熊本大学学位論文
Platinum TALEN による遺伝子破壊マウスの作製と
変異個体スクリーニング法の確立
2014
中川 佳子
本論文は学術雑誌に掲載された次の論文を基礎とするものである。
Screening methods to identify TALEN-mediated knockout mice
Yoshiko Nakagawa, Takashi Yamamoto, Ken-Ichi Suzuki, Kimi Araki, Naoki Takeda, Masaki Ohmuraya, Tetsushi Sakuma.
Exp. Anim., 63, 79-84 (2014).
Application of Oocyte Cryopreservation Technology in TALEN-Mediated Mouse Genome Editing
Yoshiko Nakagawa, Tetsushi Sakuma, Naomi Nakagata, Sho Yamasaki, Naoki Takeda, Masaki Ohmuraya, Takashi Yamamoto.
Exp. Anim., 63, 349-355 (2014).
Birth of mice from vitrified/warmed 2-cell embryos transported at a cold temperature
Toru Takeo, Takehito Kaneko, Yukie Haruguchi, Kiyoko Fukumoto, Hiromi Machida, Mik
a Koga, Yoshiko Nakagawa, Yumi Takeshita, Toyokazu Matsuguma, Shuuji Tsuchiyama, Norihiko Shimizu, Takanori Hasegawa, Motohito Goto, Hitoshi Miyachi, Masayuki Anzai, Ena Nakatsukasa, Koji Nomaru, Naomi Nakagata.
Cryobiology, 58, 196-202 (2009).
Short-term storage and transport at cold temperatures of 2-cell mouse embryos produced by cryopreserved sperm
Toru Takeo, Tomoko Kondo, Yukie Haruguchi, Kiyoko Fukumoto, Yoshiko Nakagawa, Yumi Takeshita, Yuko Nakamuta, Shuuji Tsuchiyama, Norihiko Shimizu, Takanori Hasegawa, Motohito Goto, Hitoshi Miyachi, Masayuki Anzai, Rie Fujikawa, Koji Nomaru, Takehito Kaneko, Yoshiaki Itagaki, Naomi Nakagata.
J. Am. Assoc. Lab. Anim. Sci., 49, 415-419 (2010).
その他の論文
Fertilization of C57BL/6 mouse sperm collected from cauda epididymides after preservation or transportation at 4 degrees C using laser-microdissected oocytes
Takehito Kaneko, Kiyoko Fukumoto, Yukie Haruguchi, Tomoko Kondo, Hiromi Machida, Mika Koga, Yoshiko Nakagawa, Shuuji Tsuchiyama, Kiyora Saiki, Shiho Noshiba, Naomi Nakagata.
Cryobiology, 59, 59-62 (2009).
目次
要旨・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・
1
第1章 序論・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・ 3 第2章 実験材料・・・・・・・・・・・・・・・・・・・・・・・・・・・・・ 10
2-1) Platinum TALEN
プラスミド作製2-2) SSA
アッセイ
2-3) TALEN mRNA
合成2-4) 受精卵の準備および TALEN mRNA
のマイクロインジェクション2-5) GFP
蛍光観察2-6) HMA 2-7) RFLP
解析2-8) DNA
シーケンシング2-9) 精子の凍結融解および体外受精
2-10)
受精卵の凍結融解第3章 実験方法・・・・・・・・・・・・・・・・・・・・・・・・・・・・・15
3-1) 新鮮受精卵を用いた Platinum TALEN
による遺伝子破壊マウスの作製と変異個体スクリーニング法の検討
3-1-1) Platinum TALEN
プラスミド作製3-1-2) TALEN
活性測定のためのSSA
アッセイ3-1-3) TALEN mRNA
合成3-1-4) pCAG-eGFP
マウス受精卵の準備3-1-5) TALEN mRNA
のマイクロインジェクション3-1-6) 胎仔あるいは産仔の蛍光観察
3-1-7) HMA
のためのゲノミックPCR
3-1-8) RFLP
解析3-1-9) PCR
産物の塩基配列決定3-2) 凍結融解受精卵を用いた Platinum TALEN
による遺伝子破壊マウスの作製
3-2-1) Platinum TALEN
プラスミド作製とSSA
アッセイ3-2-2) Ayu8104
マウス受精卵の準備
A)交配による受精卵の作製
B)体外受精による受精卵の作製
3-2-3) C57BL/6N
マウス受精卵の準備
3-2-4) 変異個体スクリーニングのための解析
第4章 結果・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・ 36
4-1)
新鮮受精卵を用いたPlatinum TALEN
による遺伝子破壊マウスの作製と変異個体スクリーニング法の検討
4-1-1) TALEN mRNA
使用濃度の検討(前核内インジェクション)4-1-2) 細胞質へのインジェクション
4-1-3) 変異個体スクリーニングのための解析
4-2)
凍結融解受精卵を用いたPlatinum TALEN
による遺伝子破壊マウスの作製4-2-1) Ayu8104
マウスにおける外来遺伝子の破壊4-2-2) C57BL/6N
マウスにおける内在遺伝子の破壊第5章 結論・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・ 43
5-1)
新鮮受精卵を用いたPlatinum TALEN
による遺伝子破壊マウスの作製5-2)
変異個体スクリーニング法の確立5-3)
凍結融解受精卵を用いたPlatinum TALEN
による遺伝子破壊マウスの作製第6章 図表・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・ 48 第7章 参考文献・・・・・・・・・・・・・・・・・・・・・・・・・・・・・ 69
謝辞・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・ 72
1
論文要旨
Platinum TALEN
