北海道大学大学院薬学研究院薬剤分子設計学研究室 (〒0600812 札幌市北区北 12 条西 6 丁目)
e-mail: tnakam@pharm.hokudai.ac.jp
本総説は,平成 30 年度日本薬学会奨励賞の受賞を記念 して記述したものである.
2018 The Pharmaceutical Society of Japan
―Review for award―
脂質ナノ粒子を基盤としたがん免疫療法のためのナノ DDS 開発
中 村 孝 司
Development of a Nano DDS for Cancer Immunotherapy Based on Llipid Nanoparticles
Takashi NakamuraLaboratory for Molecular Design of Pharmaceutics, Faculty of Pharmaceutical Sciences, Hokkaido University: Kita 12, Nishi 6, Kita-ku, Sapporo 0600812, Japan.
(Received July 19, 2018)
The appearance of immune checkpoint inhibitors has been a major turning point in cancer therapy. The success of immune checkpoint therapy has revolutionized the ˆeld of cancer therapy, and immunotherapy has joined the cancer treatment ranks as a pillar. To induce eŠective anti-tumor immune responses, it is necessary both to enhance the activity of immune cells and to block immune suppression by tumor cells. Carrier type drug delivery systems based on nanobiotechnology (nano DDS) represent a potentially useful technology for e‹ciently achieving both: enhancement of the activity of immune cells and blocking immune suppression. It has become clear that nano DDS can improve the prac-tical utility of a wide variety of immune functional molecules and thus regulate drug kinetics and intracellular dynamics to improve drug e‹cacy and reduce side eŠects. We have been in the process of developing a nano DDS for the enhance-ment of cancer immunotherapy. A nano DDS encapsulating an agonist of a simulated interferon gene pathway greatly enhanced the activity of the agent's antitumor immune response. To block immune suppression, we successfully devel-oped a small interfering RNA loaded into a nano DDS which regulates gene expression in immune cells. In this review, we summarize our recent eŠorts regarding cancer immunotherapy using nano DDS.
Key words―drug delivery system; lipid nanoparticle; cancer immunotherapy; adjuvant; small interfering RNA
