ペガシス/コペガス 2.5 臨床に関する概括評価 Page
1
ペガシス皮下注
90 μg
(ペグインターフェロン アルファ
-2a(遺伝子組換え))
コペガス錠
200 mg
(リバビリン)
[
C 型代償性肝硬変]
第
2部 (モジュール2):CTD の概要(サマリー)
2.5 臨床に関する概括評価
中外製薬株式会社
ペガシス/コペガス 2.5 臨床に関する概括評価 Page
2
略語一覧
略語 省略していない表記
AFP α-fetoprotein/α フェトプロテイン
ALT Alanine aminotransferase/アラニン・アミノトランスフェラーゼ(GPT)
AST Asparatate aminotransferase/ ア ス パラ ギン 酸アミ ノト ランス フェ ラーゼ
(GOT)
AUC0-t Area under the concentration-time curve from zero to time t/投与後 t 時間まで
の血中濃度-時間曲線下面積
Cmax Maximum concentration/最高血中濃度
CDS Core Data Sheet/企業中核データシート
CLss/F Total body clearance at steady state/定常状態における全身クリアランス
CMH Cochran-Mantel-Haenszel/コクランマンテルヘンツェル(検定)
CT Computerized tomography/コンピューター断層撮影法
FAS Full Analysis Set/最大の解析対象集団
γ-GTP γ-Glutamyltranspeptidase/γ-グルタミルトランスペプチダーゼ
HCV Hepatitis C virus/C 型肝炎ウイルス
HCV-RNA Hepatitis C virus-ribonucleic acid/C 型肝炎ウイルスリボ核酸
HIV Human immunodeficiency virus/ヒト免疫不全ウイルス
IFN Interferon /インターフェロン
ITT Intention to treat
KIU Kilo-international unit/キロ国際単位
KL-6 シアル化糖鎖抗原
MIU Million international unit/百万国際単位
MRI Magnetic resonance imaging/核磁気共鳴画像法
MSSA Methicillin-sensitive Staphylococcus aureus/メチシリン感受性黄色ブドウ球菌
PEG Polyethylene glycol/ポリエチレングリコール
PEG-IFNα-2a (Ro25-8310)
Peginterferon Alfa-2a(Genetical Recombination)/ペグインターフェロン ア ルファ-2a(遺伝子組換え)
PEG-IFNα-2b Peginterferon Alfa-2b(Genetical Recombination)/ペグインターフェロン ア ルファ-2b(遺伝子組換え)
RBV Ribavirin/リバビリン
PIVKA-Ⅱ Protein induced by Vitamin K absence or antagonists-Ⅱ/凝固因子プロトロンビ ンの前駆体
rIFNα-2a Interferon Alfa-2a(Genetical Recombination)/インターフェロン
アルファ-2a(遺伝子組換え)
rIFNα-2b Interferon Alfa-2b(Genetical Recombination)/インターフェロン
アルファ-2b(遺伝子組換え)
SD Standard deviation/標準偏差
ペガシス/コペガス 2.5 臨床に関する概括評価 Page
3
目次
頁 2.5 臨床に関する概括評価 ... 6 2.5.1 製品開発の根拠 ... 6 2.5.1.1 C 型代償性肝硬変に対する治療の必要性 ... 6 2.5.1.2 C 型代償性肝硬変に対する治療の現状 ... 7 2.5.1.3 C 型代償性肝硬変に対する PEG-IFNα-2a とリバビリン併用療法開発の根拠 ... 9 2.5.1.4 C 型代償性肝硬変に対する PEG-IFNα-2a とリバビリン併用療法開発の経緯 ... 15 2.5.1.5 臨床試験データパッケージ ... 19 2.5.2 生物薬剤学に関する概括評価 ... 21 2.5.3 臨床薬理に関する概括評価 ... 21 2.5.4 有効性の概括評価 ... 22 2.5.4.1 対象患者と患者背景 ... 22 2.5.4.2 C 型代償性肝硬変での効果 ... 23 2.5.4.2.1 投与終了後 24 週時のウイルス学的効果(観察群との比較) ... 23 2.5.4.2.2 ジェノタイプ別,ウイルス量別及び IFN 製剤治療歴別集計 ... 24 2.5.4.3 初回陰性化時期と投与終了後 24 週時のウイルス学的効果の関連 ... 25 2.5.4.4 部分集団解析 ... 27 2.5.4.5 PEG-IFNα-2a の用量の検討 ... 27 2.5.4.6 有効性のまとめ及び考察 ... 32 2.5.5 安全性の概括評価 ... 35 2.5.5.1 暴露量 ... 35 2.5.5.1.1 PEG-IFNα-2a 及びリバビリンの投薬率/累積投与量 ... 35 2.5.5.1.2 減量・休薬の頻度 ... 36 2.5.5.2 有害事象 ... 37 2.5.5.2.1 比較的よくみられる有害事象 ... 37 2.5.5.2.2 有害事象の重症度別集計 ... 41 2.5.5.2.3 発現時期別有害事象 ... 42 2.5.5.2.4 死亡 ... 43 2.5.5.2.5 その他の重篤な有害事象 ... 43 2.5.5.2.6 その他の重要な有害事象 ... 47 2.5.5.2.7 特別な患者集団及び状況下における安全性 ... 50 2.5.5.3 臨床検査値 ... 53 2.5.5.4 バイタルサイン,心電図 ... 59 2.5.5.5 C 型慢性肝炎との比較 ... 59 2.5.5.6 安全性のまとめ及び考察 ... 61 2.5.5.7 市販後のデータ ... 64ペガシス/コペガス 2.5 臨床に関する概括評価 Page
4
2.5.6 ベネフィットとリスクに関する結論 ... 66 2.5.6.1 用法・用量 ... 66 2.5.6.2 ベネフィット ... 66 2.5.6.3 リスク ... 68 2.5.6.4 結論 ... 68 2.5.7 参考文献 ... 69 <表一覧> 表 2.5.1.2-1 C 型代償性肝硬変における IFNβ による HCV-RNA 陰性化率 ... 9 表 2.5.1.2-2 C 型代償性肝硬変における IFNβ を20週間以上投与した場合の HCV-RNA 陰 性化率(HCV セログループ別,HCV-RNA 量別) ... 9 表 2.5.1.2-3 C 型代償性肝硬変における IFNα による HCV-RNA 陰性化率 ... 9表 2.5.1.2-4 C 型代償性肝硬変における IFNα による HCV-RNA 陰性化率(HCV-RNA 量別) ... 9 表 2.5.1.3-1 海外第Ⅲ相臨床試験の投与終了後24週時のウイルス学的効果(NV15801, ITT) ... 11 表 2.5.1.3-2 海外第Ⅲ相臨床試験の投与終了後24週時のウイルス学的効果(NV15942, ITT) ... 12 表 2.5.1.3-3 国内外で肝硬変と定義される集団の組織学的診断 ... 12 表 2.5.1.3-4 海外第Ⅲ相臨床試験の C 型代償性肝硬変患者(移行期の患者を含む)におけ る投与終了後24週時のウイルス学的効果(NV15801,ITT) ... 13 表 2.5.1.3-5 海外第Ⅲ相臨床試験の C 型代償性肝硬変患者(移行期の患者を含まない)に おける投与終了後24週時のウイルス学的効果(NV15942,ITT) ... 14 表 2.5.1.3-6 国内第Ⅲ相臨床試験の C 型慢性肝炎患者における投与終了後24週時における ウイルス学的効果(JV15725,FAS) ... 15 表 2.5.1.4-1 C 型代償性肝硬変患者(JV19595及び JV19889)における PEG-IFNα-2a 及び リバビリンの用量調整基準:C 型慢性肝炎患者(JV15725)との比較 ... 19 表 2.5.1.5-1 臨床試験データパッケージ ... 20 表 2.5.4-1 有効性評価に用いた臨床試験一覧 ... 22 表 2.5.4.2.1-1 投与終了後24週時のウイルス学的効果(FAS) ... 23 表 2.5.4.2.2-1 ジェノタイプ別及びウイルス量別の投与終了後24週時のウイルス学的効果 (FAS) ... 24 表 2.5.4.2.2-2 ジェノタイプ1かつ高ウイルス量及び「ジェノタイプ1かつ高ウイルス量」 以外の患者別投与終了後24週時のウイルス学的効果(FAS) ... 24 表 2.5.4.2.2-3 HCV-RNA 量が500 KIU/mL 以上の患者における投与終了後24週時のウイル ス学的効果(FAS) ... 25 表 2.5.4.2.2-4 IFN 製剤治療歴別の投与終了後24週時のウイルス学的効果(FAS) ... 25 表 2.5.4.2.2-5 IFN 製剤前治療が IFN 製剤単独又はリバビリン併用別の投与終了後24週時 のウイルス学的効果(FAS) ... 25 表 2.5.4.3-1 初回 HCV-RNA 陰性化時期別の投与終了後24週時のウイルス学的効果 (JV19595,FAS 通期) ... 26 表 2.5.4.3-2 初回 HCV-RNA 陰性化時期別の投与終了後24週時のウイルス学的効果 (JV19889,FAS) ... 27 表 2.5.4.5-1 投与終了後24週時のウイルス学的効果(JV19595,FAS 通期) ... 28 表 2.5.4.5-2 投与終了時のウイルス学的効果(JV19595,FAS 通期) ... 29