による遺伝子破壊マウスの作製と 変異個体スクリーニング法の確立中川 佳子
ジンクフィンガーヌクレアーゼ(zinc-finger nuclease; ZFN) や
TALE
ヌクレ アーゼ(transcription activator-like effector nuclease; TALEN) などの部位特異的 ヌクレアーゼやCRISPR/Cas (clustered regularly interspaced short palindromic
repeats; CRISPR)
システムを利用したゲノム編集は、様々な生物において、遺伝子改変法の新たなスタンダードとなりつつある。これらの技術をノックア ウトマウスの作製に適用するためには、人工ヌクレアーゼのインジェクショ ンによって得られた産仔の単純かつ容易なジェノタイピングの手法が広く求 められている。しかしながら、人工ヌクレアーゼを導入した個体群から目的 のミュータントマウスをスクリーニングする手法について、詳細に比較検討 した報告はこれまでなされていない。そこで、本研究において、
enhanced green fluorescent protein (eGFP)
遺伝子をPlatinum TALEN
によって破壊したノック アウトマウスの解析法を包括的に検討した。加えて、広島大学にて開発された
Platinum TALEN
は、培養細胞でのSSA
アッセイやアフリカツメガエルあるいはラットの受精卵へのインジェクションにおいて、従来の
TALEN
に比べ、高効率な遺伝子破壊が可能であることが報告されていたが、マウスでの使用 実績がなかったため、Platinum TALENを用いて、ノックアウトマウスを作製 するための詳細な条件検討を行った。その結果、
F0
世代にて、eGFP
の蛍光 が完全に消失した個体を作製することが可能となった。さらに、得られた産 仔において、蛍光観察、ヘテロ二重鎖移動度アッセイ、制限酵素断片長多型2
解析、および
DNA
シーケンシングを行った結果、それぞれのアッセイ法で変 異の検出感度に違いがあり、正確に変異個体を選抜するためには、これらの 手法を組み合わせることが重要であることを見いだした。また、体外受精や胚凍結、精子凍結などの生殖工学技術は、遺伝子改変マ ウスの保存、個体化、輸送に不可欠な技術であるが、これらの技術が
TALEN
などの部位特異的ヌクレアーゼを用いたゲノム編集による遺伝子改変マウス 作製にも応用可能か否かは、これまで明らかにされていない。そこで、本研究では、
Platinum TALEN
のmRNA
を受精卵へインジェクションしてゲノム改変マウスを作製する際、凍結融解受精卵を用いて外来遺伝子あるいは内在遺 伝子のターゲティングが可能か検討した。外来遺伝子の破壊においては、交 配後に採卵した凍結受精卵だけでなく、新鮮精子あるいは凍結精子を用いた 体外受精後の凍結受精卵も作製し、それぞれを融解後、インジェクションを 行った。内在遺伝子の破壊においては、
5
週齢の幼若雌マウスと性成熟後の8
~12週齢雌マウスを交配に使用し、これらの凍結受精卵を融解してインジェ クションを行った。全ての実験区から得られた産仔について、
TALEN
による 変異導入の有無を詳細に解析した結果、これらの凍結受精卵から、TALEN
を 用いて高効率に遺伝子改変マウスを作製できることが明らかとなった。本研究において使用した変異個体スクリーニング法は、特別な設備や特に 高価な試薬類、あるいは洗練されたプロトコールを必要とする実験ではなく、
様々な生物においてゲノム編集技術の導入を試みる幅広い研究者に対して、
本研究は大いに役立つものと考える。さらに、体外受精や受精卵の凍結融解 などの生殖工学技術を活かすことにより、マウスの飼育費や作業者の負担が 軽減され、次世代型の遺伝子改変技術であるゲノム編集法を用いたノックア ウト/ノックインマウス作製の利便性が大いに高まると期待される。
3
第
1
章 序論これまでのノックアウトマウス作製では、ターゲティングベクターとゲノ ム
DNA
との相同組換えを利用し、ES
細胞を介してキメラマウスが作製され てきた(Capecchi 2005)。この方法では、ターゲティングベクターの構築やES
細胞へのベクター導入、相同組換えクローンの単離・同定、キメラマウ スの作製、近交系マウスへのもどし交配など、かなりの時間と手間のかかる 作業が必要であった。しかし、近年、ジンクフィンガーヌクレアーゼ(zinc-finger nuclease; ZFN)や
TALE
ヌクレアーゼ(transcription activator-likeeffector nuclease; TALEN)などの部位特異的ヌクレアーゼや CRISPR/Cas
(clustered regularly interspaced short palindromic repeats; CRISPR)システムを 利用したゲノム編集が可能となり、ZFN、TALEN ではこれらの
mRNA
を、CRISPR/Cas
システムではCas9
のようなRNA
誘導型DNA
ヌクレアーゼ(RNA-guided nuclease; RGN)の
mRNA
とガイド鎖RNA(guide RNA)を目
的とする系統の受精卵へインジェクションする試みが様々な施設で行われ てきた。この方法を利用することにより、ES 細胞を介さず、もどし交配の 必要もない短時間かつ低費用、簡便なノックアウトマウスの作製が可能とな り、現在では、多数の報告がよせられている(Davies et al. 2013; Ma et al. 2013;Panda et al. 2013; Qiu et al. 2013; Sung et al. 2013; Wefers et al. 2013; Wu et al.
2013; Aida et al. 2014)
。今後も、これらの技術を利用したノックアウトマウ スの作製は増加すると考えられ、得られた産仔の簡便で迅速なジェノタイピ ング法の確立が急がれている。しかしながら、これらの技術を用いて作製し た個体群から目的の変異マウスをスクリーニングする手法は、現在までに 様々な方法が報告されているものの、それらについて、詳細に比較検討した4
報 告 は これ ま でな され て い ない 。 そこ で、 本 研 究で は
enhanced green fluorescent protein; eGFP
遺伝子を全身で発現するトランスジェニックマウ スを用い、eGFP 遺伝子をTALEN
によって破壊したマウスの解析法を包括 的に検討した。また、本研究で使用したPlatinum TALEN(広島大学山本研
究室にて開発)は、培養細胞でのsingle-strand annealing assay
(SSAアッセイ)やカエル、ラット受精卵へのインジェクションにおいて、従来の
TALEN
に 比べ、高効率に変異導入可能なことが確認されていたが、マウスでの使用実 績がなかった(Sakuma et al. 2013 b)。そこで、本研究では、はじめに、受精 卵へのインジェクションに用いるTALEN mRNA
の濃度検討、前核内と卵細 胞質へのインジェクションの比較検討を行い、マウスにおけるPlatinum
TALEN
の使用条件を決定した。次に、産仔への発生率や産仔の蛍光消失率が最良であった実験区の産仔を用いて、
eGFP
遺伝子が破壊されたマウスの 解析法を包括的に検討した。以下、TALEN
とPlatinum TALEN
について簡単 に説明する。TALEN
は制限酵素FokI
のDNA
切断ドメインと、植物病原細菌キサントモナス属から分泌される
TALE
タンパク質のDNA
