第 2 部(モジュール 2):CTD の概要(サマリー)
2.5 臨床に関する概括評価
目次
1. 製品開発の根拠 ... 6
1.1 概要 ... 6 1.2 多発性硬化症について ... 6 1.2.1 病因 ... 7 1.2.2 MS の診断基準 ... 7 1.2.3 MS の治療 ... 11 1.3 MS の治療におけるナタリズマブ療法 ... 11 1.3.1 科学的背景 ... 11 1.3.2 日本でナタリズマブの MS 治療薬としての開発を行う根拠 ... 12 1.3.3 臨床開発プログラムの概要 ... 12 1.3.4 臨床開発の経緯... 142. 生物薬剤学に関する概括評価 ... 16
3. 臨床薬理に関する概括評価 ... 18
3.1 用量の選択の根拠 ... 19 3.2 臨床薬理に関する結論 ... 204. 有効性の概括評価 ... 21
4.1 外国人患者における有効性 ... 21 4.2 日本人患者における有効性 ... 22 4.3 日本人患者のデータの外国人患者のデータとの比較 ... 23 4.4 本邦で承認されている他の MS 治療薬とのナタリズマブの有効性の比較 ... 27 4.5 要約及び結論 ... 295. 安全性の概括評価 ... 30
5.1 ナタリズマブの安全性プロファイルの概観 ... 30 5.1.1 日本人患者における安全性プロファイル ... 30 5.1.2 投与時反応及び過敏症反応 ... 31 5.1.3 ヘルペス感染及びその他の日和見感染 ... 31 5.1.4 肝臓に関する事象 ... 32 5.1.5 PML のリスクとその軽減 ... 32 5.2 国内で承認されたその他の MS 治療法の安全性プロファイルの概要 ... 33 5.3 要約及び結論 ... 336. リスク-ベネフィットに関する結論 ... 34
7. 参考文献 ... 36
表目次
表 1-1 MS の McDonald の診断基準 ... 9 表 1-2 今回の提出資料に含めた試験 ... 13 表 2-1 本剤の製造工程及び開発経緯の概要 ... 16 表 4-1 外国人患者の試験 AN100226-231 及び日本人患者の試験 101MS203 の 6 ヵ月目の評 価項目の要約 ... 25 表 4-2 外国人患者の第 III 相試験における主要評価項目及び副次評価項目の要約 ... 26 表 4-3 本邦で承認されている薬剤との比較 ... 28 表 5-1 危険因子により層別化した PML の発現率の推定値 ... 32略号一覧
略号 省略していない表現(英) 省略していない表現(日)
AE Adverse event 有害事象
ARR Annualized relapse rate 年間再発率
AUClast Area under the serum concentration-time curve
to the last sample timepoint
定量可能な最終時点までの血清中濃 度-時間曲線下面積
BBB Blood-brain barrier 血液脳関門
CD Crohn’s disease クローン病
CIS Clinically isolated syndrome 多発性硬化症が疑われる最初の臨床
症状
Cmax Maximum serum concentration 最高血清中濃度
CNS Central nervous system 中枢神経系
CSF Cerebrospinal fluid 脳脊髄液
DMD Disease modifying drug 病態修飾薬
DMT Disease modifying therapy 病態修飾療法
DS Drug substance 原薬
EDSS Expanded Disability Status Scale 総合障害度評価尺度
EU European Union 欧州連合
FDA Food and Drug Administration US 規制当局(米国食品医薬品局)
GA Glatiramer acetate グラチラマー酢酸塩(Copaxone®)
Gd Gadolinium ガドリニウム
HIV Human immunodeficiency virus ヒト免疫不全ウイルス
IFN Interferon インターフェロン
IFNβ Interferon beta インターフェロン ベータ
IFNβ-1a Interferon beta-1a インターフェロン ベータ-1a
(AVONEX®)
IgG Immunoglobulin G 免疫グロブリンG
IM Intramuscular 筋肉内
IV Intravenous 静脈内
JCV John Cunningham (JC) virus John Cunningham ウイルス
MRI Magnetic resonance imaging 磁気共鳴画像(法)
