タプコム配合点眼液
第 2 部(モジュール 2):CTD の概要(サマリー)
2.5 臨床に関する概括評価
目次
2.5 臨床に関する概括評価... 4 2.5.1 製品開発の根拠... 4 2.5.1.1 申請医薬品の薬理学的分類... 4 2.5.1.2 緑内障及び高眼圧症の臨床的/病態的側面... 5 2.5.1.3 本剤開発の意義... 6 2.5.1.4 臨床開発計画... 7 2.5.1.5 規制当局によるガイダンスや助言... 11 2.5.2 生物薬剤学に関する概括評価... 11 2.5.3 臨床薬理に関する概括評価... 12 2.5.3.1 01111002 試験 添付資料 5.3.3.1-001 ... 12 2.5.3.2 臨床薬理のまとめ... 12 2.5.4 有効性の概括評価... 14 2.5.4.1 有効性を評価した試験... 14 2.5.4.2 個々の試験における有効性の結果... 17 2.5.4.3 全試験を通しての結果の比較と解析... 21 2.5.4.4 用法・用量の検討... 30 2.5.4.5 有効性のまとめ... 32 2.5.5 安全性の概括評価... 33 2.5.5.1 医薬品への曝露... 33 2.5.5.2 有害事象... 36 2.5.5.3 重篤な有害事象及び他の重要な有害事象... 44 2.5.5.4 臨床検査値の評価... 45 2.5.5.5 眼科検査結果の評価... 45 2.5.5.6 安全性のまとめ... 45 2.5.6 ベネフィットとリスクに関する結論... 47 2.5.6.1 ベネフィット... 47 2.5.6.2 リスク... 51 2.5.6.3 結論... 53 2.5.7 参考文献... 55表 2.5.1-1. 臨床に関する概括評価で用いた略語一覧
略語又は用語 略さない表現又は定義
AUCinf Area under the concentration vs. time curve from 0 to infinity(投与後 0-無限 大時間の濃度-時間曲線下面積)
Cmax Maximum concentration(最高濃度)
DE-111 点眼液 有効成分としてタフルプロストを0.0015%、チモロール 0.5%相当量のチ モロールマレイン酸塩を含有する点眼液 X%/Y%DE-111 点 眼液 有効成分としてタフルプロストをX%、チモロール Y%相当量のチモロ ールマレイン酸塩を含有する点眼液。ただし、有効成分としてタフルプ ロストを0.0015%、チモロール 0.5%相当量のチモロールマレイン酸塩を 含有する点眼液は、「DE-111 点眼液」と表記
FAS Full Analysis Set(最大の解析対象集団)
ICH International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use
(日米EU 医薬品規制調和国際会議)
LOCF Last Observation Carry Forward(最終観察値をそれ以降の値に外挿する方 法)
MedDRA Medical Dictionary for Regulatory Activities(ICH で開発された国際医学用 語集)
PG Prostaglandin(プロスタグランジン)
PPS Per Protocol Set(治験実施計画書に適合した解析対象集団) QOL Quality of life(生活の質)
SD Standard Deviation(標準偏差) SE Standard Error(標準誤差) 対照薬 治験において比較の対照として用いられる薬剤 タフルプロスト DE-111 点眼液に含まれる有効成分の一つであり、プロスタグランジン F2α誘導体 タフルプロスト カルボン酸 タフルプロストの活性本体 タフルプロスト 点眼液0.0015% 有効成分としてタフルプロストを0.0015%含有する点眼液。「タプロス® 点眼液0.0015%」等として販売されている 治験薬 治験依頼者から提供される被験薬、対照薬、導入期用タフルプロスト点 眼液0.0015%、導入期用チモロール点眼液 0.5%及びプラセボ点眼液 チモロール点眼 液0.25% 有効成分としてチモロール0.25%相当量のチモロールマレイン酸塩を含 有する点眼液。「チモプトール®点眼液0.25%」等として販売されている
表 2.5.1-1. 臨床に関する概括評価で用いた略語一覧 (続き) 略語又は用語 略さない表現又は定義 チモロール点眼 液0.5% 有効成分としてチモロール0.5%相当量のチモロールマレイン酸塩を含 有する点眼液。「チモプトール®点眼液0.5%」等として販売されている チモロールマレ イン酸塩 DE-111 点眼液に含まれる有効成分の一つであり、β 遮断薬 治療期 治療期開始登録が完了した時点から、本治験で規定されたすべての検 査・観察項目が終了するまでとし、規定した検査・観察項目が実施でき ない場合は最終検査観察までとする。また、被験薬開始日を0 日とする 導入期 同意取得後、治験で規定された検査・観察項目を実施した時点から、治 療期開始登録前までとする トラフ眼圧値 朝点眼前の眼圧値 被験薬 有効性及び安全性評価の対象とされる薬剤(本治験では、DE-111 点眼液) プラセボ点眼液 DE-111 点眼液の基剤点眼液(有効成分を含有しない点眼液) 2.5 臨床に関する概括評価 2.5.1 製品開発の根拠 2.5.1.1 申請医薬品の薬理学的分類 DE-111 点眼液は、緑内障及び高眼圧症の治療を目的とした有効成分としてタフルプロスト を0.0015%、チモロール 0.5%相当量のチモロールマレイン酸塩を含有する配合点眼液である。 タフルプロストは参天製薬株式会社及び旭硝子株式会社により開発されたPGF2α誘導体で あり、日本においては緑内障・高眼圧症治療剤として2008 年 10 月に承認され、同年 12 月よ り「タプロス®点眼液0.0015%」として販売されており、タフルプロストを 0.0015%含有する 点眼液は2012 年 6 月時点で世界 51 ヵ国にて販売されている。また、チモロールマレイン酸 塩を含有する薬剤は、緑内障・高眼圧症治療剤として1978 年にアメリカで発売後、日本にお いては1980 年代より参天製薬株式会社及び MSD 株式会社によって「チモプトール®点眼液 0.25%、0.5%」や「チモプトール®XE 点眼液 0.25%、0.5%」等として販売されている。2010 年10 月時点でチモロールとして 0.25%あるいは 0.5%相当量のチモロールマレイン酸塩を含 有する点眼液は世界80 ヵ国以上、その持続性ゲル化点眼液は世界約 60 ヵ国で発売されてい る。 PGF2α誘導体であるタフルプロストは点眼後、角膜に存在するエステラーゼにより活性本 体であるタフルプロストカルボン酸に速やかに代謝され、プロスタノイドFP 受容体アゴニ ストとして作用し、ぶどう膜強膜流出路からの房水流出を促進することで眼圧下降効果を示
す。また、β 遮断薬であるチモロールは、毛様体に存在する β 受容体を遮断し、毛様体突起 での房水の産生を抑制することによって眼圧下降効果を示す。 2.5.1.2 緑内障及び高眼圧症の臨床的/病態的側面 緑内障は、視神経と視野に特徴的変化を有し、通常、眼圧を十分に下降させることにより 視神経障害を改善もしくは抑制しうる眼の機能的構造的異常を特徴とする疾患と定義されて おり、緑内障の機能的構造的異常の本態は緑内障性視神経症である(1)。緑内障は隅角所見、 眼圧上昇を来たしうる疾患(状況)の有無及び付随する要因により分類することができ、基 本的には、眼圧上昇の原因を他に求めることのできない原発緑内障(原発開放隅角緑内障と 原発閉塞隅角緑内障に大別される)、他の眼疾患や全身疾患あるいは薬物使用が原因となっ て眼圧上昇が生じる続発緑内障、胎生期の隅角発育異常により眼圧上昇をきたす発達緑内障 の3 病型に分類される。なお、原発開放隅角緑内障のうち、眼圧が常に統計学的に規定され た正常値に留まるサブタイプは正常眼圧緑内障と分類されている。一方、高眼圧症は眼圧等 房水動態の点では原発開放隅角緑内障と共通する特徴を有しながら、視神経並びに視野の特 徴的変化を欠く病型であり、原発開放隅角緑内障の前段階、あるいは視神経の眼圧抵抗性の 強い症例と考えられている。 緑内障により視野障害が進行すると、日常生活に支障が生ずるのみでなく、最終的には失 明に至る。2005 年度に報告された厚生労働科学研究(2)によると、眼疾患による失明原因とし ては緑内障が第一位であった。2000 年~2001 年に実施された詳細な緑内障疫学調査である多 治見スタディ(3)(4)において、40 歳以上の日本人における緑内障の有病率は 5.0%(原発開放隅 角緑内障3.9%、原発閉塞隅角緑内障 0.6%、続発緑内障 0.5%)、高眼圧症は 0.8%であった。 緑内障の危険因子には高眼圧、眼循環障害、視神経の脆弱性、家族歴等種々の要因が考え られているが、エビデンスに基づく唯一確実な治療法は眼圧下降であり(5)(6)(7)、眼圧下降剤に よる治療が主体となっている。現在用いられている眼圧下降剤としては、房水産生量を減少 させるβ 遮断薬や炭酸脱水酵素阻害薬、主経路(線維柱帯流出路)からの房水流出量を増加 させる副交感神経作動薬、副経路(ぶどう膜強膜流出路)からの房水流出量を増加させるPG 関連薬やα1遮断薬等がある。日本緑内障学会緑内障診療ガイドライン(第3 版)において眼 圧下降治療における薬物治療の一般的方針としては、第一選択薬として単剤での治療を開始 し、単剤で目標眼圧を達成できない場合に、薬剤の切り替えあるいは作用機序の異なる薬剤 の追加併用が推奨されている。また、多剤併用療法の際には配合点眼薬の使用により患者の アドヒアランスやQOL の向上も考慮すべきと、配合剤の意義についても述べられている。
