九州大学学術情報リポジトリ
Kyushu University Institutional Repository
新規医療用麻薬製剤の日本人健康成人における臨床 薬物動態評価及び医療用麻薬の個別化医療実現のた めの新規バイオマーカーの探索
東山, 馨
https://doi.org/10.15017/1932004
出版情報:九州大学, 2017, 博士(臨床薬学), 論文博士 バージョン:
権利関係:
新規医療用麻薬製剤の日本人健康成人における
臨床薬物動態評価及び医療用麻薬の個別化医療実現のための 新規バイオマーカーの探索
2018 年
東山 馨
1
原著論文
1) Toyama K, Uchida N, Ishizuka H, Sambe T, Kobayashi S. Single-dose evaluation of safety, tolerability and pharmacokinetics of newly formulated hydromorphone immediate-release and hydrophilic matrix extended-release tablets in healthy Japanese subjects without co-
administration of an opioid antagonist. J Clin Pharmacol 2015;55(9):975-84.
2) Toyama K, Furuie H, Kuroda K, Ishizuka H. Pharmacokinetic Bioequivalence Studies of an Extended-Release Oxycodone Hydrochloride Tablet in Healthy Japanese Subjects Under Fasting and Fed Conditions Without an Opioid Antagonist. Drugs in R&D 2017;17(3):363-370.
3) Toyama K, Kiyosawa N, Watanabe K, Ishizuka H. Identification of circulating miRNAs differentially regulated by opioid treatment. Int J Mol Sci 2017;18(9):1991.
2
目次
原著論文 ... 1
目次 ... 2
序章 ... 6
1. 本研究の目的 ... 6
2. 本研究の背景 ... 6
2.1 がん疼痛と治療の現状 ... 6
2.2 医療用麻薬の後発医薬品 ... 10
2.3 医療用麻薬の国内初期臨床開発の環境 ... 10
2.4 医療用麻薬に関するバイオマーカー ... 13
2.5 microRNAのオピオイド関連バイオマーカーとしての可能性 ... 14
2.6 本研究の概観... 15
3. 本章の引用文献 ... 16
薬物動態パラメータの略号一覧 ... 18
第一章:新規医療用麻薬ヒドロモルフォン即放錠及び徐放錠の日本人健康成人における 体内動態評価... 19
1. 本研究の目的 ... 19
2. 緒言 ... 19
3. 方法 ... 23
3.1 被験者 ... 23
3.2 試験デザイン、投与量及び投与方法 ... 24
3.3 投与量の設定根拠 ... 25
3.4 血漿中薬物濃度測定用検体の採取 ... 26
3.5 尿中薬物濃度測定用検体の採取 ... 26
3.6 バイオマーカー研究用血液の採取 ... 26
3.7 血漿中及び尿中薬物濃度の測定 ... 27
3
3.8 薬物動態解析... 27
3.9 食事の影響の検討 ... 28
3.10 即放錠と徐放錠の比較 ... 28
4. 結果 ... 29
4.1 被験者背景 ... 29
4.2 即放錠(ステップ1、2、5)の薬物動態プロファイル、用量比例性 ... 30
4.3 徐放錠(ステップ3、4)の薬物動態プロファイル ... 33
4.4 即放錠及び徐放錠の比較(ステップ2、3) ... 36
4.5 即放錠及び徐放錠それぞれにおける食事の影響(ステップ2、3) ... 36
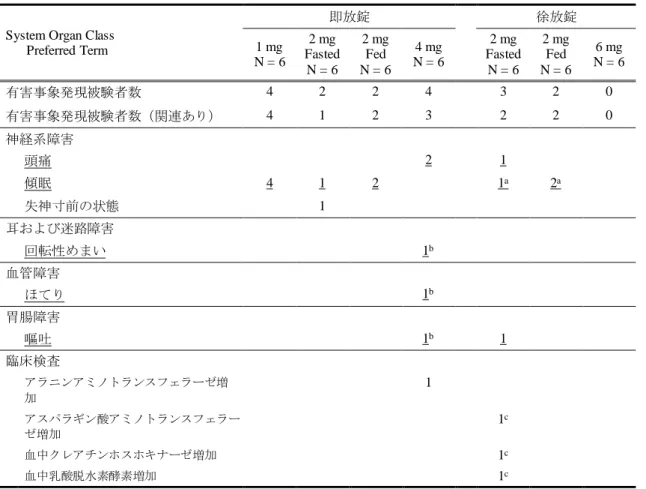
4.6 安全性 ... 38
5. 考察 ... 40
6. 小括 ... 44
7. 本章の引用文献 ... 45
第二章:新規オキシコドン製剤(即放錠及び徐放錠)の日本人健康成人における体内動態 評価 ... 48
1. 本研究の目的 ... 48
2. 緒言 ... 48
3. 方法 ... 51
3.1 被験者(DS-5058b即放錠及び徐放錠試験共通)... 51
3.2 DS-5058b即放錠の生物学的同等性試験 ... 52
3.2.1 試験デザイン、投与量及び投与方法 ... 52
3.2.2 投与量の設定根拠 ... 52
3.2.3 血漿中薬物濃度測定用検体の採取... 53
3.3 DS-5058b徐放錠の生物学的同等性試験 ... 54
3.3.1 試験デザイン、投与量及び投与方法 ... 54
3.3.2 投与量の設定根拠 ... 54
3.3.3 血漿中薬物濃度測定用検体の採取... 55
4
3.4 血漿中薬物濃度の測定(DS-5058b即放錠及び徐放錠試験共通) ... 55
3.5 薬物動態解析(DS-5058b即放錠及び徐放錠試験共通)... 56
3.6 生物学的同等性の評価(DS-5058b即放錠及び徐放錠試験共通) ... 56
3.7 バイオマーカー研究用血液の採取(DS-5058b徐放錠試験) ... 56
3.8 安全性(DS-5058b即放錠及び徐放錠試験共通)... 57
4. 結果(DS-5058b即放錠試験) ... 58
4.1 被験者背景 ... 58
4.2 薬物動態 ... 58
4.2.1 血漿中オキシコドン濃度推移 ... 58
4.2.2 生物学的同等性の検討 ... 60
4.3 安全性 ... 61
5. 結果(DS-5058b徐放錠試験) ... 62
5.1 被験者背景 ... 62
5.2 薬物動態 ... 62
5.2.1 血漿中オキシコドン濃度推移(第1試験、空腹時投与) ... 62
5.2.2 血漿中オキシコドン濃度推移(第2試験、食後投与) ... 64
5.2.3 生物学的同等性の検討 ... 66
5.3 安全性 ... 67
6. 考察 ... 69
7. 小括 ... 71
8. 本章の引用文献 ... 72
第三章: 医療用麻薬の新規バイオマーカーとしての血中miRNAの探索 ... 74
1. 本研究の目的 ... 74
2. 緒言 ... 74
