九州大学学術情報リポジトリ
Kyushu University Institutional Repository
マイクロ空間を用いたタンパク質の晶析挙動の制御 とX線回折実験への応用
真栄城, 正寿
https://doi.org/10.15017/1441287
出版情報:Kyushu University, 2013, 博士(工学), 課程博士 バージョン:
権利関係:Fulltext available.
マイクロ空間を用いたタンパク質の 晶析挙動の制御と X 線回折実験への応用
九州大学大学院 総合理工学府 物質理工学専攻 新素材開発工学講座
真栄城 正寿
目次
要旨 1
第1章 序論
1-1 はじめに 4
1-2 タンパク質の結晶化と結晶構造解析 6
1-3 マイクロ流体デバイスを用いたタンパク質の結晶化 18
1-4 研究目的 23
1-5 参考文献 25
第2章 微小空間でのタンパク質の晶析挙動の解析
2-1 序論 28
2-2 実験方法 30
2-3 結果および考察 33
2-4 結論 48
2-5 参考文献 49
第3章 微小液滴中のタンパク質結晶化過程に関する理論的検討と 臨界液滴サイズの導出
3-1 序論 51
3-2 実験方法 53
3-3 結果および考察 55
3-4 結論 62
3-5 参考文献 63
第4章 臨界サイズを基にした微小液滴による結晶化制御
4-1 序論 66
4-2 実験方法 68
4-3 結果および考察 73
4-4 結論 95
4-5 参考文献 96
第5章 In-situ X線回折実験への応用
5-1 序論 100
5-2 実験方法 102
5-3 結果および考察 107
5-4 結論 114
5-5 参考文献 115
第6章 結論
結論 118
謝辞 121
研究業績 122
要旨
タンパク質の結晶化と結晶を用いた立体構造解析は、その機能に関する生化 学的情報を与える。そのため、タンパク質の立体構造解析は、学術分野だけで なく創薬などの産業分野においても重要な研究テーマである。一般的にタンパ ク質の立体構造は、タンパク質の単結晶を用いたX線結晶構造解析によって解 析が行われている。しかしながら、良質なタンパク質の単結晶を得るためには、
最適な結晶化条件の探索が必要であり、それには膨大なサンプル量と労力を必 要とする。この点から、近年では単結晶作製のプロセス自体が立体構造解析に おけるボトルネックとなっている。また、タンパク質結晶化の制御技術は未だ に確立されておらず、複数の単結晶が合体、密集するなど結晶同士が重なり合 うと単結晶X線回折実験が困難となる。研究室レベルのX線回折装置では、結 晶構造解析において1辺が少なくとも約0.1 mmの単結晶を必要とする。それよ りも小さい20〜30 µm程度の結晶から得られる回折データは、回折強度が弱い ため高分解能で立体構造を決定することが困難である。したがって、理想的に は結晶化容器内で結晶の集合を抑制し、1個の単結晶を作製することが望ましい。
さらに、タンパク質の単結晶を得た後のX線結晶構造解析においては、抗凍結 処理などの前処理やX線回折装置へのマウントなど、熟練した結晶のハンドリ ングが必要である。このように、タンパク質の結晶化制御とX線回折実験は未 だに熟練者の勘や経験に依存する要素が多く、プロセスの改善が強く望まれて いる。一方で、マイクロ化学システムを用いたタンパク質の結晶化は、消費サ ンプル量の低減や結晶化条件スクリーニングの効率化が期待され、またIn-situ のX線結晶構造解析が可能である。さらに、微小空間は界面張力が支配的な空 間であるため宇宙空間のような微小重力場と類似した環境でのタンパク質の結 晶成長が期待される。上記の特徴から、これまでにいくつかの研究グループか
ら結晶化条件スクリーニングデバイスが報告されている。しかしながら、それ らの研究ではデバイス開発やコンセプトが報告されているのみであり、タンパ ク質結晶化の制御については明らかになっていない。
そこで、本研究では、研究室レベルのX線回折装置でも簡便、効率的なタン パク質の結晶構造解析を実現可能なツール開発を大きな研究目的とした。具体 的には、効率的なX線回折実験のために、微小液滴内で微結晶析出と結晶の集 合を抑制し、100 µm程度のタンパク質の単結晶を1個だけ作製するための微小 液滴の大きさについて実験と理論の両方から検討を行った。また、結晶成長過 程におけるタンパク質分子の物質収支から、単結晶が1個だけ析出する液滴の 大きさ(臨界サイズ)という概念を提案した。さらに、臨界サイズをもとに分 子量が大きく異なる4種類のタンパク質を用いて、過飽和度を変化させて晶析 挙動の調査を行い、最適な大きさの微小液滴を用いることで結晶化を制御でき ることを明らかにした。さらに、上記の実験を基に確立した結晶化制御技術を
用いて、In-situでのタンパク質単結晶のX線回折実験を試みた。その結果から、
結晶構造解析において、煩雑な前処理を必要としない効率的な1次スクリーニ ング方法を提案した。最後に、本研究の内容を総括し、微小液滴中でのタンパ ク質の晶析挙動の制御方法についてまとめ、将来を展望した。
第 1 章
序論
1-1 はじめに
20 世紀はバイオテクノロジーにとって著しい飛躍を遂げた世紀であった。
1953年にDNAの二重らせん構造がはじめて明らかにされ、それらの構造に基づ いて生命現象を研究する分子生物学が発展してきた[1]。そして、1998 年にはヒ トのES細胞が作製されており、2003年にはヒトゲノムの解読完了宣言がなされ た。このように、遺伝情報はほぼは明らかになっており、次の課題としてタン パク質の立体構造の解析が進められている。生物機能の直接の担い手であるタ ンパク質の立体構造と機能の相関を解明することは、プロテオミクスにおいて 必要不可欠である。タンパク質の立体構造と機能を知ることで、生命現象の解 明がさらに進み、創薬においては合理的な薬剤設計が可能となる。特にタンパ ク質の立体構造が原子レベルで分かっていれば、そのタンパク質の立体構造に 基づいて有効に働く薬剤分子を設計するSBDD(Structure Based Drug Design)が 原理的に可能であり、新薬開発の新しいアプローチとして注目されている。
このように、構造生物学はライフサイエンス全体の基盤としての役割を果た すと同時に、その研究成果が医薬開発や産業利用につながることから、欧米を はじめ世界各国で国家的な取り組みが行われている。わが国においても、平成 14年度より5年にわたって実施された文部科学省の「タンパク3000プロジェク ト」などにおいて多様なタンパク質の立体構造の解析が進められた[2]。また、そ の後も「ターゲットタンパク研究プログラム」、「創薬等支援技術基盤プラット フォーム事業」など後継プロジェクトが展開されている。しかしながら、タン パク質の立体構造の解析において、タンパク質の結晶化自体が未だに熟練者の 勘や経験に依存するところが大きい。タンパク質の結晶化は、低分子の結晶化 と原理的には同じであると考えられている。しかし、タンパク質は分子が大き く、立体構造を取ることがその結晶化を困難にしている。結晶化の試み自体は、
