著者
神澤 恒毅
内容記述
学位授与大学: Osaka Prefecture University(大阪
府立大学), 学位の種類: 博士(理学), 学位記番号:
論理第93号, 学位授与年月日: 2010-09-30, 指導教
員: 古田雅一.
博士学位論文
ニトロニルニトロキシド系安定有機
ラジカル結晶の構造と磁性に関する
研究
大阪府立大学大学院
理学系研究科物理科学専攻
神澤 恒毅
序論
目 次
1. 序論 ... - 3 - 1.1. 有機磁性体とは ... - 3 - 1.2. ニトロニルニトロキシドの構造と磁性 ... - 4 - 1.3. 有機磁性による量子スピン系の研究 ... - 8 - 1.4. 本論文の目的と内容 ... - 12 - 1.5. 引用文献 ... - 13 - 2. 実験 ... - 15 - 2.1. 物性測定 ... - 15 - X 線構造解析... - 15 - SQUID 磁束計を用いた静磁場中での磁化測定 ... - 16 - X-band EPR(電子スピン共鳴) ... - 18 - 強磁場 EPR 測定 ... - 19 - 2.2. 試料作製 ... - 20 - フッ素置換フェニルニトロニルニトロキシド ... - 20 - 2,3-dimethyl-2,3-dinitrobutane の合成 ... - 21 - 2,3-bis(hydroxyamino)-2,3-dimethylbutane の合成 ... - 21 - 2-(2,3,5-trifluorophenyl)-1,3-dihydroxy-4,4,5,5-tetramethylimidazollidine... - 22 - 2-(2,3,5,6-tetrafluorophenyl)-1,3-dihydroxy-4,4,5,5-tetramethylimidazollidine ... - 22 - 2-(2,3,4,5-tetrafluorophenyl)-1,3-dihydroxy-4,4,5,5-tetramethylimidazollidine ... - 22 - 2-(2,3,6-trifluorophenyl)-1,3-dihydroxy-4,4,5,5-tetramethylimidazollidine... - 23 - 2-(2,3,5-trifluorophenyl)-4,4,5,5-tetramethylimidazollidine-3-oxide-1-oxyl(2,3,5-F3P NN) ... - 23 - 2-(2,3,5,6-tetrafluorophenyl)-4,4,5,5-tetramethylimidazollidine-3-oxide-1-oxyl(2,3,5, 6-F4PNN) ... - 24 - 2-(2,3,4,5-tetrafluorophenyl)-4,4,5,5-tetramethylimidazollidine-3-oxide-1-oxyl(2,3,4, 5-F4PNN) ... - 24 - 2-(2,3,6-trifluorophenyl)-4,4,5,5-tetramethylimidazollidine-3-oxide-1-oxyl(2,3,6-F3P NN) ... - 24 - BIPNNBNO ... - 26 - Nitroso-t-butane ... - 27 - 3. フッ素置換フェニルニトロニルニトロキシドの構造と磁性 ... - 35 - 3.1. 概要 ... - 35 - 3.2. 2,3,5-F4PNN および 2,3,5,6-F4PNN ... - 38 -結晶構造 ... - 38 - 静磁場中での磁化測定 ... - 48 - 2,3,5-F3PNN および 2,3,5,6-F4PNN のまとめ... - 57 - 3.3. -2,3,6-F3PNN ... - 58 - 結晶構造 ... - 58 - 磁化測定 ... - 64 - -2,3,6-F3PNN のまとめ ... - 70 - 3.4. 2,3,4,5-F4PNN ... - 71 - 結晶構造 ... - 71 - 2,3,4,5-F4PNN のまとめ ... - 81 - 3.5. -2,3,6-F3PNN ... - 82 - 結晶構造 ... - 82 - 磁化測定 ... - 89 - -2,3,6-F3PNN のまとめ ... - 99 - 3.6. 分子間接触と磁気相互作用の考察 ... - 101 - 3.7. 結論 ... - 106 - 3.8. 引用文献 ... - 110 - 4. S = 1 と S = 1/2 を分子内に含む混合スピン系 BIPNNBNO の構造と磁性 ... 112 -4.1. 研究背景 ... - 112 - 4.2. 概要 ... - 113 - 4.3. 結果と考察 ... - 114 - BIPNNBNO の分子内磁気相互作用の見積もり ...- 114 - BIPNNBNO の結晶構造 ...- 116 - 磁性モデルについて ...- 119 - BIPNNBNO の静磁場中での磁化測定 ...- 121 - X-band EPR(電子スピン共鳴)の測定 ...- 123 - 強磁場 EPR の測定 ...- 126 - 4.4. 結論 ... - 147 - 引用文献 ...- 148 - 5. まとめ ... - 149 - 謝辞 ... 152
1. 序論
1.1. 有機磁性体とは
近年、分子で構成された化合物、特に C, H, N, O などの軽い典型元素のみからなる有機 化合物の結晶において磁気相互作用を発現する物質が合成され、その研究活動が盛んに行 われている。磁性を担う不対電子を持つ有機化合物は有機ラジカルと呼ばれる。一般に有 機ラジカルは反応性に富み、反応中間体としてのみ存在することも多い。室温大気中で安 定に存在し得る有機ラジカルの合成研究は 1960 年代に始まった。多くはスピンラベル試薬 の開発を目的とした溶液状態の研究であった。安定有機ラジカルの固体状態の研究は 1960 年代後半から少しずつ行われるようになった。化学結合の概念からすればラジカル分子間 には互いのスピンを反平行に揃える反強磁性相互作用が働くと予想される。実際に多くは 反強磁性相互作用を示した[1]。しかし、当時分子間に強磁性相互作用を示す物質が二例報 告され、その磁性発現機構に興味がもたれた[2]。そのうちの 1 つであるガルビノキシルの 強磁性的分子間相互作用について 1980 年代に分子論的考察が行われた[3]。ニトロニルニト ロキシドは 1970 年代初めにスピンラベル試薬として開発された安定ラジカル骨格である が[4]、この固体状態の磁性研究が 1980 年代後半から行われ、1991 年には p-NPNN の 相結 晶で三次元的強磁性磁気秩序が観測された[5]。これは純粋な有機結晶による最初の強磁性 体であり、世界に先駆け日本で発見された。これを契機として有機磁性体の研究が盛んに 行われるようになった。今や純粋有機ラジカルによる単純強磁性体は数十種類を越えた。 また、分子内に S = 1 と S = 1/2 をあわせもつ有機トリラジカル PNNBNO において有機フ ェリ磁性体も発見された[6]。フェリ磁性体は大きさの異なる 2 種類のスピン種を互いに反 平行に揃えることで自発磁化を得るものである。軽元素から構成される有機磁性体は、磁 気異方性の小さい理想的なハイゼンベルグスピン系を形成する。このようなスピン系では スピンのベクトル描像が成り立たずスピンの量子揺らぎの効果が顕著にあらわれる。有機 化合物から成るフェリ磁性体は量子効果の観点から、そのスピン状態に興味がもたれてい C H O O Galvinoxyl N N O O O2N p-NPNN N N O O N N O O PNNBNOる。従来の遷移金属化合物から構成される磁性体は少なからず磁気異方性の影響を受けて いる。有機磁性体はスピンの量子効果を研究する格好の素材であり、分子設計による様々 なスピン空間構造の構築とその物性研究による量子効果の解明が期待されている。
1.2. ニトロニルニトロキシドの構造と磁性
