2.7.6.9 有効性及び安全性試験:実薬対照(プラセボ対照なし)
2.7.6.9.1 国内第Ⅲ相試験−「Genotype 1 かつ高ウイルス量」の C 型慢性肝炎患者を対象とし
た PEG-IFN α -2b+ リバビリン併用投与試験(試験番号 C033* )
日本人における Genotype 1 かつ高ウイルス量の C 型慢性肝炎患者を対象とし, PEG-IFN α -2b + リバビリン併用時の有効性が IFN α -2b + リバビリン併用時よりも劣らないことを検証する ため,第Ⅲ相試験を本邦において実施した.試験の概要を表 2.7.6-54に示した.
表 2.7.6-54 試験の概要(C033*) (1 of 7)
項目 内容
治験課題名 「Genotype 1かつ高ウイルス量」のC型慢性肝炎患者を対象としたPEG-IFNα-2b+ リバビリン併 用投与臨床試験
目的
「Genotype 1かつ高ウイルス量」のC型慢性肝炎患者を対象とし,PEG-IFNα-2b + リバビリン併 用時の有効性がIFNα-2b + リバビリン併用時よりも劣らないことを検証する.有効性の主要評価 項目は,投与期間終了後24週時点のウイルス学的効果(HCV-RNA陰性化)とする.
治験種類
中央登録法による多施設共同,オープンラベル,無作為化,実薬対照,並行群間比較試験(検証 試験),48週間の投与期間と24週間のfollow-up期間
[治験のダイアグラム]
入院(2週)
<PEG/R群>
<IFN/R群>
中間集計
投与期間 経過観察期間
(48週) (24週)
スクリーニング期間
(90日以内)
同意 スクリーニング
IFNα-2b + リバビリン併用投与 外来(46週)
PEG-IFNα-2b + リバビリン 併用投与 無作為化(1:1)
被験者数
計画時被験者数 500名 (PEG-IFNα-2b群:250名,IFNα-2b群:250名) 登録被験者数 514名 (PEG-IFNα-2b群:258名,IFNα-2b群:256名) 投与被験者数 511名 (PEG-IFNα-2b群:257名,IFNα-2b群:254名) 解析対象被験者数
安全性 507名 (PEG-IFNα-2b群:254名,IFNα-2b群:253名) 有効性(FAS) 506名 (PEG-IFNα-2b群:254名,IFNα-2b群:252名)
被験者数設 定根拠
目標被験者数は,欧米のIFN未投与のC型慢性肝炎患者を対象としたPEG-IFNα-2b + リバビリ ン併用投与第Ⅲ相臨床試験( C034* )の試験結果を参考に設定した.この試験では,「Genotype 1bかつ高ウイルス量(100 kcopies/mL以上)」の患者に対するHCV-RNA持続陰性化率は,PEG-IFN α-2b 1.5 µg/kg + リバビリン併用群46 %,IFNα-2b 300 万IU + リバビリン併用群34 %で,持 続陰性化率の差は12 %であった.
一方,今回実施する試験ではIFNα-2bの投与量を600万IUと設定している.また,今回の試験 では対象患者にIFN前治療無効例を含んでいる.これらの点を考慮すると,本試験でのPEG-IFN α-2b + リバビリン併用群と IFNα-2b + リバビリン併用群の HCV-RNA 持続陰性化率の差は,
C034試験での差よりも小さくなると考えられる.
そこで,本試験におけるPEG-IFNα-2b + リバビリン併用群のHCV-RNA持続陰性化率は,IFNα -2b + リバビリン併用群より少なくとも5 %は上回る(IFNα-2b + リバビリン併用群41 %に対し てPEG-IFNα-2b + リバビリン併用群46 %)と仮定し,PEG-IFNα-2b + リバビリン併用群のIFN α-2b + リバビリン併用群に対する非劣性を検証するために必要な被験者数として,1群250名,
計500名を設定した(下表参照).
*;新薬承認情報提供時に置き換えた
601
表 2.7.6-54 試験の概要(C033**) (2 of 7)
項目 内容
被験者数設 定根拠
参考として,下表には優越性を検証する場合の被験者数も併記したが, C034** 試験と同様に2 群間の差が12 %となった場合,1群250名であれば,PEG-IFNα-2b + リバビリン併用群のIFNα -2b + リバビリン併用群に対する優越性は検出力78 %で検証される.
各種条件で非劣性及び優越性を検証する場合の検出力
HCV-RNA持続陰性化率 検出力 PEG-IFNα-2b
+リバビリン
IFNα-2b
+リバビリン n=250 n=200 n=150 非劣性(Δ=10 %) 46 % 41 % 92 % 85 % 74 %
優越性 46 % 34 % 78 % 68 % 56 % nは1群あたりの被験者数,有意水準は片側2.5 %
対象 「Genotype 1かつ高ウイルス量」のC型慢性肝炎患者
選択基準
• 同意取得時の年齢が20歳以上で同意能力のある患者
• スクリーニング検査時の体重が40kgを超えて100 kg以下の患者
• 治験責任(分担)医師の管理下において,治験期間中(同意取得後から経過観察期間が終了す るまで)の避妊が可能な患者(性別を問わない)
• 登録前60日以内のスクリーニング検査(集中測定)において,C型肝炎ウイルスのサブタイプ がGenotype 1(Genotype 1a及び1b)であり,かつ血中HCV-RNA量がRT-PCR法(アンプリコ アHCVモニター)で100 k IU/mL以上の患者
• 登録前60日以内のスクリーニング検査(集中測定)において,血清ALT値が基準値上限を超え る患者[血清ALT基準値上限(三菱化学ビーシーエル):45 IU/L]
• 登録前60日以内のスクリーニング時の臨床検査による以下の「慢性肝炎と肝硬変の判別式」の 計算結果が負(<0)で慢性肝炎と判断された患者
判別式:
γ-グロブリン* (%)×0.124+ヒアルロン酸* (ng/mL)×0.001 + 性別(男性=1, 女性=2)×
(-0.413) + 血小板数*(万/mm3)×(-0.075)−2.005
*同日採血分の測定結果を用い,計算に供すること
• 登録前1年(365日)以内に肝生検が施行されており(登録時点での病理診断の有無を問わない), 肝組織標本の貸出しが可能な患者
• 登録前60日以内のスクリーニング検査において,以下の基準をすべて満たす患者 ヘモグロビン ≧ 12 g/dL
好中球数 ≧ 1,500 /mm3 血小板数 ≧ 10万 /mm3
クレアチニンクリアランス(以下のCockcroftらの予測式)が51以上の患者 Ccr(男性)=(140−年齢)× 体重(kg) /( 72×血清クレアチニン濃度(mg/dL) ) Ccr(女性)=Ccr(男性)×0.85
空腹時血糖が110 mg/dL未満の患者(ただし,空腹時血糖110 mg/dL以上126 mg/dL未満の 患者については,HbA1cが6.5%未満であれば,登録可能)[糖尿病に関する薬物療法がなされ ていないときの測定結果]
• 投与開始時から,少なくとも2週間の入院が可能な患者
除外基準
• 過去にpolyethylene glycol修飾IFNあるいはリバビリンの投与を受けたことのある患者
• 過去にIFN療法の30週を超える長期投与を受けたことのある患者
• 過去にIFN治療歴がある場合は,登録の時点で,前回のIFN投与終了後90日を経過していない 患者
• 登録前60日以内のスクリーニング検査(集中測定)において,HBs抗原陽性の患者
• 登録前60日以内のスクリーニング検査(集中測定)において,抗核抗体価が160倍以上の患者
• 肝硬変,肝不全,肝癌の患者又はこれらの既往歴のある患者
• 自己免疫性肝炎,アルコール性肝障害,薬剤性肝障害等の肝疾患を合併している患者
• 肝性脳症,食道静脈瘤破裂,腹水の既往を有する患者
• うつ病あるいは精神神経障害のある患者又はこれらの既往歴のある患者
• 薬物治療を必要とするてんかん発作のある患者又はその既往歴のある患者
• 狭心症,心不全,心筋梗塞,高度の高血圧症(拡張期血圧が120 mmHg以上)あるいは高度の 不整脈(「副作用の重篤度分類」のgrade 3に示された所見)のある患者又はこれらの既往歴のあ る患者
• 慢性肺疾患の患者又はその既往歴のある患者
**;新薬承認情報提供時に置き換えた
表 2.7.6-54 試験の概要(C033*) (3 of 7)
項目 内容
除外基準
• 自己免疫疾患(クローン病,潰瘍性大腸炎,慢性関節リウマチ,特発性血小板減少性紫斑病,
全身性エリテマトーデス,自己免疫性溶血性貧血,強皮症等)のある患者又はこれらの既往歴 のある患者
• 異常ヘモグロビン症(サラセミア,鎌状赤血球性貧血)の患者
• 悪性腫瘍の患者
• 薬物療法でコントロール不能な甲状腺機能異常を有する患者
• 臓器移植を受けた患者(ただし,角膜,毛移植を除く)
• 登録前30日以内に,グリチルリチン・システイン・グリシンを含有する注射用製剤(強力ネオ ミノファーゲンC等),小柴胡湯,ウルソデスオキシコール酸,テオフィリン,アンチピリン,
ワルファリンの投与を受けた患者
• 登録前90日以内に,抗ウイルス剤又は抗腫瘍剤の投与あるいは免疫調節療法(ステロイド剤投 与,放射線療法を含む)を受けた患者[局所投与及び外用剤を除く]
• 登録前180日以内に,その他の治験薬の投与を受けた患者
• IFN製剤,ヌクレオシドアナログ又はワクチン等の生物学的製剤に対して過敏症の既往歴のある 患者
• 妊娠又は授乳中である患者(男性の場合:パートナーが妊娠中の患者),並びに同意取得時から 登録時までに測定した血清HCG測定結果より妊娠が否定されない患者
• 投与開始直前実施のプリック試験において,PEG-IFNα-2bあるいはIFNα-2bに対して,特異的 な反応の認められた患者
• その他,治験責任(分担)医師が本治験の対象として不適当と判断した患者
使用薬剤 用法用量
被験薬
一般名: peginterferon alfa-2b(INN)
製剤: 注射用凍結乾燥製剤
投与方法: 1.5 µg/kgを週1回,48週間皮下投与する.