これまでには宇宙空間のような微小重力場から電場、磁場、レーザー、そして 微小空間を用いた結晶化など様々な手法が開発されてきた[3-7]。しかしながら、
結晶化制御技術は未だに確立されておらず、どの方法も標準的手法となり得て いない。このような背景からも、タンパク質の晶析挙動を解明し、結晶化制御
技術を確立することは、タンパク質の結晶構造解析を効率化するために重要で あり、生命現象の謎を解き明かし、人類に新しい創薬をもたらす可能性を秘め ていると考えられる。
1-2 タンパク質の結晶化と結晶構造解析 1-2-1 タンパク質の結晶化
タンパク質とは、L-アミノ酸が多数重合した高分子化合物であり、生物の重 要な構成成分のひとつである。しかしながら、タンパク質は単なるアミノ酸の 重合体ではなく、固有の立体構造と機能をもつ分子である。タンパク質を構成 する天然アミノ酸は20種類存在する。タンパク質はそれぞれ固有のアミノ酸配 列を持ち、そのようなタンパク質のアミノ酸の配列順序のことを 1 次構造とい う。さらに高次構造としてαへリックスやβシートなどの 2 次構造、タンパク 質全体としての3次構造をとる。3次構造は、タンパク質の1次構造、つまりタ ンパク質を構成するアミノ酸配列に大きく依存すると考えられている。そして、
そのアミノ酸残基の配列はそれぞれのタンパク質に独自のものであるため、タ ンパク質は独特の化学的性質を持ち、特異的な機能を果たす。例えば、側鎖が タンパク質分子の外側に突き出しているアミノ酸残基は、タンパク質の全体的 な化学的性質を決定したり、タンパク質の機能に直接関わったり、他のタンパ ク質などの生体分子との相互作用に関連している。また、側鎖がタンパク質分 子の内側に突き出しているアミノ酸残基は、疎水結合などを通して、全体とし ての三次構造の維持に働いている。タンパク質は精密な三次構造を持っている ため、1個のアミノ酸残基を変化させるだけで、その三次構造が壊れたり、機能 が失われたりすることが少なくない。このように、タンパク質の立体構造はそ の機能と密接に関連があり、また活性部位の構造は創薬において重要な情報を
与える[8,9]。そのため、現在でもタンパク質の立体構造解析は、産学問わず世界
中で盛んに研究が行われている。
一般的にタンパク質の立体構造は、タンパク質の単結晶を用いたX線結晶構 造解析によって解析が行われている。結晶構造解析のプロセスは、①タンパク 質の調製、②結晶化実験、③X線回折実験、④構造解析の4つに大別すること ができる。良質なタンパク質の単結晶を得るためには、最適な結晶化条件の探 索が必要であり、現在では単結晶作製のプロセス自体が立体構造解析における ボトルネックとなっている。結晶化条件の探索項目としては、①タンパク質濃
度、②沈殿剤の種類と濃度、③緩衝液の種類と濃度、pH、④温度、⑤添加物、
⑥結晶化方法(図1-1参照)などがあげられ、これらについて網羅的に結晶化条 件探索が行われる[10,11]。マクロスケールにおいては、これらの条件探索は手動 のピペッティング操作、または自動化ロボットで行われているが、前者の場合 には莫大な労力が必要であり、後者の場合には高価な投資コスト・運転コスト が課題としてあげられる。したがって、より安価で効率的な結晶化条件探索シ ステム開発が期待される。
ここで、結晶化とは溶液中に溶解している溶質が固体として析出する相変化 である。結晶化では系内で結晶核の発生と結晶の成長という2つの現象が生じ ている。一般的に溶液中の核形成は、エンブリオ(幼核)が発生と消滅を繰り 返しており、臨界核半径を超えるサイズの核が発生した場合に、その核が結晶 成長すると考えられている。これは、低分子からタンパク質のような高分子に も共通している現象である。つまり、これらの核形成速度と結晶成長速度を制 御することが晶析操作においては重要となる。図1-2に一般的なタンパク質の相 図を示す。タンパク質の結晶化は、図1-2に示すように過飽和状態(領域Lま
たはM)の溶液中で行われる。溶液の過飽和度は結晶化の駆動力であり、高過
飽和度の溶液ではすぐに核形成が始まり結晶が析出する[12,13]。タンパク質の結 晶化において、この過飽和度を制御するための条件は、大きく分けて①タンパ ク質濃度、②沈殿剤の種類と濃度、③温度の3条件である。この3条件を適切 に調整することで、良質なタンパク質の結晶を得ることができる。図1-2の相図 ではLの領域で核形成が生じ、Mの領域は結晶成長領域である。また、Pの領 域ではアモルファスの沈殿が生じる。Sの領域は未飽和領域であり結晶は溶解す る。この相図作成は、タンパク質の結晶化において、良質で大きな単結晶を得 る上で結晶化条件探索と同様に重要な過程である。核形成領域では結晶の核形 成、準安定領域では結晶成長が優先的に進行する。また、タンパク質結晶化に おける特徴の1つは、他の低分子や無機化合物と比較して、準安定領域が大き いことがあげられる。そのために、核形成には高い過飽和比が必要となるが、
高い過飽和比の溶液からは微結晶の析出や結晶同士の集合が生じやすいため、
構造解析に適した大きさの単結晶を作製するためには試行錯誤を繰り返す必要 がある。特に、タンパク質の結晶のような回折能が低い試料において、一般的 な研究室レベルのX線回折装置を使用する場合、構造解析には1辺が少なくと
も100 µm以上の単結晶を必要とする。それよりも小さい20〜30 µm程度の結晶
から得られる回折データは、回折強度が弱いため高分解能で立体構造を決定す ることが困難である。このような微小結晶は、高輝度放射光X線によって結晶 構造解析が行われるが詳細は次節で述べる。したがって、理想的には結晶化容 器内で微結晶の析出と結晶の集合を抑制し、1個の良質で大きな単結晶を作製す ることが望ましい。しかしながら、未だにタンパク質結晶化の制御技術は確立 されておらず、熟練者の勘や経験に大きく依存している。例えば、図1-1に示す 結晶化実験のセットアップにおいて、結晶化溶液の調製方法(ピペッティング 回数や混ぜ方)、結晶化溶液の体積、セットアップ後に静置するか緩やかに混合 するかなど研究室ごとに手法が異なることも少なくない。さらに、結晶化条件 によっては図1-3に示すように、複数の単結晶が合体、密集するなど結晶同士が 集合する場合がある。このような結晶サンプルは、X線回折実験の前処理過程 で結晶に物理的損傷が生じやすい。また、構造解析においては、回折点同士の 重複が生じて正しい積分強度の見積もりができない場合や、解析自体が不可能 なデータである場合がある。そのため、結晶の集合を抑制し、1個の良質で最適 な大きな単結晶を作製する技術開発が強く望まれている。
