九州大学学術情報リポジトリ
Kyushu University Institutional Repository
非小細胞肺癌の薬剤感受性に関する研究
日下部, 大樹
http://hdl.handle.net/2324/4060102
出版情報:九州大学, 2019, 博士(臨床薬学), 課程博士 バージョン:
権利関係:
学位請求論文
非小細胞肺癌の薬剤感受性に関する研究
九州大学大学院 薬学府 生命物理化学分野
日下部 大樹
2019
目次
第1部
EGFR活性型変異陽性の非小細胞肺癌における
EGFR-TKIオシメルチニブの薬剤耐性機序解明
1-1. 略語一覧 ... 2
1-2. 序論 ... 4
1-3. 方法 ... 9
1-4. 結果 ... 14
1. EGFR-TKI耐性細胞株のオシメルチニブ感受性評価 ... 14
2. オシメルチニブ処理時の下流シグナル評価 ... 16
3. 種々の分子標的薬に対する感受性と生存シグナル評価 ... 18
4. SFKsの発現レベルと依存性評価 ... 22
5. SRC活性化因子の探索① AXLの関連評価 ... 24
6. SRC活性化因子の探索② CDCP1の関連評価 ... 28
7. オシメルチニブに耐性となった患者の腫瘍内AXLおよびCDCP1発現評価 ... 31
1-5. 考察 ... 32
1-6. 小括 ... 36
1-7. 参考文献 ... 38
第2部
KRAS活性型変異陽性の非小細胞肺癌におけるフェロトーシス感受性に関する研究
第1章 序章
1-1. 略語一覧 ... 46
1-2. はじめに ... 48
1-3. 参考文献 ... 54
第2章 リゾリン脂質アシル転移酵素を介したリン脂質組成調節とフェロトーシス感受性 2-1. 略語一覧 ... 58
2-2. 序論 ... 60
2-3. 結果 ... 64
1. FINs感受性評価... 64
2. 脂肪酸処理のFINs感受性への影響評価 ... 66
3. リン脂質組成の変動解析 ... 68
4. 脂肪酸処理時の酸化脂質検出 ... 71
5. 他のKRAS変異陽性の非小細胞肺癌細胞株における検討 ... 74
6. 脂肪酸の種類とFINs感受性評価 ... 75
7. RSL-3感受性と相関するリゾリン脂質アシル転移酵素の探索 ... 79
8. LPCAT3とFINs感受性との相関解析 ... 82
9. OA処理におけるAGPAT5またはLCLAT1発現抑制の影響評価 ... 84
10. AGPAT5の継続的な発現抑制とフェロトーシス耐性の関連評価 ... 86
11. 補助資料 ... 88
2-5. 考察 ... 90
2-6. 小括 ... 94
2-7. 参考文献 ... 96
第3章 アルデヒドデヒドロゲナーゼ3A1とフェロトーシス感受性
3-1. 略語一覧 ... 100
3-2. 序論 ... 102
3-3. 結果 ... 104
1. フェロトーシス関連因子のフェロトーシス感受性差への影響評価 ... 104
2. ALDH3A1発現量とフェロトーシス感受性差の相関解析 ... 109
3. ALDH3A1阻害時のフェロトーシス感受性評価 ... 114
4. AA処理とALDH3A1阻害剤の併用処理における細胞死評価 ... 116
3-4. 考察 ... 117
3-5. 参考文献 ... 120
第4章 総括 ... 123
第5章 実験方法 ... 127
参考文献 ... 138
発表論文 ... 139
学会発表 ... 140
謝辞 ... 142
1
第 1 部
EGFR 活性型変異陽性の非小細胞肺癌における
EGFR-TKI オシメルチニブの薬剤耐性機序解明
2
1-1. 略語一覧
BLK B Lymphoid tyrosine kinase
CDCP1 CUB Domain containing protein 1
CSK C-Terminal SRC kinase
CTNNB1 Catenin beta 1
DAG Diacylglycerol
EGFR Epidermal growth factor receptor
FAK Focal adhesion kinase
FGFR Fibroblast growth factor receptor
FOXO1 Forkhead Box O1
GSK3β Glycogen Synthase Kinase 3 Beta
HER Human Epidermal growth factor receptor
IGF-1R Insulin like growth factor 1 receptor
JAK Janus kinase
MAPK Mitogen-Activated protein kinase
MET Mesenchymal epithelial transition, receptor tyrosine kinase
mTORC1 Mammalian target of rapamycin complex 1
NSCLC Non-small cell lung cancer
PDGFR Platelet derived growth factor receptor
PFS Progression free survival
PI3K Phosphatidylinositol-4,5-bisphosphoate 3-kinase
PIK3CA PI3K Catalytic Subunit Alpha
PKC Protein kinase C
3
PLCγ Phospholipase C gamma
PTEN Phosphate and tensin homolog
RTK Receptor tyrosine kinase
SCLC Small cell lung cancer
SFK SRC family kinase
SGK Serum glucocorticoid-induced protein kinase
STAT Signal transducer and activator of transcription
TAM Tyro3-Axl-Mer
TGFα Transforming growth factor α
TKI Tyrosine kinase inhibitor
TSC2 Tuberous sclerosis complex subunit 2
VEGFR Vascular endothelial growth factor receptor
4
1-2. 序論
がん細胞の生存に依存する分子を標的とした薬剤は、分子標的薬と呼ばれ、従来の化学療 法剤とは異なる特性を有している。白金製剤やアルキル化剤などの化学療法剤は、がん細胞 の爆発的な増殖に必須のDNA複製を標的としており、増殖の盛んな細胞をアポトーシスへ 導くことができる。しかし、化学療法剤はがん細胞ばかりでなく正常細胞にも障害を与えて しまう1。一方、分子標的薬はがん細胞に特徴的な因子を標的とした阻害薬であり、がん細 胞選択的な増殖抑制効果を目的に開発されている 2。現在に至るまで、Epidermal Growth Factor Receptor (EGFR)やHuman Epidermal Growth Factor Receptor (HER2)など様々な因子に 対する分子標的薬が開発され、臨床で用いられている。
肺がんは日本におけるがんの中で最も死亡数が多く、その治療法の開発に向け活発な研 究が行われてきた。肺癌は組織型により、SCLC(小細胞肺癌)とNSCLC(非小細胞肺癌)
に区別される。東アジア人において NSCLC の約 50%は EGFR の Exon19 の欠損 (ΔL747-
P753)やL858Rなどの活性型変異を有しており、その生存や増殖をEGFR経路に依存してい
る3。このことから、EGFRを標的とした治療薬創出研究がなされ、2002年にEGFRのチロ シンキナーゼ阻害剤 (EGFR-TKI) であるゲフィチニブが日本で初めて承認された4。現在で
もEGFR-TKIの改良は続けられており、2016年には第三世代のEGFR-TKIが臨床で使用可
能となっている。
EGFRはErbBファミリータンパクに属する膜受容体型チロシンキナーゼである。EGFや Transforming growth factor α (TGFα)などのリガンドがEGFRへ結合すると構造が変化し、ホ モまたはヘテロ二量体を形成する。それに伴い、自己リン酸化と細胞内チロシンキナーゼの 活性化が誘導され、様々な生理機能を調節する5。EGFRの下流シグナルにはJAK/SRC/STAT
やPI3K/Akt、MAPK、PLCγ/PKC経路などが存在する。これらの下流因子は、細胞の生存や
増殖、血管新生、分化、癌化誘導に関与している6 (Fig. 1)。EGFRの活性型変異はチロシ
5
ンキナーゼドメインにおいて観察され、リガンド非依存性の恒常的な活性化を可能にする7。 特に、活性型EGFRに依存するNSCLCでは、恒常的なAktおよびERKシグナルの活性化 が細胞の生存や増殖を調節する重要な働きを担っている8。
PLCγ DAG
P
P P P
EGF TGFα
Growth factors
EGFR (WT) EGFR (Activating mutation)
EGFR Phosphorylation
STAT Survival Proliferation Oncogenesis
