NMR スペクトルの多変量解析による
アクリル系共重合体の一次構造解析
2011 年 3 月
目 次
第1章 序論 1 1-1 高分子材料と高分子一次構造の関係 1 1-2 従来の NMR 法による高分子の一次構造解析とその限界 2 1-3 多変量解析と高分子分析への応用 3 1-4 高分子の一次構造解析への多変量解析の適用 5 1-5 本研究の目的および概要 5 第2章 NMR の装置構成と測定条件および多変量解析の手順 15 2-1 NMR の装置構成と測定条件 15 2-2 多変量解析の方法と原理 15 2-3 NMR スペクトルへの多変量解析の適用手順 19 第3章 メタクリル酸メチル-メタクリル酸 t-ブチル二元共重合体の組成および モノマー連鎖分布の定量解析 23 3-1 緒言 23 3-2 モデル共重合体の合成 24 3-3 主成分分析 26 3-4 共重合組成の推定 35 3-5 2 連子モノマー連鎖分布の推定 37 第4章 メタクリル酸メチル-メタクリル酸 t-ブチル-メタクリル酸 2-ヒドロキ シエチル三元共重合体の組成およびモノマー連鎖分布の定量解析 47 4-1 緒言 47 4-2 モデル共重合体の合成 48 4-3 主成分分析 53 4-4 共重合組成の推定 58 4-5 2 連子モノマー連鎖分布の推定 69第5章 メタクリル酸メチル-メタクリル酸 t-ブチル二元共重合体の立体規則性 の定量解析 87 5-1 緒言 87 5-2 モデル共重合体の合成 87 5-3 主成分分析 ~単独重合体を主眼とした解析~ 91 5-4 共重合体の主成分分析と統計的二次元 NMR による考察 95 5-5 共重合体の 3 連子立体規則性の推定 105 第6章 結論 113 6-1 本研究の結論 113 6-2 今後の展望 114 本論文に関わる発表論文 116 共同研究者一覧 118 謝辞 119
第1章 序論
1-1 高分子材料と高分子一次構造の関係 高分子材料は,今や我々の社会や生活を支える基盤材料として発展を遂げている。 実用的な高分子材料の多くは多成分共重合体である。特に電子材料や光学材料には, 高速に高度化する情報通信技術社会の中で,さらなる性能の向上や新しい機能の追加 が求められている。 共重合体の重合度(分子量),組成,モノマー連鎖,立体規則性や,これらの分布, 末端基および分岐構造などの一次構造(図 1-1)は,高分子材料の性能に大きな影響 を与えている。実際,立体規則性とガラス転移温度の関係[1]や,モノマー連鎖と相溶 性の関係[2]にはじまり,半導体の微細加工用レジストに用いられる共重合体の末端基 構造とレジスト感度の関係[3]など,一次構造と材料物性との関係をテーマとした研究 は,枚挙に暇がない。このように,高分子の一次構造の精密な制御が材料性能の向上 や新しい機能の発現に直結するため,その詳細な分析や定量の重要性が近年ますます 高まっている。 図 1-1 高分子の一次構造。1-2 従来の NMR 法による高分子の一次構造解析とその限界 高分子の一次構造を解析する手段として,赤外吸収スペクトル(IR)法,熱分解ガス クロマトグラフィー-質量分析(PyGC-MS)法,マトリックス支援レーザー脱離イオン 化-質量分析(MALDI-MS)法などが知られている。中でも最も有力な手段の一つとし て,核磁気共鳴分光(NMR)法がある。高分子への NMR 法の適用は,1957 年に Gutowsky らによって初めてなされた[4]のに続き,1960 年には1H NMR スペクトルが 3 連子立 体規則性(イソタクチック mm,ヘテロタクチック mr およびシンジオタクチック rr) によって分裂することを,Nishioka ら[5],Bovey ら[6],Johnsen ら[7]がそれぞれ独立 に発見した。その後,モノマー連鎖[8-10],末端基[11-13],分岐構造[14-17]など,NMR 法による一次構造解析が進められてきた。しかし,これらは,単独重合体や構造の単 純な共重合体を対象としたものである。 図 1-2 メタクリル酸メチル-メタクリル酸 n-ブチル共重合体のカルボニル炭素の 13 C NMR スペクトル(CDCl3中 55 °C,125 MHz)[18]。
共重合体を構成するモノマー成分数が増えるにしたがって,そのNMR スペクトル は,モノマー連鎖や立体規則性を反映して,ブロードで複雑な形状になる(図 1-2)。 そのため,より高分解能なNMR 装置を用いたとしても,個々の共鳴線の帰属には多 大な時間と労力を要するうえに,そこから一次構造の定量的な情報を取得することは 難しい。 1-3 多変量解析と高分子の評価解析への応用 多変量解析は,複雑かつ大量の情報や差異が判別できないデータ群から,いくつか の有用な情報へと変換する強力な手法である。近年のコンピューターの演算能力や処 理速度の向上と,ソフトウェアの入手が容易になったことも相まって,多変量解析が 種々の問題解決に利用されている。中でも,生物の代謝産物を網羅的に解析するメタ ボロミクス(メタボローム解析)分野への応用が活発である[19]。例えば,植物のス トレス状態の解析[20],神経疾患の原因解析[21],病気の診断[22],毒物投与ラット[23] や糖尿病ラット[24]の病態分類などに利用されている。また,日本茶[25]や薬用植物 [26]の抽出物の NMR スペクトルを多変量解析し,それらの品質管理へ適用する試み も興味深い。 多変量解析を高分子の分析や物性評価へ応用した例も少なくない。1976 年に Chaurasia らは,ゼロずり粘度 η0と分子量 M の関係式(η0 = K · cα · Mβ)(K は定数,c はサンプル濃度,M は分子量)において,様々な c や M を持つ η0のデータから多変 量回帰により K,α,β を求めた[27]。同じ年に,Kullik らは 120 種類の高分子の PyGC パイログラムをクラスター分析し[28],Linnahalme らは要因分析や正準相関分析によ り,ホルムアルデヒド-フェノール共重合体から成形した化粧合板の物性と原料の種 類や使用量の関係を明らかにしている[29]。その後,河内山らが主成分分析(PCA)を初 めて応用し,高圧法ポリエチレンの製造におけるプロセス因子とポリマー構造および 物性との関連付けを行った[30]。また,Pell らは,エチレン-酢酸ビニル共重合体(EVA 樹脂)の赤外発光スペクトルから,サンプル中の酢酸ビニル組成,サンプル厚さ,測 定温度を,部分最小二乗回帰(PLSR)を用いて初めて推定した[31]。NMR スペクトルを 利用した例では,Lennholm らが固体13C NMR スペクトルと PCA を組み合わせて,パ ルプの叩解(パルプを叩き,切断された繊維が水和・膨潤・絡み合うようにする作業 工程)によるセルロース構造変化を追跡した[32]のが最初である。
高分子と多変量解析に関連した論文は,現在まで613 件に及ぶ(2010 年 8 月 11 日 時点での SciFinder による検索[33])。このうち評価解析に関する論文は 284 件であっ た(図 1-3a)。これらを使用データの種類により分類すると(重複を含む),分光分析 法によるものが60.1 %と,圧倒的な割合を占めていた(図 1-3b)。また,分光分析法 全体に対し,赤外および近赤外分光法が59.0 %,ラマン分光法が 19.5 %を占めていた (図 1-3c)。これらのスペクトルの解析に多変量解析が早くから利用されている理由 は,分子の振動スペクトルが複雑に重なり合うことや,二倍音,三倍音になるにつれ てブロードなスペクトルになるため,官能基や分子構造に関する情報が直接得られに くいためと考えられる。また,スペクトル分解能が4 – 8 cm-1程度なため,データ量 が少なく計算負荷が比較的小さいことも一因と考えられる。 図 1-3 高分子の多変量解析に関する文献調査結果。 a) 全報告数での分類,b) 「ポ リマー分析・解析」におけるデータ種類での分類,c) 「分光法」における手法での 分類。
