九州大学学術情報リポジトリ
Kyushu University Institutional Repository
キトサンおよびその誘導体の分析化学への応用
倉内, 芳秋
https://doi.org/10.11501/3150975
出版情報:Kyushu University, 1998, 博士(工学), 論文博士 バージョン:
権利関係:
第6章 βーシク口デキストリンで修飾したキトサンの分子認識
キラルな固定相を用いたガスクロマトグラフィーや高速液体クロマトグラ
フィーによるエナンチオマーの分離は, 分離対象物と固定相問での可逆的なジ アステレオマ一生成の平衡反応を利用したものである。 とれは一対の光学異性 体とキラルな固定相との間で可逆的な会合体が形成され, その会合体の安定性 の差がお互いを分離する駆動力となっている.
多糖類はその構造よりキラルな場を提供することができる. これらを固定相 に用いた光学分割は, すでに古くから知られており, セルロース, デンプンな
どの天然高分子を充填剤とする液体クロマトグラフィーにより試みられてい る1). ただし, そのままではキラル認識能は弱く, 何らかの化学修飾による相 互作用部位の認識機能改善の工夫が必要である. 例えば, セルロースを不均一 系でアセチル化して得られたセルローストリアセテートの微結品を固定相とし た芳香族化合物のキラル分離2) や, セルロースおよびアミロースのトリス(
フェニルカルバモイル)誘導体を被覆したシリカゲル固定相による芳香族化合 物やコバルト錯体のキラル分離3,4) の報告がある. また, 最近我々は, セル ロースと類似の構造を有するキトサンを化学修飾して, N-アリールメチリデ ンキトサンのフェニルカルバモイル誘導体を合成し, これをシリカゲルに担持 した固定相によるHPLCによって, 芳香族アルコールなどに対する高いキラル 認識能の発現を報告した5). キトサンは, アミノ基をグルコピラノースユニッ トに含むため化学修飾が容易で, キラル固定相として用いる場合, 分子設計が
容易な利点を持つ多糖である.
一方, 種々のシクロデキストリン (CD)誘導体をシリカゲル担体に結合し たものを固定相とするキラルHPLC分離が多数報告されている6,7,8,9,10). 例え
ば, ダンシルアミノ酸7), ジニトロベンゾイルアミン類83), フェニルアルキ ル誘導体化したアルコール類やアミン類10)などのキラル分離がそれである.
一連のサイクロボンドカラム(ASTEC Cyclobond)という商品名で市販され ているCDキラル固定相も, 主として逆相モードのクロマトグラフィーにおい て, たくさんのエナンチオマ一分離に使用されており1L12), アミノ酸の2,4
-ジニトロフェニjレ(DNP)誘導体13,14)やプロフェン類15)およびグリシルジ
ペプチド類16)のキラル分離例などがある. これらのエナンチオ分離能は, CD 空孔とキラルな分離対象物の疎水部分との包接錯体の形成, CD環の周囲にあ るヒドロキシル基との水素結合および、CD外周の置換基との立体障害などに よって発現される11,12) .
同様にCDを固定相としているが, より高度なキラル分離の例として,
Thuaudら17,18)はß-CDをエピクロロヒドリンで架橋した水溶性のポリマー
を固定相とした系を用いて, 先述のCDをスペーサーで介してシリカゲル表面 に固定したCyclobond Iカラムと異なるキラル認識能を報告している. 例え
ば, Cyclobond I で困難であったバルファリン(下図)のキラル分離が, 架橋 CDポリマーでは可能になっている17). ここで注目すべきことは, パルファリ
ンは包接可能な二つの疎水部分を分子中に 有するため, ボリマー中に局在化する ß- CDの高い濃度部位ではそれらが同時に取り 込まれた包接錯体を作っている可能性があ るとと18,19,20), そして, そのために独立し
αヌ川co町
。
てCD環が存在するCyclobond Iのゲスト認 識と異なるキラル分離を示している可能性
Warfarin
18)を示唆していることである. 同様な分離機構の可能性が, キャピラリー電
気泳動分析における塩基性化合物のキラル分離においても, 最近Ingelse等21) によって指摘されている. これらの報告を参考に
すると, ポリマー中にCD環を高密度で, さらには
規則正しく配置できるなら, 高度なキラル分離を 示すポリマーが合成できると思われる.
本章では, 化学修飾キトサンのキラル分離能に スポットを当て, ß-CDをキトサン骨格上に高密 度で分校させた修飾体を合成する(右模式図) • さらに, 得られたß-CD修飾キトサンを固定相と するDNPアミノ酸および類似化合物のHPLCによ るキラル分離について検討する.
骨格となるキトサンは 2位に反応活性なアミ
Chitosan
ノ基を有するDーグルコサミンユニットが ß-1,4結合で連なったアミノ多糖で ある. これはグリオキシル酸との反応で高置換度のN-カルボキシメチル化キ トサン(NCMC)を高収率で与える22) . NCMCと, ß- CDの6位ヒドロキン ル基のひとつにアミノ基を導入した, 6-アミノ-6ーデオキシ- ß-CD (NH2- ß
CD)との縮合によって目的のキトサン誘導体(ß -CD-NCMC)を合成する.
前駆体となるNCMCは水溶性を示すため, 水中におけるNH2- ß-CDとの縮合 も均一系での操作が可能になり, 水溶性カルボジイミドを用いることで容易に 進行するものと思われることから, ß-CD部分が高置換度で導入される可能性 が高い.
。-CD-NCMCの予想される特徴として, 規則正しい高次構造を構築できる 場合, CDとキトサンの両者に由来する認識能が相乗され, 高度なキラル認識 能の発現が期待できる. 言い換えれば, 隣接するCD環の共同効果, キトサン 骨格との立体障害および、水素結合の形成による分子認識が期待できる. また,
本修飾体の親水的なキトサンマトリックスはß-CD環の周りの環境を親水的に するため, ゲスト化合物との包接錯体形成に有利に作用するととも特徴の一つ に挙げられる.
6.1 実験
6.1 .1 試薬および装置
キトサンは加ト吉製の脱アセチル化95%のもの(100L, YJ24-M)をそのま
ま使用した. ß-CDは和光製の一級試薬, または三楽製のRINGDEX-Bをイオ ン交換水で再結晶し, 真空乾燥と粉砕を数回繰り返した後, 最終的に110"-'
1200Cで真空乾燥したものを用いた. なお, その際五酸化二リンのトラップを 使用し, 乾燥剤が湿らなくなるまで乾燥を続けた. 塩化4-トルエンスルホニル
(塩化トシル), 五酸化二リン, ヨウ素, 過塩素酸ナトリウムー水和物, 塩 酸, 酢酸, 水酸化ナトリウム, 塩化ナトリウム, 炭酸水素ナトリウム, リン酸 二水素ナトリウム二水和物, 酢酸ナトリウムは和光製特級試薬を, 酸化バリウ ム, アジ化ナトリウム, 5%パラジウムカーボン, 過塩素酸, リン酸水素二ナ トリウム, 金属ナトリウムは和光製試薬を用いた. SIGMA社製のグリオキシ ル酸一水和物と2,4-ジニトロフルオロベンゼン, Aldrich社製のシアノ水素化
...
ホウ素ナトリウム, 同仁化学研究所製の1ーエチル-3-(3-ジメチルアミノプロピ ル)カルボジイミド塩酸塩(WSC), および, 信越化学製の3-グリシドキシプ ロピルトリメトキシシランはそれぞれ市販品をそのまま使用した.
ピリジンは和光製を酸化バリウム存在下3---5時間還流した後 蒸留してモレ キュラーシーブ中に保存した. ベンゼンは金属ナトリウム存在下3時間加熱・
還流した後, 蒸留した. メタノールは蒸留した和光製の一級試薬および同仁化 学研究所製試薬(紫外部吸収スペクトル用)の2種類を必要に応じて使用し た. アセトニトリル, クロロホルム, ヘキサン, アセトンは和光製を, エタ ノールは日本アルコール販売製をそれぞれ蒸留して使用した. 和光製の1- ブタ ノール, 1-プロパノール, 1,1,2,2-テトラクロロエタン, 25%アンモニア水,
ジエチルエーテル, 酢酸エチルは購入品をそのまま使用した.
DNP-Lーアラニン, DNP-Lーパリン, DNP-L一ロイシン, DNP-L一フェニルア ラニン, DNP-L-トリフトファン, DNP-L-セリンは, SIGMA社製のものをそ のまま用いた. Dーアラニン, DL-ノルバリン, DL-ノルロイシン, DLーバリン,
DL一ロイシン, DL-α-フェニルグリシン, D-フェニルアラニン, D-トリプト ファン, DLーアスパラギン酸, DL-グルタミン酸ー水和物, DL-アスパラギンー 水和物, DL-シトルリンは和光製である. DL-2-アミノ-n-酪酸 DL-フェニル アラニンメチルエステル塩酸塩, DL一トリプトファンメチルエステル塩酸塩は SIGMA社より購入した.