2.5.1.3 本剤開発の意義 現在、緑内障・高眼圧症の治療にはPG 関連薬や β 遮断薬が第一選択薬として使用されて いるが、単剤ですべての患者に対して十分な眼圧下降を達成できない場合もあり、2 剤以上 の薬剤が併用されている患者も少なくない。2009 年に全国 30 施設で実施された緑内障患者 の治療に関する実態調査(8)によると、単剤で治療されていた患者は48.4%と半数以下であり、 2 剤併用が 24.3%、3 剤併用が 12.8%であった。単剤での治療薬としては、PG 関連薬が 65.6%、 次いでβ 遮断薬類が 30.4%を占めており、β 遮断薬のなかではチモロール類が最も多く使用 されていた。また、2 剤併用される薬剤としては、PG 関連薬を含む組み合わせが中心であり、 PG 関連薬と β 遮断薬の併用が 58.6%と過半数を占めていた。この併用においては点眼回数が 最大で1 日 3 回必要となるが、薬剤数と点眼回数の増加を負担に感じる患者は多い。実際に、 2000 年に実施された緑内障患者へのアンケート調査(9)でも、理想の点眼薬としては「少ない 点眼回数でよいこと」が最上位に挙げられており、患者のニーズは高く、配合点眼液で薬剤 数及び1 日の点眼回数が減ることによって、患者の利便性が向上すると考えられる。また、 緑内障は慢性疾患であり、長期間にわたって眼圧下降剤を継続使用する必要があるが、高齢 者の患者が多く、視野障害が高度に進行した段階や急性緑内障発作を起こしている状態を除 いて明確な自覚症状は乏しい。このため、多剤併用療法が必要な患者において複数の点眼液 をすべて規定どおりに点眼し続けることは容易ではない。規定どおりの点眼回数が守られて いなければ期待した眼圧下降効果が得られず、視野障害の進行を十分抑制できないため、良 好なアドヒアランスが得られやすい薬剤を選択することが重要である。日本緑内障学会緑内 障ガイドライン(第3 版)でも、アドヒアランスを改善するポイントの一つとして、配合点 眼液の利用により最小限の治療とすることなどが述べられている。 2 剤以上の点眼液を同一時間帯に併用する場合、先に点眼された点眼液の有効成分が眼表 面から吸収あるいは消失する前に次の点眼を行うと、先の点眼液の有効成分が希釈されたり 洗い流されたりする〔洗い流し効果(ウォッシュアウト)〕ために、薬効の減弱が懸念され る。非臨床試験で、正常眼圧サルにチモロール点眼液0.5%とタフルプロスト点眼液 0.0015% を間隔をあけずに連続点眼した結果、DE-111 点眼液より眼圧下降効果が有意に減弱すること が示された〔2.6.2.2.1 (3)〕。またラットにおいて、チモロール点眼液 0.5%とタフルプロスト 点眼液0.0015%を連続点眼した結果、チモロールの房水中濃度は、5 分間隔をあけて点眼し た場合より低下した〔2.6.4.4.1 (2)〕。このような薬効の減弱への懸念から、点眼液の添付文 書では適用上の注意として「他の点眼剤と併用する場合には、少なくとも5 分間以上の間隔 をあけて点眼すること。」等の記載があるように、臨床現場では併用に関して患者への特別 な点眼指導が必要であり、同時に服用が可能な内服と異なり点眼液の併用は患者への負担が 大きい。実際に、30%近くの患者が 1 分以内に 2 剤を点眼していたとの報告(10)もあり、洗い 流し効果による薬効の減弱が懸念される。また、点眼剤で防腐剤として汎用されているベン ザルコニウム塩化物は、眼表面の角膜細胞や結膜細胞への障害性を有することが報告されて おり(11)、長期間にわたって眼圧下降剤を継続使用する場合に、多剤併用療法は単剤治療と比
較して角結膜がベンザルコニウム塩化物に曝露される機会が増加し、角結膜上皮障害の出現 頻度が高くなることが懸念される。配合点眼液の利用で1 日 1 回の点眼治療にすることによ り、ベンザルコニウム塩化物の1 日あたりの曝露量を大幅に軽減することが可能であり、眼 表面の安全性を向上させることによる臨床的メリットも期待される。 有効性に関して、PG 関連薬と β 遮断薬の組み合わせによる 1 日 1 回点眼の配合点眼液は、 β 遮断薬の点眼回数が 1 日 2 回から 1 日 1 回に減少するため、PG 関連薬 1 日 1 回点眼と β 遮 断薬1 日 2 回点眼の併用と比べて効果減弱の懸念がある。実際に、ラタノプロスト点眼液 0.005%1 日 1 回点眼とチモロール点眼液 0.5%1 日 2 回点眼の併用と、ラタノプロストを 0.0015%、チモロール 0.5%相当量のチモロールマレイン酸塩を含有する配合点眼液 1 日 1 回 点眼の眼圧下降効果を12 週間クロスオーバーで比較した試験において、ラタノプロスト点眼 液0.005%とチモロール点眼液 0.5%の併用が配合点眼液よりも 1 mmHg 以上強い眼圧下降効 果を示したとの報告(12)もあり、単剤併用に比して効果減弱のない配合点眼液が望ましい。 DE-111 点眼液は、臨床での使用頻度が最も高い PG 関連薬と β 遮断薬の組み合わせとなる タフルプロストを0.0015%及びチモロール 0.5%相当量のチモロールマレイン酸塩を含有する 配合点眼液である。製剤設計の工夫として、両有効成分の有効性を十分発揮できるよう、タ フルプロスト及びチモロールの 低下させることなく、室温保 存可能な安定性を担保できる を選択した。利便性向上、アドヒアランス及びQOL の 改善が期待でき、緑内障の薬物療法改善に寄与できるものと考える。 2.5.1.4 臨床開発計画 2.5.1.4.1 臨床データパッケージ DE-111 点眼液の臨床データパッケージを表 2.5.1.4-1-1 に示す。 DE-111 点眼液の臨床試験は、DE-111 点眼液の健康成人男性を対象とした臨床薬理試験- 第Ⅰ相-(以下、01111002 試験)、DE-111 点眼液の原発開放隅角緑内障又は高眼圧症を対 象としたタフルプロスト点眼液及びタフルプロスト点眼液/チモロール点眼液併用との二重 盲検比較試験-第Ⅲ相、検証的試験-(以下、01111004 試験)、DE-111 点眼液の原発開放 隅角緑内障又は高眼圧症を対象としたチモロール点眼液との二重盲検比較試験-第Ⅲ相、検 証的試験-(以下、01111005 試験)及び DE-111 点眼液の開放隅角緑内障又は高眼圧症を対 象としたオープンラベルによる長期投与試験-第Ⅲ相-(以下、01111006 試験)の 4 試験で あり、いずれも日本国内において実施した。 01111002 試験は、健康成人男性を対象に DE-111 点眼液の各配合成分との安全性及び体内 薬物動態を比較検討する目的で、タフルプロスト点眼液0.0015%、チモロール点眼液 0.5%(1 日2 回点眼)及びタフルプロスト点眼液 0.0015%とチモロール点眼液 0.5%(1 日 2 回点眼) の併用を対照とし、無作為化クロスオーバー評価者盲検試験を実施した。本試験の結果、有
効成分としてタフルプロストを0.0015%、チモロール 0.5%相当量のチモロールマレイン酸塩 を含有するDE-111 点眼液は、各配合成分との比較において安全性に問題なく、配合による 薬物動態学的相互作用はないものと考えられ、第Ⅲ相試験に移行した。 01111004 試験は、原発開放隅角緑内障又は高眼圧症患者を対象に、DE-111 点眼液の配合 成分の一つであるタフルプロスト点眼液0.0015%との優越性を検証すること、及びタフルプ ロスト点眼液0.0015%とチモロール点眼液 0.5%(1 日 2 回点眼)の併用との非劣性を検証す る目的で、二重盲検並行群間比較試験にて安全性を含め比較検討した。タフルプロスト点眼 液0.0015%を 4 週間投与する導入期後に、眼圧 18 mmHg 以上の患者を DE-111 点眼液、タフ ルプロスト点眼液0.0015%、又はタフルプロスト点眼液 0.0015%とチモロール点眼液 0.5%(1 日2 回点眼)の併用のいずれかの投与群に無作為割付けし、4 週間投与の治療期を設定した。 平均日中眼圧は1 日 3 回(朝点眼前、点眼 2 時間後、点眼 8 時間後)の測定とし、治療期 0 週及び4 週に行った。この結果、DE-111 点眼液はタフルプロスト点眼液 0.0015%に対する優 越性及びタフルプロスト点眼液0.0015%とチモロール点眼液 0.5%(1 日 2 回点眼)の併用と の非劣性が検証され、安全性に問題はなかった。 01111005 試験は、原発開放隅角緑内障又は高眼圧症患者を対象に、DE-111 点眼液の配合 成分の一つであるチモロール点眼液0.5%(1 日 2 回点眼)との優越性を検証する目的で、二 重盲検並行群間比較試験にて安全性を含め比較検討した。チモロール点眼液0.5%を 4 週間投 与する導入期後に、眼圧20 mmHg 以上の患者を DE-111 点眼液、又はチモロール点眼液 0.5% (1 日 2 回点眼)のいずれかの投与群に無作為割付けし、4 週間投与の治療期を設定した。平 均日中眼圧は1 日 3 回(朝点眼前、点眼 2 時間後、点眼 8 時間後)の測定とし、治療期 0 週 及び4 週に行った。この結果、DE-111 点眼液はチモロール点眼液 0.5%(1 日 2 回点眼)に対 する優越性が検証され、安全性に問題はなかった。 01111006 試験は、開放隅角緑内障又は高眼圧症患者を対象に、オープンラベル試験にて、 安全性及び眼圧下降効果を検討した。開放隅角緑内障又は高眼圧症患者をタフルプロスト点 眼液0.0015%、チモロール点眼液 0.5%(1 日 2 回点眼)、又はタフルプロスト点眼液 0.0015% とチモロール点眼液0.5%(1 日 2 回点眼)の併用のいずれかに無作為割付けし、4 週間投与 する導入期を設定した。その後、それぞれの投与群からDE-111 点眼液に切り替え治療期と して52 週投与した。この結果、DE-111 点眼液は安定かつ良好な眼圧下降効果を示し、DE-111 点眼液の安全性に問題はなかった。 以上の試験成績より、DE-111 点眼液の有効性及び安全性が確認されたため、製造販売承認 申請することとした。
