遷移金属錯体触媒を用いるカルボン酸の高効率分子
変換反応に関する研究
著者
前谷 臣治
内容記述
学位記番号:論理第102号, 指導教員:柳日馨
遷移金属錯体触媒を用いるカルボン酸の
高効率分子変換反応に関する研究
Highly Efficient Transformations of Carboxylic Acids Catalyzed by
Transition-Metal Complexes
前谷 臣治
Shinji Maetani
大阪府立大学大学院 理学系研究科
Graduate School of Science
Osaka Prefecture University
目次 緒論………...…1 参考文献………...………6 本論 第 1 章 長鎖脂肪族カルボン酸の脱カルボニル化反応によるオレフィンの効率 合成...7 第1 節 緒言………8 第2 節 Ir 触媒を用いる脂肪族カルボン酸の脱カルボニル化反応…...……12 第3 節 反応機構について………..25 第4 節 Fe 触媒を用いる脂肪族カルボン酸の脱カルボニル化反応…….….26 第5 節 反応機構に関する考察……….…….36 第6 節 結論……….……….40 第7 節 実験の部……….……….42 第8 節 参考文献……….……….48 第 2 章 2-アリーロキシ安息香酸の脱カルボニル的環化反応によるジベンゾフ ラン類の合成……….51 第 1 節 緒言……….……….52 第 2 節 反応条件の最適化および一般性の検討……….………….57 第3 節 反応機構に関する考察………..66
第 4 節 2-フェノキシ安息香酸ペンタフルオロフェニルエステルの脱カル ボニル的環化反応……….70 第5 節 結論……….……….75 第6 節 実験の部………..76 第7 節 参考文献………..………88 第 3 章 ビアリールカルボン酸の分子内アシル化反応によるフルオレノン類の 合成……….91 第1 節 緒言………..92 第2 節 反応条件の最適化および一般性の検討………..95 第3 節 反応機構について………103 第4 節 結論………...……….104 第5 節 実験の部………105 第6 節 参考文献………110 総括………...112 論文リスト………...…114 学会発表………...…115 謝辞………...117
1 緒論 有機合成化学は、化合物の物質変換を基盤として医薬品や機能性物質などの 現代社会には欠かせない有用化合物の創出を担う化学分野の一つである。これ までに多種多様な分子変換反応が開発され、現在もなお、より高性能かつ高活 性な反応の開発を求めて研究が続けられている。さらに近年、環境保護の観点 から、環境に調和した合成手法、すなわち、廃棄物を産出しない、省エネルギ ー型の反応開発が求められている。この社会的かつ時代的な要求に応えるため に、近年、遷移金属錯体を触媒として利用する有機合成反応の潜在力が注目さ れている。これら遷移金属触媒反応には、穏和な反応条件や中性条件、合成段 階の短縮や不斉合成など、合成手法として魅力的な利点を有するものが多く開 発されており、現代有機合成における力量あるツールとして広く認識されつつ ある。 カルボン酸は容易に入手可能な化学原料であり、種々の方法により酸ハロゲ ン化物や酸無水物、アミド、エステルなどの誘導体へと変換可能なことから、 有機合成化学において極めて重要な化合物群に位置づけられている。また、カ ルボン酸は酸無水物や酸ハロゲン化物へと変換することで、遷移金属錯体と容 易に反応することが古くから知られており、近年ではその反応性を触媒反応へ と応用した例が多数報告されている1)。それらの反応は主に以下の2 つの反応形 式、すなわちカルボニル生成物を与えるものと、脱カルボニル化生成物を与え るものに分類される。 カルボン酸無水物のアシル炭素-酸素結合は、Pd や Rh、Ir などの低原子価遷 移金属錯体に容易に酸化的付加し、アシルメタル錯体を与える。この中間体を 求核剤等で捕捉することで新たなカルボニル化合物を得ることができる。一方
2 で、アシルメタル錯体は反応条件により、一酸化炭素の逆挿入、すなわち脱カ ルボニル化反応を起こし、アリールメタルやアルキルメタル錯体を与えること が知られている2)。このような同一のカルボン酸から出発し、異なる活性種を利 用する反応開発に多くの興味が持たれ、これまで多種多様な分子変換反応が報 告されて来た。以下には反応形式により分類したカルボン酸の変換反応に関す る最近の報告例をいくつか示す。 アシルメタル種を鍵活性種とした反応例 下記の報告例では、カルボン酸とボロン酸によるカップリング反応がPd 触媒 により進行し、非対称ケトンが効率的に得られることが示されている。ジメチ ルジカーボネートを添加し、系中で酸無水物を調製する方法により、様々なカ ルボン酸から反応を行うことができる3a)。 カルボン酸のアルデヒドへの還元反応が、Pd 触媒と NaH2PO4を用いることに より達成されている。酸無水物としてピバル酸無水物を添加することでカルボ ン酸から原料由来の酸無水物を調製する方法をとり、アシルメタル種の生成を 促進している3b)。
3 アリールメタル種を鍵活性種とした反応例 安息香酸無水物とスチレン類との反応を行った場合には、脱カルボニル化を 伴ったヘック型のカップリング反応が進行する。アシルパラジウム種から脱カ ルボニル化反応により、アリールパラジウム種へと変換することが本反応の鍵 過程となっている4a)。 Rh 触媒を用いることで、安息香酸無水物の脱カルボニル化を伴った C-H 活 性化反応が進行することも明らかにされている。また、酸塩化物も同様の触媒 系を用いることで適応可能とされており、新しいタイプの C-H アリール化反応 が示された4b)。 本研究では金属種による反応特性の違いに着目し、様々な遷移金属触媒によ るカルボン酸を出発基質とした効率的分子変換反応の開発を目指し検討を行う こととした。 第 1 章では、天然に豊富に存在している長鎖脂肪族カルボン酸の脱カルボニ ル化反応による末端および内部オレフィンの選択的合成について行った検討結 果について述べる。これまでに脂肪族カルボン酸からオレフィンを合成する脱 カルボニル化反応はいくつか報告例があるが、それらは末端オレフィンの選択 的合成に特化した反応例であった5)。各種合成中間体として有用な内部オレフィ ンの選択的合成を検討した結果、Ir 錯体を触媒として用いることで良好に達成さ れることを示した。また、反応条件を変えることで同様の触媒系において、末 端オレフィンの選択的合成が達成させることも明らかにした。 これまで同反応は、Pd や Rh、Ir などの遷移金属触媒を用いて検討が行われて いたが、それらは高価かつ希少な金属種であるため、実用性は低い。そこで、
4 安価な触媒としてFe に着目して種々検討を行った結果、Fe 触媒を用いても目的 の変換反応が効率的に進行することを明らかとした。さらに、Ir 触媒系の時と同 様に内部オレフィンと末端オレフィンそれぞれを選択的に合成する触媒系を見 出すことができた。Fe 触媒によるカルボニル化合物の減炭反応は、これまでに 報告のない新たな反応例である。そこで、反応機構について考察を行ったとこ ろ、本反応が一酸化炭素の脱離を伴った脱カルボニル化反応であることを明ら かとした。 第2 章では、2-アリーロキシ安息香酸の脱カルボニル的環化反応によるジベン ゾフラン合成について述べる。以前に 2-アリーロキシ安息香酸からジベンゾフ ランを合成する反応として、Pd 触媒を用いた脱炭酸的環化反応が報告されてい る6)。触媒的脱カルボニル化反応系を2-アリーロキシ安息香酸に適応した場合、 脱カルボニル化を伴った環化反応が進行し、ジベンゾフラン類が得られること を期待して検討を行った。その結果、Rh 触媒を用いることで脱カルボニル化を 伴った環化反応が進行し、ジベンゾフラン類が高収率で得られることを見出し た。また、反応機構に関する考察を行ったところ、一酸化炭素の脱離を確認し、 本反応が脱カルボニル化を伴った環化反応であることを明らかとした。 第 3 章では、ビアリールカルボン酸の分子内アシル化反応によるフルオレノ ン類の合成について行った検討結果について述べる。フルオレノンの一般的な 合成法としては、ビアリールカルボン酸の Friedel-Crafts 型分子内アシル化反応 が挙げられるが、この方法の場合、当量以上のルイス酸を必要としている7)。一
5
方、触媒的な合成法としては固体酸であるNafion®を用いた反応が知られている
が8)、遷移金属触媒を用いた反応例はこれまで報告されていない。そこで、著者
は遷移金属触媒によるビアリールカルボン酸からのフルオレノン合成を検討し
6
参考文献
1. (a) Tsuji, J.; Ohno, K. J. Am. Chem. Soc. 1966, 88, 3452. (b) Kokubo, K.; Matsumasa, K.; Miura, M.; Nomura, M. J. Org. Chem. 1996, 61, 6941. (c) Sugihara, T.; Satoh, T.; Miura, M.; Nomura, M. Angew. Chem., Int. Ed. 2003, 42, 4672. (d) Sugihara, T.; Satoh, T.; Miura, M. Tetrahedron Lett. 2005, 46, 8269. (e) Goossen, L. J.; Rodriguez, N.; Goossen, K. Angew. Chem., Int., Ed. 2008, 47, 3100.