また,NMR 分光法を用いた 14 件には,SEC-NMR 法で得られた SEC クロマトグラ ムや DOSY 法で得られた拡散係数分布のカーブフィッティングに多変量解析を用い た例[34,35],固体13C NMR スペクトルの多変量解析による架橋高分子のモルフォロジ ーおよびダイナミクスの解析[36,37],セルロース構造変化の追跡[32,38]や,植物細胞 壁の分類[39],1H NMR スペクトルの PLSR によるスチレン-ブタジエンブロック共 重合体の組成推定[40]などが含まれている。 1-4 高分子の一次構造解析への多変量解析の適用 上述した高分子の評価解析に関する論文284 件の中で,高分子の一次構造を取り扱 ったものは 30 件である。しかし,そのほとんどが単独重合体の混合物や共重合体中 の組成に関するものであった。先述した Pell らによる EVA 樹脂中の酢酸ビニル組成 の推定を皮切りに,三井らがポリエチレン(PE)とポリプロピレン(PP)の混合物の PyGC パイログラムを用いた PE 組成の推定[41],Miller らがスチレン(St)-ブタジエン(Bd) 共重合体の赤外および近赤外スペクトルを用いたBd の cis-1,4,trans-1,4,1,2 構造と St の組成推定[42],Shimoyama らが EVA 樹脂の近赤外[43]およびラマン[44]スペクト ルを用いた酢酸ビニルの組成推定について報告している。組成以外の一次構造につい ては,PCA や PLSR の生みの親の一人である Wold らによる DNA の合成プロセスと 得られた DNA のモノマー連鎖の関係を調べた例[45], PP の赤外スペクトルと 13C NMR 法で求めた立体規則性を検量データとして,アタクチック PP の立体規則性を推 定したOzzetti らによる例[46],Hughes らが PE の赤外スペクトルや示差走査熱量測定 (DSC)で得られるサーモグラムを用いた分岐度の推定を行った例[47]などが報告され ているにすぎない。 1-5 本研究の目的および概要 本研究では,種々の一次構造解析に対して最も有力な手法の1つであるNMR 法の 中で,一次構造情報がより多く反映されている 13C NMR スペクトルを多変量解析す ることで,複雑に分裂する多成分共重合体のNMR スペクトルの帰属を行うことなく, 構造因子に関する定量的な情報を得ることを目的とする。 まず,本章に続き,第2章では,本研究で用いたNMR の装置構成および測定条件 と,NMR スペクトルを多変量解析へ適用する手順を示す。
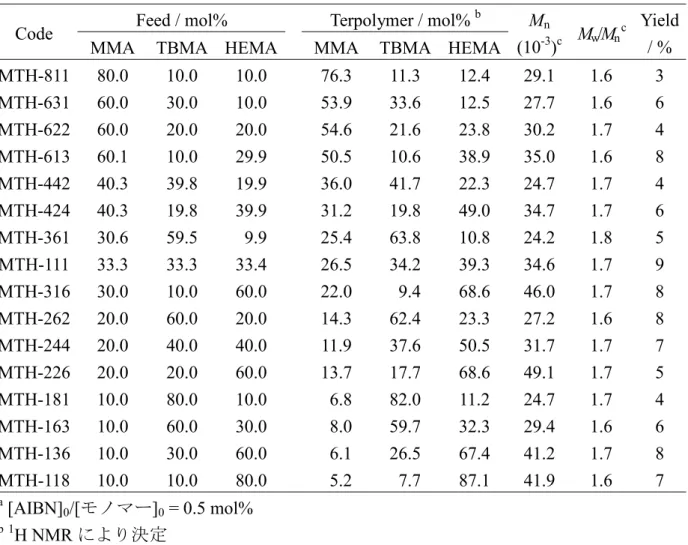
第3章では,メタクリル酸メチル(MMA)-メタクリル酸 t-ブチル(TBMA)二元共重 合体の組成およびモノマー連鎖分布の定量解析について述べる。共重合体の組成やモ ノマー連鎖は,高分子成形材料の濁り(ヘイズ)やコーティング材料の異物や不溶成 分の発生などに大きく関与しているため,それらの定量的解析が求められている。こ こで用いた二元共重合体は,重合開始剤として 2,2’-アゾビスイソブチロニトリル (AIBN),溶媒として乳酸エチルを用いたラジカル重合で得られたものであり,工業的 かつ実用的な多成分共重合体の基本となるモデルである。仕込みモノマー組成や重合 時間を種々調整することにより,組成や収率の異なる共重合体を16 種準備した。MMA とTBMA の単独重合体とこれらの混合試料 9 種を加え,合計 27 種の試料の13C NMR スペクトルのPCA を行ったところ,図 1-4 に示すように,第 1 主成分(PC1)にモノマ ー単位の組成,第 2 主成分(PC2)に 2 連子モノマー連鎖の同種・異種性がそれぞれ反 映されていることがわかった。
図 1-4 MMA と TBMA の単独重合体とそれらの混合試料 9 種,および MMA-TBMA 共重合体 16 種の13
C NMR スペクトルから得られた PCA スコアプロット。各主成分 軸の( )内は寄与率を示す。
また,単独重合体2 種とこれらの混合試料 9 種の 13C NMR スペクトルと組成を回帰 モデルとしたPLSR により,共重合体 16 種の組成を精度良く推定することができた。 さらに,収率を10 %以内に抑制した初期共重合体 9 種と単独重合体 2 種の13C NMR スペクトルと,3 種類の 2 連子モノマー連鎖分率 fMM,fMT,fTTとを回帰モデルとした PLSR により,初期共重合体 2 種を混合した未知試料の fMM,fMT,fTTを精度良く推定 することができた。併せて,初期重合体ではない共重合体7 種についても,fMM,fMT, fTTを推定できることを示した。以上のことから,13C NMR スペクトルへ多変量解析 を適用することで,スペクトルの帰属をすることなく,共重合体の組成に加えて,2 連子モノマー連鎖分布の定量的な情報が得られることを述べる。 第4章では,MMA-TBMA-メタクリル酸 2-ヒドロキシエチル(HEMA)三元共重合 体の組成およびモノマー連鎖分布の定量解析について述べる。MMA,TBMA および HEMA の単独重合体とそれらの混合試料 34 種,MMA-TBMA,TBMA-HEMA およ び HEMA-MMA の二元共重合体それぞれ 9 種,ならびに,MMA-TBMA-HEMA 三元共重合体16 種の合計 80 種の13C NMR スペクトルについて PCA を行った。その 結果,図 1-5 に示すように,主として PC1 と PC2 に三角相図の形でモノマー単位の 組成,第 3 主成分(PC3)に主として 2 連子モノマー連鎖の同種・異種性がそれぞれ反 映されていた。また,単独重合体3 種とそれらの混合試料 34 種,3 系列の二元共重合 体各9 種の13C NMR スペクトルと組成とを回帰モデルとした PLSR により,三元共重 合体16 種の組成を精度良く決定することができた。さらに,3 系列の二元初期共重合 体各9 種の13C NMR スペクトルと 6 種類の 2 連子モノマー連鎖分率 fMM,fTT,fHH,fMT, fTH,fHMと,同種および異種2 連子モノマー連鎖分率 f2-homo(=fMM+fTT+fHH),f2-hetero(=fMT +fTH+fHM)を回帰モデルとした PLSR により,種々の三元共重合体の 2 連子モノマー 連鎖分率とを,実用的な精度で推定することができた。これらの結果から,MMA- TBMA 二元共重合体での解析手法が,さらに複雑な NMR スペクトルとなる三元共重 合体へ拡張できることを示した。
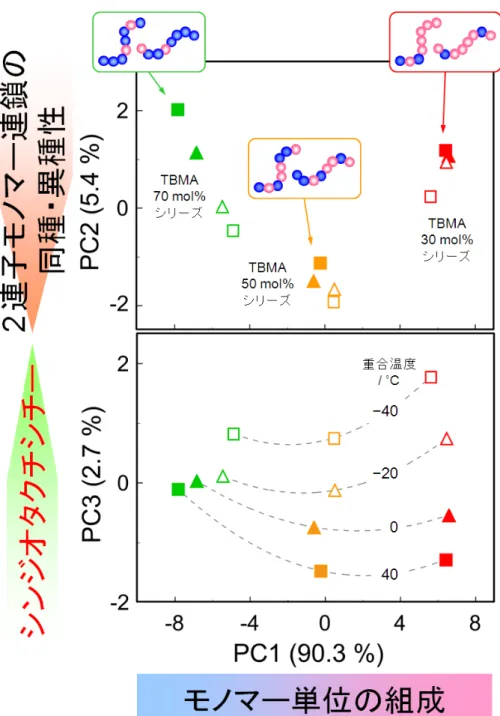
図 1-5 MMA,TBMA および HEMA の単独重合体とそれらの混合試料 34 種,MMA -TBMA,TBMA-HEMA および HEMA-MMA の二元共重合体それぞれ 9 種,およ び,MMA-TBMA-HEMA 三元共重合体 16 種,合計 80 種の13 C NMR スペクトルか ら得られた PCA スコアプロット。各主成分軸の( )内は寄与率を示す。 第5章では, MMA-TBMA 二元共重合体の立体規則性の定量解析について述べる。 NMR 法での共重合体の立体規則性の解析は,モノマー連鎖による共鳴信号の分裂が 加わるため,単独重合体と比較して格段に難しい。また,立体規則性は,高分子材料 の耐熱性を支配しているため,共重合体の立体規則性の定量的な評価手法の確立が急 務である。ここで用いた試料は,3 通りの仕込みモノマー組成と,4 通りの重合温度 で得られた共重合体 12 種である。これらの13C NMR スペクトルの PCA を行った結 果,図 1-6 に示すように,PC1 に主としてモノマー単位の組成,PC2 に主として 2 連 子モノマー連鎖の同種・異種性,PC3 に主としてシンジオタクチシチーがそれぞれ反 映されていた。
図 1-6 重合温度や仕込みモノマー組成が異なる MMA-TBMA 二元共重合体 12 種の 13 C NMR スペクトルから得られた PCA スコアプロット。各主成分軸の( )内は寄 与率を示す。 さらに,統計的二次元NMR(stat-2D NMR)法を用いて,PC2 と PC3 に反映されている 一次構造情報の詳細な把握を試みた。この検討には,共重合体 12 種とそれらを酸加 水分解とメチルエステル化して得られた12 種の PMMA を用いた。これらの試料を用 いた stat-2D NMR スペクトルを図 1-7 に示す。PMMA のシンジオタクチック 5 連子 (rrrr)に帰属される信号と,共重合体の 6 つの信号とが正の相関として表された。これ
ら6 つの信号は,rrrr の立体規則性を有する 6 種類の 3 連子モノマー連鎖(M-M-M, M-M-T,T-M-T,T-T-T,T-T-M,M-T-M)に対応すると考えられる。 図 1-7 MMA-TBMA 二元共重合体 12 種とそれらから誘導して得られた PMMA12 種とのカルボニル炭素領域における統計的二次元 NMR スペクトル。 そして,比較的組成の近い試料ごとにstat-2D NMR を行い,PCA ローディングと比 較した結果,MMA 組成が大きい場合は,PC2 に 2 連子モノマー連鎖の同種・異種性, PC3 に rrrr が反映されていた。一方で TBMA 組成が大きい場合は,rrrr かつ TBMA 単位を含むモノマー連鎖に由来する共鳴信号が PC3 だけでなく PC2 にも反映されて いることがわかった。また全ての共重合体のうち10 種の13C NMR スペクトルと立体 規則性3 連子 rr,mr,mm を回帰モデルとした PLSR により,「未知試料」とした共重 合体2 種の rr,mr,mm を精度良く推定できた。以上のことから,PCA に stat-2D NMR 法を導入することで,各主成分が持つ構造因子をより的確に把握できるとともに,本 手法が共重合体の立体規則性の推定にも適用できることを示した。 最後に,第6章ではNMR スペクトルの多変量解析による多成分共重合体の一次構 造解析の到達点と,今後の展望について述べる。
文献および注釈
[1] V. N. Tsvetkov, J. Polym. Sci., 1962, 57, 727.