3. 方法 ... 76
3.1 血液サンプル... 76
3.1.1 ヒドロモルフォン臨床試験(第一章) ... 76
5
3.1.2 オキシコドン臨床試験(第二章)... 76
3.2 miRNA抽出及び定量PCR解析 ... 77
3.2.1 RNA調製 ... 77
3.2.2 定量PCR解析 ... 77
3.2.3 データ解析 ... 78
4. 結果 ... 80
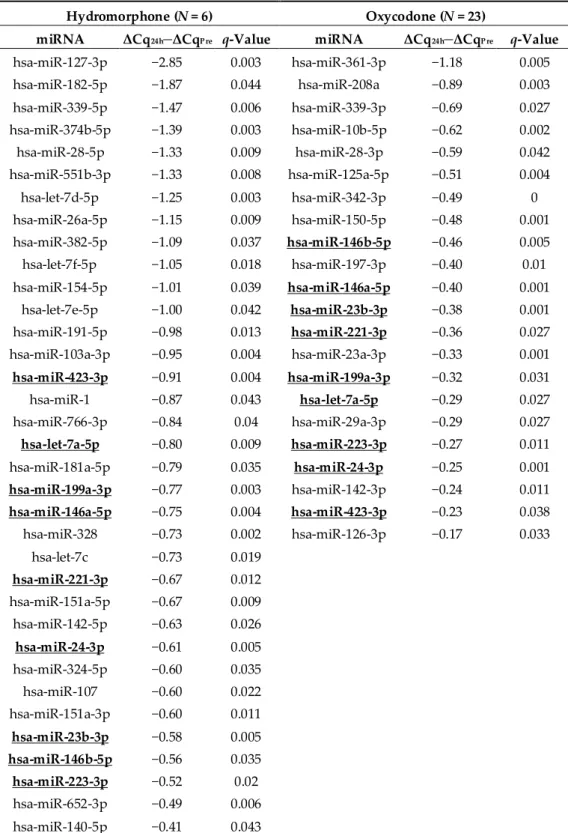
4.1 ヒドロモルフォン及びオキシコドン投与によるmiRNA変動の全体像... 80
4.2 ヒドロモルフォン及びオキシコドンにより血中レベルが増加したmiRNA ... 83
4.3 ヒドロモルフォン及びオキシコドンにより血中レベルが低下したmiRNA ... 84
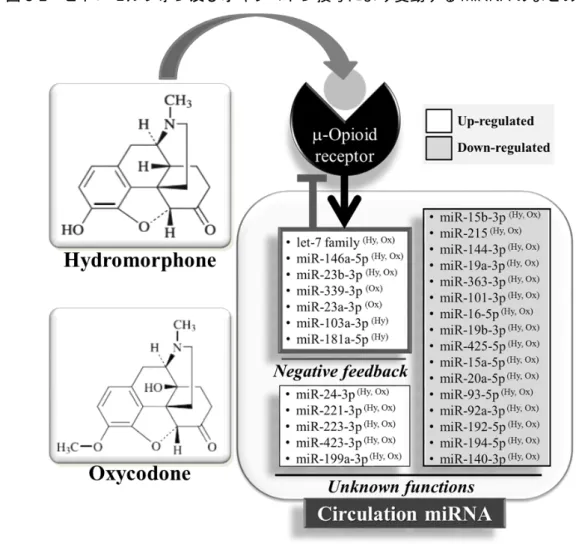
4.4 µ-オピオイド受容体(MOR)シグナルに関連するmiRNA(ヒドロモルフォン 及びオキシコドンにより血中レベルが増加したもの) ... 85
5. 考察 ... 86
6. 小括 ... 90
7. 本章の引用文献 ... 91
まとめ ... 94
謝辞 ... 96
6
序章
1. 本研究の目的
本研究は、がん疼痛治療のために新規開発された医療用麻薬の製剤特性を、健康成人被 験者を対象とし、被験者の安全性を最優先に、かつ規制要件を満たす臨床試験を計画し、
臨床薬物動態評価を行うとともに、医療用麻薬の使用対象となるがん疼痛患者の適切な選 択、オピオイドの有効性、安全性評価、及び至適用法用量設定のための客観的かつ定量的 なバイオマーカーとしての血中microRNAの活用を探索するものである。
2. 本研究の背景
2.1 がん疼痛と治療の現状
がん罹患者数は年々増加し1981年以降は日本人の死因の第1位となっており、高齢化 の進行と合わせて今後も増加していくと推測される[1]。がん罹患率が上昇する一方で診 断・治療の進歩によって生存率が大幅に向上しており、長期生存する患者に対する
Quality-of-life(QOL)向上の重要性が高まっている。がん患者は身体的、精神的な苦痛を
伴い、その一因であるがん疼痛を抱えている割合は53%~71%と報告されている[2]。がん 疼痛は、がんの早期から終末期に至るまで多くの患者が体験している痛みであり、がん患 者のQOL向上のためにはがん疼痛からの解放が必須である。国内では2007年4月にがん 対策基本法が施行され、その中で「疼痛等の緩和を目的とする医療が早期から適切に行わ れるようにすること」と明記された[3]。がん患者とその家族が可能な限り質の高い療養生 活を送ることができるようにするためには、治療の初期段階から緩和ケア及びその一部を なす疼痛治療を実施することが重要であるとされている。
世界保健機関(World Health Organization:WHO)は1986年に「がんの痛みからの解 放」の初版を発行し、WHO方式がん疼痛治療法が公表された[4]。WHO方式がん疼痛治療 法では、「薬による治療法が、がん患者の痛みのマネジメントの主役を果たす」とされて おり、鎮痛薬使用の5原則として、次の5点に要約している。
7
1 経口的に (by mouth)
2 時刻を決めて規則正しく (by the clock)
3 除痛ラダーにそって効力の順に (by the ladder)
4 患者ごとの個別的な量で (for the individual)
5 そのうえで細かい配慮を (attention to detail)
図2.1-1に示す「WHO三段階除痛ラダー」では、痛みの強さによる鎮痛薬の選択及び鎮
痛薬の段階的な使用法が示されており、軽度の痛みには非オピオイド鎮痛薬を使用し(除 痛ラダーの第1段階)、非オピオイド鎮痛薬で十分な効果が得られない、又は軽度から中 等度の痛みに対しては弱オピオイド鎮痛薬を使用し(除痛ラダーの第2段階)、弱オピオ イド鎮痛薬で効果が不十分、もしくは中等度から高度の痛みの場合は強オピオイド鎮痛薬 へ変更する方法(除痛ラダーの第3段階)が推奨されている。WHO方式がん疼痛治療法 は世界各国でがん疼痛に対する薬物療法の基本となっており、日本でもWHO方式がん疼 痛治療法をもとにがん疼痛の薬物療法に関するガイドライン(2014年版)が作成されてい る[5]。
図2.1-1 WHO三段階除痛ラダー
(WHO編. がんの痛みからの解放、第2版、金原出版、1996)
8
強オピオイド鎮痛薬はがん疼痛に最も有効な治療手段であり、効果的に疼痛のコントロ ールを行うことが可能とされている[6]。強オピオイド鎮痛薬の中ではモルヒネが第一選択 薬に、モルヒネの使用が困難な患者には代替薬として他の強オピオイド鎮痛薬が推奨され ている[4]。オピオイドによる薬物療法では、鎮痛効果が不十分もしくは副作用のために鎮 痛効果を得られるだけのオピオイドが投与できない患者に対して、投与中のオピオイドか ら他のオピオイドに変更することで疼痛をコントロールし、副作用を軽減する治療戦略が 取られている(オピオイドスイッチング、図2.1-2)[7]。オピオイドスイッチングは、強 オピオイド鎮痛薬の間で行われており、国内でWHO三段階除痛ラダーの第3段階として 使用可能な強オピオイド鎮痛薬は、モルヒネ、オキシコドン、フェンタニル、タペンタド ールに留まっている。そのため、臨床使用可能なオピオイドの選択肢の充実が治療戦略設 計のために望まれる。
図2.1-2 これまでのオピオイドスイッチング(概念図)
がん疼痛には、24時間のうち12時間以上経験される平均的な痛みである持続痛と、持 続痛の有無や程度、鎮痛薬治療の有無に関わらず発生する一過性の痛みの増強である突出 痛があり、多くの患者はこれらの性質が複合した痛みを感じている(図2.1-3)[5]。
9
図2.1-3 痛みのパターン(文献5)
がん疼痛を効果的にコントロールするためには、即放性製剤と徐放性製剤を組み合わせ て用いる必要があり、がん疼痛の薬物療法に関するガイドライン(2014年版)では、即放 性製剤と徐放性製剤の鎮痛効果と副作用に臨床的な意味のある差はなく、中等度以下かつ 安定している痛みでは即放性製剤と徐放性製剤のいずれを使用しても良いとされている [5]。持続痛の定時鎮痛薬としては、患者の利便性やアドヒアランスを考慮して服用回数の 少ない徐放性製剤を用いることが一般的である。一方、がん患者の約70%に突出痛がみら れ、特に突出痛は進行がんで発生頻度が高く、徐放性製剤で治療を行う場合でも一時的な 痛みの増強に対するレスキュー薬として即放性製剤の使用が必須とされており、また、痛 みが高度又は不安定な場合には即放性製剤や持続注射を用いることが推奨されている[5]。