表 2.5.1.4-1-1. 臨床データパッケージ 試験名 (治験実施計画書番号) 試験の 目的 デザイン 被験薬・対照薬 対象 目標 症例数 投与方法 投与 期間 総 括 報 告 書 作 成 状 況 DE-111 点眼液の健康成 人男性を対象とした臨 床薬理試験-第Ⅰ相- (01111002) 薬物動 態 忍容性 クロスオ ーバー評 価者盲検 試験 ①DE-111 点眼液 ②タフルプロス ト点眼液 0.0015% ③チモロール点 眼液0.5% ④タフルプロス ト点眼液 0.0015%とチモ ロール点眼液 0.5%併用 健康成 人男性 32 例 ①1 日 1 回 ②1 日 1 回 ③1 日 2 回 ④1 日 1 回 +1 日 2 回 点眼 期 第Ⅰ 期 7 日 + 点眼 期 第Ⅱ 期 7 日 完 了 DE-111 点眼液の原発開 放隅角緑内障又は高眼 圧症を対象としたタフ ルプロスト点眼液及び タフルプロスト点眼液 /チモロール点眼液併 用との二重盲検比較試 験-第Ⅲ相、検証的試験 - (01111004) 有効性 検証試 験(実薬 対照) 無作為化 二重盲検 比較試験 ①DE-111 点眼液 ②タフルプロス ト点眼液 0.0015% ③タフルプロス ト点眼液 0.0015%とチモ ロール点眼液 0.5%併用 原発開 放隅角 緑内障 高眼圧 症 480 例 ①1 日 1 回 ②1 日 1 回 ③1 日 1 回 +1 日 2 回 導入 期 4 週 + 治療 期 4 週 完 了
表 2.5.1.4-1-1. 臨床データパッケージ (続き) 試験名 (治験実施計画書番号) 試験の 目的 デザイン 被験薬・対照薬 対象 目標 症例数 投与方法 投与 期間 総 括 報 告 書 作 成 状 況 DE-111 点眼液の原発開 放隅角緑内障又は高眼 圧症を対象としたチモ ロール点眼液との二重 盲検比較試験-第Ⅲ相、 検証的試験- (01111005) 有効性 検証試 験(実薬 対照) 無作為化 二重盲検 比較試験 ①DE-111 点眼液 ②チモロール点 眼液0.5% 原発開 放隅角 緑内障 高眼圧 症 140 例 ①1 日 1 回 ②1 日 2 回 導入 期 4 週 + 治療 期 4 週 完 了 DE-111 点眼液の開放隅 角緑内障又は高眼圧症 を対象としたオープン ラベルによる長期投与 試験-第Ⅲ相- (01111006) 長期安 全性 オープン ラベル試 験 DE-111 点眼液 開放隅 角緑内 障 落屑緑 内障 色素緑 内障 高眼圧 症 126 例 1 日 1 回 導入 期 4 週 + 治療 期 52 週 完 了 2.5.1.4.2 外国臨床データ 外国においては参天製薬株式会社の子会社であるSanten Oy が、DE-111 点眼液と同様に有 効成分としてタフルプロストを0.0015%、チモロール 0.5%相当量のチモロールマレイン酸塩 を含有した防腐剤非含有製剤であるDE-111A 点眼液の開発を継続中である。
2.5.1.5 規制当局によるガイダンスや助言 DE-111 点眼液の臨床データパッケージは、独立行政法人医薬品医療機器総合機構の医薬品 相談( 年 月 日申込)にて、 年 月 日に開催された対面 助言での指導・助言に従って構成した。 対面助言における指導・助言の主なポイントを以下 (1) ~ (6) に示す。 (1) (2) (3) (4) (5) (6) 2.5.2 生物薬剤学に関する概括評価 該当しない。
2.5.3 臨床薬理に関する概括評価 DE-111 点眼液の薬物動態を確認する目的で、健康成人男性を対象とした第Ⅰ相試験を日本 国内にて1 試験実施した。 2.5.3.1 01111002 試験 添付資料 5.3.3.1-001 第Ⅰ相試験では、健康成人男性32 例を対象として DE-111 点眼液を 1 回 1 滴、1 日 1 回、7 日間、両眼点眼した場合の体内薬物動態について、タフルプロスト点眼液0.0015%(1 回 1 滴、1 日 1 回)、チモロール点眼液 0.5%(1 回 1 滴、1 日 2 回)、及びタフルプロスト点眼 液0.0015%(1 回 1 滴、1 日 1 回)とチモロール点眼液 0.5%(1 回 1 滴、1 日 2 回)の併用に おける7 日間、両眼点眼と比較検討した。 この結果、血漿中タフルプロストカルボン酸濃度は、DE-111 点眼液、タフルプロスト点眼 液0.0015%、タフルプロスト点眼液 0.0015%とチモロール点眼液 0.5%の併用のいずれにおい ても、点眼後0.083~0.25 時間に Cmaxを示し、その後速やかに消失して、点眼後55 分以降に は定量下限界 (0.01 ng/mL) 未満であった。DE-111 点眼液において Cmax(平均±標準偏差)は、 反復点眼1 日目及び 7 日目で、0.02480±0.00537 及び 0.02223±0.01267 ng/mL、タフルプロス ト点眼液0.0015%では 0.02493±0.00946 及び 0.02487±0.00770 ng/mL、タフルプロスト点眼液 0.0015%とチモロール点眼液 0.5%の併用では 0.03321±0.04132 及び 0.02481±0.00929 ng/mL で あった。 血漿中チモロール濃度は、DE-111 点眼液、チモロール点眼液 0.5%、タフルプロスト点眼 液0.0015%とチモロール点眼液 0.5%の併用のいずれにおいても、点眼後 0.117~2 時間に Cmax を示した。DE-111 点眼液において、Cmax(平均±標準偏差)は、反復点眼 1 日目及び 7 日目 で1.409±0.344 及び 1.293±0.551 ng/mL、チモロール点眼液 0.5%では 1.353±0.719 及び 1.544±0.690 ng/mL、タフルプロスト点眼液 0.0015%とチモロール点眼液 0.5%の併用では 1.572±0.663 及び 1.762±0.698 ng/mL となった。平均 AUCinfは、反復点眼1 日目及び 7 日目で、 DE-111 点眼液では 6.766±1.888 及び 6.449±2.774 ng・hr/mL、チモロール点眼液 0.5%では 6.348±3.004 及び 7.796±2.775 ng・hr/mL、タフルプロスト点眼液 0.0015%とチモロール点眼液 0.5%の併用では 7.361±2.560 及び 8.403±2.365 ng・hr/mL であった。 2.5.3.2 臨床薬理のまとめ 01111002 試験における体内薬物動態の検討では、すべての点眼液において血漿中タフルプ ロストカルボン酸、チモロールともに反復点眼による体内動態への影響は認められなかった。 また、DE-111 点眼液の血漿中タフルプロストカルボン酸及びチモロール濃度は、タフルプロ
スト点眼液0.0015%、チモロール点眼液 0.5%、及びタフルプロスト点眼液 0.0015%とチモロ ール点眼液0.5%の併用と比較して差異が認められなかったことから、配合による薬物動態学 的相互作用はないと考えられた。
2.5.4 有効性の概括評価 2.5.4.1 有効性を評価した試験 有効性を評価した試験の一覧を表 2.5.4.1-1 に示した。 DE-111 点眼液の緑内障及び高眼圧症に対する有効性は、導入期にタフルプロスト点眼液 0.0015%を点眼し、タフルプロスト点眼液 0.0015%及びタフルプロスト点眼液 0.0015%とチモ ロール点眼液0.5%(1 日 2 回点眼)の併用を対照とした 01111004 試験、導入期にチモロー ル点眼液0.5%を点眼し、チモロール点眼液 0.5%を対照とした 01111005 試験、並びに長期投 与試験である01111006 試験により評価した。 表 2.5.4.1-1. 有効性を評価した試験の一覧 デザイン 試験 (相) 試験目的 対象 導入期 (投与期間、導入 期用点眼液) 治療期 (投与期間、被験 薬・対照薬) 比較対照試験 (二重盲検群 間比較試験) 01111004 試験 (第Ⅲ相) タフルプロストに 対する優越性 原発開放隅角緑内 障、高眼圧症 FAS:487 例 PPS:474 例 4 週間 ・タフルプロスト 4 週間 ・DE-111 ・タフルプロスト ・併用 併用に対する非劣 性 01111005 試験 (第Ⅲ相) チモロールに対す る優越性 原発開放隅角緑内 障、高眼圧症 FAS:166 例 PPS:154 例 4 週間 ・チモロール 4 週間 ・DE-111 ・チモロール 非対照試験 (オープン試 験) 01111006 試験 (第Ⅲ相) 長期投与における 有効性 原発開放隅角緑内 障(広義)(原発 開放隅角緑内障と 正常眼圧緑内障)、 落屑緑内障、色素 緑内障、高眼圧症 :136 例 4 週間 ・タフルプロスト ・チモロール ・併用 52 週間 ・DE-111 2.5.4.1.1 有効性の評価方法 緑内障治療における真のエンドポイントは、視野障害の進行防止にあるが、その評価は長 期にわたるため、真のエンドポイントを用いることは現実的ではない。日本緑内障学会緑内 障診療ガイドライン(第3 版)(1)に「現在、緑内障に対するエビデンスに基づいた唯一確実 な治療法は眼圧を下降することである」とされているように、臨床現場における緑内障に対 する治療は眼圧下降を目的としている。また、現在までに製造販売承認を得ている緑内障治