[2] G. Kraus, C. W. Childers, J. T. Gruver, J. Appl. Polym. Sci., 1967, 11, 1581.
[3] H. Momose, S. Wakabayashi, T. Fujiwara, K. Ichimura, J. Nakauchi, SPIE Proc., 2001,
4345, 695.
[4] H. S. Gutowsky, A. Saika, M. Takeda, D. E. Woessner, J. Chem. Phys., 1957, 27, 534. [5] A. Nishioka, H. Watanabe, K. Abe, Y. Sono, J. Polym. Sci., 1960, 48, 241.
[6] F. A. Bovey, G. V. D. Tiers, J. Polym. Sci., 1960, 44, 173. [7] U. Johnsen, K. Tessmar, Colloid Polym. Sci., 1960, 168, 160. [8] A. R. Katritzky, D. E. Weiss, Chem. Br., 1976, 12, 45.
[9] 畑田耕一, 高分子, 1989, 38, 430.
[10] I. R. Herbert, NMR Spectrosc. Polym., 1993, 50. [11] 北山辰樹, 高分子, 1988, 37, 476.
[12] 柘植新, 大谷肇, 化学, 1990, 45, 214.
[13] J. C. Bevington, J. R. Ebdon, T. N. Huckerby, NMR Spectrosc. Polym., 1993, 80. [14] 藤原譲, 網屋繁俊, ぶんせき, 1978, 715.
[15] I. Ando, T. Yamanobe, T. Asakura, Prog. Nucl. Magn. Reson. Spectrosc., 1990, 22, 349.
[16] H. N. Cheng, Chem. Anal., 1991, 113, 409.
[17] T. Asakura, M. Demura, T. Hayashi, Annu. Rep. NMR Spectrosc., 1994, 29, 325. [18] T. Nishiura, T. Kitayama, K. Hatada, Int. J. Polym. Anal. Charact., 2000, 5, 401 [19] 吉田欣史, 久原とみ子, 菊池淳, ぶんせき, 2009, 371.
[20] P. Krishnan, N. J. Kruger, R. G. Ratcliffe, J. Exp. Bot., 2005, 56, 255. [21] E. Holmes, T. M. Tsang, S. J. Tabrizi, NeuroRx, 2006, 3, 358.
[22] M. Coen, E. Holmes, J. C. Lindon, J. K. Nicholson, Chem. Res. Toxicol., 2008, 21, 9. [23] E. Holmes, A. W. Nicholls, J. C. Lindon, S. Ramos, M. Spraul, P. Neidig, S. C. Connor,
J. Connelly, S. J. Damment, J. Haselden, J. K. Nicholson, NMR Biomed, 1998, 11, 235.
[25] L. Tarachiwin, K. Ute, A. Kobayashi, E. Fukusaki, J. Agric. Food Chem., 2007, 55, 9330.
[26] L. Tarachiwin, A. Katoh, K. Ute, E. Fukusaki, J. Pharm. Biomed. Anal., 2008, 48, 42. [27] N. M. Chaurasia, A. Kumar, S. K. Gupta, Polymer, 1976, 17, 570.
[28] E. Kullik, M. Kalurand, M. Keel, J. Chromatogr., 1976, 126, 249. [29] T. Linnahalme, A. Mattila, J. J. Lindberg, Pap. Puu, 1976, 58, 763.
[30] 河内山勝晴, 荒井康全, 三宅康之, 茂木義博, 化学工学, 1983, 47, 150.
[31] R. J. Pell, B. C. Erickson, R. W. Hannah, J. B. Callis, B. R. Kowalski, Anal. Chem.,
1988, 60, 2824.
[32] H. Lennholm, T. Iversen, Nord. Pulp Pap. Res. J., 1995, 10, 104.
[33] 検索式:“multivariate analysis of polymer”+“chemometrics of polymer”-[特許 文献]
[34] L. C. M. Van Gorkom, T. M. Hancewicz, J. Magn. Reson., 1998, 130, 125.
[35] K. Ute, R. Niimi, M. Matsunaga, K. Hatada, T. Kitayama, Macromol. Chem. Phys.,
2001, 202, 3081.
[36] D. E. Axelson, A. K. Nyhus, J. Polym. Sci. B: Polym. Phys., 1999, 37, 1307. [37] D. J. Harris, M. K. Alam, Polymer, 2002, 43, 5147.
[38] T. Josefsson, H. Lennholm, G. Gellerstedt, Cellulose, 2001, 8, 289. [39] H. Kim, J. Ralph, Org. Biomol. Chem., 2010, 8, 576.
[40] M. Sardashti, J. J. Gislason, X. Lai, C. A. Stewart, D. J. O'Donnell, Appl. Spectrosc.,
2001, 55, 467.
[41] 三井利幸, 肥田宗政, 藤村義和, 分析化学, 1989, 38, 349.
[42] C. E. Miller, B. E. Eichinger, T. W. Gurley, J. G. Hermiller, Anal. Chem., 1990, 62, 1778.
[43] M. Shimoyama, S. Hayano, K. Matsukawa, H. Inoue, T. Ninomiya, Y. Ozaki, J. Polym.
Sci. B: Polym. Phys., 1998, 36, 1529.
[44] M. Shimoyama, H. Maeda, K. Matsukawa, H. Inoue, T. Ninomiya, Y. Ozaki, Vib.
Spectrosc., 1997, 14, 253.
[45] S. Wold, J. Jonsson, M. Sjoestroem, M. Sandberg, S. Raennar, Anal. Chim. Acta, 1993,
[46] R. A. Ozzetti, A. Pedro de Oliveira Filho, U. Schuchardt, D. Mandelli, J. Appl. Polym.
Sci., 2002, 85, 734.
[47] J. Hughes, R. Shanks, F. Cerezo, Journal of Thermal Analysis and Calorimetry, 2004,
第2章 NMR の装置構成と測定条件および多変量解析の手順
