES アナライザSES2002 を用いている(図 2.3.1)。電子分析器システムは真空中に配置された幾 つかの電極によって構成されている。この電極に直流電圧を印加し、静電場を作ることで、 真空中を運動する電子の軌道を制御して光電子の運動エネルギーを分析する。真空中に放出 された光電子は、電子レンズによって集められアナライザの入射スリット上に集光される。 集光された電子はアナライザを通過することでエネルギーを振り分けられ検出器に到達す る。 高いエネルギー分解能を実現するためには、この静電場をいかに高精度で制御できるかが 決定的に重要である。このため、各々の電極の電位を独立した電圧印加モジュールにより制 御することで電極間の干渉を抑えている。また、各電極の内部表面はグラファイトでコーテ ィングすることで電場の均一性を保っている。更に、電極間とモジュールの全てのケーブル にシールドを施し、電磁波吸収などによるノイズを防いでいる。 電源のふらつき等による分解能の低下も考慮し、電圧系配線の整備にも注意が払われてい る。更に、固体測定の際は、試料と装置全体のアースが問題になるが、太い銅製の線をマニ ピュレータと試料チャンバに接触良く付けることにより、アースが強化されている。 静電半球型アナライザのエネルギー分解能は、半球を通過する際の電子のエネルギー(パ スエネルギー)、アナライザのスリット幅、二重半球の平均半径をそれぞれ、DE (eV)、EP (eV)、w (mm)、R (mm)とすると、近似的に (2.2) で表される。したがって原理的には R を大きくすれば幾らでも高い分解能が得られるが、 余りにも大きいと半球の加工精度が問題となる。現時点ではR = 200 mm までが経験的に最 適な大きさとして知られており、本装置ではこれを用いている。また、スリット幅を狭くす る、あるいはパスエネルギーを小さくしても高分解能が得られるが、検出される光電子強度 は減少する。実験室光源に比べて格段に高い輝度をもつ放射光を励起光とすることで、これ が補われている。 従来型のチャンネルトロンを用いた電子分析器は半球の出射面の 1 点において電子を検 出していたのに対し、本装置は同じ出射面において通過した電子の 2 次元(2D)分布を検出 できることが大きな特徴である。検出面において、電子軌道の直径方向(スリットの幅の方 向)がエネルギー分布、それと直交する方向が入射スリット上の電子分布に対応する。入射 スリット上での電子分布は電子レンズの集光モードにより分類され、光電子放出時の「空間 分布」に対応するモードと「角度分布」に対応するモードがある。特に、「角度分布」モードは 一度に±7°の角度範囲で電子を取り込むことができ、これにより高効率での角度分解測定が 可能である。 2D 検出のために、アナライザの終端には MCP と蛍光板がついている。MCP 間には 1400– 1700 V の高電圧がかけられており、MCP に到達した電子は 106倍に増幅される。MCP と蛍 光板の間にも3600 V の高電圧が印加され、MCP で増幅された電子はこの電圧で更に加速し て蛍光板に衝突し蛍光板を局地的に発光させる。この様にして入射スリット上の電子分布の イメージが、蛍光板上の光の分布に変換される。特に、MCP と蛍光板はこの装置の中で最 も繊細な部分でありゴミや水に非常に弱く、常に高真空に保っておく必要がある。この光は ビューポートを通過し、真空の外に取付けてあるCCD カメラにより検出され、それを電気 信号に変換して測定系に送る。したがって、いかに電子の2D 像を精密に取り込むかが、高 エネルギー分解能、高角度分解能にとって重要となる。そのためCCD カメラの焦点を蛍光 板上に合わせる調整においては細心の注意を要する。
図2.3.1. VG Scienta 社製の SES 2002 アナライザの外観.
2.3.2. ビームライン光学系
図2.3.2 に、本研究で使用した PF BL-2A MUSASHI の光学系配置図を示す。このビーム ラインでは、VUV(hn = 30–300 eV)用と SX(hn = 250–2000 eV)用の 2 台のアンジュレータを タンデム配置し、利用する放射光のエネルギー領域に応じてそれぞれのアンジュレータ及び 回折格子を切り替えて使用する。アンジュレータからの放射光は、平面鏡M2 と不等間隔刻 線平面回折格子により特定の波長の光が出射スリット上に集光され、出射スリットを通過し た後に平面鏡 M4 により再び集光され、試料上へ照射される。本ビームラインは可変偏角 Monk-Gillieson 型分光器を採用しており、エネルギーに応じて M2 により最適偏角を調整す ることで、回折効率やエネルギー分解能を損なうことなく広い範囲で使用エネルギーを変化 させることが可能である。 図2.3.2. PF BL-2A MUSASHI の光学系配置図.