重水は必要に応じて重水素含有率99.75%と100%の2種類を使用した. 重水 素化した水酸化ナトリウム水溶液は, 秤量した水酸化ナトリウムを重水に溶解 後凍結乾燥し, さらに一定量の重水に溶解して調製した. 重クロロホルム は和 光製を, メタノール-d4, ジメチルスルホキシド- d6, 重塩酸, 重酢酸は
Aldrich社製をそのまま使用した. 有機溶媒系の内部標準試薬には和光製のテ トラメチルシラン(TMS)を, 水溶液系の内部標準試薬にはMerck社の3-(ト リメチルシリル)-1-プロパンスルホン酸ナトリウム(DSS)をそれぞれ用い た.
薄層クロマトグラフィー用プレートは, Merck社製DC-Plastikfolien Kieselgel 60 F254 (20 X 20 cm)を適当な大きさ(7X2 cm)に切って使用 した. 担体用シリ カゲルは富士シリシア製Super Micro Bead Silica Gel (平均
�
孔径: 1037 Á, 平均粒径: 9.1μm, 表面積: 39m2g-1)を使用した. IR測定 用Merck社製臭化カリウムは120tで一晩乾燥させたものを用いた. 透析膜は SPECTRUM MEDICAL社製のSPECTRAPOR(分画分子量6000�8000)を 適当な長さ(15 cm)に切断し, イオン交換水で煮沸したものを使用した. 強 酸性カチオン交換樹脂は, オルガノ製AMBERLITE CG-120 1YPE 1を使用し た.
合成物の構造決定には主としてブルカー製ARX300NMR(300MHz) , 日本 電子製JEOL JNM-GSX400(400MHz)核磁気共鳴装置および目立製270-50 型赤外分光光度計を用いた. HPLC測定には, 東ソー製おK-GEL ODS-120T (7.8 mmIDX30 cm) , TSK-GEL SCX (6.0 mrnIDX15 cm)の2種類のカ
ラムを使用した. クロマトグラフ装置は, レオダイン社製7125型サンプルイ ンジェクタを備えた目立製 L-6000型ポンプ, 目立製L-4000型紫外可視吸光検 出器もしくは日本分光製830-RI型示差屈折計, 島津製クロマトパックC-R5A 型記録計もしくは東亜電波製CDR-11A型記録計で構成させた. また, 調製し た固定相のキラル分離能の評価には, テフロンチューブ(外径2 mmx内径0.5 mm)を分離カラムとする日本分光製Familic-100型ミクロポンプ, UVIDEC- 100型回折格子紫外分光光度計, および東亜電波製EPR-211A型記録計で構成 される装置を使用した.
6.1 .2 N-(カルボキシメチル)キトサンの合成
Chitosan
CHOCOOH
\1 !…�'ol
NaBH3CNN、‘
C HCOOH 11
Scheme 6-1 Synthesis ofNCMC
ド巴子
NH CH2COOH NCMC。-CDをキトサン骨格に導入する際の前駆体の一つであるNCMCをまず合
可..
成する必要がある. 本節ではMuzzarelliらの報告22)に従って, 水溶性が非常に 高く, またß- CD導入に有利と思われるスペーサーを有するN-(カルボキシメ チル)キトサン(NCMC)を合成した(Scheme 6-1) .
キトサン5.0g(グルコサミンユニットに換算して31.0mmol)をグリオ キシ ル酸 1 水和物7.13 g (77.5 mmol)を含む水溶液650 cm3に溶解し1.0 mol dm-3水酸化ナトリウム水溶液を少量ず、つ加えてpH5.0に調整後, 48時間室温
で撹持すると粘度が高くなりゲル状のシッフ塩基が生成した. この粘性溶液 に, シアノ水素化ホウ素ナトリウム3.90g (62.1mmol)を溶解したイオン交 換水10cm3を加えて24時間撹枠すると溶液の粘度は低下し若干白濁した. エタ ノール 1dm3を加えたところ, 白色沈殿が生じたため1時間静置し, デカン テーションにより上澄みを除去した. 次に0.1 mol dm-3NaOH水溶液 1dm3に 溶解Lたあと少量の不溶物を遠心分離で除去し, 再度エタノール1.0dm3を添 加して再沈精製した. 得られた白色ゲルはNCMCのナトリウム塩(NCMCNa) で減圧乾燥により3.37 g を得た.
さらに, 得られた生成物のうち50.0mgをイオン交換水10cm3に溶解し,
1.0mol dm-3塩酸を用いて pH1.0に調整した. 30分間携枠後, エタノール 10cm3を加えて撹枠し2 時間静置した. 生じた白色沈殿を遠心分離(
4000rpm, 5 分間)した後, 上記と同様の方法によりエタノールで2回洗浄し た最後に, 得られた白色沈殿を真空乾燥(収量: 31.9mg)し, とれを NCMCの塩 酸塩 (NCMCHCl)とした.
原料 キトサンの脱アセチル化度はlHNMRにより決定した. キトサン5.0mg を3.0%重酢酸/重水0.5cm3に溶解し凍結乾燥した. 得られた固体を再度 3.0
%重酢酸/重水0.5cm3に溶解し, lHNMRスペクトルを測定した. NCMCNa およびNCMCHClのlHNMRスペクトルは, それぞれ5.0mgを重水1.0cm3に 溶解し凍結乾燥した後, アルカリ状態での測定では0.01mol dm-3のNaOD/重 水0.5cm3に再度溶解して, また, 酸性状態での測定の場合には重水0.5cm3に 再度溶解後, 6.0mol dm-3重塩 酸を用いてpD1.0に調整し直して測定した.
13 C核磁気共鳴スペクトル測定用試料はNCMCNa 50.0mgを重水0.5cm3に 溶解し凍結乾燥した. この操作を2回繰り返した後, 0.01mol dm-3のNaOD/
重水0.5cm3に再度溶解して調製した. また, 外部標準試薬としてτ'MSを封入
�
したキャピラリーを試料管中心部に入れて用いた.
6.1 .3 モノ(6-アミノー6-デオキシ)- ß-シクロデキストリンの合成 本項では, ß-CD-NCMCのもう片方の前駆体であるNH2- ß-CDの合成に ついて述べる. これ はMelton等の報告23)を参考にして, 以下の3段階の経路 (Scheme 6-2)に従って 合成した
白 ß-CD
+ TsCI 勘ト己
+ NaN3 -v白
H21 Pd-C ートScheme
6・2
Synthesis ofNH2-ß・CD
6.1.3.1 βーシクロデキストリンのモノトシル化
白 白
白
NH2・ß-CD
。-CDの6位ヒドロキシル基と塩化トシルを反応させた後, 再結晶により モノ(6-ひp-トリル スルホニル)-ß-シクロデキストリン(モノトシル化。
CD)を得た. 本反応 はMeltonらの報告23)をはじめ, Matsuiらの報告24) 石原らの報告25)を参考に行った
乾燥ß-CD50.0 g (44.1mmol)を乾燥ピリジン500cm3に少量ずつ溶解し た. 之の操作はアルコン雰囲気中で、行った. 30分間氷冷(50C以下)した 後, 塩化トシル8. 41g (44.1mmol)を含む乾燥ピリジン溶液50cm3を1時間か
けて少量ずつ滴下した. これを20tの恒温槽に入れ 2 4時間携枠した. 反応溶
司....
液にイオン交換水50cm3を加えてエバポレーターで濃縮した. この操作をピリ ジン臭がしなくなる程度まで数回繰り返した. 水分がなくなるまで十分濃縮乾
回し, さらに粉砕して白色粉末を得た. 未反応の塩化トシルを除去するため に, この粉末をジエチルエーテjレ250cm3で洗浄し上澄みをデカンテーション で除去した. この洗浄を2回繰り返した. 最後にイオン交換水で2 回, 1 -ブタ ノール:エタノール:イオン交換水=5:4:3混合溶媒で 3 回再結品し, 得ら れた魚鱗状白色結晶を真空乾燥した. 収量:10.lg (収率:18%) .
6.1.3.2 モノ(6- 0- p-トリルスルホニル)-βーシクロデキストリンの アジド化
モノトシル化。 -CDに求核試剤であるアジ化ナトリウムを反応させて, モノ (6ーアジド-6ーデオキシ)- ß-シクロデキストリン(アジド化。-CD)を合成し
た. 本反応はMeltonらの報告23)に従った.
モノトシル化。-CD6.0g (4.65mmol)とアジ化ナトリウム6.0g (92.3 mmol)にイオン交換水600cm3を加え, 95tの湯浴中で9 0分間携枠, 還流し た. 反応溶液を室温まで放置冷却後鴻過した. その滅液をエバポレーターで 30cm3にまで濃縮した. これを遠心管2本に分け入れ, それぞれに1, 1,2,2ーテ
トラクロロエタン10cm3を加えて激しく振渥撹持した. 白濁した溶液を遠心分 離(4000 rpm, 10分間)したところ, 3層(上から水層, 白色沈殿, テトラク ロロエタン)に分離した. このうち最上層の水層を除去した. 遠心管の内容物 全てをビーカーに移し, イオン交換水を少量ずつ加えながら950Cの湯浴中で携 枠した. 白色沈殿が完全に溶解した時点で熱時水層のみを回収し一晩静置冷却 すると, 輝光性白色結晶が析出した. これをグラスフィルターで吸引滅過した 後, 真空乾燥した収量:4.88g (収率:91%) .