AKT Angiogenesis Tumorigenesis Stress sensitivity Inhibition of apoptosis
ERK Cell motility Gene expression Cell-cycle progression
PKC Transformation
Differentiation Apoptosis JAK
SRC
PI3K
Ras Raf MEK
Figure 1. The main downstream signaling pathways regulated by EGFR 4. Abbreviations, JAK; Janus kinase, STAT; Signal transducer and activator of transcription, PI3K; Phosphatidilinositol-4,5- bisphosphoate 3-kinase, PLC; Phospholipase C, DAG; Diacylglycerol, PKC; Protein kinase C
6
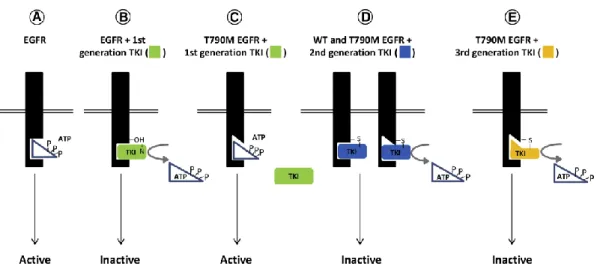
EGFR-TKIはEGFRのATP binding pocket へ結合することにより、EGFRのキナーゼ活性
を阻害する薬剤である (Fig. 2A and B)。第1世代EGFR-TKIのゲフィチニブとエルロチニブ は、可逆的にEGFR活性を阻害することで活性型EGFR変異陽性で未治療のNSCLC患者に 対し著明な治療効果を示してきた9。しかし、ほとんどの腫瘍は1 年以内に耐性を獲得し、
その際EGFRT790M耐性変異が高頻度(約60 %)で観察される10。T790M耐性変異は、EGFR
のATPへの親和性を亢進させると同時に、第一世代EGFR-TKIの結合を阻害する (Fig.2 C)。 その後、第二世代のEGFR-TKIであるアファチニブは、EGFRのC797部位への共有結合を
介してT790M耐性変異陽性のEGFR活性を阻害可能な薬剤として開発されたが (Fig. 2 D)、
野生型EGFRへの結合能が高いために患者への副作用が強く、T790M耐性肺癌腫瘍に対し ては生存期間を延長できなかった。一方、第三世代のオシメルチニブはEGFRのC797部位 への共有結合能だけでなく、活性型変異とT790M耐性変異の双方を有するEGFRに対し高 い選択性を持つことから、T790M耐性変異を克服可能な薬剤として開発された11 (Fig.2 E)。 オシメルチニブは臨床において T790M 陽性の患者に対し高い活性を示しており、現在
T790M変異を有する肺癌に用いることのできる唯一のEGFR-TKIである12。さらに、EGFR-
TKI治療歴のないEGFRのT790M耐性変異陰性および活性型変異陽性のNSCLC患者に対 してもPFSの延長が報告され13、一次治療としての使用が認められている。
Figure 2. Model depicting the effects of 1st, 2nd and 3rd generation TKIs in receptor activity of WT, mut and T790M EGFR molecules10
7
EGFR の活性型変異およびT790M耐性変異陽性の NSCLC患者に対して、オシメルチニ ブは他の分子標的薬よりも獲得耐性が生じにくく、治療有効期間が長いことが臨床および 非臨床の研究により示されている13–15。しかし、他のEGFR-TKIと同じく、オシメルチニブ でも耐性を示す腫瘍が出現する11,16。オシメルチニブ耐性で観察されるEGFR遺伝子の変容
には、C797S耐性変異やEGFR遺伝子増幅、T790M変異の消失などがある17–23。一方、EGFR
以外の遺伝子の変容も腫瘍の悪性進展を促進し、オシメルチニブ耐性腫瘍出現に関与して いる24。EGFR以外の遺伝子変異として、RASやBRAF、PI3K Catalytic subunit α (PIK3CA)、 Phosphate and tensin homolog (PTEN)、Catenin beta 1 (CTNNB1)、Tuberous sclerosis complex
subunit 2 (TSC2)の遺伝子変異は、オシメルチニブ獲得耐性に関与することが報告されてい
る14,25–27。さらに、HER2やMesenchymal epithelial transition, receptor tyrosine kinase (MET)、 Fibroblast growth factor receptor (FGFR)、Mitogen-activated protein kinase 1 (MAPK1)、Akt3、 AXLの発現上昇やSRC family kinase (SFK)/Focal adhesion kinase (FAK)経路、ヘッジホッグ経 路の活性化のオシメルチニブ耐性への関与も示されている28–32。以上より、オシメルチニブ 耐性腫瘍においては、多様な耐性メカニズムが推察される。しかし、臨床においてEGFR遺 伝子の変容とそれ以外の因子の双方またはどちらかがオシメルチニブ耐性の獲得にどの程 度関与しているか、そして、どのようにしてその耐性を克服するかについては未だ明らかと なっていない。
一方、分子標的薬の治療効果の最大化と個々の患者への適正な治療(Precision medicine) を実践するためには、治療対象となるがん細胞がどのシグナル経路に生存を依存している かについて精査し、薬物を投与する指標を明確化する必要がある33。特に、分子標的薬の初 期治療に耐性を示したがん細胞は、分子標的薬から自身を守る巧妙で複雑な耐性機序を発 達させている。この薬剤耐性機序を予測し次の治療法を考案する基礎研究として、培養がん 細胞株への分子標的薬の持続的な処理により樹立された耐性株を用いた解析が盛んに行わ
れてきた11。実際にIn vitroで出現した耐性機序が数多く臨床でも観察されている14,23,34–36。
8
本研究では、EGFR の活性型変異とT790M 耐性変異の双方を有するNSCLC細胞株であ
る H1975 株に対し、オシメルチニブに長期間暴露することでオシメルチニブ耐性株を樹立
した。この耐性株を用いた解析から、CUB domain containing protein 1 (CDCP1)とAXLによ るSRCの活性化がオシメルチニブ獲得耐性に寄与することを明らかにし、この獲得耐性に 対する治療標的分子を提示した (Fig. 3)。
Figure 3. This work: Osimertinib resistant cells were established from H1975 cells. Comparative analysis showed CDCP1/AXL/SRC axis contributed to osimertinib resistance.
5 nM 2 μM
Cultured in increasing, step-wise dose of osimertinib H1975 cells
(Sensitive)
6 months
Osimertinib Resistant cells
(OR1, OR2) Comparative analysis
CDCP1/AXL/SRC axis contributes to osimertinib resistance !
9
1-3. 方法
細胞培養およびH1975由来オシメルチニブ耐性株の樹立
H1975とHCC827株は American Type Culture Collection (ATCC)より購入した。PC-9株は 九州がんセンター一瀬幸人先生より、11-18株は近畿大学中川和彦先生より寄贈いただいた。
全ての肺癌細胞株は10 % FBSを添加したRPMI1640培地を用い、5 % CO2条件下 37 ℃で 培養した。H1975株に対し、オシメルチニブの濃度を段階的に2 μMまで上昇させながら約 6か月培養することでそれぞれ独立したオシメルチニブ耐性株OR1とOR2を樹立した。
試薬
エルロチニブはF. Hoffman-La Roche Ltdより、TPX-0005はTP Therapeutics, Inc.より寄贈 いただいた。ダサチニブはBio Visionから、ソラフェニブはToronto Research Chemicals Inc.;
より購入した。その他の阻害剤についてはSelleck Chemicalsより購入した。
Anti-HER2 および anti-pHER2抗体はUpstate Biotechnologyより購入し、anti-EGFR お よびanti-pEGFR、anti-pHER3、anti-c-Met、anti-pc-Met、anti-Erk1/2、anti-pErk1/2、anti-Akt、 anti-pAkt (T308 or S473)、anti-STAT3、anti-pSTAT3、anti-PTEN、anti-AXL、anti-CDCP1、anti- SRC、anti-Fyn、anti-Lyn、anti-Yes、anti-Lck、anti-pSFK (Y416) 抗体はCell Signaling Technology より購入した。また、anti-HER3はSanta Cruz Biotechnologyより、anti-β-actinはAbcam, Inc.