療薬は、すべて眼圧を主たるエンドポイントとして評価されていることからも、有効性のエ ンドポイントとして眼圧を用いた。 緑内障治療では、眼圧を1 日中コントロールすることが重要であることから(13)(14)(15)、 01111004 試験及び 01111005 試験では、眼圧の日内変動の確認を行うために日中に複数の眼 圧測定時点を設定し、0 週からの平均日中眼圧変化量を主要評価項目とした。測定時点は、 投与眼圧の眼圧下降効果が最も弱いトラフ眼圧値(朝点眼前)に加え、DE-111 点眼液の有効 成分(タフルプロスト及びチモロールマレイン酸塩)の点眼後の眼圧下降効果のピーク時間 を考慮して(16)(17)(18)、点眼2 時間後及び点眼 8 時間後を設定した。01111006 試験では、トラ フ眼圧値(朝点眼前)の0 週及び-4 週からの眼圧変化量及び眼圧変化率を評価した。 01111004 試験及び 01111005 試験では、有効性解析対象集団は最大の解析対象集団 (FAS) を採用した。また、治験実施計画書に適合した解析対象集団 (PPS) についても解析し、FAS との相違について考察した。なお、有効性の概括評価 (2.5.4) は FAS の結果を用いて記述し た。 2.5.4.1.2 対象患者 比較試験(01111004 試験、01111005 試験)では、原発開放隅角緑内障及び高眼圧症を対象 とした。また、配合点眼液は単剤で目標眼圧を達成できない場合における使用が想定される ことから、タフルプロスト点眼液0.0015%又はチモロール点眼液 0.5%の使用においても一定 以上の眼圧値を示す患者を対象とした。 長期投与試験(01111006 試験)では、正常眼圧緑内障が多い日本での実態をふまえ、対象 患者としてより幅広く、開放隅角緑内障(正常眼圧緑内障を含めた広義の原発開放隅角緑内 障、色素緑内障、落屑緑内障)及び高眼圧症を対象とした。 患者の年齢についても、当該疾患は高齢者に多く認められることから、下限は日本におけ る成人年齢とし、上限は設定しなかった。 2.5.4.1.3 試験デザイン 2.5.4.1.3.1 対照薬 DE-111 点眼液は、有効成分としてタフルプロストを 0.0015%、チモロール 0.5%相当量の チモロールマレイン酸塩を含有する配合点眼液であり、各有効成分の単剤からの切り替えが 想定される。このことから、タフルプロスト点眼液0.0015%及びチモロール点眼液 0.5%に対 する眼圧下降効果の優越性を検証することを目的に、各単剤を対照とした無作為化二重盲検 比較試験(01111004 試験、01111005 試験)を設定した。
また、DE-111 点眼液は、タフルプロスト点眼液 0.0015%及びチモロール点眼液 0.5%の併 用からの切り替えも想定されるため、併用との非劣性を検証することとした。なお、PG 関 連薬が1 日 1 回点眼であるのに対し、β 遮断薬のチモロール点眼液 0.5%は 1 日 2 回点眼であ ることから、これらを有効成分とする配合点眼液を1 日 1 回点眼とした場合、β 遮断薬の投 与回数減少による眼圧下降効果減弱が懸念される。実際に、PG 関連薬と β 遮断薬の既承認 配合点眼液では併用より眼圧下降効果が減弱するとの報告(12)もあり、併用との非劣性の検証 は意義あるものと考えた。この併用を対照とした非劣性試験は、タフルプロスト点眼液 0.0015%と同時比較することにより、タフルプロスト点眼液 0.0015%とチモロール点眼液 0.5% の併用の有効性について分析感度が確認できることから、併用を対照とした非劣性試験はタ フルプロスト点眼液0.0015%対照の比較試験と同一試験(01111004 試験)で実施することと した。非劣性マージンは、各配合成分であるPG 関連薬及び β 遮断薬のプラセボに対する眼 圧下降効果(PG 関連薬:-6.5~-8.4 mmHg、β 遮断薬:-5.2~-6.9 mmHg、プラセボ:-1.3~-1.6 mmHg)(19)の1/2 以下を満たし、かつ他剤眼圧下降薬の臨床試験で非劣性マージンとして設 定頻度の多い1.5 mmHg とした。 2.5.4.1.3.2 導入期 DE-111 点眼液は、有効成分としてタフルプロストを 0.0015%、チモロール 0.5%相当量の チモロールマレイン酸塩を含有する配合点眼液であり、タフルプロスト点眼液0.0015%とチ モロール点眼液0.5%の各単剤で効果不十分、あるいはタフルプロスト点眼液 0.0015%とチモ ロール点眼液0.5%の併用でアドヒアランス及び QOL の向上が望まれる患者への使用が想定 される。このため、タフルプロスト点眼液0.0015%及びタフルプロスト点眼液 0.0015%とチ モロール点眼液0.5%(1 日 2 回点眼)の併用を対照とした 01111004 試験では、タフルプロ スト点眼液0.0015%を点眼する導入期を設定した。チモロール点眼液 0.5%を対照とした 01111005 試験では、チモロール点眼液 0.5%を点眼する導入期を設定した。01111006 試験で は、タフルプロスト点眼液0.0015%、チモロール点眼液 0.5%、及びタフルプロスト点眼液 0.0015%とチモロール点眼液 0.5%(1 日 2 回点眼)の併用のいずれかを点眼する導入期を設 定した。また、各導入期薬剤別の眼圧下降効果に関する検討が可能と考えられる例数を確保 するために、1:1:1 の割合で無作為化し、非盲検で実施した。 導入期間は、前治療薬の影響を消失させ、導入期用点眼液の有効性が安定する期間として、 4 週間と設定した。 2.5.4.1.3.3 治療期 01111004 試験では、治療期開始時に DE-111 点眼液、タフルプロスト点眼液 0.0015%、又 はタフルプロスト点眼液0.0015%とチモロール点眼液 0.5%(1 日 2 回点眼)の併用のいずれ かに無作為に割付け、二重盲検下で実施した。01111005 試験では、治療期開始時に DE-111
点眼液又はチモロール点眼液0.5%(1 日 2 回点眼)のいずれかに無作為に割付け、二重盲検 下で実施した。01111006 試験では、治療期に DE-111 点眼液を点眼した。 01111004 試験及び 01111005 試験における治療期間は、これまでに実施されたタフルプロ スト点眼液0.0015%及びチモロール点眼液 0.5%の臨床試験の結果から、両点眼液の眼圧下降 効果が安定する4 週間の治療期を設定した(20)(21)。また、01111006 試験では、長期投与におけ る検討を行うため、ICH-E1 ガイドライン「致命的でない疾患に対し長期間の投与が想定され る新医薬品の治験段階において安全性を評価するために必要な症例数と投与期間」(1995 年) に準じて52 週間の治療期を設定した。 2.5.4.1.3.4 症例数 タフルプロスト点眼液0.0015%及びタフルプロスト点眼液 0.0015%とチモロール点眼液 0.5%の併用を対照とした 01111004 試験では、DE-111 点眼液とタフルプロスト点眼液 0.0015% との期待される眼圧変化量の差を1.5 mmHg、標準偏差を 4 mmHg とし、有意水準を 5%、t 検定を用いた検出力を90%とするとき、1 群の必要例数は 151 例となった。また、DE-111 点 眼液とタフルプロスト点眼液0.0015%とチモロール点眼液 0.5%の併用との期待される眼圧変 化量の差を0 mmHg、非劣性マージンを 1.5 mmHg、標準偏差を 4 mmHg とし、有意水準を 5%、t 検定を用いた検出力を 90%とするとき、1 群の必要例数は 151 例となったため、目標 症例数を1 群 160 例と設定した。 チモロール点眼液0.5%を対照とした 01111005 試験では、DE-111 点眼液とチモロール点眼 液0.5%との期待される眼圧変化量の差を 2.0 mmHg、標準偏差を 4.0 mmHg とし、有意水準 を5%、t 検定を用いた検出力を 80%とするとき、1 群の必要例数は 64 例となったため、目標 症例数を1 群 70 例と設定した。 01111006 試験では、ICH-E1 ガイドライン「致命的でない疾患に対し長期間の投与が想定 される新医薬品の治験段階において安全性を評価するために必要な症例数と投与期間」(1995 年)を満たす例数として、52 週点眼例を 100 例以上と設定し、中止率約 20%を考慮して目標 症例数を126 例と設定した。 2.5.4.2 個々の試験における有効性の結果 2.5.4.2.1 01111004 試験〔タフルプロスト点眼液 0.0015%及びタフルプロスト点眼 液0.0015%とチモロール点眼液 0.5%(1 日 2 回点眼)の併用を対照とし た試験〕 添付資料5.3.5.1-001 原発開放隅角緑内障又は高眼圧症患者における眼圧下降効果を指標として、DE-111 点眼液 (1 回 1 滴、1 日 1 回、4 週間両眼点眼)の、タフルプロスト点眼液 0.0015%(1 回 1 滴、1
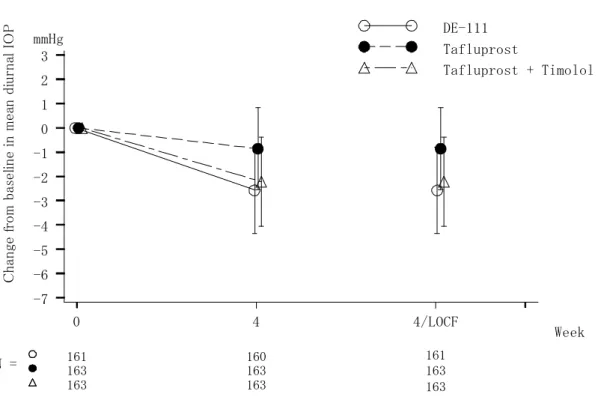