6.1 .3.3 モノ(6- アジドー6-デオキシ)- ß-シクロデキストリンの還元
パラジウムカーボンを触媒としてアジド化。-CDを接触還元した. 本反応も Meltonらの報告23)に従った.
アジド化。-CD4.80g (4.14 mmol)をイオン交換水500 cm3に加えて懸濁 し, 5%パラジウムカーボン1.41gを添加した後, 水素雰囲気下(1.0kg/cm2)
司...
室温で24時間境枠した. 反応溶液を孔径0.45μmのメンブレンフィルターを用 いて吸引滅過し, その鴻液をエバポレーターで濃縮した. 得られた淡黄白色固 体を真空乾燥した. 収量: 4.08g (収率: 87%) .
6.1.3.4 モノ(6-アミノー6-デオキシ)-βーシク口デキストリンの精製 粗アミノ化。-CDはカチオン交換樹脂を用いたイオン交換クロマトグラ
フィーにより精製した. なお樹脂はスルホン酸基を有する強酸性カチオン交換 樹脂を, 溶出に使用するアルカリ水溶液には脱塩操作を必要としないアンモニ ア水を用いた.
強酸性カチオン交換樹脂100gを 3日間アセトニトリルでソックスレー洗浄 した. 続いてイオン交換水中で撹枠・静置した後, 微粒子をデカンテーション で除去した. この操作を微粒子がなくなるまで繰り返し, φ2.6X50cmのカラ ムに充填した. これに5.0mol dm-3アンモニア水400cm3および, 溶出液が中 性になるまでイオン交換水を順次流して樹脂を脱色・洗浄した. さらに4.0mol dm-3塩酸800 cm3• 2.0mol dm-3塩化ナトリウム水溶液400 cm3, 2.0mol dm-3塩酸400cm3 イオン交換水を順次流して樹脂を H型に再生した
粗アミノ化。-CD4.00g(純度66%, アミノ化。-CDに関して2.31mmol)を イオン交換水70cm3に溶解した(pH1.8). これを上述のカラムに流し, 次い でイオン交換水で洗浄した. この際, 溶出液を200cm3ごとのフラクションに 分けて, ß-CDが検出されなくなるまで、1000cm3流した. 続いて5.0mol dm-3 アンモニア水を流してアミノ化 。-CDを溶出させた. 先と同様に, 溶出液を 200cm3ごとのフラクションに分けてアミノ化。-CDが確認されなくなるまで 2800cm3で、洗浄した. 得られた全てのアンモニア溶出液をエバポレーターで濃 縮すると淡黄色国体が析出したため, とれを真空乾燥した. 収量: 2.25g.得ら れた淡黄色粉末をイオン交換水25cm3に再度溶解し, アセトニトリjレ250cm3
(水溶液の10倍量)を加えて携枠した. 生じた淡茶白色沈殿を遠心分離(
4000rpm, 10分間)した後, 真空乾燥した. 収量: 2.24g (回収率: 86%) .
6.1.3.5
総括本合成経路は文献などですでに知られているものであるが, その収率や純度
...
は大きく実験者のテクニックに依存するため 注意を要する操作や注意点を以 下に述べる.
まず, ß-CDからトシル化。-CDを誘導する過程が重要なポイントである.
以下の点に特に留意する必要がある. (1) 反応に使用するß-CDおよびピリジ ンの乾燥を十分に行う. とくに再結晶後のß-CDは五酸化リン存在下で十分な 乾燥を行うこと. (2)ピリジンの除去は低温(40t以下)かつ迅速に行う.
(3)
ジエチルエーテルによる洗浄は, 反応物の水分を十分に除去し乾燥粉末にした 後で行う. また洗浄後可能なかぎりエーテルを除去する. これらによってある 程度の収率の再現性 (通常15�20%) が確保できた.次に, 粗アミノ化。-CDのイオン交換カラムによる精製では あらかじめ樹 脂を洗浄したにもかかわらずアンモニア溶出液に著しい着色が認められる場合 がある. これは高濃度のアンモニア水の導入に伴う中和熱の発生によって, 樹 脂からの着色成分が脱離するものである. この問題の解決策として, 低濃度の アンモニア水(約1.0mol dm-3)で溶出を始め 徐々に濃度を高くし最終的に必 要な高濃度のアンモニア水(この場合5.0mol dm-3)を溶出させるという, グラ ジエント溶出が効果的であった.
。-CDを原料とするアミノ化。-CDの合成は全体としての収率は約15%で あった.
6.1.4 ß-シク口デキストリン修飾キトサンの合成
カルボジイミドを用いて NCMCとアミノ化。-CDを縮合させ, ß-CD残基 を有するキトサン誘導体 (ß -CD-NCMC) を合成した. ペプチド合成試薬の カルボジイミドとしては 一般にジシクロヘキシルカルボジイミド (DCC ) と水溶性カルボジイミド (WSC) の2つがよく知られているが, 本研究では NCMC, アミノ化 。-CDいずれもが水に対して高い溶解性を示すこと, また 反応後の尿素誘導体がWSCを用いた場合水溶性を示し透析による除去が可能に なることなどを考慮して WSCを用いた (Scheme
6-3) .
本合成のポイントは, ポリマー主鎖にどれだけ高置換度でß-CD残基を導入 出来るかという点である. そこで反応に際しては, 以下の点に留意して高置換 度の修飾体の生成を試みた.
...
(1) 反応系の濃度
カルボジイミドによる縮合を 収率よく行うには, 不活性な 中間体であるアシル尿素の生 成を防ぐために, 試薬濃度を 高くすることが必要となる.
そのため個々の溶液は出来る 限り高濃度とし, これらを混 合し反応させた. また修飾体 の主鎖となるNCMCに対して
とH2COOH 4H2
C=O
。・CD・NCMC Scheme 6-3 Synthesis of
ß-CD・NCMC
NH2-ß-CDは3倍量, 縮合剤のWSCは60倍量を使用した(WSCは通常理論 量の10"-' 100倍が使用されている26) ) .
(2) pH
WSCを用いた反応の至適pHは4.5"-'6.0とされている26) . 一方NCMCは pH2"-'4付近では溶解性を示さない. 以上の条件を踏まえ, 本研究では比較 的中性 に近いpH6.0"-'6.5での反応を行った. なおWSCを用いた反応では,
反応初期 (特に反応開始から数時間)のpH変化が著しいことが知られてお り, この間 pHの調整をこまめに行った.
( 3 ) 反応後の処理
高濃度下での反応を行うため, 反応後の溶液には大量の低分子不純物(未反 応のNH2-ß-CDやWSC, 水溶性カルボジイミドから生じる大量の副生成物 の尿素誘導体(WSU)が含まれる. これを除去する操作としてセルロース 膜透析を行い, その後エタノールで生成物を析出させた.
6.1.4.1
アミノ化ß-シクロデキストリンと N喧カルボキシメチルキトサンの縮合
NCMCNa 127mg (カルボキシメチル基導入率84.5%, カルボキシメチル 化されたグルコサミンユニットに関して0.47mrnoDをイオン交換水10cm3に 溶解した. との時pH8.2であったため, 1.0mol dm-3塩酸を少量ずつ加えて pH6.3に調整した(溶液A). NH2-ß-CD1.76g (純度93.9%, 1.46mrnoD
...
をイオン交換水50cm3に溶解し, 微量の不溶物を除去するために遠心分離(
4000 rpm, 10分間)を行った. この時pH8.3であったため 1.0mol dm-3塩酸 を少量ずつ加えてpH6.3に調整した(溶液B). WSC 5.60g (29.2mmol)を イオン交換水20cm3に溶解した. この時pH6.7であったため 1.0mol dm-3塩 酸を少量ず、つ加えてpH6.3に調整した(溶液C ). 溶液Aに溶液Bを少量ずつ 滴下・混合した. この混合溶液を25.00Cの恒温槽に入れ, 溶液C を少量ずつ滴 下・混合した. これをアルゴン雰囲気下で24時間撹枠した. なお, 反応初期の
1 時間は10分おきにpHを調整し6.0'"'-' 6.5の範囲に保った.
24時間後, 反応溶液中に微量の不溶物が生じていたため, 4000rpmで10分 間遠心分離を行い上澄みを回収した. 1.0mol dm-3塩酸を用いてこの上澄みを pH2.0に調整し, メンブレンチューブ(分画分子量: 6000'"'-'8000)に入れて 外部液がpH2.0の塩酸中で 3 日間透析を行った(ldm3ビーカーを使用し, 定 期的に外部水溶液を交換した). 次いで1.0mol dm-3水酸化ナトリウム水溶液
を用いてpH12. 0に調整し直し, 先と同様にpH12.0の水酸化ナトリウム水溶液 中で3日間透析を行った. エバボレーターで約10cm3にまで濃縮し, 3倍量(
30 cm3)のエタノールを加えて撹持した. 生じた白色ゲル状沈殿を4000rpm で10分間遠心分離し真空乾燥した. 得られたものはß-CD-NCMCのナトリウ ム塩(ß -CD-NCMCNa)である. 収量: 142mg. 酸型のß-CD-NCMCHCl は溶解した水溶液をpH2.0に調整後, エタノールで再沈殿することによって得 た. ß -CD- NCMCNa 5.0mgを重水1.0cm3に溶解し凍結乾燥した後, 0.01 mol dm-3 NaOD/重水1.0cm3に再度溶解してlH N MRスペクトル測定用試料 とした.