より、Anti-GAPDHはPROMEGAから購入した。
10 ウエスタンブロッティング
35 mm dishで各種刺激条件において培養した細胞を2回冷PBSで洗浄し、RIPA bufferで
溶解後その上清を細胞ライセートとした。ライセートを 4 倍濃縮したサンプルバッファー と混合し、100℃で5分間加熱処理することでタンパク質を変性させた。調整したサンプル をSDS-PAGEにて分離し、Immobilon-P membranes (Millipore)へ転写後、各種抗体にてタンパ ク質を検出した。検出には抗ウサギもしくはマウス IgG の HRP 連結型 2 次抗体を用い、
HRPによる化学発光をLAS-4000にて検出した。
共免疫沈降
15 cm Dishで各種刺激した細胞を冷PBSで洗浄後、IP lysis buffer (50 mM Tris-HCl (pH 8.0), 250 mM NaCl, 1 mM EDTA, 10% glycerol, 0.3% NP-40)で溶解した。ライセートをNET-gel buffer [50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM EDTA, 0.25% gelatin, 0.02% sodium azide, 1 mM PMSF, 10 mg/ml leupeptin, and 10 mg/ml aprotinin]で希釈し、抗SRCもしくはCDCP1抗体を 添加した後、4℃で一晩撹拌した。その後、Protein A/G PLUS-Agarose beads (Santa Cruz
Biotechnology)と4 C°で1時間反応させ、ビーズと抗体、タンパク質複合体を含む溶液をNET-
gel bufferおよび0.1% NP40を含む10 mM Tris-HCl (pH 7.5)で一回ずつ洗浄した。遠心分離に より単離したBeads複合体をサンプルバッファーと混合し、100℃で5分間煮沸した後ウエ スタンブロッティングへ用いた。
11 Small interfering RNAトランスフェクション
Opti-MEM培地(Invitrogen)中でsmall interfering RNAs (siRNA)とLipofectamine RNAiMAX を混合し、複合体を形成させた。メーカーのプロトコルに従い、siRNA-Lipofectamine 複合 体混合溶液を細胞にトランスフェクションした。SRC siRNA#3, SRC siRNA#1 と CDCP1 siRNA、AXLsiRNAはInvitrogenより購入した。Non-specific (control) siRNAはQiagenより 購入した。
マイクロアレイ解析
完全相補RNAをSuperPrint G3 Human GE Microarray 8x60Kを用い、本機械のプロトコル に従い、増幅およびラベル化、ハイブリダイゼーションを行った (Agilent Technologies)。ハ イブリダイゼーション後のマイクロアレイスライドはAgilent scannerにて測定した。 目的 因子の発現強度とバックグランド強度はAgilent Feature Extraction Softwareで解析した。発 現上昇もしくは低下因子を同定するため、それぞれの因子に対するプローブの検出強度を コントロールとサンプル群との強度比で算出した。強度比は非 Log スケールで解析した。
基準値として、検出強度がコントロール群よりも2倍以上増加したものを発現上昇、0.5倍 以下に低下したものを発現低下とした。
セルカウントおよび細胞増殖評価
24 well plateに細胞を播種した翌日にsiRNAをトランスフェクションし、AXLまたはSRC、
CDCP1の発現抑制を行った。siRNA処理後、各時点において細胞をトリプシンで剥離し、
Z2 Coulter Particle Count and Size Analyzer (Beckman Coulter Inc., CA)を用いて細胞数を計測し た。各時点において、独立した検討を3回行った。
12 細胞毒性評価
96 well flat-bottom plateに細胞を播種し24時間培養後、各種薬剤を処理した。薬剤処理で
は、種々の濃度の薬剤またはSCADS (Screening committee of anticancer drugs) 阻害剤キット を処理し72時間、37 ℃および 5 % CO2環境下で培養した。刺激後に15 μLのCell Count Reagent SF (Nacalai Tesque) を各wellに添加し、2から4時間37 ℃および5 % CO2環境下で 培養した。450 nmの吸光度をiMarkTM プレートリーダーで検出し、コントロール群の吸光
度を100 %の生存率と換算して薬剤処理群の細胞生存率を算出した。それぞれの薬剤濃度に
おいて、独立した検討を3回行った。IC50値は細胞生存率が50 %となる薬剤濃度を生存曲 線から算出した。
動物
動物実験は、九州大学大学院医学研究科の動物実験倫理委員会による承認を受けた。
CLEA Inc.より雄性の無胸腺 nu/nuマウスを購入し、12時間明暗サイクル環境下マイクロア
イソレーターケージ内で飼育した。飲料水と給餌は自由に摂取できる環境で、断続的に与え 続けた。動物実験における腫瘍成長や活動、摂食の観察および苦痛の軽減は、Guidelines of the Harvard Medical Area Standing Committee on Animalsを遵守した。
担癌マウスモデル作成
約5.0 × 106のOR1株を50 %マトリゲルを含む200 µLの培地に懸濁し、マウスの右腹壁
に移植した。腫瘍径を計測し、腫瘍体積は長さlength × width2 × 0.5より近似値を算出した。
腫瘍体積が100から200 mm3に達した時、マウスを無作為に6匹ずつ群分けし、オシメル チニブ投与群(40 μg / 匹、週3回経口投与)とダサチニブ投与群(600 μg / 匹、週3回経 口投与)を作成した。投薬22日後にそれぞれ解剖し、腫瘍を回収した。腫瘍は測定までの
間、-80 ℃にて保存した。
13
EGFR-TKI治療の再発肺癌患者より回収した腫瘍切片
久留米大学病院において、EGFR-TKI治療を受けたEGFR exon19欠失変異を有する
NSCLC患者よりEGFR-TKI治療前後の腫瘍サンプルを回収した。Case1の患者ではエルロ
チニブで治療後、T790M耐性変異出現が確認されたため、その後1年間オシメルチニブで 治療した。腫瘍組織はオシメルチニブ治療前後で回収した。本研究は久留米大学病院の治 験審査委員会により承認され、患者はヘルシンキ宣言に従いインフォームドコンセントが 提供されている。
腫瘍組織の免疫学的染色
ヒト肺癌組織のパラフィン包埋組織サンプルを使用して、4μmの切片を作成し、コーテ ィングされたスライドガラスにマウントした。サンプルはepitope retrieval solution 2 (pH9.0) で処理し、Bond-IIIシステム (Leica Microsystems, Newcastle, UK)のオンボード熱誘導抗原活 性化を30分間行った。その後、抗AXL抗体 (1:100)、および抗CDCP1抗体 (1:100)と30 分間インキュベートした。染色には Horseradish peroxidase (HRP)ポリマーを二次抗体とし、
3,3’-Diaminobenzidine (DAB)を色素原として用いた。検出は Refine polymer detection system
(Leica Microsystems, Newcastle, UK)を使用した。
14
1-4. 結果
1. EGFR-TKI耐性細胞株のオシメルチニブ感受性評価
始めに、これまでに樹立されたEGFR-TKI耐性株について、EGFR-TKI世代間によりオシ メルチニブ感受性に違いがあるか否かを検討するため、ゲフィチニブとエルロチニブ、アフ ァチニブの耐性肺癌細胞株を比較した (Table 1)。EGFRのエクソン19欠失活性変異を持つ PC-9 株より樹立したゲフィチニブもしくはエルロチニブ耐性株は、PTEN の欠失 37,38また はインテグリン/SRC バイパス経路の活性化39により、それぞれ耐性を獲得している。双方 の細胞株はオシメルチニブに対し、親株の PC-9 と比較して 100 倍以上の耐性を示した。