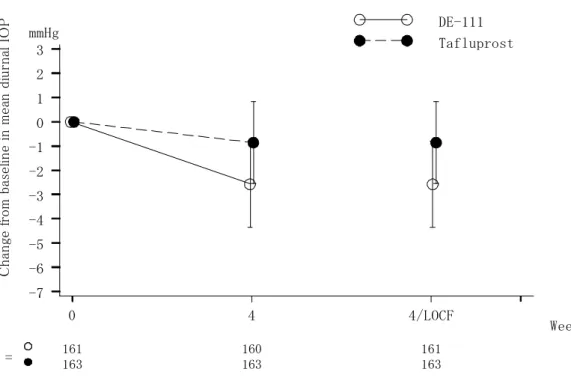
日1 回、4 週間両眼点眼)との優越性、タフルプロスト点眼液 0.0015%(1 回 1 滴、1 日 1 回、 4 週間両眼点眼)とチモロール点眼液 0.5%(1 回 1 滴、1 日 2 回、4 週間両眼点眼)の併用と の非劣性を、多施設共同無作為化二重盲検並行群間比較試験により検証した。また、安全性 についても比較検討した。 原発開放隅角緑内障又は高眼圧症患者のうち、導入期としてタフルプロスト点眼液 0.0015%を 4 週間点眼後の眼圧が 18 mmHg 以上の被験者を、置換ブロック法を用いて DE-111 群、タフルプロスト群、又は併用群に1:1:1 の割合で無作為に割付け、治療期 4 週間を二重 盲検下で実施した。 文書同意を得た症例574 例のうち、558 例に導入期のタフルプロスト点眼液 0.0015%が点 眼され、このうち489 例が無作為化割付けされ(DE-111 群 161 例、タフルプロスト群 164 例、併用群164 例)、治療期に移行した。治療期用治験薬が点眼された 488 例(DE-111 群 161 例、タフルプロスト群 164 例、併用群 163 例)のうち、0 週の眼圧測定値が 1 日分(3 時 点)利用できなかった1 例を除外した 487 例(DE-111 群 161 例、タフルプロスト群 163 例、 併用群163 例)を有効性解析対象集団 (FAS) とした。 主要評価項目である、治療期終了時(4 週又は中止時)における 0 週からの平均日中眼圧 変化量を図 2.5.4.2-1 に示した。平均日中眼圧変化量(平均値±標準偏差)は、DE-111 群で -2.6±1.8 mmHg、タフルプロスト群で-0.9±1.7 mmHg、併用群で-2.2±1.8 mmHg であった。 DE-111 点眼液とタフルプロスト点眼液 0.0015%との群間差(DE-111 群-タフルプロスト群、 平均値±標準誤差)は-1.7±0.2 mmHg、95%信頼区間は-2.1~-1.3 mmHg であり、DE-111 点眼 液のタフルプロスト点眼液0.0015%に対する優越性が検証された (P<0.001) 。また、タフル プロスト点眼液0.0015%とチモロール点眼液 0.5%の併用との群間差(DE-111 群-併用群、平 均値±標準誤差)は-0.3±0.2 mmHg、95%信頼区間は-0.7~0.1 mmHg であり、その上限値は事 前に規定した非劣性マージン1.5 mmHg を超えず、DE-111 点眼液のタフルプロスト点眼液 0.0015%とチモロール点眼液 0.5%(1 日 2 回点眼)の併用に対する非劣性が検証された。
図 2.5.4.2-1. 平均日中眼圧変化量:01111004 試験 2.5.4.2.2 01111005 試験(チモロール点眼液 0.5%を対照とした試験) 添付資料 5.3.5.1-002 原発開放隅角緑内障又は高眼圧症患者における眼圧下降効果を指標として、DE-111 点眼液 (1 回 1 滴、1 日 1 回、4 週間両眼点眼)の、チモロール点眼液 0.5%(1 回 1 滴、1 日 2 回、 4 週間両眼点眼)との優越性を、多施設共同無作為化二重盲検並行群間比較試験により検証 した。また、安全性についても比較検討した。 原発開放隅角緑内障又は高眼圧症患者のうち、導入期としてチモロール点眼液0.5%を 4 週 間点眼後の眼圧が20 mmHg 以上の被験者を、置換ブロック法を用いて DE-111 群又はチモロ ール点眼液0.5%のいずれかに 1:1 の割合で無作為に割付け、治療期 4 週間をダブルダミー法 を用いた二重盲検下で実施した。 文書同意を得た症例203 例のうち、188 例に導入期のチモロール点眼液 0.5%が点眼され、 治療期に移行した166 例(DE-111 群 82 例、チモロール群 84 例)を、有効性解析対象集団 (FAS) とした。 主要評価項目である、治療期終了時(4 週又は中止時)における 0 週からの平均日中眼圧 変化量を図 2.5.4.2-2 に示した。治療期終了時(4 週又は中止時)における 0 週からの平均日 中眼圧変化量(平均値±標準偏差)は、DE-111 点眼液で-3.2±2.1 mmHg、チモロール点眼液 0.5%で-1.7±2.1 mmHg であり、DE-111 点眼液とチモロール点眼液 0.5%との群間差(DE-111 C h an ge f ro m b as el in e in m ea n d iu rn al I O P -7 -6 -5 -4 -3 -2 -1 0 1 2 3 Week mmHg N = DE-111 Tafluprost Tafluprost + Timolol 161 160 163 163 163 163 0 4 4/LOCF 161 163 163
群-チモロール群、平均値±標準誤差)は-1.5±0.3 mmHg、95%信頼区間は-2.2~-0.9 mmHg で あり、DE-111 点眼液のチモロール点眼液 0.5%に対する優越性が検証された (P<0.001) 。 図 2.5.4.2-2. 平均日中眼圧変化量:01111005 試験 2.5.4.2.3 01111006 試験(長期投与試験) 添付資料 5.3.5.2-002 開放隅角緑内障又は高眼圧症患者におけるDE-111 点眼液(1 回 1 滴、1 日 1 回、52 週間両 眼点眼)の安全性及び眼圧下降効果をオープンラベルによる多施設共同試験により検討した。 原発開放隅角緑内障(広義)(原発開放隅角緑内障又は正常眼圧緑内障)、落屑緑内障、 色素緑内障又は高眼圧症患者を対象に、タフルプロスト点眼液0.0015%、チモロール点眼液 0.5%、又はタフルプロスト点眼液 0.0015%とチモロール点眼液 0. 5%の併用に 1:1:1 で無作為 に割付け、導入期4 週間をオープンラベル下で実施した後、治療期として DE-111 点眼液を 52 週間点眼した。 文書同意を得た症例162 例のうち、148 例が無作為割付け(タフルプロスト点眼液 0.0015% 51 例、チモロール点眼液 0.5% 49 例、タフルプロスト点眼液 0.0015%+チモロール点眼液 0.5% 48 例)された。治療期に移行し、DE-111 点眼液を点眼した 136 例を、有効性解析対象集団 とした。 眼圧値は0 週と比較し、4 週から有意に下降し、52 週までその効果は持続した。また、導 入期点眼液別では、タフルプロスト点眼液0.0015%又はチモロール点眼液 0.5%から DE-111 C h an ge f ro m b as el in e in m ea n d iu rn al I O P -7 -6 -5 -4 -3 -2 -1 0 1 2 3 Week mmHg N = DE-111 Timolol 82 79 84 83 0 4 4/LOCF 82 84
点眼液への切り替えでは、眼圧値は0 週と比較し 4 週から有意な眼圧下降を示し、52 週まで その効果は持続した。タフルプロスト点眼液0.0015%とチモロール点眼液 0.5%の併用から DE-111 点眼液への切り替えでは、すべての測定時期において、0 週と比較し眼圧値の有意な 変化は認められず、安定した眼圧下降効果を示した。 2.5.4.3 全試験を通しての結果の比較と解析 2.5.4.3.1 試験対象集団 2.5.4.3.1.1 被験者背景 有効性を評価したすべての試験で、緑内障及び高眼圧症を対象とした。01111004 試験及び 01111005 試験では、原発開放隅角緑内障及び高眼圧症が、約半数ずつ組入れられた。01111006 試験では、原発開放隅角緑内障及び高眼圧症に加え、正常眼圧緑内障が35.3%組入れられた。 平均年齢は、61.7~64.0 歳であり、高齢者(65 歳以上)の割合は、46.6~54.4%と約半数を 占めた。緑内障前治療薬ありの割合は、80.7~85.3%と 80%以上を占めた。 2.5.4.3.1.2 有効性の評価対象集団と市販後に使用が予想される患者集団との差異 緑内障は高齢者で多く認められている疾患であり、今回実施した試験はいずれも高齢者が 約半数組入れられた。また、比較試験(01111004 試験、01111005 試験)での原発開放隅角緑 内障及び高眼圧症に加え、長期投与試験(01111006 試験)では、日本人に多い正常眼圧緑内 障も組入れられており、市販後に使用が予想される患者集団との差異はないと考えられた。 2011 年に、日本緑内障学会緑内障診療ガイドラ イン(第3 版)に改訂され、配合点眼液に関する記載が追記された。この中で、配合点眼液 は、単剤で目標眼圧が達成されない場合や、多剤併用療法時のアドヒアランス及びQOL 向 上のために使用されることが記載されており、DE-111 点眼液の臨床試験においても、タフル プロスト点眼液0.0015%又はチモロール点眼液 0.5%単剤で一定以上の眼圧を示す患者や、タ フルプロスト点眼液0.0015%とチモロール点眼液 0.5%の併用から切り替えた患者を対象とし ていることから、市販後に使用が予想される患者集団との差異はないと考えられた。
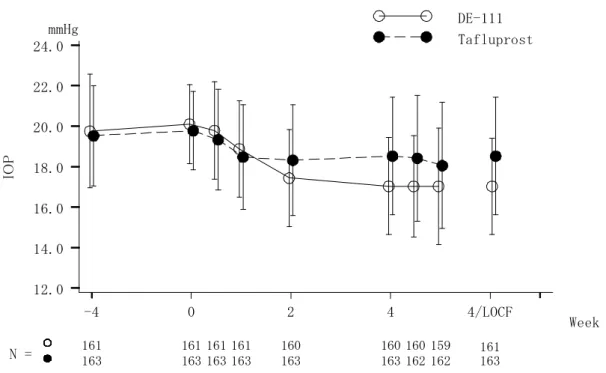