対照薬
一般名: インターフェロンアルファ−2b(遺伝子組換え)(JAN)
名称(略称):IFNα-2b
製剤: 注射用凍結乾燥製剤
投与方法: 600万IUを投与開始初期の2週間は週6回,その後の46週間は週3回,筋肉内投 与する.
併用薬
一般名: リバビリン (JAN) 製剤: カプセル製剤
投与方法: 被験者の登録時の体重に応じて,規定された投与量(600〜1,000 mg/日)を1日2 回(朝夕食後)48週間,毎日服用する.
投与スケジュール
<PEG/R群> PEG-IFN α -2b + リバビリン併用投与
< IFN/R 群> IFN α -2b + リバビリン併用投与
( 6 回 / 週) ( 3 回/週)
2 週間 46 週間
リバビリン 600 , 800 又は 1000 mg/ 日 経口投与(毎日)
PEG-IFN α -2b 1.5 μ g/kg 皮下投与
( 1 回 / 週)
リバビリン 600 , 800 又は 1000 mg/ 日 経口投与(毎日)
48週間
IFN α -2b 600 万 IU 筋肉内投与
*;新薬承認情報提供時に置き換えた
603
表 2.7.6-54 試験の概要(C033*) (4 of 7)
項目 内容
併用禁止薬
登録後から治験薬投与終了後の観察期間終了時までの全期間併用禁止薬剤
• 局所投与及び外用剤以外の抗ウイルス剤(抗HIV薬を含む),抗腫瘍剤,免疫調節作用を有す る薬剤(IFN,グルココルチコイド,IL-2等)
• グリチルリチン・システイン・グリシンを含有する注射用製剤(強力ネオミノファーゲン C 等)
• ウルソデスオキシコール酸
• ウルソデスオキシコール酸以外の肝臓疾患治療薬(経口肝臓疾患治療薬は,同意取得前から 投与を行っている場合に限り,同意取得後に増量しないことを条件に併用可能)
• 小柴胡湯
• 他の治験薬
投与期間中の併用禁止薬剤
テオフィリン,アンチピリン,ワルファリン
観察・測定 スケジュー
ル
観察・測定スケジュールの概略
スクリーニング (日)
-90 1 2 3 4 8 12 16 20 24 28 32 36 40 44 48 4 8 12 24
基準日(Day)1) -90 1 8 15 22 29 57 85 113 141 169 197 225 253 281 309 337 365 393 421 505
同意取得 2) ○
入院3)/外来
背景調査 ○
体重 ○
体温4) 併用薬の調査 リバビリン服薬状況調査
肝生検 5) ○
腹部CT又は超音波検査 ○
胸部X線 ○
心電図 ○
眼底検査 ○
プリック試験 ○
被験者日誌 有害事象等の調査
血液学的検査a) ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○
血液生化学検査(空腹時)b) ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ●
甲状腺機能検査c) ● ● ● ● ● ● ● ● ● ●
妊娠検査(血清HCG;女性のみ) ● ● ● ● ● ● ● ● ● ●
HCV-RNA定量d) ● ● ● ● ● ● ● ● ● ●
HCV-RNA定性e) ● ● ● ● ● ● ● ● ●
HCV genotype ●
HBs抗原 ●
抗核抗体 ●
腫瘍マーカーf) ●
必要採血量(mL) 29 14 8 19 8 19 8 19 8 8 19 8 8 19 8 8 19 19 8 19 19
1)投与開始日をDay1として各日付を算出 a) 白血球数、赤血球数、ヘモグロビン、ヘマトクリット、血小板数、白血球分画、網赤血球比率
2)スクリーニング検査を実施する前に同意取得 b)ALT、AST、γ-GTP、ALP、LDH、総ビリルビン(直接/間接ビリルビン含む)、総蛋白、アルブミン、
3)14日以上 クレアチニン、尿酸、BUN、空腹時血糖、HbA1C*、総コレステロール、CRP、γ-グロブリン*、ヒアルロン酸*
4)投与14日目まで測定 c)TSH、FT3、FT4、TRAb*、TPOAb*
5)登録前365日以内に実施 d)branched DNA probe法*、アンプリコアHCVモニター
投与中止時には「投与開始後48週時点」の調査を実施 e) アンプリコア定性
●:三菱化学ビーシーエルによる集中測定 f)AFP、PIVKA-II 安全性に問題が生じた場合は、規定外にも調査を実施 * スクリーニング時のみ
# 投与開始前の臨床検査はDay-14〜投与開始直前に実施 --●#--
--●#--
投与終了後(週)
PEG/R群(週1回来院)・IFN/R群(週3回来院)
投 与 開 始 前
--○#-- --●#-- 開 始 直 前
入院・投与
投与開始後(週)
評価基準
有効性
主要評価項目
1. ウイルス学的効果[投与期間終了後24週時点(経過観察終了時点)]
投与期間終了後24週時点における血中HCV-RNA定性値(RT-PCR法:アンプリコア定性)
を指標として以下の基準で判定する.
陰性: 投与期間終了後24週時点における血中HCV-RNA定性値が陰性である被験者 陽性: 投与期間終了後24週時点における血中HCV-RNA定性値が陽性である被験者 判定不能:データ欠損のため判定できない被験者
治験薬投与中止後IFN製剤及び/又はリバビリンを投与した被験者 副次的評価項目
1. ウイルス学的効果[投与開始後24週時点]
投与開始後24週時点における血中HCV-RNA定性値(RT-PCR法:アンプリコア定性)を指 標として以下の基準で判定する.
陰性: 投与開始後24週時点における血中HCV-RNA定性値が陰性である被験者 陽性: 投与開始後24週時点における血中HCV-RNA定性値が陽性である被験者 判定不能:データ欠損のため判定できない被験者
2. ウイルス学的効果[投与期間終了時点(投与中止時点)]
投与開始後24週時点における血中HCV-RNA定性値(RT-PCR法:アンプリコア定性)を指 標として以下の基準で判定する.
陰性: 投与期間終了時点における血中HCV-RNA定性値が陰性である被験者 陽性: 投与期間終了時点における血中HCV-RNA定性値が陽性である被験者 判定不能:データ欠損のため判定できない被験者
*;新薬承認情報提供時に置き換えた
表 2.7.6-54 試験の概要(C019*) (5 of 7)
項目 内容
評価基準
3. ウイルス学的効果[投与期間終了後12週時点]
投与期間終了後12週時点における血中HCV-RNA定性値(RT-PCR法:アンプリコア定性)
を指標として以下の基準で判定する.
陰性: 投与期間終了後12週時点における血中HCV-RNA定性値が陰性である被験者 陽性: 投与期間終了後12週時点における血中HCV-RNA定性値が陽性である被験者 判定不能:データ欠損のため判定できない被験者
治験薬投与中止後IFN製剤及び/又はリバビリンを投与した被験者 4. ALT正常化[投与期間終了時点(投与中止時点)]
投与期間終了時点における血清ALT値を指標として,以下の基準で判定する.
有効: 投与期間終了時点における血清ALT値が基準値上限以下である被験者 無効: 投与期間終了時点における血清ALT値が基準値上限を超える被験者 判定不能:データの欠損のため判定できない被験者
投与開始前の血清ALT値が基準値上限以下である被験者 5. ALT正常化[投与期間終了後24週時点(経過観察終了時点)]
投与期間終了後24週時点における血清ALT値を指標として,以下の基準で判定する.
有効: 投与期間終了後24週時点における血清ALT値が基準値上限以下である被験者 無効: 投与期間終了後24週時点における血清ALT値が基準値上限を超える被験者 判定不能:データの欠損のため判定できない被験者
投与開始前の血清ALT値が基準値上限以下である被験者
治験薬投与中止後IFN製剤及び/又はリバビリンを投与した被験者 安全性
自他覚症状
治験責任(分担)医師は,投与期間中及び経過観察期間中を通じて,問診により自覚症状・
他覚所見の有無を調査する.