ペガシス/コペガス 2.5 臨床に関する概括評価 Page
5
表 2.5.4.5-3 投与終了時ウイルス陰性例のウイルス再燃率(JV19595,FAS 通期) ... 29
表 2.5.4.6-1 ジェノタイプ別ウイルス量別の投与終了後24週時のウイルス学的効果:PEG-IFNα-2a とリバビリン併用療法,IFNβ 製剤単独療法及び IFNα 製剤単独療法 の比較 ... 34 表 2.5.5.1.1-1 PEG-IFNα-2a の投薬率/累積投与量(JV19595,FAS 通期) ... 35 表 2.5.5.1.1-2 PEG-IFNα-2a の投薬率/累積投与量(JV19889,FAS) ... 36 表 2.5.5.1.1-3 リバビリンの投薬率/累積投与量(JV19595,FAS 通期) ... 36 表 2.5.5.1.1-4 リバビリンの投薬率/累積投与量(JV19889,FAS) ... 36 表 2.5.5.2.1-1 発現率が10%以上の有害事象(JV19595,SAFETY) ... 39 表 2.5.5.2.1-2 発現率の群間差が10%以上の有害事象(JV19595,SAFETY) ... 40 表 2.5.5.2.1-3 発現率が10%以上の有害事象(JV19889,SAFETY) ... 41 表 2.5.5.2.5-1 重篤な有害事象発現例数(JV19595,SAFETY) ... 45 表 2.5.5.2.5-2 重篤な有害事象発現例数(JV19889,SAFETY) ... 47 表 2.5.5.2.6-1 投与中止に至った有害事象発現例数(JV19595,SAFETY) ... 48 表 2.5.5.2.6-2 投与中止に至った有害事象発現例数(JV19889,SAFETY) ... 49 表 2.5.5.2.6-3 治験薬の減量・休薬に至った有害事象(10%以上)(JV19595,SAFETY) 50 表 2.5.5.2.6-4 治験薬の減量・休薬に至った有害事象(10%以上)(JV19889,SAFETY) 50 表 2.5.5.3-1 いずれかの投与群で20%以上認められた臨床検査値異常(JV19595,SAFETY) ... 54 表 2.5.5.3-2 20%以上認められた臨床検査値異常(JV19889,SAFETY) ... 54 表 2.5.5.3-3 好中球数の投与後最小値(JV19595,SAFETY) ... 56 表 2.5.5.3-4 血小板数の投与後最小値(JV19595,SAFETY) ... 57 表 2.5.5.3-5 ヘモグロビン濃度の投与後最小値(JV19595,SAFETY) ... 59 <図一覧> 図 2.5.4.5-1 HCV-RNA 陰性化率(95%信頼区間)の推移(JV19595,FAS 通期) ... 30 図 2.5.4.5-2 ジェノタイプ1かつ高ウイルス量の患者における HCV-RNA 陰性化率(95%信 頼区間)の推移(JV19595,FAS 通期) ... 30 図 2.5.4.5-3 ウイルス変化量の推移(平均値± SD)(JV19595,FAS 通期) ... 31 図 2.5.4.5-4 ジェノタイプ1かつ高ウイルス量の患者におけるウイルス変化量の推移(平 均値 ± SD)(JV19595,FAS 通期) ... 32 図 2.5.5.3-1 好中球数の推移(中央値)(JV19595,SAFETY) ... 55 図 2.5.5.3-2 血小板数の推移(中央値)(JV19595,SAFETY) ... 57 図 2.5.5.3-3 ヘモグロビン濃度の推移(中央値)(JV19595,SAFETY) ... 58
ペガシス/コペガス 2.5 臨床に関する概括評価 Page
6
2.5 臨床に関する概括評価
2.5.1 製品開発の根拠
ペグインターフェロン アルファ-2a(遺伝子組換え)(治験成分記号:Ro25-8310,以下 PEG-IFNα-2a)はインターフェロン(以下 IFN)療法の有効性及び投与方法を改善する目的で インターフェロン アルファ-2a(遺伝子組換え)(以下 rIFNα-2a)に分子量約40KD の分枝メ トキシポリエチレングリコール1分子を共有結合させたたんぱく質である。PEG-IFNα-2a は, rIFNα-2a に比し薬物動態と薬力学の改善が認められ,C 型慢性肝炎患者に対して,週1回投与 による臨床的有効性及び安全性が確認されたことから,国内では,まず単独療法において「C 型慢性肝炎におけるウイルス血症の改善」の効能で2003年10月に承認された。 リバビリンは抗ウイルス剤として開発されたヌクレオシドアナログで,リバビリン単独療法 ではC 型慢性肝炎に対する十分な効果が認められていないが,IFN 製剤との併用により IFN の 単独療法と比較して有効性が改善することから,PEG-IFNα-2a とリバビリン(治験成分番号: Ro20-9963)の併用療法は2007年1月に C 型慢性肝炎におけるウイルス血症の改善を適応とし 承認を取得している。 PEG-IFNα-2a とリバビリンの併用療法の C 型代償性肝硬変に対する適応は,海外では C 型慢 性肝炎に含めた適応として2002年6月に EU,2002年12月に米国で承認を取得し,以降カナダ, ニュージーランド,オーストラリアなど世界110カ国以上で承認されている。現在,米国にお ける治療ガイドライン1)ではPEG-IFNα-2a とリバビリンの併用療法は C 型代償性肝硬変を含む C 型慢性肝炎での標準的な治療法となっている。一方,国内においても PEG-IFN とリバビリ ン併用療法は,C 型慢性肝炎に対する標準治療となっており,現在最も高い効果が期待できる 治療法であるが,C 型代償性肝硬変に対する適応は得られていない。 今回,国内で実施した臨床試験(JV19595及び JV19889)より,C 型代償性肝硬変に対する PEG-IFNα-2a とリバビリンの併用療法の有効性及び安全性が確認されたことから,ペガシス皮 下注90 μg 及びコペガス錠200 mg の C 型代償性肝硬変に対する新効能追加に係る承認事項一部 変更承認申請を行うものである。2.5.1.1
C 型代償性肝硬変に対する治療の必要性
(1) 代償性肝硬変の病態及び C 型代償性肝硬変の成因 肝硬変は肝本来の小葉構造が破壊され,代わりに偽小葉と線維化がびまん性に置き換えられ た病変である。慢性肝炎の病態は,肝組織の線維化の程度を表す staging と炎症所見を表す grading に分けて評価されており,国内で広く用いられている新犬山分類では staging の F4と分 類された場合を肝硬変と判定している2)。臨床的には,黄疸,腹水,肝性脳症などの肝不全症 状を呈する非代償性肝硬変とこれらを認めない代償性肝硬変に分けられる3)。肝硬変は,最終 的に肝不全・肝細胞癌に至る重篤な転帰をたどる疾患である。 肝硬変のうちC 型代償性肝硬変は,C 型肝炎ウイルス(以下,HCV)の感染に起因する慢性 炎症(C 型慢性肝炎)において壊死した肝細胞が線維に置き換えられ肝硬変に発展したもので ある。 (2) C 型代償性肝硬変の疫学及び治療の必要性 国内では肝硬変の成因のうち,76%がウイルス肝炎に由来していると報告されている。特に, C 型慢性肝炎由来の肝硬変は60%以上に上ると報告され,肝硬変の病態を呈する患者において HCV 感染は最大の原因と考えられている4)。 近年,HCV は世界的な保健・衛生上の問題とされている。全世界で1億7,000万人が HCV の 慢性キャリアであると推定され5),先進国では慢性肝炎の70%が HCV によるものと考えられて いる。現在,国内の HCV のキャリアは約200万人と推定されており,これらの感染経路は輸ペガシス/コペガス 2.5 臨床に関する概括評価 Page
7