結合ドメインを融合させ た人工ヌクレアーゼである。DNA結合ドメインは自然界のTALE
で高度に 保存されたモジュールの繰り返し構造をとっている。1 モジュールは約34
アミノ酸で構成され、各モジュールの12,13
番目のアミノ酸残基(repeatvariable di-residue; RVD)が標的 DNA
の1
塩基を認識し、安定的な結合を行 うために重要である。標的DNA
の塩基配列に対応するモジュールをつなぎ 合わせ、FokI
のDNA
切断ドメイン配列をもつベクターに組み込むことによ り、目的とする遺伝子の配列に作用するTALEN
の作製が可能となる。二組の
TALEN
が標的配列に結合すると、FokIのDNA
切断ドメインが二5
量体を形成し、スペーサー配列に
DNA
二本鎖切断(double-strand break; DSB)を引き起こす。
DSB
は相同組み換え修復(homology-directed repair; HDR)よ りもむしろ非相同末端結合(non-homologous end-joining; NHEJ)によって修 復されることが多く、その過程でランダムな塩基の欠失や挿入が起こり、フ レームシフトによって遺伝子機能が破壊される。広島大学山本研究室では
ZFN
やTALEN
の有用性に早くから着目し、ゲノ ム編集コンソーシアムの運営をはじめ、ゲノム編集研究会や人工ヌクレアー ゼ作製講習会を通してゲノム編集に関する情報交換やゲノム編集基盤技術 の開発・発展に尽力されている。特にTALEN
については、Daniel Voytasら が開発した10
モジュールのアセンブリ法を用いるGolden Gate
法でのTALEN
プラスミド構築法(Cermak et al. 2011)に改良を加え(Sakuma et al.2013 a)、高速かつ簡便に、高活性な TALEN
を作製することに成功し、Platinum TALEN
とそのプラスミド構築法を開発した(Sakuma et al. 2013 b)。 主な改良・開発点は、作製したTALEN
プラスミドの活性評価が哺乳動物培 養細胞で行え、かつそのままmRNA
合成に利用できるようCMV
とT7
プロ モーターをもつdestination vector
を構築したこと、モジュールのアセンブリ で失敗することがないよう6
あるいは4
モジュールのアセンブリ法を開発し たこと、TALENの切断活性を上昇させることが知られているTALE
のN
末 端・C末端欠失(NC型)を導入したこと、34アミノ酸からなるTALE
リピ ートの4
番目と32
番目のアミノ酸残基に多様性を持たせ、細胞毒性を出さずに
TALEN
の切断活性を上昇させたことである。様々な動物受精卵への
TALEN
インジェクションでは、TALEN
のプラスミ ドDNA
ではなく、これらをもとに合成したmRNA
を用いることにより、作 製した変異個体において、ゲノムDNA
へのプラスミドDNA
の挿入が回避6
でき(トランスジェニック個体にならずに)、かつ、迅速に
TALEN
タンパ ク質の合成が行われ、胚発生の極めて初期に標的遺伝子が破壊されることか ら、F0世代での完全なノックアウト個体作製が可能となる利点がある。目的とする培養細胞や生物個体に
TALEN
を作用させた後は、変異体の同 定が必要となる。以下に述べるように、変異体同定法は、これまで様々な方 法が報告されている。変異導入により形成されたミスマッチなヘテロ二本鎖 を特異的に切断するSurveyor
ヌクレアーゼやT7
エンドヌクレアーゼI
を利 用する方法(Watanabe et al. 2012; Mussolino et al. 2011; Tesson et al. 2011)、DNA
融解曲線を利用してPCR
産物のヘテロ二本鎖を検出する高解像度融解 曲線分析(high resolution melt analysis; HRMA)(Dahlemet al. 2012)、
pBluescript II
プラスミドのマルチクローニングサイトを標的配列が含まれるPCR
産物に置換し、野生型ゲノムがインフレームでlacZα遺伝子に挿入で
きるよう設計したblue/white selection
を利用するlacZα遺伝子破壊測定法
(LacZ assay)(Hisano et al. 2013)、標的配列を含む
PCR
産物を電気泳動 し 泳 動 度 の 遅 い バ ン ド を 検 出 す る ヘ テ ロ 二 重 鎖 移 動 度 ア ッ セ イ(heteroduplex mobility assay; HMA)(Ota et al. 2013)、スペーサー配列内の塩 基配列を特異的に認識切断する制限酵素を利用した制限酵素断片長多型解 析(restriction fragment length polymorphism; RFLP analysis)(Ochiai et al. 2010;
Ansai et al. 2013; Suzuki et al. 2013)
、標的配列を含むPCR
産物をサブクロー ニングして配列を読むDNA
シーケンシング解析などである。得られたキメラマウスの毛色により解析が容易な
ES
細胞を介したノック アウトマウス作製と異なり、TALEN
を用いたノックアウトマウス作製では、明らかな表現型が出ない限り、ファウンダーマウス(
F0
)での野生型個体と 変異型個体の判別が困難である。そこで、本研究では、上記の様々な変異体7
同定法のうち、感度や再現性に問題がなく、特別な機器や高価な試薬、洗練 されたプロトコールを必要としない
HMA、RFLP
解析、DNAシーケンシン グとeGFP
の蛍光観察を組み合わせることにより、TALEN
を用いたeGFP
遺 伝子の破壊について詳細に解析した。また、熊本大学生命資源研究・支援センター中潟研究室では、マウス胚・
精子の凍結バンク業務を行うと共に、新たなマウス生殖工学技術の開発、改 良、確立を目的として研究を行っており、中潟研究室にて確立された生殖工 学技術の多くは世界的にも注目されている(Guan et al. 2012)。精子の凍結 保存と融解法の開発、C57BL/6 マウスの凍結融解精子を用いた体外受精や
129、 BALB/c、ICR
など低受精率系統を用いた体外受精において受精率を飛躍的に向上させたこと、冷蔵保存あるいは冷蔵輸送されたマウス精巣上体尾 部を用いた効率的な体外受精法の開発、
2
細胞胚の冷蔵保存と冷蔵輸送法の 開発などがその例である。私は、主に、体外受精により得られた2
細胞胚を 凍結保存後、融解し、冷蔵で胚を輸送する方法の開発を行い、複数の輸送先 施設にて、冷蔵輸送された2
細胞胚の個体化を確認することができた。この 輸送法は、M2メディウムで満たしたエッペンチューブに2
細胞胚を浸し、保冷剤とともに発砲スチロール容器に梱包後、一般的な宅配便で送る簡便な ものであり、ドライシッパーは必要ない。また、輸送先では
2
細胞胚を回収 する簡単な作業だけで、偽妊娠雌マウスへの移植作業を開始でき、冷蔵輸送 胚の産仔への発生率も良好であったため、現在では、施設間のマウスの授受 に利用されている。そこで、これまでに習得した生殖工学技術を活かしてゲ ノム編集を行うことにより、さらに高速、低コスト、簡便なノックアウトマ ウスの作製が可能になるのではないかと考え、以下の研究を行うこととした。受精卵へのインジェクションによるゲノム編集技術を用いてノックアウ
8
トマウスやノックインマウスを作製している施設では、一般的に、過排卵処 理をした雌マウスを個別飼育した雄マウスと交配させ、膣栓が確認された雌 マウスから受精卵を採卵し、インジェクションに使用している(Davies et al.