臨床検査値
原則として「臨床検査値異常変動の判定」に従い,異常変動の有無を判定する.
異常変動「有」と判定した項目については有害事象として取扱い,症例報告書の臨床検査値 異常変動欄に詳細を記載する.
有害事象
1. 有害事象及び副作用の定義
本治験では有害事象,副作用及び予測できない副作用を以下のように定義する.
1) 有害事象
治験薬を投与された被験者に生じたあらゆる好ましくない医療上のできごと.必ずしも当 該治験薬の投与との因果関係が明らかなもののみを示すものではない.
すなわち,有害事象とは,治験薬が投与された際に起こる,あらゆる好ましくないあるい は意図しない徴候(臨床検査値の異常変動を含む),症状又は病気のことであり,当該治 験薬との因果関係の有無は問わない.
2) 副作用
投与量にかかわらず,投与された治験薬に対するあらゆる有害で意図しない反応(臨床検 査値の異常変動を含む),すなわち,当該治験薬と有害事象との間の因果関係について,
少なくとも合理的な可能性があり,因果関係を否定できない反応を指す.
3) 予測できない副作用
副作用のうち,治験薬概要書に記載されていないもの,あるいは記載されていてもその性 質,症状の程度又は発生数,発生頻度,発生条件等の発生傾向が記載内容と一致しないも の,治験実施医療機関に報告していないものをいう.
2. 有害事象等の監視
治験責任(分担)医師は,有害事象の発現について,慎重に監視し,有害事象が発現した 場合は適切な処置を行い,有害事象が消失,軽快又は治験薬投与前の状態に復するまで追 跡観察を行う.有害事象が消失,軽快又は治験薬投与前の状態に復していない時点で,治 験責任(分担)医師がさらなる追跡観察は不必要と判断し追跡観察を終了した場合,又は 被験者が拒否したために追跡検査の継続が不可能となった場合には,その理由を症例報告 書に記入する.また,発現したすべての有害事象について,事象名,発現日,程度,重篤 度,治験薬の処置,その他の処置,治験薬との因果関係,転帰確認日,転帰を症例報告書 に記入する.
*;新薬承認情報提供時に置き換えた
605
表 2.7.6-54 試験の概要(C033*) (6 of 7)
項目 内容
評価基準
3. 有害事象の程度
有害事象の発現が確認された場合,治験責任(分担)医師は以下の基準を参照して「軽度」
「中等度」「高度」の3段階に分類する(参考資料1:「副作用の重篤度分類」参照). 軽 度 徴候又は症状が認められるが,日常的活動が妨げられず処置を要さないもの 中 等 度 不快感のため日常的活動が妨げられる,又は臨床状態に影響が認められるもの
で,処置を要するもの
高 度 日常的活動が不能となる,又は臨床状態に重大な影響が認められるもの 4. 治験薬との因果関係
発現した有害事象と治験薬との因果関係の判定は次の4段階で行い,1〜3を副作用として扱 う.「関連なし」と判断した場合,その判定理由について症例報告書に記載する.
0. 関連なし: 治験薬以外の要因により明瞭に説明できるもの,あるいは治 験薬とその有害事象との間に時間的関連性の面で妥当性のな いもの
1. 関連あるかもしれない: 治験薬以外のことが原因で発現したと考えられるが,治験薬 との関連が否定できないもの
2. 多分関連あり: 治験薬以外のことが原因で発現した可能性が低いもの 3. 関連あり: 治験薬の投与からその有害事象の発現までの時間的関連性に
妥当性があり,治験薬以外の原因から説明できないもの
統計手法
比較可能性 [FASとPPSの両方で集計する.ただし,PPSは集計のみで検定は行わない.]
下記の検定法を用いて比較可能性を吟味する.なお,IFN治療歴については検定を実施しない.
計量尺度データ:Wilcoxon-Mann-Whitney test 順序分類尺度データ:拡張Mantel-Haenszel test 分類尺度データ:Fisher’s exact test
1. 被験者背景
性別,年齢,体重,罹病期間,合併症の有無,IFN治療歴(未治療例,再燃例,無効例),肝 生検(国際分類),リバビリン投与量
2. 投与開始前の臨床検査値
HCV-RNA量(RT-PCR法:アンプリコアHCVモニター),ALT,ヘモグロビン,好中球数,
血小板数 3. 実施状況
治験の中止,PEG-IFNα-2b・IFNα-2bの減量・投与中止,リバビリンの減量・投与中止 有効性
主要評価項目:ウイルス学的効果(投与期間終了後24週時点のHCV-RNA陰性化率)
1. 主解析(FAS)
陰性化率について,投与群間の差(PEG/R群−IFN/R群)とその信頼区間を算出する.非劣 性を示す場合は下側同等限界を-10%とし,信頼区間の下限が限界値を上回ったときにPEG/R 群はIFN/R群に対して劣らないと推論する.
なお,陰性化率は,判定不能を陽性に含めて算出する.
2. 感度分析
PPSでの解析:主解析と同様の解析を,PPSで行う.
共変量を考慮した解析:副解析として,IFN 治療歴を共変量にして投与群間の差とその信頼 区間を算出する.
3. 予後因子の検討
ウイルス学的効果(投与期間終了後24週時点)に関連する下記項目に示す予後因子を,変数 選択型logistic regression analysisで探索する.
IFN治療歴(未治療例,再燃例,無効例),肝生検(国際分類),投与開始前の HCV-RNA量
(RT-PCR法:アンプリコアHCVモニター),投与開始前のALT 4. 部分集団での検討
ウイルス学的効果(投与期間終了後24週時点)について,探索的に,下記の項目で分類した 部分集団において,投与群間の差とその信頼区間を算出する.
IFN治療歴(未治療例,再燃例,無効例),肝生検(国際分類),投与開始前の HCV-RNA量
(RT-PCR法:アンプリコアHCVモニター),投与開始前のALT,高齢・非高齢の別
*;新薬承認情報提供時に置き換えた
表 2.7.6-54 試験の概要(C033*) (7 of 7)
項目 内容
統計手法
副次的評価項目
以下の項目に対して,投与群間の差(PEG/R群−IFN/R 群)とその信頼区間を算出する.な お,ウイルス学的効果では判定不能を陽性,ALT正常化では判定不能を無効に含めて算出す る.
ウイルス学的効果(投与開始後24週時点,投与期間終了時点,投与期間終了後12週時点)
ALT正常化(投与期間終了時点,投与期間終了後24週時点)
HCV-RNA定性値の最初の陰性化時期
投与期間中のHCV-RNA定性値とウイルス学的効果(投与期間終了後24週時点)を対応させ,
最初に陰性化した時期について,投与群別に頻度を集計する.
HCV-RNA定性値の再燃率
HCV-RNA定性値について,投与期間終了時点で陰性化している被験者が投与期間終了後24 週時点で陽性になった割合(再燃率)を投与群別に算出する.
HCV-RNA定性値について,投与開始後24週時点で陰性化している被験者が投与期間終了後 24週時点で陽性になった割合を投与群別に算出し,再燃率と比較検討する.
HCV-RNA定性値及びALT正異判定の経時変化
HCV-RNA定性値について,各調査時点における陰性化率を投与群別に算出する.
ALT正異判定について,各調査時点における正常化率を投与群別に算出する.
安全性 投与期間
投与群別に,投与期間(2,4,8,12,24,36,48週間以下及び48週間超過)毎に被験者数 を集計する.
有害事象及び副作用
投与開始以降の有害事象及び副作用それぞれについて投与群別の発現被験者数及び発現率を 示すとともに,器官別大分類別及び各事象(症状及び所見)別の発現被験者数及び発現率を 集計する.各事象については,程度で分類した集計も行う.また,重篤な症状についても投 与群別に発現被験者数及び発現率を集計する.以上について,投与期間と経過観察期間のそ れぞれで集計を行う.また,各事象別の集計は,全期間でも行う.
副作用用語は,MedDRA収載用語を使用する.
臨床検査値(血液学的検査,血液生化学検査及び甲状腺機能検査)
1. 測定値の集計
計量尺度データ: 投与群別に,調査時期毎に,記述統計量(被験者数,平均値,標準偏 差,skewness,kurtosis,最小値,25%点,中央値,75%点及び最大値)
を算出する.なお,白血球分画の集計は,白血球と分画%より算出し た実数を使用する.ヘモグロビン,好中球数及び血小板数の投与開始 から経過観察が終了するまでの最低値について,副作用の重篤度分類 に基づくgrade に分類し,その頻度を投与群別に集計する.
順序分類尺度データ:投与群別に,調査時期毎に頻度を集計する.
2. 異常値の集計
「臨床検査値異常変動の判定」に基づいた分類について,投与群別に,調査時期毎に頻度を 集計する.