次に、タンパク質の結晶成長過程について述べる。核形成した結晶の成長は、
①界面律速型の結晶成長、②拡散律速型の結晶成長に分けられる。界面律速型 の結晶成長とは、結晶表面への溶質分子の取り込みが結晶成長の律速段階であ る成長様式である。一方で、拡散律速型の結晶成長とは、結晶表面での溶質分 子の取り込みが速く、溶質分子の結晶近傍への拡散が遅い場合の結晶成長様式 である。ここで、一般的にタンパク質の結晶は、タンパク質分子の結晶表面へ の取り組みが遅い、界面律速型の結晶成長であると言われている。それは、分 子サイズが大きく、分子の非対称性が高いために結晶成長面のキンクに取り込 まれにくいためであると考えられている[12]。この点は、低分子の結晶化と大き
く異なる点である。一方で、宇宙空間のような微小重力場ではタンパク質の結 晶成長は拡散律速型であると報告されている[14]。微小重力場では密度差対流が 抑制されており、タンパク質分子の結晶近傍への輸送過程が拡散のみとなる。
その結果、結晶周辺に拡散場が形成されて結晶表面のタンパク質濃度が低下し、
拡散律速で結晶成長が進行する。微小重力場のような対流が抑制された環境で 作製された結晶は、地上で作製した結晶と比較してモザイシティが低い良質な 結晶が得られやすい傾向にある[15]。しかしながら、その結晶成長に関する詳細 なメカニズムは十分に明らかになっておらず、未だに研究が続けられている。
また、タンパク質結晶の大きな特徴に1つが、結晶格子中に多量の水和水を 含有していることである。水溶液中のタンパク質はその立体構造を保ったまま、
分子表面のわずかな領域が接触することによって結晶となる。接触面の相互作 用はほとんどの場合、疎水結合をはじめとする極めて弱い結合である。タンパ ク質の結晶は、このように弱い結合によって形成されているため、その物理的 強度も大変低い。例えば、ピペットチップの先端などで突くと結晶は簡単に破 損する。このため、タンパク質の結晶の取り扱いには熟練したハンドリングを 必要とする。この特徴は、図1-3のような結晶同士が集合した場合に大変不利に 働く。詳細は次節で述べるが、結晶化溶液内で析出した結晶は、溶液内から拾 い上げられて前処理を施した後にX線回折実験が行われる。この前処理操作に おいて、結晶同士が集合すると、脆くて壊れやすいタンパク質の結晶を1個拾 い上げる操作が困難となる。これらの理由から、結晶化溶液内での結晶同士の 集合を抑制して、簡便に効率よく1個の単結晶を作製できる技術開発が望まれ ている。
Figure 1-1. Traditional crystallization methods. (a) Hanging drop vapor diffusion method. (b) Sitting drop vapor diffusion method. (c) Micro batch method.
Figure 1-2. Phase diagram of protein crystallization. There are four main types of phases: the precipitant zone [P], the labile zone [L], the metastable zone [M], and the unsaturated zone [S].
Figure 1-3. Photographs of protein crystals.
1-2-2 タンパク質の結晶構造解析
現在、タンパク質の立体構造は大きく分けて 2 つの手法で解析が行われてい る。このうち、タンパク質の立体構造の多くは、X 線結晶構造解析によって決 定されている。これまでに決定されたタンパク質の立体構造のうち、80%以上が X線結晶解析によるものである[12,16]。残り十数%はNMRによるもので、この方 法は溶液状態で構造を決定できるという利点がある。しかしながら、対象とな るタンパク質の分子量に制限がある。NMRで決定されたタンパク質は、そのほ とんどが2~3万程度の分子量である。X線結晶構造解析は、タンパク質の良質 で大きな単結晶を作製する必要がある。一般的な研究室レベルの X 線回折装置 では、1辺が少なくとも100 µm以上の単結晶を必要とする。しかしながら、前 節で述べたように大きなタンパク質の単結晶の作製は容易ではない。そのため、
様々な条件を探索しても 20〜30 µm程度の微結晶しか得られない場合も多い。
タンパク質結晶からの回折強度は、結晶の体積と入射 X 線の強度に比例する。
また、研究室レベルの X 線回折装置は、ビームの収束が難しく、微結晶の測定 ではバックグラウンドが大きくなる問題がある。このような理由によって、20 µm程度の微結晶は、立体構造解析に不向きであると考えられる。
この課題に対して、近年ではシンクロトロン光施設の発展と高輝度 X 線の普 及によって、10 µm程度の微結晶であっても短時間で立体構造解析が可能となっ てきた。我が国においては、兵庫県にある SPring-8 やつくばの高エネルギー加 速器研究機構・物質構造科学研究所のフォトンファクトリーなどの利用によっ て、これまで解析が困難であったタンパク質の立体構造が決定されている。さ らに、2006 年から理化学研究所が整備を進めてきた X 線自由電子レーザー
(XFEL)施設「SACLA」が 2012 年より共用利用を開始し、タンパク質の立体 構造解析はさらに加速されると予想される。特に、SACLAの利用は、将来的に 1 分子のタンパク質からでも立体構造解析を解析できる手法になると大きな期 待が寄せられている。このように、高輝度な放射光 X 線を用いたタンパク質の 結晶構造解析は、強力な解析方法であるが、一方で課題も少なくない。その課 題の1つが、タンパク質結晶の放射線損傷である。これは、結晶に照射されたX
線によって結晶中に多量に含まれる水分子から酸素ラジカルが発生し、そのラ ジカルが立体構造を破壊しながら拡散する現象である。したがって、放射線損 傷によって測定中に回折強度の低下やモザイシティの上昇が観測され、場合に よっては回折パターンが消失する。