幾つか知られる安定ラジカル骨格の中でニトロニルニトロキシド(NN)誘導体は最も広 く結晶構造と磁性が調べられている。この節では NN の結晶構造と磁性の報告例を紹介す る。 p-CF3PNN における分子間反強磁性相互作用 [7] まず、分子間反強磁性相互作用を持つ NN 類縁化合物の例として p-CF3PNN について示 す。p-CF3PNN の結晶構造は、晶系 Triclinic、空間群 Pī であり、a 軸方向に NO 基同士の接 近(N···O 3.70 Å, 3.76 Å)による交互一次元鎖を形成している。その磁化率の温度依存性を Fig. 1(a)に示す。磁化率の温度依存性から、反強磁性相互作用が働き温度低下に伴い磁化率 はゼロに向かって減少していることが分かる。これは 2J/kB = 20.8 K, = 0.1 の交互一次元 鎖モデルによって磁化率挙動が再現されている。また、40 T までの磁場で測定した磁化曲 線を Fig. 1(b)に示す。実線が = 0, 0.2, 0.4, 1 のときの絶対零度における計算結果である。 測定点は = 0 と 0.2 の間に入り、磁化率の解析結果と対応していることが分かる。結晶構 造と磁気の結果を説明できる磁性モデルとの対応から、p-CF3PNN 結晶が示す分子間反強 磁性相互作用は、NO 基同士の接近によってと対応づけられる。 (a) (b)Fig. 1. (a) p-CF
3PNN の磁化率の温度依存性 (b) p-CF
3PNN の磁化曲線
p-FPNN における分子間強磁性相互作用[8]
NN 系化合物で分子間強磁性相互作用を示す一つとして p-FPNN について紹介する。
p-FPNN の磁化率と温度の積(pT)の温度依存性を Fig. 2(a)示す。室温ではpT の値は 0.375
emu K mol1であり、S = 1/2 が 1 mol 存在するときの Curie 定数に等しい。温度低下ととも に値は上昇し、強磁性相互作用が働いていることが分かる。2 K の磁化曲線は 2 つの S = 1/2 が強磁性的に結合して S = 1 が形成されることを示した。S = 1 が 1/2 mol 形成されるとpT 値の値は 0.5 emu K mol1をとると予想され、実験は 4 K 以下でこの値を越えている。この ことは S = 1 種の間にも弱い強磁性相互作用が働くことを示している。pT の温度依存性を 強磁性的ダイマー形成とダイマー間に弱い強磁性相互作用が働くモデルで解析し、ダイマ ー内強磁性相互作用 2J/kB = +10.0 K、ダイマー間強磁性相互作用 2J’/kB = +0.04 K と見積も
られた。Fig. 2(a)の実線は、このモデルによる計算値である。結晶構造は晶系 tetragonal、
空間群 I41で、NO 基とフェニル基の接近(O···C 3.12 Å)による二量体構造が特徴的である。 二量体間には NO 基の酸素原子と NN 基(O-N-C-N-O)中心炭素間に接近(O···C 3.54 Å)がみら れる。結晶構造の模式図を Fig. 2(b)に示す。丸は二量体を棒は二量体間の相互作用を表す。 結晶構造と磁性モデルの対応から 2J/kB = 10 K, 0.04 K の強磁性相互作用はそれぞれ NO 基 とフェニル基の接近および NO 基と O-N-C-N-O の C 原子との接近に帰属される。 磁気相互作用発現の機構の分子論的考察 NN ラジカルが示す磁気相互作用発現には、分子間でのラジカル部位の相対配置が重要 な役割を果たしていることが上記の例から分かる。フェニルニトロニルニトロキシド
Fig. 2. (a) p-FPNN の
pT の温度依存性 (b) 結晶構造模式図
(a) (b)(PNN)について中性子回折実験によって得られたスピ ン密度分布を Fig. 3 に示す[9]。実線で表されるのが正 のスピン密度、破線で表されるのが負のスピン密度で、 分子内において正と負のスピン密度が交互に配列して い る の が 分 か る ( ス ピ ン 分 極 ) 。 特 に NN 部 位 (O-N-C-N-O)にスピン密度が集中しており、酸素およ び窒素原子上に正の大きなスピン密度((O) = (N) = 0.27)が、中心の炭素原子には負のスピン密度((C) = 0.11)が分布している。スピン密度が分子全体に分布 していることが有機磁性体の特徴であり、磁気相互作用と結晶構造の対応を複雑なものに している。 ラジカル分子間の反強磁性相互作用は化学結合の概念から容易に理解できるが、強磁性 相互作用発現の機構として、1960 年代に McConnell は 2 つの条件を提案した。 原理 1: 隣接分子間での正のスピン密度と負のスピン密度の相対[11] 原理 2: 三重項電荷移動励起状態との共鳴[12] まず、原理 1 は正のスピン密度同士、あるいは負のスピン密度同士の接近は反強磁性相互 作用を、正のスピン密度と負のスピン密度の接近は強磁性相互作用が発現するというもの である。NO 基同士の接近、NO 基と O-N-C-N-O の中心 C 原子の接近がそれぞれ反強磁性 および強磁性相互作用を発現することは、この原理で理解される。一方、原理 2 は Fig. 4 で表すラジカル二量体中の電荷移動励起配置の図で説明される。このときの電荷移動は分 子 A から B へ起きるものとし、それぞれのラジカル分子の SOMO と NHOMO のみが寄与 することを仮定する。この場合、電子配置 としては図に示す 3 種類が存在する。T1は 三重項状態で、他の 2 つは一重項状態であ る。分子 A の NHOMO のスピン電子が分子 B の SOMO に移動して出来る T1は、共鳴 によって強磁性的な三重項基底状態を約 t2/U だけ安定化させる。U は T1の配置エ ネルギーであり、t は T1をもたらすトラン ス フ ァ ー 積 分 で 、 こ の 場 合 分 子 A の NHOMO と分子 B の SOMO の重なり積分
Fig. 3. PNN のスピン密度分布
Fig. 4. 様々な電荷移動励起配置
[10]にほぼ比例する。従って、U を小さくする意味での T1の安定化と、t を大きくする NHOMO と SOMO の間の大きな重なりは強磁性相互作用の獲得に有利に働く。しかし、T1, S0, S1の うち最もエネルギーが低いのは、SOMOSOMO の分子間相互作用から得られる S0である。 通常の電荷移動相互作用で考えられる配置は S0のみであり、これは分子間に常に反強磁性 相互作用をもたらす。大半の有機ラジカルが反強磁性相互作用を示すのもこのためだと考 えられる。従って、強磁性的分子間力を獲得するための第 1 条件は、S0を与えるトランス ファー積分を小さくすること、つまり SOMOSOMO が直交する、あるいはそれに極めて 近い分子間配置の獲得ということになる。S1は NHOMOSOMO の相互作用から得られる 一重項電子配置で T1と等しいトランスファー積分を持つ。S1のエネルギーは、一重項状態 内の電子間反発が対応する三重項状態内のそれに比べて大きいことから、T1より常にエネ ルギーは高いが、T1によってもたらされる強磁性的状態の安定化は、残念なことに S1によ ってかなり相殺される。この点も強磁性的な有機ラジカル分子が少ない原因である。 以上の McConnell の原理 2 から強磁性発現のための条件は 2 つある。 (a) S0, S1の不安定化と T1の安定化
(b) SOMOSOMO の直交と SOMONHOMO の大きな重なり(SOMOSOMO の重なり は反強磁性相互作用をもたらす) 分子軌道計算により、フェニルニトロニルニトロキシドの SOMO は NO 基上に分布し、 NHOMO、NLUMO はフェニル基上に分布することが分かっている。p-FPNN のダイマー形 成においてみられた NO 基とフェニル基を接近させた分子間配置では SOMOSOMO の重 なりが無視できる。一方で SOMONHOMO、SOMONLUMO の重なりをもつので、強磁 性相互作用を発現すると理解される。 上述した p-FPNN と p-CF3PNN で観測された強磁性および反強磁性相互作用をもたらす
Fig. 5. ニトロニルニトロキシドにおける分子間配置と磁気相互作用の関係
(a) NO 基とフェニル基との接近 (b) NO 基と O-N-C-N-O の NN 中心炭素原子との接近 (c) NO 基同士の接近 反強磁性相互作用 強磁性相互作用 (a) (b) (c)分子配置を Fig. 5 にまとめる。これらの磁気相互作用発現は定性的には McConnell の提案 で理解できる。定量的な理解は、特に強磁性相互作用については困難であり、理論的な研 究が行われているものの、磁気力は遠達力であり、観測される磁気相互作用もあまり大き くないので実験が先行する形で進められている。分子内スピン密度分布は隣接原子間で正 負が交互に表れるので、磁気相互作用は分子パッキングのずれに敏感に変化する。特に NO 基と O-N-C-N-O の中心 C 原子の接近は、分子間相対配置のずれに敏感である。ピリミジ ニルニトロニルニトロキシドでは 2.92 Å の接近に対して 2J/kB = 18.2 K の相互作用が報告 されているが p-FPNN のダイマー間相互作用が非常に小さい。磁気作用を定量的に理解す るためには系統的な実験データの蓄積が必要である。しかし、NN の置換基を変えると結 晶構造はしばしば大きく変化し、これまで系統的な研究はほとんどなされていない。