医学専門家
治験調整 医師
医学統計ア ドバイザー 実施医療
機関 他 施設
公表文献 未定
治験期間 年 月〜 年 月
*;新薬承認情報提供時に置き換えた
607
同意取得から治験完了までの被験者の内訳を図 2.7.6-8に,経過観察期間の被験者の内訳を 図 2.7.6-9に,無作為化後から経過観察期間終了までに治験を中止した被験者を表 2.7.6-55 に示す.
同意を取得した被験者は 786 名で,このうちの 272 名は治験薬割付けに至らなかった.治験 薬が割付けられなかった被験者の内訳は,スクリーニングでの不適: 233 名,同意撤回: 15 名,
登録締切: 2 名及びその他: 22 名(契約解除の 8 名を含む)であった.
登録がなされて治験薬が割付けられた被験者は 514 名( PEG/R 群: 258 名, IFN/R 群 256 名)
で,治験薬投与が開始された被験者は 511 名( PEG/R 群: 257 名, IFN/R 群: 254 名)であった.
すなわち,無作為化から投与開始前までに治験を中止した被験者は 3 名[PEG/R 群:1 名(投 与開始前のヘモグロビン値が中止基準に抵触) , IFN/R 群: 2 名(同意の撤回: 1 名,契約解除:
1 名) ]であった.
治験薬投与が開始された被験者 511 名(PEG/R 群:257 名,IFN/R 群:254 名)のうち, 48 週間の投与を完了できなかった, すなわち投与を中止したのは PEG/R 群で 20.2% (52 名/257 名) , IFN/R 群で 21.7%(55 名/254 名)であった.その主な投与中止理由はヘモグロビン/血小板数/
好中球数の減少による中止が PEG/R 群で 8.9% ( 23 名 /257 名) , IFN/R 群で 8.3% ( 21 名 /254 名) , その他の有害事象による中止が PEG/R 群で 8.9%(23 名/257 名) ,IFN/R 群で 9.8%(25 名/254 名)であった.
投与終了(中止)後の経過観察期間対象被験者は 511 名( PEG/R 群: 257 名, IFN/R 群: 254 名)であった.この 511 名のうち 24 週間の経過観察を完了できなかったのは,19 名(PEG/R 群: 6 名, IFN/R 群: 13 名)であった.その主な観察中止理由は同意撤回による中止が PEG/R 群で 2.3% ( 6 名 /257 名) , IFN/R 群で 3.9% ( 10 名 /254 名) ,有害事象(ヘモグロビン / 血小板数 / 好中球数の減少を除く)による中止が PEG/R 群で 0.0% ( 0 名 /257 名) , IFN/R 群で 0.4% ( 1 名 /254 名)であった.
経過観察期間の中止被験者数及び理由を含めた,本試験における両群の中止には差はみられ
なかった.
*:契約解除した治験実施医療機関における実施分の9名(うち,文書同意未取得の5名)を含む
図 2.7.6-8 被験者の内訳(同意取得〜投与終了時点)
経過観察終了 被験者( 251 名)
経過観察中止被験者(6名)
・同意撤回: 6名
経過観察中止被験者(13名)
・同意撤回: 10名
・有害事象: 1名
・その他: 2名
経過観察終了
被験者( 241 名)
IFN α -2b +リバビリン併用投与群 経過観察対象被験者
**(254 名) PEG-IFN α -2b +リバビリン併用投与群
経過観察対象被験者
**(257 名)
**:治験薬を1度でも投与した被験者では,投与終了後24週間の経過観察期間を設けた。
図 2.7.6-9 被験者の内訳(経過観察期間)
233
同意取得被験者(786 名
*)
同意取得後無作為化されなかった 未登録被験者(272 名)
・同意撤回: 15名
・スクリーニング不適: 233名
・登録締切り: 2名
・その他: 22名
無作為化された被験者
(514 名)
PEG-IFNα-2b+リバビリン併用投与群
(258 名)
治験薬投与 開始被験者
(257 名)
治験薬 48 週 投与終了被験者
( 205 名)
投与開始後中止被験者(52名)
・有害事象(Hb/PLT /Neu減少): 23名
・有害事象(Hb/PLT /Neu減少を除く): 23名
・誤投与(市販薬): 1名
・パートナーの妊娠: 1名
・選択/除外基準違反: 1名
・患者希望: 2名
・他の治験実施計画 違反: 1名 無作為化後から投与開始まで の中止被験者(1名)
・投与開始基準に抵触: 1名
IFNα-2b+リバビリン併用投与群
( 256 名)
投与開始後中止被験者(55名)
・有害事象(Hb/PLT /Neu減少): 21名
・有害事象(Hb/PLT /Neu減少を除く): 25名
・誤投与(市販薬): 4名
・パートナーの妊娠: 2名
・選択/除外基準違反: 1名
・同意撤回: 1名
・プリック陽性: 1名 無作為化後から投与開始まで の中止被験者(2名)
・同意の撤回: 1名
・治験契約解除*: 1名
治験薬投与
開始被験者
(254 名)
治験薬 48 週 投与終了被験者
( 199 名)
609
表 2.7.6-55 中止及び中止理由一覧(C033*)
試験名(番号)
項目
第Ⅲ相試験 (C033*)
投与群 PEG/R群 IFN/R群
PEG-IFNα-2b/ IFNα-2b
用法・用量 1.5 µg/kg×1回/週×48週 6MIU×6 回/週 ×2 週 → 6MIU×3回/週×46週 リバビリン用法用量 600-1,000 mg/日 600-1,000 mg/日
投与期間 48週間 48週間
被験者数 257 254
治験薬が投与された被験者の合計 257 (100.0%) 254 (100.0%)
投与開始から投与開始後24週までの中止被験者 30 (11.7%) 39 (15.4%)
同意の撤回 0 (0.0%) 1 (0.4%)
ヘモグロビン/血小板/好中球の減少 16 (6.2%) 17 (6.7%)
有害事象(ヘモグロビン/血小板/好中球の減少
を除く)の発現 11 (4.3%) 14 (5.5%)
その他の治験実施計画逸脱 1 (0.4%) 0 (0.0%)
市販薬(レベトール)の誤投与 1 (0.4%) 0 (0.0%)
パートナーの妊娠判明 1 (0.4%) 2 (0.8%)
プリック試験陽性のため 0 (0.0%) 1 (0.4%)
市販薬(イントロンA)の誤投与 0 (0.0%) 4 (1.6%)
投与開始から24週までの完了被験者 227 (88.3%) 215 (84.6%)
投与開始後24週から投与開始後48週までの中止
被験者 22 (8.6%) 16 (6.3%)
ヘモグロビン/血小板/好中球の減少 7 (2.7%) 4 (1.6%)
有害事象(ヘモグロビン/血小板/好中球の減少
を除く)の発現 12 (4.7%) 11 (4.3%)
選択/除外基準違反 1 (0.4%) 1 (0.4%)
被験者からの中止の申し入れ 2 (0.8%) 0 (0.0%)
48週間の投与を完了した被験者 205 (79.8%) 199 (78.3%)
経過観察期間対象被験者 257 (100.0%) 254 (100.0%)
経過観察期間24週までの中止被験者 6 (2.3%) 13 (5.1%)
同意の撤回 6 (2.3%) 10 (3.9%)
転勤の為 0 (0.0%) 1 (0.4%)
来院せず 0 (0.0%) 1 (0.4%)
有害事象(ヘモグロビン/血小板/好中球の減少
を除く)の発現 0 (0.0%) 1 (0.4%)
経過観察期間24週までの完了被験者 251 (97.7%) 241 (94.9%)
(CHC500:CSR14_1_1_4)
本治験では登録され無作為化がなされた被験者を all randomized subjects 解析対象集団(無作 為化された全ての被験者, ARS 解析対象集団)とし,安全性を評価した被験者を安全性解析対 象集団とし,有効性を評価した被験者を Full analysis set 解析対象集団(基本解析対象集団, FAS 解析対象集団)とした. FAS 解析対象集団のうち治験実施計画書の規定に違反した被験者を除 いた Per Plotocol Set 解析対象集団(治験実施計画書に適合した有効性解析対象集団, PPS 解析 対象集団)とした.
本試験において安全性の解析対象集団は安全性解析対象集団とし,有効性の解析対象集団は FAS 解析対象集団とした。ただし,主要評価項目に関する主解析の感度分析では PPS 解析対象 集団あるいは ARS 解析対象集団を対象とした解析及び共変量(インターフェロン治療歴)を考 慮した解析(FAS 解析対象集団)も行い,主解析の結果に対する安定性を確認した。
本治験に登録され無作為化がなされた被験者(ARS 解析対象集団)は 514 名(PEG/R 群:
258 名, IFN/R 群: 256 名)であった.