2. (a) Blake, D. M.; Shields, S.; Wyman, L. Inorg. Chem. 1974, 13, 1595. (b) Miller, J. A.; Nelson, J. A. Organometallics 1991, 10, 2958. (c) Nagayama, K.; Kawataka, F.; Sakamoto, M.; Shimizu, I.; Yamamoto, A. Chem. Lett. 1995, 367.
3. (a) Nagayama, K.; Shimizu, I.; Yamamoto, A. Chem. Lett. 1998, 1143. (b) Kakino, R.; Narahashi, H.; Shimizu, I.; Yamamoto, A. Chem. Lett. 2001, 1242. (c) Goossen, L. J.; Ghosh, K. Chem. Commun. 2002, 836.
4. (a) Stephan, M. S.; Teunissen, A. J. J. M.; Verzijl, G. K. M.; de Vries, J. G. Angew.
Chem. Int., Ed. 1998, 37. 662. (b) O’Brien, E. M.; Bercot, E. A.; Rovis, T. J. Am. Chem. Soc. 2003, 125, 10498. (c) Goosen, L. J.; Paetzolda, J. Adv. Synth. Catal. 2004, 346, 1665. (d) Kajita, Y.; Kurahashi, T.: Matsubara, S. J. Am. Chem. Soc. 2008, 130, 17226. (e) Jin, W.; Yu, Z.; He, W.; Ye, W.; Xiao, W.-J. Org. Lett.
2009, 11, 1317. (f) Ye, W.; Luo, N.; Yu, Z. Organometallics 2010, 29, 1049. (g)
Havlik, S. E.; Simmons, J. M.; Winton, V. J.; Johnson, J. B. J. Org. Chem. 2011, 76,
3588. (h) Ochi, Y.; Kurahashi, T.; Matsubara, S. Org. Lett. 2011, 13, 1374.
5. (a) Fenton, D. M. U.S. Patent 3,530,198, 1970. (b) Miller, J. A.; Nelson, J. A.; Byrne, M. P. U.S. Patent 5,077,447, 1991. (c) Miller, J. A.; Nelson, J. A.; Byrne, M. P. J. Org. Chem. 1993, 58, 18. (d) Goossen, L. J.; Rodríguez, N. Chem. Commun.
2004, 724. (e) Nôtre, J. L.; Scott, E. L.; Franssen, M. C. R.; Sanders, J. P. M. Tetrahedron Lett. 2010, 51, 3712. (f) Miranda, M. O.; Pietrangelo, A.; Hillmyer, M.
A.; Tolman, W. B. Green Chem. 2012, 14, 490.A
6. (a) Voutchkova, A.; Coplin, A.; Leadbeaterb, N. E.; Crabtree, R. H. Chem.
Commun. 2008, 6312. (b) Wang, C.; Piel, I.; Glorius, F. J. Am. Chem. Soc. 2009, 131, 4194.
7. (a) Denney, B.; Klemchuk, P. P. J. Am. Chem. Soc. 1958, 80, 6014. (b) Wade, L. G.; Acker, K. J.; Earl, R. A.; Osteryoung, R. A. J. Org. Chem. 1979, 44, 3724. (c) Ladd, D. L.; Weinstock, J.; Wise, M.; Gessner, G. W.; Sawyer, J. L.; Flaim, K. E. J.
Med. Chem. 1986, 29, 1904.
7
第
1 章
長鎖脂肪族カルボン酸の
8 第1 節 緒言 オレフィンは、有機合成化学において欠くことのできない合成中間体であり、 かつ化学工業における基礎原料としても用いられている極めて重要な化合物で ある1)。工業的なオレフィンの合成法として、エチレンなどの低鎖長オレフィン のオリゴマー化による方法が実施されているが 2)、原料は石油由来であるため、 いずれ枯渇する可能性がある。したがって、将来的なオレフィンの安定供給の ためには、再生可能原料を用いた合成法が必要とされている3)。そのような方法 の一つに、植物由来の長鎖脂肪族カルボン酸から遷移金属触媒を用いてオレフ ィンを合成する脱カルボニル化反応が知られている。以下にその例を示すが、 これまでの報告例ではPd や Rh 触媒を用いた末端オレフィンの選択的合成に焦 点が置かれていた4)。 例えば、初期のFenton らによる報告例では Pd 触媒を用いて炭素数 8 の直鎖脂 肪族カルボン酸の脱カルボニル化反応を 200 ℃以上の高温下行うことで、一炭 素減炭されたオレフィンを得ている(Scheme 1-1)4a)。この時、生成物としては 末端オレフィンとその異性化体である内部オレフィンの異性体混合物を得てい るが、本触媒系においては末端オレフィンがより多く得られている(Scheme 中 のt は末端オレフィン、i は内部オレフィンを示す)。 Scheme 1-1 また、Miller らは Pd や Rh 触媒による脂肪族カルボン酸の脱カルボニル化反 応において、無水酢酸を添加することで反応が効率的に進行することを報告し ている(Scheme 1-2)4b,4c)。本系においては、生成物を蒸留しながら反応を行う ことでオレフィンの異性化を防ぎ、末端オレフィンを選択的に得ている。なお、
本論文にはオレフィン収率は記載されておらず、TON(Turn Over Number: 触媒
9 Scheme 1-2 その後、Goossen らにより、蒸留法を必要としない Pd 触媒による末端オレフ ィンの選択的合成が報告された 4d)。ここでは、触媒系として PdCl2/DPE-phos、 添加剤としてピバル酸無水物が有効なことが示されている(Scheme 1-3)。本系 においては、反応温度 110 ℃の比較的低温下で反応を進行させることで異性化 を防ぎ、末端オレフィンを選択的に得ているものと考えられる。 Scheme 1-3 最近、Goossenらの系とほぼ同様の触媒系を用いた脱カルボニル化反応がScott らによって報告された。この報告では、酸無水物に加えてNEt3を添加すると反
応効率が向上することが示されている(Scheme 1-4)4e)。また、Tolmanらは
PdCl2/Xanthphos触媒系を用いて、スチレンやアクリル酸誘導体が対応する脂肪
10 Scheme 1-4 Scheme 1-5 末端オレフィンを選択的に合成している例は前述したとおり、多くの報告が なされているのに対し、内部オレフィンの選択的合成は未だ報告されておらず、 以 下 のRh触媒を用いた反応例における選択性77%が最も良い結果である (Scheme 1-6)5)。近年、内部オレフィンの異性体混合物は、潤滑油や界面活性 剤などの合成中間体として有用であることが明らかにされており、これに伴っ て効率的かつ簡便な内部オレフィンの合成法の開発が望まれている6)。 Scheme 1-6
11 本研究では、長鎖脂肪族カルボン酸の脱カルボニル化反応において、末端オ レフィン、そして内部オレフィンを選択的に合成可能とする触媒系の開発を目 的とし検討を行った。その結果、Ir触媒を用いた場合に、期待した反応が良好に 進行し、内部オレフィンの選択的合成が可能なことを明らかとした。また、反 応条件を変えることで末端オレフィンの選択的合成が可能なことも明らかにし た。さらに、より安価な触媒系の開発を行った結果、Feを触媒として用いた場 合にも脂肪族カルボン酸の脱カルボニル化反応が効率良く進行し、内部オレフ ィン、末端オレフィンそれぞれを選択的に得られる条件を見出した(Scheme 1-7)。 以下、第2節では、Ir触媒を用いたオレフィン合成に関する反応条件の最適化お よび一般性の検討結果について述べ、第3節では、その反応機構について述べる。 そして、第4節では、Fe触媒を用いたオレフィン合成に関する反応条件の最適化 および一般性の検討結果について述べ、第5節では、その反応機構に関する考察 について述べる。 Scheme 1-7