血,血液製剤の使用,薬物乱用者間の注射器の使いまわし,鍼治療,刺青及び母子感染等によ るものと考えられている。 C 型慢性肝炎患者の約40%は10~15年のうちに肝硬変に進行し,約25%は30年で肝細胞癌に 進展することが知られており6),特に肝硬変まで進展した場合は発現年率で7~8%が肝細胞癌 に進展すると報告されている7)。また,2007年人口動態統計によると国内における肝及び肝内 胆管の悪性新生物による死亡者数は,1975年以降増加の一途をたどっており,近年はわずかな がら減少傾向であるものの,2009年度の死亡者数は32,725人であった8)。更に,1990年代以降 に報告された肝細胞癌発症例では,その約70%が HCV の持続感染に由来していることが報告 されている9)。 国内では HCV 感染者の年齢層は海外に比べて高く,HCV 感染者における肝細胞癌死亡率の 増加が問題となっており10),特に,C 型肝硬変の平均年齢は66.5歳と高く70歳前後が最も多い 10)。このような高齢者は肝細胞癌の好発年齢であることから,治療の緊急性が非常に高い集団 と考えられている。 ウイルスを駆除することにより肝細胞癌を予防できるとの報告は多数あり7),11)-13),「科学的 根拠に基づく肝癌診療ガイドライン(2009年版)」においても,C 型代償性肝硬変の発癌予防 に,インターフェロンを中心としたウイルス駆除療法を推奨している14)。更に,日本肝臓病患 者団体協議会からも「C 型代償性肝硬変に対するインターフェロン治療を早期に保険適用とす ること」との緊急要望も厚労省大臣官房に提出されている(2006年4月20日)。 こうした状況を踏まえ,厚生労働省は2002年度から「C 型肝炎等緊急総合対策」を実施して おり,2005年度には検査や治療面の見直しを目的として,「C 型肝炎対策等に関する専門家会 議」による報告書が取り纏められ,2008年には肝炎治療の改善につながる基本方針として「肝 炎研究7カ年戦略」が取り纏められた15)。また,2010年には肝炎対策基本法の施行により C 型 慢性肝炎治療の促進に向けた国家的対策が積極的に進められている。 以上,C 型慢性肝炎から連続した病態で,肝細胞癌に至る主な原因である C 型代償性肝硬変 について,欧米に比しペグ化 IFN 製剤とリバビリンの併用療法が使用できない等のドラッグ ラグがあり,わが国の C 型代償性肝硬変患者には高齢者が多いことも鑑み,緊急に欧米と同 じ治療法を導入する必要性が極めて高い疾患と考えられる。2.5.1.2
C 型代償性肝硬変に対する治療の現状
(1) 海外における治療の現状 海外ではC 型慢性肝炎と C 型代償性肝硬変を区別せずに治療薬の開発が進められている。現 在 C 型代償性肝硬変を含む C 型慢性肝炎を適応とする抗ウイルス療法として,インターフェロンアルファ(以下,IFNα)等の従来型 IFN 製剤,PEG-IFNα-2a 等のペグ化 IFN 製剤,及び
これらの IFN 製剤とリバビリンの併用療法が承認されており,米国国立衛生研究所(NIH)16) 及び欧州肝臓学会(EASL)17)のコンセンサス声明では C 型代償性肝硬変も IFN 療法の適応と されている。また,米国肝臓学会(AASLD)1)のガイドラインでも,ペグ化IFN 製剤とリバビ リンの併用療法がC 型代償性肝硬変の標準治療として推奨されている。 (2) 国内における治療の現状 国内では,C 型代償性肝硬変に対する治療としては,HCV の駆除を目的とした抗ウイルス療 法及び肝炎沈静化を目的とした対症療法である肝庇護療法が行われている。 本承認申請時点では,C 型代償性肝硬変患者における抗ウイルス療法として,インターフェ ロンベータ(以下,IFNβ)製剤が2006年4月に,天然型 IFNα 製剤が2008年10月にそれぞれ単 独療法による適応を取得しているのみで,これらの承認されている IFN 製剤はいずれもジェ ノタイプ及びウイルス量の分類ですべての患者を適応に含んでいるわけではない。すなわち, IFNβ 製剤はセログループ1かつ高ウイルス量(1 Meq/mL 以上)の C 型代償性肝硬変患者を含 まない臨床試験を実施したものであり,IFNα 製剤はセログループ1かつ血中ウイルス量が高い
ペガシス/コペガス 2.5 臨床に関する概括評価 Page
8
患者(HCV-RNA がアンプリコア法で500 KIU/mL 以上)に対して投与終了後24週時のウイル ス陰性化効果が十分でなく適応から除外されている。したがって,セログループ1の血中 HCV-RNA が高い C 型代償性肝硬変患者に対し肝線維化の進展抑制及び ALT の上昇を抑える など肝炎沈静化を目的とした肝庇護療法などの対症療法に依存しているのが現状である。また, これらの既承認の IFN 製剤は週3回以上の投与が必要であり利便性が低いことが課題となって いる。 C 型代償性肝硬変患者に対し,海外で標準療法として使用されている PEG-IFN とリバビリン の併用療法は,国内既存の IFN 製剤無効・再燃例やセログループ1かつ高ウイルス量の患者に 対する有効性が期待され,更に既存の IFN 製剤に比べ週1回投与での治療が可能となり利便性 も高いことから,早期のドラッグラグ解消が望まれている。 以下に,肝庇護療法及びIFN 単独療法の現状について記載した。 1) 肝庇護療法 肝庇護療法薬として,主にウルソデスオキシコール酸,グリチルリチン・グリシン・システ イン配合剤が使用されている。 ウルソデスオキシコール酸は,経口投与による治療が可能で,患者への負担が少ない薬剤で ある。「C 型慢性肝疾患における肝機能の改善」の効能・効果に対し,用法・用量は1日600 mg を3回に分割投与で,最大投与量は900 mg とされている18)。C 型慢性肝炎患者を対象とした 臨床試験において,ウルソデスオキシコール酸600 mg/日及び900 mg/日を24週間投与した際の ALT のベースラインに対する低下率は,600 mg/日(198例)で29.2%,900 mg/日(193例)で 36.2%であった19)。また,C 型慢性肝炎患者にウルソデスオキシコール酸600 mg/日(257例) (必用に応じ900 mg/日へ増量)を1年以上投与したところ,ベースラインに対する ALT の低 下率(中央値)は43.4%であったと報告されている18)。 グリチルリチン・グリシン・システイン配合剤はウルソデスオキシコール酸より肝機能改善 効果が高いと考えられている。用法・用量は「慢性肝疾患における肝機能異常の改善」に対し 1日1回40~60 mL を静脈内に注射又は点滴静注することとされている20)。C 型慢性肝炎及び肝 硬変患者(178例)にグリチルリチン・グリシン・システイン配合剤40 mL/日3週間連続静脈内 投与後,2週目の ALT が正常上限値の1.5倍以下に改善しなかった患者(93例)に,40 mL/日継 続投与群と100 mL/日継続投与群の有効性を比較したところ,ALT が正常上限値の1.5倍以下に なった患者は,40 mL/日継続投与群で26.1%(12/46例),100 mL/日投与群で52.3%(23/44例) であり,増量の有用性も報告されている21)。 2) IFN 単独療法 ① IFNβ22),23) 国内では初めての C 型代償性肝硬変に対する抗ウイルス療法として,2006年4月に天然型 IFNβ 製剤が「C 型代償性肝硬変におけるウイルス血症の改善(セログループ1の血中 HCV-RNA 量が高い場合を除く)」の効能・効果で承認された。C 型代償性肝硬変に対する IFNβ 製 剤の標準的な治療方法は,1日600万国際単位(以下,100万国際単位を MIU と標記)で投与を 開始し,投与後6週間までは1日3~6 MIU を連日投与する。以後1日3 MIU を週3回の静脈内投 与あるいは点滴静注する。 国内で実施された C 型代償性肝硬変患者(セログループ1かつ高ウイルス量の患者は除外) を対象とした臨床試験の有効性成績を表 2.5.1.2-1及び表 2.5.1.2-2に示す。投与終了後6カ月目 の HCV-RNA 陰性化率は,IFNβ の投与期間とともに高くなり,20~22週及び34~36週投与の 患者でそれぞれ28.9%及び38.8%であった。また,セログループ1以外の血中 HCV-RNA 量が高 い患者に対する持続性ウイルス学的効果は,10.0%と低かった。 本試験では,セログループ1かつ高ウイルス量の患者は組み入れられておらず,IFNβ の有効ペガシス/コペガス 2.5 臨床に関する概括評価 Page
9
性は確認されていない。 表 2.5.1.2-1 C 型代償性肝硬変における IFNβ による HCV-RNA 陰性化率 A 群 B 群 C 群 HCV-RNA 陰性化率 (投与終了6カ月後) 7/48(14.6%) 13/45(28.9%) 19/49(38.8%)• A 群:IFNβ を1日6 MIU を1週間連日投与後,3 MIU で連日又は週6回投与により計6~7週間投与 • B 群:A 群の投与方法に更に IFNβ を3 MIU 週3回投与を継続して計20~22週間投与
• C 群:A 群の投与方法に更に IFNβ を3 MIU 週3回投与を継続して計34~36週間投与
[文献22)より作成] 表 2.5.1.2-2 C 型代償性肝硬変における IFNβ を20週間以上投与した場合の HCV-RNA 陰性化率 (HCV セログループ別,HCV-RNA 量別) HCV セログループ セログループ1 セログループ1以外 HCV-RNA 量(Meq/mL) 1未満 1未満 1以上 HCV-RNA 陰性化率 (投与終了6カ月後) 10/28(35.7%) 16/33(48.5%) 3/30(10.0%) [文献22)より作成] ② IFNα(NAMALWA)24),25) 2008年10月に天然型 IFNα 製剤が「C 型代償性肝硬変におけるウイルス血症の改善(セログ ループ1の血中 HCV-RNA 量が高い場合を除く)」の効能・効果で,承認された。C 型代償性 肝硬変に対する IFNα 製剤の標準的な治療方法は,1日6 MIU で投与を開始し,投与後2週間ま では連日,その後1日3~6 MIU を週3回皮下又は筋肉内投与する。 国内で実施された臨床試験の有効性成績を表 2.5.1.2-3及び表 2.5.1.2-4に示す。C 型代償性肝 硬変における IFNα による HCV 持続陰性化率は,最も効果の高かった46週間投与で32.1% (9/28例)であった。また,セログループ1かつウイルス量が500 KIU/mL 以上の患者において 陰性化した患者は認められなかったため,セログループ1かつウイルス量が500 KIU/mL 以上の 患者は適応から除外された。 表 2.5.1.2-3 C 型代償性肝硬変における IFNα による HCV-RNA 陰性化率 3S 群 3L 群 6S 群 HCV-RNA 陰性化率 (投与終了6カ月後) 7/31(22.6%) 9/28(32.1%) 9/30(30.0%)