さらに、オピオイドスイッチングの初期治療や痛みの評価に基づく定時投与量の調節時に は、短い期間で適切な用量を設定する目的で即放性製剤が用いられる。医療用麻薬適正使 用ガイダンスでは、持続痛の定時鎮痛薬として用いる徐放性製剤とレスキュー薬として用 いる即放性製剤は同一有効成分の薬剤を準備することが記載されている[7]。このようにオ ピオイドによるがん疼痛治療では、オピオイドの選択肢を充実させるとともに、同一の有 効成分を有する多様な製剤を揃えることが重要である。
10
2.2 医療用麻薬の後発医薬品
医療用医薬品には、先発医薬品及び後発医薬品があり、先発医薬品は新薬として医薬品 メーカーによって独占的に製造・販売できる特許期間等がある。一方、後発医薬品は、先 発医薬品の特許期間満了後に販売される医薬品のことで、ジェネリック医薬品とも呼ばれ ている[8]。
先発医薬品の開発には、臨床試験だけでも初期第I相試験から後期臨床試験を実施する 必要があり、多くの場合、長期の開発期間と多額の投資が必要である。一方、後発医薬品 の開発では、品質、有効性、及び安全性が先発医薬品と同等であることを証明する必要が あるが、臨床試験としては一般的には健康成人を対象とした生物学的同等性試験の実施の みでよく、生物学的同等性が検証されることで先発医薬品と治療学的に同等であるものと して製造販売が承認される。そのため、後発医薬品では開発期間が新薬より短く、また、
研究開発に要する費用が低く抑えられることから、先発医薬品に比べて薬の価格が安い。
後発医薬品の使用を促進することは、患者の経済負担及び医療費圧迫軽減の観点から重要 であり、医療用麻薬についても他の医療用医薬品と同様に、今後の需要増に伴う医療費増 大の観点から後発医薬品の開発が望まれる。
2.3 医療用麻薬の国内初期臨床開発の環境
新有効成分の医薬品の臨床開発では、通常、健康成人を対象とした臨床第I相試験によ り、ヒトでの安全性、忍容性、及び薬物動態を評価した後に、その疾患を有する少数の患 者を対象として有効性、安全性、及び至適用法用量を検討する臨床第II相を実施し、さら に検証試験として臨床第III相試験を実施する(図2.3-1)。
11
図2.3-1 一般的な新規医薬品の臨床開発
医療用麻薬は、国内では麻薬及び向精神薬取締法で指定されている薬物であり、様々な 規制要件及び倫理的観点から健康成人を対象とした臨床第I相試験を国内で実施すること は容易ではなく、放射標識体を用いたヒトのマスバランス試験と同様に、海外で健康成人 を対象とした臨床試験を計画、実施せざるを得ないと考えられてきた。例えば、既に上市 されている医療用麻薬の一つであるオキシコドン製剤では、海外で健康成人を対象とした 試験が実施され、安全性や薬物動態が評価されている。国内では患者を対象とした臨床試 験のみが実施され、海外の臨床試験結果とあわせて国内における承認申請が行われている [9]。前述のように、医療用麻薬では同一の有効成分を有する徐放性製剤、即放性製剤等の 多様な製剤を揃える必要があり、新規に開発した各製剤がそれぞれの使用目的に適ったヒ ト体内動態特性を有するかを適切に評価する必要がある。一般に、新規開発製剤のヒト体
内動態をin vitro試験で得られる溶出特性や動物の薬物動態試験等から精度よく予測するこ
臨床試験
(治験) 第 I相試験:臨床薬理試験
少数の健康被験者を対象に、薬の安全性及び薬物動態を 明らかにする。
第 II相試験:探索的試験
少数の患者を対象に、有効かつ安全な薬の至適用法用量 等を決定する。
第 III相試験:検証的試験
多数の患者を対象に、有効性及び安全性を既に発売さ れている薬等と比較する。また、長期間使用したときの 有効性及び安全性を調べる。
厚生労働省による承認
(医薬品としての製造・販売を許可する)
12
とには限界があり、作製された製剤の体内動態特性は、臨床試験で評価することが多い。
製剤評価のために実施する臨床薬理試験は、通常健康成人を対象とする。健康成人を対象 とするのは、患者を対象とした試験と比較して、年齢、体重、性別、健康状態等の被験者 背景因子が類似した被験者を同時期に多数募集可能であることや薬物動態の詳細な評価の ために必要な頻回採血が可能であることによる。また、このような健康成人を対象とした 臨床試験を実施するために専門の設備やスタッフを備えた専門治験施設があるため、新規 開発製剤の製剤特性を比較的少数の被験者によって精度よく評価することが可能である。
逆に、医療用麻薬の投与対象として想定される被験者集団(がん性疼痛を有する患者)で は、年齢、体重、病態等が類似した被験者を薬物動態評価に必要な例数集めることは容易 ではなく、これらの背景要因が類似している多数の被験者を一度に集めて実施する健康成 人を対象とした場合よりも、薬物動態の個人差が大きく生じる可能性がある。また、被験 者の病態を考慮すると薬物動態評価のための採血や検査回数等を健康成人よりも制限する 必要がある。さらに、患者の治療と並行して臨床試験を実施する必要があるため、健康成 人対象試験のような治験専門施設で実施することは難しい。そのため、患者を対象とした 臨床薬理試験では、新規開発製剤が目的とする体内動態特性を示すか否かを精度よく評価 することは容易ではない。
また、新規開発製剤が食事により服薬時にどのような影響を受けるかを評価するために 実施する臨床薬理試験も通常健康成人を対象とする。食事が、新たに開発した製剤の薬物 動態にどのような影響を及ぼすかは、服薬時の食事制限(空腹時投与、食後投与等)設定 に重要であり、食事による体内動態変動が有効性及び安全性にどの程度の影響を与えるか を精査するための重要な情報の一つである。薬物動態に対する食事の影響の評価は、多く の場合、図2.3-2のようなクロスオーバー試験で検討が行われるが、がん疼痛をオピオイ ドでコントロールする必要がある患者を対象としてこのような試験を実施することは、上 述した理由に加え、治験期間が比較的長期にわたること、他の治療薬の使用及び食事条件 を長期間制限する必要があること、さらに薬剤の休薬期間を設ける必要があること等、被 験者保護の観点で臨床試験の実施が難しい。これらの点は、後発医薬品開発で実施する健 康成人を対象としたするクロスオーバー試験による生物学的同等性試験でも同様である。
13
さらに、麻薬の輸出入は関連法規により厳しく制限されており、国内で製造した医療用麻 薬を海外に輸出して、臨床試験を実施することは難しい。そのため、医療用麻薬の新規製 剤開発では、後発医薬品の開発を含め、初期臨床開発において、国内で健康成人被験者を 対象とした医療用麻薬の臨床試験の実施を検討する必要がある。