1.3. 有機磁性による量子スピン系の研究
軽元素のみから構成される有機ラジカルは磁気異方性の小さい理想的なハイゼンベルグ スピン系を形成し、スピンの量子ゆらぎの効果が顕著に現れる系である。この節では有機 ラジカルを用いた量子スピン系の報告例を示す。 カチオンラジカル塩の静電引力を利用した S = 1 のカゴメ格子カチオンラジカル m-N-alkylpyridinium -nitronyl nitroxide(m-AlkylPYNN)と BF4 、Iな どの対アニオンからなる塩は、カゴメ格子反強磁性体とみなすことができる初めての有機 物質である。カゴメ格子は三角形が頂点を共有する「籠目」状構造を指し、各頂点に存在 するスピン間に反強磁性相互作用が働くとき、隣接スピン間の相互作用は競合し、幾何学 的にスピンフラストレーションを生じる。このような磁性体においては強い量子揺らぎの 効果によって、量子スピン液体状態という特異な状態をとることが理論的に予測されてい る。m-AlkylPYNN塩の結晶構造は、 NO ラジカルとカチオン部位であ るピリジン環の接近によって強磁 性的二量体(S = 1)が形成される。 二量体間にも NO 基同士の接近に よる反強磁性相互作用が存在する。 m-AlkylPYNN塩の磁気相互作用
Fig. 6. (a) 分子配置を簡略化したモデル図(○
が一つのラジカル分子) (b) ダイマーを辺の
中点に置くと、S = 1 のカゴメ格子を形成
(a) (b) orを考慮した分子配置のモデルを Fig. 6 に示す。二量体内には強磁性相互作用 J1により、ラ
ジカルのペアが低温で S = 1 を形成する。さらに二量体間には弱い反強磁性相互作用 J2が
働き、この系は全体として S = 1 のカゴメ反強磁性体とみなせる。また、この系の特徴の 一つは、ピリジン環に付加されているアルキル基の長さによって結晶格子が異なる。メチ ル基をもつ m-MePYNN·BF4·1/3(acetone)や m-MePYNN·ClO4·1/3(acetone)は三方晶で歪みの
ないカゴメ格子を形成する[14]。一方、エチル基をもつ m-EtPYNN·I·(H 2O)1.5は、結晶格子が 三方晶から 0.1 ~ 0.5°歪んでいることから、歪んだカゴメ格子反強磁性体とみなせ、スピ ンフラストレーションが解消していると考えられる。 分子平面のねじれを利用した次近接相互作用をもつフラストレート磁性体 分子間での NO ラジカル接近は磁性を決める上で非常に重要な位置づけにある。その分 子間接近において、もう一つ重要な要素であるのが分子内でのねじれである。有機物であ るが故の分子内に存在するねじれは、分子間でのラジカル接近による新たな磁気的ネット ワークの構築に大きく寄与し、それは次元性の向上へと繋がる。その例として F2PIMNH がある[15]。F 2PIMNH は分子内に NN ラジカルと t-BuNH の 2 つのユニットから構成される。 その結晶構造は空間群 Pca21となっている。c 軸方向には NO ラジカルと t-BuNH による水 素結合を形成し、ここでジグザグ鎖をつくる(O···H 2.254 Å)。注目すべきはこの最近接鎖 に加え、分子内のねじれに起因する NO ラジカルと t-BuNH の窒素原子間にも接近がみら れ(O···N 5.372(9) Å)、次近接鎖を形成 することにある(Fig. 7 (a))。これをモデ ル化すると Fig. 7(b)のようになり、点 線が最近接鎖、破線が次近接鎖を表す。 このように F2PIMNH は分子内のねじ れによって最近接鎖と次近接鎖の 2 種 類の分子間接近を有し、擬二次元的な railroad trestl モデルで表されるような ス ピ ン ネ ッ ト ワ ー ク を 形 成 す る 。 F2PIMNH は railroad trestle ハイゼンベ
ルグスピン系でスピンギャップをもつ 最初の例である。
(a) (b)
railroad trestle
Fig. 7. (a) F
2PIMNH の結晶構造
(b) 結晶構造模式図
分子内磁気相互作用を利用した S = 1 種の反強磁性格子 磁気異方性の小さいハイゼンベルグ型の一次元反強磁性体は、スピン量子数が S = 1/2 の 場合磁気的な性質は厳密に議論され、またスピン量子数の大きな場合には古典スピン系と して扱われてきた。その両者は共通の現象が観測され、その磁気的性質を決定するのは次 元性、相互作用の対称性と自由度と考えられてきた。しかし、1983 年に Haldane が系の磁 気的性質を決めるのはスピンの大きさであると発表した[16]。一次元反強磁性体において整 数スピンのときは基底状態と第一励起状態の間にエネルギーギャップが存在し、半整数の ときにはギャップは存在しないという予想から S = 1 の系において精力的に研究が行われ、 量子磁性に興味が持たれるようになった。 その発展系として分子内に S = 1/2 を 2 つ含み、それらが強磁性ダイマーを形成し、さら に強磁性ダイマー間には反強磁性相互作用させたスピン格子系が注目された。その例とし て共役系有機ラジカル F2PNNNO がある[17]。分子性化合物の特徴として、分子内磁気相互 作用はトポロジーで決定されるため、分子内に S = 1/2 を 2 つ配置し、それらを強磁性相互 作用させた F2PNNNO は、室温以下で分子内に S = 1 を形成する。また、その構造は分子内 にフッ素原子の導入により、ラジカル平面をねじれさせ、二次元的なスピン格子を実現し た。F2PNNNO は分子内で形成された強磁性ダイマーS = 1 種とそのダイマー間に働く反強 磁性相互作用による歪んだ蜂の巣格子を形成し(Fig. 8(a))、その静磁化率の温度依存性から 基底状態は非磁性であった。磁化曲線は磁化が飽和磁化の半分の値で一定値をとる磁化プ ラトーを示し、磁化の量子化現象として興味がもたれている。また、3 kbar の圧力下では Fig. 8(b)に示されるように磁化率はゼロに落ち込まないエネルギーギャップが観測されて いる。これは圧力効果によって J’の磁気相互作用が抑制された結果である。
Fig. 8. (a) F
2PNNNO 結晶構造模式図 (b) 静磁化率の温度依存性
△: 0 kbar, ○: 3 kbar
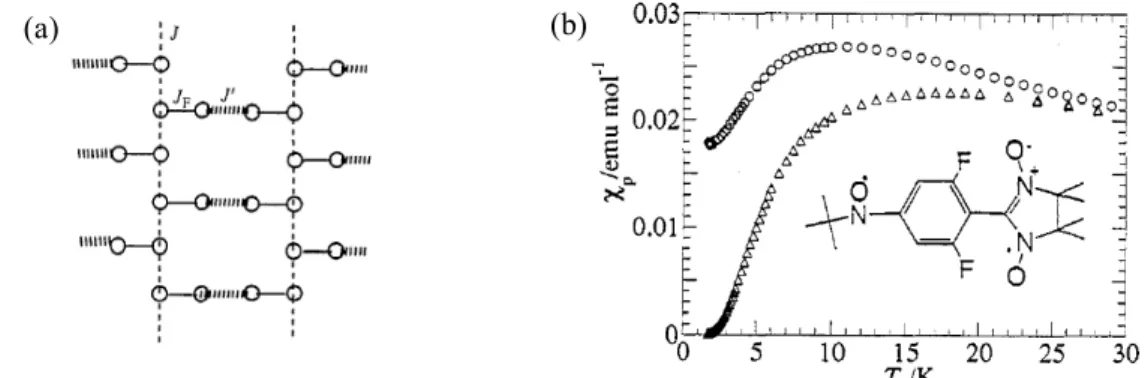
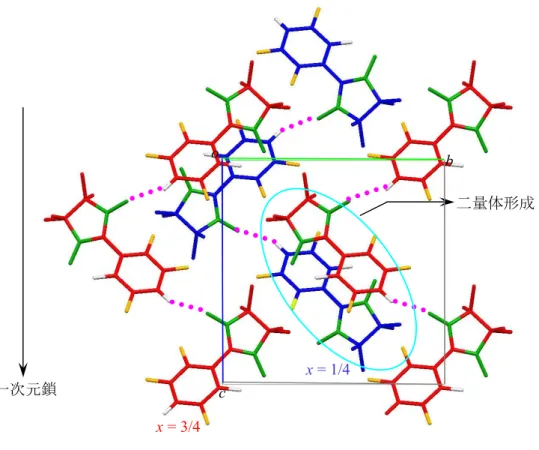
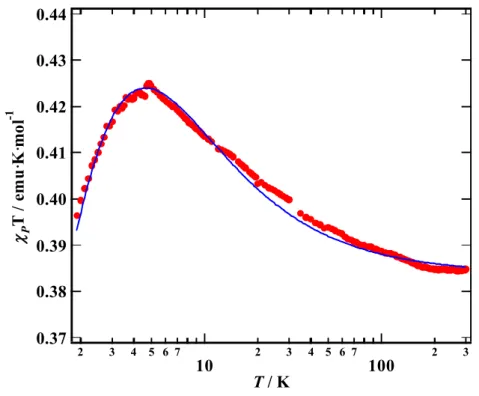
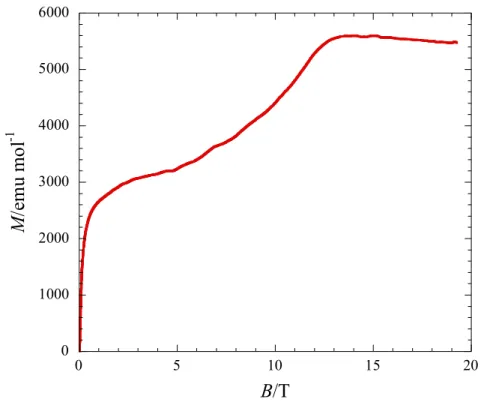
フェリ磁性体[6] PNNBNO は分子内に 3 つのラジ カル基を含むトリラジカルであり、 分子内磁気相互作用の符合はトポ ロジーで決定される。BNO ユニッ ト内ではスピンが強磁性結合し、S = 1 を形成している[18]。BNO ユニ ットと S = 1/2 を持つニトロニル ニトロキシド(NN)間には反強磁 性相互作用が働く。分子内では S = 1 と S = 1/2 の反強磁性ダイマーを形成している(Fig. 9)。 PNNBNO の結晶構造は、空間群 Pnma で平面性分子 PNNBNO は b 軸方向に head-to-tail で分子が積層する構造を示す(Fig. 10)。このときの NN ラジカルと NO ラジカルの接近は 4.906(2) Å でフェリ磁性梯子鎖構造を形成する。S = 1 と S = 1/2 を結ぶ分子内反強磁性相互 作用が梯子の桁の部分になる。梯子の足に相当する分子間反強磁性相互作用が S = 1 と S = 1/2 の間に働いている。 PNNBNO の静磁化率の温度依存性は、温度低下とともにpT 値は減少して行き、反強磁 性的な挙動を示す(Fig. 11)。50 K 付近でpT 値は一定値をとり、このときの値は 0.375 emu K mol1に近づく。分子内に S = 1 と S = 1/2 が反強磁性ダイマーを形成したときの値がこれに 相当する。このため 30 K 以上は分子内磁気相互作用が支配している。pT が一定値をとっ
Fig. 10. PNNBNO のフェリ磁性鎖構造
S=1/2 S = 1 S=1/2 S=1/2 S=1/2 JF JAF JAF JAF N N O O PNNBNO JF JAF JAF J F>>JAF 2JF/kB = 860 K 2JAF/kB = -216 K N N O OFig. 9. PNNBNO の分子内相互作用