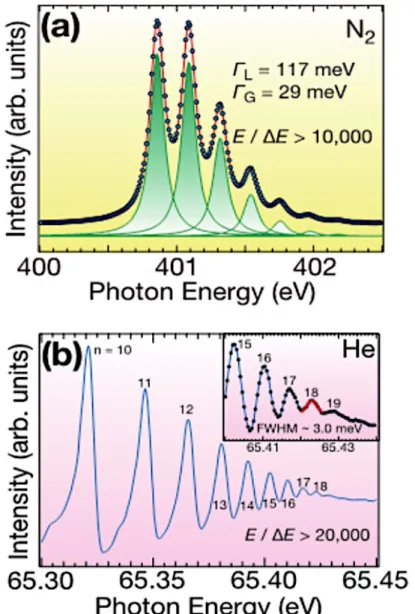
図2.3.3 に本ビームラインで得られた、ガスの吸収スペクトルの例を示す。図 2.3.3(a)の N2ガスの吸収スペクトル(~400 eV)においては、自然幅のローレンツ関数とビームラインエ ネルギー分解能のガウス関数との畳み込みの関数でフィッティングを行った結果、エネルギ ー分解能 29 meV という値が得られている。また、図 2.3.3(b)の He ガスの吸収スペクトル (~65 eV)においては、自然幅が無視できるほど小さい(~1 meV)ため、ピークの FWHM が直 接エネルギー分解能に対応すると考えられる。このEWHM から、エネルギー分解能 3 meV という値が得られている。この様に、VUV、SX どちらのエネルギー領域においても、非常 に高いエネルギー分解能が達成できていることが分かる。 図2.3.3. N2ガス及びHe ガスの吸収スペクトル.ピークの FWHM から,(a)SX 領域,(b)VUV 領域 における光のエネルギー分解能をそれぞれ見積もることができる.
2.4. 酸化物薄膜の作製
本研究では、表面・界面を原子レベルで制御した酸化物エピタキシャル薄膜試料を作製す る必要がある。表面の平坦たん性・結晶性、及びヘテロ界面の急 峻しゅん性に優れた高品質な薄膜試料 を必要とする。当研究室では、これらの条件を満たす酸化物薄膜の作製技術を開発・発展さ せてきた。以下に、酸化物薄膜の作製技術と実験装置について説明する。2.4.1. パルスレーザー堆積法
本研究では、酸化物薄膜試料はパルスレーザー堆積(PLD)法を用いて作製している。PLD 法は、原料としたターゲットの組成をほぼそのままに堆積できる手法であることから、半導 体薄膜、有機薄膜、そして本研究で取扱う酸化物薄膜の作製に広く用いられる手法である。 この手法は、真空チャンバ内に設置した原料ターゲット(高密度焼結体ペレット)に試料作製 チャンバの外部からレーザー光を照射することで物質の化学結合を分断し、分解気化による 引き剥がしを行い、ターゲットに対向する基板に薄膜を堆積させることができると考えられ ている[6]。 ほかの代表的な薄膜作製手法であるMBE 法や有機金属気相成長法に比べると、PLD 法で は以下の長所が挙げられる。 l 高融点材料を蒸発させるための十分なエネルギーが得られるため、多様な固体材料の 薄膜を作製できる。 l 蒸発させるためのエネルギー源(レーザー)が薄膜作製チャンバの外にあり、フィラメ ントなどの原料加熱部を内部にもたないため、酸素分圧に依存しない薄膜堆積が可能 である。 l 成長速度がレーザーのエネルギー密度([エネルギー強度]/[照射面積])やパルスレーザ ーの照射周期を変えることで制御できる。 l 非平衡な薄膜成長過程であるため、熱力学的平衡条件に限定されない薄膜成長ができ る。 l 高真空中で後にのべる反射高速電子線回折装置と組合せることで、その場観察による 膜厚制御が可能である。この様な反射高速電子線回折観察とPLD 法を組合せた薄膜成 長法は特にレーザーMBE 法と呼ばれている[6]。 短所としては、数平方ミリメートル程度の面積でしか均一な薄膜が得られないという点が 挙げられる。ただし、本研究で行った電子分光測定における入射光のスポット径は500 × 100µm2程度であり、上記の面積は実験を行う上では十分に広く、本研究遂行上の問題はない。 PLD 法における主な薄膜作製パラメータは、基板温度、酸素分圧、レーザーのエネルギー 密度や照射面積などのレーザーパラメータである。ここで、基板温度は成長時の熱力学的条 件や吸着原子の基板表面における拡散に対して支配的な要素である。また、酸素分圧は熱力 学的条件のみならず、雰囲気酸素とアブレーションされた前駆体との間の相互作用によって 決まる吸着原子の動力学的条件に影響する。レーザーパラメータは、吸着原子の運動エネル ギーや、前駆体の化学量論組成を変化させる。 この様に、PLD 法は多くのパラメータをもつことから、非常に複雑な薄膜作製手法であ る。しかしながら裏を返すと、多くの自由度を巧みに駆使することで、熱力学的平衡状態の 観点からバルク体では実現しにくい化学量論組成や、不安定な価数を有する薄膜試料の合成 を可能にする手法である。
2.4.2. レーザーMBE 装置