12 第2 節 Ir触媒を用いる脂肪族カルボン酸の脱カルボニル化反応 1. 内部オレフィン合成における反応条件の最適化 ステアリン酸(1a)を基質として用い、反応温度250 ℃、反応時間3時間とし た条件下でRhおよびIr錯体を用いて検討を行った。結果をTable 1-1に示す。 Table 1-1 触媒として[RhCl(CO)2]2、配位子にPPh3を用いて検討を行ったところ、低収率 ながらヘプタデセンが得られた。この時、末端オレフィン2aと内部オレフィン 3aの選択性はほぼ1対1の比であった(entry 1)。バスカ錯体IrCl(CO)(PPh3)27)を 触媒に用いた場合、脱カルボニル化反応が良好に進行し、収率68%でオレフィン 得られた。さらに、生成物の選択性は2a/3a = 4/96となり、内部オレフィンが主
生成物であった(entry 2)。[IrCl(cod)]2/PPh3やIr(CO)3Cl/PPh3触媒系においても同
様に反応が進行し、内部オレフィンが主生成物として得られた(entry 3-4)。
13 触媒検討の結果、Ir触媒を用いることで期待した脂肪族カルボン酸の脱カルボ ニル化反応が良好に進行し、内部オレフィンが主生成物として得られることが 明らかとなった。なお、Ir触媒による脂肪族カルボン酸の脱カルボニル化は前述 したMillerらの報告中に一例見られるが、そこでは蒸留法による末端オレフィン の選択的合成のみが検討されている(Scheme 1-8)4b)。したがって、脂肪族カル ボン酸の脱カルボニル化反応におけるIr触媒の反応性は未知な部分が多く、本研 究によりそれを明らかにすることとした。 Scheme 1-8
14 続いて、Ir触媒系において反応の効率化を図るため、添加剤の検討を行った。 触媒量、反応温度、反応時間は固定。結果をTable 1-2に示す。 Table 1-2 IrCl(CO)(PPh3)2触媒系において、Millerらの報告例を参考に無水酢酸(以下、 Ac2Oと略)を添加したところ、収率に向上が見られた(entry 2)。興味深いこ とにヨウ化カリウム(以下、KIと略)を触媒に対して10当量用いた場合、Ac2O 添加系と同様の収率で、かつ完全な選択性で内部オレフィンのみが得られた (entry 3)。その他のカリウム塩としてKBrやKClを添加して検討を行ったが、 反応効率に低下が見られた(entry 4-5)。本検討で見出したKIの添加効果は、そ の他のIr触媒系においても確認された(entry 6-9)。 検討の結果、IrCl(CO)(PPh3)2触媒を用い、KIを添加した場合、内部オレフィン がほぼ完全な選択性で得られることが明らかとなったことから、entry 3を内部オ
15
レフィン合成における最適条件とした。KIの効果については、KIを添加しない
系においてIrI(CO)(PPh3)2を触媒として用いたところ、IrCl(CO)(PPh3)2/KI系と同
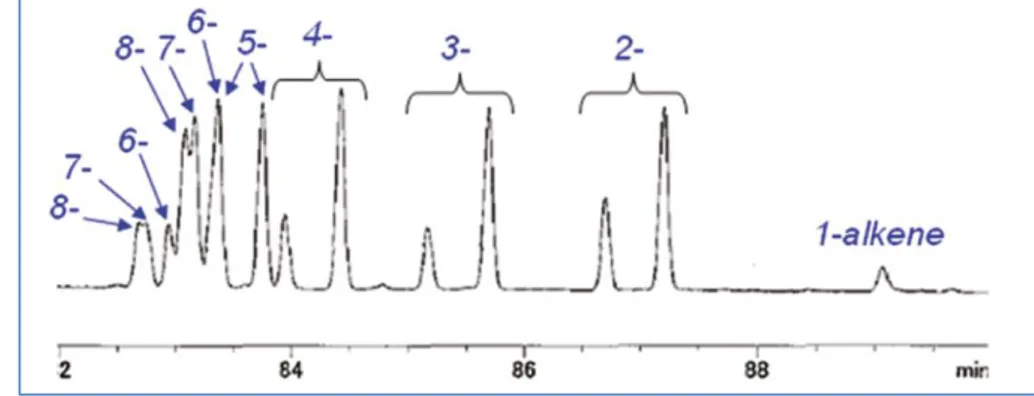
様の結果が得られたことから、KIによって触媒の配位子交換が起こり8)、触媒活 性を高めているものと考えられる(Scheme 1-9)。 Scheme 1-9 本反応において得られた内部オレフィン3aは、二重結合の位置に分布のある 異性体混合物である。このような化合物の二重結合の位置を決定する方法とし て、ジメチルジスルフィド化による誘導体処理後にガスクロマトグラフ質量分 析を行う方法が知られている9)。付加した位置で開裂したフラグメントイオンが 強く検出されるため、元の二重結合の位置を決定することができ、ガスクロマ トグラフィーを分析することで異性体の生成比を明らかにすることができる (Scheme 1-10)。 Scheme 1-10
16
既存の方法を用いて内部オレフィン3aのジメチルジスルフィド化を行った後
に、付加体をガスクロマトグラフィーおよびガスクロマトグラフ質量分析にて
解析した結果、2-ヘプタデセンから8-ヘプタデセンまで、位置異性体がほぼ均等
に生成していることが明らかとなった(Scheme 1-11、Figure 1-1、Figure 1-2)。
Scheme 1-11
Figure 1-1: ガスクロマトグラフィーの分析結果(数字は二重結合の位置)
17
2. 内部オレフィン合成における基質一般性
最適条件(Table 1-2, entry 3)を用い、様々な脂肪族カルボン酸からの内部オ
レフィン合成を行った。結果をTable 1-3に示す。
18 ステアリン酸よりも炭素鎖の短い脂肪酸1b-1eを用いた場合にも、対応する内 部オレフィン3b-3eが選択的に得られた(entry 1-4)。ステアリン酸よりも炭素 鎖の長い脂肪酸1f, 1gにおいても良好に反応は進行し、同様に内部オレフィンを 選択的に与えた(entry 5-6)。第2級の脂肪族カルボン酸1h, 1iを用いた場合、5 時間反応を行うことで対応する内部オレフィン3b, 3cをそれぞれ良好な収率で 得ることができた(entry 7-8)。環状の脂肪族カルボン酸1jにも本触媒系は適応 可能であり、期待した環状アルケン4を合成することができた(entry 9)。 続いて、本触媒系をα,β-不飽和カルボン酸および芳香族カルボン酸へ適応 した。これらの反応では脱炭酸型の反応が進行したものと考えられる10)。炭素数 16のα,β-不飽和カルボン酸1kを基質とした場合には、ペンタデセンが77%の 収率で得られた。生成物の選択性は内部オレフィンが93%となり、脂肪族カルボ ン酸の場合より若干低下する結果であった(Scheme 1-12)。また、ナフタレン カルボン酸1lを用いた場合には、ナフタレン5が93%の収率で得られた(Scheme 1-13)。 Scheme 1-12 Scheme 1-13
19 ヒドロキシカルボン酸の脱カルボニル化反応についても検討を行った。β位 にヒドロキシル基を有するカルボン酸1mを用いて、[IrCl(cod)]2/PPh3触媒系にて 検討を行ったところ、原料から一炭素減炭されたケトン6が83%の収率で得られ た(Scheme 1-14)。これは、カルボン酸の脱カルボニル化反応が進行してエノ ールが生成した後に、異性化することで得られたものと考えられる。また、副 生成物としてオレフィン3cが確認されたが、これは1mの分子内脱水反応が進行 してα,β-不飽和カルボン酸が生成した後に、先に述べた脱炭酸型反応が進行し て得られたものと考えている。 Scheme 1-14 また、12位にヒドロキシル基を有するカルボン酸1nを用いた場合には、脱カ ルボニル化に続いて、二重結合の異性化がヒドロキシル基炭素まで進行して生 成したと考えられるケトン7が低収率ながら得られた(Scheme 1-15)。なお、こ の場合には分子内脱水と脱カルボニル化反応により生成すると考えられるジエ ン3a’が主生成物として確認されたことから、ケトンの選択的な合成にはより温 和な条件が必要と考えられる。 Scheme 1-15
20 一方、α位にヒドロキシル基を有するカルボン酸1oを用いて検討を行った場 合、期待したアルデヒドは得られず、原料から2炭素減炭されたアルカン8が64% の収率で得られた(Scheme 1-16)。この反応は、カルボン酸の脱カルボニル化 によるエノールの生成、ケトエノール互変異性の後にアルデヒドが生成するも のの、アルデヒドの脱カルボニル化反応が逐次的に進行したことでアルカンが 得られたと考えている11)。 Scheme 1-16
21 3. 末端オレフィン合成における反応条件の最適化 内部オレフィン合成の反応系において、反応温度の低温化を検討したところ、 末端オレフィンの生成が優先することが明らかとなった。そこで、末端オレフ ィンの選択的合成を目指し、検討を行うこととした。以下、条件検討の結果を Table 1-4に示す。 Table 1-4 内部オレフィンが選択的に得られる条件において、反応温度を200 ℃に下げ 検討を行った場合、反応は全く進行せず、原料が回収された(entry 2)。添加剤 としてAc2Oを1当量用いたところ、オレフィンが低収率ながら得られ、末端オレ
フィンが主生成物として確認された(entry 3)。興味深いことに、KIとAc2Oを
併用した場合には収率に向上が見られた(entry 4)。さらに反応温度を下げ160 ℃
で検討を行ったところ、オレフィン収率は低下したが、末端オレフィンの選択
22
収率と末端オレフィンの選択性が共に向上した(entry 6)。Ac2Oの当量数を2当
量に増やして同様に検討を行ったところ、選択性にほぼ変化はなく、オレフィ ンが72%の収率で得られた(entry 7)。反応時間を5時間に延長することで、収 率は84%にまで向上した(entry 8)。 以上の検討の結果、KIとAc2Oを添加し、反応温度を下げて脂肪族カルボン酸 の脱カルボニル化反応を行うことで末端オレフィンが選択的に合成可能となる ことを明らかにした。
23
4. 末端オレフィン合成における基質一般性
最適条件(Table 1-4, entry 8)を用い、様々な脂肪族カルボン酸からの末端オ
レフィン合成を検討した。結果をTable 1-5に示す。