た後、値は低温で上昇するような挙動を示 し、フェリ磁性相互作用が発現したことを 示 す (2JF/kB = 860 K, 2JAF/kB = 216 K, 2J’AF/kB = 0.6 K)。フェリ磁性鎖間の分子間 接近も S = 1 と S = 1/2 部位の交互配列が実 現されており、比熱測定から Tc = 0.28 K の 三次元フェリ磁性秩序が観測された。
1.4. 本論文の目的と内容
有機磁性研究は最近 20 年間で大きく進展した。特に分子設計、合成技術の向上に伴って、 多種多様な有機ラジカルが合成され、その磁性が議論されてきた。今や有機分子の多様性 に着目し、スピン空間構造を制御することが期待されている。有機ラジカルは理想的な Heisenberg スピン系を形成し、スピンの量子効果が顕著に現れる系であることに注目する と、強磁性および反強磁性相互作用を制御することは重要である。分子間相互作用に関し ては、ニトロニルニトロキシド系有機ラジカルは結晶構造と磁性が多く報告されているも のの、置換基の変化によって結晶構造が大きく変化するために、これまで系統的な研究は ほとんどなされてこなかった。 本研究では、有機モノラジカルを用いた分子間磁気相互作用と分子間配置の相関関係に 関する研究、および、分子内磁気相互作用制御による高スピン種を含む磁性体の開発、さ らに単結晶を用いた電子スピン共鳴の実験から、分子間磁気相互作用について考察をした。 モノラジカル結晶については、フッ素原子が小さい原子半径を持ち電子数に富むことに注 目し、フェニル基の水素原子をフッ素原子で複数個置換することで類似の結晶構造を実現 し、構造と磁性と比較検討することで分子間配置と磁気相互作用の相関関係について新た な知見を得た。また、分子内磁気相互作用の符号(強磁性/反強磁性)を制御することで、 S = 1 と S = 1/2 の二量体構造を持つ分子を合成した。ラジカル平面のねじれによって複雑 な分子間反強磁性ネットワークが形成されるが、単結晶を用いた強磁場中での電子スピン 共鳴実験から、分子間磁気相互作用について考察した。 本論文内容は 5 章から構成される。第 1 章では有機磁性体の研究背景を述べ、研究目的 を論じた。第 2 章では、本研究で行った実験について述べる。Fig. 11. PNNBNO の
pT の温度依存性
第 3 章「フッ素置換フェニルニトロニルニトロキシドの構造と磁性」では 4 種類のフッ 素置換フェニルニトロニルニトロキシド(F4PNN、F3PNN)の結晶構造と磁性を報告し、分子 パッキングと磁気相互作用の相関について比較検討を行った。2,3,5-F3NN、2,3,6-F3PNN、 2,3,4,5-F4PNN および 2,3,5,6-F4PNN の新規化合物を合成した。これら 4 種すべての結晶構 造解析と磁化測定を行った。結晶構造に基づくモデルで磁化挙動を解析し、特定の分子間 配置に磁気相互作用を帰属した。また、これら 4 種類の化合物における分子間配置を分類 し、構造と磁性の関係について考察した。 次に第 4 章では、分子内に 3 つのラジカル基を導入した π 共役ヘテロスピン系有機トリ ラジカル BIPNNBNO の結晶構造と磁化および電子スピ ン共鳴実験を行った。BIPNNBNO は S = 1/2 と S = 1 の反 強磁性二量体を形成する。結晶中では分子間に 3 種類の ラジカル間接近がみられた。単結晶を用いた電子スピン 共鳴実験により、磁気相互作用について考察した。 第 5 章では、本研究のまとめを述べる。
1.5. 引用文献
[1] J. Yamauchi, Bull. Chem. Soc. Jpn., 44, 2301 (1971). J.Yamauchi, T. Fujito, E. Ando, Y. Deguchi, J. Phys. Soc. Jpn. 25, 1558 (1968). H. Lemaire, P. Rey, A. Rassat, A. D. Combareu, J. C. Michel, Mol. Phys. 14, 201 (1968).
[2] K. Mukai, H. Nishiguchi, and Y. Deguchi, J. Phys. Soc. Jpn., 23, 125 (1967). K. Mukai, Bull. Chem. Soc. Jpn., 42, 40 (1962). C. Veyret, A. Blaise, and A. Blaise, Mol. Phys., 25, 873 (1973). G. Chouteau and Cl. Veyret-Jeandey, J. Phys. (Paris), 42, 1441 (1981). A. Benoit, J. Flouquet, B. Gillon, and J. Schweizer, J.
Magn. Magn. Mater., 31-34, 1155 (1983).
[3] K. Awaga, T. Sugano, M. Kinoshita, Chem. Phys. Lett., 14, 540 (1987).
R = F F F F F F F F F F F F F F R N N O O 2,3,5-F3PNN 2,3,5,6-F4PNN 2,3,4,5-F4PNN 2,3,6-F3PNN N N N N O O O O BIPNNBNO
[4] Ullman E. F., Osiecki J. H., Boocock D. G. B. and Darcy R., J. Am. Chem.
Soc., 94, 7049 (1972).
[5] M. Tamura, Y. Nakazawa, D. Shiomi, K. Nozawa, Y. Hosokoshi, M. Ishikawa, M. Takahasi, and M. Kinoshita, Chem. Phys. Lett., 186, 401 (1991). Y. Nakazawa, M. Tamura, N. Shirakawa, D. Shiomi, M. Takahashi, M. Kinoshita, and M. Ishikawa, Phys. Rev. B, 46, 8906 (1992).
[6] Y.Hosokoshi, K. Katoh, Y. Nakazawa, H. Nakano, and K. Inoue, J. Am.
Chem. Soc,.123, 7921(2001).
[7] K. Nozawa, Y. Hosokoshi, D. Shiomi, M. Tamura, N. Iwasawa, H. Sawa, R. Kato, and M. Kinoshita, Synth. Met., 56, 3323(1993).
[8] Y. Hosokoshi, M. Tamura, M. Kinoshita, H. Sawa, R. Kato, Y. Fujiwara, and Y. Ueda, J. Mat. Chem., 4, 1219 (1994).
[9] Andrei Zheludev, Vinenzo Barone, Michel Bonnet, Bernard Delley, Andre Grand, Eric Ressouche, Paul Rey, Robert Subra, and Jacques Schweizer, J. Am.
Chem. Soc, 116, 2019 (1994).
[10] 季刊化学総説電子系有機固体日本化学会編 No.35, p175185 (1998).
[11] H.M. McConnell, J. Chem. Phys., 39, 1910 (1963).
[12] H. M. McConnell, Proc. R. A. Welch Found. Chem. Res., 11, 144 (1967). [13] Fabrice Lanfranc de Panthou, Dominique Luneau, Jean Laugier, and Paul Rey, J. Am. Chem. Soc. 115, 9095 (1993).
[14] Kunio Awaga, Tsunehisa Okuno, Akira Yamaguchi, and Morikuni Hasegawa, Phys. Rev. B, 49, 3975 (1994).
[15] Yuko Hosokoshi, Keiichi Katoh, Katsuya Inoue, and Tsuneaki Goto, J.
Phys. Soc. Jp., 68, 2910 (1999).
[16] F. D. M. Haldane, Phys. Rev. Lett., 50, 1153 (1983). [17] Y. Hosokoshi, K. Inoue, Synth. Met., 103, 2323 (1999).