2-1 NMR の装置構成と測定条件 本研究で用いたNMR 装置は,10 mmφ TH プローブを装備した日本電子製 ECX400 である。試料濃度は8 wt/vol%とし,測定温度は 55 °C とした。測定溶媒は,MMA- TBMA 二元共重合体の解析では CDCl3,MMA-TBMA-HEMA 三元共重合体の解析 ではCDCl3/DMSO-d6 [4/6 (mol/mol)]をそれぞれ用いた。パルス幅,繰り返し時間およ び積算回数は,それぞれ,1H 核測定では 8.5 μs (45 °),8.90 s,16 回とし,1H 広帯域 デカップリングをによる13C 核測定では 7.5 μs (45 °),2.73 s,10,000 回(二元共重合 体の組成およびモノマー連鎖分布の定量解析;第3章)あるいは5,000 回(三元共重 合体の組成およびモノマー連鎖分布の定量解析;第4章,二元共重合体の立体規則性 の定量解析;第5章)とした。 31,250Hz の観測幅に対して 32,768 のデータポイントで自由誘導減衰(FID)を取得し た後,二度のゼロフィリングによってデータポイントを4 倍し,フーリエ変換するこ とで 13C NMR スペクトルを得た。フーリエ変換時のウィンドウ関数は指数関数とア ポダイゼーション関数を組み合わせて用い,ブロードニングファクター(BF)は,第3 章では3.0 Hz,第4章および第5章では 2.0 Hz とした。13C NMR スペクトルでは,内 部標準としてCDCl3の化学シフトを77.0 ppm と設定した。 2-2 多変量解析の方法と原理 多変量解析は,図 2-1 に示すように,比較や定性を目的とする方法(目的変量なし) と,予測や推定・定量を目的とする方法(目的変量あり)に大別できる。前者は,ス ペクトルなどの化学データ(説明変量)だけで試料間の類似性を比較することができ る点が特徴であり,主成分分析(PCA)[1,2]がよく知られている。一方,後者は,化学 データに加えて,特性値などの検量データ(目的変量)が必要であるが,これらのデ ータ群により構築した回帰モデルを用い,未知試料の特性値の予測あるいは推定・定 量が可能な点が特徴である。回帰モデルとしては,部分最小二乗回帰(PLSR)[3]がよく 知られている。 本研究では,種々の重合条件で合成したモデル共重合体の 13C NMR スペクトルに 対してPCA および PLSR を適用し,構造因子の定性および定量分析を行った。また,主成分分析のローディング(後述)を考察するため,統計的二次元NMR(stat-2D NMR) 法も用いた。以下に,PCA,PLSR,stat-2D NMR 法の原理の概略を示す。 図 2-1 多変量解析の分類[4]。 主成分分析(PCA) PCA では,試料数 n,1 つのサンプルあたりのデータ数 r からなるデータの集合で ある行列 X(n 行 r 列)を,式(2-1)のようにスコア行列 T とローディング行列 P に分 解する。 X i i 2 2 1 1 X t p t p t p R R P T X= + = + ++ + (2-1) ここで,RXは残差行列であり,tiと piはそれぞれ i 番目のスコアとローディングであ る。1 番目のローディングは行列 X の分散が最大となるように表現されたベクトルで あり,この p1の方向が第 1 主成分軸となる。一方,スコア t1は,第1 主成分軸への 射影量となる。NMR スペクトルをデータとして取り扱う場合,r はスペクトルのデー
タポイント数(あるいはスペクトルを一定間隔で短冊状に分けたバケット数)に,集 合 X でまとめられたデータは各化学シフトにおける信号強度(あるいは積分強度)に それぞれ相当する。また,p1は行列 X の特徴が最も表現されている擬似的な NMR ス ペクトルであり,t1は擬似的なNMR スペクトル p1の含有量を表している。 2 番目のローディング p2(=第2 主成分)は,行列 X から t1と p1の積で表された 行列を差し引いた残差行列 RXの分散が最大となるように表現されたベクトル(主成 分軸)であり,その軸への射影量がスコア t2となる。したがって,サンプル数と同じ 第 n 主成分まで tiと piへの分解を繰り返すと,行列 X は n 個の tp の和で完全に表す ことができる。しかし,通常は,最初の数成分までが意味のあるベクトルであり,主 成分数の大きいものは,測定装置や測定条件に由来するノイズとみなされる。 データの集合行列 X を分解して得られた各主成分が持つ意味の度合いは,寄与率と して表される。取り扱うデータの量や質にも依存するが,一般的に寄与率の合計(累 積寄与率)が90 – 99 %を超える主成分数となれば,行列 X を十分に説明できている とみなすことができる。 部分最小二乗回帰(PLSR) PLSR では,まず回帰モデルを作成する必要がある。特性値の集合である行列 Y も, PCA により式(2-2)の通りに,スコア行列 U とローディング行列 Q に分解する。 Y i i 2 2 1 1 Y u q u q u q R R Q U Y= + = + ++ + (2-2) ここで,RYは残差行列であり,uiと qiはそれぞれ i 番目のスコアとローディングであ る。q1は行列 Y の特徴が最も表現されている擬似的な特性であり,u1は擬似的な特 性値を表していると言える。PLSR では,一旦 PCA で求めた擬似的な NMR スペクト ルの含有量を表す t1(説明変量側のスコア)と,擬似的な特性値 u1(目的変量側のス コア)との相関が最大になるように,それぞれのローディングも含めた t1,u1,p1, q1を調整する。このため p1はPCA での第 1 主成分とは少し異なるベクトルとなるた め,第 1 潜在変数(LV1)と呼ばれている。次に,調整された t1,u1,p1,q1を用いて, 式(2-3)および(2-4)の残差行列 RX,RYを計算する。
1 1 X X t p R = - (2-3) 1 1 Y Y u q R = - (2-4) 残差行列 RX,RYを再びスコア ti,uiとローディング pi,qiに分解し,同様の手順で tiと uiの相関が常に最大になるように,2 番目以降のスコアとローディングを計算す る。このようにして作成された回帰モデルを用いて,未知試料の特性値を予測する。 PCA や PLSR の詳細は,いくつかの総説[5,6]に記載されている。 統計的二次元 NMR(stat-2D NMR)法 本研究で提案するstat-2D NMR 法は,Barton II らによって提案された統計的二次元 相関スペクトルの原理[7]を NMR スペクトルへ応用したものである[8]。同一の試料を 起源とし,異なる測定核種で得られたNMR スペクトル群や,高分子反応により異な る構造へ変化させた試料の NMR スペクトル群など,2 種類の NMR スペクトル群で 構成される行列の共分散行列を求め,それぞれのNMR スペクトル群における信号強 度の増減の同調・非同調を調べる手法である。図 2-2 に示すように,stat-2D NMR の 相関が正の場合は,元の信号強度の増減が同調(いずれも増加あるいは減少)してお り,逆に負の相関の場合は,元の信号強度の増減は非同調(一方が増加すると,もう 一方は減少)であることがわかる。 試料数を n,ある条件で測定した全試料の NMR スペクトルの信号強度あるいは積 分強度からなるデータの集合を行列 A,別の条件で測定した NMR スペクトルの信号 強度あるいは積分強度からなるデータの集合を行列 B とする。なお,行列 A および B のデータポイント数をそれぞれ d および e とする。そのため行列 A は n 行 d 列,行列 B は n 行 e 列となる。次に,行列 B の転置行列 BTと行列 A の積から,式(2-5)の共分 散行列 Z(e 行 d 列)を求める。この共分散行列 Z が stat-2D NMR スペクトルである。 A B Z= 1 T • n (2-5) 統計的二次元相関スペクトルの原理の詳細は,文献[9]に記載されている。
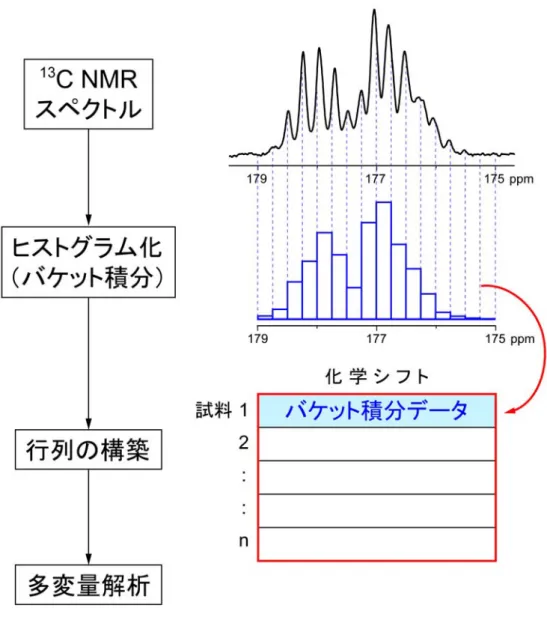
図 2-2 本研究の場合を例にした統計的二次元 NMR の概要。 2-3 NMR スペクトルへの多変量解析の適用手順 具体的な手順を図 2-3 に示す。まず,先述の条件で測定された 13C NMR スペクト ルのデータ量を圧縮するため,解析に用いる共鳴領域を一定間隔で r 分割されたヒス トグラムへ変換する。ヒストグラムの高さは,分割されたスペクトルの面積(積分強 度)し,バケット積分と呼ぶ。この操作を行うソフトウェアとして,日本電子製Alice2 ver.5 for metabolome ver.1.6 を用いた。このようにして,1 行 r 列のバケット積分行列 を得る。
続いて,同様の操作で,解析に用いる全試料(試料数 n)のスペクトルを処理した 後,バケット積分データを配列した n 行 r 列の行列を構築する。
PCA および PLSR の原理に基づき,ローディングとスコアが得られる。PCA および PLSR を行うソフトウェアとして Pattern Recognition Systems 製 Sirius ver.7.0 を用いた。
文献
[1] B. R. Kowalski, C. F. Bender, J. Am. Chem. Soc., 1972, 94, 5632. [2] B. R. Kowalski, C. F. Bender, J. Am. Chem. Soc., 1973, 95, 686. [3] S. Wold, Technometrics, 1974, 16, 1.
[4] 三井利幸, ケモメトリックスの基礎と応用; アイピーシー: 東京, 2003, 46. [5] T. M. Alam, M. K. Alam, G. A. Webb In Annual Reports on NMR Spectroscopy;
Academic Press: 2005; Vol. 54, p 41.
[6] P. Geladi, B. R. Kowalski, Anal. Chim. Acta, 1986, 185, 1.
[7] F. E. Barton, II, D. S. Himmelsbach, J. H. Duckworth, M. J. Smith, Appl. Spectrosc.,
1992, 46, 420.
[8] T. Hirano, H. Momose, T. Maeda, T. Naono, S. Asakawa, Y. Katsumoto, K. Ute, to be
submitted.