6.1.5 β-CDシク口デキストリン修飾キ卜サンのシリカゲルへの固定
本項ではß-CD- NCMCをHPLC固定相として用いる場合の調製法について 述べる. 通常CDやその誘導体を固定相として利用する場合, スペーサーと呼 ばれる有機シランを介して 化学結合で CDをシリカゲルに固定化する方法に よってカラム充填剤が調製されており737,28), 本研究でもこの方法に従って
。-CD-NCMCをシリカゲル表面に共有結合で固定化した. なお担持に使用す るスペーサーとしては (1)窒素原子を含むものは被覆率が低く, 化学的に不
『・w
安定であると言われている11)ため窒素原子を含まないこと, (2) ß-CD-
NCMC中に多数存在するヒドロキシル基との反応が可能であることから, その 末端にオキシラン環を有する3-グリシドキシプロピルトリメトキシシランを 用いシラン化した後ß-CD-NCMCを共有結合させたCScheme
6-4) .
+w- pC H釘0・CH ペア 2
\-ーイO
…h
O
トO-Siー(CH?b-{)・CHっCH一一CH。
Scheme
6-4Modification of silica geI with ß-CD・NCMC
6.1.5.1
シリカゲルへのオキシラン環の導入大孔径シリカゲ、ル1.00gに2.0mol dm-3の塩酸20cm3を加えて900Cの油浴中 で2時間撰搾・還流した. 室温まで放置冷却後, グラスフィルターで吸引滅過 した. イオン交換水20cm3による洗浄とグラスフィルターでの吸引鴻過を5回 繰り返した後, 1200Cの乾燥器中で一晩乾燥させた. セプタムキャップを付け た三つ口フラスコにこのシリカゲルを入れ, 真空ポンプで10分間減圧した後,
120Lの油浴中で再度3時間真空乾燥した. 減圧状態で10%の3ーグリシドキ シフロピルトリメトキシシラン/乾燥ベンゼン溶液20cm3をセフ。タムキャップ
�
からシリンジ針で注入し, 窒素雰囲気下850Cで12時間携枠・還流した. 室温 まで放置冷却後, グラスフィルターで吸引猿過した. メタノ-)レ20cm3による 洗浄とグラスフィルターでの吸引鴻過を5回繰り返した後, 真空乾燥を行った (収量 : O.86g) .
6.1 .5.2 βーシク口デキストリン修飾キトサンのシリカゲルへの固定お よびカラムの作製
。-CD-NCMCNa20.0mgをO.lmol dm-3水酸化ナトリウム水溶液1.0cm3に 溶解し, 先のシラン化シリカゲル100mgに少量ずつ加えた. これを超音波で懸 濁した後, 400Cの湯浴中で48時間撹持した. この際, シリカゲルを傷めない ようにするため撹枠子は使用せず, 20cm3のナス型反応容器全体をゆっくりと
回転させた. 反応後, 遠心分離(4000rpm, 10分間)を行い 上澄みを除去し た. イオン交換水5.0cm3での洗浄と遠心分離を上澄みが中性になるまで繰り 返した後, 真空乾燥し白色粉末を得た(収量: 88.0mg) .
得られたß-CD-NCMC担持シリカゲjレ30mgをメタノ-)レ0.3cm3に懸濁さ せ, Familic-100型ミクロポンフを用いてテフロンチューブに充填し, 約15cm の長さのHPLC用カラムとした.
6.1 .6 アミノ酸および類似化合物の2,4-ジニトロフェニル誘導体化
N02
R-CH-COOH
('1N02
弘、/グ
F〈UY
N02
Scheme 6・5 Synthesis of 2,4・DNP derivatives
。-CD-NCMC担持シリカゲル固定相によるキラル分離対象試料として, ア ミノ酸およびその類似化合物のDNP誘導体を用いた. 市販されていないものに
ついては, ラセミ体または一方のエナンチオマーのDNP誘導体化を行った. 反
--
応はSanger試薬と呼ばれる2,4-ジ、ニトロフルオロベンゼンを用い, 遊離のア ミノ酸また は類似化合物を誘導体化した(Scheme 6-5) . 本研究ではLiらの 報告14)を参考にして行った. 原料によって, 精製操作を含めて大きく4種類 の反応操作に分類できたので以下にその代表反応操作例を述べる. 全ての誘導 体 は1HNMRによって同定した.
6.1 .6.1 D-アラニンの2,4-ジニトロフェニル誘導体化
D-アラニン890mg(10.0mmol)を1.0mol dm-3炭酸水素ナトリウム水溶液 20cm3に溶解し, これに2,4ージニトロフルオロベンゼ、ン1.40cm3(10.0 mmol )を含むエタノール溶液20cm3を加えて, 密栓ナスフラスコ中室温で3時間携 枠した. エバポレーターで濃縮した後, その残留物をイオン交換水10cm3に溶 解した. 未反応の2,4-ジニトロフルオロベンゼンを除去するためにジエチル エーテjレ30cm3で6回抽出を行った後, 水層を6.0mol dm-3塩酸でpH1.5に調
整した. 水層が透明になるまで、ジエチルエーテjレ30cm3で抽出を行いエーテル 層全てを 濃縮し, 得られた淡黄色結品を真空乾燥した. 収量: 1.96g (収率:
77%) , 融点: 176.5-179.30C.
同様な 操作で以下の2,4ージニトロフェニル(DNP)誘導体を得た.
DNP一DL-2-アミノ-n-酪酸;
収量: 2.25g (収率: 84%) , 融点: 148.7 -150.30C.
DNP一DL-ノjレノてリン;
収量: 2.28g (収率: 81%) , 融点: 141.6-144.20C.
DNP一DL-ノノレロイシン;
収量: 2.24g (収率: 75%) 融点: 95.8-100.20C.
DNP一 DL-パリン;
収量: 2.50g (収率: 88%) , 融点: 185.1-188.30C.
DNP一DL-ロイシン;
収量: 2.50g (収率: 84%) , 融点: 123.0-125.40C.
DNP一DL一α-フェニルグリシン;
収量: 2.14g (収率: 68%) , 融点: 180.1-181.90C.
DNP-D-フェニルアラニン;
...
収量: 1.62g (収率: 49%) , 融点: 188.4-189.9t.
DNP-D-トリフ。トファン;
収量: 1.65g (収率: 89%) , 融点: 212.7-215.7t.
DNP-D-セリン;
収量: 2.39g (収率: 88%) , 融点: 179.7-182.3t.
DNP一DLーアスパラギン酸;
収量: 1.26g (収率: 42%) , 融点: 186.6-189.0t.
DNP- DL-グルタミン酸;
収量: 1.23g (収率: 39%) , 融点: 74.8-77.8t.
DNP一DL-3-アミノ-3-フェニルフ。ロピオン酸;
収量: 473mg (収率: 57%) , 融点: 67.5-69.9t.
6.1 .6.2 DL-フェニルアラニンメチルエステルの2,4-ジニトロフェニ ル誘導体化
DLーフェニルアラニンメチルエステル塩酸塩1.08g (5.0mmol)を1.0mol dm-3炭酸水素ナトリウム水溶液20cm3に溶解し, これに2,4ージニトロフルオ
ロベンゼ、ンO.70cm3 (5.0mmol)を含むエタノール溶液20cm3を加えて, 密栓 ナスフラスコ中室温で 3 時間撹持した. エパポレーターで濃縮した後, 残留物 にイオン交換水10cm3を添加した. ジエチルエーテjレ30cm3で抽出を試みたと
ころ黄色沈殿が生じ, これは水層, エーテル層いずれにもほとんど溶解しな かったのでグラスフィルターで鴻別して一旦真空乾燥した後, クロロホルム:
ヘキサン=1: 1混合溶媒で再結品した. 得られた濃黄色結晶を吸引鴻過し真空 乾燥した. 収量: 464mg (収率: 27%) , 融点: 120.1-120.90C.
同様の操作でDNP一DLートリプトファンメチルエステルを得た. 収量:
300mg (収率: 31%) . 融点: 187.7 -188.50C.
6.1.6.3 DL-シトルリンの2,4-ジニトロフェニル誘導体化
DL-シトルリン438mg (2.5mmol)を1.0mol dm-3炭酸水素ナトリウム水溶 液5cm3に溶解し, これに2,4ージニトロフルオロベンゼ、ンO.35cm3 (2.5江田101) を含むエタノール溶液 5 cm3を加えて, 密栓ナスフラスコ中室温で3時間撹枠
.,.r
した. エバポレーターで濃縮した後, その残留物にイオン交換水2.5cm3を添 加した. 未反応の2,4ージニトロフルオロベンゼンを除去するためにジエチル エーテjレ10cm3で6回抽出を行った後, 水層を6.0mol dm-3塩酸でpH1.5に調
整した. 再度ジエチルエーテル10cm3で抽出を試みたところ黄色沈殿が生じ,
これは水層, エーテル層いずれにもほとんど溶解しなかった. そこでこの沈殿 をグラスフィルターで漉別し真空乾燥し黄色結晶を得た. 収量: 503mg (収
率: 59%) , 融点: 180.7-182.50C.