EGFRのL858R活性変異を持つ11-18株より樹立したゲフィチニブ耐性株では、活性型EGFR
アレルの欠失が生じており40、オシメルチニブに対して約10倍の耐性を示した。EGFRの エクソン19欠失変異を持つHCC827株より樹立したアファチニブ耐性株では、SRC/FAK経 路が活性化している41。この株はオシメルチニブに対して100倍以上の耐性を示した。これ
Cell lines EGFR
mutations Selected drug
IC50 (µmol/L)1)
Resistant mechanism Erlotinib Afatinib Osimertinib
PC-9 Del19 0.016 (1.00) 0.00059 (1.00) 0.0049 (1.00)
PC-9/GEF1-1 Gefitinib 7.38 (452) 1.83 (3083) 1.64 (333) PTEN loss PC-9/GEF2-1 Gefitinib >10 (>613) 2.18 (3664) 2.28 (462)
PC-9/ER2-2 Erlotinib 7.96 (488) 2.10 (3533) 0.967 (196) Integrin/SRC activation PC-9/ER2-3 Erlotinib 6.97 (427) 1.49 (2503) 0.784 (159)
0.046 (1.00)
11-18 L858R 0.095 (1.00) 0.0072 (1.00)
11-18/GEF10-1 Gefitinib 0.69 (7.25) 0.24 (33.5) 0.63 (13.9) mutant EGFR loss 11-18/GEF20-1 Gefitinib 0.47 (4.93) 0.23 (31.7) 1.48 (32.5)
11-18/ER1-7 Erlotinib 4.67 (49.2) 0.89 (125) 1.49 (32.7) mutant EGFR loss 11-18/ER2-1 Erlotinib 1.91 (20.1) 0.92 (128) 1.41 (31.0)
]
HCC827 Del19 0.0234 (1.00) 0.00381 (1.00) 0.00597 (1.00)
HCC827/BR1-8 Afatinib >10 (>428) 3.83 (1005) 1.95 (326) SRC/FAK activation HCC827/BR2-3 Afatinib >10 (>428) 3.58 (940) 4.10 (686)
H1975 L858R
T790M 5.48 (1.00) 0.21 (1.00) 0.014 (1.00)
H1975/OR1 Osimertinib 6.30 (1.15) 0.94 (4.48) 4.0 (286) This study H1975/OR2 Osimertinib 6.97 (1.27) 0.66 (3.14) 4.04 (289)
1) T he relative resistance, defined as the IC50 value divided by the IC50 value of the parental cells, is shown in parentheses.
Table 1. Lung cancer cell lines resistant to First and Second generation EGFR-TKIs show cross- resistance to Third generation TKI osimertinib.
15
らの耐性株は既報のEGFR-TKI耐性機序であるEGFRのT790M耐性変異を出現させること なく、オシメルチニブに交差耐性を示した。
次に、これまでの報告と同様の手法を用いて37,39、EGFRのT790M耐性変異とL858R活 性変異を持つNSCLC細胞株H1975に対し、オシメルチニブの濃度を段階的に2μMまで上 昇させながら約6か月培養することで、オシメルチニブ耐性株H1975/OR1とH1975/OR2を それぞれ独立に樹立した。親株であるH1975と比較して耐性株OR1とOR2は共にオシメ ルチニブに対し約300倍耐性であり、エルロチニブとアファチニブに対してはH1975と同 等の耐性を示した (Fig. 4)。オシメルチニブへの獲得耐性機序として知られる EGFR の
C797S 耐性変異は 23、今回樹立したオシメルチニブ耐性株では観察されなかった (data not
shown)。
T790M耐性変異を持たず、EGFRの活性型変異のみを持つ3つの肺癌細胞株 (PC-9, 11-18,
HCC827)とH1975 株のエルロチニブまたはアファチニブの感受性をIC50 で比較した。そ
の結果、H1975は他の3株と比較して50倍以上の耐性を示したが、これはT790M耐性変
異を有するためであると考えられる (Table 1)。 0
20 40 60 80 100 120
0.0001 0.01 1 erlotinib (μM) 0
50 100 150
0.0001 0.01 1 osimertinib (μM)
H1975 OR1 OR2
0 20 40 60 80 100 120
0.000001 0.001 1 afatinib (μM)
Cell viability(%)
Figure 4. Dose response curves of H1975, OR1 and OR2 to osimertinib, erlotinib and afatinib when continuously exposed to drugs for 3 days. Each value is the mean ±SD of triplicate dishes.
16 2. オシメルチニブ処理時の下流シグナル評価
続いて、樹立したオシメルチニブ耐性株の耐性機序を推察するため、受容体および下流シ グナル因子の発現量を細胞株間で比較した。H1975と比較してOR1とOR2では、EGFRと HER2、HER3、MET の発現量と、それらのリン酸化体の発現量が減少していた。また、下 流シグナル因子の中でもpAkt/Akt発現はOR1とOR2で上昇していたが、pSTAT3/STAT3は 低下していた (Fig. 5A)。
次に、各細胞株にオシメルチニブを 6時間処理したときのEGFRおよびEGFR下流シグ ナル因子の発現量を測定した。オシメルチニブはH1975と同様にOR1とOR2のEGFRお よびERKのリン酸化を阻害した。一方、Aktのリン酸化はH1975のみで阻害され、オシメ ルチニブ耐性株では維持されていた (Fig. 5B)。
以上の結果から、OR1とOR2のオシメルチニブ耐性機序にはAktシグナルの活性化の維 持が関与していることが示唆された。耐性株において、EGFRからERKへのシグナル伝達 は保持されているが、細胞の生存には影響しないと考えられる。
17
A
osimertinib (μM) 0
H1975
B
0.05 0.5 5 pEGFR (Y1058)
EGFR pAkt (T308) pAkt (S473) Akt pErk Erk GAPDH
0 OR1
0.05 0.5 5 0
OR2 0.05 0.5 5 pEGFR
EGFR pHER2 HER2 pHER3 HER3 pc-Met c-Met
H1975 OR1
OR2
pSTAT3 STAT3 pAkt (T308) pAkt (S473) Akt pPTEN PTEN pErk1/2 Erk1/2 GAPDH
H1975 OR1
OR2
Figure 5. (A) Western blot analysis of EGFR and other molecules in cell lysates of H1975, OR1 and OR2. (B) Expression levels of EGFR and other molecules are analyzed by western blot analysis when treated with various doses of osimertinib for 6 hr.