*;新薬承認情報提供時に置き換えた
本治験に登録され無作為化がなされた被験者(ARS 解析対象集団)は 514 名(PEG/R 群:
258 名,IFN/R 群:256 名)のうち,安全性の評価できるか否かに関して検討した結果,安全性 解析対象集団は 507 名であり,その内訳は PEG/R 群 254 名, IFN/R 群 253 名となった.安全性 解析対象集団として除外した 7 名(PEG/R 群:4 名,IFN/R 群:3 名)の内訳は重大な GCP 違 反 5 名( PEG/R 群: 3 名, IFN/R 群: 2 名) ,治験薬未投与 2 名( PEG/R 群: 1 名, IFN/R 群: 1 名)であった.重大な GCP 違反 5 名の内容は同意未取得で複数の被験者に対して治験を実施し ていたことにより,治験契約解除になった治験実施医療機関の被験者が IFN/R 群で 1 名(治験 薬は未投与 / 文書同意は取得) ,有効期限切れの治験薬投与が, PEG/R 群及び IFN/R 群で,それ ぞれ 3 名(有効期限切れの PEG-IFNα-2b を投与)及び 1 名(有効期限切れの IFNα-2b を投与)
であった。治験薬未投与 2 名の内容は患者都合による治験薬投与開始前の脱落(同意撤回)が IFN/R 群で 1 名,投与開始前の血液学的検査でヘモグロビンが中止基準に抵触した被験者が PEG/R 群で 1 名であった.
本治験に登録され無作為化がなされた被験者(ARS 解析対象集団)514 名(PEG/R 群:258 名, IFN/R 群:256 名)のうち,有効性の評価できるか否かに関して検討した結果, FAS 解析対 象集団は 506 名となり,その内訳は PEG/R 群: 254 名, IFN/R 群: 252 名となった. FAS 解析 対象集団として除外した 8 名(PEG/R 群:4 名,IFN/R 群:4 名)の内訳は先に示した安全性解 析対象集団として除外した重大な GCP 違反 5 名(PEG/R 群:3 名,IFN/R 群:2 名) ,治験薬未 投与 2 名( PEG/R 群: 1 名, IFN/R 群: 1 名)に加え, IFN α -2b 製剤未投与 1 名( IFN/R 群: 1 名)であった.IFNα-2b 製剤未投与 1 名の内容は投与開始日の朝分のリバビリン投与を治験実 施計画書から逸脱して,プリック試験を実施する前に服用していたが,結果としてプリック試 験が陽性であったため,中止規定に従い治験薬の投与を中止した被験者が IFN/R 群で 1 名であ った.
FAS 解析対象集団は 506 名( PEG/R 群: 254 名, IFN/R 群: 252 名)のうち, PPS 解析対象 集団は 429 名であり,その内訳は PEG/R 群 219 名,IFN/R 群 210 名となった.PPS 解析対象集 団として除外した 77 名( PEG/R 群: 35 名, IFN/R 群: 42 名)の内訳は対象疾患外 8 名( PEG/R 群:4 名,IFN/R 群:4 名) ,除外基準違反 2 名(PEG/R 群:1 名,IFN/R 群:1 名) ,中止基準 違反 3 名(PEG/R 群:1 名,IFN/R 群:2 名) ,併用薬違反 17 名(PEG/R 群:6 名,IFN/R 群:
11 名) ,治験薬投与規定違反 5 名(PEG/R 群: 1 名, IFN/R 群: 4 名) ,投与量不十分 70 名(PEG/R 群:32 名,IFN/R 群:38 名)であった.
本項において安全性に係わる成績は安全性解析対象集団における成績を記載し,有効性に係 わる記載を FAS 解析対象集団とした。主要評価項目に関する主解析の感度分析において PPS 解 析対象集団あるいは ARS 解析対象集団を対象とした解析を実施したが特記すべき事項は認め られなかったため割愛した.
安全性解析対象集団 507 名(PEG/R 群 254 名,IFN/R 群 253 名)について,治験薬投与期間
と観察期間ごとの被験者数を表 2.7.6-56に示す.投与期間が 24 週に達した被験者は PEG/R 群
87.8 %( 223 名 /254 名) , IFN/R 群 84.6% ( 214 名 /253 名)であり,両群とも 80% 以上の被験者に
おいて 24 週間以上の投与がなされた.また, 48 週間の投与を完了した被験者数は PEG/R 群
79.5 %( 202 名 /254 名) , IFN/R 群 78.3% ( 198 名 /253 名)であり, 2 群間でほぼ同程度であった.
611
表 2.7.6-56 投与期間別被験者数及び割合(C033*)
投与期間 PEG/R群(n= 254) IFN/R群(n= 253)
2週
(1〜14日) 250(98.4%) 243(96.0%)
4週
(15〜28日) 245(96.5%) 236(93.3%)
8週
(29〜56日) 240(94.5%) 232(91.7%)
12週
(57〜84日) 233(91.7%) 226(89.3%)
24週
(85〜168日) 223(87.8%) 214(84.6%)
36週
(169〜252日) 210(82.7%) 205(81.0%)
48週
(253〜投与終了時点) 202(79.5%) 198(78.3%)
(CSR T01)
初回投与量別投与期間別被験者数及び割合を表 2.7.6-57に示す.
投与期間が 24 週に達した被験者は PEG/R 群でリバビリン 600 mg 群 87.2 %( 82 名 /94 名) , 同 800 mg 群 88.0%(125 名/142 名) ,同 1,000 mg 群 88.9%(16 名/18 名)であった.また IFN/R 群ではリバビリン 600 mg 群 81.1%(86 名/106 名) ,同 800mg 群 86.3%(113 名/131 名) ,同 1,000 mg 群 93.8 %( 15 名 /16 名)であった.
PEG/R 群で投与期間が 48 週間の投与を完了した被験者数はリバビリン 600 mg 群 78.7% (74 名 /94 名) ,リバビリン 800 mg 群 78.9 %( 112 名 /142 名) ,リバビリン 1,000 mg 群 88.9 %( 16 名 /18 名)であった.また IFN/R 群で投与期間が 48 週間の投与を完了した被験者数はリバビリン 600 mg 群 72.6 %( 77 名 /106 名) ,リバビリン 800 mg 群 82.4 %( 108 名 /131 名) ,リバビリン 1,000 mg 群 81.3 %( 13 名 /16 名)であった.
表 2.7.6-57 初回投与量別投与期間別被験者数及び割合(C033*)
投与群 PEG/R群(n=254) IFN/R群(n=253)
PEG-IFNα-2b/IFNα-2b
用法・用量 1.5 µg/kg 1.5 µg/kg 1.5 µg/kg 600MIU 600MIU 600MIU リバビリン
用法・用量 600mg 800mg 1,000mg 600mg 800mg 1,000mg 投与期間\被験者数 94 142 18 106 131 16 2週
(1〜14日)
93
(98.9%)
139
(97.9%)
18
(100.0%)
100
(94.3%)
127
(96.9%)
16
(100.0%)
4週
(15〜28日)
91
(96.8%)
137
(96.5%)
17
(94.4%)
96
(90.6%)
124
(94.7%)
16
(100.0%)
8週
(29〜56日)
89
(94.7%)
135
(95.1%)
16
(88.9%)
93
(87.7%)
123
(93.9%)
16
(100.0%)
12週
(57〜84日)
85
(90.4%)
132
(93.0%)
16
(88.9%)
92
(86.8%)
119
(90.8%)
15
(93.8%)
24週
(85〜168日)
82
(87.2%)
125
(88.0%)
16
(88.9%)
86
(81.1%)
113
(86.3%)
15
(93.8%)
36週
(169〜252日)
77
(81.9%)
117
(82.4%)
16
(88.9%)
80
(75.5%)
110
(84.0%)
15
(93.8%)
48週
(253〜投与終了時点)
74
(78.7%)
112
(78.9%)
16
(88.9%)
77
(72.6%)
108
(82.4%)
13
(81.3%)
(CSR T02)
*;新薬承認情報提供時に置き換えた
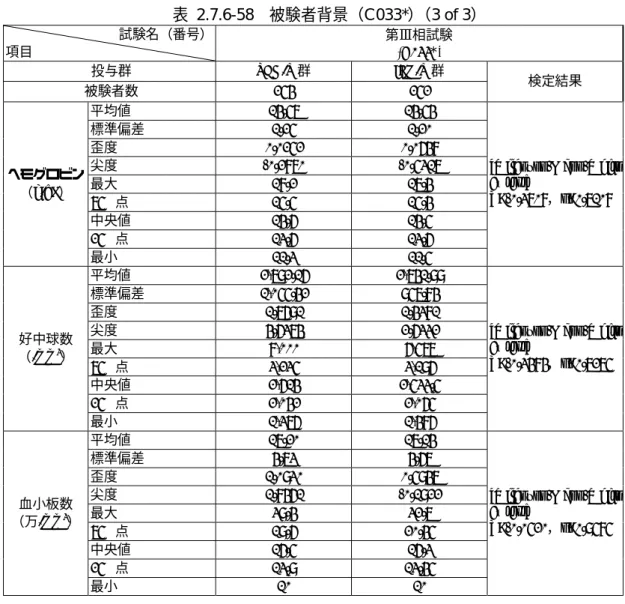
FAS 対象集団 506 名(PEG/R 群 254 名, IFN/R 群 252 名)の被験者背景を表 2.7.6-58に示し た.