6.1.6.4 (R)ー(+)-2-アミノー3-フェニルー1-プ口パノールの2,4-ジ ニトロフェニル誘導化
(R)-(+)-2ーアミノ-3-フェニル-1 -プロパノ�)レ378mg (2.5mmol)を1.0 mol dm-3炭酸水素ナトリウム水溶液10cm3に溶解し, これに2.4 -ジニトロフ ルオロベンゼン0.35cm3 (2.5mmol)を含むエタノール溶液10cm3を加えて,
密栓ナスフラスコ中室温で3時間携枠した. エバポレーターで濃縮した後, そ の残留物をイオン交換水10cm3に溶解した. ジエチルエーテル30cm3で抽出を 行ったところ, エーテル層に著しい着色が認められたため, 水層が透明になる までこの操作を繰り返した. エーテル層全てを濃縮し油状の生成物を得た. こ れをメタノール5.0cm3に溶解し, 冷凍庫(OOC以下)で1週間静置した 生じ た結晶を回収するため冷却時に上澄みを除去し, そのまま凍結乾燥した (常温 では溶解する恐れがあるため, 鴻過操作は行わなかった). このメタノールで の再結晶を 2 回繰り返し, 最終的に吸湿性の黄澄色結品を得た. 収量: 336mg
(収率: 42%) 融点: 48.4-51.60C.
同様の操作でDNP-(S)-(一)-2 -アミノ-3ーフェニル -1-プロパノールを得た.
収量: 414mg (収率: 52%) , 融点: 48.0-51.0oC.
6.2 結果および考察
6.2.1 キトサンおよびN-カルボキシメチルキトサンの構造
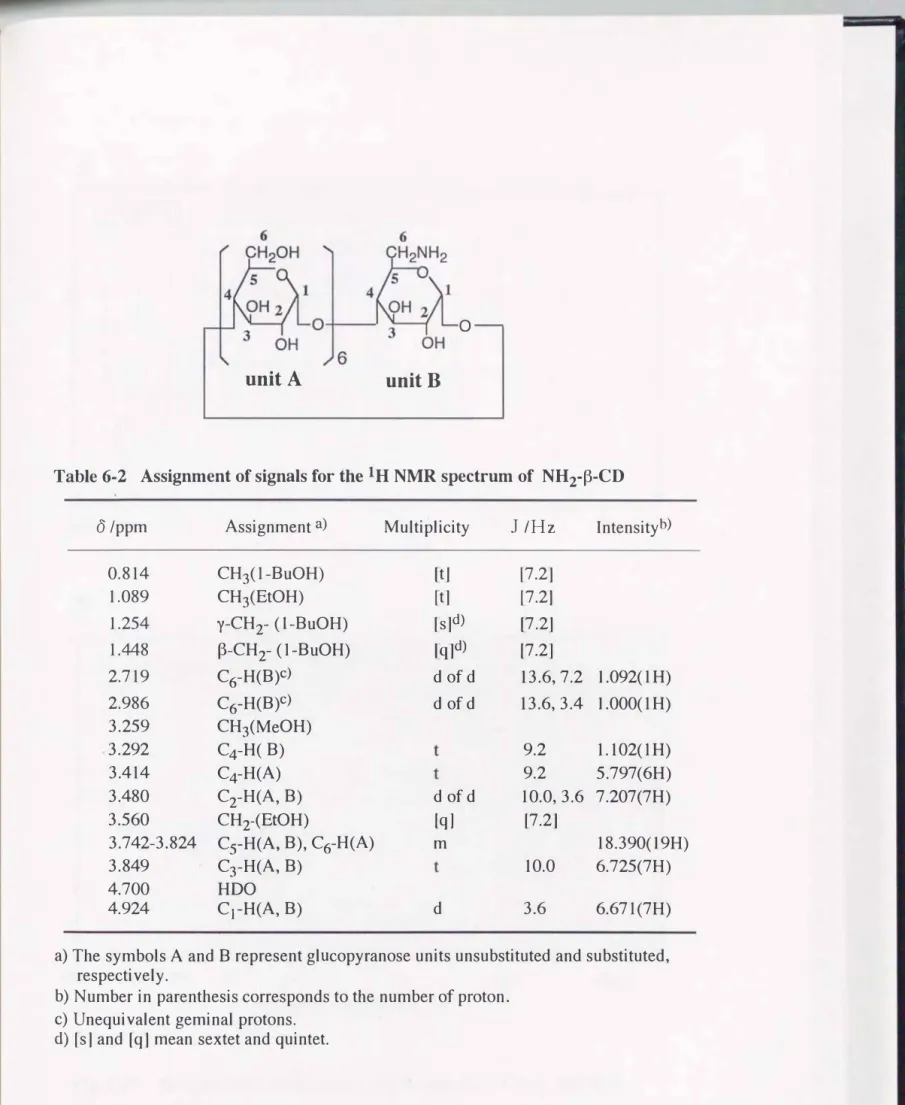
臭化カリウム錠剤法による赤外吸収スペクトルおよびlHNMRスペクトルを それぞれFig. 6 -1, Fig. 6-2に示す. Fig. 6 -1のIRスペクトルには1612cm-1の アミノ基由来の吸収以外に, 1662cm-1にN-アセチル 基のC=o伸縮振動の吸
--
2,�,凶'" I , 10 11 12 IJ l' 15 20 25 30 ・η
! '1:
1 '1 i1': 1: 1:1,:',:úlid
"11i・'dUù',J"I'1'I1rlrflll'rI
M��I,\!
rl'd111!':',I;'lï,lm \\'; ]'1\ '1\1,' ; i � ! Ili\\'Oi,':fl'iii""':,''Ii' ,1・
'1'
rl-1.1.i 4出 |出 i4ifii i
i!llllli
斗1ーー・1
+I �-II
I!日I�H
' ー
�
1: I J !- 1111 111U
!.;!IIIWlli!
! L�_ t
ーIt
.匠‘比-lrp. 十if↓
ヨl r-[iJ ',I'ttt !皆, ��ifltH' lHI
Illí rm !11i IfHf fI
ム L
�� 信防 l立 ii
,,� じ Il tl\
四�
以l|!i
!間
k
i‘ ー
1147
�() ーT「 7L. ・111
tl
UI1m 1i 間!!i !出 |出 lHl!| H U l;|1ii|
1'�lt
ト十+時 将開|出It 附ii
11.> 』 " I�ム=I� .�t-"
40 00 3500 3000 2500 2000 1500 Wavenumber / cm・1 Fig.6・1 IR spectrum of chitosan
5
10 9 8 7 6 5 4
ö/ppm
i iii
'i!!T
I ï
mm,ì
�111.lii
1000
3 2
jilli!lii
� i i!l1
i11i1181i
t11aI ll lil ιi ' 1 1 』'''1, 'I,! 'I!
ι | l l:;
←附 3
! : ! |L
ilJ
; iIii!
il
d'! I
i:';I
i � i!P
(i!iiiiii!;
5ÙO
。
Fig.6-2 400 MHz lH NMR spectrum of chitosan. The spectrum on the upper side shows an enlarged spectrum. Chitosan was dissolved泊30/0
CD3COOD 1D20. The chemical shift was calibrated by HDO signal at ö=4.70.
『司'
収が認められた. 本研究で使用したキトサンは"100%脱アセチル化"と表示さ れたものであるが, 完全に脱アセチル化されていないためにこのような吸収が 見られたものと思われる. このことはさらにlHNMRスペクトルFig.6-2にお いてはっきりと確認できる. Ô 1.985にアセチル基に由来するメチルシグナル が検出され(それより若干高磁場のÔ 1.948のシグナルは重酢酸溶媒に微量含 まれる酢酸のアセチル基である), このシグナルとDーグルコサミンユニットお よびNーアセチルーDーグルコサミンユニットのC2-HがÔ 3.0 92に共有するシグ ナルを比較すると, 94.6%脱アセチル化されたものであり, 高分子の糖ユニッ ト20個あたり約1 個にアセチル基が残存している構造を有することが示唆され た. 本研究ではこの" 94.6%脱アセチル化キトサン"をそのまま使用することと した.