[9] I. Noda, Y. Ozaki, Two-Dimensional Correlation Spectroscopy - Applications in
第3章 メタクリル酸メチル-メタクリル酸 t-ブチル二元共重合体の組
成およびモノマー連鎖分布の定量解析
3-1 緒言 工業的に製造されている高分子材料の多くは,数種類のモノマーからなる共重合体 である。中でも,メタクリル酸メチル(MMA)を代表とするメタクリル酸エステルを用 いた共重合体には,屋外広告用の看板,自動車用のランプカバー,コンパクトディス クやブルーレイディスクの最表層用コーティング材料,半導体リソグラフィー用のレ ジスト材料,塩化ビニル樹脂やポリプロピレン樹脂の成形加工用添加剤など,幅広い 用途がある。ところが,共重合体の組成やモノマー連鎖は,看板やランプカバーなど の成形材料の濁り(ヘイズ)やコーティング材料の異物・不溶成分の発生と密接に関 係している。このように,材料の性能向上や新機能の発現には,共重合組成,モノマ ー連鎖やそれらの分布といった高分子の一次構造を詳細に解析する必要がある。 共重合体の解析をする手法の 1 つとして NMR 分光法がある。Brar らは,MMA- メタクリル酸n-ブチル(nBuMA)共重合体のモノマー連鎖解析において,仕込みモノマ ー組成が異なる初期共重合体を合成し,モノマー反応性比から得られるモノマー連鎖 分布の理論値を 13 C NMR スペクトルの信号強度の増減に照らし合わせることによっ て,帰属を試みている[1]。また,Nishiura らも,同じく MMA-nBuMA 共重合体につ いて,立体特異性重合技術を駆使して合成したシンジオタクチックおよびイソタクチ ック共重合体の 13 C NMR スペクトルの帰属情報をもとに,アタクチック共重合体の 13 C NMR スペクトルでの 3 連子モノマー連鎖と 5 連子立体規則性の帰属を行っている [2]。しかし,モノマー連鎖や立体規則性による共鳴信号の分裂が重なり合うため,共 重合体の NMR スペクトルは複雑になり,それぞれの信号を帰属することは一般的に は難しい。また,共重合体のモノマー種が増えるにつれて,NMR スペクトルが幅広 になると同時に,さらに複雑なスペクトルとなる。 多変量解析は,複雑な情報や違いの判別が難しい情報から,有用な情報へと変換で きる強力な手法である。高分子の組成分析では比較的多くの報告例がある。例えば, 主成分分析(PCA)や部分最小二乗回帰(PLSR)を赤外発光スペクトル[3],ラマンスペク トル[4],近赤外スペクトル[5]へそれぞれ適用し,エチレン-酢酸ビニル共重合体の 識別や,酢酸ビニル組成の推定がなされている。また PLSR を熱分解ガスクロマトグラフィーで得られたパイログラムへ適用し,ポリエチレン/ポリプロピレン混合試料 の組成が推定されている[6]。さらに,スチレン-ブタジエン共重合体の赤外および近 赤外スペクトル[7]あるいは 1 H NMR スペクトル[8]へ PLSR を適用し,ブタジエンと スチレンの組成が推定されている。しかしながら,我々の知る限りでは,多変量解析 を合成高分子の NMR スペクトルへ適用し,共重合組成とモノマー連鎖に関する解析 を行った事例はない。 そこで本研究では,工業的かつ実用的な多成分共重合体の最も基本となる二元共重 合体のモデルとして,MMA-メタクリル酸 t-ブチル(TBMA)共重合体を選択した。仕 込みモノマー組成や重合時間を種々調整して得られた組成や収率の異なる共重合体 16 種と,MMA および TBMA 単独重合体,およびそれらの混合試料 9 種の13C NMR スペクトルの PCA および PLSR を行い,共重合体の組成およびモノマー連鎖分布に 関する定量的な情報の取得を試みた。 3-2 モデル共重合体の合成 試薬 MMA および TBMA(三菱レイヨン)は,減圧蒸留により精製して用いた。2,2’-ア ゾビスイソブチロニトリル(AIBN)(和光純薬工業)は,メタノール中での再結晶によ り精製して用いた。乳酸エチルおよびメタノール(キシダ化学)は,精製せずに用い た。 重合 モノマー混合物の 20 wt%乳酸エチル溶液中,80 °C,窒素雰囲気下で,フリーラジ カル重合を行った。重合開始から 3 ~ 7 分後に反応溶液を室温(20 – 25 °C)まで急 冷し,大量のメタノール/水混合溶液(3/7 vol/vol)へ注ぎ,白色沈澱を得た。この 沈澱物をろ取した後,真空中 60 °C にて乾燥し,重合率を 10 %未満に抑えた初期共重 合体を得た。また,重合時間を 4 時間とした以外は同様の操作で,MMA 単独重合体 (PMMA),TBMA 単独重合体(PTBMA)と,重合率が 10 %以上の後期共重合体も合成 した。各試料の収率は重量法で求めた。
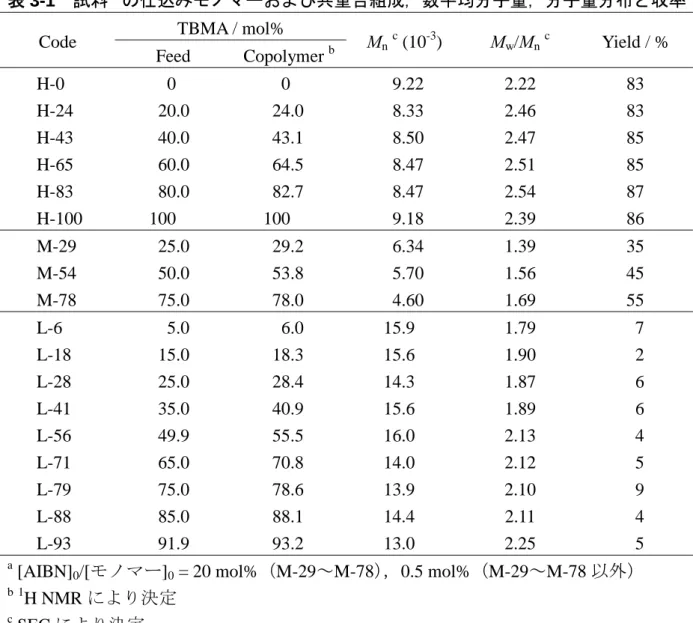
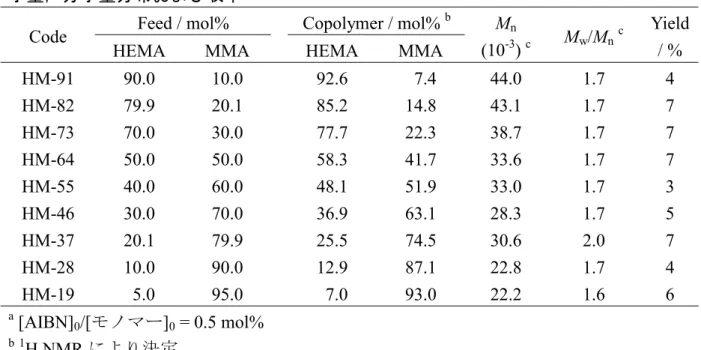
表 3-1 試料aの仕込みモノマーおよび共重合組成,数平均分子量,分子量分布と収率 TBMA / mol% Code Feed Copolymer b Mn c (10-3) Mw/Mnc Yield / % H-0 H-24 H-43 H-65 H-83 H-100 0 20.0 40.0 60.0 80.0 100 0 24.0 43.1 64.5 82.7 100 9.22 8.33 8.50 8.47 8.47 9.18 2.22 2.46 2.47 2.51 2.54 2.39 83 83 85 85 87 86 M-29 M-54 M-78 25.0 50.0 75.0 29.2 53.8 78.0 6.34 5.70 4.60 1.39 1.56 1.69 35 45 55 L-6 L-18 L-28 L-41 L-56 L-71 L-79 L-88 L-93 5.0 15.0 25.0 35.0 49.9 65.0 75.0 85.0 91.9 6.0 18.3 28.4 40.9 55.5 70.8 78.6 88.1 93.2 15.9 15.6 14.3 15.6 16.0 14.0 13.9 14.4 13.0 1.79 1.90 1.87 1.89 2.13 2.12 2.10 2.11 2.25 7 2 6 6 4 5 9 4 5 a
[AIBN]0/[モノマー]0 = 20 mol%(M-29~M-78),0.5 mol%(M-29~M-78 以外) b1 H NMR により決定 c SEC により決定 測定 試料の数平均分子量Mnと分子量分布Mw/Mnは,PMMA を標準試料としたサイズ排 除クロマトグラフィー(SEC)により測定した。SEC 装置は,分析カラムとして昭和電 工製 Shodex GPC K-805L(内径 8.0 mm×長さ 30 cm)を 3 本直列に接続し,検出器と して示差屈折率計を装備した東ソー製 HLC-8220 を用いた。溶離液はテトラヒドロフ ラン(THF)を用い,測定温度を 40 °C,流速を 1.0 mL/min とした。試料濃度は 5.0 mg/mL, 試料注入量は 0.1 mL とした。本研究で用いた試料の TBMA 単位の仕込みモノマーお よび共重合組成,Mn,Mw/Mn,および,収率を表 3-1 に示す。 第2章と同じ測定条件で得られた13
C NMR スペクトルを日本電子製 Alice2 ver.5 for metabolome ver.1.6 を用いて,次に示す共鳴領域を 0.25 ppm 間隔でバケット積分した。
・ 15.1 – 23.1 ppm(α-メチル炭素) ・ 26.0 – 29.0 ppm(TBMA 単位の側鎖 t-ブチル基のメチル炭素) ・ 44.1 – 48.1 ppm(主鎖 4 級炭素) ・ 44.2 – 58.2 ppm(MMA 単位の側鎖メチル炭素と,主鎖メチレン炭素) ・ 79.5 – 83.0 ppm(TBMA 単位の側鎖 t-ブチル基の 4 級炭素) ・ 175.0 – 179.0 ppm(カルボニル炭素)
各共鳴領域での積分強度を 100 に規格化した後,Pattern Recognition Systems 製 Sirius ver.7.0 を用いて,PCA および PLSR を実行した。なお,Sirius ver.7.0 でのデータ解析 中に,各バケット範囲での積分強度の平均化および中心化が自動で行われる。
単独重合体混合試料の調製
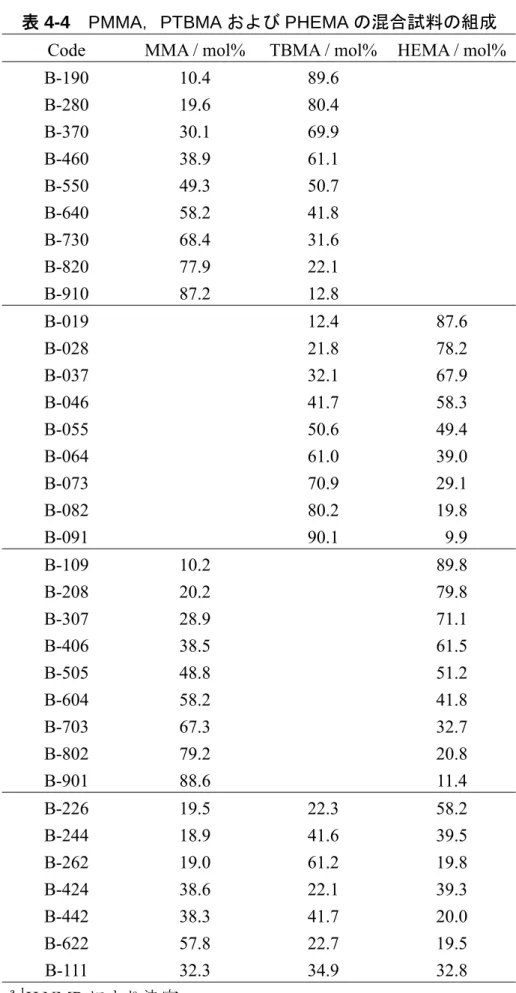
PCA あるいは PLSR での標準試料として用いるために,PMMA と PTBMA の混合 試料 9 種を調製し,それらの組成を表 3-2 に示す。
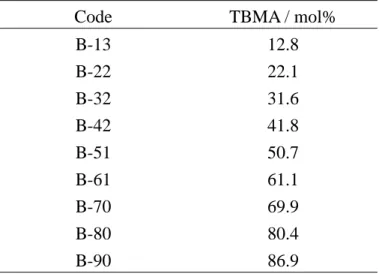