1. はじめに がん免疫療法は外科療法,化学療法,放射線療法 につぐ第 4 のがん治療法として期待されてきた一方 で,懐疑的な位置づけに長い間甘んじていた.しか しながら,最近の免疫チェックポイント阻害療法や キメラ抗原受容体導入 T 細胞療法(CAR-T 療法) の臨床での成功は,がん治療に革命を起こし,免疫 療法に対する認識を一変させた.そして,がん免疫 療法は今や最も積極的に医薬品開発が進められてい る分野の 1 つになっている.現在,がん免疫応答は 「がん免疫サイクル」と呼ばれる時空間的なダイナ ミクスとして捉えられており,このサイクルを正常 にまわすことが治療の鍵となっている.1)がん組織 における抗原の放出に始まり,樹状細胞などの抗原 提示細胞による抗原提示,リンパ節における T 細 胞の活性化,活性化 T 細胞のがん組織への遊走,T 細胞によるがん細胞の認識と排除,それに伴うがん 抗原の放出によりサイクルが繰り返される.しかし ながら,がん患者ではサイクルの一部若しくは複数 のプロセスが阻害されているため,阻害されている プロセスを改善するための様々な治療法を組み合わ せた複合がん免疫療法の開発が積極的に進められて いる.そのような中,drug delivery system(DDS) 技術,特にナノバイオテクノロジーを駆使したキャ リア型 DDS(ナノ DDS)に大きな期待が寄せられ ている.がん抗原,ペプチド,難溶性アジュバン ト,核酸など,免疫応答を制御するための分子は多 種多様である.しかしながら,それらの分子は分解 や細胞親和性などの問題のために単体では機能しな い.また,がん免疫療法における標的は 1 つではな く,様々な免疫組織や免疫細胞へと機能性分子を効 率的に送達することが求められる.筆者はがん免疫 応 答 を 強 化 す る た め の 脂 質 ナ ノ 粒 子 ( lipid nanoparticle; LNP)を基盤としたナノ DDS 開発を 進めており,本総説では最近の成果を中心に紹介す
中村孝司 2010年北海道大学大学院生命科学院博 士後期課程修了,博士(生命科学)取 得,20072010 年日本学術振興会特別 研究員.博士研究員を経て,2011 年 1 月より現職,助教.2015 年日本薬学会 北海道支部奨励賞,2016 年日本薬剤学 会奨励賞,2018 年日本薬学会奨励賞を 受賞. やアジュバントなどの免疫活性化分子を樹状細胞に 代表される抗原提示細胞へと効率的に送達しなけれ ばならない.がん抗原はタンパク質やペプチドであ るため,それ単体では分解や細胞取り込みが障壁と なり機能を発揮することはできない.また,アジュ バントに関しても難溶性,易分解性などの物理学的 性質が障壁となり標的細胞への送達効率が課題と なっている.筆者は標的細胞へ効率的に免疫活性化 分子を送達する独自のナノ DDS として,R8 修飾 LNP(R8-LNP)を開発し,抗原を効率的に樹状細 胞内へと送達,がん免疫応答に重要な MHC クラ ス I(MHC-I)分子への抗原提示を特異的に促進す ることに成功した.2)この特異的な MHC-I 抗原提 示が R8-LNP によるプロテアソームを介した抗原 プロセッシング過程の促進によるものであるという 興味深い知見を初めて見い出すこともできた.3)抗 原に加え,アジュバントとして 2 本鎖 RNA を搭載 した R8-LNP を用いてアジュバントの細胞内認識 におけるトポロジー制御の重要性を初めて提唱し た.4)また,強力ながん免疫応答を誘導する natural killer T(NKT)細胞を活性化させる糖脂質アジュ バントとして a-ガラクトシルセラミドが注目され ているが,5)難溶性の脂質分子であるため抗原提示 細胞へと効率的に送達できる技術が必要であった. そこで筆者は R8-LNP の脂質膜に a- ガラクトシル セラミドを搭載することで抗原提示細胞に効率的に 送達し,LNP を用いたがん免疫療法例として初め て報告した.68)このような抗原やアジュバントの 送達における R8-LNP の高い有用性は,がん免疫 療法以外にも結核ワクチンへと応用することができ た.杉田昌彦教授(京都大学ウィルス・再生医科学 研究所)との共同研究の成果として,結核菌の脂質 抗原のナノ DDS 化・抗原提示細胞への送達に初め て成功し,その有用性をモルモットやアカゲザルで 実証した.913) 3. BCG菌体成分搭載 R8-LNP による膀胱がん 免疫療法の開発 表在性膀胱がんの外科的手術後の再発防止や膀胱 上皮内がんの治療のために,BCG 生菌の膀胱内注 のがん免疫療法剤の開発が強く望まれてきた.この ような状況下,BCG 生菌の免疫活性化中心である
BCG細胞壁骨格(BCG cell wall skeleton;
BCG-CWS)は BCG 生菌に代わる非感染性の製剤候補と して期待されているが,数十数百 mm の巨大分子 かつ難溶性であるため,細胞への親和性が極端に低 いという問題を抱えていた.この長年の問題を打破 す る た め に , 筆 者 は BCG-CWS を ナ ノ サ イ ズ の LNP へと搭載する新規パッケージング法として, Liposome Evaporatedvia emulsiˆed lipid(LEEL)