表 2.7.6-58 被験者背景( C033* ) ( 1 of 3 )
試験名(番号)項目
第Ⅲ相試験 (C033*) 投与群 PEG/R群 IFN/R群
被験者数 254 252 検定結果
男性 165 (65.0%) 164 (65.1%) 性別 女性 89 (35.0%) 88 (34.9%)
[Fisher's exact test]
p =1.0000
平均値 52.17 50.73
標準偏差 10.26 bae5
歪度 -0.6956 -0.4596
尖度 -0.1444 -0.0339
最大 70 70
75%点 60 59
中央値 54 51
25%点 46 44
最小 22 20
[Wilcoxon–Mann–Whit ney test]
Z=-1.8359,p=0.0664
65歳未満 237 (93.3%) 229 (90.9%) 年齢
(歳)
65歳以上 17 (6.7%) 23 (9.1%)
[Fisher's exact test]
p =0.3275
平均値 63.97 63.68
標準偏差 10.23 11.11
歪度 0.3130 0.3460
尖度 -0.2691 -0.1980
最大 96 97.2
75%点 70.9 70.95
中央値 64 64.2
25%点 56 55.1
最小 43.5 40.5
[Wilcoxon-Mann-Whitn ey test]
Z=-0.3238,p=0.7461
60kg以下 94 (37.0%) 106 (42.1%) 60kg超過80kg以下 142 (55.9%) 130 (51.6%) 体重
(kg)
80kg超過 18 (7.1%) 16 (6.3%)
[拡張Mantel-Haenszel test]
χ2=1.2895,df= 1,
p=0.2561
平均値 10.11 9.31
標準偏差 7.62 7.30
歪度 1.3043 1.2192
尖度 1.8355 1.5863
最大 40 38
75%点 13 12
中央値 9 8
25%点 5 4
最小 0 0
[Wilcoxon-Mann-Whitn ey test]
Z=-1.3991,p=0.1618
1年未満 5 (2.0%) 7 (2.8%) 1年以上5年未満 50 (19.7%) 66 (26.2%) 5年以上10年未満 89 (35.0%) 81 (32.1%) 罹病期間
(年)
10年以上 110 (43.3%) 98 (38.9%)
[拡張Mantel-Haenszel test]
χ2=2.5bae,df= 1,
p=0.1131 なし 55 (21.7%) 43 (17.1%)
合併症の
有無 あり 199 (78.3%) 209 (82.9%)
[Fisher's exact test]
p =0.2162 未治療例 137 (53.9%) 139 (55.2%)
再燃例 91 (35.8%) 81 (32.1%) 無効例 26 (10.2%) 31 (12.3%) IFN治療歴
不明 0 (0.0%) 1 (0.4%)
[Fisher's exact test]
p =0.6160 1a 2 (0.8%) 0 (0.0%)
1b 252 (99.2%) 252 (100.0%) Genotype
1a,1b以外 0 (0.0%) 0 (0.0%)
[Fisher's exact test]
p =0.4990
*;新薬承認情報提供時に置き換えた
613
表 2.7.6-58 被験者背景(C033*) (2 of 3)
試験名(番号)
項目
第Ⅲ相試験 (C033*) 投与群 PEG/R群 IFN/R群
被験者数 254 252 検定結果
no fibrosis 0 (0.0%) 0 (0.0%) mild fibrosis 107 (42.1%) bae (41.3%) moderate fibrosis 109 (42.9%) 114 (45.2%) severe fibrosis 34 (13.4%) 30 (11.9%) cirrhosis 1 (0.4%) 3 (1.2%) 肝生検
(国際分類)
staging
読影不能 3 (1.2%) 1 (0.4%)
[拡張Mantel-Haenszel test]
χ2=0.0211,df= 1,
p=0.8844 None 0 (0.0%) 0 (0.0%)
Minimal chronic
hepatitis 48 (18.9%) 49 (19.4%) Mild chronic hepatitis 88 (34.6%) 89 (35.3%) Moderate chronic
hepatitis 102 (40.2%) 94 (37.3%) Severe chronic
hepatitis 13 (5.1%) 19 (7.5%) 肝生検
(国際分類)
grading
読影不能 3 (1.2%) 1 (0.4%)
[拡張Mantel-Haenszel test]
χ2=0.0032,df= 1,
p=0.9547
平均値 689.13 693.93
標準偏差 212.93 208.89
歪度 -1.0699 -1.1072
尖度 -0.1378 -0.0336
最大 850 850
75%点 850 850
中央値 850 850
25%点 550 540
最小 130 110
[Wilcoxon-Mann-Whitn ey test]
Z=0.1848,p=0.8534
300未満 22 (8.7%) 16 (6.3%) 300以上500未満 31 (12.2%) 38 (15.1%) 500以上850未満 71 (28.0%) 68 (27.0%) HCV-RNA
定量
[RT-PCR法:
アンプリコア HCV モニター]
(KIU/mL)
850以上 130 (51.2%) 130 (51.6%)
[拡張Mantel-Haenszel test]
χ2=0.0151,df= 1,
p=0.9023
平均値 114.92 11bae
標準偏差 70.44 63.56
歪度 1.7202 1.5080
尖度 3.6565 2.8100
最大 433 409
75%点 146 136
中央値 93.5 95
25%点 64 66
最小 32 28
[Wilcoxon-Mann-Whitn ey test]
Z=-0.2186,p=0.8269
45未満 16 (6.3%) 16 (6.3%) 45以上100未満 121 (47.6%) 117 (46.4%) 100以上200未満 88 (34.6%) 96 (38.1%) 血清中ALT
濃度
(IU/L)
200以上 29 (11.4%) 23 (9.1%)
[拡張Mantel-Haenszel test]
χ2=0.0007,df= 1,
p=0.9784
*;新薬承認情報提供時に置き換えた
表 2.7.6-58 被験者背景(C033*) (3 of 3)
試験名(番号)
項目
第Ⅲ相試験 (C033*) 投与群 PEG/R群 IFN/R群
被験者数 254 252 検定結果
平均値 14.57 14.54
標準偏差 1.25 1.20
歪度 0.0252 0.0648
尖度 -0.2870 -0.5317
最大 18.2 17.4
75%点 15.5 15.4
中央値 14.6 14.5
25%点 13.6 13.6
ヘモグロビン
(g/dL)
最小 11.3 11.5
[Wilcoxon-Mann-Whitn ey test]
Z=-0.3708,p=0.7108
平均値 2,792.16 2,741.99
標準偏差 1,055.42 957.84
歪度 1.7691 1.4381
尖度 4.6374 2.6332
最大 8,000 6,577
75%点 3,235 3,196
中央値 2,614 2,533.5
25%点 2,042 2,065
好中球数
(/mm3)
最小 1,376 1,486
[Wilcoxon-Mann-Whitn ey test]
Z=-0.3484,p=0.7275
平均値 17.20 17.14
標準偏差 4.73 4.67
歪度 1.0930 0.5947
尖度 1.8461 -0.1922
最大 39.4 32.7
75%点 19.6 20.45
中央値 16.5 16.3
25%点 13.9 13.45
血小板数
(万/mm3)
最小 10 10
[Wilcoxon-Mann-Whitn ey test]
Z=-0.0520,p=0.9585
( CHC500 : CSR14_1_4_1&CSR14_1_4_6 )
両群間に不均衡がみられたのは,年齢( Wilcoxon-Mann-Whitney test , p=0.0664 ) ,罹病期間
(拡張 Mantel-Haenszel test , p=0.1131 )であった.不均衡の認められた項目に関して,これら 2 因子を共変量として加えたロジスティックモデルにより,不均衡を調整した薬剤群間の投与終 了時,投与終了後 12 週時,投与終了後 24 週時の HCV-RNA 陰性化率及び ALT 正常化率に影響 は及ぼさなかった.その他の項目では両群間に不均衡は認められなかった.
*;新薬承認情報提供時に置き換えた
615
投与 24 週時,投与終了時,投与終了後 12 週時,投与終了後 24 週時の HCV-RNA 陰性化率を 表 2.7.6-59に示す.