2. 実験
2.1. 物性測定
X 線構造解析
室温での測定は Rigaku 社、イメージング プレート単結晶自動 X 線構造解析装置 R-AXIS RAPID-F(Fig. 12)を用いて各単結晶 における構造解析を行った。キャピラリー の先に単結晶を接着させた。接着剤には2 液混合タイプの無溶剤系エポキシ樹脂接着 剤を用いた。接着にもちいるガラス棒や接 着剤は非晶質であるため単結晶のような X 線干渉模様を与えないため、測定には影響 されない(室温、線源 Mo)。この単結晶を接 着させたキャピラリーをゴニオメーターヘ ッドにセットし、これをゴニオに装着した後、測定を行った。 単結晶に X 線を照射し、散乱を二次元 X 線検出器で記録する。この回折強度の測定から、 格子定数、空間群、密度や単位格子内の分子数を決定する。検出されるブラッグスポット は逆空間上での方位、即ち逆格子ベクトル(h, k, l)で特定されるそれぞれの位置での X 線強 度情報を持っている。ここでの方位と強度は、結晶構造因子と呼ばれる量の絶対値の二乗 に相当する積分強度である。構造因子が求まれば、位相情報を補う手法として直接法を導 入し、最小二乗計算、フーリエ変換を行い実空間での電子密度を計算することで分子構造 を決定できる。 100 K での測定は名古屋大学大学院理学研究科阿波賀邦夫教授の協力のもと行われたも のである。Rigaku 社、CCD 単結晶自動 X 線構造解析装置 Saturn 724 を用いて 2,3,4,5-F4PNN および 2,3,5,6-F4PNN の単結晶において構造解析を行った。単結晶をパラトンオイルで付着 させ、液体窒素気流の中で測定を行った(100 K、線源 Mo)。 それぞれの測定結果および解析結果は第 3 章に示す。Fig. 12. Rigaku 社イメージングプレ
ー ト 単 結 晶 自 動 X 線 解 析 装 置
R-AXIS RAPID-F
SQUID 磁束計を用いた静磁場中での磁化測定
磁化率の温度依存性および磁化の磁場依存性の測定には Quantum Design 社 MPMS SQUID 磁束計を用いて行った(Fig. 13(a))。SQUID 磁束計では試料の磁化を捕らえるため、 ジョセフソン接合を有する超伝導検出コイルを用いている。検出コイルは 4 重巻きから成 っており、この検出コイルの中を試料が走査する。このようにして試料の磁化に相当する 電圧信号を検出することができる(Fig. 13(b))。それぞれ多結晶試料をラップに包み、それ をストローに詰め込み磁束計に導入した。印加磁場は試料や温度によって異なり、それら は以下に示されるとおりである。また、測定に際して一度試料を 1.9 K まで冷却した後、 300 K まで温度を上昇させながら測定を行った。得られた生データは、ラップの反磁性成 分および Pascal の分子反磁性成分を補正して解析を行った。それぞれの測定結果および解 析結果は第 3 章、4 章で示される。 2,3,5-F3PNN: 試料多結晶(ブロック状結晶)、試料重量 16.860 mg、ラップ重量 8.386 mg、 測定磁場 0.1 T(1.9 K~30 K), 1 T(35 K~60 K), 4 T(60 K~300 K)、反磁性成分dia = 1.505104 emu mol1 2,3,5,6-F4PNN: 試料多結晶(ブロック状結晶)、試料重量 14.472 mg、ラップ重量 8.273 mg、 測定磁場 0.1 T(1.9 K~30 K), 1 T(35 K~60 K), 4 T(60 K~300 K)、反磁性成分dia = 1.555104 emu mol1
Fig. 13. (a) MPMS SQUID 磁束計 (b) SQUID 磁束計概略図
2,3,4,5-F4PNN: 試料多結晶(板状結晶)、試料重量 18.007 mg、ラップ重量 9.754 mg、測 定磁場 0.1 T(1.9 K~30 K), 1 T(35 K~60 K), 4 T(60 K~300 K)、反磁性成分dia = 1.825104 emu mol1 -2,3,6-F3PNN: 試料多結晶(ブロック状結晶)、試料重量 18.944 mg、ラップ重量 7.133 mg、 測定磁場 0.1 T(1.9 K~30 K), 1 T(35 K~60 K), 4 T(60 K~300 K)、反磁性成分dia = 1.505104 emu mol1 -2,3,6-F3PNN: 試料多結晶(ブロック状)、試料重量 15.831 mg、ラップ重量 7.187 mg、測
定磁場 0.1 T(1.9 K~30 K), 1 T(35 K~60 K), 4 T(60 K~300 K)、dia = 1.505104 emu mol1
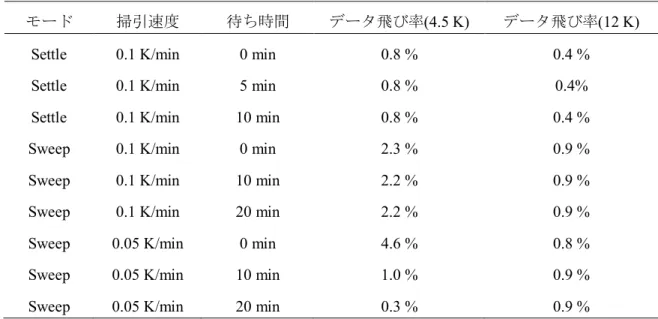
BIPNNBNO: 試料単結晶(菱形板状結晶)、試料重量 5.41 mg、ラップ重量 15.25 mg、測 定磁場 0.1 T(1.9 K~30 K), 1 T(30 K~300 K) MPMS SQUID 磁束計の 4 K および 12 K 近傍での温度安定性について また、SQUID 磁束計による測定において、液体ヘリウムの沸点領域(4.2 K)と装置内部の 温度計が切り替わる 12 K 付近で数%のデータのとびが観測された。そのため Quantum Design 社の MPMS SQUID 磁束計の温度安定性を評価するため、S = 3/2 常磁性体である明 礬(CrK(SO4)4·12H2O)を用いて磁化率測定条件の最適化を試みた。まず、MPMS SQUID 磁
束計には 2 つの温度コントロールモードが存在し、1 つは温度をリニアに変化させながら 測定を行う Sweep モード、もう 1 つはターゲット温度に安定させながら測定を行う Settle モードの 2 種類がある。この 2 つのモードの比較と掃引速度、更には 4.2 K と 12 K で待ち 時間を設けて測定を行った。
Fig. 14、Table 1 の結果をみると、Sweep モードで測定を行ったとき掃引速度 0.05 K/min で比較的データの飛びは抑えられた。しかし、Settle モードで測定を行った場合、4.5 K、 12 K 共にデータの飛びは抑えられ、また掃引速度に依存しない結果となった。この結果か ら、Settle モードで掃引速度 0.1 K/min、待ち時間無しとしたときが MPMS SQUID 磁束計
の性能を最大限に活かせると評価した。この測定条件をもとに、F3PNN および F4PNN の
Table 1. 測定条件
モード 掃引速度 待ち時間 データ飛び率(4.5 K) データ飛び率(12 K)
Settle 0.1 K/min 0 min 0.8 % 0.4 %
Settle 0.1 K/min 5 min 0.8 % 0.4%
Settle 0.1 K/min 10 min 0.8 % 0.4 %
Sweep 0.1 K/min 0 min 2.3 % 0.9 %
Sweep 0.1 K/min 10 min 2.2 % 0.9 %
Sweep 0.1 K/min 20 min 2.2 % 0.9 %
Sweep 0.05 K/min 0 min 4.6 % 0.8 %
Sweep 0.05 K/min 10 min 1.0 % 0.9 %
Sweep 0.05 K/min 20 min 0.3 % 0.9 %
X-band EPR(電子スピン共鳴)
X-band EPR は分子科学研究所 Bruker E300 を用いて測定が行われた。EPR 分光装置は試 料形状(液体、固体)に影響されずに非破壊でフリーラジカルを検出できる手段である。EPR 分光装置に用いられる電磁波帯には、L-band(1,000 MHz)、X-band(9,500 MHz)、Q-band(35,000 MHz)、W-band(95,000 MHz)がある。通常は高感度である X-band 帯を用いた EPR が用いら
れることが多い。 EPR は共振器の中に試料を設置し、一定の出力のマイクロ波を試料に照射する。その後 磁場を変化させるとエネルギー準位に分裂(ゼーマン分裂)を生じ、そのエネルギー間に相 当するマイクロ波を吸収することで観測される。信号が観測される周波数と磁場 H が実 験的に求まると、共鳴条件から g 因子が得られる。この g 因子は不対電子に対する拘束力 を表す指標であり、自由電子の g = 2.0023 から開きが大きいほど、その拘束力が大きいこ とを示す。また、EPR スペクトルは通常一次微分形として得られ、そのピーク to ピーク線 幅(Hpp)からラジカル間相互作用を議論することが可能である。 本実験では 2 ~ 300 K の温度範囲で X-band EPR の測定を行い、得られたスペクトルから 線幅と g 因子の温度依存性について議論する。
強磁場 EPR 測定
BIPNNBNO の単結晶試料における強磁場 EPR 測定は、東北大学金属材料研究所野尻浩 之教授の指導のもと行われたものである。測定は予めテフロン試料台を結晶が同軸方向に 整列できるように加工しておいた。BIPNNBNO 単結晶を 3 つテフロン試料台に乗せ、非磁 性アピエゾン真空用グリースを用いて固定し、また磁場マーカーである Ruby も同様に固 定した。サンプル台には同じくテフロン製のカバーをかぶせて試料を固定した。これに各 結晶軸方向に平行にパルス磁場を加えてその信号を読み取った。測定結果および解析結果 は第 4 章に示す。Fig. 15. EPR スペクトル
共鳴条件:
h = g
BH
h: プランク定数 B: ボーア磁子 g: g 因子2.2. 試料作製
本研究における対象物質である F3PNN、F4PNN および BIPNNBNO は以下に示す手順で 合成を行った。フッ素置換フェニルニトロニルニトロキシド
フッ素置換フェニルニトロニルニトロキシドラジカルを 4 種類合成した。常法に従って 出発原料であるアルデヒド体からビスヒドロキシルアミンによる脱水縮合反応により環状 ヒドロキシルアミンを得た。これを塩化メチレンに溶かし、NaIO4 によって酸化反応を行 い、フッ素置換フェニルニトロニルニトロキシドラジカルとした(Scheme 1)。 R = F F F F F F F F F F F F F F NHOH NHOH R N N HO HO CH2Cl2, H2O R N N O O 1 2 R-PNN R-CHO MeOH NaIO4Scheme 1