18
3. 種々の分子標的薬に対する感受性と生存シグナル評価
オシメルチニブ耐性株における生存シグナルを明らかにするため、種々の分子標的薬に 対する感受性を検討した (Table 2)。オシメルチニブ耐性株はMET/ALKやInsulin like growth factor 1 receptor (IGF-1R)、Platelet derived growth factor receptor (PDGFR) 、MEK1/2、JNK、 Serum glucocorticoid-induced protein kinase (SGK)の阻害剤に対してはH1975株と同程度の感 受性を示した。その一方で、オシメルチニブ耐性株はH1975株と比較して、SRC family kinase
(SFK)阻害剤のダサチニブに対して10から50倍の、同じくSFK阻害剤のPP1とサラカチ
ニブに対して3から6倍の高感受性を示した。また、AZD8055やLY294002などのPI3K/Akt
Drugs Targets IC50, μM1)
H1975 OR1 OR2
Crizotinib MET, ALK 1.82 (1) 1.38 (0.76) 1.20 (0.66)
PPP IGF-1R 0.65 (1) 0.54 (0.83) 0.74 (1.14)
Sorafenib RAF, VEGFR2
PDGFR 4.74 (1) 5.32 (1.12) 5.21 (1.10)
Dasatinib SRC, ABL, KIT,
EPH 0.23 (1) 0.0049 (0.021) 0.0207 (0.09)
PP1 SRC, LCK, FYN,
KIT, EGFR 9.60 (1) 1.88 (0.20) 3.07 (0.32)
Saracatinib SRC, LCK, FYN,
KIT, EGFR 3.65 (1) 0.55 (0.15) 0.53 (0.15)
AZD8055 mTORC1, C2 0.048 (1) 0.015 (0.31) 0.016 (0.33)
GSK650394 SGK1/2 65.24 (1) 20.59 (0.32) 53.78 (0.82)
LY294002 PI3K 21.35 (1) 9.79 (0.46) 17.10 (0.80)
AKT inhibitor Ⅳ AKT 0.19 (1) 0.21 (1.11) 0.28 (1.47)
U0126 MEK1/2 ND2) ND2) ND2)
SP600125 JNK1/2
Aurora A 22.30 (1) 51.76 (2.32) 26.83 (1.20) 1) IC50values (μM) are presented as average values of triplicate experiments. The relative drug sensitivity
for each drug is presented in parenthesis as the IC50 of OR1/2 normalized to the IC50of H1975 cells.
2) ND: not detectable, no cell toxicity is seen by the drug up to 30 μM
Table 2. Osimertinib resistant cells show collateral sensitivity to inhibitors of SFK
19
シグナル因子阻害剤に対しても、耐性株で感受性が亢進していた。従って、オシメルチニブ 耐性株ではSRCおよびAktシグナルが生存に関与していることが推察された。
そこで次に、H1975とオシメルチニブ耐性株において、ダサチニブによる細胞内シグナル 阻害効果について比較検討した。0.5および5 μMのダサチニブ処理により、オシメルチニ ブ耐性株選択的にAktのリン酸化が阻害された。このとき、EGFRやERKのリン酸化に変 化は見られなかった (Fig. 6)。
Dasatinib (nM) 0
H1975
0.05 0.5 5 0
OR1
0.05 0.5 5 0
OR2
0.05 0.5 5
pEGFR (Y1068) EGFR pSFK (Y416)
pAkt (T308) pAkt (S473) Akt pErk1/2 Erk1/2 GAPDH Src
Figure 6. Expression levels of EGFR and other molecules are analyzed when treated with various doses of dasatinib for 6 hr.
20
続いて、ダサチニブのオシメルチニブ耐性株に対する増殖阻害効果をin vivo で検証した
(Fig. 7)。腫瘍体積が100~200 mm3に達した後ダサチニブを経口投与したところ、ダサチニ
ブはOR1の腫瘍に対し、腫瘍の増大を抑制した。従って、オシメルチニブ耐性株はin vivo
とin vitro両系において、ダサチニブに随伴感受性を示すことがわかった。
0 100 200 300 400 500 600
0 5 10 15 20 25
Control Osimertinib Dasatinib
Tumor growth (mm3)
days
OR1
**
Figure 7. Animals bearing OR1 xenografts were treated with osimertinib and dasatinib. Results are shown as mean tumor volumes and SD at each time-point. Results are shown as mean tumor volumes and SEM at each time-point. **P<0.01, two-tailed Student t-test.
21
TPX-0005はSRC/FAK/JAK2の阻害剤であり、現在進行した固形腫瘍に対するphase1/2臨
床治験が行われている 42,43。SRC シグナルのオシメルチニブ耐性株への寄与を評価するた
め、TPX-0005の細胞毒性を比較したところ、オシメルチニブ耐性株はH1975株と比較して
TPX-0005に高い感受性を示した (Fig. 8A)。また、全株のSFKと FAKのリン酸化はTPX-
0005により同程度抑制された。その一方で、10 μMのTPX-0005処理は、オシメルチニブ耐 性株特異的にAktおよびERKのリン酸化を抑制した (Fig. 8B)。
0 20 40 60 80 100 120
0.001 0.01 0.1 1 10
H1975 OR1 OR2
TPX-0005 (µM)
Cell viability (%)
A B
TPX-0005(µM)
H1975 OR1 OR2
pSFK (Y416)
Erk1/2 pErk1/2 Akt SRC
pAkt (S473) FAK pFAK (Y397)
α-tubulin
0 0.1 1 10 0 0.1 1 10 0 0.1 1 10
Figure 8. (A) Dose response curves of three cell lines to TPX-0005 when treated for 3 days. Each value is the mean ±SD of triplicate dishes. (B) Expression levels of pSFK, pFAK, pAkt and pERK in H1975, OR1 and OR2 are analyzed in the presence of TPX-0005 for 6 hr.
22 4. SFKsの発現レベルと依存性評価
次に、オシメルチニブ耐性株とH1975株で、SFKsの発現量について評価した。マイクロ アレイを用い、10種類のSFKについてmRNA発現量を比較したところ、FGRとSRCの発 現量がオシメルチニブ耐性株で増加していた (Fig. 9A)。また、ウエスタンブロットでタン パク発現量を測定したところ、H1975 株と比較してオシメルチニブ耐性株では、SRC タン パクの発現レベルが上昇していたが、FYNやLYN、LCK、YES、CSKに関しては耐性株で の上昇は認められず、本結果はマイクロアレイ結果と一致した (Fig. 9B)。一方、FGRタン パクは検出することができなかった (data not shown)。
H1975 OR1 OR2
B
OR1 OR2
SFK genes ratio Z-score ratio Z-score
BLK N.D. N.D. N.D. N.D.
CSK 1.139 0.21 0.977 -0.062
FGR 3.896 1.04 7.183 1.584
FYN 0.824 -0.35 0.097 -3.516
HCK N.D. N.D. N.D. N.D.
LCK 0.019 -2.96 0.058 -3.495
LCK 0.018 -4.42 0.023 -4.593
LYN 0.795 -0.50 1.015 0.020
SRC 1.862 1.07 1.398 0.507
YES1 0.927 -0.15 1.265 0.426
LYN
CSK FYN
GAPDH SRC
YES LCK
A
Figure 9. (A) mRNA expression levels of 10 SFK genes are determined by Microarray analysis.
Relative fold changes (OR1 or OR2 vs H1975) are presented when expression level of each gene in H1975 is normalized as 1.0. N.D., not detected. (B) Protein expression levels of 6 SFK genes in three cell lines by Western blot analysis.
23
続いて、SRC のオシメルチニブ耐性株の増殖能への関与を検討するため、siRNA を用い てSRCの発現を抑制した。その結果、OR1とOR2の細胞増殖はsiSRC処理により約50 % に抑制された。一方、H1975に対するsiSRCによる抑制効果は10から20 %であった (Fig.
10A)。また、SRCの発現抑制はH1975と比較してOR1とOR2でAktおよびSFKのリン酸 化を顕著に抑制した (Fig. 10B)。
0 1 2 3 4
0 1 2 3 4
A
Cell number (x105)
H1975
0 1 2 3 4 5
OR2 OR1
siSRC#3 siSRC#1
sicontrol
pAKT(T308)
pERK1/2 ERK1/2 AKT SRC
GAPDH pSFK(Y416)
B
H1975pAKT(S473)
siSRC#3 siSRC#1
sicontrol siSRC#3 siSRC#1
sicontrol
OR1 OR2
*
**
**
**
**
**
Figure 10. (A) SRC knockdown induced cell growth inhibition at differential levels by H1975, OR1, and OR2 when treated with siRNAs for 4 days. Each bar shows the mean ± SD of triplicate wells,
*P<0.05, **P<0.01, two-tailed Student t-test. (B) Effects of SRC siRNA on activation of SFK and Akt are analyzed when treated with SRC siRNAs for 3 days. GAPDH served as loading control.