2013; Ma et al. 2013; Panda et al. 2013; Qiu et al. 2013; Sung et al. 2013; Wefers et al. 2013; Wu et al. 2013; Aida et al. 2014)
。この方法では、個別飼育した多く の雄マウスを常に準備しておかなければならず、飼育スペースの占有や飼育 費の負担により、実験者を圧迫することが多い。さらに、実験日にはその都 度、雌マウスを準備し、受精卵の採卵を行わなければならない。しかしなが ら、凍結融解受精卵をインジェクションに用いることができれば、常時個別 飼育した雄マウスを飼育する必要がなくなるため、飼育費の負担が軽減され る。また、実験日に採卵する必要がなくなるため、作業の効率化が可能とな る。さらに、体外受精により作製した受精卵を凍結融解し、インジェクショ ンに用いることができれば、極めて効率よく作業を進めることが可能となる。体外受精や精子および受精卵の凍結融解などの生殖工学技術がトランス ジェニックマウスの作製に応用可能であることは、これまでの研究により明 らかとなっていたが(Nakagata N. 1996)、TALENなどの部位特異的ヌク レアーゼを用いたゲノム編集による遺伝子改変マウス作製にも応用可能か 否かは、これまで明らかにされていない。そのため、本研究では、交配によ り得られた受精卵、新鮮精子あるいは凍結融解精子を用いた体外受精により 得られた受精卵を凍結融解し、
TALEN
を用いた遺伝子破壊マウスの作製に 使用可能か検討した。TALEN
による遺伝子破壊が、マウスの個体発生に影 響を与えないよう、マウスでは発現しないβ-ラクタマーゼ(β-lactamase; bL)をコードする 1 コピーの外来遺伝子をもつ
Ayu8104
マウスを用い(Imaizumi
et al. 1999)、bL
遺伝子の破壊を試みた。さらに、通常、交配による受精卵9
の採取には、様々な施設において、幼若雌マウスと性成熟後の雌マウスのど ちらも使用されている。そのため、
5
週齢あるいは8-12
週齢の雌マウスを交 配および採卵に用いたC57BL/6
系統の受精卵を凍結融解し、C
型レクチンド メインファミリーb1遺伝子(C-type lectin domain family 4, member b1; Clec4b1)の
TALEN mRNA
をインジェクションすることにより、採卵に使用する雌マウスの週齢に左右されることなく、内在遺伝子の破壊が可能か否かを調べた。
bL
遺伝子、Clec4b1 遺伝子の破壊についても、eGFP遺伝子の破壊時と同様 に、得られた産仔について、変異個体のスクリーニングを行い、変異導入効 率を調べた。10
第2章 実験材料
ここでは、実験を進める上で重要な試薬やキット、機器について記載する。
DNA
ポリメラーゼ、大腸菌培養試薬など一般的な遺伝子工学試薬は、通常 の実験室で用いているものを使用して問題ない。また、電気泳動装置やゲル 撮影装置、インキュベーター、サーマルサイクラー、遠心機などの一般的な 機器も通常の研究室で使用しているもので問題ない。2-1)Platinum TALEN
プラスミド作製・Platinum Gate TALEN Kit (Addgene cat.#1000000043)
Kit
には以下のプラスミドが含まれる。P1-4HD, p1-4NG, p1-4NI,p1-4NN
計16
個のModule plasmids
(アンピシリン 耐性)。pFUS2_a1a, a2a, a2b, a3a, a3b, a4a, a4b, b1, b2, b3, b4
計11
個の第1
段階の4
モジュールアセンブリに使用するIntermediate array plasmids(スペクチノ
マイシン耐性)。ptCMV-136/63-VR-HD, NG, NI, NN
、ptCMV-153/47-VR-HD, NG, NI, NN
計8
個のDestination vectors(アンピシリン耐性)
。・BsaI-HF (NEB cat.#R3535S)
・Esp3I (Thermo Scientific cat.#ER0452)
・10×T4 DNA Ligase Reaction Buffer (NEB cat.#B0202S)
・
Quick Ligation Kit (NEB cat.#M2200S)
・Wizard SV Gel and PCR Clean-up System (Promega cat.#A9281)
・
ChargeSwitch-Pro Plasmid Mini Kit (Life Technologies cat.#CS30250)
11
・PCRプライマー
pCR8 F1: 5′- TTGATGCCTGGCAGTTCCCT-3′
pCR8 R1: 5′- CGAACCGAACAGGCTTATGT-3′
TALE-F: 5′-GAGCACCCCTCAACCTGACCCCAG-3′
TALE-R: 5′- CTCGAAAGCTGGGCCACGATTG-3′
2-2)SSA
アッセイ・pGL4-SSA vector (Addgene cat.#42962)
・ChargeSwitch-Pro Plasmid Mini Kit (Life Technologies cat.#CS30250)
・KpnI (NEB cat.#R0142S)
・HEK293T細胞
・Lipofectamine LTX (invitrogen cat#15338-100)
・Dual-Glo Luciferase Assay System (Promega cat#E2920)
・TriStar LB 941 plate reader (Berthold Technologies)
2-3)TALEN mRNA
合成・SmaI (TAKARA cat.#1085A)
・QIAquick PCR purification kit (Qiagen cat.#28104)
・mMESSAGE mMACHINE T7 Ultra kit (Life technologies cat.#AM1345)
・RNeasy Mini kit (Qiagen cat.#74104)
2-4)受精卵の準備および TALEN mRNA
のマイクロインジェクション・
pregnant mare serum gonadotropin (PMSG; Serotropin; Aska Pharmaceutical)
・human chorionic gonadotropin (hCG; Veterinary Puberogen; Novartis Animal
12
Health)
・Hyaluronidase (SIGMA cat.#H-3506)
・potassium simplex optimized medium with amino acids (KSOM-AA; 自家作製)
・流動パラフィン(ナカライテスクcat.#26137-85)
・流動パラフィン軽質(ナカライテスクcat.#26144-85)
・10×RNase-free PBS (Life technologies cat.#AM9624)
・pCAG-eGFPホモマウス(B6;D2-Tg (CAGEGFP)49SImeg; CARD ID: 267;
http://cardb.cc.kumamoto-u.ac.jp/transgenic/strainsDetailAction.do?strainId=26 7)
・採卵用雌マウス(pCAG-eGFPマウス用)
C57BL/6NCrlCrlj (Charles river Japan)
・Ayu 8104ホモマウス(Imaizumi et al. 1999)
・採卵用雌マウス(Ayu 8104マウス用)