Fig. 16. (a) EPR 試料の固定 (b) EPR 概略図
BIPNNBNO (単結晶) 磁場マーカー (Ruby) テフロン試料台 テフロンカバー (a) (b)
2,3-dimethyl-2,3-dinitrobutane の合成
NO2 H NaOH NaI, I2 H2O, EtOH NO2 NO2Scheme 2
1 L ナスフラスコに H2O 125 mL 入れ、NaOH 40.50 g(1.01 mol)を発熱に注意しながらゆっ くり加えた。溶液の温度が常温に戻ってから EtOH 450 mL 加えた。これに 2-ニトロプロパ ン 89.91 g(1.01 mol)を加え、次に NaI 50.94 g(0.34 mol)を加えて溶かした。この溶液に I2100.49 g(0.39 mol)を約 30 分かけて加えた(黄色)。オイルバスを 125℃ににし、1 時間還流し た。その後、室温まで戻した後、冷蔵庫で一晩冷却した。生じた沈殿を吸引濾過し、H2O 500 mL で洗液の色が無色になるまで良く洗浄した。白色固体 64.87 g(0.37 mol)を得た。収率は 95%であった。
2,3-dihydroxylamino-2,3-dimethylbutane の合成
NO2 NO2 NH4Cl, Zn H2O, THF NHOH NHOHScheme 3
1 L ビーカーに NH4Cl 21.63 g(0.404 mol)はかりとり、更に 2,3-dimethyl-2,3-dinitrobutane 17.69 g(0.10 mol)加えた。これを THF 300 mL に溶かし、攪拌を始めた。溶液の温度を 15~20℃ にし、Zn 39.99 g(0.61 mol)を約 4 時間かけて加えた。温度を保ちながら、更に 1 時間攪拌 した。これを吸引濾過し、濾液を減圧濃縮すると白色固体が得られた。白色固体に炭酸ナ トリウムを加え、乳鉢ですりつぶした。これをクロロホルムで抽出し、減圧濃縮した。Et2O を少量加え、冷蔵庫で冷やし、沈殿を吸引濾過した。白色粉末 8.3883 g(0.057 mol)を得た。 収率 57%であった。2-fluorophenyl-1,3-dihydroxy-4,4,5,5-tetramethylimidazolidine(2)の合成
Scheme 4
4 種 類の fluorobenzaldehyde(1) を そ れぞ れメ タ ノー ル に 溶か し攪 拌 をし た 。こ れ に 2,3-dihydroxylamino-2,3-dimethylbutane を加え、脱水縮合反応によって環化した。このとき 常温で約 2 日攪拌を行った。2,3,6-trifluorobenzaldehyde は室温における収率が低いため、 40℃で約 4 時間加熱しながら攪拌を行った。その後、約 1 日攪拌を行ったところ環状ヒド ロキシルアミン(2)が白色沈殿として得られた。 【合成手順の詳細】2-(2,3,5-trifluorophenyl)-1,3-dihydroxy-4,4,5,5-tetramethylimidazolidine
(2, R = 2,3,5-F
3Ph)
30 mL ナスフラスコに 2,3,5-triflulrobenzaldehyde 210.2 mg(1.31 mmol)をはかりとった。メ タノール 1.8 mL に溶かし、攪拌を始めた。これに 2,3-dihydroxylamino-2,3-dimethylbutane 232.44 mg(1.56 mmol)を加え、室温で 4 時間攪拌した。白色沈殿を生じたので吸引濾過をし、 白色粉末 295.70 mg(1.02 mmol)を得た。収率 78%であった。2-(2,3,5,6-tetrafluorophenyl)-1,3-dihydroxy-4,4,5,5-tetramethylimidazolidine
(2, R = 2,3,5,6-F
4Ph)
30 mL ナスフラスコに 2,3,5,6-tetrafluorobenzaldehyde 400.9 mg(2.25 mmol)をはかりとった。 これにメタノール 3.3 mL を加え、攪拌を始めた。次に 2,3-dihydroxylamino-2,3-dimethylbutane 268.2 mg(1.81 mmol)を加え室温で 2 日間攪拌すると、白色沈殿を生じた。沈殿を吸引濾過 し、白色粉末 477.40 mg(1.55 mmol)を得た。収率 69%であった。 R CHO NHOH NHOH R N N HO HO H 1 2 MeOH2-(2,3,4,5-tetrafluorophenyl)-1,3-dihydroxy-4,4,5,5-tetramethylimidazolidine
(2, R = 2,3,4,5-F
4Ph)
30 mL ナスフラスコに 2,3,4,5-tetrafluorobenzaldehyde 400.4 mg(2.25 mmol)はかりとった。 これにメタノール 3.3 mL を加え、攪拌を始めた。2,3-dihydroxylamino-2,3-dimethylbutane 273.4 mg(1.84 mg)をはかりとり加え、室温で 2 日間攪拌した。白色沈殿を生じ、これを吸 引濾過すると白色粉末 420.3 mg(1.36 mmol)を得た。収率 61%であった。2-(2,3,6-trifluorophenyl)-1,3-dihydroxy-4,4,5,5-tetramethylimidazolidine(2, R =
2,3,6-F
3Ph)
30 mL ナスフラスコに 2,3,6-trifluorobenzaldehyde 199.34 mg(1.24 mmol)はかりとった。こ れをメタノール 1.7 mL に溶かし、攪拌を始めた。2,3-dihydroxylamino-2,3-dimethylbutane 228.16 mg(1.54 mmol)を加え、40℃で 4 時間攪拌した後、室温で 25 時間攪拌した。白色沈 殿を生じ、これを吸引濾過した。白色粉末 311.20 mg(1.07mmol)を得た。収率 87%であった。2-fluorophenyl-4,4,5,5-tetramethyl-4,5-dihydro-1H-imidazol-1-oxyl
3-N-oxide(R-PNN)の合成
Scheme 5
得られた環状ヒドロキシルアミン(2)をそれぞれジクロロメタンに溶かし攪拌を始めた。 このとき溶液は懸濁液であった。この溶液に過ヨウ素酸ナトリウム水溶液を環状ヒドロキ シルアミンに対して 3/8 当量加え酸化させた。このとき水溶液はゆっくり滴下していった。 溶液の色は濃紫色へと変化していった。反応進行状況は薄層クロマトグラフィー(TLC)で 確認し、十分に反応が行われるまで攪拌を行った。反応の際に生じた不純物はカラムクロ マトグラフィー(CHCl3/SiO2)で分離精製した。F4PNN においては収率 50~60%と低収率であ ったが F3PNN では約 80%と高収率であった。F4PNN については温度や攪拌時間など、反 応条件を見直す必要があるだろう。 R N N HO HO CH2Cl2, H2O R N N O O 2 R-PNN NaIO4【合成手順の詳細】
2-(2,3,5-trifluorophenyl)-4,4,5,5-tetramethyl-4,5-dihydro-1H-imidazol -1-oxyl
3-N-oxide (2,3,5-F
3PNN)
200 mL ナスフラスコに化合物(2, R = 2,3,5-F3Ph)を 295.70 mg(1.02 mmol)はかりとった。 塩化メチレン 100 mL を加え攪拌を始めた(白色懸濁液)。過ヨウ素酸ナトリウム 81.9 mg(0.38 mmol)/H2O 4 mL 水溶液をゆっくり加え、一晩攪拌すると紫色に変化した(TLC, CHCl3, Rf = 0, 0.2, 0.8)。H2O 20 mL を加え、分液ロートを用いて有機層を分けた。これを炭 酸ナトリウムで乾燥した後、吸引濾過した。溶液を減圧濃縮した後、カラムクロマトグラ フィー(CHCl3)によって分離精製した。得られた溶液を減圧濃縮すると、紫色固体が 190.62 mg(0.66 mmol)得られた。収率 64%であった。2-(2,3,5,6-tetrafluorophenyl)-4,4,5,5-tetramethyl-4,5-dihydro-1H-imidazol -1-oxyl
3-N-oxide (2,3,5,6-F
4PNN)
300 mL ナスフラスコに化合物(2, R = 2,3,5,6-F4Ph) 477.40 mg(1.55 mmol)はかりとった。塩 化メチレン 200 mL を加え、攪拌を始めた(白色懸濁液)。これに NaIO4 118.4 mg(0.55 mmol)/H2O 8 mL をゆっくり加えた後、室温で 3 時間攪拌すると紫色溶液へと変化した(TLC, Et2O, Rf = 0, 0.6, 0.9)。H2O 40 mL を加え、有機層を分液ロートで分けた。溶液を炭酸ナト リウムで乾燥した後、吸引濾過した。溶液を減圧濃縮し、カラムクロマトグラフィー(Et2O) を用いて分離精製した。得られた溶液を減圧濃縮し、紫色微結晶 280.9 mg(0.92 mmol)を得 た。収率 59%であった。2-(2,3,4,5-tetrafluorophenyl)-4,4,5,5-tetramethyl-4,5-dihydro-1H-imidazol -1-oxyl
3-N-oxide (2,3,4,5-F
4PNN)
300 mL ナスフラスコに化合物(2, R = 2,3,4,5-F4Ph)を 420.3 mg(1.36 mmol)をはかりとった。 これを塩化メチレン 200 mL に溶かし、攪拌を始めた(白色懸濁液)。NaIO4 120.5 mg(0.56 mmol)/H2O 10 mL 水溶液をゆっくり加え、室温で 3 時間攪拌した。溶液の色は紫色に変化 した(TLC, Et2O, Rf = 0, 0.6, 0.9)。H2O 60 mL 加え、有機層を分液ロートで分け、炭酸ナトリ ウムで乾燥した。吸引濾過し、溶液を減圧濃縮した。カラムクロマトグラフィー(Et2O)を 用いて分離精製し、得られた溶液を減圧濃縮した。紫色微結晶 202.0 mg(0.66 mmol)を得た。 収率 48%であった。2-(2,3,6-trifluorophenyl)-4,4,5,5-tetramethyl-4,5-dihydro-1H-imidazol -1-oxyl