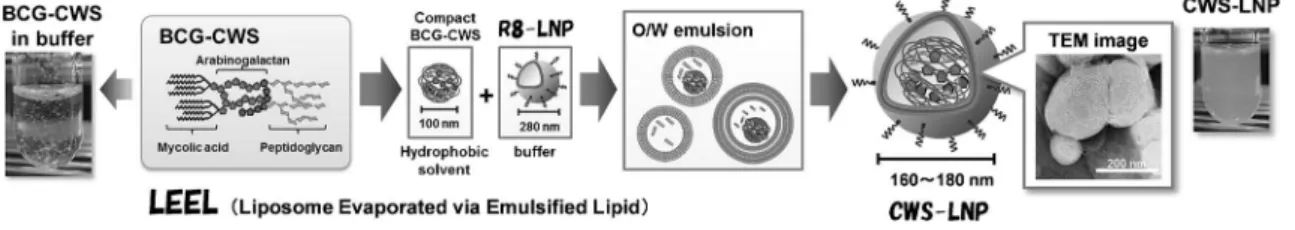
法を開発した(Fig. 1).14)本方法は BCG-CWS が コンパクトな構造をとる疎水環境を維持した状態で LNPに内封することが可能であるため,凝集がな く高い均一性を有する水性懸濁剤を調製することが で き る . BCG-CWS を 搭 載 し た R8-LNP ( CWS-LNP)は R8 ペプチドによる高い細胞親和性によ り,マウス膀胱がん細胞(MBT-2)やマウス骨髄 由来樹状細胞(bone marrow derived dendritic cell; BMDC)に 95%以上の効率で取り込まれ,マウス 膀胱がん皮下移植モデルやラット膀胱がん自然発症 モデ ル にお い て, 強力 な 抗膀 胱が ん 作用 を示 し た.14)さらに,CWS-LNP による抗膀胱がん作用の 誘導メカニズムは BCG 生菌と同様のメカニズムで あることが明らかになった.15)これらの結果は, CWS-LNPが BCG 生菌の代替え製剤として高いポ テンシャルを有していることを示している.また製 剤学的観点から LNP の尿中での安定性を向上させ る研究も進めており,カチオン性 LNP にポリエチ レングリコール(polyethylene glycol; PEG)化脂質 を導入することでヒト尿中での凝集を防ぐことがで
きることを報告している.16)興味深いことに,PEG
Fig. 1. Schema of Liposomes Produced by Evaporatedvia the Emulsiˆed Lipid (LEEL) Method
BCG-CWS forms compact particles in hydrophobic solvents. The BCG-CWS included hydrophobic solution is then loaded on a buŠer containing R8-LNP. The loaded solution is then emulsiˆed by the means of a probe-type sonicator to form an oil in water (O/W) emulsion. Finally, the hydrophobic solvent is removed by evaporation to form a CWS-LNP. 込みに大きく影響することを見い出した. 4. pH 応答性 LNP による stimulator of interfer-on genes(STING)アゴニスト送達 がん免疫サイクルの開始には,樹状細胞によるが ん抗原の提示と成熟化が必要である.樹状細胞の成 熟化は,病原体由来や内因性のアジュバント成分を 自然免疫受容体が認識することで誘導される.がん 免疫サイクルでは,樹状細胞が細胞質 DNA セン サーである cyclic-GMP-AMP(cGAMP)synthase (cGAS)を介して,がん細胞由来の DNA を認識す ることで下流の STING 経路を活性化し,成熟化す ると考えられている.17)また,免疫原性が高い腫瘍 に対するがん免疫応答には cGAS-STING 経路が必 須であり,他の自然免疫受容体の関与はほとんどな いことが報告された.18)それゆえ,cGAS-STING 経路を活性化させるアゴニスト(STING アゴニス ト)は真のがんアジュバントになると期待されてい る.筆者は STING アゴニストとして,環状ジヌク レオチドである cyclic-di-GMP(cdGMP)を選択し た.cdGMP は,細胞質の DNA センサーの 1 つで ある DDX41 に結合し,STING と複合体を形成す ることで,tank binding kinase 1 (TBK-1)-interferon
regulator factor 3(IRF3)を介したシグナル伝達を
活性化,I 型 interferon(IFN)を産生する.19,20)し かしながら,DNA や環状ジヌクレオチドは負電荷 を有する親水性の核酸分子であるため,それ自身で は細胞膜を透過することができず,受容体が存在す る細胞質への送達ががんアジュバントとしての開発 の大きな障壁となっていた.この問題を解決するた めには,単に細胞へ到達するだけではなく,「細胞 質」へと STING リガンドを届けるように細胞内動 態を厳密に制御可能なナノ DDS が必要であった. 通常,ナノ粒子などの異物はエンドサイトーシス により細胞に取り込また後,ライソソームと融合し 分解されるため,STING アゴニストを細胞質へと 届けるためにはエンドソームから細胞質へと脱出し なければならない.筆者が所属する北海道大学大学 院薬学研究院・薬剤分子設計学研究室(原島秀吉教 授),以下当研究室では搭載分子を細胞質へと送達 するための pH 応答性脂質の開発を進めている.第 1 世代の YSK05 は血液中などの中性環境では電荷 を持たず,エンドソームなどの酸性環境に応答して 正に荷電する pH 応答能と膜融合性の高い脂肪酸鎖 を有する.21)YSK05を含む LNP に cdGMP を搭載