• 3S 群:IFNα 6 MIU を2週間連日投与後,3 MIU を週3回23週間筋肉内投与 • 3L 群:IFNα 6 MIU を2週間連日投与後,3 MIU を週3回46週間筋肉内投与 • 6S 群:IFNα 6 MIU を2週間連日投与後,6 MIU を週3回23週間筋肉内投与
[文献24)より作成]
表 2.5.1.2-4 C 型代償性肝硬変における IFNα による HCV-RNA 陰性化率(HCV-RNA 量別)
HCV セログループ 1 2 HCV-RNA 量(KIU/mL) 100未満 100以上 500未満 500以上 100未満 100以上 500未満 500以上 HCV-RNA 陰性化率 (投与終了6カ月後) 4/5 (80.0%) 4/18 (22.2%) 0/9 (0.0%) 11/16 (68.8%) 5/24 (20.8%) 1/17 (5.9%) [文献24)より作成]
2.5.1.3
C 型代償性肝硬変に対する PEG-IFNα-2a とリバビリン併用療法開発の根
拠
C 型慢性肝炎患者を対象とした海外の臨床試験(NV15801及びNV15942)において,PEG-ペガシス/コペガス 2.5 臨床に関する概括評価 Page
10
IFNα-2a とリバビリン併用療法が,従来型の IFN とリバビリンの併用療法と比べ高い有効性を 示すことが,C 型慢性肝炎患者及びそれらの患者のうちジェノタイプ1かつ高ウイルスの患者 においても示された(「(1)海外における C 型慢性肝炎に対する PEG-IFNα-2a とリバビリン併 用療法の成績」に詳細を記載)。また,同じ海外試験に組み込まれた C 型代償性肝硬変患者 での成績を解析したところ,抗ウイルス治療の有効性は慢性肝炎よりやや低くなる傾向が認め られたものの,PEG-IFNα-2a とリバビリン併用療法において良好な有効性を示した(「(2)海外 における C 型代償性肝硬変に対する PEG-IFNα-2a とリバビリン併用療法の成績」に詳細を記 載)。安全性については,これらの海外試験の成績から,程度が高度な有害事象及び重篤な有 害事象発現率はC 型慢性肝炎患者に比べ,C 型代償性肝硬変患者において高い傾向が認められ たが,C 型代償性肝硬変患者での安全性に起因する投与中止率は8.9~22.9%であり,治療効果 を得るために重要な課題である治療の継続は可能と考えられた。 また,すでに確認されている通り,国内で実施したジェノタイプ1b の C 型慢性肝炎患者を 対象とした試験(JV15725)においても,PEG-IFNα-2a とリバビリン併用療法が PEG-IFNα-2a 単独療法と比べ高い有効性を示した(「(3)国内における C 型慢性肝炎に対する PEG-IFNα-2a とリバビリン併用療法の成績」に詳細を記載)。 以上のことから,日本人のC 型代償性肝硬変患者においても PEG-IFNα-2a とリバビリン併用 療法は,既承認の IFN 単独療法による無効例及び IFN 単独療法が適応となっていないジェノ タイプ1かつ高ウイルスの患者での高い有効性が期待できる。 なお,前述のように,C 型代償性肝硬変は肝細胞癌の主要原因であり,治療の緊急性及び必 要性が高く,効果の高い PEG-IFNα-2a とリバビリン併用療法が日本肝臓病患者団体協議会を 始めとする医療現場から切望されている。このような要望に応えるためにも,PEG-IFNα-2a と リバビリンの併用療法については早期承認取得を目指している。 また,独立行政法人医薬品医療機器総合機構(以降,機構)により,C 型代償性肝硬変は, 高率で肝細胞癌を発症する生命に重大な影響のある疾患に該当し,海外臨床試験(NV15801) の成績を基に PEG-IFNα-2a とリバビリン併用療法が①肝硬変移行期の患者も含む C 型代償性 肝硬変患者に対しての成績ではあるが C 型慢性肝炎患者に大きく劣らない持続性ウイルス学 的効果が認められたこと,②C 型代償性肝硬変に対して承認されている IFNβ 製剤の適応対象 外であるジェノタイプ1かつ高ウイルス量の C 型代償性肝硬変患者に対しても持続性ウイルス 学的効果が認められていることから,既存薬に対し高い有用性が期待されると判断され,20 年 月 日優先対面助言品目に指定された。 (1) 海外における C 型慢性肝炎に対する PEG-IFNα-2a とリバビリン併用療法の成績 米国においては,C 型慢性肝炎を対象とした第Ⅲ相試験が2試験(NV15801及び NV15942) 実施された。 第Ⅲ相臨床試験(NV15801)では,代償性肝硬変患者を含む C 型慢性肝炎患者を対象として PEG-IFNα-2a(180 μg,週1回皮下投与)単独群,PEG-IFNα-2a(180 μg,週1回皮下投与)とリ バビリン(1,000~1,200 mg/日,1日2回経口投与)併用群,rIFNα-2b(3 MIU,週3回皮下投与) とリバビリン(1,000~1,200 mg/日,1日2回経口投与)併用群の3群比較で実施された。その結 果,PEG-IFNα-2a + リバビリン併用群におけるウイルス学的効果は PEG-IFNα-2a 単独群, rIFNα-2b + リバビリン併用群に比して有意に優っており(P=0.001,P=0.004),ジェノタイプ 1の患者においても,42%(129/305例)のウイルス学的効果が認められた(表 2.5.1.3-1)。 もう一方の第Ⅲ相臨床試験(NV15942)では,PEG-IFNα-2a とリバビリンを併用した場合の 投与期間(24週間又は48週間)及びリバビリンの臨床用量(800 mg 又は1,000/1,200 mg)を検 討した。ウイルス学的効果は投与期間が24週間のグループよりも48週間のグループの方が有意 に高く(P=0.0393),またリバビリン投与量が1,000 mg 又は1,200 mg のグループの方が800 mg のグループよりも有意に高かった(P=0.0177)。ジェノタイプ別にみると,ジェノタイプ1以 外の患者においてはリバビリンの用量・投与期間に係わらずウイルス学的効果は70%~80%とペガシス/コペガス 2.5 臨床に関する概括評価 Page
11
高い効果が認められた。一方ジェノタイプ1の患者においてはリバビリンの用量が1,000又は 1,200 mg かつ投与期間が48週間の群で,50%(136/271例)と最も高い効果が得られた(表 2.5.1.3-2)。 安全性については,上述の第Ⅲ相臨床試験(NV15801)で高頻度に認められた有害事象は, 疲労,頭痛,発熱,筋痛等,いずれも IFN 投与にて発現する既知のものであった。また, PEG-IFNα-2a に関連した臨床検査値異常として好中球数減少及び血小板数減少が,リバビリン に関連した臨床検査値異常としてヘモグロビン減少が観察された。有害事象/臨床検査値異常による投与中止率は PEG-IFNα-2a とリバビリンの併用,rIFNα-2b
とリバビリンの併用,PEG-IFNα-2a 単独でそれぞれ10%,11%及び7%であった。また,治験薬との因果関係が否定できな い重篤な有害事象発現率は,各群とも4%と同程度であった。 以上,PEG-IFNα-2a とリバビリン併用療法が,従来型の IFN とリバビリンの併用療法と比べ 高い有効性を示すことが,C 型慢性肝炎患者及びそれらの患者のうちジェノタイプ1の患者に おいても示された。 表 2.5.1.3-1 海外第Ⅲ相臨床試験の投与終了後24週時のウイルス学的効果(NV15801,ITT) PEG-IFNα-2a 単独群 (P 群) PEG-IFNα-2a + リバビリン群 (P+R 群) rIFNα-2b + リバビリン群 (I+R 群) 全例 62/227(27%) 234/465(50%) 190/457(42%) 群間比較 オッズ比*(97.5%信頼区間) P 値** P+R 群 vs I+R 群 1.49 (1.09, 2.05) 0.004 P+R 群 vs P 群 2.92 (1.94, 4.41) 0.001 I+R 群 vs P 群 1.94 (1.29, 2.92) 0.001 ジェノタイプ 1 27/146 (18%) 129/305 (42%) 100/292 (34%) 群間比較 オッズ比*(97.5%信頼区間) P 値** P+R 群 vs I+R 群 1.43 (0.96, 2.11) 0.042 P+R 群 vs P 群 3.37 (1.94, 5.85) 0.001 I+R 群 vs P 群 2.35 (1.34, 4.12) 0.001 ジェノタイプ 1以外 35/81 (43%) 105/160 (66%) 90/165 (55%) 群間比較 オッズ比*(97.5%信頼区間) P 値** P+R 群 vs I+R 群 1.63 (0.95, 2.78) 0.042 P+R 群 vs P 群 2.41 (1.31, 4.47) 0.001 I+R 群 vs P 群 1.53 (0.83, 2.79) 0.117 登録された全患者を対象とした。 * 一つの群における反応のオッズに対するもう一つの群における反応のオッズの比。 ** 地域とジェノタイプを層としたCochran-Mantel-Haenszel検定により評価した。