MS Multiple sclerosis 多発性硬化症
MSFC Multiple sclerosis functional composite 多発性硬化症機能評価
NA Not applicable/Not available 該当なし/データなし
NS Not significant 有意差なし
PBMC Peripheral blood mononuclear cell 末梢血単核球
PD Pharmacodynamic (s) 薬力学(的)
略号 省略していない表現(英) 省略していない表現(日) PMDA Pharmaceuticals and Medical Devices Agency 医薬品医療機器総合機構 PML Progressive multifocal leukoencephalopathy 進行性多巣性白質脳症
QD Once daily 1 日 1 回
RMS Relapsing form of multiple sclerosis 再発性多発性硬化症 RRMS Relapsing-remitting multiple sclerosis 再発寛解型多発性硬化症
SAE Serious adverse event 重篤な有害事象
SC Subcutaneous 皮下
SPMS Secondary progressive multiple sclerosis 二次性進行型多発性硬化症
UK United Kingdom 英国
US United States 米国
VAS Visual analog scale 視覚的評価法
1. 製品開発の根拠
1.1 概要 ナタリズマブ(又は本薬/本剤)は、白血球上のα4 インテグリンに結合し、白血球の血液脳関 門(BBB)の通過を阻害することにより中枢神経系(CNS)の炎症を抑制するモノクローナル抗 体である。日本人患者を対象とした本剤の試験(101MS203)の有効性データから、再発性多発性 硬化症(RMS)患者に対する身体機能における障害進行の抑制と臨床的増悪回数の減少を目的と した本剤単剤投与の妥当性が裏付けられている。 国内外で実施した臨床試験から、本剤を 300 mg の固定用量で 4 週間に 1 回点滴静注する療法は、 多発性硬化症(MS)再発率の予防、疾患進行の抑制、並びにガドリニウム(Gd)造影病巣数、新 規又は新規に拡大した T2 高信号病巣数、及び新規活動性病巣数の減少を含む MS 疾患活動性に関 する磁気共鳴画像(MRI)評価項目によって裏付けられる非常に有効な MS 治療法であることが 確認されている。日本人 MS 患者で確認された有効性は、外国人 MS 患者で確認された有効性と 同程度である。試験間でベースライン時の身体機能障害度、前治療及び罹病期間に差はみられる ものの、本薬の有効性プロファイルはいずれの試験でも良好であり、インターフェロン ベータ (IFNβ)製剤やフィンゴリモド(ジレニア®/イムセラ®)などの既存療法で報告されている有効 性よりも一貫して高い有効性を示している。 また、α4 インテグリン飽和度、リンパ球数増加などの本薬の活性に関する薬物動態学的(PK) 指標及び薬力学的(PD)指標は日本人患者と外国人患者で同程度であり、これらの指標から、固 定用量 300 mg の 4 週間に 1 回の点滴静注が支持される。 本剤は 2004 年以降、世界各国で上市されており、曝露量は 20 万人年を超えている。本剤の安 全性プロファイルも確立されている。日本人患者における臨床試験での安全性プロファイルは外 国人患者で認められたものと類似している。投与時反応、過敏症反応、及び抗ナタリズマブ抗体 の産生が本剤の安全性に関する重要な懸念ではあるが、これらは通常の臨床使用環境において管 理可能であり、致死的アナフィラキシーの症例は報告されていない。ヘルペス感染及び肝障害も 本剤の安全性に関する懸念であり、これらは市販後調査において初めて確認された。しかしなが ら、有害事象(AE)の発現率は非常に低い。本剤のリスク-ベネフィットにかかわる最も重要な AE は、進行性多巣性白質脳症(PML)の発症である。一方、この 7 年間で、抗 JCV 抗体の陽性、 本剤の投与期間(特に、2 年を超える場合)、及び免疫抑制療法の治療歴に基づいて患者の PML 発症リスクを評価するアルゴリズムを開発した。 MS の背景情報、並びに本薬の開発と製造工程の概要、有効性及び安全性に関する基本情報を 以下に示す。PK、PD、安全性及び有効性のデータに基づき、本剤は 4 週間に 1 回 300 mg を点滴 静注する用法で MS 患者の治療に有用である。 1.2 多発性硬化症について MS は、炎症、脱髄(神経を覆う髄鞘の破壊)及びオリゴデンドロサイトや神経細胞などの CNS 細胞の喪失を病理学的特徴とする CNS の慢性的な自己免疫性神経変性疾患である。