した cdGMP/YSK05-LNP [Fig. 2(A)]は,粒子径 が約 170 nm,z 電位が-4.3 mV とほぼ中性で均一 の高い粒子であった.22)通常,RAW264.7 細胞に cdGMP 単独で処理した場合,I 型 IFN の産生は認 められないが,cdGMP/YSK05-LNP を用いた場合 は,高い I 型 IFN の産生が認められ,市販のトラ ンスフェクション試薬よりも 7 倍高いものであっ た.この結果は,YSK05 による効率的な細胞質デ リ バ リ ー を 反 映 し て い る . 続 い て , ovalbumin (OVA)をモデル抗原として抗原特異的がん免疫応 答を評価した.cdGMP/YSK05-LNP を皮下投与し
たマウス群では,OVA 特異的な cytotoxic T
lympho-cyte(CTL)の強い誘導が認められ,E.G7-OVA 腫 瘍 に 対 す る 強 力 な 抗 腫 瘍 活 性 を 示 し た [ Fig. 2 (B)].22,23)一方で,がん種によっては MHC-I の発 現を低下・消失させることで CTL による免疫監視 を逃れているが,そのようながんに対しては NK 細 胞を中心とした MHC-I 非拘束性の免疫応答が重要 である.マウス尾静脈からの cdGMP/YSK05-LNP を投与すると,強力な I 型 IFN 産生を誘導し[Fig. 2(C)],脾臓 NK 細胞の活性化マーカー(NKG2D と CD69)の発現を顕著に増加させたことから, cdGMP/YSK05-LNP は NK 細胞を活性化させるこ とが明らかになった.23)続いて,MHC-I 陰性の腫
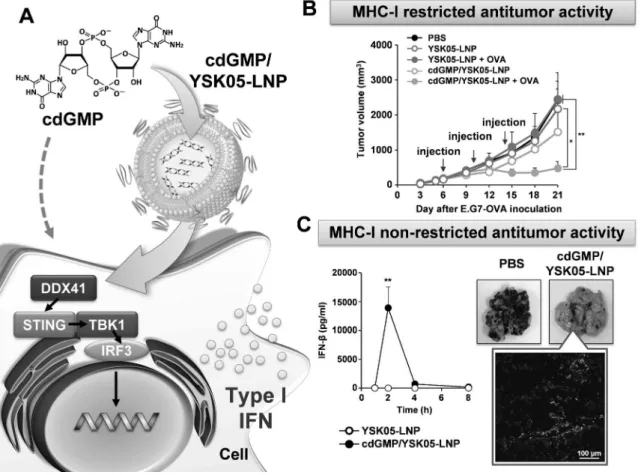
Fig. 2. Cancer Immunotherapy Using STING Agonist Loaded LNP
(A) Conceptual image of cdGMP/YSK05-LNP. (B) MHC class I (HMC-I) restricted antitumor eŠect by cdGMP/YSK05-LNP. Mice bearing E.G7-OVA tumors were subcutaneously injected with the cdGMP/YSK05-LNP and OVA (mean±S.E.M., n=5, p<0.01, p<0.05). (C) MHC-I non-restricted antitumor eŠect by the cdGMP/YSK05-LNP. Left: the IFN-b production after the treatment of cdGMP/YSK05-LNP (mean±S.E.M., n=3, p<0.01). Right: mice with B16-F10 melanomas were intravenously treated with cdGMP/YSK05-LNP. The tumor colonies in lungs were observed on day 21. The inˆltration of NK cells was observed in the lungs of mice with B16-F10 on day 9. White shows NK cells. Bar=100 mm. Reproduced from a part of our previous report.25)Copyright 2016
Else-vier B. V. 瘍モデルとして B16-F10(マウスメラノーマ)をマ ウス尾静脈から投与して作製した肺転移モデルを用 いて抗腫瘍活性を評価した結果,コントロール群と 比較して有意な腫瘍コロニーの減少が認められた [Fig. 2(C)].この抗腫瘍活性はアシアロ GM1 抗 体処理により NK 細胞を枯渇させた条件下では有意 に減少すること,肺の転移巣に NK 細胞が浸潤して いたことから[Fig. 2(C)],NK 細胞が主なエフェ クター細胞となって誘導されたことが示唆された. 以上のことから,cdGMP/YSK05-LNP は抗原特 異的及び非特異的ながん免疫応答の両方を効率的に 活性化できることから,幅広いがん種に対応可能な アジュバントシステムになることが期待される.現 在は免疫チェックポイント阻害剤を含めた他のがん 治療法との複合がん免疫療法における有用性評価を 進めている.