ペガシス/コペガス 2.5 臨床に関する概括評価 Page
12
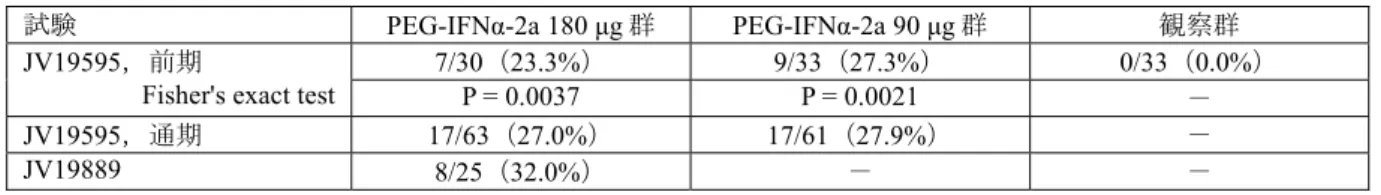
表 2.5.1.3-2 海外第Ⅲ相臨床試験の投与終了後24週時のウイルス学的効果(NV15942,ITT) ジェノ タイプ 投与前ウイルス 量 (×106 copies/mL) 24週間 PEG-IFNα-2a 180 μg リバビリン 800 mg 24週間 PEG-IFNα-2a 180 μg リバビリン 1,000-1,200 mg 48週間 PEG-IFNα-2a 180 μg リバビリン 800 mg 48週間 PEG-IFNα-2a 180 μg リバビリン 1,000-1,200 mg 全例 計 高ウイルス量*** 低ウイルス量 112/207(54%) 63/117(54%) 49/90(54%) 177/280(63%) 93/148(63%) 84/132(64%) 180/361(50%) 116/260(45%) 64/101(63%) 259/436(59%) 163/294(55%) 96/142(68%) 1 計 高ウイルス量*** 低ウイルス量 29/101(29%) 8/50(16%) 21/51(41%) 48/118(41%) 12/47(26%) 36/71(51%) 97/250(39%) 66/190(35%) 31/60(52%) 136/271(50%) 85/186(46%) 51/85(60%) 1以外 計 高ウイルス量*** 低ウイルス量 83/106(78%) 55/67(82%) 28/39(72%) 129/162(80%) 81/101(80%) 48/61(79%) 83/111(75%) 50/70(71%) 33/41(80%) 123/165(75%) 78/108(72%) 45/57(79%) 群間比較 オッズ比*(95%信頼区間) P 値** 48週間投与 vs 24週間投与 1.32(1.01, 1.73) 0.0393 1,000/1,200 mg vs 800 mg 1.35(1.05, 1.73) 0.0177 * 一つの群における反応のオッズに対するもう一つの群における反応のオッズ比 ** 地域,ジェノタイプ,ウイルス量及び投与期間若しくはリバビリン投与量を層としたCochran-Mantel-Haenszel検定により評価した。 *** 海外臨床試験では高ウイルス量の定義を > 2.0×106 copies/mLとしており,国内における高ウイルス量 の定義 ≥ 100 KIU/mL(2.7×105 copies/mLに相当)とは異なる。 [5.3.5.1-3 Table 14,Table 15より作成] (2) 海外における C 型代償性肝硬変に対する PEG-IFNα-2a とリバビリン併用療法の成績 海外においては,C 型慢性肝炎に対する PEG-IFNα-2a とリバビリン併用の臨床試験に,代償 性肝硬変の患者を含めて2つの第Ⅲ相臨床試験(NV15801及び NV15942)を実施し,その結果 をもって代償性肝硬変を含む C 型慢性肝炎患者に対する適応を取得している。代償性肝硬変 の診断基準は,一般的に肝機能評価と組織学的診断により行われる。肝機能を評価する基準は, 国内・海外ともに,Child-Pugh 分類を用い A~C と分類し,A を代償性肝硬変と診断している。 組織学的診断については,肝組織の線維化を評価しており,国内においては,新犬山分類を用 いF0~F4と分類し,「F4」と分類された場合,肝硬変と診断している2)。一方,海外における PEG-IFNα-2a の第Ⅲ相臨床試験(NV15801及び NV15942)においては,組織学的診断を,「慢 性肝炎」,「肝硬変移行期(国内では F3に相当)」,「肝硬変(国内では F4に相当)」に分 類し,肝硬変も含めて臨床試験を実施し(表 2.5.1.3-3),C 型慢性肝炎のみならず C 型代償性 肝硬変の適応症について,単独及びリバビリンとの併用で承認されている。 表 2.5.1.3-3 国内外で肝硬変と定義される集団の組織学的診断 地域 組織学的診断 海外 慢性肝炎 肝硬変移行期* 肝硬変* 国内 慢性肝炎 肝硬変移行期 肝硬変* * 各々の地域で肝硬変として扱われている集団 1) NV15801試験における C 型代償性肝硬変患者での成績 NV15801試験における C 型代償性肝硬変患者での投与終了後24週時のウイルス学的効果を表 2.5.1.3-4に示す。本試験では肝硬変移行期の患者を含む C 型代償性肝硬変患者として集計した。 C 型代償性肝硬変患者に対する投与終了後24週時のウイルス学的効果は,PEG-IFNα-2a + リ バビリン群:39.7%(23/58例),rIFNα-2b + リバビリン群:33.3%(18/54例),PEG-IFNα-2a 群:17.1%(6/35例)であり,PEG-IFNα-2a + リバビリン群で最も高い効果が認められた。ま た,国内で最も患者の比率が高いと予想される,ジェノタイプ1かつ高ウイルス(≥ 2 × 106ペガシス/コペガス 2.5 臨床に関する概括評価 Page
13
copies/mL)の患者に対しては,PEG-IFNα-2a + リバビリン群:28.6%(6/21例),rIFNα-2b + リバビリン群:16.7%(3/18例),PEG-IFNα-2a 群:6.3%(1/16例)であり,PEG-IFNα-2a + リ バビリン群での高い効果が認められた。 安全性については,中止を要する有害事象,重篤な有害事象,程度が高度である有害事象の 発現頻度は C 型慢性肝炎患者と比較し,やや高いものの,自覚的な有害事象の発現頻度は C 型慢性肝炎患者と同様であった。また,代償性肝硬変患者のみに特異的に認められる有害事象, 既存のIFN から予測できない有害事象は認められなかった。 C 型代償性肝硬変患者では投与開始前の血小板数が C 型慢性肝炎患者と比較し低いため, PEG-IFNα-2a + リバビリン群でグレード3(<50,000/mm3)以上の血小板数減少発現率が高かっ たが,休薬・減量にて治療は継続可能であった。 表 2.5.1.3-4 海外第Ⅲ相臨床試験の C 型代償性肝硬変患者(移行期の患者を含む)における 投与終了後24週時のウイルス学的効果(NV15801,ITT) PEG-IFNα-2a 単独群 (N=35) PEG-IFNα-2a + リバビリン併用群 (N=58) rIFNα-2b + リバビリン併用群 (N=54) 全例 6/35(17.1%) 23/58(39.7%) 18/54(33.3%) ジェノタイプ1 3/25(12.0%) 11/40(27.5%) 8/32(25.0%) 高ウイルス量* 1/16(6.3%) 6/21(28.6%) 3/18(16.7%) 低ウイルス量 2/8(25.0%) 5/15(33.3%) 5/14(35.7%) ウイルス量欠損 0/1(0.0%) 0/4(0.0%) 0/0 ジェノタイプ1以外 3/10(30.0%) 12/18(66.7%) 10/22(45.5%) 高ウイルス量* 2/8(25.0%) 11/16(68.8%) 6/15(40.0%) 低ウイルス量 1/2(50.0%) 1/2(50.0%) 4/7(57.1%) * 海外臨床試験では高ウイルス量の定義を > 2.0×106 copies/mLとしており,国内における高ウイルス量の定義 ≧ 100 KIU/mL(2.7×105 copies/mLに相当)とは異なる。 [5.3.5.3-2 表 3-1より作成] 2) NV15942試験における C 型代償性肝硬変患者での成績 NV15942試験における C 型代償性肝硬変患者での投与終了後24週時のウイルス学的効果を表 2.5.1.3-5に示す。本試験では肝硬変移行期の患者を含まない C 型代償性肝硬変患者として集計 した。 PEG-IFNα-2a 180 μg とリバビリン1,000~1,200 mg が投与された C 型慢性肝炎患者のうち代 償性肝硬変患者では31.4%(11/35例)のウイルス学的効果が認められた。また,ジェノタイプ 1かつ高ウイルスの患者においても25.0%(5/20例)のウイルス学的効果が得られた。 安全性については,程度が高度である有害事象,血小板数減少及びヘモグロビン減少は,慢 性肝炎患者と比較して代償性肝硬変で多く認められたものの,重篤な有害事象発現率は慢性肝 炎と大きく異ならなかった。また,休薬・減量率及び安全性の理由による投与中止率は慢性肝 炎に比べ代償性肝硬変で高かったが,中止理由に特定の傾向は認められず,多くの患者は減 量・休薬を行なうことにより投与継続が可能であった。 これらの成績より,PEG-IFNα-2a とリバビリンの併用療法は C 型代償性肝硬変を含む C 型慢 性肝炎を適応として2002年6月に欧州,2002年12月に米国で承認された。 以上,PEG-IFNα-2a とリバビリンの併用療法は,従来の IFN 療法で治療困難とされ,国内で 多数を占めるジェノタイプ1かつ高ウイルスの C 型代償性肝硬変に対して優れた効果を示すこ とが期待された。ペガシス/コペガス 2.5 臨床に関する概括評価 Page
14