24 5. SRC活性化因子の探索① AXLの関連評価
SRCは非受容体型のチロシンキナーゼであり、SH2およびSH3ドメインを介した膜受容 体や糖タンパクとの相互作用を介して下流因子を活性化する44。オシメルチニブ耐性株にお いて、何らかのReceptor tyrosine kinase (RTK)もしくは糖タンパクによるSRCの活性化が耐 性誘導に関与することが考えられる。そこで、H1975を比較対象としてマイクロアレイ解析 すると、277因子がOR1 とOR2で共通して発現上昇していることがわかった。その中で、
RTKもしくは糖タンパクに分類される因子は、AXLとPDGFRβ、TGFβR3、CDCP1の4因 子であった。これらの因子のオシメルチニブ耐性株における発現量は、H1975の4から10 倍増加していた (Table 3)。
Fold change from H1975
OR1 OR2
Gene Symbol Description ratio1) Z-score ratio1) Z-score
AXL AXL receptor tyrosine kinase(AXL) 3.7 2.7 4.2 3.3
PDGFRB platelet derived growth factor receptor beta(PDGFRB) 7.2 2.2 8.3 2.6 TGFBR3 transforming growth factor beta receptor 3(TGFBR3) 9.9 2.5 8.1 2.6 CDCP1 CUB domain containing protein 1(CDCP1) 11.2 3.4 9.5 3.4 1) Relative fold changes of OR1 or OR2 vs H1975 are presented.
Table 3. mRNA expression levels of AXL, PDGFRβ TGFβR2 and CDCP1 in three cell lines are determined by microarray analysis.
25
これら 4つの候補分子の中、PDGFRβとTGFβR3 について阻害剤の増殖阻害活性を検討 した。文部科学省新学術領域研究「がん研究分野の特性等を踏まえた支援活動」化学療法基 盤支援活動事業の標準阻害剤キットを利用したところ、PDGFR キナーゼ阻害剤である AG1296とSU11652、PDGFR Kinase inhibitor Ⅳ、PDGFR Kinase inhibitor Ⅴ に対して、全て の細胞株で同程度の感受性を示した (data not shown)。この結果は、PDGFRとRAF、Vascular endothelial growth factor receptor 2 (VEGFR2)を標的とするソラフェニブと一致した (Table 2)。 一方、pan-TGFβR阻害剤であるSB431542とTGFβ1阻害剤であるTGFβR1 Kinase inhibitor Ⅱ への感受性も3株で差は認められなかった (data not shown)。以上のことから、オシメルチ ニブ耐性株で確認されたPDGFRβとTGFβR3の発現上昇は、オシメルチニブ耐性機序とは 無関係であると考えられた。
26
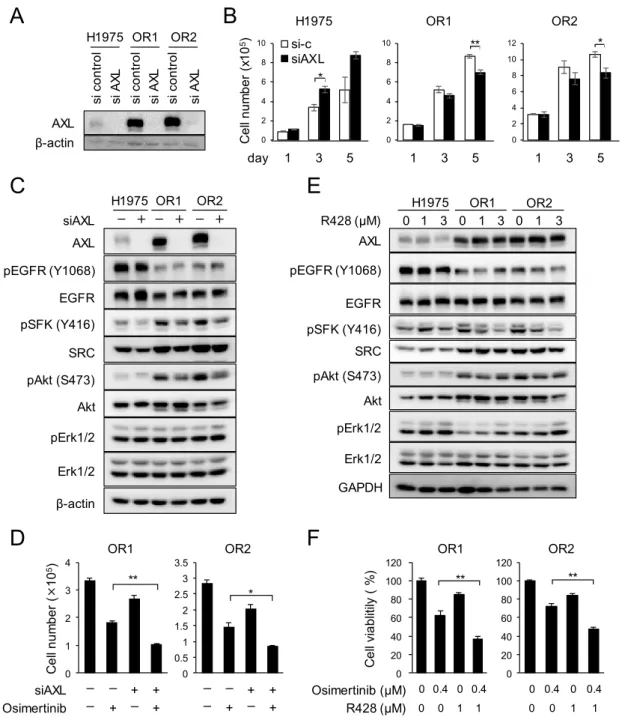
次に、AXLのオシメルチニブ耐性への関与を検討した。AXLはTYRO3/AXL/MER (TAM) 受容体の1つであり、AXLの活性化は肺がんのEGFR-TKI耐性を誘導することが既に報告 されている45。マイクロアレイの結果 (Table 3) と一致して、本研究で樹立したオシメルチ ニブ耐性株はタンパクレベルでも、H1975株と比較して非常に高いAXL発現を示した (Fig.
11A)。そこで、siRNAを用いてAXL発現を抑制したところ、OR1とOR2では細胞増殖が 約20 %抑制され、H1975では抑制効果は見られなかった (Fig. 11B)。また、OR1とOR2で はAXLの発現抑制により、AktとともにSFKのリン酸化が一部抑制された (Fig. 11C) 。さ
らに、AXLのsiRNAとオシメルチニブを併用処理すると、オシメルチニブ耐性株のオシメ
ルチニブ感受性が亢進した (Fig. 11D) 。
AXLの阻害剤であるR428でも同様に、OR1とOR2におけるSFKのリン酸化は阻害され
るが、H1975では阻害されなかった。Aktのリン酸化に関しても、わずかではあるがOR1と
OR2でのみ阻害された (Fig. 11E)。また、siRNAでの検討と同様に、R428の併用によりOR1 とOR2に対するオシメルチニブ感受性が亢進した (Fig. 11F)。以上の結果から、AXLはSFK を介してAktを活性化し、一部オシメルチニブ獲得耐性へ関与することを示した。
27
C
0 2 4 6 8 10
H1975 si-c siAXL
0 2 4 6 8 10
OR1
0 2 4 6 8 10 12
B
OR2*
** *
sicontrol siAXL
H1975 OR1 OR2
sicontrol siAXL sicontrol siAXL
A
Osimertinib (µM) R428 (µM)
0 0
0.4 0.4 0
0 1 1
0 0
0.4 0.4 0
0
1 1
Cell viablitily( %)
D
0 0.5 1 1.5 2 2.5 3 3.5
0 1 2 3 4
**
*
siAXL Osimertinib
+ + +
- +
OR1 OR2
Cell number (×105)
F
0 20 40 60 80 100 120
**
0 20 40 60 80 100 120
**
OR1 OR2
EGFR pEGFR (Y1068)
β-actin pSFK (Y416)
pAkt (S473)
Erk1/2 pErk1/2 SRC AXL
Akt siAXL
H1975
- + OR1
- + OR2
- + AXL
β-actin 5Cell number (x10)
day 1 3 5 1 3 5 1 3 5
- -
-
+ + +
- +
- -
-
3 1 0
H1975
3 1 0
OR1
3 1 0
OR2 R428 (µM)
EGFR pEGFR (Y1068)
GAPDH pSFK (Y416)
pAkt (S473)
Erk1/2 pErk1/2 SRC AXL
Akt
E
Figure 11. (A) AXL protein expression levels in three cell lines treated with control or AXL siRNA for 3 days. (B) Effects of AXL knockdown on cell growth of three cell lines. (C) Effects of AXL knockdown on expression of pSFK, pAkt and pERK in the three cell lines. Cell were treated with an AXL siRNA for 3 days and followed by Western blot analysis. (D) Combination of osimertinib and AXL siRNA on cell growth of OR1 and OR2. Cells are exposed to osimertinib at 0.1μM and/or AXL siRNA for 3 days. (E) Expression levels of pSFK, pAkt and pERK are analyzed when treated with various doses of R428 for 2 days. (F) Sensitivity to osimertinib with or without R428. Cells were exposed for 3days and subjected to a WST assay. Each value is the mean ±SD of triplicate dishes.