レーザーMBE 装置の模式図を図 2.4.1 に示す。また、以下に主要機器とサンプルホルダ について解説する。 ■ ターゲットアブレーション用パルスレーザー Nd:YAG レーザー(中心周波数l = 1064 nm)の 3 倍波(l = 355 nm)を使用している。エネル ギー及びパルス間隔をプログラムにより制御する。 ■ 基板加熱用連続発振(CW)レーザー 半導体ダイオードレーザー(l = 808 nm)を用いており、放射温度計で計測した基板温度を フィードバックすることで温度のプログラム制御を可能としている。およそ500–1450 K の 範囲で基板を加熱できる。 ■ 高純度酸素ガスライン バリアブルリークバルブによりチャンバ内の酸素分圧を10-6 Pa から~100 kPa の範囲で調 整できる。 ■ 反射高速電子線回折測定用の電子銃及びスクリーンで薄膜の成長速度を観察することもできる。 ■ サンプルホルダ 本装置用のサンプルホルダはインコネル®製で、レーザーMBE 装置には裏返した状態でセ ットする。図2.4.2 にその断面図を示す。加熱用レーザーはサンプルホルダ中央に開いた穴 を通して基板裏側のNi ブロック(表面を酸化して濃緑色の NiO にしてある)を加熱する。基 板は金属製のクランプとネジにより Pt ペーストを介して Ni ブロックに押さえつけられて おり、Ni ブロックからの熱伝導により加熱される。 図2.4.1. レーザーMBE 装置の模式図. 図2.4.2. サンプルホルダの断面図.
![図 1.3.1. (a)VO 2 がチャネル材料として組み込まれた EDLT(VO 2 -EDLT)の模式図[18].(b)VO 2 - -EDLT 構造における電界印加による MIT 制御.On/Off スイング(S 値)は約 100mV/decade である](https://thumb-ap.123doks.com/thumbv2/123deta/5888697.1047695/13.892.122.748.451.708/チャネル材料として組み込まEDLT模式EDLT構造おけるによるスイングS.webp)
![図 1.3.2. (a)VO 2 /TiO 2 (001)薄膜における電気抵抗率 r の温度依存性( r -T)[23],及び単斜晶系絶縁 体相とルチル型金属相 VO 2 の結晶構造の模式図[3].MIT に伴い V イオンが c R 軸に沿って二量化す る.この急激な電子相転移を利用した次世代デバイスの開発が進められている.(b)VO 2 バルク単結 晶における温度依存 MIT に伴う価電子帯スペクトルの変化 [10,12] . V 3d 状態が O 2p バンドと分離し てフェルミ準位近傍に存在する.](https://thumb-ap.123doks.com/thumbv2/123deta/5888697.1047695/14.892.136.792.119.418/おけるルチルイオン次世代デバイス価電子帯スペクトルフェルミ.webp)
![図 1.3.5. (a)t = 2.5–10 nm の VO 2 /TiO 2 (001)極薄膜における r -T 特性の膜厚依存性[23,29].膜厚 の減少に伴い MIT が抑制される様子が観測されている.(b)大気下で測定された t = 1 及び 7.5 nm の VO 2 /TiO 2 (001)極薄膜におけるV 2p 内殻準位の硬X 線光電子スペクトル.表面過酸化層(~0.5 nm) の形成を示唆する V 5+ 成分に由来するピーク構造が明瞭に観測されている[33].(c)VO 2 /TiO 2](https://thumb-ap.123doks.com/thumbv2/123deta/5888697.1047695/19.892.102.746.121.703/=TiO薄膜おける特性膜厚依存膜厚大気下おけるスペクトルピーク.webp)


![図 3.4.9. VO 2 /TiO 2 (001) 薄膜の r -T 特性.ここで, T MIT は熱ヒステリシス・ループの中心(降温及び昇 温過程における各 r -T 曲線の変曲点の平均)と定義した[6]. 3.4.5](https://thumb-ap.123doks.com/thumbv2/123deta/5888697.1047695/66.892.238.688.130.501/VOTiO薄膜特性ここヒステリシスループ中心降温及び温過おける曲線.webp)