Jcl: ICR (CLEA Japan)
・C57BL/6系統の受精卵採取のために使用する雌雄マウスC57BL/6NCrlCrlj (Charles river Japan)
・偽妊娠雌マウス
Jcl: ICR (CLEA Japan)
・実体顕微鏡
・倒立顕微鏡
・マイクロマニピュレーター一式(ナリシゲ)
2-5
)GFP
蛍光観察・蛍光顕微鏡
・
UV-LED
ライト13
2-6)HMA
・DNeasy Blood & Tissue Kit (Qiagen cat.#69504)
・LA Taq DNA polymerase (TAKARA cat.#RR002A)
・PCRプライマー
eGFP-F: 5′- CCTCGTGACCACCCTGACCTAC-3′
eGFP-R: 5′-CTGTTGTAGTTGTACTCCAGCTTGTGC-3′
bL-F: 5′-GTATTCAACATTTCCGTGTCGCC-3′
bL-R: 5′-GATAATACCGCGCCACATAGCAG-3′
Clec4b1-F: 5′-GGCTATCTCTGTGGTATTTAGCTC-3′
Clec4b1-R: 5′-CCACTTGTCTGCATGGTTAC-3′
2-7)RFLP
解析・Wizard SV Gel and PCR Clean-up System (Promega cat.#A9281)
・AccII (TAKARA cat.#1002A)
・HphI (New England Biolabs cat.#R0158S)
・Hpy188III (New England Biolabs cat.#R0622S)
2-8)DNA
シーケンシング・pGEM -T Easy Vector Systems (Promega cat.#A1360)
・Mini plus Plasmid DNA Extraction system (VIOGENE cat.#GF2002) ・
EcoRI (NIPPON GENE cat.#314-00112)
・T7 primer (5′-TAATACGACTCACTATAGGG-3′)
・
SP6 primer (5′-CATACGATTTAGGTGACACTATAG-3′)
・BigDye Terminator Cycle Sequencing Kit v1.1 (Life Technologies
14
cat.#4337450)
・ABI PRISM 3130 Genetic Analyzer (Life Technologies)
2-9)精子の凍結融解および体外受精
・pregnant mare serum gonadotropin (PMSG; Serotropin; Aska Pharmaceutical)
・human chorionic gonadotropin (hCG; Veterinary Puberogen; Novartis Animal
Health)
・CARD MEDIUM体外受精用培地(九動)
・FERTIUPマウス精子前培養培地(九動)
・FERTIUPマウス精子凍結保存液(九動)
・0.25ml Plastic straw (IMV)
・Human tubal fluid (HTF) 培地(自家作製)
・流動パラフィン(ナカライテスク
cat.#26137-85)
2-10)受精卵の凍結融解
・1M DMSO 溶液(1 M dimethyl sulfoxide in PB1; 自家作製)
・DAP213 溶液
(2 M DMSO, 1 M acetamide, 3 M propylene glycol in PB1; 自家作製)
・0.25M sucrose 溶液 (0.25M sucrose in PB1; 自家作製)
・KSOM-AA
・流動パラフィン(ナカライテスク
cat.#26137-85)
15
第3章 実験方法
3-1)
新鮮受精卵を用いたPlatinum TALEN
による遺伝子破壊マウスの作製と 変異個体スクリーニング法の検討3-1-1)Platinum TALEN
プラスミド作製Platinum TALEN
プラスミド作製は、広島大学のPlatinum TALEN
プラス ミド構築法に基づいて行われ、TALEN
活性測定のためのSSA
アッセイも 山本研究室のプロトコールに従って行われた。Platinum TALENの構築法 やSSA
アッセイの手順はaddgene
のホームページにも公開されている(Platinum TALENの構築法
https://www.addgene.org/TALEN/PlatinumGate/)
(SSA アッセイ
http://www.addgene.org/TALEN/Yamamotolab/)
。使用する プラスミドベクターは全てaddgene
(https://www.addgene.org/)から購入可 能である。1.
コーネル大学が運営するTALEN Targeter
(https://tale-net.cac.cornell.edu/) を用いてeGFP TALEN
の標的配列を決定した。eGFP Platinum TALEN
の 模式図を図1
に示す。2.
標的配列に対応するRVD
を決定した。Cに対してはHD、A
に対して はNI、T
に対してはNG、G
に対してはNN(NG)を使用する。ラス
トリピートはdestination vector
に付加されているので、ここでは連結 しない。Left TALEN (20
塩基認識) 5’-CTTCAAGGACGACGGCAACT-3’pFUS2 a4a a4b a4c a4d b3
HD NG NG HD / NI NI NN NN / NI HD NN NI / HD NN NN HD / NI NI HD
16
Right TALEN (18
塩基認識) 5’-CGCCCTCGAACTTCACCT-3’pFUS2 a4a a4b a4c a4d b1 HD NN HD HD / HD NG HD NN / NI NI HD NG / NG HD NI HD / HD
3.
第1のアセンブリGolden Gate
法は、各モジュールのプラスミドとベクタープラスミドを混合し、制限酵素による切断とライゲースによる連結反応を繰り返す ことにより、モジュールを順番通りに連結し、ベクターに組み込むこ とができる画期的な方法である。この
Golden Gate
反応を利用して、Left TALEN
においてはpFU2a
の4
モジュールとpFU2b
の3
モジュー ル、Right TALEN
においてはpFU2a
の4
モジュールとpFU2b
の1
モジ ュールのアセンブリ(モジュールの連結)を以下の反応液組成、条件 にて行った。1 module 3 modules 4 modules 25ng/µl pFU2 vector 0.3µl
50ng/µl module
0.3µl×1 0.3µl×3 0.3µl×4 10×T4 DNA ligase buffer 0.2µl
Quick ligase 0.1µl BsaI-HF 0.1µl
SDW 1µl 0.4µl (0.1µl) Total 2µl
37℃ 5
分間→16℃ 10分間1
セットの反応を3
セット行い、最後に4℃
にて反応を終了した。
さらに、10×NEBuffer 4 0.25µl, 10×BSA 0.25µl, BsaI-HF 0.1µlを 加え
50
℃で30
分間、80
℃で5
分間インキュベートし、4
℃にて反応を 終了した。17
4.
ライゲーション反応液0.5µl
とXL1-Blue
コンピテントセル10µl
を用 いて形質転換を行った。100ng/µl Spectinomycinを含むLB
寒天培地にX-gal
とIPTG
存在下で播種し、37℃にて一晩培養した。5.