表 2.5.1.3-5 海外第Ⅲ相臨床試験の C 型代償性肝硬変患者(移行期の患者を含まない)にお ける投与終了後24週時のウイルス学的効果(NV15942,ITT) 24週間 PEG-IFNα-2a 180 μg リバビリン 800 mg 24週間 PEG-IFNα-2a 180 μg リバビリン 1,000-1,200 mg 48週間 PEG-IFNα-2a 180 μg リバビリン 800 mg 48週間 PEG-IFNα-2a 180 μg リバビリン 1,000-1,200 mg 全例 全体 5/10 (50.0%) 8/20 (40.0%) 8/25 (32.0%) 11/35 (31.4%) 高ウイルス量* 4/7 (57.1%) 5/14 (35.7%) 5/17 (29.4%) 7/24 (29.2%) 低ウイルス量 1/3 (33.3%) 3/6 (50.0%) 3/8 (37.5%) 3/11 (27.3%) ジェノタイプ1 全体 2/5 (40.0%) 2/9 (22.2%) 5/20 (25.0%) 9/31 (29.0%) 高ウイルス量* 1/3 (33.3%) 1/5 (20.0%) 5/16 (31.2%) 5/20 (25.0%) 低ウイルス量 1/2 (50.0%) 1/4 (25.0%) 0/4 (0.0%) 4/11 (36.4%) ジェノタイプ1以外 全体 3/5 (60.0%) 6/11 (54.5%) 3/5 (60.0%) 2/4 (50.0%) 高ウイルス量* 3/4 (75.0%) 4/9 (44.4%) 0/1 (0.0%) 2/4 (50.0%) 低ウイルス量 0/1 (0.0%) 2/2 (100.0%) 3/4 (75.0%) - - * 海外臨床試験では高ウイルス量の定義を > 2.0×106 copies/mL としており,国内における高ウイルス量の定義 ≥ 100 KIU/mL(2.7×105 copies/mL に相当)とは異なる。 [5.3.5.3-2 表 3-3より作成] (3) 国内における C 型慢性肝炎に対する PEG-IFNα-2a とリバビリン併用療法の成績 国内第Ⅲ相試験(JV15725)において,インターフェロン未治療のジェノタイプ1b の C 型慢 性肝炎患者を対象としたPEG-IFNα-2a 単独(180 μg,週1回皮下投与)群と PEG-IFNα-2a(180 μg,週1回皮下投与)+リバビリン(600~1,000 mg/日,1日2回経口投与)併用群を二重盲検法 にて比較した。また,インターフェロン既治療の C 型慢性肝炎患者(すべてのジェノタイプ) における PEG-IFNα-2a(180 μg,週1回皮下投与)+リバビリン(600~1,000 mg/日,1日2回経 口投与)併用の有効性及び安全性を検討した。 その結果,PEG-IFNα-2a+リバビリン併用群におけるウイルス学的効果は60.6%(60/99例)で あり,PEG-IFNα-2a 単独群の25.7%(26/101例)に比べ高い効果が認められた(CMH 検定:P < 0.001)。またインターフェロン既治療の患者に対しても,54.0%(54/100例)のウイルス学 的効果が認められた(表 2.5.1.3-6)。 安全性については,高頻度に認められた有害事象は,発熱,倦怠感,頭痛及び脱毛等,いず れも IFN 投与にて発現する既知のものであった。PEG-IFNα-2a+リバビリン併用群では,PEG-IFNα-2a 単独群に比べ,湿疹,全身そう痒症等の皮膚症状が多く認められた。また,いずれの 投与群も,白血球数減少,好中球数減少及び血小板数減少等が認められたが,併用群において は,赤血球数減少及びヘモグロビン減少が多く認められた。重篤な有害事象発現率は,PEG-IFNα-2a+リバビリン併用群と PEG-IFNα-2a 単独群で大きく異ならなかった。また,PEG-IFNα-2a 単独群において,脳出血による死亡例が1例認められたが,脳出血発現時の急激な血圧変動 や血小板数の減少は無かった。有害事象/臨床検査値異常による投与中止率は,PEG-IFNα-2a+ リバビリン併用群とPEG-IFNα-2a 単独群で大きく異ならなかった。 これらのことから,日本人でジェノタイプ1b の C 型慢性肝炎患者においても,海外同様 PEG-IFNα-2a とリバビリン併用群が最も高い有効性が得られることが示された。 これらの結果を踏まえ,国内のC 型代償性肝硬変の開発を行うこととした。ペガシス/コペガス 2.5 臨床に関する概括評価 Page
15
表 2.5.1.3-6 国内第Ⅲ相臨床試験の C 型慢性肝炎患者における投与終了後24週時におけるウ イルス学的効果(JV15725,FAS)
IFN 製剤未治療 IFN 製剤既治療
PEG-IFNα-2a PEG-IFNα-2a +リバビリン PEG-IFNα-2a +リバビリン
HCV-RNA 陰性化率 26/101 (25.7%) 60/99 (60.6%) 54/100 (54.0%) オッズ比 4.55 ( 2.48, 8.37 ) - P 値* <0.001 - * 登録時 HCV-RNA 量を層とした Cochran-Mantel-Heanszel 検定 [5.3.5.1-4 表 11.4.1.1-1,表11.4.1.1-2,表11.4.1.2-1より作成]
2.5.1.4
C 型代償性肝硬変に対する PEG-IFNα-2a とリバビリン併用療法開発の経
緯
PEG-IFNα-2a とリバビリン併用療法は C 型慢性肝炎患者を対象とした第Ⅲ相臨床試験 (JV15725)の成績より,C 型慢性肝炎で①セログループ1(ジェノタイプ1a 又は1b)で HCV-RNA 量が高値の患者又は②インターフェロン単独療法で無効又はインターフェロン単独療法 後再燃した患者におけるウイルス血症の改善として効能・効果を2007年1月に取得した。 PEG-IFNα-2a とリバビリン併用による C 型代償性肝硬変の新効能追加に係る承認申請を目指 した第Ⅲ相臨床試験を計画するにあたり,20 年 月 日に機構との治験相談( 相談)を実施した。その結果,抗ウイルス療法を実施しない観察群に対し,「投与 終了後24週時のウイルス学的効果(HCV-RNA 陰性化率)」を主要評価項目とし優越性を検証 する第Ⅱ/Ⅲ相臨床試験(JV19595)及びこの試験の観察群に対し PEG-IFNα-2a とリバビリン併 用投与する一般臨床試験(JV19889)の2試験を実施し,これらの試験において,C 型代償性肝 硬変患者に対する有効性及び安全性を確認することにより承認申請可能と考えた。 (1) 治験実施計画 相談において,以下の申請者からの提示内容1)~3) について機構か ら助言があり,これらの助言に対する対応は第Ⅱ/Ⅲ相臨床試験(JV19595)の を考慮した上で決定し治験実施計画書を確定した。以下に機構の助言と検討結果を記載す る。 1) すべてのジェノタイプを組み入れること 【機構助言】 C 型代償性肝硬変を対象とした PEG-IFNα-2a とリバビリン併用療法に関しては, を実施するべ きである。 が困難なのであれば, を対象とすることを勧める。 【検討結果】 ジェノタイプ2a 及び2b の患者は,C 型慢性肝炎では PEG-IFNα-2a 単独療法による治療効 果が十分高いと考えられたため,リバビリン併用療法の治験では対象に含めなかった。し かしながら,①治験計画作成時にはC 型代償性肝硬変に対し IFN 製剤による治療が承認さ れていなかったこと,②海外第Ⅲ相臨床試験(NV15801)でにおいて,PEG-IFNα-2a は C 型慢性肝炎に比し C 型代償性肝硬変で低い有効性しか得られず,ジェノタイプ1以外の代 償性肝硬変患者においても,PEG-IFNα-2a 単独投与で十分なウイルス学的効果が得られな かったこと,③国内でもIFN 製剤は C 型慢性肝炎に比し C 型代償性肝硬変で効果が低いと いう多くの報告があったこと10)から,国内の C 型代償性肝硬変患者に対してもリバビリンペガシス/コペガス 2.5 臨床に関する概括評価 Page
16
との併用療法が必要であると判断した。したがって,本治験ではすべてのジェノタイプの 患者を対象とすることとした。 2) PEG-IFNα-2a の用量を90 μg と180 μg で比較検討すること 【機構助言】 90 μg については,これまで併用療法での試験成績もなく,設定根拠が不明であるため, 有効性及び安全性の両面から,及び もふまえて90 μg を設定することの 妥当性を十分に説明する必要がある。 【検討結果】 C 型代償性肝硬変は C 型慢性肝炎からの連続した疾患であり,海外臨床試験成績から C 型 代償性肝硬変に対する PEG-IFNα-2a+リバビリン併用投与の有効性は慢性肝炎よりも低く, 有効性を維持するためには,180 μg の投与が必要であると考える。一方で,C 型代償性肝 硬変患者の血小板数,白血球数,ヘモグロビン濃度は低く10),国内 C 型代償性肝硬変患者 の高齢化10)を考慮すると,180 μg の投与では休薬・減量を要する患者が多くなることが懸 念され,低用量で投与を開始した場合と PEG-IFNα-2a の総投与量が大きく異ならない可能 性も考えられた。このことから,本試験においてPEG-IFNα-2a 180 μg と低用量の有効性・ 安全性について探索的な比較を行うことにより,限られた患者数ではあるが用量反応性に ついても検討することができると考えた。