図2.3-2 クロスオーバー試験
2.4 医療用麻薬に関するバイオマーカー
オピオイドはがん疼痛治療に有用であるが、その投与量やオピオイドの選択は、患者の 年齢、性別、体重、腫瘍の大きさや転移部位、病期などによって決められるものではな く、その有効性及び副作用には大きな個人差が存在する[7]。一般に、鎮痛効果の判定は主 観的なものであり、Numerical Rating Scale(NRS)やVisual Analogue Scale(VAS)等の評 価スケールに患者がスコアリングした数値を基にした疼痛強度の評価や、患者への問診、
日誌、アンケート等が用いられる。WHOの鎮痛薬使用の5原則の中に「患者ごとの個別 的な量で」とあるが、これはオピオイドを投与して患者の状態を観察しながら医師がオピ オイドの種類や量を調整しているものであり、がん疼痛の痛みの評価は、患者の痛みの訴 えを信じることが基本であるとも言われている[7]。上述のオピオイドスイッチングについ ても客観的な指標はなく、どのタイミングで治療薬を変更するかの判断は、患者の状態を 観察しながら医師が主観的に行う。このように、現在までにオピオイドによる疼痛治療を 要する患者の層別化の手段はなく、どういった背景の患者に対して、どのオピオイドを選
14
択し、どのような用法用量を設定すべきか等の治療戦略最適化を可能にする客観的な指標 は確立されていない。そのため、治療戦略を構築する方策を確立可能にする実用的なバイ オマーカーが望まれる。
2.5 microRNA のオピオイド関連バイオマーカーとしての可能性
microRNA(miRNA)は、タンパク質をコードしない22塩基程度の長さの1本鎖RNA
であり、遺伝子の発現調節に関与するとともに、発生、細胞増殖及び細胞分化、アポトー シス又は代謝といった様々な生命現象に重要な役割を担っている[10]。また、miRNAの変 化や異常により、特定のがんや心血管疾患、精神分裂症や薬物中毒などの中枢神経系障害 が生じることが報告されており[11, 12]、様々な病態にも重要な役割を担っている。miRNA は血液中で極めて安定であり、さらに、採血によりmiRNAサンプルとして採取した場合 でも、pH変動、温熱、複数回の凍結融解の繰り返し、長期保存等の過酷な条件下でも分解 せず安定であることから[13]、miRNAの機能的な重要性のみならず、近年、バイオマーカ ーとしての有用性が注目されている[14]。
疼痛に関してこれまでに、様々なmiRNAがµ-オピオイド受容体(MOR)と鎮痛のシグ ナル伝達に関与していることが、細胞系あるいは実験動物を用いた検討で報告されている
[15]。オピオイド投与によるMOR活性化によって血液中を循環するmiRNAプロファイル
が変化する可能性が考えられ、臨床において、オピオイド刺激によって変動する血中
miRNAが同定できれば、オピオイド治療に関連するバイオマーカーとしての活用が期待で
きる。しかし、このようなオピオイド刺激によるmiRNA変動について、臨床での検討は 行われていない。
15
2.6 本研究の概観
本研究は、がん疼痛治療のために新規開発された医療用麻薬であるヒドロモルフォン及 びオキシコドンの製剤特性を、健康成人被験者に対する安全性を最優先に、かつ規制要件 を満たす臨床試験を計画し、臨床評価するのものである(第一章及び第二章)。また、こ れら臨床試験の中で、医療用麻薬のバイオマーカー探索研究のための血液検体を採取し、
オピオイド投与により変動する血中miRNAを探索し、バイオマーカーとしての活用可能 性を検討するものである(第三章)。
その結果、新有効成分を有する医療用麻薬としてヒドロモルフォン製剤(即放錠及び徐 放錠)、及び服薬利便性の高い錠剤を含めたオキシコドン製剤の後発医薬品(即放錠及び 徐放錠)が見いだされ、がん疼痛コントロールの治療戦略をより充実させるとともに、患 者の経済負担及び医療費圧迫の軽減に寄与するものと考えられた。さらにオピオイド投与 により変動する血漿中miRNAが初めて見いだされ、客観的かつ定量的な新規バイオマー カーの候補として、医療用麻薬の使用対象となるがん疼痛患者の適切な選択、有効性、安 全性評価、及び至適用法用量設定等により、オピオイドの個別化医療をサポートし、医療 用麻薬の適性使用への活用につながることが期待された。
16
3. 本章の引用文献
1) 厚生労働省 第2部 現下の政策課題への対応:がん・生活習慣病(NCDs(非感染 性疾患)対策の総合的かつ計画的な推進) 平成26年度版厚生労働白書
2) Yamaguchi T, Narita M, Morita T, et al. Recent developments in the management of cancer pain in Japan: education, clinical guidelines and basic research. Jpn J Clin Oncol. 2012;42(12):1120- 7.
3) 厚生労働省 がん対策推進基本計画 平成24年6月 4) WHO. Cancer Pain Relief. 2nd ed. 1996.
5) 日本緩和医療学会緩和医療ガイドライン委員会編 がん疼痛の薬物療法に関するガイ ドライン2014年版 金原出版 2014年
6) Zech DF, Grond S, Lynch J, et al. Validation of World Health Organization Guidelines for cancer pain relief: a 10-year prospective study. Pain. 1995;63(1):65-76.
7) 的場元弘, 山本弘史, 赤木徹, 他.医療用麻薬適正使用ガイダンス. がん疼痛治療及び 慢性疼痛における医療用麻薬の使用と管理のガイダンス.厚生労働省医薬・生活衛生 局 監視指導・麻薬対策課. 平成29年4月
8) 後発医薬品(ジェネリック医薬品)の使用促進について 厚生労働省
http://www.mhlw.go.jp/stf/seisakunitsuite/bunya/kenkou_iryou/iryou/kouhatu-iyaku/index.html [cited 2017 Dec].
9) オキシコンチン錠5 mg、同10 mg、同20 mg、及び同40 mg. 審査報告書(2003年4 月16日).
10) Ma C, Liu Y, He L. microRNAs - powerful repression comes from small RNAs. Sci China C Life Sci. 2009 Apr; 52(4): 323–330.