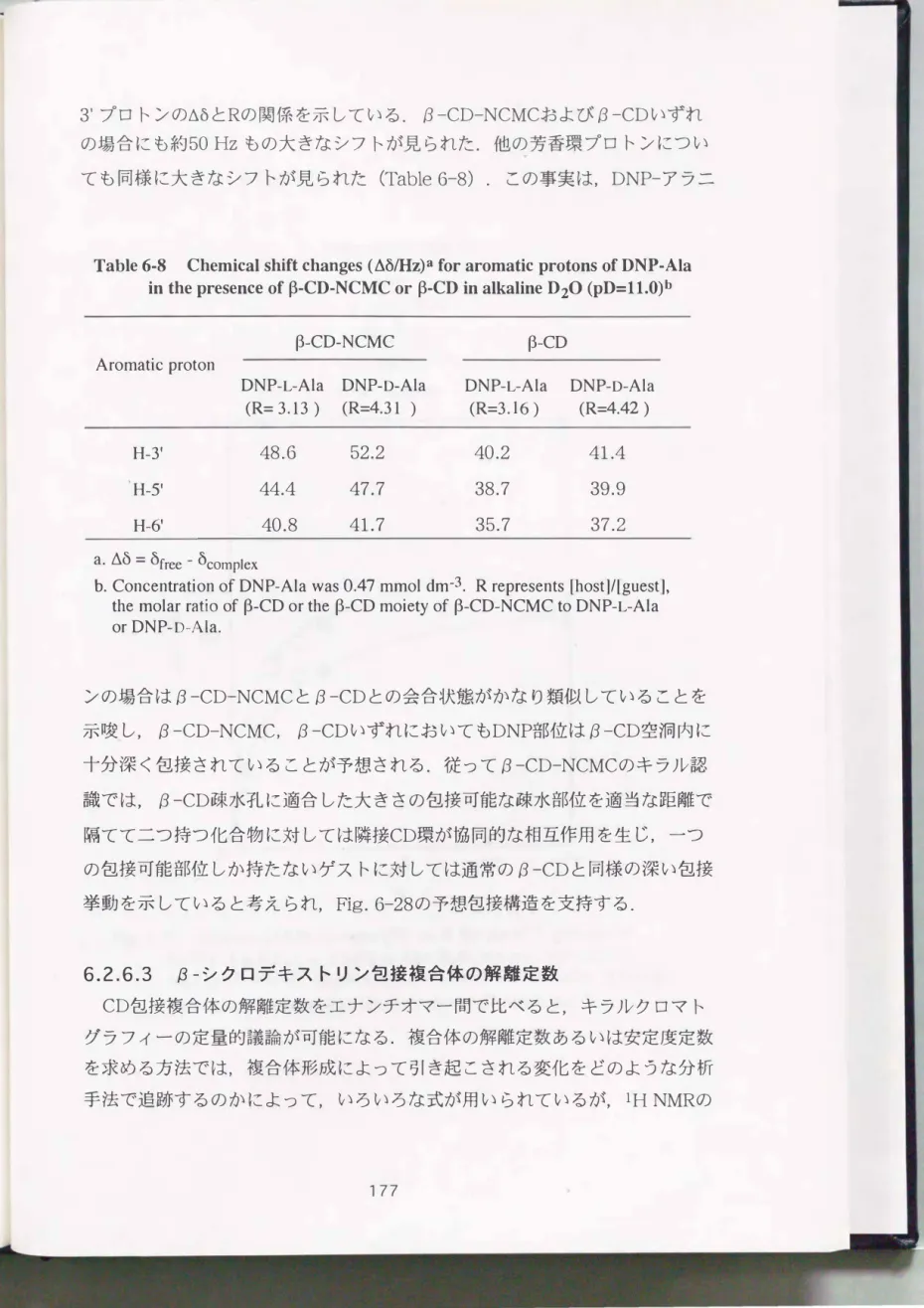
NCMCNaのIRスペクトル(Fig.6- 3)は1600cm-1付近にカルボキシラート アニオンのcoo-逆対称伸縮振動吸収帯が 1410cm-1イ寸近に同じくカルボキ シラートアニオンの対称伸縮振動吸収帯が顕著に現われている. これらは原料 キトサン(Fig.6-1)には見られなかった吸収であり, カルボキシメチル基の 導入を示唆している. また酸性条件下で析出させたNCMCHClのIRスペクトル (Fig.6- 4)では, 1740cm-1イ寸近にカルボキシル基のC=o伸縮振動, 2500--- 3300cm-1の広範囲にわたってカルボキシル基のO-H伸縮振動の吸収が認めら れる. これらもカルボキシメチル基の存在を支持している.
反応原料であるキトサンのlHNMRスペクトル(Fig.6- 2)と比較すると,
NCMCNaのスペクトル(Fig.6- 5)ではÔ 3.3付近に, NCMCHClのスペクト ル(Fig.6- 6)ではÔ 4.1付近に, それぞれ新たなシグナルが確認できる. これ らはいずれもカルボキシメチル基のメチレンプロトンに帰属され, 隣接するア ミノ基がプロトン化することで, 0.8ppmもの低磁場シフトを引き起こしてい ることがわかる.
NCMCNaのÔ 2.7付近にはショルダーとして小さなシグナルが見られるが,
これは, 修飾されなかったグルコサミンユニットのC2-Hか, またはRinaudo 等29)が報告しているようにN,Nージカルボキシメチル化されたユニットが重 なっているものと思われる. また ここには示していないが, 水溶性の50%脱 アセチル化キトサンのC2一日がÔ 3.61にシグナルを示すことから, N-アセチル
....,r
.
4000 3500
Fig.6・3
15 IOC
?O
80
10
4000 3500
Fig.6・4
Wavenumber / cm・1 IR spectrum of NCMCNa
3000 2500 2000 1500
Wavenumber / cm・1 IR spectrum of NCMCHCI
,。 11 U 13 1・15
1000
20 7� '0 ・n
500
司司...
4 3 2
10 ‘ 9 8 7 6 5 4 3 2 1 0
ö/ppm
Fig.6・5 400 MHz lH NMR spectrum of NCMCNa. The spectrum on the upper side shows an enlarged spectrum. NCMC was dissolved in alkaline D20 (pD=12). The chemical shift was calibrated by HDO signal at δ=4.70.
5 4
10 9 8 7 6 5 4
ö/ppm
3 2 。
Fig.6・6 400 MHz lH NMR spectrum of NCMCHCI. The spectrum on the upper side shows an enlarged spectrum. NCMC was dissolved in acidic D20 (pD=1.0). The chemical shift was calibrated by HDO signal at ö=4.70.
『冒.
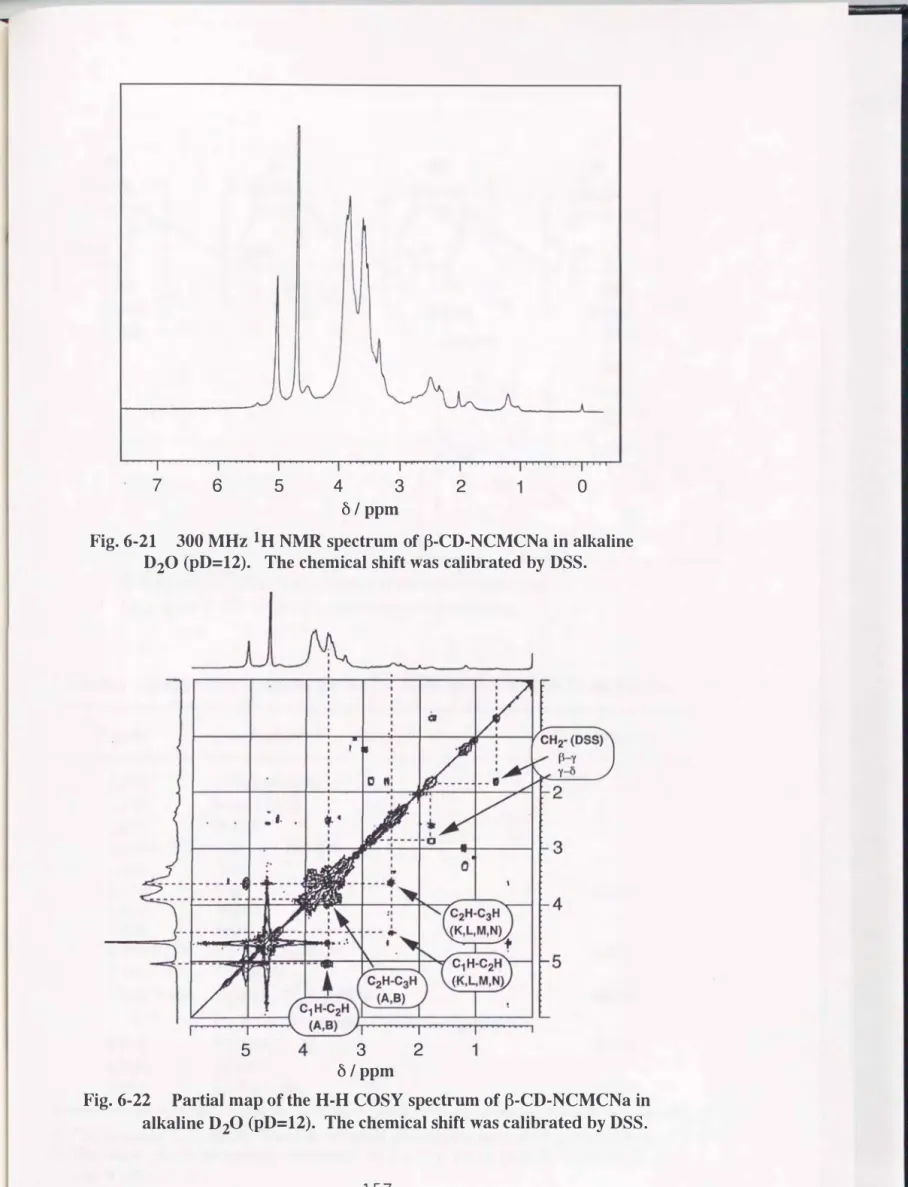
グルコサミンユニットのC2-Hは, C3-HからC6-Hのシグナル(ô 3.3-4.2) に隠れていることが予想された. これらのことについてはFig.6- 7の日-H COSYスペクトルで確認することが出来た.