そこで、放射線損傷対策として1990年以降に100 Kの低温下での回折実験法 が開発された。この方法は、100 Kの極低温の窒素ガスを結晶に吹き付けながら 測定を行う手法である。極低温下での回折実験法の開発は、室温下では放射線 損傷による結晶の劣化が観測されたタンパク質結晶であっても、良好な回折デ ータの取得を可能とした。このように、近年の極低温下での測定は X 線回折実 験において必要不可欠な手法となっている。結晶を冷却する場合には、その前 処理として抗凍結処理を施す必要がある。抗凍結処理とは、タンパク質内部の 水分子や結晶に付着している溶媒をアモルファス状に瞬間凍結させるための処 理である。この前処理が不十分な場合は、タンパク質結晶の内部に六方晶の氷 が生じる。その結果、タンパク質の立体構造の破壊や回折像に氷の回折パター ンが観測され、良質な回折データを取得することができない。したがって、抗 凍結処理は重要な前処理過程であるが、その処理具合は実験者の熟練度に大き く依存する。また、Spring-8 のビームライン「BL32XU」や SACLAなどの超高 輝度な X 線ビームを用いた場合には、抗凍結処理を施しても次第に結晶の劣化 が観測される。放射光施設の高輝度 X 線の利用は、微小結晶からでも短時間で 立体構造の解析を可能としたが、現在でも経験者の試行錯誤が繰り広げられて いる。
図1-4に結晶構造解析手順の概略図を示す。前節に記述した通り、タンパク質 の結晶化は莫大な数の結晶化条件探索が必要である。そして、その中から結晶 が得られるとX線構造解析が行われる。タンパク質のX線回折実験では、まず 結晶作製後にクライオループで結晶化溶液中から顕微鏡下で結晶を 1 個だけ拾 い上げる。続いて、結晶の抗凍結処理を行い、X 線回折装置に結晶のマウント を行う。その後、試料台を回転させて照射する X 線と結晶の軸がずれないよう に位置を合わせて測定を開始する。X 線回折実験から得られた回折データは、
HKL2000 や XDS、Mosfilmなどの解析ソフトを用いてデータ処理される。その 後、指数付けや精密化、分子置換などの処理を行って結晶学的パラメータ(格 子状数、空間群、晶系など)が決定されて、立体構造が解かれる。しかしなが ら、最初に得られた結晶から完全な回折データセットが得られて結晶構造が解 かれるわけではない。まずは、良質な回折像が得られた、あるいは条件の最適 化によって得られる可能性がある結晶化条件を絞り込み、それらについてさら に条件を最適化して立体構造が解かれる。そのため、結晶化と X 線回折実験、
最適化を繰り返す必要がある。このような理由から、結晶構造解析の初期段階 では、結晶化やX線回折実験の自動化が強く望まれている[17]。
ここで、前節で述べたとおり、タンパク質の結晶は水分子を内部に少なくと
も 50%程度含んでいるため、非常に脆くて壊れやすい。したがって、結晶化溶
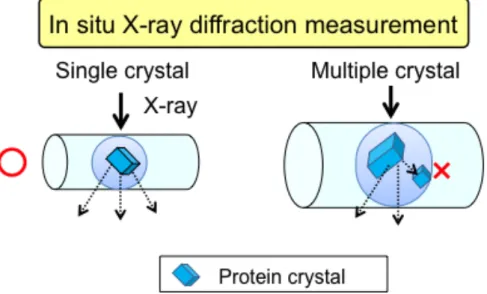
液から結晶を取り出す操作と抗凍結処理などの前処理に伴う結晶のハンドリン グはタンパク質の結晶に物理的な損傷を与える可能性が考えられる。これらの 操作を簡便にするために、クライオループなどの改良が施されてきたが、未だ に根本的な解決方法は見出されていない。この課題を解決する方法の 1 つとし て、結晶化に使用した結晶化容器(キャピラリーなど)、あるいはデバイスから 取り出すことなくX線回折実験を行う手法が期待されている。
また、その他の放射光利用に関する課題としては、現場への結晶の輸送があ げられる。タンパク質の結晶は、物理的な損傷に弱く、温度変化も結晶性を低 下させるため好ましくない。したがって、実験室で結晶化実験から X 線回折実 験まで行える環境が、タンパク質の立体構造解析において理想的である。しか しながら、膜タンパク質のような 10 µm程度の微結晶しか得られないものも少 なくない。したがって、これからのタンパク質の立体構造解析は、得られる結 晶サイズに応じてビームの選択が行われると予想される。微小結晶しか得られ ないサンプルは、放射光施設で超高輝度な微小ビーム(1 x 1 µmビームなど)を 利用した測定が望ましい。一方で、100 µm程度の結晶が得られる場合には、研 究室レベルの100~200 µmのビーム径や、放射光施設であっても結晶サイズに適 したビーム径の利用によって効率的な結晶構造解析が実現できると考えられる。
そのため、後者の場合には、結晶化溶液内での微結晶の析出や結晶同士の集合 を抑制して、100 µm程度の1個の単結晶を作製できる技術開発が必要となる。
また、その後のダメージレスなX線回折実験手法の確立も強く望まれる。
Figure 1-4. Schematic procedure of crystal structure analysis.
1-3 マイクロ流体デバイスを用いたタンパク質の結晶化
創薬や生化学などにおいてハイスループットスクリーニングは,膨大な数の サンプルを短時間で評価できるなどの利点があり、その重要性が高まっている
[18-20]。しかし、従来のハイスループットスクリーニングシステムは、マクロス
ケールの装置をそのまま自動化するものであり、膨大な初期投資や運転コスト が必要である。また、このようなシステムで網羅的に探索を行う場合、網羅的 な条件探索によって必要となるサンプル量が増加する課題もある。前節までに 述べたように、タンパク質の結晶化条件探索はタンパク質濃度や沈殿剤の種類 や濃度など、様々な条件を探索する必要がある。そのため、膨大な量のタンパ ク質サンプルを調製しなければならない。しかしながら、一般的にタンパク質 サンプルの調製には、時間とコストを必要とする。比較的に発現量が多い大腸 菌発現系を用いた場合であっても、1Lの培養液から1 mg程度のタンパク質しか 得られないことも多い。さらに、膜タンパク質では、1 mgのサンプルを調製す ること自体が困難な場合もある。そのため、できる限り少ないサンプル量消費 量で多くの条件探索が可能なシステム開発が期待されている。
2000年以降、マイクロ流体技術を用いた様々なハイスループットスクリーニ ングデバイスが報告されている。その中でも微小液滴は、使用するサンプル量 が少ない、層流流れと比較して液-液界面が増えるため比表面積が高いなどの特 徴がある。そのため、タンパク質結晶化をはじめとして[21,22]、微粒子合成[23,24]、 高分子合成[25]、水相-有機相間[26]での抽出などに広く応用されている。さらに、
マイクロ流体技術は単分散で任意の大きさの微小液滴を再現性よく大量に生成 することが可能である。
マイクロ流体技術を用いたタンパク質の結晶化方法として、微小液滴、界面 自由拡散、蒸気拡散法などの結晶化条件探索デバイスが報告されている。また、
X線回折実験や膜タンパク質結晶化への応用も試みられている[27-34]。マイクロ 流体デバイスを用いる利点としては、消費サンプル量の大幅な低減(nL/1条件)、 結晶化条件探索の効率化、労力の低減などがあげられる。さらに、デバイスの 材質や構造によっては、結晶をデバイスから取り出すことなくX線回折実験を