11) Im I, Kenny J. MicroRNAs in neuronal function and dysfunction. Trends Neurosci. 2012, 35, 325–334.
12) Wang J, Chen J, Sen S. MicroRNA as biomarkers and diagnostics. J. Cell. Physiol. 2016, 231, 25–30.
13) Brase C, Wuttig D, Kuner R, Sultmann H. Serum microRNAs as non-invasive biomarkers for
17 cancer. Mol. Cancer 2010, 9, 306.
14) Lopez P, Diallo A, Cruceanu C, Fiori M, Laboissiere S, Guillet I, Fontaine J, Ragoussis J, Benes V, Turecki G. et al. Biomarker discovery: Quantification of microRNAs and other small non-coding RNAs using next generation sequencing. BMC Med. Genom. 2015, 8, 35.
15) Barbierato M, Zusso M, Skaper D, Giusti P. MicroRNAs: Emerging role in the endogenous mu opioid system. CNS Neurol. Disord. Drug Targets 2015, 14, 239–250.
18
薬物動態パラメータの略号一覧
略号
省略していない表現
英語 日本語
AUC area under the plasma concentration-time
curve 血漿中濃度-時間曲線下面積
AUCinf area under the plasma concentration-time curve up to infinity
無限大時間までの血漿中濃度-時間曲線 下面積
AUClast area under the plasma concentration-time curve up to the last quantifiable time
定量可能な最終時点までの血漿中濃度-
時間曲線下面積 AUCt area under the plasma concentration-time curve
up to the last sampling time
最終採血時点までの血漿中濃度-時間曲 線下面積
Cmax maximum plasma concentration 最高血漿中濃度
CL/F apparent total body clearance 見かけの全身クリアランス
Fe cumulative fraction excreted into urine 累積尿中排泄率
T1/2 terminal elimination half-life 終末相の消失半減期
Tmax time to reach maximum plasma concentration 最高血漿中濃度到達時間
Vz/F apparent distribution volume 見かけの分布容積
19
第一章: 新規医療用麻薬ヒドロモルフォン即放錠及び徐放錠の日本人健康成人 における体内動態評価
1. 本研究の目的
本研究は、がん疼痛治療のために国内で新規に開発されたヒドロモルフォンの即放錠及 び徐放錠について、日本人健康成人における安全性、忍容性及び薬物動態を、健康成人被 験者に対する安全性を最優先に、かつ規制要件を満たす臨床試験を計画し、臨床評価する ものである。また、ヒドロモルフォンの体内動態に及ぼす食事の影響を検討する。あわせ て、第三章に示す医療用麻薬の個別化医療に関連するバイオマーカー探索に供する血液検 体を採取する。
2. 緒言
医療用麻薬であるヒドロモルフォン(図2-1)は、1920年代にドイツで合成された薬剤 で、選択的µ-オピオイド受容体(MOR)作動性の強オピオイドに分類される。モルヒネと 比べてMORへの親和性が高く、約5倍から8倍の鎮痛効果を有する[1-3]。ヒドロモルフ ォンの主代謝経路は、3位水酸基のグルクロン酸抱合によるヒドロモルフォン-3-グルクロ ニド(hydromorphone-3-glucuronide、H3G)への代謝である。代謝物H3GのMORに対す るアゴニスト活性は未変化体の約1/2,280と低く、モルヒネとは異なり、代謝物は鎮痛効 果には寄与しない[4]。
図2-1 ヒドロモルフォンの構造式
20
ヒドロモルフォンは、WHOガイドライン、欧州緩和ケア学会(EAPC)、欧州臨床腫瘍 学会(ESMO)、米国NCCNのガイドライン等において、モルヒネの代替薬として使用さ れる標準的薬剤と位置づけられており[5-8]、徐放性及び即放性の経口剤、注射剤として、
中等度から高度のがん又は非がん疼痛患者に対して使用されている。このようにヒドロモ ルフォンは海外ではがん疼痛治療に欠かせない標準的薬剤の一つとして広く使用されてい るものの、国内では未承認であるため、海外と比べ、疼痛コントロール及びオピオイドス イッチングを含めたがん疼痛治療戦略が限定されていた。
オピオイドによる疼痛治療では、多様な製剤がその使用目的に応じて使い分けされてい る。即放性製剤及び注射剤は、がん疼痛を効果的に治療するために欠かせない剤形であ り、徐放性製剤は、持続痛の定時鎮痛薬として重要である。さらに、一時的な痛みの増強 に対するレスキュー薬として即放性製剤が使われるが、徐放性製剤と同じ成分の製剤を準 備することが推奨されていることを考慮すると、同一の有効成分を有する多様な製剤を揃 えることが重要である。
また、国内で使用可能なオピオイド鎮痛薬の経口の徐放性製剤には、モルヒネ、オキシ コドン、タペンタドールがあるが、このうちオキシコドン、タペンタドールは、いずれも 1日2回投与なのに対し、海外で上市されているヒドロモルフォン徐放性製剤は1日1回 投与であり、1日1回投与のヒドロモルフォン徐放性製剤は投与回数の削減による患者の 利便性や服薬アドヒアランスの向上が期待できる。
このような状況を踏まえ、疼痛コントロールのために使用可能なオピオイドの選択肢を 増やし、がん疼痛コントロールの治療戦略をより充実させるために、新有効成分であるヒ ドロモルフォンの新規製剤を検討した。剤形は、がん疼痛を効果的にコントロールするた めに必要な剤形として即放錠及び徐放錠の2製剤とした。
即放性製剤については、レスキュー薬や定時投与量の調節に使用されることから短時間 での効果発現が必要となる。ヒドロモルフォン即放性製剤については、速やかな溶出性を 有する即放錠を設計した。
徐放性製剤については、親水性マトリックス製剤を検討した。徐放性製剤は持続痛に定 時鎮痛薬として使用されることから、安定した鎮痛効果及び服薬コンプライアンスが重要
21