5. small interfering RNA(siRNA)搭載 LNP を
用いた免疫細胞の遺伝子発現制御 免疫チェックポイント阻害療法が登場するまで, がん抗原やアジュバントを用いた免疫活性化が主流 であったが,免疫チェックポイント阻害療法の臨床 での成功により免疫抑制解除戦略へのパラダイムシ フトが起こっている.免疫チェックポイント分子や 抑制性細胞による免疫抑制の解除には抗体や低分子 薬物が使用されているが,標的の制限,薬効の切れ 味,腫瘍関連微小環境における免疫抑制機構の複雑 性への対応などの課題を抱えている.一方で,si-RNA は理論上すべての遺伝子を標的とできるため, siRNA による免疫細胞の遺伝子発現制御は次世代 の免疫抑制解除戦略として期待できる.しかしなが ら,免疫細胞への siRNA 導入は非常に困難であ り,多くの場合,ウイルスベクターが使用されてい る.当研究室では遺伝子・核酸送達のための非ウイ
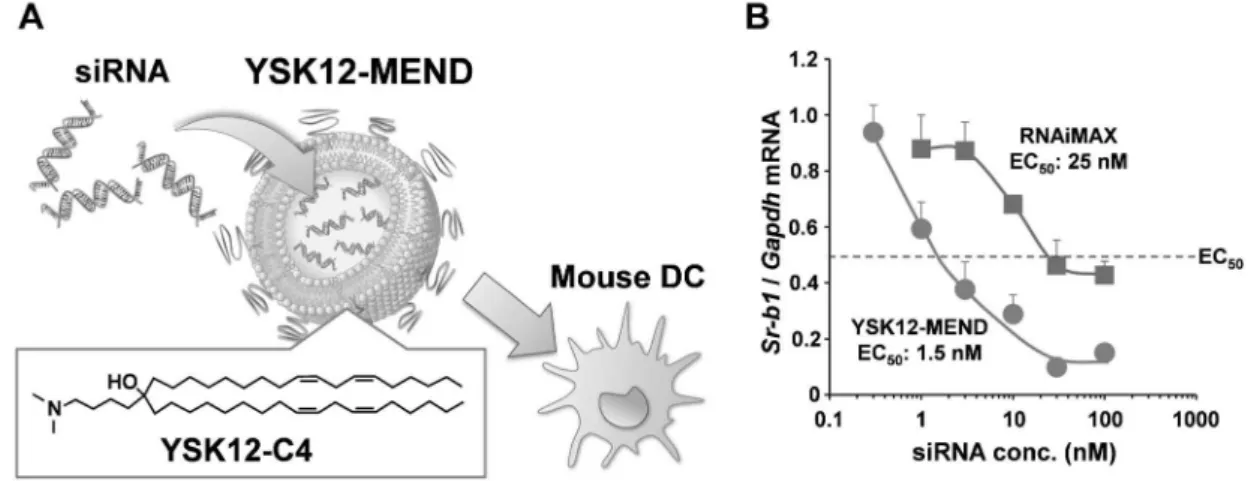
Fig. 3. Delivering siRNA to Mouse Dendritic Cells (DCs) by the YSK12-MEND
(A) Conceptual image of the MEND mediated siRNA delivery to mouse DCs. (B) Dose response curve for gene silencing e‹ciency by the YSK12-MEND in mouse DCs. Target gene was the scavenger receptor class B type 1 (Sr-b1). The vertical axis shows the relative Sr-b1/Gapdh mRNA level, in which the mean for the no treatment mouse DCs was assumed to be 1.0. Data are the mean±S.E.M. (n=35). Reproduced from a part of our previous report.25)Copyright