24 ステアリン酸よりも炭素鎖の短いカルボン酸1b-1eを用いた場合も、反応は問 題なく進行し、対応する末端オレフィン2b-2eが良好な収率で得られた(entry 1-4)。ステアリン酸よりも炭素鎖の長い基質1f, 1gにおいても結果は同様であっ た(entry 5-6)。末端にフェニル基を有する基質1p, 1q、およびシクロヘキシル 基を有する基質1rにおいても本触媒系は適応可能であった(entry 7-9)。末端に エステル基を有するカルボン酸1sの場合、エステル基は保持され、カルボン酸側 でのみ反応が進行した末端オレフィン2sが得られた(entry 10)。 本触媒系においては、第二級カルボン酸の脱カルボニル化反応は進行しなか った(Scheme 1-17)。メチル基の立体障害により、酸化的付加段階が阻害され たため、反応が進行しなかったことが考えられる。 Scheme 1-17 また、ヒドロキシカルボン酸を本触媒反応の条件においた時には、脱水反応 によるα,β-不飽和カルボン酸の生成が優先した(Scheme 1-18)。 Scheme 1-18
25 第3節 反応機構について 本反応の反応機構は以下のように考えられる(Scheme 1-19)。まず、2分子の カルボン酸、あるいはカルボン酸とAc2Oとが縮合し、原料由来の酸無水物が生 成する。続いて、酸無水物のカルボニル炭素-酸素結合がIrへ酸化的付加し、ア シルイリジウム種Aが生成後、カルボニル基が一酸化炭素として脱離し、アルキ ルイリジウム種Bが生成する12)。ついで、β-水素脱離により末端オレフィンが 生成し、同時にイリジウムヒドリド種Cが生成後、還元的脱離によりカルボン酸 が生成し、元の触媒が再生する。ここで、生成したイリジウムヒドリド種Cは異 性化触媒としても機能する。すなわち、生成したα-オレフィンにCが付加し、 アルキルイリジウム種Dが生成後、再びβ水素脱離が起こることで異性化が進行 し、内部オレフィンを与える13)。一方で、α,β-不飽和カルボン酸や芳香族カ ルボン酸を用いた場合には、脱離可能なβ水素が存在していないため、アシル イリジウム種カルボン酸による加水分解を受けて中間体Cが生成後、脱炭酸、続 く還元的脱離により生成物が得られるものと考えられる。 Scheme 1-19
26 第4 節 Fe 触媒を用いる脂肪族カルボン酸の脱カルボニル化反応 脂肪族カルボン酸の脱カルボニル化反応は、緒言で示したようにPd や Rh 触 媒を用いた反応例がほとんどであり、その他の遷移金属触媒による検討はこれ までにほとんど行われていなかった。第2 節では、Ir 触媒が脂肪族カルボン酸の 脱カルボニル化反応を効率的に進行させる効果的な触媒であることを示したが、 これらの貴金属触媒は希少かつ高価な化合物であるため、本反応の実用化を考 えた場合、より安価な触媒系の開発が必要となってくる。このような背景の下、 著者は遷移金属触媒の中でも最も豊富かつ安価なFe に着目した。近年、Fe 触媒 に特有の新規合成反応や、希少金属触媒の代替としてのFe 触媒反応が注目を集 めており、元素戦略の観点からもFe による触媒反応の開発は重要な研究課題と なっている。以下にFe 触媒を用いた反応例をいくつか示す14)-18)。
Scheme 1-20: 熊田-玉尾-Corriu 型クロスカップリング反応17a)
Scheme 1-21: 根岸型クロスカップリング反応17b)
27 前述した反応例は、従来Pd や Ni 触媒が用いられている反応系を Fe 触媒で置 き換えた例であり、Fe 触媒系においてもクロスカップリング反応や C-H アリ ール化反応が進行することが示されている。一方、Fe 触媒による減炭型の反応 例は、カルボン酸やその誘導体も含めこれまでに全く達成されていない。そこ で、著者は脂肪族カルボン酸の脱カルボニル化反応において、Fe 触媒による効 率的反応系の開発を目的とし検討を行うこととした。以下に検討結果を述べる。 1. 内部オレフィン合成における反応条件の最適化 ステアリン酸(1a)を基質とし、触媒に FeCl2、配位子にPPh3を用い、250 ℃、 3 時間の条件において添加剤の検討を行った。結果を Table 1-6 に示す。 Table 1-6 PPh3を20 mol %用いた系において添加剤を何も用いない場合、反応は全く進 行しなかった(entry 1)。KI を触媒に対して 10 当量添加したところ、低収率で はあるがオレフィンが得られ、脱カルボニル化反応がFe 触媒でも進行すること がわかった。なお、この時生成物の選択性は内部オレフィンが優先した(entry 2)。 この系において反応時間を10 時間に延長したところ、内部選択性に向上が見ら
28 れたが、オレフィン収率は低収率に留まった(entry 3)。PPh3を40 mol %用い た場合、触媒活性が大きく向上し、収率は67%、内部選択性は 97%となった(entry 4)。さらに Ac2O を添加して検討を行ったところ、オレフィン収率は 81%に向 上したが、内部選択性に低下が見られた(entry 5)。KI、Ac2O 添加系において は、ホスフィン配位子なしでも反応は中程度の収率で進行することを確認した (entry 6)。 以上、検討の結果、Fe 触媒系においても脂肪族カルボン酸の脱カルボニル化 反応は進行し、内部オレフィンが最高 97%の選択性で得られることが明らかと なった。また、Ir 触媒系と同様に本系においても KI の添加効果が見られた。続 いて、さらなる反応の効率化を図るべく、触媒検討を行った。結果を Table 1-7 に示す。 Table 1-7
FeCl2に代えFeI2を触媒として用いた場合、Ac2O のみの添加で entry 1 と同様
29 ことが考えられる(entry 1-3)19)。ハロゲン配位子を有していない錯体として種々 のカルボニル錯体を検討したが、触媒活性を示さなかった(entry 4-6)。ヨウ素 配位子を有するカルボニル錯体を用いた場合、活性はFeCl2触媒系には劣るもの の、KI を添加せずとも反応は進行した(entry 7-8)。 検討の結果、FeCl2 よりも活性の高い触媒系は見出すことができなかったが、 ヨウ素配位子を有するカルボニル錯体でも脱カルボニル化反応が進行すること が明らかとなった。そこで、FeCl2/PPh3触媒系において系中でカルボニル錯体を 発生させるべく、一酸化炭素加圧条件にて反応を検討した。結果をTable 1-8 に 示す。 Table 1-8 オートクレーブを用い、一酸化炭素を5 atm 加圧して検討を行ったところ、触 媒活性が向上し、オレフィンがほぼ定量的に得られた。また、これまでの系に おいては内部オレフィンの生成が優先したが、本系においては興味深いことに 末端オレフィンの生成比が増加した(entry 1-2)。反応時間を短縮したところ、 反応は10 分間でほぼ終了していることがわかり、時間の短縮により末端オレフ
ィンの選択性に向上が見られた(entry 3)。CO の圧力を 10、20 atm と上げるに
30 これらの検討の結果、一酸化炭素を加圧することで触媒活性が飛躍的に向上 することが明らかとなった 20)。さらに、本系においては末端オレフィンの生成 が優先することも明らかとなり、詳細な条件検討によりFe 触媒による末端オレ フィンの選択的合成が可能になるものと期待できる。そこで、さらなる検討を 行った。 2. 末端オレフィン合成における反応条件の最適化および一般性 FeCl2/PPh3触媒系においてCO の圧力、反応温度、反応時間について検討を行 った。結果をTable 1-9 に示す。 Table 1-9
31 CO を 20 atm に加圧した系において反応温度を 250 ℃より下げた場合には、 オレフィン収率に大きな低下が見られた(entry 2-3)。一方、末端オレフィンの 選択性は反応温度を下げるにつれ向上し、反応温度 220 ℃の条件においては 99%以上の選択性で末端オレフィンのみが得られたが、収率はわずか 11%であっ た(entry 4)。 反応温度 220 ℃において、収率向上のため反応時間の検討を 行ったところ、反応時間を延長した系においても選択性はほぼ維持されたがオ レフィン収率は中程度に留まった(entry 5-6)。反応温度 220 ℃の系で CO 圧を 10 atm に下げて検討を行ったところ、収率が 94%に向上したが、末端選択性は 低下した(entry 7)。 反応温度を 200 ℃に下げた場合、1 時間の反応ではオレ フィン収率は中程度であったが、末端選択性は97%と良好であった(entry 8)。 反応時間を延長すると収率は大きく向上したが、末端選択性は 89%に低下した (entry 9)。180 ℃で検討を行った場合、10 時間反応を行ってもオレフィンは
低収率であった(entry 11)。CO 圧を 2 atm まで下げた場合、160 ℃でも反応は
良好に進行したが、長時間の反応のために内部オレフィンの生成比が高くなっ た(entry 12-13)。
32 PPh3 を配位子とした本触媒系では、末端オレフィンを収率良くかつ選択的に 得ることは困難であると判断し、次に異なるホスフィン配位子の検討を行った。 結果をTable 1-10 に示す。 Table 1-10 単座のホスフィン配位子としてPCy3を用いた場合、反応は全く進行しなかっ た(entry 2)。P(o-Tol)3配位子の場合には、低収率であるが末端オレフィンの生 成が優先した(entry 3)。二座ホスフィン配位子として DPPM を用いた場合、 反応時間10 分ではオレフィンが得られなかったため、反応時間を 3 時間に延長 したところ、オレフィンが中程度の収率で得られた。また、この系においても