であり、海外では様々な徐放機構を用いて検討され、実際に臨床使用されている。浸透圧 による薬剤放出制御(osmotic-control release oral delivery system、OROS)はその一つであ り、製剤表面上から内部に水分が流入し、その圧によって製剤中の薬剤が一定速度で外部 に放出され、徐放化が達成される技術である。OROSは、ヒドロモルフォン、ニフェジピ ン、オキシブチニン等の様々な薬剤に応用され、臨床使用されている[9]。これとは異なる 製剤化技術として親水性マトリックスを用いた徐放化技術も経口徐放性製剤の開発に汎用 されている。この製剤では、製剤作製に用いる親水性のポリマーが水分と接することでゲ ル層を形成し、薬剤の放出が制御されることで徐放化が行なわれる。親水性マトリックス 製剤の最大のメリットは、比較的作製が簡単であり、製造工程に多額のコストがかから ず、様々な薬剤の放出プロファイルが設計可能な点にある[10]。そこで、ヒドロモルフォ ン徐放性製剤として、親水性マトリックスを用いた徐放錠を検討し、海外のヒドロモルフ ォン徐放製製剤と類似したin vitro溶出挙動を示す製剤を設計した。ヒドロモルフォン徐放 錠は、原薬と2種類の高分子を組み合わせることで、薬剤溶出が消化管内の幅広いpH及 び蠕動運動の影響を受け難く、投与後、消化管の広範囲で薬物を放出させるマトリックス 構造とした(図2-2)[11]。
図2-2 ヒドロモルフォン徐放化メカニズム(文献11)
22
新規製剤のヒト体内動態を、in vitro溶出特性や動物での薬物動態試験等から精度よく予 測することは難しく、特に、様々な工夫により製剤からの薬剤放出を制御している徐放性 製剤はさらに予測精度は低くなる。よって、新規に開発されたヒドロモルフォン製剤(即 放錠及び徐放錠)が想定した体内挙動をヒトで示すかは臨床薬物動態試験により検討する 必要がある。ヒドロモルフォンは麻薬であることから、健康被験者への投与は可能な限り 回避することが望ましいものの、患者を対象とした臨床薬物動態試験で精度よく製剤評価 を行うことは難しく、また海外で健康成人を対象として臨床試験を実施することも難し い。そこで、新規に開発したヒドロモルフォン製剤について、健康成人を対象として薬物 動態を検討する国内臨床試験を検討した。
医療用麻薬の有効性が望めない健康成人を対象とした臨床試験では、被験者の安全性確 保が最重要である。海外で実施される健康成人を対象としたオピオイドの臨床薬理試験で は多くの場合、健康成人に対する安全性確保のため、麻薬拮抗薬である経口ナルトレキソ ンを前投与及び併用投与することで中枢性の副作用を回避する。しかし、ナルトレキソン は国内未承認であり国内治験に使用することが容易ではない。ナルトレキソンとは別の麻 薬拮抗薬として、国内ではナロキソン注射剤が承認されているが、経口投与可能なナロキ ソン製剤はない。ナロキソンを静脈内投与した場合の体内消失は非常に早く[12]、経口ナ ルトレキソンと同様に健康成人に対する中枢性の副作用回避を目的としてナロキソン注射 剤を臨床薬理試験で使用するためには、頻回の静脈内投与や持続静脈内投与が考えられる ものの、このようなナロキソンの使用方法は現実的ではない。そこで麻薬拮抗薬を併用し なくとも被験者の安全性に問題が生じない臨床試験を計画した。
23
3. 方法
本治験はヘルシンキ宣言に基づく倫理的原則、「医薬品の臨床試験の実施の基準(GCP)
に関する省令」(平成9年3月27日厚生省令第28号)を遵守し、実施医療機関の治験審 査委員会(IRB)での審議・承認を経た後に実施した。また、治験実施計画書の変更、説 明文書及び同意文書の変更についてもIRBでの審査を受け、承認を得た。説明文書及び同 意文書は、治験開始前にIRBの承認を得た。治験実施計画書に規定された検査を実施する 前に、治験の内容・被験者の権利などを被験者本人に十分説明し、治験への参加について 被験者自らの自由意思による同意を文書によって被験者本人から得た。本治験は、昭和大 学 臨床薬理研究センターにて行なわれた。
本治験は、臨床試験情報JapicCTI-132167として登録した。
3.1 被験者
被験者の選択基準は、日本人男性で、同意取得時に20歳以上45歳以下の者、かつスク リーニング検査時に体格指数(Body Mass Index, BMI)が18.5以上25.0未満の者とした。
スクリーニング検査時の被験者の除外基準として以下を設定した。
1) 重篤な疾患の既往歴を有し、治験の実施が被験者の安全性確保上問題がある者 2) 薬物などに対する過敏症又は特異体質がある者
3) アルコール又は薬物依存者
4) 感染症検査の結果、異常が認められた者 5) 尿中薬物検査の結果、異常が認められた者
6) スクリーニング検査前1年以内に合計1200 mL以上の全血採血を行った者、
又は84日以内に合計400 mL以上の全血採血を行った者、
又は28日以内に合計200 mL以上の全血採血を行った者、
又は14日以内に成分採血を行った者
7) スクリーニング検査前120日以内に他の治験に参加し治験薬の投与を受けた者 8) 過去に医療用麻薬の臨床試験に参加した者
24
9) スクリーニング検査において、治験責任医師等により臨床的に問題となる自覚症状・
他覚所見を認めた者、心電図検査において異常を認めた者、あるいは実施医療機関 の臨床検査基準値を逸脱した者(ただし、治験への参加に問題ない場合は除く)
10) 治験期間中にコンドーム等を用いた適切な避妊を行う意思のない者 11) その他、治験責任医師等が本治験参加に不適当と判断した者
3.2 試験デザイン、投与量及び投与方法
本治験は、5つのステップで構成した。被験者数は、計30名(6名×5ステップ)とし た。治験デザインとして、ステップ1、4、及び5は単一施設、非盲検、非対照、単回投与 試験、ステップ2及び3は単一施設、非盲検、無作為化、2-wayクロスオーバー試験とし
た。表3.2-1に、各ステップの投与量及び投与方法に示す。
表3.2-1 投与量及び投与方法
ステップ 投与量 投与方法 被験者数
1 即放錠1 mg 単回投与(空腹時投与) 6名
2 即放錠2 mg 単回投与
(2-wayクロスオーバー) 6名
3 徐放錠2 mg 単回投与
(2-wayクロスオーバー) 6名
4 徐放錠6 mg 単回投与(空腹時投与) 6名
5 即放錠4 mg 単回投与(空腹時投与) 6名
薬物動態に及ぼす食事の影響は表3.2-2に示す投与方法でヒドロモルフォン錠を投与 し、ステップ2及び3で検討した。各期の休薬期間は6~8日間とした。
表3.2-2 ステップ2及び3における投与方法
投与期
投与群 第I期 第II期
A群 朝食後投与 空腹時投与
B群 空腹時投与 朝食後投与
25
各ステップにおける投与方法及び食事条件を表3.2-3に示す。
表3.2-2 各ステップにおける投与方法及び食事条件
ステップ 投与方法 食事内容 食事条件
1~5 空腹時投与 - 投与前日より10時間以上絶食
2
朝食後投与
低脂肪食
(700 kcal以下、かつ、総エネルギーに対する 脂質のエネルギーの占める割合は20%以下)
朝食を20分以内に摂り、終了 30分後に治験薬投与
3
高脂肪食
(900 kcal以上、かつ、総エネルギーに対する 脂質のエネルギーの占める割合は35%以上)
朝食を20分以内に摂り、終了 10分後に治験薬投与 後発医薬品の生物学的同等性試験ガイドライン[13] を参考に、薬物動態に及ぼす食事の影響を検討するステップ2、3 での食事内容、食事時間、及び治験薬の投与時間を設定した。
3.3 投与量の設定根拠
投与量は、即放錠1 mg、2 mg、4 mg、徐放錠2 mg、6 mgとし、即放錠2 mg及び徐放錠 2 mgについては、空腹時投与又は朝食後投与の2-wayクロスオーバー試験とした。
最低用量(1 mg)は、先に海外で実施した第I相試験[14]で忍容性及び安全性が確認さ れたヒドロモルフォン即放カプセル剤の投与量(1.3 mg)を超えない用量を設定した。