2016 Elsevier B. V.
ルスベクターとして,多機能性エンベロープ型ナノ 構造体(multifunctional envelope-type nano device;
MEND)を提唱しており,24,25)MEND を用いた樹
状細胞への siRNA 導入に長年取り組んできた.26,27)
最近,筆者は高い膜融合性を有するカチオン性脂質 YSK12-C4 を合成し,YSK12-C4 を含む MEND を 構築することで,マウス樹状細胞への効率的な
si-RNA 導入に成功した[Fig. 3(A)].28)siRNA を搭載
した YSK12-C4 を含む MEND(YSK12-MEND)は, 粒子径 180 nm,z 電位 5.8 mV, siRNA の封入率は 94%の均一な粒子であった.まず scavenger recep-tor class B member 1(SR-B1)に対する siRNA を マウス樹状細胞へと導入し,50%の遺伝子ノックダ ウンを実現するために必要な siRNA 濃度(eŠective concentration; EC50)を算出した.最も強力な si-RNA 導 入 試 薬 で あ る Lipofectaminesi-RNAiMAX ( RNAiMAX ) を 用 い た 場 合 の EC50 は 25 nMで あったのに対し,YSK12-MEND を用いた場合の EC50 は 1.5 nMと 約 17 倍 効 率 的 に 標 的 遺 伝 子 を ノックダウン可能であった[Fig. 3(B)].さらに RNAiMAX の場合は,60%ほどのノックダウン率 で頭打ちになるのに対し,YSK12-MEND は 90% 以上のノックダウン率を示した.この高いノックダ ウン効率は,YSK12-MEND の高いエンドソーム膜 破壊能によるエンドソーム脱出促進によることが示 唆されている.続いて,サイトカインシグナル経路 のネガティブ・フィードバック因子である suppres-sor of cytokine signaling 1(SOCS1)を標的とし,
ノックダウン効率を評価した.330 nMの siRNA 濃度において,80%以上のノックダウンが認められ, SOCS1 の発現低下に伴うサイトカイン産生能力の 増強も観察された.さらに SOCS1 をノックダウン したマウス樹状細胞を用いて,E.G7-OVA 腫瘍を 皮下移植したマウスに対して樹状細胞療法を試み た.その結果,YSK12-MEND を用いて SOCS1 を ノックダウンしたマウス樹状細胞を投与したマウス 群においてのみ,コントロール群と比較して有意な 腫瘍増殖抑制効果が認められた.この結果は,樹状 細胞投与に起因するがん免疫応答を促進し,抗腫瘍 活性を示すのに十分な抑制性因子のノックダウンを YSK12-MEND が実現できることを示している. 次のステージとして,ヒト免疫細胞への siRNA 導入 を検 証 した . まず ,ヒ ト 免疫 細胞 株 であ る Jurkat(ヒト T 細胞),THP-1(ヒト単球),KG-1 (ヒトマクロファージ),NK-92(ヒト NK 細胞)を 用いて検証を行った.29)これらの細胞は,文献情報 から,非ウイルスベクターによる siRNA 導入が特 に困難とされている細胞株である.130 nMの si-RNA 濃度でトランスフェクションした結果,対照 として使用した RNAiMAX は,一部の細胞の 30 nM の濃度で有意な遺伝子ノックダウンが認められた が,その効率は 4060%程度であった.一方で, YSK12-MENDは,Jurkat, THP-1, KG-1 において, 10 nMの siRNA 濃度であれば,90%の遺伝子ノッ クダウンを実現できることが明らかになった.これ らの細胞は,いずれも浮遊細胞であり,非ウイルス
細 胞 株 へ の 取 り 込 み を 調 べ た と こ ろ , YSK12-MENDはすべての細胞に均一に取り込まれていた のに対し,RNAiMAX はヘテロな取り込み,若し くは取り込まれていないことが明らかとなった.こ の細胞取り込みの大きな違いは,YSK12-MEND は 培地中でも安定かつ分散していたのに対し,RNAi-MAX は時間経過とともに凝集し,粒子径が増大す ることに起因すると考えられた.一方で,NK-92 に関しては,70%程度の遺伝子ノックダウンが可能 であったが,同時に毒性が強く発現するという問題 が残された.