33 末端オレフィンが主生成物として得られた(entry 4-5)。様々な二座配位子を用 いて検討を行ったところ、DPPPent を用いた場合に最も触媒活性が向上し、収率 は91%、さらに末端選択性は 92%に向上した(entry 6-10)。DPPPent を用いた 系において、反応温度 240 ℃で検討を行った場合、収率に若干の低下が見られ たが、末端選択性は97%に向上した(entry 11)。バイトアングルの大きな二座 配位子としてDPE-phos を用いた系においても選択性は良好であったが、収率は 中程度に留まった(entry 12)。なお、ホスフィン配位子を用いない場合におい ても本反応は進行することがわかったが、末端選択性は低下した。 検討の結果、配位子に DPPPent を用いた場合、反応が良好に進行し、末端オ レフィンが97%の選択性で得られることが明らかとなった。そこで、entry 11 を 最適条件とし、基質一般性を確認することとした。
34
3. 末端オレフィン合成における基質一般性
最適条件を用い、様々な脂肪族カルボン酸からの末端オレフィン合成を検討
した。結果をTable 1-11 に示す。
35 炭素鎖の長さにかかわらず、対応する末端オレフィン2b-2g を高選択性で合成 することができた(entry 1-6)。フェニル基を有する基質 1p, 1q の場合、3 時間 反応を行ったところ、対応する末端オレフィンの収率は中程度であったが、反 応時間を延長することで収率に改善が見られた。しかし、反応時間の延長に伴 い、選択性に若干の低下が見られた(entry 7-10)。β位にメチル基を有するカ ルボン酸1t を用いて検討を行ったところ、二置換の末端オレフィン 2t とこれが 異性化した三置換オレフィン3tのみが得られ、生成比は92/8であった(entry 11)。 第2 級カルボン酸も適応可能であった(entry 12)。Ir 触媒系と同様に本触媒系 においてもエステル基は許容された(entry 13)。末端にヒドロキシル基を有す る基質 1u の場合、Ac2O を二当量用いたところ、アセチル化されたオレフィン 2u が良好な収率、末端選択性で得られた(entry 14)。チオール部位を有する基 質においてもアセチル化されたオレフィン 2v が得られたが、低収率であった (entry 15)。
36 第5 節 反応機構に関する考察 Fe 錯体を用いた触媒反応において、しばしば Fe 錯体ではなく、錯体に含まれ ている不純物が真の触媒種として機能することが報告されている 21)。そこで、 本触媒系においてもその確認を行った(Scheme 1-23)。 Scheme 1-23 これまで用いていた純度98%の FeCl2に代えて、純度99.998%の FeCl2を用い てCO 加圧系にて検討を行ったところ、収率、選択性ともに同様の結果を与えた。 触媒を用いない条件において、反応は全く進行しなかったことから KI や DPPPent などに含まれている不純物が反応を触媒している可能性も否定された。 これにより、本反応はFe 触媒によって進行していることを明らかとした。 また、Fe 触媒を用いる反応においては、しばしばラジカル機構が提唱される。 そこで、本系におけるラジカル種の関与を確認すべく、ラジカル捕捉剤を添加 して検討を行った(Scheme 1-24)。
37 Scheme 1-24 ラジカル捕捉剤としてTEMPO、Galvinoxyl、dilylbenzene を添加して前述の条 件にて検討を行ったところ、反応は阻害されず良好に進行した。よって、本反 応はラジカル機構を経たものではないと考えられる。 次に、酸無水物からの反応を検討した。ステアリン酸無水物1a’を基質として 用いて無水酢酸を添加しない条件にて検討を行ったところ、オレフィンが 94% の収率で得られ、同時にステアリン酸が 93%の収率で生成していることがわか った(Scheme 1-25)。この結果は、本反応の真の反応基質が酸無水物であるこ と、さらにはオレフィンに相当する量のカルボン酸が生成して反応が進行して いることを示している。 Scheme 1-25
38 本反応おいて発生したガス成分について、ガスクロマトグラフィーによる分 析を行った。その結果、15 mmol のステアリン酸から、12.8 mmol の一酸化炭 素と少量のメタンおよび水素が発生していることがわかり、二酸化炭素は全く 検出されなかった(Scheme 1-26)。これにより、本反応が脱炭酸反応ではなく、 一酸化炭素の脱離を経た脱カルボニル化反応であると結論される。 Scheme 1-26 以上の検討結果より、反応機構を次のように推定した(Scheme 1-27)。まず、 カルボン酸1とAc2Oとが縮合し、原料由来の酸無水物1'が生成後、FeCl2、配位子、 KI、COより生成した活性種Aと1'とが反応し、アシルFe錯体Bが生成する。続い て、脱カルボニル化反応によりアルキルFe錯体Cが生成後22), 23)、β水素脱離に より、末端オレフィン2とFeヒドリド錯体Dが生成する。最後にAcOHの還元的 脱離により、活性種Aが再生し、触媒サイクルが完結する。なお、異性化段階は Feヒドリド錯体により進行しているものと考えられる。すなわち、Feヒドリド がオレフィンの二重結合に付加し、再びβ水素脱離が起こることで異性化が進 行するのだが、本系においては一酸化炭素がFe中心に配位することでオレフィ ンの付加が妨げられ、その結果異性化反応が抑制されていると考えている。ま た、一酸化炭素加圧系において、反応終了後に赤外吸収スペクトルを測定した ところ、金属カルボニルと思われるピークを確認した(1941, 2001, 069 cm-1)。 このことから系中ではFeカルボニル錯体が生成しているものと考えられるが、 現在のところ錯体の構造決定には至っていない。
39
40 第6節 結論 長鎖脂肪族カルボン酸の脱カルボニル化反応によるオレフィン合成に関して 検討を行った結果、以下に示す結果を得ることができた。 1. Ir触媒を用いることで内部オレフィンの選択的合成が効率よく達成されるこ とを明らかとした。本反応においてはKIの添加が有効であり、その効果とし ては配位子交換による触媒活性の向上が考えられる。また、α-ヒドロキシ カルボン酸やβ-ヒドロキシカルボン酸を基質として用いた場合には、二炭 素減炭反応によるアルカン合成や一炭素減炭反応によるケトン合成が可能 なことも明らかとした。 2. Ir錯体を触媒とし添加剤としてKIとAc2Oを用いた場合には、より低温下で脱 カルボニル化反応を行うことが可能となった。そして、これにより末端オレ フィンの選択的合成が達成されることも明らかとした。Ir触媒による末端オ レフィン合成法としては、蒸留法による合成例が一例のみ報告されていたが、 本系においてはそのような特殊な実験操作は必要なく、より簡便な方法で末 端オレフィンを得ることができる(Scheme 1-28)。 3. 反応機構としては、まず脂肪族カルボン酸から系中で酸無水物が生成し、こ れがイリジウムへ酸化的付加することでアシル錯体が生成する。続いて、脱 カルボニル化によりアルキル錯体が生成し、β水素脱離、還元的脱離が起こ ることで反応が進行しているものと考えられる。 4. 脂肪族カルボン酸の脱カルボニル化反応が遷移金属触媒の中でも最も安価 なFe触媒により進行することを新たに見出した。本系においてもIr触媒系と 同様に、KIの添加が有効であった。さらに、Fe触媒系においても内部オレフ ィンと末端オレフィンの選択的合成が可能であり、特に末端オレフィン合成 については一酸化炭素加圧の加圧が効果的であることを新たに見出した (Scheme 1-29)。 5. Fe触媒系において、反応機構解明に関する検討を行った結果、本反応がラジ カル機構ではないこと、また、一酸化炭素の脱離を伴った脱カルボニル化反
41 応であることを証明した。カルボン酸に限らず、酸塩化物などのカルボン酸 誘導体も含め、Fe触媒による脱カルボニル化反応はこれまでに報告されてお らず、本研究によりFe触媒の新たな反応性を見出すことができた。 Scheme 1-28 Scheme 1-29
42
第7節 実験の部
Ir 触媒を用いる脂肪族カルボン酸からの内部オレフィン合成
5 mL のネジ付き試験官に撹拌子、ステアリン酸 (1a, 142.2 mg, 0.50 mmol)、 IrCl(CO)(PPh3)2(7.8 mg, 0.01 mmol)、 KI(16.6 mg, 0.10 mmol)を加え、窒素置
換を3 回行った後、窒素下にて 250 C で 3 時間加熱撹拌を行った。反応終了後、 室温まで冷却し、ろ過、減圧濃縮により粗製を得た。これをシリカゲルカラム クロマトグラフィー(展開溶媒: hexane)により精製することで、ヘプタデセン (108.5 mg, 91%, 2a/3a = 1/>99)を得た。 Ir 触媒を用いる脂肪族カルボン酸からの末端オレフィン合成 5 mL のネジ付き試験官に撹拌子、ステアリン酸 (1a, 143.1 mg, 0.50 mmol)、 IrCl(CO)(PPh3)2(20.0 mg, 0.026 mmol)、KI(16.6 mg, 0.25 mmol)、Ac2O(104.0