2.5.4.3.2 有効性の結果 2.5.4.3.2.1 単剤治療患者に対する有効性 (1) タフルプロスト点眼液 0.0015%との比較 01111004 試験では、タフルプロスト点眼液 0.0015%を導入期として 4 週間点眼後、DE-111 点眼液に切り替えて治療期として4 週間点眼した群(DE-111 群)と、タフルプロスト点眼液 0.0015%を継続して 4 週間点眼した群(タフルプロスト群)を比較した。 DE-111 群及びタフルプロスト群の平均日中眼圧変化量を図 2.5.4.3-1、表 2.5.4.3-1 に示し た。DE-111 群の 4 週/中止時のベースライン(0 週)からの平均日中眼圧変化量(平均値±標 準偏差)は-2.6±1.8 mmHg であり、タフルプロスト群の変化量(平均値±標準偏差)は-0.9±1.7 mmHg であった。変化量の群間差は-1.7 mmHg と有意に優れた眼圧下降効果を示し (P<0.001) 、DE-111 点眼液のタフルプロスト点眼液 0.0015%に対する優越性が検証された。 治療期2 週(朝点眼前)及び治療期 4 週の朝点眼前、点眼 2 時間後及び 8 時間後の測定ポ イントにおける眼圧の推移を図 2.5.4.3-2 に示した。DE-111 点眼液は、トラフ値(朝点眼前) を含むすべての測定ポイントにおいて、タフルプロスト点眼液0.0015%に対する優越性を示 した (P<0.001) 。 以上、DE-111 点眼液は、点眼 2 週間後から、また、1 日を通じて、タフルプロスト点眼液 0.0015%より優れた眼圧下降効果を示すことが確認された。
図 2.5.4.3-1. タフルプロスト点眼液と比較した試験の平均日中眼圧変化量: 01111004 試験 表 2.5.4.3-1. タフルプロスト点眼液と比較した試験の平均日中眼圧変化量: 01111004 試験 平均日中眼圧 (mmHg) 群間比較 (mmHg) ベースライン (Mean±SD) 4 週/中止時 (Mean±SD) 変化量 (Mean±SD) 群間差 (Mean±SE) 95%信頼 区間 P 値 DE-111 (N=161) 19.6±2.0 17.0±2.4 -2.6±1.8 -1.7±0.2 -2.1~-1.3 <0.001 タフルプロスト (N=163) 19.2±2.1 18.3±2.8 -0.9±1.7 群間比較はベースラインを共変量とした共分散分析に基づく結果 C h an ge f ro m b as el in e in m ea n d iu rn al I O P -7 -6 -5 -4 -3 -2 -1 0 1 2 3 Week mmHg N = DE-111 Tafluprost 161 160 163 163 0 4 4/LOCF 161 163
図 2.5.4.3-2. タフルプロスト点眼液と比較した試験の眼圧の推移:01111004 試験 (2) タフルプロスト点眼液 0.0015%から DE-111 点眼液への切り替え 導入期にタフルプロスト点眼液0.0015%を点眼し治療期に DE-111 点眼液に切り替えた試 験(01111004 試験、01111006 試験)の眼圧変化量(朝点眼前)を表 2.5.4.3-2 に示した。01111004 試験の治療期4 週時のベースライン(0 週)からの眼圧変化量(朝点眼前)(平均値±標準偏 差)は-3.1±2.2 mmHg、01111006 試験の眼圧変化量(朝点眼前)(平均値±標準偏差)は、-1.7±2.3 mmHg であり、いずれも 0 週からの有意な眼圧下降を示した。2 試験間の眼圧変化量の違い は、ベースライン眼圧の影響と考えられた。 表 2.5.4.3-2. タフルプロスト点眼液から切り替えた試験の眼圧変化量 導入期 眼圧(朝点眼前)(mmHg) ベースライン (0 週) (Mean±SD) 4 週/中止時 (Mean±SD) 変化量 (Mean±SD) 01111004 試験 (N=161) タフルプロスト点眼液0.0015% 4 週間 20.1±1.9 17.0±2.4 -3.1±2.2 01111006 試験 (N=48) タフルプロスト点眼液0.0015% 4 週間 17.0±2.4 15.3±2.8 -1.7±2.3 IO P 12.0 14.0 16.0 18.0 20.0 22.0 24.0 Week mmHg N = DE-111 Tafluprost 161 161 161 161 160 160 160 159 163 163 163 163 163 163 162 162 -4 0 2 4 4/LOCF 161 163
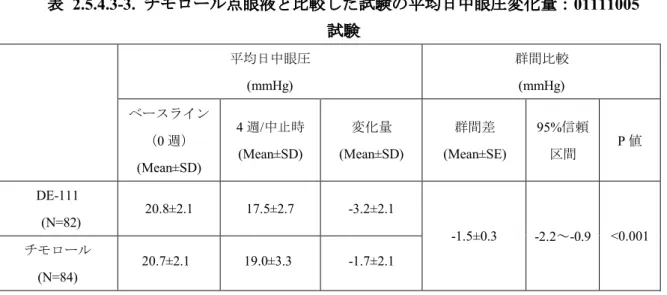
(3) チモロール点眼液 0.5%との比較 01111005 試験では、チモロール点眼液 0.5%を導入期として 4 週間点眼後、DE-111 点眼液 に切り替えて治療期として4 週間点眼した群(DE-111 群)と、チモロール点眼液 0.5%を継 続して4 週間点眼した群(チモロール群)を比較した。DE-111 群及びチモロール群の平均日 中眼圧変化量を図 2.5.4.3-3、表 2.5.4.3-3 に示した。DE-111 群の 4 週/中止時のベースライン (0 週)からの平均日中眼圧変化量(平均値±標準偏差)は-3.2±2.1 mmHg であり、チモロー ル群の変化量(平均値±標準偏差)は-1.7±2.1 mmHg であった。変化量の群間差は-1.5 mmHg と有意に優れた眼圧下降効果を示し (P<0.001) 、DE-111 点眼液のチモロール点眼液 0.5%に 対する優越性が検証された。 また、治療期2 週(朝点眼前)及び治療期 4 週の朝点眼前、点眼 2 時間後及び 8 時間後の 測定ポイントにおける眼圧の推移を図 2.5.4.3-4 に示した。DE-111 点眼液は、トラフ値(朝 点眼前)を含むすべての測定ポイントにおいて、チモロール点眼液0.5%に対する優越性を示 した (P<0.01) 。 以上、DE-111 点眼液は、点眼 2 週間後から、また、1 日を通じて、チモロール点眼液 0.5% より優れた眼圧下降効果を示すことが確認された。 図 2.5.4.3-3. チモロール点眼液と比較した試験の平均日中眼圧変化量:01111005 試験 C h an ge f ro m b as el in e in m ea n d iu rn al I O P -7 -6 -5 -4 -3 -2 -1 0 1 2 3 Week mmHg N = DE-111 Timolol 82 79 84 83 0 4 4/LOCF 82 84
表 2.5.4.3-3. チモロール点眼液と比較した試験の平均日中眼圧変化量:01111005 試験 平均日中眼圧 (mmHg) 群間比較 (mmHg) ベースライン (0 週) (Mean±SD) 4 週/中止時 (Mean±SD) 変化量 (Mean±SD) 群間差 (Mean±SE) 95%信頼 区間 P 値 DE-111 (N=82) 20.8±2.1 17.5±2.7 -3.2±2.1 -1.5±0.3 -2.2~-0.9 <0.001 チモロール (N=84) 20.7±2.1 19.0±3.3 -1.7±2.1 群間比較はベースラインを共変量とした共分散分析に基づく結果 図 2.5.4.3-4. チモロール点眼液と比較した試験の眼圧の推移:01111005 試験 (4) チモロール点眼液 0.5%からの切り替え 導入期にチモロール点眼液0.5%を点眼し、治療期に DE-111 点眼液に切り替えた試験 (01111005 試験、01111006 試験)の眼圧変化量(朝点眼前)を表 2.5.4.3-4 に示した。01111005 IO P 12.0 14.0 16.0 18.0 20.0 22.0 24.0 Week mmHg N = DE-111 Timolol 82 82 82 82 82 79 77 77 84 84 84 84 84 83 82 81 -4 0 2 4 4/LOCF 82 84
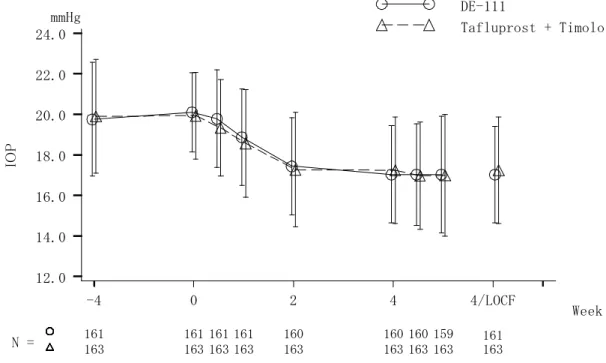
試験の治療期4 週/中止時のベースライン(0 週)からの眼圧変化量(朝点眼前)は-4.0±2.2 mmHg、01111006 試験の眼圧変化量は(朝点眼前)は、-2.1±2.2 mmHg であり、いずれも 0 週からの有意な眼圧下降を示した。2 試験での眼圧変化量の違いは、ベースライン眼圧の影 響と考えられた。 表 2.5.4.3-4. チモロール点眼液から切り替えた試験の眼圧変化量 導入期 眼圧(朝点眼前)(mmHg) ベースライン (0 週) (Mean±SD) 4 週/中止時 (Mean±SD) 変化量 (Mean±SD) 01111005 試験 (N= 82) チモロール点眼液0.5% 4 週間 21.7±1.8 17.6±2.7 -4.0±2.2 01111006 試験 (N=45) チモロール点眼液0.5% 4 週間 17.1±2.7 15.0±3.0 -2.1±2.2 2.5.4.3.2.2 併用治療患者に対する有効性 (1) タフルプロスト点眼液 0.0015%とチモロール点眼液 0.5%の併用との比較 01111004 試験では、タフルプロスト点眼液 0.0015%を導入期として 4 週間点眼後、DE-111 点眼液に切り替えて治療期として4 週間点眼した群(DE-111 群)と、タフルプロスト点眼液 0.0015%とチモロール点眼液 0.5%の併用に切り替えた群(併用群)を比較した。 DE-111 群及び併用群の平均日中眼圧変化量を図 2.5.4.3-5、表 2.5.4.3-5 に示した。DE-111 群の4 週/中止時のベースライン(0 週)からの平均日中眼圧変化量(平均値±標準偏差)は -2.6±1.8 mmHg、併用群の変化量(平均値±標準偏差)は-2.2±1.8 mmHg であり、DE-111 群の 眼圧下降効果が数値的に上回った。変化量の群間差は-0.3 mmHg であり、95%信頼区間は-0.7 ~0.1 mmHg と、事前に規定した非劣性マージン 1.5 mmHg を超えなかったことから、DE-111 点眼液のタフルプロスト点眼液0.0015%とチモロール点眼液 0.5%の併用に対する非劣性が検 証された。 また、治療期2 週(朝点眼前)、治療期 4 週の朝点眼前、点眼 2 時間後及び 8 時間後の測 定ポイントにおける眼圧変化量を図 2.5.4.3-6 に示した。DE-111 点眼液は、トラフ値(朝点 眼前)を含むすべての測定ポイントにおいて、タフルプロスト点眼液0.0015%とチモロール 点眼液0.5%の併用に対する非劣性の基準を満たした。 なお、01111004 試験において、併用群の眼圧下降効果はタフルプロスト群よりも有意に強 かったことから、非劣性について、十分な分析感度を有することが確認された。