そこで NK-92 における毒性軽減を目 的とし,さらに研究を進めた.30)YSK12-MEND に 含まれる YSK12-C4 によるエンドソーム膜破壊過 程が NK-92 に対する毒性発現に関与していると考 え,YSK12-MEND に含まれる YSK12-C4 量(総脂 質量)を軽減する戦略を立案した.YSK12-C4 はエ ンドソーム脱出時に機能するだけではなく,そのカ チオンと siRNA との静電的相互作用により,si-RNA を効率的に封入する過程に利用されている. siRNAを封入する際のカウンターカチオンとして ポリカチオンであるプロタミンを使用し,プロタミ ン/siRNA コアを形成させ,YSK12-C4 量を減少さ せ る こ と に 成 功 し た . 従 来 型 YSK12-MEND の YSK12-C4 と siRNA の電荷比は 16.9 であったが, プロタミン/siRNA コアを導入することで電荷比を 5まで下げた場合,有意な毒性軽減が認められた. 一方で,興味深いことに遺伝子ノックダウン活性は 同等の活性を維持していた.この原因を細胞内動態 の観点から解析した結果,プロタミン/siRNA コア を導入した YSK12-MEND がエンドソーム脱出以 降の過程を促進している可能性が示唆された. YSK12-MEND を用いた免疫細胞の遺伝子発現制 御の研究は in vitro レベルではあるが,実用的なレ ベルの遺伝子ノックダウン活性を示せたと考えてい る.現在は,がん免疫サイクルに関連する抑制性因 子を標的としたマウスレベルでの検証を進めてお り,近い将来報告したい. 6. おわりに 本総説では,抗原やアジュバントを搭載したナノ に相互作用するがん免疫サイクルを制御するために は,ナノ DDS 技術が不可欠であり,今後ますます 需要が高まってくると予想される.一方で,がん免 疫療法におけるナノ DDS 開発戦略のパラダイムシ フトが必要である.現在のがん免疫療法の開発にお いて,がん免疫サイクルを正常に回すことを目指 し,患者の腫瘍関連微小環境の免疫状態に応じた治 療が求められている.31,32)それゆえ,ナノ DDS 開 発も腫瘍関連微小環境の免疫状態に基づいた最適な 設計を行う必要があるだろう.33) 謝辞 このたびは日本薬学会奨励賞という名誉 ある賞を頂き,選考委員の先生方に厚く御礼申し上 げます.本研究は,北海道大学大学院薬学研究院・ 原島秀吉教授の主宰される薬剤分子設計学研究室に おいて遂行されました.ご指導・ご鞭撻を賜りまし た原島教授,並びに,いつもご協力頂いております 同研究室のスタッフ・学生の皆様に心より感謝申し 上げます.また多大なるご協力を賜りました共同研 究者の先生方に厚く御礼申し上げます.本研究は, 科研費(基盤研究 B,若手研究 A,若手研究 B,挑 戦的萌芽研究),財団(秋山記念生命科学振興財団, 持田記念医学薬学振興財団)の支援に基づくもので あり,この場を借りて感謝申し上げます. 利益相反 開示すべき利益相反はない. REFERENCES
1) Chen D. S., Mellman I., Immunity, 39, 110 (2013).
2) Nakamura T., Moriguchi R., Kogure K.,
Shastri N., Harashima H., Mol. Ther., 16, 15071514 (2008).
3) Nakamura T., Ono K., Suzuki Y., Moriguchi R., Kogure K., Harashima H., Mol. Pharm., 11, 27872795 (2014).
4) Nakamura T., Moriguchi R., Kogure K.,
Harashima H., Int. J. Pharm., 441, 476481 (2013).
Immunol., 4, 11641165(2003).
6) Nakamura T., Yamazaki D., Yamauchi J.,
Harashima H., J. Control. Release, 171, 216 224 (2013).
7) Nakamura T., Kuroi M., Harashima H.,Mol. Pharm., 12, 27912799 (2015).
8) Abdelmegeed H., Nakamura T., Harashima
H., J. Pharm. Sci., 105, 250256 (2016).
9) Komori T., Nakamura T., Matsunaga I.,
Morita D., Hattori Y., Kuwata H., Fujiwara N., Hiromatsu K., Harashima H., Sugita M., J. Biol. Chem., 286, 1680016806 (2011). 10) Hattori Y., Matsunaga I., Komori T.,
Uraka-wa T., Nakamura T., FujiUraka-wara N., Hiromatsu K., Harashima H., Sugita M., Biochem.