3-N-oxide (2,3,6-F
3PNN)
200 mL ナスフラスコに化合物(2, R = 2,3,6-F3Ph) 311.20 mg(1.07 mg)をはかりとった。こ れを塩化メチレン 120 mL に溶かし、攪拌を始めた(白色懸濁液)。NaIO4 84.67 mg(0.39 mmol)/H2O 4.5 mL 水溶液をゆっくり加え、室温で 8 時間攪拌すると溶液の色は紫色に変化 した(TLC, CHCl3, Rf = 0, 0.1, 0.2, 0.7)。H2O 50 mL 加え、分液ロートを用いて有機層を分け た。溶液を炭酸ナトリウムで乾燥した後、吸引濾過した。濾液を減圧濃縮し、カラムクロ マトグラフィー(CHCl3)で分離精製した。得られた溶液を減圧濃縮し、紫色固体 234.2 mg(0.81 mmol)を得た。収率 76%であった。BIPNNBNO
BIPNNBNO の 全 反 応 行 程 を Scheme 6 に 示 す 。 BIPNNBNO の 合 成 は 1,3,5-tribromobenzene(3)を出発原料とし、t-BuLi を用いてハロゲンをリチオ化した。これに ニトロソ-t-ブタンを加え、80℃の条件下で反応させ、4 を合成した。BIPNNBNO の合成 を目指して 4 の化合物のヒドロキシル基を保護し、化合物 5 とした。この化合物 5 を用い て予め合成しておいた 4-iodobenzaldehyde(6)と根岸カップリング反応により、7 を合成した。 化合物 7 は 2,3-dihydroxylamino-2,3-dimethylbutane との脱水縮合反応により環化し、ニトロ ニルニトロキシドラジカルの前駆体である環状ヒドロキシルアミンとした(8)。この環状ヒ ドロキシルアミンを過ヨウ素酸ナトリウムによって酸化し、ニトロニルニトロキシドラジ カルを生成した(9)。次に n-Bu4NF3H2O によって保護基をはずし 10 とした。これを酸化銀 によって酸化し、BIPNNBNO を合成した。 Br Br Br NO Br N N OH OH N HN Br N N O O R R
n-BuLi ZnCl2 i-Bu2AlH PdCl2(PPh3)2 I CHO CHO O O R R NHOH NHOH N N O O R R HO HO H CH2Cl2, H2O N N O O R R O O n-Bu4NF 3H2O N N HO HO O O N N O O O O I I H2O2 Na2WO4 NH2 H NO2 NaOH NaI, I2 H2O, EtOH NH4Cl, Zn H2O, THF R-Cl = Si Cl 3 4 5 6 7 8 9 10 t-BuLi Et2O R-Cl DMF THF MeOH NaIO4 THF Ag2O CH2Cl2 n-BuLi DMF Et2O H2O BIPNNBNO
Scheme 6
Nitroso-t-butane
NH2 Na2WO4 H2O2 H2O NO 2Scheme 7
1 L 三口フラスコにタングステン酸ナトリウム 10.07 g(34.3 mmol)はかりとり H2O 80 mL に溶かし、攪拌を始めた。これに t-ブチルアミン 100 mL(0.94 mol)を加えた。溶液を氷浴し、 15℃に保ちながら H2O2(30%) 195 mL(1.67 mol)を 7 時間かけて滴下した。溶液は青色に変化 し、青色沈殿も生じた。15℃を保ちながら、更に 1 時間攪拌した。その後、氷浴で 3 時間 冷却し、沈殿を吸引濾過した。これを少量のヘキサンで洗浄し、白色粉末 26.59 g(0.36 mol) 得た。1-bromo-3,5-bis[N-t-butyl-N-(hydroxyl)amino]benzene(4)
Scheme 8
1,3,5-トリブロモベンゼンを出発原料(3)としてジエチルエーテルに溶かし、80℃の窒素 雰囲気またはアルゴン雰囲気下で t-BuLi/ペンタン溶液を二等量用いることによってリチ オ化反応を行った。禁水反応であるため予め反応系を十分脱水しておいた。ここにニトロ ソ-t-ブタンを反応させることで 1-bromo-3,5-bis[N-t-butyl-N-(hydroxyl)amino]benzene(4)を合 成した。不純物を除去するため、最後にヘキサン少量で洗浄、またはカラムクロマトグラ フィーで精製した。収率が安定しないケースがみられたが、これは反応系の脱水が不十分 であるためである。 【合成手順の詳細】化合物 4 の合成は禁水反応であるので 300 mL 三口フラスコを加熱真 空乾燥しておき、これをアルゴン置換した。これに 1,3,5-tribromobenzene(3) 3.03 g (9.62 mmol)をはかりとり加えた。これに脱水ジエチルエーテル 120 mL を加え、攪拌を始めた。 L-N2/アセトンで80℃に保ちながら、t-BuLi (1.6 M)/ヘキサン溶液 26 mL (41.3 mmol)を滴下 Br Br Br Br N N OH OH NO 3 4 (314.80) (331.25) t-BuLi Et2Oロートでゆっくり滴下した(淡赤色)。このとき温度が上がらないように注意しながら約 1 時間攪拌をした。ニトロソ-t-ブタン 2.96 g (34.0mmol)/ジエチルエーテル 13 mL を 15 分か けて滴下し、ゆっくり室温に戻しながら一晩攪拌した(黄緑色)。一晩攪拌した後、NH4Cl 水溶液を 50 mL 加え約 10 分攪拌した。分液ロートを用いてジエチルエーテルで抽出した。 有機層を 20%NaCl 水溶液で洗浄した。分液した有機層を硫酸マグネシウムで乾燥し、吸引 濾過して硫酸マグネシウムを取り除いた。濾液を濃縮すると橙色固体を得た。不純物を取 り除くため、カラムクロマトグラフィーで分離精製した(Hexane : Et2O = 1 : 1)。溶液を濃縮 して、淡黄色粉末 2.753 g (8.31 mmol)を得た。収率は 86%であった。
1-bromo-3,5-bis[N-t-butyl-N-(t-butyldimethylsiloxy)amino]benzene(5)
Scheme 9
1-bromo-3,5-bis[N-t-butyl-N-(hydroxyl)amino]benzene(4)を予め減圧蒸留を行ったジメチル ホルムアミドに溶かし、イミダゾールと保護剤である t-ブチルジメチルクロロシランを用 いてヒドロキシル基を保護した。反応は窒素またはアルゴン雰囲気下で行い、反応温度は 50℃で一晩攪拌した。不純物の除去はカラムクロマトグラフィーを行った。ここで溶媒で ある DMF が除去されていない状態でカラムクロマトグラフィーを行うと分離精製が困難 である。また Scheme 11 に示す反応において湿りやすい DMF の存在は避けるべきであり、 分液ロートを用いて H2O 洗浄をすることで DMF を取り除いた。反応は比較的高収率(80 ~ 90%)で 1-bromo-3,5-bis[N-t-butyl-N-(t-butyldimethylsiloxy)amino]benzene(5)を得ることができ た。 【合成手順の詳細】100 mL 三口フラスコを加熱真空乾燥し、これをアルゴン置換した。そ こに 1-bromo-3,5-bis[N-t-butyl-N-(hydroxyl)amino]benzene(4) 1.919 g (5.76 mmol)をはかりと った。これにイミダゾール 2.42 g (31.9 mmol)を加え、更に t-butyldimethylchlorosilane 4.65 g (18.4 mmol)をはかりとり、加えた。これを減圧蒸留した DMF 45 mL に溶かし、45℃で一 晩攪拌した(淡黄色)。一晩攪拌後、H2O 20 mL 加えて分液ロートを用いてベンゼンで抽出 Br N N OH OH 4 Si Cl N HN Br N N O O Si Si 5 (331.25) (559.77) DMFした。その後、20%NaCl 水溶液で洗浄し、硫酸マグネシウムで乾燥した。乾燥後、硫酸マ グネシウムを吸引濾過で取り除き、濾液を減圧濃縮した(褐色オイル)。不純物を除くため、 得られた褐色オイルをカラムクロマトグラフィーで分離精製した(Hexane)。得られた溶液 を減圧濃縮し、無色透明オイルを 2.63 g (4.69 mmol)得た。収率は 81%であった。
4-iodobenzaldehyde(6)
Scheme 10
化合物 5 とのクロスカップリング材料として、4-iodobenzaldehyde(6)を合成した。出発原 料として、1,4-diiodobenzene から n-BuLi を用いて80℃でリチオ化反応を行い、DMF によ ってホルミル化した。禁水反応であるため、予め反応系を脱水しておいた。比較的収率は 安定しており、70 ~80%であった。 【合成手順の詳細】化合物 4 の合成は禁水反応であり、200 mL 三口フラスコを加熱真空乾 燥しておき、これをアルゴン置換した。これに p-diiodobenzene 2.01 g (6.10 mmol)をはかり とり加えた。予め蒸留しておいたジエチルエーテル 100 mL に溶かし、L-N2/アセトンを用 いて80℃とした。n-BuLi (1.6 M)ヘキサン溶液 3.8 mL (6.10 mmol)を約 10 分で加えた。温 度を保ちながら一時間攪拌した。ジメチルホルムアミド(DMF) 0.78 mL (10.1 mmol)を加え て、室温に戻しながら一時間半攪拌した(淡黄色)。その後、反応溶液に NH4Cl 水溶液 50 mL を加え攪拌した後、ベンゼンで抽出した。H2O 50 mL を加え洗浄し、硫酸マグネシウムで 乾燥した。乾燥後、硫酸マグネシウムを取り除くため吸引濾過をし、濾液を減圧濃縮した(白 色粉末)。原料および不純物を除くため、カラムクロマトグラフィーで分離精製した (Hexane : Et2O = 3 : 1)。溶液を減圧濃縮して白色微結晶 1.2026 g (5.20 mmol)を得た。収率 85 %であった。 I I n-BuLi, DMF I CHO 6 (329.90) (232.02) Et2O3’,5’-bis[N-t-butyl-N-(t-butyldimethylsiloxy)amino]-4-formyl biphenyl (7)