行うことが可能である。表1-1に主な研究グループとデバイスの特徴についてま とめた。Ismagilovらのグループは、微小液滴を用いた結晶化条件探索システム を考案した。微小液滴の体積は~20 nL程度であり、従来法と比較すると1条件 あたり100倍以上の消費サンプル量削減が可能である。また、結晶化デバイス は、一般的なリソグラフィー技術によって容易に作製できる。しかしながら、
実験システムは多少複雑である。さらに、タンパク質サンプルはデバイスに非 特異的に吸着しやすいため、サンプルによっては安定した液滴生成が困難とな る。最近ではSlip Chipと呼ばれる界面自由拡散型のデバイスを報告した。Slip Chipは、微小液滴法よりも実験システムは単純であるが、デバイス作製に高度 な技術が必要である。Quakeらのグループは、高度なリソグラフィー技術を用い てバルブを組み込んだマイクロ流体デバイスを報告した。結晶化方法は、界面 自由拡散法であり、デバイス中のマイクロウェルで結晶化を行う。1条件あたり の消費サンプル量は20 nL程度であり、実験システムも単純であるが、デバイス 作製には高度な技術を必要とする。Kenisらのグループは、Poly (dimethyl) siloxane
(PDMS)とCyclic olefin copolymer (COC)で作製した結晶化デバイスを報告した。
コンセプト自体はQuakeらと同じだが、より簡単にデバイス作製が可能となっ た。このように2002年以降様々なコンセプトのタンパク質結晶化条件探索デバ イスが報告されている。一方で、これらの研究は、結晶化条件探索デバイスの 開発に主眼が置かれており、従来の結晶化方法をそのまま小型化した手法であ る。したがって、それぞれの系内(微小液滴内やマイクロウェル内)で結晶化 の制御は行われておらず、100 µm程度の結晶は得られているが複数の結晶が析 出し得る環境となっている。しかしながら、研究室レベルのX線回折装置やX 線の集光が不十分な放射光施設、具体的にはビーム径が約100~200 µmのX線に よって、デバイスのままX線回折実験を行うためには系内に1個の単結晶を作 製する必要がある。そして、そのためには微小空間で微結晶析出や結晶同士の 集合を抑制し、100 µm程度の単結晶を作製するための結晶化制御技術の確立が 求められる(図1-5)。このような、系内に1個の結晶を作製する方法は、いく つかの報告がされている[35,36]。タンパク質の結晶化に関しては、Veeslerらのグ
ループが、モデルタンパク質としてリゾチームの晶析挙動を報告している。結 晶化実験には、500 x 500 x 1000 µmの液滴が用いられた。このサイズの液滴内に は、実験開始後数時間で複数の結晶が析出し、1個の結晶を作製することは困難 であることが報告された。このように、従来法を含めて微小空間でのタンパク 質の結晶化制御方法も未だに確立されておらず、より詳細な晶析挙動の解析が 必要である。
Table 1-1. Summary of protein crystallization by using a microfluidic device.
Figure 1-5. An illustration of In situ X-ray diffraction measurement by using microfluidic device.
1-4 本研究の目的
前節までに述べたように、タンパク質の結晶化制御とX線回折実験は未だに 熟練者の勘や経験に頼る部分が多い。タンパク質の結晶化制御においては、結 晶化溶液内での微結晶析出と結晶同士の集合を抑制する技術の確立が望まれる。
また、X線回折実験においては、脆くて壊れやすいタンパク質の結晶をダメー ジレスで簡便に測定可能な手法の確立が求められている。系内に100 µm程度の 単結晶を1個作製し、その結晶をそのまま測定することができれば、研究室レ ベルのX線回折装置であっても効率的な立体構造解析が可能となると考えられ る。また、これらの技術の確立によって、タンパク質の結晶化とその後の立体 構造解析を加速することができ、新規薬剤の開発をはじめとするライフサイエ ンス分野に大きな貢献を果たすことができると考えられる。
そこで、以上の背景を踏まえて本研究は、研究室レベルのX線回折装置でも 簡便、効率的なタンパク質の結晶構造解析を実現可能なツール開発を大きな研 究目的とした。この課題に対して本研究では、これまでは検討されていなかっ た微小液滴を“結晶化容器”として考え、微小液滴を用いたタンパク質の結晶 化制御を試みた。具体的には、効率的なX線回折実験のために、微小液滴内で 微結晶析出と結晶の集合を抑制し、100 µm程度のタンパク質の単結晶を1個だ け作製するための結晶化容器(微小液滴の大きさ)の条件について実験と理論 の両方から検討を行った。その過程において、1個の単結晶を作製するための“臨 界サイズ”という概念を導入した。また、確立した結晶化制御技術を基に、デ バイスから結晶を取り出さずに直接X線回折実験を行って、良質な回折像を得 た。各章の内容は以下の通りである。
第2章では、本研究で基盤となるマイクロ流体デバイスを用いてタンパク質 の微小液滴を連続的に生成する手法、実験系を確立した。そして、作製したデ バイスを用いて微小液滴の体積や形状がタンパク質の核形成や結晶成長に与え る影響について調査した。
第3章では、前章の晶析挙動の解析結果を基に、マイクロ空間中の微小液滴 内におけるタンパク質結晶の成長過程に着目し、タンパク質分子の物質収支か
ら微小液滴内で単結晶が1個だけ析出する液滴の大きさ(臨界サイズ)を理論 的に導入し、実験結果との比較を行った。
第4章では、前章で提案した臨界サイズを基にしたタンパク質結晶化の制御 を分子量が大きく異なる4種類のタンパク質を用いて調査した。さらに、核形 成における重要なパラメータである過飽和度を変化させて晶析挙動の調査を行 った。
第5章では、前章までに見出した微小液滴を用いた結晶化制御技術を利用し て、テフロンキャピラリー、およびガラスキャピラリー内の微小液滴中でタン パク質単結晶を1個だけ析出させ、In-situでのX線回折実験を試みた。
第6章では、本研究の内容を総括し、微小液滴中でのタンパク質の晶析挙動 の制御方法についてまとめ、将来を展望した。
1-5 参考文献
[1] J. D. Watson, F. H. C. Crick, Nature, 1953, 171, 737.
[2] タンパク質3000プロジェクト
[3] 田仲広明, 日本マイクログラビティ応用学会誌, 2003, 20, 105.
[4] S. K. Chung, E. H. Trinh, J. Cryst. Growth, 1998, 194, 384.
[5] M. Ataka, E. Katoh, N. I. Wakayama, J.Cryst. Growth, 1997, 173, 592.