即放錠の最高用量(4 mg)については、ヒドロモルフォンの生物学的利用率が25.7%で あることから、即放錠4 mgの血漿中濃度下面積値(AUC)が注射剤1 mg投与時のAUC と同程度であることが想定された。注射剤1 mg投与時の安全性は、海外で実施した第I相 試験[14]で確認されているため、即放錠4 mgを即放錠の最高用量に設定した。
徐放錠の最高用量(6 mg)は、先に海外で実施した第I相試験の反復投与時の最大総投 与量(6.5 mg)を超えない投与量とした。ヒドロモルフォン徐放性製剤の外国人健康成人 を対象とした海外の試験では、8~64 mgを単回投与した結果、頻脈、悪心、頭痛、酸素飽 和度低下等の有害事象が認められたが、重篤な問題は生じていない[15]。これより、徐放 錠の最高用量(6 mg)の設定に問題はないと考えた。
また、薬物動態に及ぼす食事の影響について、異なる製剤間の同一用量(2 mg)で比較 できるよう、即放錠及び徐放錠それぞれ2 mgの投与量で検討することとした。ヒドロモ ルフォン製剤の薬物動態に及ぼす食事の影響について、海外で市販されている即放性製剤
26
では、空腹時投与に比べて食後投与で、ヒドロモルフォンのCmax(平均値)は25%低 下、AUCinf は24%増加した[16]。徐放性製剤では、AUClastに対する食事の影響はないも のの、Cmaxは食後投与時に約1.2倍上昇した[17, 18]。よって、食後投与によりヒドロモ ルフォンの体内動態が変動した場合でも、即放錠及び徐放錠の最高用量(それぞれ4 mg
及び6 mg)のAUC及びCmaxを超えない投与量であり、問題はないと考えた。
3.4 血漿中薬物濃度測定用検体の採取
被験者前腕の皮静脈より、1回3 mL又は5 mLの静脈血をEDTA-2K加真空採血管に採 取した。採血後速やかに転倒混和した後氷冷し、30分以内に遠心分離(4°C、3000 rpm、
10分間)した。遠心分離によって得られた血漿を保存容器に分取して凍結し、冷凍保存
(設定温度: −20°C以下)した。
採血時期は、投与前、投与0.25、0.5、1、1.5、2、2.5、3、4、5、6、8、10、12、24、
36、48時間後とした。ただし、投与0.25時間後の採血はステップ1、2、及び5(即放
錠)のみ、投与5時間後の採血はステップ3及び4(徐放錠)のみとした。
3.5 尿中薬物濃度測定用検体の採取
採尿時期は、投与前、投与0~4、4~8、8~12、12~24、24~36、36~48時間後とし た。尿サンプルは冷凍保存(設定温度: −20°C以下)した後、測定に供した。
3.6 バイオマーカー研究用血液の採取
ステップ4において、バイオマーカー研究の同意が得られた被験者を対象とした。被験 者前腕の皮静脈より、1回3 mLの静脈血をEDTA-2K加真空採血管に採取し、速やかに転 倒混和した後氷冷し遠心分離(4°C、3000 rpm、10分間)した。遠心分離により得られた 血漿を凍結し、冷凍保存(設定温度:−20°C以下)、循環microRNA(miRNA)の探索的評 価に供した。採血時期は、投与前、投与4、8、12、24時間後(ステップ4のみ)とし た。
27
3.7 血漿中及び尿中薬物濃度の測定
血漿中及び尿中のヒドロモルフォン及び代謝物(H3G)濃度は、固相抽出により前処理 を行い、重水素体を内部標準物質として、液体クロマトグラフィー-タンデムマススペク トロメトリー法(LC-MS/MS法、AB SCIEX 5500 QTRAP、MA、USA)で測定した。
分析カラムには、Inertsil amide analytical column(3 μm、100 mm x 2.1 mm id、GL
Sciences、東京)を用い、移動相には、7.5 mM酢酸アンモニウム溶液(0.1%酢酸含有)と
アセトニトリルのグラジエント、流速は0.8 mL/minとした。検出には、ヒドロモルフォン 及びH3Gでそれぞれ、m/z 286−185及び462−185を用いた。
血漿中濃度の定量下限は、ヒドロモルフォン及びH3Gで0.05及び0.5 ng/mLで、検量線 の直線領域は、0.05−50及び0.5−100 ng/mLであった。日内変動の変動係数(CV)は、ヒ ドロモルフォン及びH3Gでそれぞれ1.8%~7.0%及び2.5%~4.0%であった。日間変動の CVは、3.4%~9.3%及び2.8%~9.2%であった。
尿中濃度の定量下限は、ヒドロモルフォン及びH3Gで2及び10 ng/mLで、検量線の直 線領域は、2−1000及び10−2000 ng/mLであった。日内変動のCVは、ヒドロモルフォン及 びH3Gでそれぞれ2.8%~9.0%及び3.8%~8.5%であった。日間変動のCVは、4.9%~9.1%
及び7.3%~13.0%であった。
3.8 薬物動態解析
血漿中及び尿中のヒドロモルフォン及び代謝物(H3G)濃度から、モデルに依存しない 方法によって以下の薬物動態パラメータを算出した。
Cmax、Tmax、AUClast、AUCinf、T1/2、Fe
ここで、Cmaxは最高血漿中濃度、Tmaxは最高血漿中濃度到達時間、AUClastは定量可 能な最終時点までのAUC、AUCinfは無限大時間までのAUC、Feは累積尿中排泄率を示 す。
薬物動態の解析には、WinNonlin Ver.6.2(Pharsight社)及びSAS(version 9.2, SAS
28 Institute Japan)を用いた。
3.9 食事の影響の検討
ステップ2及び3のヒドロモルフォンの薬物動態パラメータCmax、AUClast、AUCinf、
T1/2の自然対数変換値及びTmaxの無変換値を反応変数として、投与群、投与期、食事条 件(空腹時、食後)を固定効果、被験者を変量効果とした、線型混合モデルを用いた解析 を行い、各食事条件の最小二乗平均、食事条件間の差の最小二乗平均、並びにそれらの両
側90%信頼区間を算出した。
3.10 即放錠と徐放錠の比較
ステップ2及び3の空腹時のヒドロモルフォンの薬物動態パラメータCmax及びAUCinf の幾何平均値の比とその両側95%信頼区間を算出した。
29
4. 結果
4.1 被験者背景
被験者背景について、投与群間で大きな違いはなかった(表4.1-1)。
表4.1-1 被験者背景
IR 1 mg IR 2 mg IR 4 mg ER 2 mg ER 6 mg All
N = 6 N = 6 N = 6 N = 6 N = 6 N = 30
Age (years)
mean 39.5 36.0 40.0 35.2 35.8 37.3
SD 4.37 6.23 3.03 4.67 6.40 5.16
min 33 26 35 29 25 25
median 39.5 38.0 40.0 37.0 37.0 39.0
max 45 42 44 39 43 45
Height (cm)
mean 176.07 174.97 172.92 170.10 170.78 172.97
SD 5.695 5.665 5.472 5.022 5.778 5.647
min 168.2 164.7 166.5 160.5 162.2 160.5
median 176.35 176.85 172.75 171.80 173.00 173.50
max 185.5 179.5 179.5 174.0 176.0 185.5
Weight (kg)
mean 65.92 66.05 66.05 63.15 64.27 65.09
SD 5.700 6.788 4.838 7.253 5.840 5.829
min 58.1 55.7 61.0 55.0 55.7 55.0
median 67.35 66.95 65.00 62.60 63.80 65.35
max 73.2 75.6 75.1 71.8 71.3 75.6