投与終了後 24 週時の HCV-RNA 陰性化率は, PEG/R 群 47.6 % ( 121 名 /254 名) , IFN/R 群 44.8 %
(113 名/252 名)となり, 2 群間の差(PEG/R 群−IFN/R 群)の両側 95%信頼区間は-5.9%〜11.5%
であった.また,副次的評価項目である投与 24 週時の HCV-RNA 陰性化率は, PEG/R 群 66.9 %
( 170 名 /254 名) , IFN/R 群 65.5 %( 165 名 /252 名)となり, 2 群間の差( PEG/R 群− IFN/R 群)
の両側 95 %信頼区間は -6.8 %〜 9.7 %であり,投与終了時の HCV-RNA 陰性化率は, PEG/R 群 71.7 %( 182/254 名) , IFN/R 群 69.4 %( 175 名 /252 名)となり, 2 群間の差( PEG/R 群− IFN/R 群)の両側 95%信頼区間は-5.7%〜10.2%であり,投与終了後 12 週時の HCV-RNA 陰性化率は,
PEG/R 群 47.6 %( 121 名 /254 名) , IFN/R 群 45.6 %( 115 名 /252 名)となり, 2 群間の差( PEG/R 群−IFN/R 群)の両側 95%信頼区間は-6.7%〜10.7%となった.したがって,HCV-RNA 陰性化 率の群間(PEG/R 群−IFN/R 群)の差の 95%両側信頼区間は下側同等性限界を信頼区間の下限 が上回ったことから,それぞれの時期で IFN/R 群に対する PEG/R 群の非劣性が検証され,
PEG-IFNα-2b+リバビリン併用での有効性は IFNα-2b+リバビリン併用と比べて劣ることはな いと判断した.
なお,感度分析として PPS 及び ARS を解析対象集団とした場合及び FAS を解析対象集団と して IFN 治療歴を共変量としたロジスティック回帰分析について主解析と同様の解析を実施し た.その結果, HCV-RNA 持続陰性化率の群間の差( PEG/R 群− IFN/R 群)の差の 95% 両側信 頼区間は PPS で-7.0%〜11.9%,ARS で-5.9%〜11.4%及び IFN 治療歴を共変量とした場合で -6.9 %〜 10.1% といずれも下側同等性限界を信頼区間の下限が上回っており,感度分析において も主解析と同様の結果が得られ,主解析の頑強性が示された.
表 2.7.6-59 HCV-RNA 陰性化率(C033**)
HCV-RNA定性 PEG/R群
(n= 254 )
IFN/R群
(n= 252 )
HCV-RNA陰性化率の群間 の差*の95%信頼区間 投与24週時 (n= 254 ) (n= 252 )
陰性 170 (66.9%) 165 (65.5%) -6.8% 〜 9.7%
陽性 57 (22.4%) 53 (21.0%) 判定不能 27 (10.6%) 34 (13.5%) 投与終了時 (n= 254 ) (n= 252 )
陰性 182 (71.7%) 175 (69.4%) -5.7% 〜 10.2%
陽性 69 (27.2%) 70 (27.8%) 判定不能 3 ( 1.2%) 7 ( 2.8%) 投与終了後12週時 (n= 254 ) (n= 252 )
陰性 121 (47.6%) 115 (45.6%) -6.7% 〜 10.7%
陽性 122 (48.0%) 122 (48.4%) 判定不能 11 ( 4.3%) 15 ( 6.0%) 投与終了後24週時 (n= 254 ) (n= 252 )
陰性 121 (47.6%) 113 (44.8%) -5.9% 〜 11.5%
陽性 122 (48.0%) 120 (47.6%) 判定不能 11 ( 4.3%) 19 ( 7.5%)
*PEG/R群-IFN/R群(CSR 14_2_2_1)
投与開始前の主要な背景因子[肝生検 staging (国際分類) ,肝生検 grading (国際分類) ,投
**;新薬承認情報提供時に置き換えた
与開始前の HCV-RNA 量(RT-PCR 法) ,投与開始前の ALT,年齢(高齢/非高齢で分類)及び性 別]で層別したサブグループ解析の結果を表 2.7.6-60に示した.
投与終了後 24 週時の HCV-RNA 陰性化率において, IFN 治療歴未治療例では, PEG/R 群 43.1 %
(59 名/137 名) , IFN/R 群 46.8%(65 名/139 名) ,再燃例では,PEG/R 群 62.6%(57 名/91 名) , IFN/R 群 51.9 %( 42 名 /81 名)となり,無効例では, PEG/R 群 19.2 %( 5 名 /26 名) , IFN/R 群 19.4 %
( 6 名 /31 名)であり, IFN 治療歴における有効性が無効例であった集団では,投与終了後 24 週時 HCV-RNA 陰性化が低くなる結果となった.
表 2.7.6-60 HCV-RNA 陰性化率−被験者背景のサブグループ解析−(C033**)
項目/投与群 PEG/R群 IFN/R群
被験者数 254 252
HCV-RNA陰性化率の群 間の差*の95%信頼区間 未治療 59/137 (43.1%) 65/139 (46.8%) -15.4%〜8.0%
再燃 57/91 (62.6%) 42/81 (51.9%) -4.0%〜25.5%
無効 5/26 (19.2%) 6/31 (19.4%) -20.7%〜20.4%
IFN治療歴
不明 0/0 (0.0%) 0/1 (0.0%) − no fibrosis 0/0 (0.0%) 0/0 (0.0%) − mild fibrosis 60/107 (56.1%) 55/bae (52.9%) -10.2%〜 16.6%
moderate fibrosis 50/109 (45.9%) 44/114 (38.6%) -5.7%〜 20.2%
severe fibrosis 11/34 (32.4%) 13/30 (43.3%) -34.7%〜 12.7%
cirrhosis 0/1 (0.0%) 0/3 (0.0%) − 肝生検
(Staging)
読影不能 0/3 (0.0%) 1/1 (100.0%) -100.0%〜-100.0%
none 0/0 (0.0%) 0/0 (0.0%) − minimal chronic
hepatitis 20/48 (41.7%) 20/49 (40.8%) -18.7%〜 20.4%
mild chronic hepatitis 52/88 (59.1%) 46/89 (51.7%) -7.2%〜 22.0%
moderate chronic
hepatitis 45/102 (44.1%) 41/94 (43.6%) -13.4%〜 14.4%
severe chronic
hepatitis 4/13 (30.8%) 5/19 (26.3%) -27.5%〜 36.4%
肝生検
(Grading)
読影不能 0/3 (0.0%) 1/1 (100.0%) -100.0%〜-100.0%
300未満 13/22 (59.1%) 9/16 (56.3%) -29.0%〜 34.7%
300以上500未満 16/31 (51.6%) 15/38 (39.5%) -11.3%〜 35.6%
500以上850未満 27/71 (38.0%) 22/68 (32.4%) -10.2%〜 21.5%
投与開始前 のHCV-RNA
量(RT-PCR
法:KIU/mL) 850以上 65/130 (50.0%) 67/130 (51.5%) -13.7%〜 10.6%
10超11以下 26/72 (36.1%) 39/87 (44.8%) -24.0%〜6.5%
11超12以下 48/93 (51.6%) 35/85 (41.2%) -4.1%〜25.0%
12超13以下 40/73 (54.8%) 32/65 (49.2%) -11.1%〜22.2%
13超14以下 7/16 (43.8%) 6/12 (50.0%) -43.5%〜31.0%
リバビリン 初回投与量
(mg/kg)
14超15以下 0/0 (0.0%) 1/3 (33.3%) − 45未満 5/16 (31.3%) 8/16 (50.0%) -52.2%〜 14.7%
45以上100未満 53/121 (43.8%) 54/117 (46.2%) -15.0%〜 10.3%
100以上200未満 45/88 (51.1%) 42/96 (43.8%) -7.0%〜 21.8%
投与開始前 のALT
(IU/mL)
200以上 18/29 (62.1%) 9/23 (39.1%) -3.7%〜 49.6%
65以上 6/17 (35.3%) 7/23 (30.4%) -24.6%〜 34.3%
年齢(歳)
65未満 115/237 (48.5%) 106/229 (46.3%) -6.8%〜 11.3%
男性 94/165 (57.0%) 82/164 (50.0%) -3.8%〜 17.7%
性別 女性 27/89 (30.3%) 31/88 (35.2%) -18.7%〜 8.9%
Caucasian 0/1 (0.0%) 0/0 (0.0%) − 人種 Asian 121/253 (47.8%) 113/252 (44.8%) -5.7%〜11.7%
40以上60未満 33/94 (35.1%) 45/106 (42.5%) -20.8%〜 6.1%
60以上80未満 82/142 (57.7%) 63/130 (48.5%) -2.5%〜 21.1%
投与開始前 体重(kg)
80以上100未満 6/18 (33.3%) 5/16 (31.3%) -29.4%〜 33.5%
*PEG/R群-IFN/R群(CSR14_2_2_3〜14_2_3_7,14_2_5_1)
**;新薬承認情報提供時に置き換えた
617
投与群ごとに,各予後因子について単変量ロジスティック回帰分析を実施した.また,各予 後因子の同時影響について検討する目的で,投与群ごとに,変数増減法(stepwise 法)で変数 選択(有意水準:両側 20% )をした後に選択されたモデルを用いて,多変量ロジスティック回 帰分析を実施した.
多変量ロジスティック回帰分析(stepwise 法)の結果を表 2.7.6-61(1)及び(2)に示す.