[6] K. Nakamura, Y. Sora, H. Y. Yoshikawa, Y. Hosokawa, R. Murai, H. Adachi, Y.
Mori, T. Sasaki, H. Masuhara, Appl. Surf. Sci, 2007, 253, 6425.
[7] L. Li, D. Mustafi, Q. Fu, V. Tereshko, D. L. Chen, J. D. Tice, R. F. Ismagilov, Proc.
Natl. Acad. Sci. USA., 2006, 103, 19243.
[8] I. Tickle, A. Sharff, M. Vinkovic, J. Yon, H. Jhoti, Chem. Soc. Rev. 2004, 33, 558–
565.
[9] G. Scapin, Curr. Pharm. Design 2006, 12, 2087.
[10]A. McPherson, Methods 2004, 34, 254.
[11] R. C. Stevens, Curr. Opin. Struct. Biol. 2000, 10, 558.
[12] 坂部知平, 相原茂夫, タンパク質の結晶化, 京都大学学術出版会, 2005.
[13] 佐藤清隆, 溶液からの結晶成長, 共立出版株式会社, 2002.
[14] A. McPherson, Crystallization of Biological Macromolecule, Cold Spring Harbor Laboratory Press, 1999.
[15] Z. Kam, H. B. Shore, G. Feher, J. Mol. Biol., 1978, 123, 539.
[16] J. Drenth, et al., タンパク質のX線結晶構造解析法, Springer, 1998.
[17] M. Hiraki, S. Watanabe, N. Honda, Y. Yamada, N. Matsugaki, N. Igarashi, Y.
Gaponov, S. Wakatsuki, J.Synchrotron Rad. , 2008, 15, 300.
[18] P. A. Johnston, P. A. Johnston, Drug Discov. Today, 2002, 7, 353.
[19] J. A. Frearson, I. T. Collie, Drug Discov. Today, 2009, 14, 1150.
[20] R. C. Stevens, Curr. Opin. Struc., Biol., 2000, 10, 558.
[21] B. Zheng, L. S. Roach, R. F. Ismagilov, J. Am. Chem. Soc. , 2003, 125, 11170.
[22] C. J. Gerdts, M. Elliott, S. Lovell, M. B. Mixon, A. J. Napuli, B. L. Staker,
[23] E. M. Chan, A. P. Alivisatos, R. A. Mathies, J. Am. Chem. Soc. , 2005, 127, 13854.
[24] T. Nisisako, T. Torii, T. Takahashi, Y. Takizawa, Adv. Mater. 2006, 18, 1152.
[25] W. Li, H. H. Pham, Z. Nie, B. MacDonald, A. Guenther, E. Kumacheva , J. Am. Chem. Soc., 2008, 130, 9935.
[26] P. Mary, V. Studer, P. Tabeling, Anal. Chem., 2008, 80, 2680.
[27] L. Li, R. F. Ismagilov, Annu. Rev. Biophys. 2010, 39, 139.
[28] J. Leng, J. B. Salmon, Lab Chip 2009, 9, 24.
[29] L. Li, D. Mustafi, Q. Fu, V. Tereshko, D. L. Chen, J. D. Tice, R. F. Ismagilov, Proc. Natl. Acad. U. S. A. 2006, 103, 19243.
[30] W. Du, L. Li, K. P. Nichols, R. F. Ismagilov, Lab Chip 2009, 9, 2286.
[31] C. L. Hansen, E. Skordalakes, J. M. Berger, S. R. Quake, Proc. Natl. Acad. U. S. A.
2002, 99, 16531.
[32] K. Dhouib, C. K. Malek, W. Pfleging, B. G. Manuel, R. Duffait, G. Thuillier, R.
Ferrigno, L. Jacquamet, J. Ohana, J. L. Ferrer, A. T. Dietrich, R. Giege, B. Lorber, C.
Sauter, Lab Chip 2009, 9, 1412.
[33] S. Guha, S. L. Perry, A. S. Pawate, P. J. A. Kenis, Sens. Actuat. B. 2012, 174, 1.
[34] S. L. Perry, G. W. Roberts, J. D. Tice, R. B. Gennis, P. J. A. Kenis, Cryst. Growth Des. 2009, 9, 2566.
[35] D. Kashchiev, D. Verdoes, G. M. Van Rosmalen, J. Cryst. Growth 1991, 110, 373.
[36] M. Ildefonso, N. Candoni, S. Veesler, Cryst. Growth Des. 2011, 11, 1527.
第 2 章
微小空間でのタンパク質の
晶析挙動の解析
第
2
章 微小空間でのタンパク質の 晶析挙動の解析2-1 序論
タンパク質の機能はその立体構造と密接に関係している。そのため、医薬品 開発においてタンパク質の立体構造の情報を基にした薬剤設計などが期待され ている。タンパク質の立体構造は、主にタンパク質の単結晶を用いた単結晶 X 線構造解析によって決定されている。タンパク質の結晶は、回折強度が弱いた め研究室レベルのX線回折装置で測定を行うためには、1辺が100 µm程度の単 結晶が必要である。また、X 線回折実験においては、成長させた 1 個の単結晶 をクライオループを用いて、X 線回折装置にマウントする必要がある。一般的 に、タンパク質の結晶はその結晶格子中に 30~50%の水分子を含んでいるため、
脆くて壊れやすい特徴を持つ。したがって、このマウント操作には経験と結晶 の熟練したハンドリング技術が必要となる。そこで、近年タンパク質結晶構造 解析のさらなる効率化を目的として、タンパク質結晶化と X 線回折実験の自動 化が進められている。しかしながら、それぞれの自動化システムは独立してい るため、それらを統合したシステムの開発が望まれている[1-3]。その1つとして、