Fig.6-7において, Nーカルボキシメチル化グルコサミンユニットとN-アセ チル化グルコサミンユニットのC2-Hはそれぞ、れ, Ô 2.51とÔ 3.76に, またそ れらとの交差ピークより, これら二つのユニットのC1-Hがô4.5 3と4.5 7のわ ずかな違いではあるが, 別々のケミカルシフトを有していることも分かる. ま た, 図中のAとA'の矢印で示した交差ピークはグルコサミンユニットまたは,
N, N-ジカルボキシメチル化されたユニットのC2-HとC1-Hおよび, C2-Hと C3-Hとの相関を示している. また, 矢印BはA, A'に帰属されない方のユ ニットのC2一日とC3-Hとの相関が現れているものと解釈される. これらがいず れに相当するのかについては特定できなかった.
このように, 得られたNCMCは目的以外の少量のユニットを含んでいるが,
それらの割合を酸性状態でのスペクトル(Fig.6-6)のC2-Hシグナルで決定し た. この場合も, グルコサミンユニットまたは, N,Nージカルボキシメチル化 されたユニットはÔ 3.1付近に別々に小さな 2 つのシグナルを示しており, ど ちらかの特定は困難で、あるが, ほぼ同面積のピークを示しているため同量含ま れると仮定した. とれと, Ô 2.06のN-アセチル基の面積の3分の 1および目 的物のN-カルボキシメチル化されたユニットのÔ 3.36のC2-Hシグナル面積の 比を計算した. その結果, Nーカルボキシメチル化ユニットリv-アセチル化ユ ニット: N,Nージカルボキシメチル化ユニット:グルコサミンユニットは 0.84 : 0.06 : 0.0 5 : 0.0 5の比で含まれることが分かった(Fig.6-1 0の構造式).
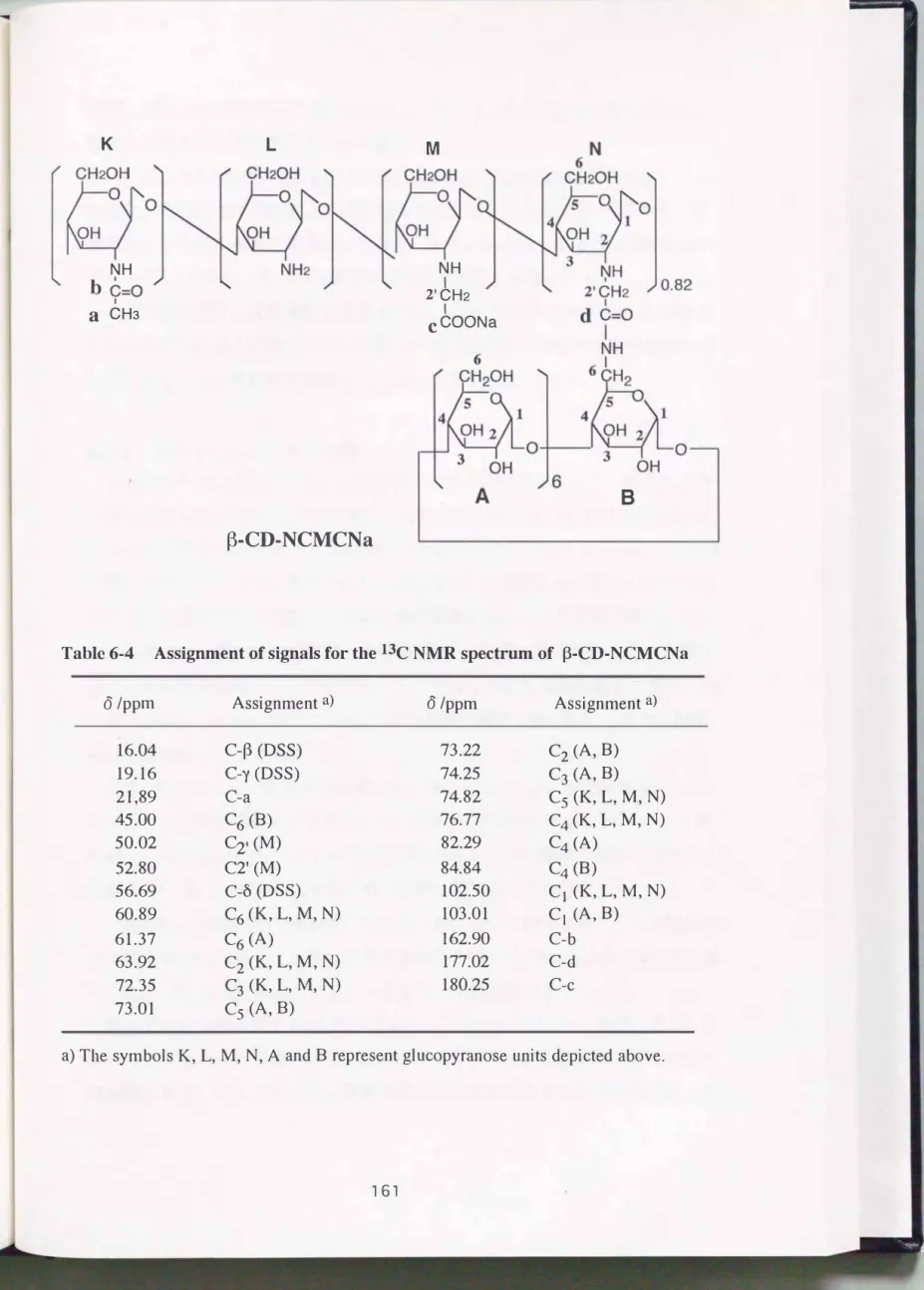
13C NMRスペクトル, lH- 13C異種核相関分光スペクトル(H-CCOSYスペ クトル)をそれぞれFig.6- 8, Fig. 6-9およびFig.6-10に示す.
13C NMRスペクトル(Fig.6- 8)のÔ 102.0に見られるシグナルは, 糖骨格 中で最も低磁場に現われる C1アノメリック炭素のものであり, H-CCOSYス ペクトル(Fig.6-9)におけるlH側の C1-Hとの交差ピークによって確認でき た. δ51.6とÔ 62.8の2つのシグナルについても, 同様にH一CCOSYスペクト ルにおけるlHとの相関から, それぞれ C2' メチレン炭素と C 2メチン炭素に帰 属される. Ô 6 0�8 0の残りのシグナルは, キトサンおよびその誘導体の 13C
司・・町
εaa\。
。
2 3 4 5 6
Vぺ 1/
/
• •
レ〆
v
• C2H・C3H一一十一一J
c 1H-C2H以/
.----レ/ レ/
a
7
2.5
3.0
3.5
4.0
4.5
8
。
A'
2 3 4 5 7 6
8
b
一一�
3.0 2.5 4.0 3.5
4.5
ö/ppm
Fig.6・7 (a) Entire and
(b)
partial H-H COSY spectrum of NCMCNa in alkaline D20 (pD=12).--r
200 180 160 140 120 1 00 80 60 40 20 õ/ppm
Fig.6・8 75 MHz 13C NMR spectra of NCMCNa in alkaline D20 (pD=12).
The chemical shift was calibrated by TMS external standard.
f州
-千千千干干干I干
・・・ " ーマ::ア i
:二ミ- --� I---l---F--l---'I
180 160 140 120 100 80 60 40 20 る/ppm
2 3 4 5
6 7
Fig.6・9 H-C COSY spectrum of NCMCNa in alkaline D20 (pD=12).
The chemical shift was calibrated by DSS.
『ーー『
90
N
HっC/\CHq(ノ ,
(c')COONa COONa
NCMC
80 70
る/ppm'
C21
60 50
Fig.6-10 Partial map of the H・C COSY spectrum ofNCMCNa in alkaline D20 (pD=12). The chemical shift was calibrated by DSS .
2.0
3.0
3.5
4.0
4.5
NMRスペクトルの帰属30)を参考にして, 高磁場側から C61 C31 C5, C4と 考えられる. 残存のアセチル基に由来するシグナルもÔ 21.9 (メチル基)と6 161.1 (カルボニル)に検出されている.
Fig. 6-8の中で最も低磁場にケミカルシフトを有するこつのシグナルはカル ボキシラートアニオンのものと考えられ, モノカルボキシメチル誘導体由来の 炭素シグナルをÔ 179.0に, またδ179.67のシグナルをジカルボキシメチル誘 導体のカルボキシル炭素に帰属した.
ところで糖骨格を形成する C1�C6はいず、れも若干幅を有するが単一のシグ ナルとして得られており(Fig. 6-8) , 4種類のユニット問での明確な相違は 確認されなかった. また1H 側との相関(Fig. 6-10の各交差ピーク)を見た場 合, C3-HやC4-Hといった各プロトンのケミカルシフトは求められるもの の, 現段階でユニットによる違いを認識するのは非常に困難である. その解析 には別のパルス系を用いた測定法, 例えば修飾シクロデキストリンのNMR分 析31)に用いられる, HOHAHA (Homonuclear Hartmann-Hahn)などの磁 化移動を利用した方法が有効であると思われる.