モジュールが正しく連結されたクローンを選別するため、コロニーPCR
を行った。10×buffer for Hybripol 0.8µl 100mM dNTP mix 0.64µl 10µM primer (pCR8 F1) 0.16µl 10µM primer (pCR8 R1) 0.16µl 50mM MgCl
20.24µl Hybripol DNA polymerase (BIOLINE) 0.04µl SDW 5.96µl Total 8µl
95℃ 30s
↓ 95℃ 15s
55℃ 15s 27cycles 72℃ 15s
↓ 72℃ 15s
↓ 4℃ ∞
各コロニーのレプリカプレートを作製した。
PCR
反応液を3
%アガロースゲルで電気泳動した。1
モジュールの挿入時、約350bp
のバンドが1
本検出され、1モジュ ール増えるごとにバンドサイズが約100bp
ずつ大きくなる。連結した モジュール数に相当する目的のサイズに、強いバンドが1
本とその下18
に約
100bp
間隔のうすいラダーバンドが検出できたクローンを正しくモジュールの連結ができたクローンと判定し、このクローンの大腸菌 を
100µg/ml Spectinomycin
を含むLB
液体培地2ml
で37℃、一晩培養
した。ChargeSwitch-Pro Plasmid Mini Kit
を用いてプラスミドを抽出・精製し、濃度を測定後
50ng/µl
に調製した。6.
第2
のアセンブリ各々連結されたモジュール(a4a-a4dと
b3
あるいはb1)
をさらにGolden Gate
反応により連結し、destination vector(ptCMV-136/63-VR-NG)に 組み込んだ。50ng/µl pFUS2 a 0.6µl×4 50ng/µl pFUS2 b 0.6µl 50ng/µl ptCMV vector 0.3µl 10×T4 DNA ligase buffer 0.4µl Esp3I 0.2µl Quick ligase 0.2µl Total 4µl
37℃ 5
分間→16℃ 10分間1
セットの反応を6
セット行い、最後に4℃
にて反応を終了した。
さらに、10×Tango buffer 0.5 µl, 10mM DTT 0.5 µl, Esp3I 0.2 µlを加え
37℃で 60
分間、80℃で5
分間インキュベートし、4℃にて反応を終了 した。7.
ライゲーション反応液2µl
とXL1-Blue
コンピテントセル20µl
を用い て形質転換を行った。100µg/ml Ampicillinを含むLB
寒天培地にX-gal
とIPTG
存在下で播種し、37
℃にて一晩培養した。19
8.
モジュールが正しく連結されたクローンを選別するため、コロニーPCR
を行った。10×buffer for Hybripol 0.8µl 100mM dNTP mix 0.64µl 10µM primer (TALE-F) 0.16µl 10µM primer (TALE-R) 0.16µl 50mM MgCl
20.24µl Hybripol DNA polymerase (BIOLINE) 0.04µl SDW 5.96µl Total 8µl
95℃ 30s
↓ 95℃ 15s
66℃ 15s 27cycles 72℃ 50s
↓ 72℃ 50s
↓ 4℃ ∞
各コロニーのレプリカプレートを作製した。
反応液を
1%アガロースゲルで電気泳動した。
第
1
のアセンブリと同様、連結したモジュール数に相当する目的のサ イズに強いバンドが1
本とその下に約100bp
間隔のうすいラダーバン ドが検出できたクローンを正しくモジュールが連結できたクローン と判定し、100µg/ml Ampicillin
を含むLB
液体培地2ml
で一晩培養し た。20
ChargeSwitch-Pro Plasmid Mini Kit
を用いてプラスミドを抽出・精製し、定量後
200ng/µl
の濃度に調製した。3-1-2) TALEN
活性測定のためのSSA
アッセイ作製した
TALEN
の活性を評価するため、SSA
アッセイを行った(Ochiaiet al. 2010)
。SSAアッセイでは、まず、TALENの標的配列を含むオリゴヌクレオチドを作製し、アニールさせた
2
本鎖のオリゴヌクレオチドをル シフェラーゼレポーターベクター内のクローニングサイトに組み込む。そ の後、このルシフェラーゼレポータープラスミドと上記で作製したTALEN
発現プラスミドをHEK293T
細胞に共導入し、ルシフェラーゼの酵素活性をもとに
TALEN
による切断活性を測定する。SSA
アッセイに用いるpGL4-SSA vector
内のTALEN
標的配列クローニ ングサイトは以下のような配列となっている。
BsaI BsaI
5’-CTAGGGTCTCT/GTCGTGCCCGGGTACTGATGT/ACCGTGAGACCTAGGA-3’
3’-GATCCCAGAGACAGC/ACGGGCCCATGACTACATGGC/ACTCTGGATCCT-5’
1. pGL4-SSA vector
をBsaI
処理し、直鎖状プラスミドにした後、-20℃にて保存した。(BsaI 処理の後、ベクターの脱リン酸化は行わない。)
BsaI
処理後は、上記、網掛けにした4
塩基の配列が突出末端となって いる。2.
以下のオリゴヌクレオチドをグライナージャパンに発注した。Sense
鎖 5’-gtcggat cttcaaggacgacggcaactacaagacccgcgccgaggtgaagttcgagggcg aggt-3’Antisense
鎖 5’-cggtacct cgccctcgaacttcacctcggcgcgggtcttgtagttgccgtcgtccttgaag atc-3’21
アニール後、網掛けにした
4
塩基の配列により突出末端が出現する。黒字はそれぞれが相補的な配列となっている。
3.
以下の条件にてオリゴヌクレオチドをアニールさせた。10×buffer 1µl 50µM Sense oligonucleotides 1µl 50µM Antiense oligonucleotides 1µl SDW 7µl Total 10µl
10×buffer
の組成は400mM Tris-HCl (pH 8), 200mM MgCl2, 500mM NaCl
である。0.2ml
チューブに上記の液を混合し、95℃で 5
分間インキュベート後、90
分ほどで25℃まで徐々に温度を下げる。
4. 1
で作製したpGL4-SSA vector
と3
で作製した2
本鎖オリゴヌクレオ チドをライゲーションし、ライゲーション反応液2µl
とXL1-Blue
コ ンピテントセル20µl
を用いて形質転換を行った。100µg/ml Ampicillin を含むLB
寒天培地に播種し、37℃にて一晩培養した。5.
コロニーをピックアップし、LB-Amp 液体培地で37℃一晩培養後、
ChargeSwitch-Pro Plasmid Mini Kit
にてプラスミドを抽出し、定量後150ng/µl
の濃度に調製した。6.