mg, 1.02 mmol)を加え、窒素置換を 3 回行った後、窒素下にて 160 C で 5 時間 加熱撹拌を行った。反応終了後、室温まで冷却し、ろ過、減圧濃縮により粗製 を得た。これをシリカゲルカラムクロマトグラフィー(展開溶媒: hexane)によ り精製することで、ヘプタデセン(100.5 mg, 84%, 2a/3a = 98/2)を得た。 内部オレフィン3a の二重結合位置の決定9) 5 mL のネジ付き試験官に撹拌子、ヘプタデセン(3a, 60.3 mg, 0.25 mmol)、I2 (29.8 mg, 0.12 mmol)、ジメチルジスルフィド (1 mL)を加え、窒素置換を行 った後に、窒素下にて室温で 2 時間撹拌を行った。反応終了後、30% NaHSO3 水溶液を加え、I2 の還元した後に、hexane/Et2O(1/1)で抽出した。そして、有機
層の分析をガスクロマトグラフィー(initial temperature 60 C, initial time 10 min, rate 2 C/min, final temperature 350 C, final time 15 min) とガスクロマトグラフ質 量分析計(initial temperature 60 C, rate 2 C/min, final temperature 300 C, final time 5 min)を用いて行った。
Fe 触媒を用いる脂肪族カルボン酸からの内部オレフィン合成
5 mL のネジ付き試験官に撹拌子、ステアリン酸 (1a, 143.5 mg, 0.50 mmol)、
FeCl2(6.6 mg, 0.05 mmol)、KI(83.3 mg, 0.50 mmol)を加え、窒素置換を 3 回
行った後、窒素下にて250 C で 10 時間加熱撹拌を行った。反応終了後、室温ま
43
トグラフィー(展開溶媒: hexane)により精製することで、ヘプタデセン(80.1 mg,
67%, 2a/3a = 3/97)を得た。
Fe 触媒を用いる脂肪族カルボン酸からの末端オレフィン合成
30 mLのステンレス製オートクレーブへ撹拌子、ステアリン酸(1a, 285.0 mg, 1.0 mmol), FeCl2(13.0 mg, 0.10 mmol), DPPPent(88.4 mg, 0.20 mmol), KI(167.2
mg, 1.0 mmol), Ac2O(103.4 mg, 1.0 mmol)加え、一酸化炭素(10 atm)で3回置
換した後、20 atmまで加圧し、240 ℃で3時間加熱撹拌を行った。反応終了後、 室温まで冷却して一酸化炭素を抜き、セライトろ過、減圧濃縮により粗製を得 た。これをシリカゲルクロマトグラフィー(展開溶媒: hexane)により精製する ことでヘプタデセン(176.6 mg, 74%, 2a/3a = 97/3)を得た。 スペクトルデータ Heptadecenes (3a)
Obtained as an inseparable mixture (terminal/internal = 1/>99); colorless oil. 1H NMR (500 MHz, CDCl3): 5.38-5.47 (m. 2H), 1.95-2.05 (m, 4H), 1.26-1.38 (m, 22H),
0.87-0.98 (m, 6H).
Hexadecenes (3b)
Obtained as an inseparable mixture (terminal/internal = 1/>99); colorless oil. 1H NMR (500 MHz, CDCl3): 5.32-5.47 (m. 2H), 1.95-2.03 (m, 4H), 1.25-1.40 (m, 20H),
0.88-0.97 (m, 6H).
Pentadecenes (3c)
Obtained as an inseparable mixture (terminal/internal = 1/99); colorless oil. 1H NMR (500 MHz, CDCl3): 5.32-5.47 (m. 2H), 1.95-2.03 (m, 4H), 1.25-1.38 (m, 18H),
44
0.88-0.98 (m, 6H).
Tetradecenes (3d)
Obtained as an inseparable mixture (terminal/internal = 1/99); colorless oil. 1H NMR (500 MHz, CDCl3): 5.32-5.47 (m. 2H), 1.95-2.05 (m, 4H), 1.26-1.40 (m, 16H),
0.87-0.98 (m, 6H).
Tridecenes (3e)
Obtained as an inseparable mixture (terminal/internal = 1/>99); colorless oil. 1H NMR (500 MHz, CDCl3): 5.35-5.45 (m. 2H), 1.96-2.02 (m, 4H), 1.26-1.38 (m, 14H),
0.87-0.98 (m, 6H).
Nonadecenes (3f)
Obtained as an inseparable mixture (terminal/internal = 1/>99); colorless oil. 1H NMR (500 MHz, CDCl3): 5.32-5.41 (m. 2H), 1.96-2.02 (m, 4H), 1.25-1.36 (m, 26H),
0.86-0.98 (m, 6H).
Henicosenes (3g)
Obtained as an inseparable mixture (terminal/internal = 1/>99); colorless oil. 1H NMR (500 MHz, CDCl3): 5.32-5.45 (m. 2H), 1.95-2.03 (m, 4H), 1.25-1.38 (m, 30H),
45
Cyclododecene (4)24
Obtained as an inseparable mixture (E/Z = 78/22); colorless oil. E isomer: 1H NMR (500 MHz, CDCl3): 1.25-1.36 (m, 12H), 1.40-1.47 (m, 4H), 2.05-2.06 (m, 4H), 5.37 (m, 2H); 13C NMR (125 MHz, CDCl 3):131.40, 32.13, 26.24, 25.60, 24.95, 24.60. Z isomer: 1H NMR (500 MHz, CDCl 3): 1.25-1.39 (m, 12H), 1.40-1.49 (m, 4H), 2.09-2.13 (m, 4H), 5.32 (m, 2H); 13C NMR (125 MHz, CDCl3):130.34, 26.95. 24.35, 23.93, 22.05. 2-Pentadecanone (5)
white solid. mp 39.0 C (lit.25 mp 38.9 C); 1H NMR (500 MHz, CDCl
3): 0.88 (t, J =
6.9 Hz, 3H), 2.41 (t, J = 7.8 Hz, 2H), 2.13 (s, 3H), 1.55-1.58 (m, 2H), 1.25-1.31 (m, 20H). The NMR data were identical with those of a commercial sample of 2-pentadecanone.
1-Heptadecene (2a)26
Obtained as an inseparable mixture (2a/3a = 97/3); colorless oil. 1H NMR (500 MHz,
CDCl3): 5.77-5.86 (m, 1H), 4.92-5.01 (m, 2H), 2.02-2.06 (m, 2H), 1.25-1.39 (m,
26H), 0.88 (t, J = 6.9 Hz, 3H); 13C NMR (125 MHz, CDCl
3): 139.19, 114.08, 33.90,
32.01, 29.78, 29.60, 29.46, 29.24, 29.03, 22.76, 14.14.
1-Hexadecene (2b)
Obtained as an inseparable mixture (2b/3b = 98/2); colorless oil. 1H NMR (500 MHz,
CDCl3): 5.77-5.86 (m, 1H), 4.91-5.01 (m, 2H), 2.01-2.08 (m, 2H), 1.26-1.39 (m,
24H), 0.88 (t, J = 6.9 Hz, 3H); 13C NMR (125 MHz, CDCl
3): 139.24, 114.06, 33.84,
46
with those of a commercial sample of 1-hexadecene.