BMI (kg/m2)
mean 21.15 21.45 22.23 21.80 22.00 21.73
SD 1.657 1.806 1.106 1.796 1.173 1.481
min 18.8 19.5 20.9 19.2 20.2 18.8
median 21.45 21.30 22.50 21.90 22.20 22.00
max 23.2 24.0 23.8 23.9 23.4 24.0
IR: 即放錠、ER: 徐放錠
30
4.2 即放錠(ステップ 1、2、5)の薬物動態プロファイル、用量比例性
即放錠1 mg、2 mg、及び4 mgの空腹時投与について、ヒドロモルフォン及びH3Gの平
均血漿中薬物濃度推移をそれぞれ、図4.2-1及び図4.2-2に、薬物動態パラメータを表4.2-
1及び表4.2-2に示す。累積尿中排泄率を図4.2-3及び図4.2-4に示す。
即放錠1 mg、2 mg、及び4 mgの空腹時投与における平均血漿中ヒドロモルフォン濃度
は、概ね投与量の増加に伴い上昇した。ヒドロモルフォンのCmax、AUClastも概ね投与量 の増加に伴い上昇した。T1/2の算術平均値は、即放錠1 mg、2 mg、及び4 mgでそれぞれ
約5、9、及び18時間であった。ヒドロモルフォンの尿中排泄率は、48時間までの回収
で、投与量の約3%であった。H3Gの尿中排泄率は、投与量の約30%であった。
図4.2-1 即放錠1 mg、2 mg、4 mgの空腹時平均血漿中ヒドロモルフォン濃度推移
---: 定量下限(0.05 ng/mL)、IR: 即放錠
表4.2-1 即放錠のヒドロモルフォン薬物動態パラメータ(空腹時)
即放錠
1 mg 2 mg 4 mg
AUCinf (ng·h/mL) 2.29 (0.676) 5.23 (1.18) 12.6 (2.97)
AUClast (ng·h/mL) 1.80 (0.583) 4.05 (0.949) 10.3 (3.31)
Cmax (ng/mL) 0.664 (0.302) 0.980 (0.352) 1.95 (0.563)
Tmax (h) 0.50 (0.25, 1.00) 0.76 (0.25, 1.50) 1.00 (0.50, 1.02)
T1/2 (h) 5.26 (3.35) 9.24 (5.88) 18.3 (11.7)
算術平均値(標準偏差)、n = 6、Tmaxは中央値(最小値、最大値)
Plasma Concentration (ng/mL)
0 1 2 3
Time (h)
Mean ± SD
IR 1mg N=6 IR 2mg (Fasted) N=6 IR 4mg N=6
0 2 4 6 8 10 12 24 36 48
31
図4.2-2 即放錠1 mg、2 mg、4 mgの空腹時平均血漿中H3G濃度推移
---: 定量下限(0.05 ng/mL)、IR: 即放錠
表4.2-2 即放錠のH3G薬物動態パラメータ(空腹時)
即放錠
1 mg 2 mg 4 mg
AUCinf (ng·h/mL) 114 (34.6) 202 (41.0) 384 (73.6)
AUClast (ng·h/mL) 87.0 (10.4) 188 (30.1) 360 (73.3)
Cmax (ng/mL) 19.4 (2.98) 35.2 (7.38) 56.0 (11.7)
Tmax (h) 1.00 (1.00, 1.50) 1.25 (1.00, 1.50) 1.00 (1.00, 1.50)
T1/2 (h) 17.2 (17.0) 10.8 (3.02) 13.0 (2.30)
算術平均値(標準偏差)、n = 6、Tmaxは中央値(最小値、最大値)
Plasma Concentration (ng/mL)
0 20 40 60 80
Time (h)
Mean ± SD
IR 1mg N=6 IR 2mg (Fasted) N=6 IR 4mg N=6
0 2 4 6 8 10 12 24 36 48
32
図4.2-3 即放錠1 mg、2 mg、4 mgの空腹時ヒドロモルフォン累積尿中排泄率推移
図4.2-4 即放錠1 mg、2 mg、4 mgの空腹時H3G累積尿中排泄率推移
Cumulative Urinary Excretion Rate (%)
0 1 2 3 4
Time (h)
Mean ± SD
IR 1mg N=6 IR 2mg (Fasted) N=6 IR 4mg N=6
0 4 8 12 24 36 48
Cumulative Urinary Excretion Rate (%)
0 10 20 30 40
Time (h)
Mean ± SD
IR 1mg N=6 IR 2mg (Fasted) N=6 IR 4mg N=6
0 4 8 12 24 36 48
33
4.3 徐放錠(ステップ 3、4)の薬物動態プロファイル
徐放錠2 mg、及び6 mgの空腹時投与について、ヒドロモルフォン及びH3Gの平均血漿
中薬物濃度推移をそれぞれ、図4.3-1及び図4.3-2に、薬物動態パラメータを表4.3-1及び
表4.3-2に示す。累積尿中排泄率を図4.3-3及び図4.3-4に示す。
徐放錠2 mg及び6 mgの空腹時投与における平均血漿中ヒドロモルフォン濃度は、投与 量の増加に伴い上昇した。ヒドロモルフォンの尿中排泄率は、48時間までの回収で、投与
量の約3%であった。H3Gの薬物動態も、同様の傾向を示した。なお、H3Gの尿中排泄率
は、48時間までの回収で、投与量の約30%~35%であった。
図4.3-1 徐放錠2 mg、6 mgの空腹時平均血漿中ヒドロモルフォン濃度推移
---: 定量下限(0.05 ng/mL)、ER: 徐放錠
表4.3-1 徐放錠のヒドロモルフォン薬物動態パラメータ(空腹時)
徐放錠
2 mg 6 mg
AUCinf (ng·h/mL) 5.65 (1.05) 22.2 (4.43)
AUClast (ng·h/mL) 5.31 (1.64) 19.2 (4.99)
Cmax (ng/mL) 0.356 (0.115) 1.09 (0.434)
Tmax (h) 5.00 (1.00, 10.00) 3.25 (1.00, 8.00)
T1/2 (h) 8.88 (2.25) 16.8 (6.69)
算術平均値(標準偏差)、n = 6、Tmaxは中央値(最小値、最大値)
Plasma Concentration (ng/mL)
0.0 0.5 1.0 1.5
Time (h)
Mean ± SD
ER 2mg (Fasted) N=6 ER 6mg N=6
0 2 4 6 8 10 12 24 36 48
34
図4.3-2 徐放錠2 mg、6 mgの空腹時平均血漿中H3G濃度推移
---: 定量下限(0.05 ng/mL)、ER: 徐放錠
表4.3-2 徐放錠のH3G薬物動態パラメータ(空腹時)
徐放錠
2 mg 6 mg
AUCinf (ng·h/mL) 228 (34.8) 702 (97.1)
AUClast (ng·h/mL) 217 (33.5) 643 (118)
Cmax (ng/mL) 12.4 (2.06) 39.7 (7.90)
Tmax (h) 4.00 (2.00, 10.00) 2.00 (1.50, 6.00)
T1/2 (h) 8.63 (2.23) 13.4 (3.60)
算術平均値(標準偏差)、n = 6、Tmaxは中央値(最小値、最大値)
Plasma Concentration (ng/mL)
0 20 40 60
Time (h)
Mean ± SD
ER 2mg (Fasted) N=6 ER 6mg N=6
0 2 4 6 8 10 12 24 36 48