以上、DE-111 点眼液は、点眼 2 週間後から、また、1 日を通じて、タフルプロスト点眼液 0.0015%とチモロール点眼液 0.5%(1 日 2 回点眼)の併用に劣らない眼圧下降効果を示すこ とが確認された。 図 2.5.4.3-5. 併用と比較した試験の平均日中眼圧変化量:01111004 試験 表 2.5.4.3-5. 併用と比較した試験の平均日中眼圧変化量:01111004 試験 平均日中眼圧 (mmHg) 群間比較 (mmHg) ベースライン (0 週) (Mean±SD) 4 週/中止時 (Mean±SD) 変化量 (Mean±SD) 群間差 (Mean±SE) 95%信頼 区間 P 値 DE-111 (N=161) 19.6±2.0 17.0±2.4 -2.6±1.8 -0.3±0.2 -0.7~0.1 0.098 併用 (N=163) 19.3±2.2 17.1±2.5 -2.2±1.8 群間比較はベースラインを共変量とした共分散分析に基づく結果 C h an ge f ro m b as el in e in m ea n d iu rn al I O P -7 -6 -5 -4 -3 -2 -1 0 1 2 3 Week mmHg N = DE-111 Tafluprost + Timolol 161 160 163 163 0 4 4/LOCF 161 163
図 2.5.4.3-6. 併用と比較した試験の眼圧の推移:01111004 試験 (2) タフルプロスト点眼液 0.0015%とチモロール点眼液 0.5%の併用からの切り替え 導入期にタフルプロスト点眼液0.0015%とチモロール点眼液 0.5%を併用し、治療期で DE-111 点眼液に切り替えた試験(01111006 試験)の眼圧変化量(朝点眼前)を表 2.5.4.3-6 に示した。01111006 試験の治療期 4 週時のベースライン(0 週)からの眼圧変化量(朝点眼 前)(平均値±標準偏差)は-0.4±2.1 mmHg であり、有意差はなく (P=0.196) 、タフルプロス ト点眼液0.0015%とチモロール点眼液 0.5%の併用から切り替えた場合でも、眼圧下降効果が 安定して維持された。 表 2.5.4.3-6.併用から切り替えた試験の眼圧変化量 導入期 眼圧(朝点眼前)(mmHg) ベースライン (0 週) (Mean±SD) 4 週/中止時 (Mean±SD) 変化量 (Mean±SD) 01111006 試験 (N=43) タフルプロスト点眼液0.0015% +チモロール点眼液0.5% 4 週間 15.8±3.0 15.4±3.4 -0.4±2.1 IO P 12.0 14.0 16.0 18.0 20.0 22.0 24.0 Week mmHg N = DE-111 Tafluprost + Timolol 161 161 161 161 160 160 160 159 163 163 163 163 163 163 163 163 -4 0 2 4 4/LOCF 161 163
2.5.4.3.2.3 長期点眼時の有効性 DE-111 点眼液を長期間(52 週間)点眼した試験(01111006 試験)において、眼圧値(朝 点眼前、全群)はすべての測定時期でベースライン(0 週)より有意に下降し、その効果は 52 週間にわたり減弱しなかった。このことから、DE-111 点眼液の有効性は長期間持続する ことが確認された。 2.5.4.3.3 部分集団における結果の比較 2.5.4.3.3.1 診断名の影響 全試験について、診断名別(原発開放隅角緑内障、高眼圧症、正常眼圧緑内障、落屑緑内 障)に分けて比較した結果、DE-111 点眼液の眼圧下降効果はいずれの集団においても認めら れた。原発開放隅角緑内障、高眼圧症、正常眼圧緑内障の集団で、眼圧下降効果の差は認め られなかった。落屑緑内障については、症例数が少なかったため詳細な検討はできなかった。 2.5.4.3.3.2 年齢の影響 全試験について、年齢を65 歳未満(非高齢者)と 65 歳以上(高齢者)に分けて比較した 結果、DE-111 点眼液の眼圧下降効果はいずれの集団においても認められ、その程度に差は認 められなかった。 2.5.4.3.3.3 性別の影響 全試験について、男女に分けて比較した結果、DE-111 点眼液の眼圧下降効果はいずれの集 団においても認められ、その程度に差は認められなかった。 2.5.4.3.3.4 ベースライン眼圧値の影響 全試験について、ベースラインの眼圧値(朝点眼前)を、21 mmHg 未満、21 mmHg 以上 に分けて比較した結果、DE-111 点眼液の眼圧下降効果は、いずれの集団においても認められ た。ベースライン眼圧が高い集団ほど大きな眼圧下降効果を示した。 2.5.4.4 用法・用量の検討 2.5.4.4.1 推奨用法・用量 DE-111 点眼液は、有効成分としてタフルプロストを 0.0015%、チモロール 0.5%相当量の チモロールマレイン酸塩を含有する配合点眼液である。
用量について、タフルプロストは有効性及び安全性が確認されている既承認の0.0015%と した。チモロールは0.25%及び 0.5%の 2 濃度で承認されているが、チモロールは濃度依存的 に眼圧下降効果を示すことが確認されており(22)(23)(24)、0.5%との組み合わせの方がより強い眼 圧下降効果を示すと推察された。非臨床試験において、チモロール点眼液0.25%、チモロー ル点眼液0.5%、0.0015%/0.25%DE-111 点眼液及び DE-111 点眼液について、正常眼圧サルを 用いて眼圧下降効果を比較検討した結果、単剤及び配合点眼液のいずれも0.5%チモロールの 方が0.25%チモロールより強い眼圧下降効果を示したことから、0.0015%タフルプロスト及び 0.5%チモロール相当量のチモロールマレイン酸塩の組み合わせを選択した〔2.6.2.2.1 (2)〕。 臨床試験では、DE-111 点眼液を有効成分の各単剤と比較した試験(01111004 試験、01111005 試験)において優越性が検証され、併用と比較した試験(01111004 試験)では非劣性が検証 された。安全性に関しては、サルを用いた反復投与毒性試験において、DE-111 点眼液及び各 有効成分を3 倍濃度とした 0.0045%/1.5%DE-111 点眼液の忍容性が確認され、また配合によ る影響は認められなかった (2.6.6.3.1) 。臨床試験では、健康成人を対象とした反復投与試験 (01111002 試験)で、DE-111 点眼液の血漿中タフルプロストカルボン酸及びチモロール濃 度は、タフルプロスト点眼液0.0015%、チモロール点眼液 0.5%、及びタフルプロスト点眼液 0.0015%とチモロール点眼液 0.5%の併用と比較して差異がなく、配合による薬物動態学的相 互作用はないと考えられ、安全性にも問題は認められなかった。また、患者を対象とした DE-111 点眼液の各単剤及び併用と比較した試験(01111004 試験、01111005 試験)、また、 長期投与試験(01111006 試験)の結果においても、安全性について特段の懸念は認められな かった。 以上のことから、0.0015%タフルプロストと 0.5%チモロール相当量のチモロールマレイン 酸塩の配合が妥当であることが確認された。なお、配合剤は単剤で十分な治療効果が得られ ない患者に使用されることが想定され、強い眼圧下降効果が必要とされること、また、実際 の臨床現場においてもPG 関連薬とチモロール点眼液 0.5%が頻用されていることから、 0.0015%タフルプロストと 0.5%チモロール相当量のチモロールマレイン酸塩の配合は医療ニ ーズに適していると考えられた。 用法について、チモロール点眼液0.5%が 1 日 2 回点眼であるが、タフルプロスト点眼液 0.0015%が 1 日 1 回点眼であることに加え、患者のアドヒアランス向上を考え、1 日 1 回点眼 と設定した。この際、配合剤ではチモロール点眼液の点眼回数が減少するため、タフルプロ スト点眼液0.0015%とチモロール点眼液 0.5%の併用に比べ眼圧下降効果の減弱が懸念される。 このため、両有効成分の有効性を十分発揮できるよう、タフルプロスト及びチモロールの 低下させることのないよう製剤設計を工夫した。01111004 試験 では、1 日 1 回点眼の DE-111 点眼液の眼圧下降効果を、タフルプロスト点眼液 0.0015%(1 日1 回点眼)とチモロール点眼液 0.5%(1 日 2 回点眼)の併用と比較した結果、非劣性が検 証された。