キャピラリー中の微小液滴内でタンパク質の結晶化を行い、キャピラリーごとX 線回折装置にマウントし、測定を行うシステムが考えられる。これまでに、キ ャピラリーやマイクロ流体デバイスを用いてタンパク質の結晶化から X 線回折 実験までを試みた研究は報告されている[4-6]。しかしながら、これらのデバイス は主に結晶化条件の探索を試みているものであり、従来法のスケールダウンを 行ったにすぎない。そのため、系内に複数の結晶が析出する場合があり、結晶 化の制御は行われていない。系内に複数の結晶が析出すると、構造解析におい て回折点同士の重複が生じて正しい積分強度の見積もりができない場合や、解 析自体が不可能なデータである場合がある。また、複数の核形成が生じた場合 は、その核形成の分だけタンパク質分子が消費されて、得られる結晶サイズが 小さくなる。このような理由から、デバイスから結晶を取り出さずに X 線回折
実験を行うためには、微小液滴中やデバイスの系内で結晶化を制御して、100 µm 程度の大きさの1個の単結晶を作製する必要がある。そのためには、微小空間・
微小液滴中でのタンパク質の晶析挙動に関する知見が必要となるが、マイクロ 空間でのタンパク質の詳細な晶析挙動は解明されていない。特に、微小空間で は体積力よりも表面張力が支配的な環境であり、それらが結晶成長過程に影響 を与えている可能性も考えられる。そこで、本章では微小液滴の体積、形状が タンパク質結晶の核形成、結晶成長に与える影響について調査した。
2-2 実験方法 2-2-1 溶液調製
モデルタンパク質にはThaumatococcus daniellii由来のソーマチンを用いた。ソ ーマチンは和光純薬工業から購入し、再精製などは行わず結晶化実験に使用し
た[7-8]。ソーマチン溶液は、適量のソーマチンを100 mM N-(2-acetamido)
iminodiacetic acid (ADA) 緩衝液(pH 6.5、シグマアルドリッチ)に溶解し、30
mg/mLの濃度に調製した。調製した溶液は、13200 rpmで遠心分離を行い、上清
を回収した。ソーマチンの正確な濃度は、ナノドロップ(Nano Drop 1000、サー モフィッシャー・サイエンティフィック)によって280 nm の吸光度を測定し、
A280 nm = 1.25 = 1 mg/mLとして算出した。その後、ソーマチンストック溶液は0.1
M ADA緩衝液(pH 6.5)によって希釈し、20 mg/mLソーマチン溶液を得た。沈
殿剤は、1.6 M酒石酸カリウムナトリウム(和光純薬工業)、50 mM
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid(HEPES)緩衝液(pH 7.0、和光 純薬工業)を用いた。すべての溶液は、調製後に0.45 µmのシリンジフィルター
(Minisart RC25; Sartorius Stedium Biotech)を通した。
2-2-2 マイクロ流体デバイスの作製
マイクロ流体デバイスは、ポリジメチルシロキサン(PDMS)(SILPOT 184 W/C
Dow Corning Toray)を用いて作製した。デバイス作製には以前報告した
Replica-molding法を用いた[9]。また、体積1 nLの液滴を生成する場合には、一
般的なリソグラフィー法でデバイスを作製した[10]。まず、基本形状のチップの 母型となるPMMAチップを微細加工で作製し、これを用いてPDMSマスターを 作製した。続けて、このPDMSマスターを用いてPDMSレプリカを作製し、別 途作製したPDMSプレートとプラズマ処理(160 W, 40 SCCM, 30 s)(CUTE-1 MP、
Femto Science)後、貼り合わせてマイクロ流体デバイスを作製した。また、プラ ズマ処理を行う前に、それぞれのPDMSは100 °Cで2時間以上加熱した。
貼り合わせ後、作製したマイクロ流体デバイスの流路は、trichloro(1H, 1H, 2H, 2H-perfluorooctyl)silane(シグマアルドリッチ)を用いて気相法で表面修飾を施
した[11,12]。表面修飾を施したマイクロ流体デバイスは、フロリナートFC-40(3 M) と1 H, 1 H, 2 H, 2 H- perfuluoro-1-octanol(PFO)(シグマアルドリッチ)の混合液
(10:1、 v/v) を流路内に流入し、3時間ほど50 °Cで乾燥させた。その後、液
滴を回収するためのperfluoroalkoxy alkane (PFA)キャピラリー(Upchurch
Scientific、Zeus)をPDMSデバイスの出口に接続し、PDMSで間隙を塞いだ。
液滴形状を制御するために3種類の内径が異なるPFAキャピラリー(I. D. 130、
200、360 µm)を用いた。また、PDMSデバイスへ送液を行うために、Poly (ether
ether ketone)(PEEK)キャピラリー(Upchurch Scientific)を流路の入口に接続し た。使用したPDMSチップと液滴生成部の拡大図を図2-1に示す。
2-2-2 結晶化実験
マクロスケールの結晶化実験は、マイクロバッチ法によって行った。まず、
10 µLのソーマチン溶液と10 µLの沈殿剤をマイクロチューブで混合した。その
後、混合した結晶化溶液を3 µL分取し、マイクロプレートのウェルにセットし た。結晶化溶液の蒸発を防ぐために、上から80 µLのミネラルオイル(シグマ アルドリッチ)で結晶化溶液を覆った。結晶化実験は4 ºCの低温室で行い、光 学顕微鏡(Eclipse TS100, Nikon)によって観測を行った。液滴を生成するための 連続相(油相)には、フロリナートFC40とPFOの混合液(10:1 v/v)を用いた。
分散相(水相)には、20 mg/mLのソーマチン溶液と1.6 M酒石酸カリウムナト リウム(和光純薬工業)、50 mM HEPES緩衝液(pH 7.0)を用いた。それぞれの 溶液は、ガラスシリンジ(GASTIGHT 1001、Hamilton)に充填し、シリンジポ
ンプ(Model 100、BAS)でPDMSデバイスへ送液した。微小液滴は、PDMSデ
バイスで連続的に生成し、それらを内径が異なるPFAキャピラリーに回収した。
出口に接続しているキャピラリーは、全体に液滴が充填されていることを確認 した後、デバイスから切り離した。その後、切断したキャピラリーは、両端を キャピラリーワックス(Hampton Research)で封止した。結晶化実験は、4 ºCの 低温室で静置することで行った。
Figure 2-1. Photographs of microfluidic chip.
(a) A photograph of PDMS chip. This dimension was easy to form more than 40 nL volume of droplet. (b) An enlarged illustration of Figure 2-1 a. (c) A photograph of PDMS chip. This dimension was easy to form less than 20 nL volume of droplet. (d) An enlarged illustration of Figure 2-1 c.