6.2.2 アミノ化βーシクロデキストリンの合成と同定
6.2.2.1 βーシクロデキストリンのモノトシル化
Fig. 6-12のIRスペクトルを原料のß-CDのそれ(Fig. 6-11)と比較する と, 新たに1372cm-1にスルホン酸エステルのs=O逆対称伸縮振動, 1182
cm-1にスルホン酸エステルのs=O 対称伸縮振動の吸収が また1604cm-1に はベンゼン環骨格の伸縮振動の吸収が認められ, CD環へのトシル基の導入を 示唆している.
薄層クロマトグラフィ一分析(展開溶媒; 25%アンモニア水:イオン交換 水:酢酸エチル: 1-フロパノール=1 : 3 : 2 : 5, ヨウ素検出法)の結果 は, ß-CDの約2倍の移動率を有する単一のスポット(RrO.35, Rc=2.25)が 検出された. ここで, �値は標準物質に対する試料の移動率を表す数値であ り, R c値=(β-CD誘導体の Rf値)/(ß-CDのRtfi直)とした場合, 文献値23) (Rc=2.1)とほぼ同様であった. 検出されたスポットはモノトシル化。-CDの ものと考えられる. また単一のスポットが得られていることから, 生成物中の
lnHHB柱ttr huunt HR持竹川口付円句uun
リ回日刊村川H付ぷ け品川け11 ηリぷ川村れ Hn村UHU吋川口 ηunH
υrTよiよItT上主主、÷lふlよjL+・lT・子1 主主 干↓JT主主宰
川vi
-
--7・4::+Il--tγ
!一ji--一-ai--:一ih--7i
1τi
1・
了
ぶ一-・τT
1よ
+t-IT
--T1よ与
.iil!.ム
÷÷?
-ナ?7
.;i;.::.;.
ム÷
一
?7了
.ijj
l!!.:
.J J ; ;
':!一
十
一?.;ij.:j.JJJ4i.iii!.占ナ?了了了
1!.;L.Lトv「「.;i!0!!.:
ム
0-?了了
l!.ナI!,l-主
4
1Ti
l-
ZT
ats
宇
IT---一・i-:;一;i・ど
l;l・一iji--一l-rIT-
--}γよ1Ll÷
i一11江主米中pnmFlE出足品川
県副TT rI J市WM保市一山怖 い山 剤 甘いFN刊日い い 川Uいい u u日刊州い MMm 川 ア一 噂 村正 市 11山小汁正小山市川河川市山山 市 … ー ' 一J B 1斗 リ ヰ北斗川川リリjバパ川汁川U MM 川 刊 竹川 明|
い什L K 甘
いιドトトいいι
れいikル
門川ハ川Vμ
!T叫いいれいけ比日 比
ー-Ti-----iE-l
J 可 「L
:a--:hpI?!:l+
I.f-- ふji---γlTl甲
-!
↑
!?14・li--7;一
--J-eH・Pao---TITI--feli--?ート
14
MI-- --e-af--juli--i-、--;L;:
一
'・
!
、 l l l 件 '|
・IFいいLトト圃ゆ1lr寸 叫4 1 4 l$4
1 刷
内川開川附山川間附川山川閉 山川山川相川山山川川山川川州出
AU品川HHHAN川村HMMH村HMHUHHHNUUN川け川以hHHHAHHHHHHHHnHHHH明
日汁HH川HH川11111HHHHUHHμれHU-EIHUH
川
dw甘 lilt-HHHHHHHH川バ川町門MME
什
HHN
U肘川甘品
川口H村 田川 11111
4000 3500 3000 2500 2000 1500 1000 500
Wavenumber / cm・1 Fig.6・11 IR spectrum of ß..CD
•
'- tit -
' ::Jai--、,aE闘t----恥副・・
.•
,.,.ι
:
aa l
: 111111141 1
11:
-lelili--li-
lt-lii d』,h 111111111til -
--ia I1 『
11
ア; l - d i l l - --:
ifiJ,--Il-FllLAH・::
・ i : : ;
l
l1
e l :
t1111111Il--1lil--li1l--1i111111111111111M11111EtIli--t1111111ill-iLI--『BE!
JFIll-圃11111'』れe- - ' - JI- -: : : : lil i - - - i - jo lli-- l i e -
'1・・1・11ltili--l・;・tif--jlla-t‘Eta--11E11111{'ote町11411let--t'tB」
ヲi il i - - i ! ー :fil- ! ー
・JITIstilli--1114li--,、1111』14116'11・・ Il---illit--噌
i tJ ・1 J
ST'』」守,fS4
6,aド
・1・・'s,B』i''h'I,tt910・0E'41Eth・'ataEgtaEtaBEEB4ヨ,aleム守Il---ll'tla--tilt--'1114111111It--'; 11 (
ー !
?i
lJ
; ! ;
・・白'E・E・
-t圃E‘t』itst'噌ltE1'・.,et'a,,,,thF』94コ
i ! i l
- -
J l
園
; HHEqg li--a l I P 。lE1 rill-』r
l llitF 哩 r
i--al111ir---li - - l j
, •
•
-T,.--et・418』taaE『・2EE'S
4
,thl&創刊hEE't'EE4Ji l i i
l i --巴 門 川川叶1111 11jllu 河 川川川川町a
i -
- J
i l l i i
J1 1
守 +
l i l-
- tti
l u圃 7 1tIot 4 ・
'14守
JElli--E・E・-tEE・I・-11a・I
l l -
l i
--・・・困問
i l i l i l i l
! i i : : i J : : 川榊山山惜川併以 … 宮川市川肝 町川川m出川貯川川 山山凶州 昭 一閉山以山川相川 町mmm m山 リ+ lγ 1主十ト寸 J オlTJすIT - -TI--Ti l -TITlf4TI--十1
巳じい枠札H甘い材什いいμHHHHいけUいけいいわいいけいいμUHHム
1 1 1
1十}ーヰl
i- - ー1I
TI
ll-+i l i-
-e 上l
is -
f-?:
凶 川川 川川 川附川川叶川川川川川川削川川川川 法白 ・ 以| 市川出 川山リ川叩 市 山川 古川叶市 山 知正山 下 品市 -十 ! li
E刊
-'ar--
7
・l-l了
!1・上i・・・:ヶ・・l占11Ifill--L・I・L
a--i
s
- -J上 空 州市川市川市中川中山正化 山j 市 地
Vl-
-1-B一-TT
li
li--7s・l-パ レ
tlliけdili--,.,Till合 ー
μじK
πい什札いいNUNドUトいいいいわれいUHKU-
ー申キ !
!Jrレdl l jd e- - 山町 1 4 , 唱 . 4 . 4 ;i
4000 3500 3000 2500 2000 1500
Wavenumber / cm・1
1000 500
Fig.6・12 IR spectrum of Ts・ß..CD
143
『可
未反応。-CD含量は極めて少ないと推測される.
一方, τ"'SK-GEL ODS-120Tカラムを用いたHPLCで生成物を分析し, その 結果をFig. 6-13に示す. イオン交換水による再結晶2回後の生成物のクロマト
グラム(Fig.6-13(a))では, 2.58分に反応副生成物であるpートルエンスルホ ン酸のピークが, 5.93分にはモノトシル化。-CDのピークが検出されている.
また, さらに大きな保持時間を有するピークが20.1分と33.7分に認められる が, これらはß-CDの6位ヒドロキシル基が 2つ以上トシル化されたH多置換体
"と考えられる. 本反応はß-CDの 6 位ヒドロキシル基の一つを選択的にトシ ル化する目的で行ったが, 必ずしもモノトシル体のみを生成する反応ではない ということがこの結果から明らかである. 報告23)によれば, 室温40分間の反
応では67%のモノトシル体, 33%の未反応。-CD, および若干量の多置換体 が生じるとされている. 従って, 反応後に未反応。-CDと多置換体を除去する
操作が不可欠であった.
本研究では精製操作としてイオン交換水および混合溶媒での再結晶25)を用い て, 未反応。-CDおよび多置換体を除去した. 前者では, ß-CDの方が 水に対 する溶解性が高いために, トシル化。-CD (モノ 体, 多置換体いずれも )が優 先的に結品化する. 一方後者は比較的極性の低い溶媒であり トシル基をより 多く有する誘導体の方が溶解度が大きい. すなわちモノトシル体が優先的に結
晶 化する. 先のTLC分析の結果から, 最終生成物中の未反応β-CD含量は非常 に少なく, イオン交換水による 2回の再結晶で十分に除去されていると思われ る. これに対して多置換体は, 混合溶媒による再結晶で除去を行うと同時に HPLC分析で随時その含有量を測定した. 混合溶媒での再結晶を繰り返すこと で生成物中の多置換体量は徐々に減少し, 3回再結品後には痕跡程度にまで除
去されており(Fig. 6-13(b)) , ほぼ純粋なモノトシル体が得られた.
とのモノトシル化。-CDのlHNMRスペクトルをFig.6-14に示す. 芳香環プ ロトン領域の 2つの二重線(ô 7.433とÔ 7. 7 53 )およびメチルシグナル(ô 2.430 )の存在によって, ß-CD環へのトシル基の導入が確認出来た. またそ の シグナル強度をß-CD骨格のプロトンと比較することでモノトシル体である ことが分かる. トシル基のついた グルコピラノースユニット( ユニットB)の C1-Hは, 導入されたベンゼン環の環電流効果もしくはトシル基が置換したこ