オリゴヌクレオチドの末端には1
ヶ所KpnI
の制限酵素サイトが設計 されている。そのため、正しくオリゴヌクレオチドが挿入されると、このプラスミドを
KpnI
処理した際、2
ヶ所で切断されることとなり、22
3,800bp
と1,800bp
のDNA
断片が確認できる。このような原理により、KpnI
処理によるインサートチェックを行った。7. Lipofectamine LTX
を用いて、以下の手順によりHEK293T
細胞へのト ランスフェクションを行った。・10cm dishに
70-80% confluent
のHEK293T
細胞を準備する。・以下の混合液を作製し、6µlを
DNA
溶液として使用する。200ng/µl left TALEN expression vector 1.5 µl 200ng/µl right TALEN expression vector 1.5 µl 150ng/µl SSA reporter vector 1 µl 30ng/µl pRL-CMV 1 µl Nuclease-free water 4 µl
・DNA希釈用と
Lipofectamine LTX
希釈用の無血清DMEM
を25µl×
サンプル数量準備する。
・96wellプレートの
1well
にDNA
希釈用無血清DMEM
を25µl
いれ、DNA
溶液6µl
を加える。よくピペッティングする。・Lipofectamine LTX希釈用の無血清
DMEM
に1well
あたり0.7µl
とな るようLTX
を加え、均一になるよう、よくピペッティングする。LTX
を加えてから5
分以内に1well
あたり25µl
を加え、よくピペッティン グする。・HEK293T 細胞を培養している
10cm dish
からメディウムを除去し、15% FBS-DMEM
を加え、よくピペッティングして細胞を懸濁する。細胞数を計測し、
6×10
5cells/ml
となるよう調製する。・LTXを
96well
プレートに加えて30
分が経過したら、準備した細胞を
1well
あたり100µl
加え、37
℃、5% CO
2下でインキュベートする。23
・トランスフェクションの
24
時間後にルシフェラーゼの活性測定を行 う。8. Dual-Glo Luciferase Assay System
とTriStar LB 941 plate reader
を用いて ルシフェラーゼアッセイを行った。3-1-3)TALEN mRNA
合成1. SSA
アッセイにて高い切断活性が確認されたTALEN
プラスミドをSmaI
処理し、直鎖化した。2. QIAquick PCR purification kit
を用いて制限酵素反応液を精製し、50µl のRNase-free water
で溶出し、DNAの濃度を測定した。3.
直鎖化したTALEN
プラスミドはT7 RNA polymerase promoter
を含むた め、mMESSAGE mMACHINE T7 Ultra kitを用いて転写反応を行った。T7 2×NTP/ARCA 10μl 10×T7 Reaction Buffer 2μl DNA template (0.1μg) 6μl T7 Enzyme mix 2μl Total 20μl
混合液を
37℃で 2
時間インキュベートした。この反応で5’末端にキャ
ップ構造をもつmRNA
が合成される。反応後、
TURBO DNase
を1μl
加え、37℃で 15
分間インキュベートし、DNA
を分解した。4. mMESSAGE mMACHINE T7 Ultra kit
を用いてPoly (A)
の付加を行った。24 上記反応液
20μl
Nuclease-free water 36μl 5×E-PAP buffer 20 μl 25mM MnCl
210μl ATP solution 10μl Total 96μl
上記の溶液を混合した後、E-PAP を
4μl
加え、やさしくピペッティン グし、37℃で45
分間インキュベートした。5. RNeasy Mini kit
を用いて、合成mRNA
の精製を行った。精製後のmRNA
は濃度を測定し、0.5mlチューブに1μg
ずつ分注後、-80℃にて保存し た。3-1-4)pCAG-eGFP
マウス受精卵の準備
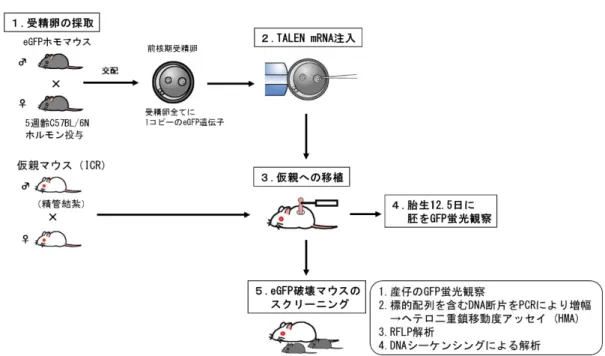
TALEN
によるeGFP
遺伝子破壊マウス作製の概要を図2
に示す。1.
発情前期の10
週齢Jcl: ICR
雌マウスを精管結紮雄マウスと交配させ、翌日、膣栓が確認できた雌マウスを偽妊娠雌マウスとして
TALEN mRNA
インジェクション後の受精卵移植に用いた。2. 6cm dish (IWAKI cat.#1010-060)
に1mg/ml
ヒアルロニダーゼを含むKSOM-AA
の500μl
ドロップを1
つとKSOM-AA
の100μl
ドロップを3
つつくり、流動パラフィンでおおい、37℃、5% CO
2インキュベータ ー内に静置した。3. TALEN mRNA
を イン ジェ クシ ョン する た め、 以下 の手 順に よ りpCAG-eGFP
マウスの受精卵を採卵した。PMSGとhCGを48時間間隔で
25
各
5単位ずつ腹腔内投与することにより、過排卵処理した5週齢の
C57BL/6NCrlCrlj
雌マウスと個飼いにしたpCAG-eGFP 雄マウスを交 配し、翌日、膣栓が確認できた雌マウスを選別した。4.
プラグが確認できた雌マウスを頸椎脱臼にて安楽死させ、子宮、卵管、卵巣をとりだした。3で準備した6cm dishのヒアルロニダーゼを含む
KSOM-AAドロップへ、卵管膨大部から卵子塊を導入した。卵丘細胞
が除去された卵子をKSOM-AAドロップで3回洗浄した。5.
雌性前核と雄性前核が確認された受精卵を選別し、TALEN mRNAのイ
ンジェクションに使用した。3-1-5)TALEN mRNA
のマイクロインジェクション1. 9cm
ガラスディッシュ(IWAKI)にフェノールレッドを含まない
KSOM-AA
ドロップをつくり、軽質流動パラフィンでおおい、37℃、5% CO
2インキュベーター内に静置した。2.
各々のTALEN mRNA を1×RNase-free PBSで10、100、 150ng/µl
となる よう希釈し、等量ずつ混合して、インジェクション針に充填した。ガ ラスディッシュのKSOM-AAドロップに受精卵をうつし、倒立顕微鏡 下でマニピュレーターを用いて、受精卵の前核内あるいは細胞質へTALEN mRNAをインジェクションした(図 3)
。3.
インジェクション後の受精卵を37℃、5% CO
2インキュベーター内に26
て
1
時間静置した。4.