Biophys. Res. Commun., 409, 304307
(2011).
11) Morita D., Hattori Y., Nakamura T., Igarashi T., Harashima H., Sugita M., Infect. Immun., 81, 311316 (2013).
12) Morita D., Miyamoto A., Hattori Y., Komori T., Nakamura T., Igarashi T., Harashima H., Sugita M., Biochem. Biophys. Res. Com-mun., 441, 108113 (2013).
13) Hattori Y., Morita D., Fujiwara N., Mori D., Nakamura T., Harashima H., Yamasaki S., Sugita M., J. Biol. Chem., 289, 1540515412 (2014).
14) Nakamura T., Fukiage M., Higuchi M.,
Nakaya A., Yano I., Miyazaki J., Nishiyama H., Akaza H., Ito T., Hosokawa H., Nakaya-ma T., HarashiNakaya-ma H., J. Control. Release, 176, 4453(2014).
15) Nakamura T., Fukiage M., Suzuki Y., Yano
I., Miyazaki J., Nishiyama H., Akaza H., Harashima H., J. Control. Release, 196, 161 167(2014).
16) Nakamura T., Noma Y., Sakurai Y., Harashi-ma H., Biol. Pharm. Bull., 40, 234237 (2017).
17) Spranger S., Int. Immunol., 28, 383391 (2016).
18) Woo S. R., Fuertes M. B., Corrales L., Spran-ger S., Furdyna M. J., Leung M. Y., Duggan R., Wang Y., Barber G. N., Fitzgerald K. A., Alegre M. L., Gajewski T. F., Immunity, 41, 830842 (2014).
19) Burdette D. L., Monroe K. M., Sotelo-Troha
K., Iwig J. S., Eckert B., Hyodo M., Hayaka-wa Y., Vance R. E., Nature, 478, 515518 (2011).
20) Parvatiyar K., Zhang Z., Teles R. M., Ouyang S., Jiang Y., Iyer S. S., Zaver S. A., Schenk M., Zeng S., Zhong W., Liu Z. J., Modlin R. L., Liu Y. J., Cheng G., Nat. Immunol., 13, 11551161(2012).
21) Sato Y., Hatakeyama H., Sakurai Y., Hyodo M., Akita H., Harashima H., J. Control. Release, 163, 267276 (2012).
22) Miyabe H., Hyodo M., Nakamura T., Sato
Y., Hayakawa Y., Harashima H., J. Control. Release, 184, 2027 (2014).
23) Nakamura T., Miyabe H., Hyodo M., Sato
Y., Hayakawa Y., Harashima H., J. Control. Release, 216, 149157 (2015).
24) Kogure K., Akita H., Yamada Y., Harashima H., Adv. Drug Deliv. Rev., 60, 559571 (2008).
25) Sato Y., Nakamura T., Yamada Y., Harashi-ma H., J. Control. Release, 244, 194204 (2016).
26) Akita H., Kogure K., Moriguchi R.,
Nakamura Y., Higashi T., Nakamura T., Ser-ada S., Fujimoto M., Naka T., Futaki S., Harashima H., J. Control. Release, 143, 311 317 (2010).
27) Warashina S., Nakamura T., Harashima H., Biol. Pharm. Bull., 34, 13481351(2011).
28) Warashina S., Nakamura T., Sato Y.,
Fujiwara Y., Hyodo M., Hatakeyama H., Harashima H., J. Control. Release, 225, 183 191 (2016).
29) Nakamura T., Kuroi M., Fujiwara Y.,
Warashina S., Sato Y., Harashima H., Sci. Rep., 6, 37849 (2016).
30) Nakamura T., Yamada K., Fujiwara Y., Sato Y., Harashima H., Mol. Pharm., 15, 2142 2150(2018).
31) Blank C. U., Haanen J. B., Ribas A.,
Schumacher T. N., Science, 352, 658660 (2016).
32) Chen D. S., Mellman I.,Nature, 541, 321330 (2017).
33) Nakamura T., Harashima H.,Ther. Deliv., 8, 9871000(2017).