以上のことから、DE-111 点眼液の用法は、タフルプロスト点眼液 0.0015%とチモロール点 眼液0.5%(1 日 2 回点眼)の併用に劣らない眼圧下降効果を示した 1 日 1 回点眼が妥当であ ることが確認された。 2.5.4.5 有効性のまとめ 1. DE-111 点眼液の眼圧下降効果について、タフルプロスト点眼液 0.0015%に対する優越 性が検証された。 2. DE-111 点眼液の眼圧下降効果について、チモロール点眼液 0.5%に対する優越性が検証 された。 3. DE-111 点眼液の眼圧下降効果について、タフルプロスト点眼液 0.0015%とチモロール 点眼液0.5%(1 日 2 回点眼)の併用に対する非劣性が検証された。 4. DE-111 点眼液の眼圧下降効果について、投与後 2 週以降、また日中すべての眼圧測定 時点において、タフルプロスト点眼液0.0015%及びチモロール点眼液 0.5%との優越性、 並びにタフルプロスト点眼液0.0015%とチモロール点眼液 0.5%の併用との非劣性が示 され、眼圧が1 日中コントロール可能であることが確認された。 5. DE-111 点眼液は、タフルプロスト点眼液 0.0015%並びにチモロール点眼液 0.5%からの 切り替えにおいて、さらなる眼圧下降効果を示した。また、タフルプロスト点眼液 0.0015%とチモロール点眼液 0.5%の併用からの切り替えにおいても、変わらない安定し た眼圧下降効果を示した。 6. DE-111 点眼液の眼圧下降効果は、52 週間の長期投与において減弱しなかった。 7. DE-111 点眼液の眼圧下降効果は、診断名、高齢者/非高齢者、性別に影響されなかっ た。
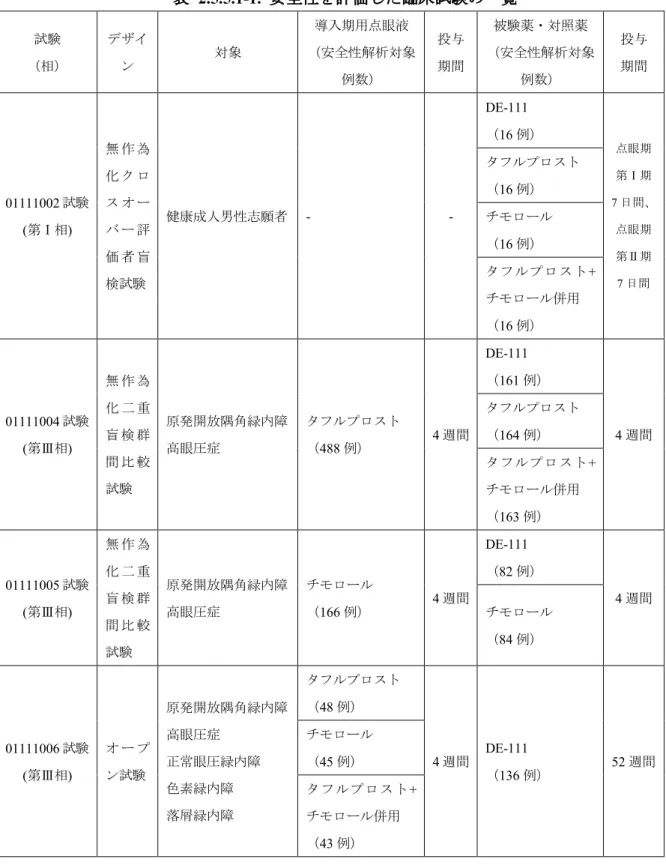
2.5.5 安全性の概括評価 2.5.5.1 医薬品への曝露 2.5.5.1.1 安全性を評価した試験 DE-111 点眼液は有効成分としてタフルプロストを 0.0015%、チモロール 0.5%相当量のチ モロールマレイン酸塩を含有する配合点眼液である。各有効成分単剤の効能・効果及び DE-111 点眼液の承認申請における効能・効果は「緑内障・高眼圧症」である。第Ⅰ相試験 (01111002 試験)では健康成人男性志願者を、第Ⅲ相試験(01111004 試験及び 01111005 試 験)では原発開放隅角緑内障及び高眼圧症を、また、第Ⅲ相長期投与試験(01111006 試験) では、正常眼圧緑内障が多い日本での実態をふまえ、開放隅角緑内障(正常眼圧緑内障を含 めた広義の原発開放隅角緑内障、色素緑内障、落屑緑内障)及び高眼圧症を対象とした。 DE-111 点眼液の用法用量は 1 回 1 滴、1 日 1 回とし、投与期間はそれぞれの試験の目的に 応じ、表 2.5.5.1-1 のとおり設定した。また、DE-111 点眼液は、タフルプロスト点眼液 0.0015% とチモロール点眼液0.5%の各単剤で効果不十分、あるいはタフルプロスト点眼液 0.0015%と チモロール点眼液0.5%の併用でアドヒアランス及び QOL の向上が望まれる患者への使用が 想定される。このため、タフルプロスト点眼液0.0015%及びタフルプロスト点眼液 0.0015% とチモロール点眼液0.5%(1 日 2 回点眼)の併用を対照とした 01111004 試験では、タフル プロスト点眼液0.0015%を点眼する導入期を設定した。チモロール点眼液 0.5%(1 日 2 回点 眼)を対照とした01111005 試験では、チモロール点眼液 0.5%を点眼する導入期を設定した。 また、01111006 試験では、各単剤のみならず併用からの切り替えにおける有効性及び安全性 を同一試験内で比較検討するため、タフルプロスト点眼液0.0015%、チモロール点眼液 0.5%、 又はタフルプロスト点眼液0.0015%とチモロール点眼液 0.5%(1 日 2 回点眼)の併用のいず れかを点眼する導入期を設定し、無作為割付け後、非盲検で実施した。 導入期間は、前治療薬の影響を消失させ、導入期用点眼液の有効性が一定となる期間とし て、4 週間と設定した。 DE-111 点眼液の安全性解析対象となった例数は、全試験で 395 例であった。また、患者を 対象とした01111004 試験、01111005 試験及び 01111006 試験で 379 例であり、これらについ て併合解析を実施した。 安全性を評価した臨床試験の一覧を表 2.5.5.1-1 に示した。
表 2.5.5.1-1. 安全性を評価した臨床試験の一覧 試験 (相) デザイ ン 対象 導入期用点眼液 (安全性解析対象 例数) 投与 期間 被験薬・対照薬 (安全性解析対象 例数) 投与 期間 01111002 試験 (第Ⅰ相) 無 作 為 化 ク ロ ス オ ー バ ー 評 価 者 盲 検試験 健康成人男性志願者 - -DE-111 (16 例) 点眼期 第Ⅰ期 7 日間、 点眼期 第Ⅱ期 7 日間 タフルプロスト (16 例) チモロール (16 例) タフルプロスト+ チモロール併用 (16 例) 01111004 試験 (第Ⅲ相) 無 作 為 化 二 重 盲 検 群 間 比 較 試験 原発開放隅角緑内障 高眼圧症 タフルプロスト (488 例) 4 週間 DE-111 (161 例) 4 週間 タフルプロスト (164 例) タフルプロスト+ チモロール併用 (163 例) 01111005 試験 (第Ⅲ相) 無 作 為 化 二 重 盲 検 群 間 比 較 試験 原発開放隅角緑内障 高眼圧症 チモロール (166 例) 4 週間 DE-111 (82 例) 4 週間 チモロール (84 例) 01111006 試験 (第Ⅲ相) オ ー プ ン試験 原発開放隅角緑内障 高眼圧症 正常眼圧緑内障 色素緑内障 落屑緑内障 タフルプロスト (48 例) 4 週間 DE-111 (136 例) 52 週間 チモロール (45 例) タフルプロスト+ チモロール併用 (43 例)
2.5.5.1.2 安全性の評価方法 DE-111 点眼液は有効成分として、タフルプロストを 0.0015%と 0.5%相当量のチモロール マレイン酸塩を含有する配合点眼液である。タフルプロストは薬理学上PGF2α 誘導体に分類 される。タフルプロスト(販売名:タプロス®点眼液0.0015%)の添付文書(25)では、承認時の 副作用(臨床検査値異常変動を含む)は総症例483 例中 326 例 (65.7%) に認められた。主な 副作用は、結膜充血151 件 (31.3%) 、睫毛の異常 93 件 (19.3%) 、そう痒感 85 件 (17.6%) 、 眼刺激感65 件 (13.5%) 、虹彩色素沈着 39 件 (8.1%) 等であった。また、特定使用成績調査 (第5 回安全性定期報告時)の副作用は総症例 3,260 例中 396 例 (12.1%) であった。主な副 作用は、眼瞼色素沈着93 件 (2.9%) 、結膜充血 74 件 (2.3%) 、角膜びらん等の角膜上皮障害 58 件 (1.8%) 、眼瞼の多毛症 40 件 (1.2%) 、睫毛の異常 39 件 (1.2%) 等であった。重大な副 作用として虹彩色素沈着が8.1%発現した。一方、チモロールマレイン酸塩(販売名:チモプ トール®点眼液0.25%、チモプトール®点眼液0.5%)の添付文書(26)では、臨床試験における副 作用は総症例818 例中 148 例(18.09%)であった。主な副作用は、眼局所では眼刺激症状 81 件 (9.90%)、角膜炎・角膜びらん等の角膜障害 36 件 (4.40%) 、霧視・視力低下等の視力障害 22 件 (2.69%) であり、全身では徐脈等の不整脈 8 件 (0.98%) 、頭痛 6 件 (0.73%) であった。 使用成績調査(再審査終了時)における副作用は総症例5,617 例中 266 例 (4.74%) であった。 主な副作用は眼局所では角膜炎・角膜びらん等の角膜障害80 件 (1.42%) 、眼刺激症状 53 件 (0.94%)、霧視・視力低下等の視力障害 21 件 (0.37%) であり、全身では徐脈等の不整脈 23 件 (0.41%) 、頭痛 13 件 (0.23%) であった。以上のとおり、両成分ともに全身よりも眼局所 性事象の発現頻度が高かった。これらを踏まえ、01111004 試験及び 01111005 試験において、 有害事象、臨床検査、血圧・脈拍数及び眼科的検査(細隙灯顕微鏡検査、視力検査)を、01111006 試験において、有害事象、臨床検査、血圧・脈拍数及び眼科的検査(細隙灯顕微鏡検査、視 力検査、視野検査)を安全性の評価項目とした。また、タフルプロスト点眼液0.0015%で特 徴的に認められた眼瞼色素沈着、眼瞼の多毛症、睫毛の異常等については臨床試験において 点眼期間が4 週間の試験では認められず長期試験でのみ認められ、3 ヵ月後頃から 6 ヵ月後 頃に発現率が高かった(27)ことから、01111006 試験においては DE-111 点眼液点眼開始時に写 真撮影を行い、虹彩及び眼瞼の色調、並びに睫毛について変化の有無を確認し、有害事象の 精査に努めた。 治験薬点眼開始から最終日検査又は治験中止時までに観察されたすべての症状の発現・悪 化及び治験責任医師・治験分担医師が医学的に有害と判断した自覚症状以外の所見の発現・ 悪化を有害事象(あらゆる医療上の好ましくない、あるいは意図しない疾病または徴候:被 験者にとって有害・不快な症状・所見)とし、治験薬との因果関係の有無は問わなかった。 なお、治験薬との因果関係が明確に否定できないものを副作用とした。