表 3-2 PMMA と PTBMA の混合試料の組成 Code TBMA / mol%
B-13 B-22 B-32 B-42 B-51 B-61 B-70 B-80 B-90 12.8 22.1 31.6 41.8 50.7 61.1 69.9 80.4 86.9 a1 H NMR により決定 3-3 主成分分析
PMMA[H-0],PTBMA[H-100],PMMA と PTBMA の混合試料[B-42]MMA- TBMA 共重合体[L-41]の13C NMR スペクトルを図 3-1 に示す。PMMA と PTBMA の両方に共通な構造である α-メチル炭素 1,主鎖 4 級炭素 3,主鎖メチレン炭素 5,
て複雑なスペクトルとなった。また,PTBMA の側鎖 t-ブチル基の 4 級炭素 6 に帰属 される共鳴領域も同様に,立体規則性によって複雑なスペクトルとなった。一方,共 重合体の上記の共鳴領域は,同程度の組成とした PMMA と PTBMA の混合試料の共 鳴領域と比較して,さらに複雑なスペクトルとなった。この原因は,立体規則性に加 えて,モノマー連鎖構造に起因する共鳴信号の分裂が重なり合ったためである[9]。 図 3-1 各共鳴領域における13 C NMR スペクトルの比較(100 MHz,CDCl3中 55 °C)。
a) PMMA[H-0],b) MMA-TBMA 共重合体[L-41],c) PMMA と PTBMA の混合試 料[B-42],d) PTBMA[H-100] 1:α-メチル炭素,2:TBMA 単位の側鎖 t-ブチル基のメチル炭素,3:主鎖 4 級炭素, 4:MMA 単位の側鎖メチル炭素,5:主鎖メチレン炭素,6:TBMA 単位の側鎖 t-ブチ ル基の 4 級炭素,7:カルボニル炭素 まず,図 3-1 の 6 つの共鳴領域について,各試料のバケット積分値から構成したデ ータ群を用いて,それぞれ独立に PCA を行った。各共鳴領域の第 1 主成分(PC1)と第 2 主成分(PC2)のスコアプロットを図 3-2 に示す。カッコ内は各主成分の寄与率である。 全てのスコアプロットにおいて,PC1 と PC2 の寄与率の合計が 96 %を超えていたた め,これらのスコアプロットは各共鳴領域のスペクトル情報の大部分を説明できてい
ると言える。図 3-2a~c に示すように,カルボニル炭素,主鎖 4 級炭素,そして α-メチル炭素のスペクトル情報が反映されたスコアプロットは,ほぼ同じ形状となった。 これは,これらの炭素原子が MMA と TBMA の両方に共通な構造のためである。一 方,図 3-2e,f に示す TBMA 単位の側鎖 t-ブチル基の 4 級炭素とメチル炭素領域のス コアプロットは,いずれも PMMA だけが大きく外れた形状となり,他の試料と容易 に判別できた。これらの共鳴領域では,PMMA の共鳴信号が観測されないからであ る。また,主鎖メチレン炭素と MMA 単位の側鎖メチル炭素領域のスコアプロット(図 3-2d)の形状は,カルボニル炭素,主鎖 4 級炭素,α-メチル炭素のスコアプロット(図 3-2a~c)と似た形状となったが,PC1 が大きくなるにつれて,混合試料の PC1 のプ ロット間隔が狭くなった。この理由は,MMA 単位にしかない側鎖メチル炭素の共鳴 信号が,主鎖メチレン炭素の共鳴信号に重なったためと考えられる。仮に,MMA 単 位の側鎖メチル炭素の共鳴信号を主鎖メチレン炭素の共鳴信号から分離できたとす ると,主鎖メチレン炭素の PCA スコアプロットは,カルボニル炭素,主鎖 4 級炭素, α-メチル炭素のスコアプロット(図 3-2a~c)と同じ形状になると推測される。 13 C NMR スペクトルに含まれる共重合体の組成とモノマー連鎖の情報を無駄にし ないために,PMMA と PTBMA の両方に共通な構造であるカルボニル炭素,主鎖 4 級炭素,α-メチル炭素の共鳴領域について,バケット積分のデータ群を結合し,再度 PCA を行った。上記 3 つの共鳴領域を結合したデータ群(データ群 A)の PC1 と PC2 のスコアプロットを図 3-3 に示す。PC1 と PC2 の寄与率は,それぞれ 77.1 %と 20.7 % となり,2 つの主成分でデータ群 A の情報をほぼ網羅できたと言える。なお,以後の 検討には,データ群 A を用いることとした。
図 3-2 27 試料の13 C NMR スペクトルの各共鳴領域での第 1 主成分-第 2 主成分ス コアプロット。 a) カルボニル炭素領域,b) 主鎖 4 級炭素領域,c) α-メチル炭素領域,d) MMA 単位 の側鎖メチル炭素と主鎖メチレン炭素領域,e) TBMA 単位の側鎖t-ブチル基の 4 級炭 素領域,f) TBMA 単位の側鎖t-ブチル基のメチル炭素領域
◆:PMMA,◆:PTBMA,◇:PMMA と PTBMA の混合試料,□:初期共重合体,■: 後期共重合体
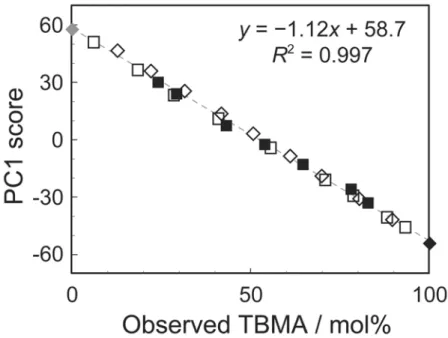
まず,PC1 について考える。PMMA(◆),PTBMA(◆)とそれらの混合試料(◇) の PC1 のスコアは,TBMA 組成の増加とともに単調に減少した。また共重合体(□, ■)の PC1 のスコアも同様の傾向を示した。PCA では,スコアプロットの他に,ロ ーディング(各主成分を表す疑似的なスペクトル)が得られる。カルボニル炭素,主 鎖 4 級炭素,および α-メチル炭素の13 C NMR スペクトルと,対応するローディング を図 3-4 に示す。PC1 ローディングの正側が PMMA の共鳴信号に,負側が PTBMA の共鳴信号をそれぞれ捉えていることがわかる。つまり,PC1 では,MMA 単位と TBMA 単位を判別していると言える。図 3-3 のスコアプロットの結果を考慮すると, PC1 には試料の組成が反映されていることが示唆された。そこで 27 種の試料全てに ついて,PC1 の値と試料中の TBMA 組成との関係を調べた。その結果,図 3-5 に示 すように,両者の相関係数R2は 0.998 となり,非常に良い相関があることが確認でき た。 図 3-3 27 試料の13 C NMR スペクトルのカルボニル炭素,主鎖 4 級炭素,α-メチル 炭素領域を結合したデータ群 A の第 1 主成分-第 2 主成分スコアプロット(記号は図 3-2 と同じ)。
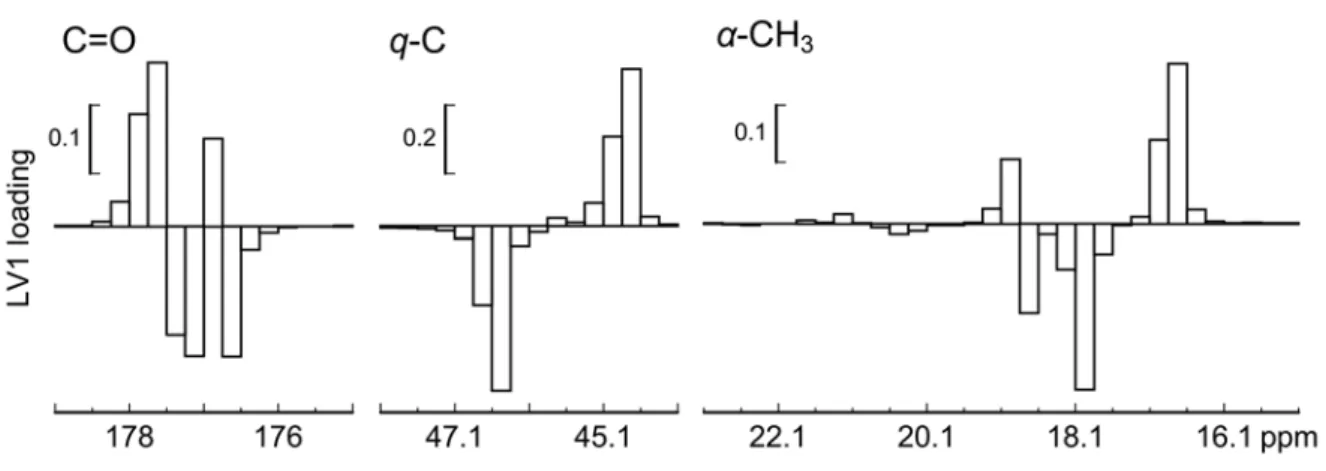
図 3-4 カルボニル炭素,主鎖 4 級炭素,α-メチル炭素領域の13
C NMR スペクトルと, データ群 A の第 1 主成分と第 2 主成分ローディング d)。
図 3-5 27 試料の PC1 のスコアと TBMA 組成の関係(記号は図 3-2 と同じ)。 次に,PC2 について考える。図 3-3 では,共重合体のスコアプロットは,収率や分 子量に関係なく,逆放物線を示した。また,共重合体(□,■)と混合試料(◇)の PC2 のスコアの差は,等モル組成において最大となった。図 3-4 の PC2 ローディング では,正側が 2 つの単独重合体の共鳴信号に,負側が共重合体に特有な共鳴信号(例 えば,175.75 – 176.5 ppm や 178.0 – 179.0 ppm)に,それぞれ対応していた。これらの 結果から,PC2 には試料中の 2 連子モノマー連鎖の同種・異種性が反映されていると 考えられた。そこで,2 連子モノマー連鎖の同種・異種性を定量的に表す指標として, MMA 単位と TBMA 単位で構成される 2 連子の異種モノマー連鎖分率 fMTを導入した。 一般に,メタクリル酸エステルのラジカル共重合は,ターミナルモデルに従うことが 知られている[10]。モノマーM1と M2から構成される共重合体中の M1-M2連鎖分率f12 は,式(3-1)で表される[11]。 21 12 21 12 12 2 P P P P f (3-1) ここで,P12と P21は,それぞれ M1末端ラジカルに M2モノマーが付加する確率と, M2末端ラジカルに M1モノマーが付加する確率である。重合初期では,式(3-2)および
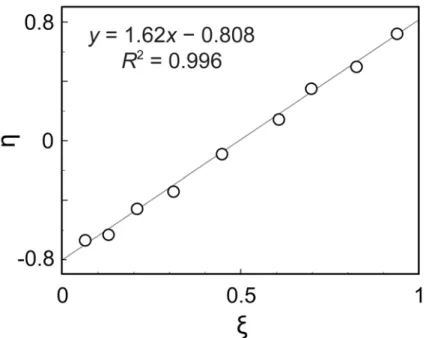