*P<0.05, **P<0.01, two-tailed Student t test.
28 6. SRC活性化因子の探索① CDCP1の関連評価
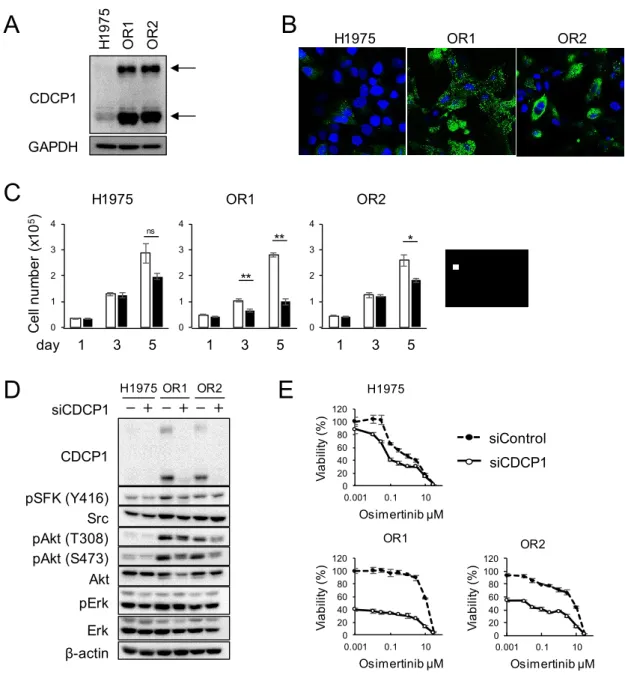
続いて、CDCP1がオシメルチニブ耐性に関与するか検討した。CDCP1はプロテアーゼの 一種であるプラスミンにより断片化された後、ヘテロまたはホモダイマーを形成すること で、がん細胞の転移や浸潤に関与する46,47。また、CDCP1はSFKを介して、RAF/MEK/ERK
や PI3K/Akt とは独立した経路で薬剤耐性に関与することも報告されている 48。そこで
CDCP1の発現について検討した。マイクロアレイの結果 (Table 3)と同様に、CDCP1の全長
(140 kDa)と断片化体 (80 kDa)の発現量はH1975と比較してOR1とOR2で顕著に増加して
いた (Fig. 12A)。蛍光免疫染色によりCDCP1の発現と局在を確認すると、CDCP1はOR1と
OR2の細胞質と形質膜上で発現が上昇していることがわかった (Fig. 12B)。そこで、CDCP1 の発現上昇がオシメルチニブ耐性に関与するか検討した。siRNA による発現抑制を行った 結果、OR1とOR2に対して30から70%の増殖抑制効果を示したが、H1975に対して有意 な効果は観察されなかった (Fig. 12C)。siCDCP1処理はオシメルチニブ耐性株のSFKとAkt のリン酸化を抑制した (Fig. 12D)。さらに、CDCP1 の発現抑制とオシメルチニブとの併用 効果について検討した結果、OR1とOR2の細胞生存率はCDCP1の発現抑制により顕著に 抑制されたが、オシメルチニブを併用した際の相乗効果は観察されなかった (Fig. 12E)。
29
A
**
**
0 1 2 3 4
H1975
B
D
0 1 2 3 4
OR1
0 1 2 3 4
OR2
E
0 20 40 60 80 100 120
0.001 0.1 10
Viability (%)
Osimertinib μM H1975
0 20 40 60 80 100 120
0.001 0.1 10
Viability (%)
Osimertinib μM OR1
0 20 40 60 80 100 120
0.001 0.1 10
Viability (%)
Osimertinib μM OR2 CDCP1
pSFK (Y416) siCDCP1
Src
Akt
Erk β-actin pAkt (S473) pAkt (T308)
pErk
H1975 OR1 OR2
- + - + - +
siControl siCDCP1
C
H1975 OR1 OR2
H1975
CDCP1
OR1 OR2
GAPDH
Cell number (x105)
day 1 3 5 1 3 5 1 3 5
ns *
Figure 12. (A) Expression levels of CDCP1 protein in three cell lines by western blot analysis. Arrows indicate two forms (140kDa and 80 kDa) of CDCP1 protein. (B) Immunofluorescence analysis of CDCP1 in H1975, OR1 and OR2 cell. (C) Effects of CDCP1 knockdown on cell growth of three cell lines. Each value is the mean ± SD of triplicate dishes. *P<0.05, **P<0.01, two-tailed Student t-test.
(D) Effects of CDCP1 knockdown on activation of SFK (pSFK) and Akt (pAkt) by Western blot analysis when treated with siRNA for 4 days. (E) Effects of combination of osimertinib and CDCP1 knockdown on cell growth in three cell lines. Cells were exposed to various doses of osimertinib with or without CDCP1 siRNA for 5 days, and followed by WST assays. Each value is the mean ±SD of triplicate dishes.
30
CDCP1はSRCシグナルの伝達における、足場タンパクとしての役割を持つ49,50。そこで、
オシメルチニブ耐性における CDCP1 と SRC の相互作用を共免疫沈降法で評価した。OR1
とOR2では、抗CDCP1抗体を用いた免疫沈降によりSRCとの相互作用が観察された (Fig.
13A)。また、抗SRC抗体による免疫沈降においてもCDCP1との相互作用が観察された(Fig.
13B)。以上の結果から、CDCP1はSRCとの相互作用を介してAktを活性化し、オシメルチ ニブ獲得耐性に関与していることが示唆された。
A
H1975 OR1 OR2
Input IP: IgG IP: CDCP1
CDCP1 SRC
H1975 OR1 OR2 H1975 OR1 OR2
B
H1975 OR1 OR2
Input IP: IgG IP: SRC
CDCP1
H1975 OR1 OR2 H1975 OR1 OR2
N.S.
SRC
Figure 13. Co-immunoprecipitation assays to determine interaction of SRC with CDCP1. SRC was detected after immunoprecipitation with anti-CDCP1 antibody (A), and CDCP1 was detected after immunoprecipitation with anti-SRC antibody (B).
31
7. オシメルチニブに耐性となった患者の腫瘍内AXLおよびCDCP1発現評価
最後に、オシメルチニブへ耐性を示したNSCLC患者の腫瘍において、AXLおよびCDCP1 の発現レベルの上昇が観察されるか否か、検証した。久留米大学病院病理部河原明彦先生と の共同研究より、EGFR活性型変異陽性かつ T790M耐性変異陽性である NSCLC患者のオ シメルチニブ治療前と耐性出現後の腫瘍サンプルについて、AXL および CDCP1 の免疫染 色を試みた。オシメルチニブ治療前では、AXLおよびCDCP1の発現はほとんど観察されな かった。一方、再発腫瘍内では、AXLとCDCP1双方の発現レベルががん細胞の細胞膜付近 で顕著に上昇していた (Fig. 14)。
Pre-osimertinib
tumor Refractory
tumor
CDCP1
AXL
Figure 14. Immunohistochemically analysis of AXL or CDCP1 expression in patients' refractory to osimertinib treatment.