1-Pentadecene (2c)26
Obtained as an inseparable mixture (2c/3c = 97/3); colorless oil. 1H NMR (500 MHz, CDCl3): 5.78-5.86 (m, 1H), 4.92-5.01 (m, 2H), 2.02-2.06 (m, 2H), 1.26-1.39 (m,
22H), 0.88 (t, J = 6.9 Hz, 3H); 13C NMR (125 MHz, CDCl
3): 139.25, 114.06, 33.85,
31.95, 29.70, 29.53, 29.38, 29.18, 28.97, 22.71, 14.12.
1-Tetradecene (2d)27
Obtained as an inseparable mixture (2d/3d = 97/3); colorless oil. 1H NMR (500 MHz, CDCl3): 5.78-5.86 (m, 1H), 4.92-5.01 (m, 2H), 2.01-2.06 (m, 2H), 1.26-1.39 (m,
20H), 0.88 (t, J = 6.9 Hz, 3H); 13C NMR (125 MHz, CDCl
3): 139.15, 114.08, 33.91,
32.03, 29.77, 29.63, 29.47, 29.27, 29.05, 22.77, 14.12.
1-Tridecene (2e)28
Obtained as an inseparable mixture (2e/3e = 97/3); colorless oil. 1H NMR (500 MHz,
CDCl3): 5.77-5.86 (m, 1H), 4.91-5.01 (m, 2H), 2.02-2.06 (m, 2H), 1.26-1.39 (m,
18H), 0.88 (t, J = 6.9 Hz, 3H); 13C NMR (125 MHz, CDCl
3):139.23, 114.07, 33.87,
31.97, 29.69, 29.57, 29.40, 29.21, 29.01, 22.73, 14.12.
1-Nonadecene (2f)29
Obtained as an inseparable mixture (2f/3f = 98/2); colorless oil. 1H NMR (500 MHz,
CDCl3): 5.77-5.86 (m, 1H), 4.91-5.01 (m, 2H), 2.01-2.06 (m, 2H), 1.25-1.39 (m,
30H), 0.88 (t, J = 6.9 Hz, 3H); 13C NMR (125 MHz, CDCl
3): 139.20, 114.07, 33.87,
47
1-Henicosene (2g)30
Obtained as an inseparable mixture (2g/3g = 98/2); white solid. mp 32.3 C (lit.31 mp
35.5 C); 1H NMR (500 MHz, CDCl3): 5.77-5.86 (m, 1H), 4.91-5.01 (m, 2H), 2.01
-2.06 (m, 2H), 1.25-1.39 (m, 34H), 0.88 (t, J = 6.9 Hz, 3H); 13C NMR (125 MHz, CDCl3):139.23, 114.07, 33.86, 31.96, 29.73, 29.55, 29.41, 29.20, 28.99, 22.72, 14.13.
Methyl 8-nonenate (2s)32
Obtained as an inseparable mixture (2s/3s = 95/5); colorless oil. 1H NMR (500 MHz, CDCl3): 5.76-5.84 (m, 1H), 4.92-5.01 (m, 2H), 3.66 (s, 3H), 2.30 (t, J = 7.3 Hz,
2H), 2.01-2.06 (m, 2H), 1.57-1.65 (m, 2H), 1.31-1.41 (m, 6H); 13C NMR (125 MHz, CDCl3):174.24, 138.96, 114.24, 51.41, 34.04, 33.66, 28.94, 28.66, 24.86.
2-Methyl-l-tetradecene (2t)33
Obtained as an inseparable mixture (2t/3t = 92/8); colorless oil. 1H NMR (500 MHz,
CDCl3): 5.77-5.86 (m, 1H), 4.91-5.01 (m, 2H), 2.02-2.06 (m, 2H), 1.26-1.39 (m,
18H), 0.88 (t, J = 6.9 Hz, 3H); 13C NMR (125 MHz, CDCl
3):139.23, 114.07, 33.87,
31.97, 29.69, 29.57, 29.40, 29.21, 29.01, 22.73, 14.12.
13-Tetradecenyl acetate (2u)34
Obtained as an inseparable mixture (2u/3u = 96/4); colorless oil. 1H NMR (500 MHz,
CDCl3): 5.76-5.84 (m, 1H), 4.91-5.01 (m, 2H), 3.66 (s, 3H), 2.30 (t, J = 7.6 Hz,
2H), 2.01-2.08 (m, 2H), 1.59-1.65 (m, 2H), 1.31-1.41 (m, 6H); 13C NMR (125
MHz, CDCl3): 171.18, 139.19, 114.05, 64.62, 33.96, 29.57, 29.48, 29.23, 29.12, 28.92,
48
第8 節 参考文献
1. Vogt, D. in Applied Homogeneous Catalysis with Organometallic Compounds, ed. B. Cornils and W. A. Herrmann, Wiley-VCH, Weinheim, 1996, p. 245.
2. (a) Skupińska, J. Chem. Rev. 1991, 91, 613. (b) Mecking, S. Angew. Chem., Int. Ed.
2001, 40, 534.
3. (a) Biermann, U.; Friedt, W.; Lang, S; Lühs, W.; Machmüller, G.; Metzger, J. O.; Klaas, M. R.; Schäfer, H. J..; Schneider, M. P. Angew. Chem., Int. Ed. 2000, 39, 2206. (b) Hill, K. Pure Appl. Chem. 2000, 72, 1255. (c) Eissen, M.; Metzger, J. O.; Schmidt, E.; Schneidewind, U. Angew. Chem., Int. Ed. 2002, 41, 414. (d) Demirbaş, A. Energy Convers. Manage. 2003, 44, 2093. (e) Dyer, J. M.; Stymne, S.; Green, A. G.; Carlsson, A. S. Plant J. 2008, 54, 640.
4. (a) Fenton, D. M. U.S. Patent 3,530,198, 1970. (b) Miller, J. A.; Nelson, J. A.; Byrne, M. P. U.S. Patent 5,077,447, 1991. (c) Miller, J. A.; Nelson, J. A.; Byrne, M. P. J. Org. Chem. 1993, 58, 18. (d) Goossen, L. J.; Rodríguez, N. Chem. Commun.
2004, 724. (e) Nôtre, J. L.; Scott, E. L.; Franssen, M. C. R.; Sanders, J. P. M. Tetrahedron Lett. 2010, 51, 3712. (f) Miranda, M. O.; Pietrangelo, A.; Hillmyer, M.
A.; Tolman, W. B. Green Chem. 2012, 14, 490.
5. Foglia, T. A.; Barr, P. A. J. Am. Oil Chem. Soc. 1976, 53, 737.
6. (a) Zhang, J. J.; Lai, S.-M. U.S. Patent 6,348,132, 2002. (b) Brown, D. S.; Doll, M. J. PCT Int. Appl.WO2005031066, 2005. (c) Baralt, E. J.; King, C. A. U.S. Patent 6,054,629, 2000. (d) Brown, D. S.; Doll, M. J. U. S. Pat. Appl. Publ. 20050070747, 2005. (e) Saruwatari, T.; Fujikawa, N.; Yamane, H. U.S. Patent 7,718,580, 2010. 7. Vaska, L.; DiLuzio, J. W. J. Am. Chem. Soc. 1961, 83, 2784.
8. (a) Chock, P. B.; Halpern, J. J. Am. Chem. Soc. 1966, 88, 3511. (b) McGinnety, J. A.; Doedens, R. J.; Ibers, J. A. Inorg, Chem. 1967, 6, 2243. (c) Clark, G. R.; Lu, G.-L.; Roper, W. R.; Wright, L. J. Organometallics 2007, 26, 2167.
9. Buser, H.-R.; Arn, H.; Guerin, P.; Rauscher, S. Anal. Chem. 1983, 55, 818.
10. For transition-metal-catalyzed decarboxylation of carboxylic acids, see the following. Pd: (a) Dickstein, J. S.; Mulrooney, C. A.; O’Brien, E. M.; Morgan, B. J.; Kozlowski, M. C. Org. Lett. 2007, 9, 2441. Cu: (b) Goossen, L. J.; Thiel, W. R.; Rodríguez, N.; Linder, C.; Melzer, B. Adv. Synth. Catal. 2007, 349, 2241. Ag: (c) Goossen, L. J.; Linder, C.; Rodríguez, N.; Lange, P. P.; Fromm, A. Chem. Commun.
49
2009, 7173.
11. For Ir-catalyzed decarbonylation of aldehydes, see: Iwai, T.; Fujihara, T.; Tsuji, Y.
Chem. Commun. 2008, 6215.