低用量の選択にあたり,PEG-IFNα-2a 90 μg は, 国内第Ⅱ相試験(JV15724)において,180 μg とともに有効性・安全性が検討されており, 180 μg に比し有効性はやや劣るものの投与中の血小板数(中央値)は180 μg より減少率が 低い傾向が認められており,本試験の低用量群として90 μg が適切な用量であると考えた。 すなわち,PEG-IFNα-2a 180 μg で投与を開始した場合に90 μg で開始するより,たとえ休 薬・減量の頻度が多くても高い総投与量が確保でき,安全に投与できるなら180 μg が臨床 用量として推奨されると考えた。以上のことから,C 型代償性肝硬変に対する第Ⅱ/Ⅲ相臨 床試験の低用量群として90 μg 群を設定した。 3) 肝細胞癌既往を有する患者を組み入れること 【機構助言】 肝細胞癌既往を有する患者を組み入れることについては,肝細胞癌術後及び局所療法後, どのくらいの時期から組み入れ可能であるのか現時点では明確な基準及びその妥当性が示 されていないことから,再発の可能性が高い肝細胞癌既往を有する患者をウイルス血症の 改善を目的とする本試験に組み入れることは,患者の安全性の担保及び本試験からの脱落 による試験の質の低下等の観点から勧められない。 【検討結果】 肝細胞癌治療後の代償性肝硬変においてはその後の肝細胞癌再発率も高く,死亡につなが る緊急性からインターフェロン治療がより強く求められており,代償性肝硬変患者の中で も特にウイルス駆除の意義は大きいと考えられること,肝細胞癌既往のある患者は,実際 の医療現場の実態を反映した患者集団であり,市販後の有効性,安全性を正確に判断する データを得るためにも必要であると判断した。 また,肝細胞癌の既往のある患者の安全性担保及び治験の質確保のため以下の①~④の方 策を講じることとした。 ① 肝細胞癌既往の有無で発癌リスクをできる限り大きく異ならないように症例登録する組 入れ基準を設定する。 肝細胞癌既往を有する患者の再発が多くなる理由として,治療病変の残存による再発 (局所再発),治療時に画像診断では検出されなかった肝内転移巣の増大による再発 (肝内転移),肝内の新病巣出現による再発(多中心性発生)の三つの再発様式がある ためと考えられている。その内,局所再発と肝内転移は治療後2年までに起こると考えペガシス/コペガス 2.5 臨床に関する概括評価 Page
17
られており26),治療後2年間無再発の患者を対象とすることで,全体の再発率を下げる ことは可能である。その結果,治験実施計画書の肝細胞癌既往を有する患者の選択基準 として,「治療後2年間,肝細胞癌の再発のない患者」を設定した。 ② 投与中の発癌症例の安全性確保の取扱いをあらかじめ決めておく。 治験実施計画書の中止基準として,「肝細胞癌と確定診断された場合」を設定した。 ③ 治験実施中に肝細胞癌をできる限り迅速に発見できるための検査スケジュールを設定す る。 治験実施計画書の検査項目として,「腫瘍マーカー(AFP,PIVKA-Ⅱ,AFP-L3):4週 ごとに測定」,「腹部超音波,CT 又は MRI:12週ごとに測定」を設定した。 ④ 肝細胞癌既往がない患者の C 型代償性肝硬変患者のみでも有効性・安全性を検証可能と する。 目標症例数60例に対し,肝細胞癌既往を有する患者は最大30例まで登録可能とし,肝細 胞癌既往がない患者については,検出力80%以上を確保可能とした。 (2) 目標症例数の変更 観察群の目標症例数については,試験実施中の20 年 月 日に60例から30例に変更した。 それに伴い解析方針について,有効性・安全性を含む,観察群と180 μg 群,90 μg 群の比較は, 前期登録症例(目標症例数各群30例)を対象とした解析を本試験の主要な解析とすることとし た。以下にその経緯を示す。 試験計画立案時においては,肝細胞癌既往を有する患者についての治療ニーズが高く,その 患者が多く登録されると予想した。また,肝細胞癌既往を有する患者は,肝細胞癌既往がない 患者と比べ発癌リスクは 倍程度高いとされていたことから,発癌による途中脱落のリスクを 考慮し,肝細胞癌既往を有する患者の割合を,各群最大50%までと制限し,最も多く組み入れ られた場合でも,肝細胞癌既往がない患者集団のみで検出力を保つことが可能な例数を確保す るために,目標例数を各群60例と設定した。 しかしながら,治験依頼者及び治験責任医師又は治験分担医師とは独立して設立された効果 安全性評価委員会による安全性検討会(20 年 月 日,30例(投与群20例,観察群10例)が 週を終了した時点で実施)が開催され,この時点で肝細胞癌患者の登録例は想定より少なく, 肝細胞癌既往を有する患者の組み入れ基準を,再発患者も可能とすることが決定された。更に, 委員から「今後も肝細胞癌患者の登録が大幅に増加するとは想定されず,目標症例数の変更も 考えておく必要はないか。」との助言をいただいた。 その後も肝細胞癌既往を有する患者の登録数の増加は認められず,登録開始1年後の段階で, わずか 例(3.5%: 例)のみであった。 以上のように,当初の想定とは大きく異なり,肝細胞癌既往を有する患者の登録は少数であ り,登録を続けても大幅に増加する可能性は低いと予想され,全解析集団における肝細胞癌既 往を有する患者を含めた集団を主要な解析対象集団としても,投与群と観察群との有効性比較 については,各群30例で80%以上の検出力は確保できると考えられた。また,観察群について は,抗ウイルス療法が使用できないことから,HCV-RNA の消失がほとんど望めず,被験者へ の倫理的配慮も考慮し,本試験の主要な解析対象の目標症例数を30例に変更することとし,観 察群(非盲験)の目標症例数を30例に変更した。一方,投与群については,安全性の検討にお いて,発現率 %の有害事象を見逃す確率が %未満となるためには 例以上必要となることを 考慮して,当初の計画通り目標症例数を各群60例とした。このように,観察群と投与群で目標 症例数が異なるが,主要評価等において投与群と観察群との比較を行う場合には,投与群も観 察群の登録締切りまでに登録された患者(前期登録例)を解析対象とした。 なお,当該治験実施計画書変更については, 審議され20 年 月 日に承認された。その後, にて承認された。ペガシス/コペガス 2.5 臨床に関する概括評価 Page
18
(3) 用量調整基準の設定 20 年 月治験実施計画書検討会にて医学専門家及び効果安全性評価委員との協議の結果, C 型代償性肝硬変患者の平均年齢は高いことが予想され,C 型慢性肝炎と同様の用量調整基準 のまま PEG-IFNα-2a とリバビリンの併用療法を行う場合,好中球数減少,血小板数減少及び ヘモグロビン減少により多くの中止・脱落例が出ることが危惧されたことから,治療完遂例が できる限り多くなるよう表 2.5.1.4-1に示す用量調整基準を設定することとした。 ① PEG-IFNα-2a より早期から治験薬の減量を行うことができるよう,減量基準を慢性肝炎の基準より厳 しくすることとした。また,投与初期に大きく好中球数が減少する症例があるため, 750 /mm3未満となってからの減量では中止基準まで下がってしまう可能性があることか ら,1,000 /mm3未満となったときから早めに減量を行うよう設定した。 ② リバビリン C 型慢性肝炎を対象とした第Ⅲ相臨床試験(JV15725)においてヘモグロビン減少によ る中止が併用群において11例(5.5%)であったことから,ヘモグロビン減少に関しては 10 g/dL 未満8.5 g/dL 以上になった場合はリバビリンを半量以下に減量し,10 g/dL 以上に 回復した場合は増量可とした。これはヘモグロビン減少による中止例を減らし,リバビ リンの継続投与を可能とするため設定した。 また,治療完遂例ができる限り多くなるよう,リバビリンの割付用量が800 mg 及び 1,000 mg の患者に関しても200 mg まで減量可能とした。 また,C 型代償性肝硬変では,投与初期にヘモグロビン濃度が大きく減少する患者が多 いことが推測され,投与4週時までは,ヘモグロビン濃度が11 g/dL 未満となった場合, リバビリンの減量を開始することとした。ペガシス/コペガス 2.5 臨床に関する概括評価 Page
19
表 2.5.1.4-1 C 型代償性肝硬変患者(JV19595及び JV19889)における PEG-IFNα-2a 及びリバ ビリンの用量調整基準:C 型慢性肝炎患者(JV15725)との比較 PEG-IFNα-2a リバビリン C 型代償性肝硬変 C 型慢性肝炎 C 型代償性肝硬変 C 型慢性肝炎 好中球数 (/mm3) < 1,000 1/2量に減量 ≥ 1,000 に 回 復 後 全量で投与再開 規定なし 変更なし 規定なし < 750 1/4量に減量 ≥ 750に回復後1/2 量で投与再開 1/2量に減量 ≥ 750に回復後全 量で投与再開 変更なし 変更なし < 500 休薬 ≥ 500 に回復後1/4 量で投与再開 休薬 ≥ 500 に 回 復 後 1/2量で投与再開 休薬 ≥ 500 に回復後全量 で投与再開 休薬 ≥ 500に回復後全 量で投与再開 < 250 中止 中止 中止 中止 血小板数 (/mm3) < 50,000 休薬 ≥ 50,000に回復後 1/2量で投与再開 休薬 ≥ 50,000 に 回 復 後1/2量で投与再 開 休薬 ≥ 50,000に回復後全 量で投与再開 休薬 ≥ 50,000に回復全 量で後投与再開 < 35,000 休薬 ≥ 50,000に回復後 1/4量で投与再開 < 25,000 中止 中止 中止 中止 ヘモグロビン 濃度 (g/dL) 1~4週 < 11 変更なし 変更なし 減量* > 11に回復後増量可 投与時期に関わら ず< 10で減量* (≥ 10に回復後も 増量なし) 5~48週 < 10 変更なし 減量* > 10に回復後増量可 < 8.5 中止 中止 中止 中止 * リバビリンの減量・回復後投与量 規定投与量 C 型代償性肝硬変 C 型慢性肝炎 減量 回復後投与量 減量 回復後投与量 1,000 mg/日 400 mg/日 ヘモグロビン濃度の回復がみら れない場合は200 mg/日に減量可 600 mg/日に増 量可 600 mg/日 600 mg/日 (増量なし) 800 mg/日 600 mg/日 200 mg/日 400 mg/日に増 量可 400 mg/日 400 mg/日 (増量なし)2.5.1.5