(3-3)が成り立つ[12]。 2 1 2 1 2 12 · ) 1 · 4( + 1 + 1 · 2 = F F r r F P - (3-2) 2 1 2 1 1 21 · ) 1 · 4( + 1 + 1 · 2 = F F r r F P - (3-3) ここで,r1と r2は,それぞれモノマーM1と M2のモノマー反応性比である。F1と F2 は,それぞれ初期重合体の M1単位と M2単位のモル組成であり,F1 + F2 = 1 である。 よって,初期重合体のf12は,式(3-4)で与えられる。 2 1 2 1 2 1 12 · ) 1 · 4( + 1 + 1 · · 4 = F F r r F F f - (3-4) 式(3-4)が示すように,f12は共重合組成に依存する。共重合組成が等モル(F1 = F2 = 0.5) のとき,f12は最大となる。ベルヌーイ統計に従うようなモノマー連鎖分布となる共重 合系(r1 = r2 = 1)では,F1 = F2 = 0.5 のとき f12 = 0.5 となる。一方,単独重合体と交 互共重合体の f12は,それぞれ 0 と 1 となる。なお,f12は,Harwood らが定義したラ ンナンバー(RN) [13]と同じ意味を持つ(RN = 100 f12)。 本研究における MMA と TBMA のモノマー反応性比 rMと rTは,表 3-1 に示した L-6 ~ L-93 の 初 期 重 合 体 9 種 の 仕 込 み モ ノ マ ー 組 成 と 共 重 合 組 成 を 用 い て , Kelen-Tüdõs 法[14]により,rM = 0.81 ± 0.06 と rT = 1.26 ± 0.03 と算出された(図 3-6)。 この値は,既報と近い値(rM = 0.96,rT = 1.35:Yuki ら[10];rM = 0.68,rT = 1.29:Zhao[15] ら)である。そこで,初期共重合体 9 種と単独重合体 2 種について,PC2 のスコアと fMTの関係を調べた結果,図 3-7 に示すように,両者の相関係数 R2は 0.996 となり, 非常に良い相関があることが確認できた。
図 3-6 MMA-TBMA 共重合系での Kelen-Tüdõs プロット。
図 3-7 初期共重合体 9 種と単独重合体 2 種の PC2 のスコアと MMA-TBMA モノマー 連鎖分率 ƒMTの関係(記号は図 3-2 と同じ)。
3-4 共重合組成の推定 まず,PMMA,PTBMA とこれらの混合試料 9 種のカルボニル炭素,主鎖 4 級炭素 およびα-メチル炭素の13 C NMR スペクトルを説明変量とし,1H NMR スペクトルか ら求めた TBMA 組成(表 3-1)を目的変量として作成した部分最小二乗回帰(PLSR) モデルの精度および正確さを確認するため,leave-one-out 法[16]によるクロスバリデ ーションを行った。回帰モデルに用いた潜在変数は 1(LV1)とし(寄与率 99.5 %),そ のローディングを図 3-8 に示す。このローディングの形状は,PCA での PC1 ローデ ィングとほぼ同じであったため,LV1 は組成推定用の PLSR モデルとして妥当である ことが確認できた。組成推定用 PLSR モデルにおいて,1 H NMR スペクトルから求め た TBMA 組成と,PLSR で推定した TBMA 組成との関係は,図 3-9 に示すように, 相対標準偏差RSD = 1.6 %,R2 = 0.999 で一致した。この PLSR モデルは,高い精度と 正確さで混合試料の組成を推定できると言える。 図 3-8 PMMA,PTBMA とこれらの混合試料 9 種からなる TBMA 組成推定用 PLSR モデルの第 1 潜在変数ローディング。
図 3-9 PMMA,PTBMA とこれらの混合試料 9 種からなる TBMA 組成推定用 PLSR モデルのクロスバリデーション(記号は図 3-2 と同じ)。 そこで,この PLSR モデルを用いた PLSR により,共重合体 16 種のカルボニル炭素, 主鎖 4 級炭素および α-メチル炭素の13 C NMR スペクトルから,これらの試料の TBMA 組成の推定を試みた。その結果,図 3-10 に示すように,共重合体の13 C NMR スペク トルの PLSR で推定した TBMA 組成は,1 H NMR スペクトルから求めた TBMA 組成 に対して,RSD = 3.4 %,R2 = 0.997 で一致した。このことから,単独重合体とそれら の混合試料の13 C NMR スペクトルと組成から構築した PLSR モデルを用いれば,スペ クトルの帰属をすることなく,高い精度と正確さで共重合体の組成が推定できること が明らかになった。
図 3-10 PLSR モデルで推定した共重合体 16 種の TBMA 組成と1 H NMR による実測 値との相関(記号は図 3-2 と同じ)。 3-5 2 連子モノマー連鎖分布の推定 3-3節の PCA における PC2 の正側のローディング結果や,共重合体の PC2 の値 と fMTとの相関関係から,同種モノマー連鎖分率 fMM(MMA-MMA 連鎖)および fTT (TBMA-TBMA 連鎖)を含めた 2 連子モノマー連鎖分布を推定できると考えた。 初期共重合体のfMMとfTTは,式(3-5)と(3-6)として,それぞれ表される[11]。 ) · ) 1 · 4( + 1 + 1 · 2 (1 · = T M T M T M TM MT MM TM MM r r F F F F P P P P f - -= + (3-5) ) · ) 1 · 4( + 1 + 1 · 2 (1 · = T M T M M T TM MT TT MT TT r r F F F F P P P P f - -= + (3-6) ここで,PMMは MMA 末端ラジカルに MMA モノマーが付加する確率であり,PTTは TBMA 末端ラジカルに TBMA モノマーが付加する確率である[12]。また,PMM = 1 – PMT, PTT = 1 – PTMである。初期共重合体 9 種について,式(3-4),(3-5)および(3-6)を用いて 3 種類の 2 連子モノマー連鎖分率 fMM,fMT,fTTを計算し,表 3-3 にまとめた。単独重
合体の混合試料の 2 連子モノマー連鎖分率は,混合試料中の MMA および TBMA 組 成をそれぞれfMMおよびfTTとし,fMT = 0 とした。 表 3-3 理論式から計算された初期共重合体 9 種の 2 連子モノマー連鎖分布 Code fMM / % fMT / % fTT / % L-6 L-18 L-28 L-41 L-56 L-71 L-79 L-88 L-93 88.4 66.7 51.3 35.0 19.9 8.6 4.6 1.4 0.5 11.2 29.9 40.6 48.2 49.2 41.3 33.5 21.0 12.7 0.4 3.4 8.1 16.8 30.9 50.1 61.8 77.5 86.8 PMMA,PTBMA とこれらの混合試料 9 種,初期共重合体 9 種のカルボニル炭素, 主鎖 4 級炭素および α-メチル炭素の13 C NMR スペクトルを説明変量とし,理論式か ら求めたfMM,fMT,fTT(表 3-3)を目的変量として作成した PLSR モデルの精度およ び正確さを確認するため,leave-one-out 法を用いたクロスバリデーションを行った。 これらの PLSR モデルでは,第 1 潜在変数(LV1)と第 2 潜在変数(LV2)の寄与率は,表 3-4 に示す値となり,それぞれの累積寄与率は 99 %を越えた。 表 3-4 2 連子モノマー連鎖量推定用 PLSR モデルの寄与率 LV fMM / % fMT / % fTT / % LV1 LV2 94.7 4.9 97.4 2.3 93.4 6.4 Total 99.6 99.7 99.8 fMM推定用 PLSR モデルでの LV1 ローディングは,PMMA のスペクトルが正,PTBMA のスペクトルが負で抽出され,LV2 ローディングは共重合体に特有な共鳴信号が抽出 されていた(図 3-10a)。これらは正負の符合反転はあるものの,PCA での PC1 と PC2 のローディングとほぼ同じ形状となった。つまり,LV1 は MMA の 2 連子,LV2 は異 種 2 連子を,それぞれ捉えていると言える。一方,fTT推定用 PLSR モデルでの LV1
ローディングは,fMM推定用 PLSR モデルとは逆に PTBMA のスペクトルが正,PMMA のスペクトルが負で抽出されたが,LV2 ローディングは fMM推定用 PLSR モデルと同 じ形状となった。このことから,LV1 は TBMA の 2 連子,LV2 は異種 2 連子を,そ れぞれ捉えていると言える。 図 3-11 PMMA,PTBMA とこれらの混合試料 9 種,初期共重合体 9 種からなる 2 連子モノマー連鎖量推定用回帰モデルの第 1 および第 2 潜在変数ローディング。