Scheme 11
化 合 物 5 と 化 合 物 6 を 用 い て ク ロ ス カ ッ プ リ ン グ 反 応 で 3’,5’-bis[N-t-butyl-N-(t-butyldimethylsiloxy)amino]-4-formyl biphenyl(7)を合成した。まず、化 合物 3 を80℃において n-BuLi を等量モル用いてリチオ化し、ZnCl2を加えた。禁水反応で あるので、予め脱水した反応系を準備した。カップリングに際して触媒として PdCl2(PPh3)2 溶液に i-Bu2AlH を加えて用いた。この触媒溶液に化合物 5 と化合物 6 の溶液を加えて室温 で反応させていった。収率は安定して得ることができず、脱水が不十分と考えられる。 【合成手順の詳細】化合物 7 の合成は禁水反応であるので 200 mL 三口フラスコを加熱真 空 乾 燥 し て お き 、 ア ル ゴ ン 置 換 し た 。 こ れ に 1-bromo-3,5-bis[N-t-butyl-N-(t-butyldimethylsiloxy)amino]benzene(5)を 0.60 g (1.07 mmol)はか りとり加えた。予め蒸留しておいたテトラヒドロフラン(THF) 20 mL に溶かし攪拌を始め、 L-N2/アセトンを用いて80℃とした(無色透明)。これに n-BuLi (1.6 M)ヘキサン溶液 0.90 mL (1.46 mmol)をゆっくり加えた。30 分攪拌をすると、溶液の色は淡黄色へと変化した。 これに予め 150℃で一晩加熱真空乾燥しておいた ZnCl2 160 mg (1.17 mmol)を THF 20 mL に 溶かして滴下ロートに入れ、一時間半かけてゆっくり滴下した。その後、L-N2/アセトンバ スからフラスコをはずし、1 時間かけて室温に戻していった[A]。 溶液 A を室温に戻す間に 100 mL 三口フラスコを加熱真空乾燥しておき、アルゴン置換 した。PdCl2(PPh3)2 16 mg をはかりとり加えた。THF 3 mL を加え、攪拌を始めた(黄色)。こ れに i-Bu2AlH 0.1 mL をマイクロシリンジで加えると溶液の色は黄色から黒色へと変化し た[B]。 溶液[B]の作製と同時に 30 mL 三口フラスコを加熱真空乾燥しておき、アルゴン置換した。 これに 4-iodobenzaldehyde(6) 253.13 mg (1.09 mmol)を加えて、THF 10mL に溶かした[C]。 フラスコ[B]の滴下ロートに溶液[A]と[C]2 種類を加えた。この溶液を 15 分かけて滴下し、 さらに室温で一時間半攪拌した(淡黄色)。その後、H2O 20 mL 加え約 10 分攪拌した後、分 Br N N O O Si Si 5n-BuLi, ZnCl2, i-Bu2AlH PdCl2(PPh3)2 I CHO CHO N N O O Si Si 7 (559.77) (584.98) THF
液ロートを用いてジエチルエーテルで抽出した。有機層を H2O で洗浄し、硫酸マグネシウ ムで乾燥した。乾燥後吸引濾過をし、硫酸マグネシウムを取り除き、濾液を減圧濃縮した。 原料および不純物を取り除くため、これをカラムクロマトグラフィーによって分離精製し た(Hexane : Et2O = 3 : 1)。溶液を減圧濃縮することで無色透明オイル 150.89 mg (0.26 mmol) を得た。収率 24%であった。
3’,5’-bis{N-t-butyl-N-(t-butyldimethylsiloxy)amino}-4-(1,3-dihydroxy-4,4,5,5-tetra
methyl imidazolidine-2-yl biphenyl(8)
Scheme 12
3’,5’-bis[N-t-butyl-N-(t-butyldimethylsiloxy)amino]-4-formyl biphenyl (7)をメタノールに溶か し、ここに 2,3-dihydroxylamino-2,3-dimethylbutane を加え、脱水縮合反応によって環状ヒド ロキシルアミン化合物(8)を得た。 【合成手順の詳細】 30 mL ナスフラスコに 3’,5’-bis[N-t-butyl-N-(t-butyldimethylsiloxy)amino]-4-formyl biphenyl (7)を 120 mg (0.21 mmol)はかりとった。これをメタノール 1.2 mL を加え、攪拌を始めた。 更に 2,3-dihydroxylamino-2,3-dimethylbutane 57.08 mg (0.38 mmol)をはかりとり、加えた。こ れを室温で 2 日間攪拌すると、白色沈殿が生じた。沈殿を吸引濾過し、白色粉末 122 mg (0.17 mmol)を得た。収率 81%であった。 CHO N N O O Si Si 7 (584.38) NHOH NHOH N N O O Si Si N N HO HO H 8 (715.17) MeOH3’,5’-bis{N-t-butyl-N-(t-butyldimethylsiloxy)amino}-4-(1-oxyl-3-oxide-4,4,5,5-tetra
methyl imidazolin-2-yl)biphenyl(9)
Scheme 13
環状ヒドロキシルアミン(8)をジクロロメタンに溶かし、過ヨウ素酸ナトリウム水溶液を 滴下して加え、酸化反応を行った。過ヨウ素酸ナトリウム水溶液をゆっくり加えていくと、 溶液の色は濃い青色となった。収率は 60%程度であり、比較的低収率であった。これは酸 化の際、ニトロニルニトロキシドラジカルの他にイミノニトロキシドラジカルが生成され ているためである。 【合成手順の詳細】 3’,5’-bis{N-t-butyl-N-(t-butyldimethylsiloxy)amino}-4-(1,3-dihydroxy-4,4,5,5-tetramethylimidazolidine-2-yl biphenyl(8)を 210.11 mg (0.29 mmol)はかりとり、200 mL ナスフラスコに 入れた。これを予め蒸留しておいたジクロロメタン 150 mL に溶かし、攪拌を始めた(白色 懸濁液)。溶液に酸化剤である NaIO4 24.5 mg/H2O 5 mL をゆっくり加えて約 2 時間攪拌した (濃青色)。分液ロートを用いて有機層を分け、H2O で洗浄をした。硫酸マグネシウムで乾 燥した後、吸引濾過をし、硫酸マグネシウムを取り除いて溶液を減圧濃縮した(濃青色オイ ル)。得られた濃青色オイルに含まれる不純物を取り除くためにカラムクロマトグラフィー で分離精製した(Hexane : Et2O = 1 : 2)。溶液を減圧濃縮すると青色オイル 130 mg (0.18 mmol)を得た。収率 61%であった。 N N O O Si Si N N HO HO H CH2Cl2, H2O N N O O Si Si N N O O 8 9 (715.17) (712.14) NaIO4
3’,5’-bis(N-t-butyl-N-hydoxylamino)-4-(1-oxyl-3-oxide-4,4,5,5-tetramethyl
imidazolin-2-yl)biphenyl (10)
Scheme 14
化合物 9 を THF に溶かし、n-Bu4NF·3H2O を加え、脱保護反応を行った。溶液は青色か ら濃緑色へと変化し、3’,5’-bis(N-t-butyl-N-hydoxylamino)-4-(1-oxyl-3-oxide-4,4,5,5-tetramethy l imidazolin-2-yl)biphenyl (10)を固体として得た。反応は不純物を含みカラムクロマトグラ フィーで分離精製を行った。 【合成手順の詳細】 3’,5’-bis{N-t-butyl-N-(t-butyldimethylsiloxy)amino}-4-(1-oxyl-3-oxide-4,4,5,5-tetramethyl imidazolin-2-yl)biphenyl(9)を 130 mg (0.18 mmol)はかり、50 mL ナスフラスコに入れた。予 め蒸留しておいた THF 13 mL 加えて溶かし、室温で攪拌を始めた(濃青色)。これに n-Bu4NF·3H2O 256.11 mg (0.76 mmol)を加え、2 時間攪拌した(濃緑色)。反応が完結したのを 確認し、反応溶液に H2O 10 mL を加えて分液ロートを用いて Et2O で抽出した。有機層を 硫酸マグネシウムで乾燥した後、吸引濾過して溶液を減圧濃縮すると濃緑色固体 81.2 mg (0.17 mmol)を得た。収率 94%であった。3’,5’-bis(N-t-butyl-N-oxylamino)-4-(1-oxyl-3-oxide-4,4,5,5-tetramethyl
imidazolin-2-yl)biphenyl (BIPNNBNO)
Scheme 15
N N O O Si Si N N O O n-Bu4NF 3H2O N N HO HO N N O O 9 10 (712.14) (483.62) THF N N HO HO N N O O N N O O N N O O 10 (483.62) (481.61) Ag2O CH2Cl2 BIPNNBNO化合物 10 をジクロロメタンに溶かして、酸化銀(Ag2O)を用いて酸化した。濃緑色固体と して得られ、収率は 60%程度であった。酸化中は、TLC で反応を確認しながら、分解物が 生じたときは直ちに吸引濾過を行った。 【合成手順の詳細】 3’,5’-bis(N-t-butyl-N-hydoxylamino)-4-(1-oxyl-3-oxide-4,4,5,5-tetramethyl imidazolin-2-yl)biphenyl (10)を 81.6 mg (0.17 mmol)をはかりとり、30 mL ナスフラスコに加 えた。これにジクロロメタン 4.5 mL を加えて溶かした。溶液を氷浴で 0℃に冷却し、攪拌 を始めた。この溶液に Ag2O 184.15 mg (0.80 mmol)を約 10 分で加え、温度を保ちながら約 1 時間攪拌した(濃緑色)。吸引濾過をし、濾液を減圧濃縮した。不純物を除くためにカラム クロマトグラフィーで分離精製した(Et2O)。溶液を減圧濃縮し、濃緑色固体 49.51 mg (0.10 mmol)を得た。収率 58%であった。