12. For oxidative addition of carboxylic acid anhydrides to transition-metal complexes to generate acyl metals, see the following. Ir: (a) Blake, D. M.; Shields, S.; Wyman, L. Inorg. Chem. 1974, 13, 1595. Rh: (b) Miller, J. A.; Nelson, J. A.
Organometallics 1991, 10, 2958. Pd: (c) Nagayama, K.; Kawataka, F.; Sakamoto,
M.; Shimizu, I.; Yamamoto, A. Chem. Lett. 1995, 367.
13. For IrH-catalyzed isomerization of carbon-carbon double bonds, see the following. (a) Bingham, D.; Webster, D. E.; Wells, P. B. J. Chem. Soc., Dalton Trans. 1974, 1514. (b) Chin, C. S.; Park, J.; Kim, C.; Lee, S. Y.; Shin, J. H.; Kim, J. B. Catal.
Lett. 1988, 1, 203. (c) Cooper, A. C.; Caulton, K. G. Inorg. Chim. Acta 1996, 251,
41. (d) Yamamoto, Y.; Miyairi, T.; Ohmura, T.; Miyaura, N. J. Org. Chem. 1999,
64, 296.
14. (a) Bolm, C.; Legros, J.; Paih, J. L.; Zani, L. Chem. Rev. 2004, 104, 6217. (b) Enthaler, S.; Junge, K.; Beller, M. Angew. Chem. Int. Ed. 2008, 47, 3317; (c) Correa, A.; Manchenô, O. G.; Bolm, C. Chem. Soc. Rev. 2008, 37, 1108. (d) Sherry, B.; Fürstner, A. Acc. Chem. Res. 2008, 41, 1500. (e) Czaplik, W. M.; Mayer, M.; Cvengroš, J.; von Wangelin, A. J. ChemSusChem, 2009, 2, 396.
15. For selected examples of iron-catalyzed reduction, see: (a) Bart, S. C.; Lobkovsky, E.; Chirik, P. J. J. Am. Chem. Soc. 2004, 126, 13794. (b) Casey, C. P.; Guan, H. J.
Am. Chem. Soc. 2007, 129, 5816. (c) Enthaler, S.; Hagemann, B.; Erre, G.; Junge,
K.; Beller, M. Chem.–Asian J. 2006, 1, 598. (d) Nishiyama, H.; Furuta, A. Chem.
Commun. 2007, 760.
16. For selected examples of iron-catalyzed oxidation, see: (a) Legros, J.; Bolm, C.
Angew. Chem., Int. Ed. 2003, 42, 5487. (b) Bukowski, M. R.; Comba, P.; Lienke,
A.; Limberg, C.; de Laorden, C. L.; Mas-Ballesté, R.; Merz, M.; Que, Jr. L. Angew.
Chem., Int. Ed. 2006, 45, 3446. (c) Chen, M. S.; White, M. C. Science. 2007, 318,
783. (d) Gelalcha, F. G.; Bitterlich, B.; Anilkumar, G.; Tse, M. K.; Beller, M.
Angew. Chem., Int. Ed. 2007, 46, 7293. (e) Chen, M. S.; White, M. C. Science. 2010, 327, 566.
17. For selected examples of iron-catalyzed cross-coupling reactions, see: (a) Fürstner, A.; Leitner, A.; Méndez, M.; Krause, H. J. Am. Chem. Soc. 2002, 124, 13856. (b) Nakamura, M.; Ito, S; Matsuo, K.; Nakamura, E. Synlett 2005, 1794. (c) Carril, M.; Correa, A.; Bolm, C. Angew. Chem., Int. Ed. 2008, 47, 4862. (d) Loska, R.; Volla, C. M. R.; Vogel, P. Adv. Synth. Catal. 2008, 350, 2859. (e) Hatakeyama, T.;
50
Hashimoto, T.; Kondo, Y.; Fujiwara, Y.; Seike, H.; Takaya, H.; Tamada, Y.; Ono, T.; Nakamura, M. J. Am. Chem. Soc. 2010, 132, 10674. (f) Liu, W.; Cao, H.; Lei, A.
Amgew. Chem. Int. Ed. 2010, 49, 2004.
18. For selected examples of iron-catalyzed carbonylation, see: (a) Driller, K. M.; Klein, H.; Jackstell, R.; Beller, M. Angew. Chem., Int. Ed. 2009, 48, 6041. (b) Driller, K. M.; Prateeptongkum, S.; Jackstell, R.; Beller, M. Angew. Chem., Int. Ed. 2011, 50, 537. (c) Pizzetti, M.; Russo, A.; Petricci, E. Chem.–Eur. J. 2011, 17, 4523.
19. (a) Booth, G.; Chat, J. J. Chem. Soc. 1962, 2099. (b) Roger, C.; Hamon, P.; Toupet, L.; Rabaâ, H.; Saillard, J.-Y.; Hamon, J.-R.; Lapinte, C. Organometallics 1991, 10, 1045.
20. (a) Chatani, N.; Ie, Y.; Kakiuchi, F.; Murai, S. J. Am. Chem. Soc. 1999, 121, 8645. (b) Chatani, N.; Inoue, H.; Morimoto, T.; Muto, T.; Murai, S. J. Org. Chem. 2001,
66, 4433.
21. Buchwald, S. L.; Bolm, C. Angew. Chem., Int. Ed. 2009, 48, 5586.
22. Decarbonylation of thioanhydrides by iron complex, see: Trost, B. M.; Chen, F.
Tetrahedron Lett. 1971, 12, 2603.
23. Decarbonylation of esters by iron complex, see: Trovitch, R. J.; Lobkovsky, E.; Bouwkamp, M. W.; Chirik, P. J. Organometallics 2008, 27, 6264.
24. Maercker, A.; Girreser, U. Tetrahedron 1994, 50, 8019.
25. Bailey, A. V.; Mitcham, D.; Skau, E. L. J. Chem. Eng. Data 1970, 15, 542.
26. Barton, D. H. R.; Boivin, J.; Crepon, E.; Sarma, J.; Togo, H.; Zard, S. Z.
Tetrahedron 1991, 47, 7091.
27. Burns, D. H.; Miller, J. D.; Chan, H.-K.; Delaney, M. O. J. Am. Chem. Soc. 1997,
119, 2125.
28. Rojas, G.; Wagener, K. B. J. Org. Chem. 2008, 73, 4962.
29. Biellmann, J.-F.; d’Orchymont, H.; Schmitt, J.-L. J. Am. Chem. Soc. 1979, 101, 3283.
30. Feldhues, M.; Schafer, H. J. Tetrahedron 1985, 41, 4213.
31. Schmidt, A. W.; Schoeller, V.; Eberlein, K. Chem. Ber. 1941, 74, 1313.
32. Zhu, H.; Wickenden, J. G.; Campbell, N. E.; Leung, J. C. T.; Johnson, K. M.; Sammis, G. M. Org. Lett. 2009, 11, 2019.
33. Pearlaman, B. A.; Putt, S. R.; Fleming, J. A. J. Org. Chem. 1985, 50, 3625. 34. Lin, J.; Liu, F.; Wang, Y.; Liu, M. Synth. Commun. 1995, 25, 3457.
51
第
2 章
2-アリーロキシ安息香酸の脱カルボニル的環化反応
52 第1 節 緒言 ジベンゾフラン骨格1)は、様々な生物活性物質や機能性分子の部分構造として 多く見られることから、その効率的な合成法に関する研究が盛んに行われてい る2)。中でも最も効率的なジベンゾフラン骨格の構築法としては、Pd触媒や銅触 媒を用いた種々の反応基質からの分子内環化反応による方法が知られている。 以下に基質の構造により分類した過去の反応例を示す。 ・1-ハロ-2-アリーロキシベンゼンの脱ハロゲン化型分子内環化反応 オルト位にアリーロキシ基を有するハロゲン化アリールを基質とし、Pd 触媒 存在下、脱ハロゲン化水素型の環化反応を行うことでジベンゾフラン骨格を構 築している。初期の例では、ハロゲン化アリールとしてヨウ素化物のみの反応 に限定されていたが、近年では工業的にも有用な塩素化物が適応可能となった
(Scheme 2-1, 2-2)3a, 3b)。また、Larock らは系中にてアリーロキシヨードベンゼ
ンを調製し、Pd 触媒による環化反応を行うワンポット型の反応系を報告してい
る(Scheme 2-3)3c)。 Scheme 2-1
53
Scheme 2-3
・ジアリールエーテルの脱水素型環化反応
Pd 触媒によるジアリールエーテルの脱水素を伴った酸化的環化反応による合
成法も検討されている。Itatani らの報告例は遷移金属触媒によるジベンゾフラン
合成の最初の例である(Scheme 2-4)4a)。その後Fagnou らにより改良型の触媒
系が報告されている(Scheme 2-5)4b)。
Scheme 2-4