32
1-5. 考察
肺癌におけるオシメルチニブ耐性は遺伝子変異や発現量変化など、多様なメカニズムに よって誘導されることが報告されている17-27。本研究では、H1975株より樹立したオシメル チニブ耐性株が以下の新しい性質を獲得していることを明らかにした。オシメルチニブ耐 性株は、① EGFR の発現が低下しており、オシメルチニブ処理条件下においてもAktの活 性化が維持されていた (Fig. 5)。② in vitroとin vivoの両方において、ダサチニブやPP1、サ ラカニチニブなどのSFK阻害剤に対して高い感受性を示した (Fig. 7 and Table 2)。③ SRC の発現抑制により細胞増殖と Akt の活性化が抑制された (Fig. 10)。これはダサチニブや TPX-0005のSFK標的阻害剤でも同様の結果を示した (Fig. 6 and 8) 。④ AXLとCDCP1の 発現量が著に増加しており、これら因子の発現抑制は耐性株の細胞増殖を有意に抑制した
(Fig. 11 and 12) 。以上のことから、本研究ではオシメルチニブ耐性にAXLとCDCP1の発
現レベルの上昇とSRCの活性化によるEGFR非依存的なAkt活性化が新たに関与すること を明らかにした。
AktシグナルはFOXO1やGSK3β、mTORC1を介して、細胞の生存や細胞周期、タンパ ク質合成を制御する、細胞の生存に重要な経路である。肺癌や乳癌、胃癌、グリオーマなど 様々ながん種に対して、Aktシグナル阻害剤が腫瘍退縮効果を示し、抗がん治療標的となり 得る可能性が示されている 51。薬剤耐性細胞株において Akt シグナルへの生存の依存性が 亢進することは多数報告されている 38,39,41,52–54。すなわち、Akt シグナル経路は、がん細胞 が薬剤耐性獲得に利用するシグナル因子の1つと考えられる。
オシメルチニブを含むEGFR-TKI耐性細胞では、EGFRや他のRTKのタンパク発現が消 失または低下した際にSFK遺伝子が活性化し、バイパスシグナルとして働くことが報告さ
れている 11,16。また、無作為な遺伝子の発現亢進による ORF based screening では、活性型
EGFR変異を持つ肺癌細胞のEGFR への依存性に影響を与える因子として、18種類のキナ
33
ーゼもしくはキナーゼ関連因子を同定している55。興味深いことに、それら18種類中7種 類がSFKであった。
さらに、EGFR-TKI耐性肺癌細胞において、EGFRに非依存的な細胞の生存と増殖にSRC
の活性化は重要な役割を担っており、EGFRのT790M 耐性変異に対して、アファチニブと ダサチニブの併用処理は効果を示すことが報告されている56,57。さらに、質量分析を利用し た定量的プロテオミクス解析を用いた、オシメルチニブ耐性誘導による細胞内分子の変動 を観察した報告においても、ダサチニブとオシメルチニブの併用処理が耐性を克服しうる 可能性が示唆されている 58。本研究室においても EGFR-TKI への耐性を獲得したがん細胞 株はSRCの活性化に生存を依存することを示した39,41 (Table 1 and 2, Fig. 6)。以上より、SFK 阻害剤の併用は、オシメルチニブ耐性を克服しうる有用な治療方法となる可能性がある。
本研究では、EGFRの発現が著しく低下した場合、SRC経路が生存シグナルの代替経路と して活性化する可能性を示した。このSRC活性化の要因の1つとして、AXL因子の活性化 が挙げられる。AXLはTAM受容体チロシンキナーゼの1つであり、EGFR活性型変異陽性 の肺癌細胞株において、しばしば発現上昇が認められている42,59。また、AXLの発現抑制も しくは阻害剤の投与がin vivoとin vitroの両系において、オシメルチニブ耐性を克服可能で あったことも示されている60。このことから、オシメルチニブ処理とAXL分子との間に何 らかの関連が推察される。本検討で用いたオシメルチニブ耐性株では、親株と比較してAXL の発現量が増加していた (Fig. 13)。さらに、AXLの発現抑制および阻害剤R428の処理は、
オシメルチニブ耐性株特異的に細胞増殖およびAktとSFKのリン酸化を抑制したことから
(Fig. 11)、AXL は耐性誘導に関連するSRC/Akt シグナルの活性化に関与する可能性が示さ
れた。
34
一方、AXLとオシメルチニブ耐性に関する報告として、STC2発現上昇によるAXL発現 の誘導がある61。しかし、本研究のオシメルチニブ耐性株では、マイクロアレイ解析の結果 からSTC2発現の上昇は認められておらず、この経路の耐性誘導への関与は低いと考えられ る (data not shown)。
以上より、AXLの発現上昇はがん細胞がEGFR-TKIから免れる代替シグナルとしての役 割を持つことが考えられる。AXL 標的型薬剤の併用処理は、少なくとも部分的には、オシ メルチニブ耐性を克服できる可能性がある。
本研究ではCDCP1についてもオシメルチニブ耐性株で親株と比較して発現量が増加して
いた (Fig. 13)。また、肺癌患者組織においてもオシメルチニブ治療後の再発腫瘍でCDCP1
の発現増加が観察された (Fig. 14)。CDCP1が高発現している肺癌患者は、低発現の患者と 比較して予後不良であることが知られている 62。また、CDCP1 は足場非依存的な増殖にも 関与しており63、腫瘍の悪性化に必要であることが示唆されている。これまでにCDCP1の 強制発現によって、がん細胞のSFKとAktリン酸化が誘導されることが確認されているこ
とから50,64,65、CDCP1/SFK/Akt経路と腫瘍の悪性進展には密接な関連があることが示唆され
ている。本研究でもCDCP1の発現抑制は、オシメルチニブ耐性株の細胞増殖ならびにSFK およびAktのリン酸化を抑制した。さらに、オシメルチニブ耐性株を用いた共免疫沈降法に
よりCDCP1 と SRCの相互作用が確認された。以上の結果から、オシメルチニブ耐性株に
おいては、CDCP1の発現上昇と相関してSRCの活性化が誘導された結果、その下流シグナ ルであるAktがリン酸化され生存シグナルを伝達していることが推察される。
35
36
1-6. 小括
本研究では、オシメルチニブ耐性獲得メカニズムとして、EGFRシグナルが消失する代わ りにSRC シグナルが活性化すること、そして SRCの活性化は Aktシグナル経路へと繋が り、耐性株の生存に寄与していることを明らかにした。さらに、CDCP1およびAXLの発現 上昇は SRC の活性化と関連したことから、CDCP1-AXL/SRC/Akt 経路がオシメルチニブ耐 性へ関与することを示した (Fig. 15)。しかしながら、オシメルチニブ耐性誘導により発現上
昇した CDCP1 と AXL がどのように SRC を活性化しているか、また、どのような機序で
CDCP1やAXLの発現が亢進するのかについては、さらなる検討が必要である。
以上より本研究では、オシメルチニブ獲得耐性メカニズムとしてCDCP1-AXL/SRC/Akt代 替経路の活性化を示すとともに、耐性克服治療法としてオシメルチニブとCDCP1もしくは AXL、SRC阻害剤の併用投与が有用である可能性を提示した。
37
AKT EGFR L858R/T790M
cell growth/survival
Osimertinib treatment HER2
HER3
ERK
EGFRm T790M
A
EGFR L858R/T790M
Inhibition of cell growth/survival by
osimertinib HER2
HER3
osimertinib
AKT ERK
ERK AKT
EGFR
L858R/T790M CDCP1 AXL
Cell growth/survival SFK
Osimertinib
treatment ERK AKT
EGFR
L858R/T790M CDCP1 AXL
Cell growth/survival osimertinib SFK
B
In sensitive cells
In resistant cells
Figure 15. Hypothetical model how osimertinib resistance is acquired. (A) In osimertinib sensitive cells, PI3K/Akt pathway driven by EGFR harboring EGFRm and T790M is highly susceptible to osimertinib, resulting in cell growth/survival suppression. (B) By contrast, in osimertinib resistant cells, expression of the driver EGFR is reduced, accompanied by SRC activation together with enhanced expression of AXL and CDCP1. Enhanced expression of AXL activates SFK and Akt signaling pathway, resulting in promotion of cell growth and survival. Further enhanced expression of CDCP1 also activates Akt pathway in collaboration with SFK, promoting cell growth and survival.
Compensatory activation of cell growth/survival signaling pathway by SFK and/or AXL or CDCP1 could be targetable for overcoming therapeutics of osimertinib resistance.