臨床試験データパッケージ
今回の承認申請においては,国内で実施された第Ⅱ/Ⅲ相臨床試験(JV19595)及び一般臨床 試験(JV19889)を評価資料とした。なお,国内で実施した C 型慢性肝炎対象の第Ⅲ相臨床試 験(JV15725)及び海外で実施された代償性肝硬変を含む C 型慢性肝炎を対象とした第Ⅲ相臨 床試験の2試験(NV15801,NV15942)を参考資料とした。 以下に,今回の承認申請における臨床試験データパッケージを示す(表 2.5.1.5-1)。ペガシス/コペガス 2.5 臨床に関する概括評価 Page
20
表 2.5.1.5-1 臨床試験データパッケージ 国内臨床試験 試験 試験内容 対象 投与例数 治験期間 試験期間 第Ⅱ/Ⅲ相臨床試験 JV19595 [5.3.5.1-1] 評価資料 有効性・安全性・薬 物動態検討 C 型代償性 肝硬変 157例 投与期間:48週間 経過観察期間:24週間 (観察群は投与期間の み) 20 年 月- 20 年 月 一般臨床試験 JV19889 [5.3.5.2-1] 評価資料 有効性・安全性 C 型代償性 肝硬変 25例 投与期間:48週間 経過観察期間:24週間 20 年 月- 20 年 月 第Ⅲ相臨床試験 JV15725 [5.3.5.1-4] 参考資料 有効性・安全性・薬 物動態検討 C 型慢性肝炎 300例 投与期間:48週間 経過観察期間:24週間 20 年 月- 20 年 月 海外臨床試験 試験 試験内容 対象 投与例数 投与期間 試験期間 第Ⅲ相臨床試験 NV15801 [5.3.5.1-2] 参考資料 PEG-IFNα-2a /リバビ リ ン ,PEG-IFNα-2a 単独,rIFNα-2b / リ バビリンとの比較 C 型慢性肝炎 (C 型代償性肝 硬変含む) 1,125例 C 型 代 償 性 肝 硬 変 患 者 ( 移 行 期 含む) 58例* 投与期間:48週間 経過観察期間:24週間 19 年 月- 20 年 月 第Ⅲ相臨床試験 NV15942 [5.3.5.1-3] 参考資料 PEG-IFNα-2a /リバビ リン投与量及び投与 期間の検討 C 型慢性肝炎 (C 型代償性肝 硬変含む) 1,284例 C 型 代 償 性 肝 硬 変 患 者 ( 移 行 期 含まない) 35例** 投与期間:24又は48週 間 経過観察期間:24週間 19 年 月- 20 年 月 * PEG-IFNα-2a 180 μg 及びリバビリン併用群 ** PEG-IFNα-2a 180 μg 及びリバビリン1,000-1,200 mg 併用,48週間投与群 以上の国内における臨床試験は,ヘルシンキ宣言,薬事法14条第3項及び第80条の2並びに厚 生省令第28号(平成9年3月27日)「医薬品の臨床試験の実施の基準(GCP)に関する省令」及 び厚生労働省令第106号(平成15年6月12日)「医薬品の臨床試験の実施の基準に関する省令の 一部を改正する省令」を遵守した。また,海外で実施された臨床試験も GCP 並びにヘルシン キ宣言を遵守した。ペガシス/コペガス 2.5 臨床に関する概括評価 Page
21
2.5.2
生物薬剤学に関する概括評価
本申請では製剤及び生物学的同等性試験について新たに追加する資料はない。なお,本申請 に含まれる第Ⅱ/Ⅲ相臨床試験(JV19595)におけるリバビリンの薬物動態の検討に用いた薬物 濃度定量法に変更があったので2.7.1.1項に記載した。2.5.3 臨床薬理に関する概括評価
(1) PEG-IFNα-2a の薬物動態 C 型代償性肝硬変患者を対象とした第Ⅱ/Ⅲ相臨床試験(JV19595)の12週時の血清中 PEG-IFNα-2a 濃度を用いて薬物動態パラメータを算出した。PEG-PEG-IFNα-2a の同一用量が,12週時及 びその直前に4週間以上投与された被験者は,90 μg が5例及び180 μg が2例で,それぞれの12週 時のPEG-IFNα-2a の Cmax(平均値,以下同様)は12.4 ng/mL 及び24.9 ng/mL,AUC0-168hは1850ng・h/mL 及び3650 ng・h/mL,Tmaxは52.2 h 及び83.9 h,CLss/F は0.0519 L/h 及び0.0513 L/h であっ た。 (2) リバビリンの薬物動態 C 型代償性肝硬変患者を対象とした第Ⅱ/Ⅲ相臨床試験(JV19595)の12週時の血漿中リバビ リン濃度を用いて薬物動態パラメータを算出した。パラメータの算出は,試験登録時に体重に よって割り付けられた投与量が12週時及びその直前に4週間以上投与された被験者(14例)の ものを用いた。リバビリンのCmax(平均値 ± SD,以下同様)は2550 ± 454 ng/mL,AUC0-12hは 24500 ± 4900 ng・h/mL,Tmaxは2.64 ± 1.27 h,CLss/F は17.3 ± 4.21 L/h であった。 (3) C 型代償性肝硬変患者と C 型慢性肝炎患者の薬物動態の比較 C 型代償性肝硬変患者を対象とした第Ⅱ/Ⅲ相臨床試験(JV19595)における PEG-IFNα-2a 180 μg 投与時の12週時の各測定時点の血清中 PEG-IFNα-2a 濃度は,C 型慢性肝炎患者を対象と した第Ⅲ相臨床試験(JV15725)で得られた濃度と同程度で同様の推移を示した。また,C 型 代償性肝硬変患者及び C 型慢性肝炎患者で得られた PEG-IFNα-2a の Cmax はそれぞれ24.9 ng/mL 及び32.4 ± 14.2 ng/mL で,AUC0-168hはそれぞれ3650 ng・h/mL 及び4390 ± 1730 ng・h/mL であり,いずれも両試験間で同程度の値を示した。以上のことから,C 型代償性肝硬変患者に おけるPEG-IFNα-2a の薬物動態は C 型慢性肝炎患者と大きく異ならないと考えられた。 C 型代償性肝硬変患者における血漿中リバビリン濃度は C 型慢性肝炎患者におけるリバビリ ン濃度と同様の推移を示した。C 型代償性肝硬変患者及び C 型慢性肝炎患者のそれぞれの Cmax(平均値 ± SD,以下同様)は2550 ± 454 ng/mL 及び2710 ± 989 ng/mL,AUC0-12hは24500 ± 4900 ng・h/mL 及び25800 ± 9260 ng・h/mL であり,いずれも両試験間で同程度であった。以上の ことから,C 型代償性肝硬変患者におけるリバビリンの薬物動態は,C 型慢性肝炎患者と大き く異ならないと考えられた。
ペガシス/コペガス 2.5 臨床に関する概括評価 Page
22
2.5.4
有効性の概括評価
有効性評価に用いた臨床試験一覧を表 2.5.4-1に示す。
C 型代償性肝硬変に対する PEG-IFNα-2a とリバビリン併用による有効性は,国内で実施され た第Ⅱ/Ⅲ相臨床試験(JV19595)及び一般臨床試験(JV19889)の成績に基づいて評価した。
有効性の主な解析対象は,Full Analysis Set(FAS)(治験薬が1回でも投与され,かつ投与後 の有効性に関する観察が1回でも行われた患者集団)とした。第Ⅱ/Ⅲ相臨床試験及び一般臨床 試験のFAS 対象例数は表 2.5.4-1に示した通りである。 第Ⅱ/Ⅲ相臨床試験は,観察群の目標症例数を試験途中で変更したため,観察群の登録締切 りまでに登録された患者(前期登録例,以降「前期」)と全登録期間で集積された患者(通期 登録例,以降「通期」)の2つの解析集団に分けられる。投与群と観察群との比較を行う場合 には,前期を主要な解析集団とし,PEG-IFNα-2a の用量の探索的な検討は通期にて実施した。 表 2.5.4-1 有効性評価に用いた臨床試験一覧 試験名 治験実施計画 書番号 [資料番号] デザイン 対象 投与期間 観察期間 投与群 有 効 性 評 価 例数(FAS) 国内試験(評価資料) 第 Ⅱ/ Ⅲ 相 臨 床試験 JV19595 [5.3.5.1-1] 中央登録方式によ る多施設共同,無 作為化,部分盲検 並行群間比較試験 C 型 代 償 性 肝 硬変 観察群:48週間 治験薬投与群 投与期間:48週間 観察期間:24週間 観察群 33(前期) PEG-IFNα-2a 90 μg 群 33(前期) 61(通期) PEG-IFNα-2a 180 μg 群 30(前期) 63(通期) 一般臨床試験 JV19889 [5.3.5.2-1] 中央登録方式によ る多施設共同,一 般臨床試験 C 型 代 償 性 肝 硬変 投与期間:48週間 観察期間:24週間 PEG-IFNα-2a 180 μg 群 25