(a) (b)
400 µm 200 µm
200 µm 200 µm
1 mm
(c) (d)
200 µm 100 µm
100 µm 1 mm
2-3 結果および考察
PDMSデバイスの流路構造と送液量によって、微小液滴の体積は制御するこ とが可能であった。また、微小液滴の形状は回収するキャピラリーの内径によ って制御可能であった。ここで、同体積の液滴を内径が異なるキャピラリーに 回収した場合、内径が小さいキャピラリーでは、液滴の形状が細長くなる。一 方で、内径が大きいキャピラリーに回収した場合には、液滴の形状は楕円状と なる。例として、図2-2 (a)、(b)に50 nLの微小液滴を直径200、360 µmの キャピラリーに回収した写真を示す。微小液滴の外側は、油相として用いたFC40 とPFOの混合液である。これらの溶液は、水分子を全く透過しないので結晶化 実験中に液滴が濃縮されることはない。図2-2から分かるように、(a)および(b)
で体積は同じ50 nLであるが、その形状は大きく異なっていた。これらの微小液 滴でソーマチンの結晶化を行った結果、実験開始後、数時間で2〜3個の結晶が 析出した。図2-2 (c)、(d)に静置後24時間で微小液滴中に析出したソーマチ ン結晶の写真を示す。まず、微小液滴の体積がソーマチンの結晶化に与える影 響について調査した。そこで、PDMSデバイスで1 nL〜70 nLまでの様々な体積 の液滴を生成し、結晶化実験を行った。図2-3に微小液滴の体積と析出した結晶 数の関係を示す。図2-3から分かるように、3種類のチューブ全てにおいて液滴 体積の増加に伴い、液滴内で析出する結晶数が増加する傾向がみられた。そし て、液滴の体積が減少するほど、析出する結晶数のばらつきが小さくなる傾向 がみられた。これは、液滴体積が大きいほど液滴内部に多くのソーマチン分子 を含有している。したがって、液滴内で結晶が1個析出した後も核形成が起こ り得る過飽和度が維持され、さらに結晶が析出したと考えられる。また、液滴 体積が小さいほど核生成、結晶成長時に過飽和度が大きく低下するため、析出 する結晶数が少なくなり、結晶数のばらつきが小さくなると考えられる。一方 で、マイクロバッチ法の場合、結晶化開始から1〜2時間程度で3 µL結晶化液 滴中にソーマチンの結晶が析出することを確認した。図2-4にマイクロバッチ法 での結晶化結果を示す。さらに、24時間静置後には図に示すように結晶化液滴 中に多数のソーマチン結晶が析出していた。
次に、液滴形状がソーマチンの結晶化に与える影響について調査した。その 結果、液滴の体積が30 nL以上の場合は、チューブ内径が小さくなるほど1つの 液滴内で多くの結晶が析出した。また、液滴体積が20 nL以下の場合は、チュー ブ内径によらずにほぼ1個の結晶が析出した。また、真球状の液滴の場合には、
微小液滴中で1個の結晶のみが析出する傾向が示唆された(図2-5)。これらの 結果から、マイクロ空間では液滴の体積、形状を調整することによって、液滴 内で析出する結晶数の制御が可能であると考えられる。これは、図2-4のように 従来のバッチ法などでは困難である。また、析出した結晶の大きさは約60〜150 µmであった。この大きさの結晶であれば、シンクロトロン光のみならず研究室 レベルのX線回折装置でも回折データの取得が可能である。したがって、本技 術は脆くて壊れやすいタンパク質の結晶をダメージレスで簡便に測定可能な手 法になると考えられる。
Figure 2-2. Photographs of 50 nL volume of droplets collected on PFA capillary. (a) The capillary diameter is 200 µm. Droplet shape became elongate. (b) The capillary diameter is 360 µm. The droplet shape became elliptical. (c and d) Thaumatin crystal within a nanodroplet after incubation for 24 hr. Each scale bar is 200 µm.
(b)
(d) (c)
(a)
Figure 2-3. Relationship between droplet volume and number of crystals. The error bar for each plot was calculated from ~ 200 droplets. Each plot represents the number of crystals within a nanodroplet at a capillary diameter of 360 µm (!), 200 µm ("), 130 µm (#), respectively.
Figure 2-4. Thaumatin crystals within a 3 µL droplet (micro batch method)
Figure 2-5. Photographs of spherical droplets collected in PFA capillary. (a) The capillary diameter is 200 µm. (b) The capillary diameter is 360 µm.
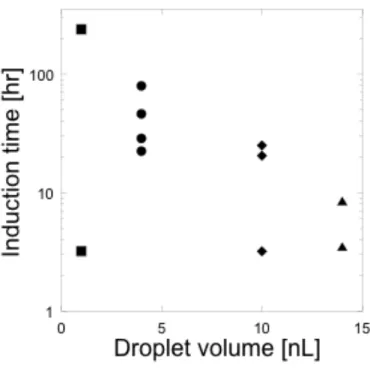
最後に、微小液滴中でのソーマチンの結晶成長過程の解析を行った。バッチ 法では結晶化初期段階で複数の結晶が析出してしまうため、1個の結晶に注目し た晶析挙動の解析は困難であった。しかしながら、タンパク質の結晶の成長機 構を解析するには、1個の結晶に注目して解析を行う必要がある。マイクロ空間 の微小液滴中では、結晶の析出を制御することができるため、図2-6のように微 小液滴中に析出した1個の結晶に注目して解析を行うことが可能であった。図 2-7にソーマチンの結晶成長速度の測定結果を示す。結晶成長速度は、液滴体積 に関わらずに約10-20 µm/hrであった。しかしながら、結晶が析出するまでの時 間である誘導期は液滴体積に大きく依存する傾向が明らかとなった。体積14 nL の液滴の場合、ソーマチンの結晶化が静置後数時間で開始された(図2-7 a)。次 に、液滴体積が10 nLの場合、静置後12時間以内に結晶が確認できる液滴が多 かった。しかしながら、24時間以上の静置で結晶が得られる液滴数がわずかに 増加する傾向があり、誘導期にばらつきが生じはじめた。そこで、体積4 nLの 微小液滴で晶析挙動の解析を行った。結晶成長速度については、他の体積の液 滴とほぼ一定だが、誘導期には大きなばらつきが生じた(図2-7 b)。図2-7(a)
の結果と比較すると誘導期がはるかに長くなっていることが分かる。さらに、
液滴体積が1 nLの場合でもソーマチンの結晶を得ることが可能であった。この 場合には4 ˚Cに静置後、数時間で結晶が析出している液滴を確認したが、静置 後10日以上で結晶が析出する液滴も確認された。図2-8に液滴体積と誘導期の 関係を示す。液滴体積の減少に伴って誘導期が長くなるとともに、その時間に 大きなばらつきが生じている。そこで、誘導期のばらつきの要因について検討 を行った。図2-7(a、b)のプロットのX切片の時間において、液滴中に初めて 臨界核を超える大きさの核が生成していると考えられる。液滴中では臨界半径 以下の不安定な核が生成、溶解を繰り返している。このとき、ある確率で臨界 半径を超える大きさの核が生成する場合がある。それが安定な核となり結晶成 長へと進行する。この核形成、成長を物理化学的に解析する手法の1つとして アブラミの式が用いられている[13,14]。
C = 1 - exp (- k tn) (1)
![Figure 1-2. Phase diagram of protein crystallization. There are four main types of phases: the precipitant zone [P], the labile zone [L], the metastable zone [M], and the unsaturated zone [S]](https://thumb-ap.123doks.com/thumbv2/123deta/9920547.1920725/15.892.269.646.226.622/figure-phase-diagram-protein-crystallization-precipitant-metastable-unsaturated.webp)