生存卵を偽妊娠雌マウス1匹あたり、平均23個(16~35個/匹)ずつ、偽妊娠1日目ICR雌マウスの卵管へ移植した。
3-1-6)胎仔あるいは産仔の蛍光観察
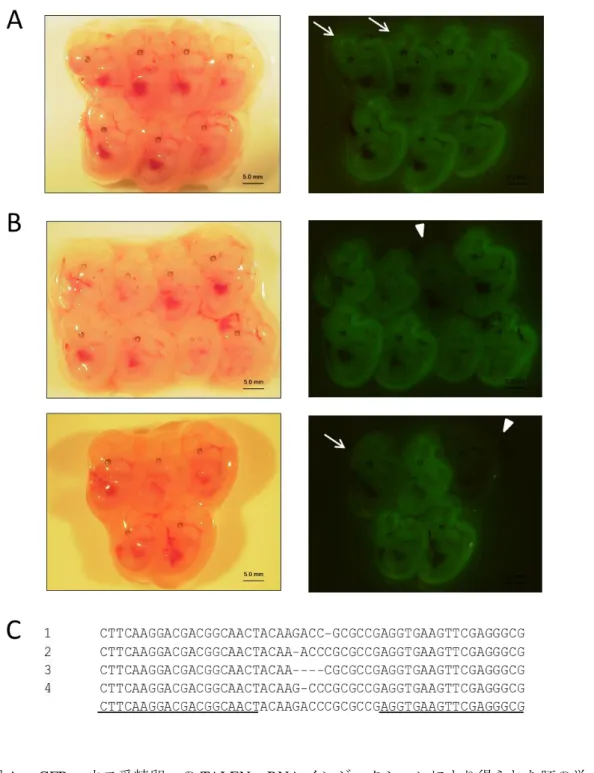
12.5
日胚は蛍光顕微鏡にて観察を行った。産仔は生誕日あるいはその翌日に
UV-LED
ライトをあて、蛍光観察を行った。3-1-7)HMA
のためのゲノミックPCR
1.
離乳後あるいは生後1
日目の産仔から各個体の尾を採取し、DNeasyBlood & Tissue Kit
を用いてゲノムDNA
を抽出した。育児放棄や喰殺 による死亡個体も解析に用いるため、できる限り回収し、ゲノムDNA
を抽出した。2. eGFP TALEN
の標的配列を含むDNA
断片を増幅するゲノミックPCR
を行った。Genomic DNA (250-500ng) 5µl
10× buffer for La Taq 5µl
dNTP mix (2.5mM each)
7.5µl
10µM primer (eGFP –F) 2.5µl
10µM primer (eGFP –R) 2.5µl
25mM MgCl
25µl
LA Taq DNA polymerase 0.5µl
SDW 22µl
Total 50µl
27
94℃ 2m
↓ 94℃ 30s
64℃ 30s 38cycles 72℃ 20s
↓ 72℃ 5m
↓ 4℃ ∞
3. PCR
産物3µl
を3%アガロースゲルで電気泳動し、エチジウムブロマ
イドにより染色した。UVを照射し、ゲルを撮影した。残りの
PCR
産 物はRFLP
解析と塩基配列の決定に使用した。3-1-8)RFLP
解析1.
上記のPCR
産物をWizard SV Gel and PCR Clean-up System
を用いて精 製した。スペーサー配列内の1ヶ所を切断するAccII
を用いて、精製 したPCR
産物を処理することにより、スペーサー内での変異導入の有 無を確認した。Purified PCR product 3µl 10×M buffer 1µl AccII 0.15µl SDW 5.85µl Total 10µl
上記の反応液を
37℃で一晩インキュベートした。
2. 3%アガロースゲルで全量を電気泳動し、エチジウムブロマイドにより
染色した。
UV
を照射し、ゲルを撮影した。28
3-1-9)PCR
産物の塩基配列決定1.
上記のPCR
産物をpGEM -T Easy vector
のクローニングサイトに挿入 するため、ライゲーション反応を行った。2×Rapid Ligation buffer 2.5µl pGEM -T Easy vector 0.1µl PCR product 0.17µl T4 DNA Ligase 0.5µl SDW 1.73µl Total 5µl
上記の反応液を
16℃で 1
時間インキュベートした。2.
ライゲーション反応液1.25µl
とXL-1 Blue
コンピテントセル10µl
を 用いて形質転換を行い、LB-Amp 寒天培地に播種後、37℃で一晩培養 した。3.
コロニーをピックアップし、2ml のLB-Amp
液体培地で、37℃にて一 晩培養した。4. Mini plus Plasmid DNA Extraction system
を用いて大腸菌からプラスミ ドを抽出した。5.
目的のサイズのDNA
断片が挿入されていることを確認するため、プ ラスミドをEcoRI
処理した。Plasmid 5µl
10×H buffer 1µl
EcoRI 0.3µl
SDW 3.7µl
Total 10µl
29
37℃で 30
分間インキュベートした。6. 2%アガロースゲルで電気泳動し、エチジウムブロマイドにより染色し
た。UVを照射し、ゲルを撮影した。
7.
目的のサイズのDNA
断片が挿入されていたクローンを用いて、BigDye Terminator Cycle Sequencing Kit
を用いてシークエンス反応を 行った。Ready Reaction Premix 2µl 5×BigDye Sequencing buffer 4µl T7 or SP6 primer (1.6pmol) 1.6µl Plasmid (80-150ng) 1µl SDW 11.4µl Total 20µl
96℃ 1m
↓ 96℃ 10s
50℃ 5s 25cycles 60℃ 2m
↓ 4℃ ∞
8.
各サンプルに100%エタノール 50µl
と3M
酢酸ナトリウム (pH5.2) 2µl を加え、よく混合し、氷上で5
分静置した。14,500rpmで15
分間遠心 し、上清を除去後、70%
エタノールを200µl
加えた。5
分間遠心し、上 清を除いて、よく乾燥させた。これをHi-Di Formamide
に溶解し、95℃
で
2
分インキュベートし、氷上に静置した。これらのサンプルをABI
PRISM 3130 Genetic Analyzer
にて解析し、塩基配列を決定した。30
3-2)凍結融解受精卵を用いた Platinum TALEN
による遺伝子破壊マウスの作製
基本的に、前述の
eGFP
遺伝子の破壊と同様の手順により、実験を行っ た。TALEN mRNA の合成や受精卵へのマイクロインジェクション、DNA シーケンシングなど重複する部分については、ここでは記載しない。3-2-1)Platinum TALEN
プラスミド作製とSSA
アッセイeGFP Platinum TALEN
プラスミドと同様にbL
またはClec4b1
のPlatinum TALEN
プラスミドを作製した。今回、使用したdestination vector
は、ptCMV-153/47-VR
である。bL
およびClec4b1 Platinum TALEN
の模式図を図10
に示す。SSA
アッセイでは、bLおよびClec4b1 TALEN
の標的配列を含むオリゴ ヌクレオチドを作製し、アニールさせた2
本鎖のオリゴヌクレオチドをそ れぞれpGL4-SSA vector
内のTALEN
標的配列クローニングサイトに組み込んだ。
HEK293T
細胞へのトランスフェクションおよびルシフェラーゼアッセイは、前述と同様の方法で行った。
3-2-2)Ayu8104
マウス受精卵の準備Ayu8104
マウスはbL
外来遺伝子をもつジーントラップマウスである(Imaizumi