HCV-RNA 持続陰性化率に影響をおよぼす有意な予後因子としては, PEG/R 群では,インター フェロン治療歴,肝生検 staging (国際分類) ,性別及び投与期間が同定され(有意水準:両側 5% ) , IFN/R 群では,インターフェロン治療歴,肝生検 staging (国際分類) ,肝生検 grading (国 際分類) ,HCV-RNA 量(RT-PCR 法)及び投与期間が同定されたが,性別は要因として同定さ れなかった(有意水準:両側 5% ) .
表 2.7.6-61 ( 1 )多変量ロジスティック回帰分析の結果( PEG/R 群) [ FAS ]
R 14*無作為化後の変更を反映したもの.
表中にはstepwise法で変数選択された因子のみを示した.
表 2.7.6-60(2)多変量ロジスティック回帰分析の結果(IFN/R 群) [FAS]
信頼区間
下限 上限
インターフェロン治療歴* 0.0099
再燃/未治療 0.963 0.519 1.787 0.9046
無効/未治療 0.210 0.075 0.587 0.0029
肝生検staging(国際分類) 0.479 0.269 0.853 0.0124
肝生検 grading(国際分類) 1.702 1.061 2.731 0.0275
HCVRNA量(RT‑PCR法) 0.0245
300KIU/mL以上500KIU/mL未満/300KIU/mL未満 0.719 0.201 2.578 0.6129 500KIU/mL以上850KIU/mL未満/300KIU/mL未満 0.414 0.125 1.366 0.1476 850KIU/mL以上/300KIU/mL未満 1.216 0.395 3.745 0.7329 投与期間% 80%以上/80%未満 18.229 5.919 56.145 <.0001
因子 オッズ比 P値
*無作為化後の変更を反映したもの.
表中にはstepwise法で変数選択された因子のみを示した.
HCV-RNA 陰性化率の経時的変化を表 2.7.6-62及び図 2.7.6-10に示す.両群とも投与開始後 2 週目より HCV-RNA が陰性化し,投与開始後 4 週目から 12 週目にかけて陰性化を示す被験者が 多くなる.その後,緩やかに陰性化した被験者が増加し,投与開始後 36 週目にほぼ定常状態に 達することが認められた.なお,各測定時点のデータが欠測あるいは「判定不能」の場合は評 価母数から省いた.
信頼区間
下限 上限
インターフェロン治療歴* 0.0020
再燃/未治療 1.757 0.919 3.357 0.0882
無効/未治療 0.213 0.067 0.678 0.0088
肝生検staging (国際分類) 0.541 0.346 0.844 0.0068
ALT 0.0801
45IU/L超過100IU/L未満/45IU/L以下 1.007 0.284 3.570 0.9916 100IU/L以上200IU/L未満/45IU/L以下 1.815 0.480 6.861 0.3797 200IU/L以上/45IU/L以下 3.684 0.774 17.541 0.1015
体重 0.0548
60kgを超え80kg以下/40kgを超え60kg以下 1.635 0.759 3.521 0.2091 80kgを超え100kg以下/40kgを超え60kg以下 0.424 0.111 1.620 0.2095
性別 女性/男性 0.419 0.192 0.914 0.0289
投与期間(%) 80%以上/80%未満 15.750 5.362 46.261 <.0001
因子 オッズ比 P値
618
表 2.7.6-62 HCV-RNA 陰性化率の経時変化(C033*)
測定時期 PEG/R群
(n=254)
IFN/R群
(n=252)
投与開始後2週 5/253 (2.0%) 16/249 ( 6.4%) 投与開始後4週 23/249 (9.2%) 31/238 (13.0%)
投与開始後12週 144/238 (60.5%) 124/232 (53.4%) 投与開始後24週 170/227 (74.9%) 165/218 (75.7%) 投与開始後36週 171/218 (78.4%) 162/208 (77.9%) 投与開始後48週 164/206 (79.6%) 155/201 (77.1%) 投与終了時 182/251 (72.5%) 175/245 (71.4%) 投与終了後4週 131/245 (53.5%) 127/239 (53.1%) 投与終了後12週 121/243 (49.8%) 115/237 (48.5%) 投与終了後24週 121/243 (49.8%) 113/233 (48.5%)
観察終了時 121/244 (49.6%) 114/237 (48.1%) (CSR 14_2_6_2)
0.0 20.0 40.0 60.0 80.0 100.0
0 12 24 36 48 60 72 84 96
投与及び観察時期(週)
HCV‑RNA陰性化率(%)
PEG/R群 IFN/R群
投与期間 観察期間 観察終了
投与終了時 12 24
図 2.7.6-10 HCV-RNA 陰性化率の経時変化(C033*)
619
HCV-RNA が最初に陰性化した時期と,HCV-RNA 持続陰性化率の関係を表 2.7.6-63に示し た.投与開始後 12 週時点までで HCV-RNA(RT-PCR 法:定性)が,初めて陰性化した被験者 では,両群ともに 75%以上の被験者で HCV-RNA 持続陰性化が得られた.すなわち,投与開始 後 2,4 及び 12 週時点に初めて HCV-RNA が陰性化した被験者で HCV-RNA 持続陰性化が得ら れた被験者は, PEG/R 群で, 100% ( 5 名 /5 名) , 100% ( 18 名 /18 名)及び 71.1% ( 86 名 /121 名) , IFN/R 群で 75.0%(12 名/16 名) ,84.2%(16 名/19 名)及び 69.9%(65 名/93 名)であった.投 与開始後 24 週で初めて陰性化した被験者のうち,HCV-RNA 持続陰性化が得られた被験者は,
PEG/R 群及び IFN/R 群で,それぞれ 36.4% ( 12 名 /33 名)及び 40.8% ( 20 名 /49 名)であった.
なお,投与開始後 24 週より後に HCV-RNA が初めて陰性化した被験者で,HCV-RNA 持続陰性 化が得られた被験者は認められなかった.
表 2.7.6-63 HCV-RNA が最初に陰性化した時期と HCV-RNA 持続陰性化率(C033*)
時期 HCV-RNA定性 PEG/R群(n=254) IFN/R群(n=252)
被験者数 n=5 n=16 陰性 5 (100.0%) 12 (75.0%) 陽性 0 (0.0%) 1 (6.3%) 投与開始後2週時
判定不能 0 (0.0%) 3 (18.8%) 被験者数 n=18 n=19
陰性 18 (100.0%) 16 (84.2%) 陽性 0 (0.0%) 3 (15.8%) 投与開始後4週時
判定不能 0 (0.0%) 0 (0.0%) 被験者数 n=121 n=93
陰性 86 (71.1%) 65 (69.9%) 陽性 34 (28.1%) 24 (25.8%) 投与開始後12週時
判定不能 1 (0.8%) 4 (4.3%) 被験者数 n=33 n=49
陰性 12 (36.4%) 20 (40.8%) 陽性 21 (63.6%) 28 (57.1%) 投与開始後24週時
判定不能 0 (0.0%) 1 (2.0%)
被験者数 n=11 n=6
陰性 0 (0.0%) 0 (0.0%) 陽性 11 (100.0%) 6 (100.0%) 投与開始後36週時
判定不能 0 (0.0%) 0 (0.0%)
被験者数 n=4 n=2
陰性 0 (0.0%) 0 (0.0%) 陽性 4 (100.0%) 2 (100.0%) 投与開始後48週時
判定不能 0 (0.0%) 0 (0.0%)
被験者数 n=1 n=1
陰性 0 (0.0%) 0 (0.0%) 陽性 1 (100.0%) 1 (100.0%) 投与中止時
判定不能 0 (0.0%) 0 (0.0%) (CSR
14_2_6_3_2)
投与終了時点で, HCV-RNA (RT-PCR 法:定性)が陰性であった 357 名(PEG/R 群:182 名,
IFN/R 群: 175 名)のうち,投与終了後 24 週時点のデータが利用可能(判定不能を除く)な 353 名( PEG/R 群: 181 名, IFN/R 群: 172 名)について,投与終了後 24 週時点での HCV-RNA ( RT-PCR 法:定性)の測定結果から,再燃率を算出した.その結果,投与終了後 24 週時点での再燃率は PEG/R 群及び IFN/R 群で,それぞれ 33.7% ( 61 名 /181 名)及び 34.3% ( 59 名 /172 名)と両群で ほぼ同様の結果であった.投与終了後 12 週時点で HCV-RNA が陰性であった被験者のうち,投
*;新薬承認情報提供時に置き換えた

![表 2.7.6-54 試験の概要(C019*) (5 of 7) 項目 内容 評価基準 3. ウイルス学的効果[投与期間終了後 12 週時点] 投与期間終了後12週時点における血中 HCV-RNA 定性値(RT-PCR 法:アンプリコア定性)を指標として以下の基準で判定する. 陰性: 投与期間終了後12週時点における血中HCV-RNA定性値が陰性である被験者陽性: 投与期間終了後12週時点における血中HCV-RNA定性値が陽性である被験者判定不能:データ欠損のため判定できない被験者 治験薬投与中止後](https://thumb-ap.123doks.com/thumbv2/123deta/7623712.2548745/5.892.109.790.97.1100/試験ウイルス週時点おけるアンプリコアとしておけるおけるデータ.webp)