fMT推定用 PLSR モデルの LV1 ローディングは,正負の符合反転や強度の違いはあ るものの,fMMおよびfTT推定用 PLSR モデルの LV2 ローディングと同じ形状となった。 また,fMT推定用 PLSR モデルの LV2 ローディングは,fMM推定用 PLSR モデルの LV1 ローディングと同じ形状となった。つまり,LV1 は異種 2 連子,LV2 は MMA の 2 連 子を,それぞれ捉えていると言える。これらの結果から, 2 つの潜在変数 LV1 と LV2 はfMM,fMT,fTT推定用 PLSR モデルとして妥当であることが確認できた。 図 3-12 PMMA,PTBMA とこれらの混合試料 9 種,初期共重合体 9 種からなる 2 連子モノマー連鎖分布推定用 PLSR モデルのクロスバリデーション(記号は図 3-2 と 同じ)。
2 連子モノマー連鎖分布推定用 PLSR モデルにおいて,理論式から求めた fMM,fMT, fTTと,PLSR で推定した fMM,fMT,fTTとは,図 3-12 に示すように,非常に良い一致 を示した。この PLSR モデルにより,2 連子モノマー連鎖分布が高い精度と正確さで 推定できることがわかる。 図 3-12 で構築した PLSR モデルの適用範囲を確認するため,2 連子モノマー連鎖分 布が大きく異なる初期共重合体 2 種を混合した試料(CB-1,CB-2)について,2 連子モ ノマー連鎖分布の推定を試みた。元の初期共重合体の試料コードと混合試料 CB-1, CB-2 の TBMA 組成を表 3-5 に示す。この PLSR モデルを用いた PLSR により,CB-1, CB-2 のカルボニル炭素,主鎖 4 級炭素および α-メチル炭素の13C NMR スペクトルか ら,これら混合試料のfMM,fMT,fTTを推定した。その結果,図 3-13 に示すように, CB-1 と CB-2 の 13C NMR スペクトルの PLSR で推定した fMM,fMT,fTTは,CB-1 と CB-2 の混合比から計算で求めた fMM,fMT,fTTに対して,RSD = 2.6%,R2 = 0.995 で一 致した。CB-1 と CB-2 の 2 連子モノマー連鎖分布は,図 3-14 に示すように,初期共 重合体の組成とモノマー反応性比から算出した理論曲線から大きく外れているにも 関わらず,図 3-12 で構築した PLSR モデルによって,高い精度と正確さで推定でき た。 表 3-5 初期共重合体の混合試料 CB-1,CB-2 Original copolymer Code I II TBMA / mol% a CB-1 CB-2 L-71 L-6 L-28 L-93 47.0 48.4 a1 H NMR により決定
図 3-13 2 連子モノマー連鎖分布推定用 PLSR モデルを用いた初期共重合体混合試料 CB-1,CB-2 の 2 連子モノマー連鎖分布の推定。 ●:ƒMM,●:ƒTT,○:ƒMT 図 3-14 初期共重合体混合試料と元の初期共重合体の 2 連子モノマー連鎖分布の比 較(---は,初期共重合体の組成とモノマー反応性比から算出した理論曲線)。 a) CB-1,b) CB-2 ●:混合試料の ƒMM,●:混合試料の ƒTT,○:混合試料の ƒMT ■:初期共重合体の ƒMM,■:初期共重合体の ƒTT,□:初期共重合体の ƒMT
さらに,上述の PLSR モデルを用いて推定した後期共重合体 7 種(H-24~H-83, M-29~M-79)の 2 連子モノマー連鎖分布を図 3-15 に示す。後期共重合体の fMM,fMT, fTTは,初期重合体の組成とモノマー反応性比から算出した理論曲線から,わずかに外 れていることがわかった。理想共重合(r1 = r2 = 1)でない限り,重合反応中の未反応 モノマー組成は刻々と変化するため,ある瞬間に生成する共重合体の組成と 2 連子モ ノマー連鎖分率も変化する。この現象を理論式で表した Spinner の方法[17]を用いて, ある瞬間に生成する共重合体の fMTの変化と,生成した共重合体全体の平均 fMTを計 算した。その結果,図 3-16 に示すように,PLSR により推定した後期共重合体の fMT は,重合率 100 %における共重合体の平均 fMT(計算値)と同じ傾向を示した。この ことから,PLSR により推定した後期共重合体の 2 連子モノマー連鎖分布が妥当であ ることが確認できた。 図 3-15 後期共重合体の 2 連子モノマー連鎖分布(---は,初期共重合体の組成とモ ノマー反応性比から算出した理論曲線)。 ■:ƒMM,■:ƒTT,□:ƒMT
図 3-16 重合率の違いによる MMA-TBMA 共重合体の平均異種 2 連子モノマー連鎖 分率 a),ならびに,重合率の変化に伴う生成共重合体の組成変化と異種 2 連子モノ マー連鎖分率の変化 b)-d)。Spinner の方法により算出。 仕込みモノマー組成(MMA/TBMA mol%):b) 80/20,c) 50/50,d) 20/80 本章では,初期共重合体の 13 C NMR スペクトルと理論式から算出した 2 連子モノ マー連鎖分率の PLSR モデルを構築し,それを用いて未知試料の 2 連子モノマー連鎖 分布を精度良く推定できた。理論式(3-1)~(3-6)を組み合わせて連鎖分率を算出し,初 期共重合体の13 C NMR スペクトルとともに PLSR モデルを構築すれば,3 連子などの より長いモノマー連鎖分率も,PLSR によって推定できるはずである。 実際の高分子材料は,共重合体の精製工程や混合工程を経る場合が多いため,共重 合体の 2 連子モノマー連鎖分布は,理論式から算出されたものと異なる可能性がある。 しかし,図 3-12 の PLSR モデルのように,初期共重合体の 2 連子モノマー連鎖分布 と13 C NMR スペクトルを用いた PLSR モデルを構築すれば,どんな操作や工程を経た 共重合体であろうとも,正確で精度の高い 2 連子モノマー連鎖分布を推定することが できる。
文献
[1] A. S. Brar, G. S. Kapur, Polym. J., 1988, 20, 371.
[2] T. Nishiura, T. Kitayama, K. Hatada, Int. J. Polymer Anal. Charact., 2000, 5, 401 [3] R. J. Pell, B. C. Erickson, R. W. Hannah, J. B. Callis, B. R. Kowalski, Anal. Chem.,
1988, 60, 2824.
[4] M. Shimoyama, H. Maeda, K. Matsukawa, H. Inoue, T. Ninomiya, Y. Ozaki, Vib.
Spectrosc., 1997, 14, 253.
[5] M. Shimoyama, S. Hayano, K. Matsukawa, H. Inoue, T. Ninomiya, Y. Ozaki, J. Polym.
Sci. B: Polym. Phys., 1998, 36, 1529.
[6] 三井利幸, 肥田宗政, 藤村義和, 分析化学, 1989, 38, 349.
[7] C. E. Miller, B. E. Eichinger, T. W. Gurley, J. G. Hermiller, Anal. Chem., 1990, 62, 1778.
[8] M. Sardashti, J. J. Gislason, X. Lai, C. A. Stewart, D. J. O'Donnell, Appl. Spectrosc.,
2001, 55, 467.
[9] K. Hatada, T. Kitayama, NMR Spectroscopy of Polymers; Springer, 2004.
[10] H. Yuki, Y. Okamoto, Y. Shimada, K. Ohta, K. Hatada, J. Polym. Sci. Polym. Chem.
Ed., 1979, 17, 1215.
[11] K. Ito, Y. Yamashita, J. Polym. Sci. A: General Papers, 1965, 3, 2165. [12] C. Tosi, Makromol. Chem., 1967, 108, 307.
[13] H. J. Harwood, W. M. Ritchey, J. Polym. Sci. B: Polym. Lett., 1964, 2, 601. [14] T. Kelen, F. Tüdõs, J. Macromol. Sci., Chem., 1975, 9, 1
[15] Y. Zhao, Y. W. Luo, C. Ye, B. G. Li, S. Zhu, J. Polym. Sci. A: Polym. Chem., 2009, 47, 69.
[16] A. Lorber, B. R. Kowalski, Appl. Spectrosc., 1990, 44, 1464.