CNS の脱髄性 疾患としては最も多くみられ、患者数は全世界で約 250 万人である。有病率は白人が最も高く、 その中でも北米、欧州、オーストラリア及びニュージーランドでの報告率が高い(Noseworthy et al., 2000、Rosati 2001)。日本では年々、患者数が増加しており、過去 10 年間で 2 倍近い数字になっ ている(Houzen et al., 2012)。 MS は厚生労働省の特定疾患に指定されている。2004 年に日本で実施された全国疫学調査によ ると、MS の患者数は MS の疑いのある患者まで含めると 12400 人、MS の疑いのある患者を含め ないと 9900 例であり、有病率は 7.7 人/10 万人と推計された(Kira 2006、Kira et al., 2005)。別のら、2006 年には 13.1/10 万人人、2011 年には 16.2 人/10 万人に上昇していることが確認されて いる(Houzen et al., 2012、Nakamura et al., 2009)。
全世界で最も多くみられる MS の病型は再発寛解型多発性硬化症(RRMS)である。欧米諸国、 日本とも MS 患者の約 70~90%が RRMS を呈し(Saida 2002、Weinshenker et al., 1989)、残りの患
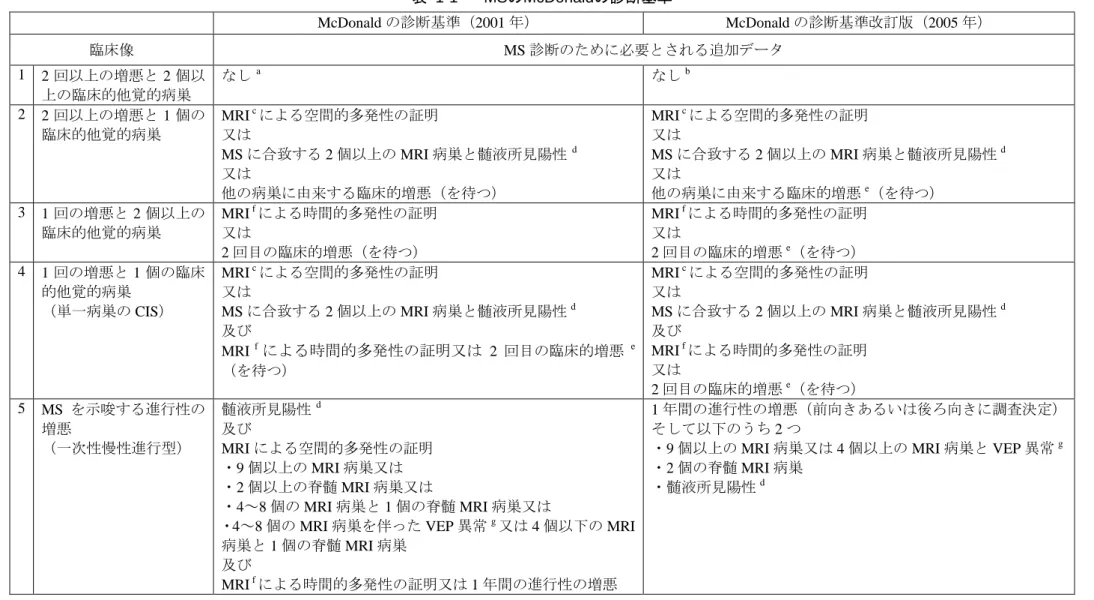
者は進行型 MS を呈する。 RRMS では、神経学的症状の悪化(急性増悪/再発)と症状が比較的安定する時期を繰り返す。 神経学的症状の悪化の症状は 1 日をとおして発現する可能性があり、視力喪失又は複視、四肢の しびれ又はピリピリ感、筋力低下、不明瞭な発語、協調運動困難、膀胱機能障害のほか、認知障 害、心理的変化などがみられる。RRMS の初期には、再発してもこれらの症状は数日又は数週間 で完全に消失する傾向にあるが、疾患の経過とともに完全な回復は難しくなる。最終的に、RRMS 患者の約 70%が、明らかな再発の有無にかかわらず、神経学的症状が進行性に悪化する病型(二 次性進行型多発性硬化症[SPMS])に移行する。RRMS から SPMS へと進行するにつれ、再発と は無関係に身体的障害が進行しやすくなる。病態修飾療法(DMT)を施行しない場合、全 MS 患 者の約半数が最初の診断から 15 年以内に補助具なしでは歩行不可能になる(Weinshenker 1995)。 適切な治療を受けている患者では、寿命が短縮することはまれであるが、MS は生涯のうちで活 動的かつ生産的な時期における疾病負担が大きく、この負担は徐々に増加していく。このことは、 MS 患者の生活の質に関する研究で毎回報告されている。また、MS は患者だけでなく、家族や介 護者の生活にも多大な負担をかける。 1.2.1 病因 MS の原因となる病的変化は、活性化 T リンパ球が BBB を通過し、内皮細胞の活性化、更なる リンパ球及び単球の動員、炎症誘発性サイトカインの放出に至る一連の反応が開始されるために 出現すると考えられている。MS 病巣は CNS 全体にみられる。MS 病巣の形成により炎症、浮腫 及び脱髄が生じ、最終的にオリゴデンドロサイト細胞死や軸索切断などの不可逆的損傷に至る。 このような病態生理学的過程の進行に伴い、一過性の臨床症状として再発が生じることも多く、 次第に MS の後期にみられる持続的な身体的障害があらわれるようになる。SPMS では通常、CNS の変性過程が持続すると報告されているが、二次性進行期の MS 患者でも多くの場合、活動性炎 症が引き続きみられる。炎症カスケードがどのように開始されるのか完全には解明されていない が、初期の重要な段階として、炎症細胞が接着し、循環血から BBB を通過して CNS へと経内皮 移動すると考えられている。 1.2.2 MSの診断基準 「多発性硬化症治療ガイドライン 2010」によると、MS の診断基準としては、McDonald の診断 基準が国際的に用いられている(多発性硬化症治療ガイドライン委員会 2010)。 診断基準は 1960 年代に作成され、長年にわたる改訂により新たな診断ツールが取り入れられて きた。McDonald の診断基準はまず、2001 年にインターナショナルパネルにより発表され (McDonald et al., 2001)、2005 年に改訂が加えられたあと(Polman et al., 2005)(表 1-1)、2010 年
に更に改訂された(Polman et al., 2011)。McDonald の診断基準は主に、臨床像又は早期の MRI 検
査により、異なる時点及び CNS の異なる場所に脱髄性病巣が 2 個確認され、臨床症状と臨床検査 値から他疾患の可能性が除外されていることをもって MS と診断する。MRI 検査が改良されたこ とを考慮して、McDonald の診断基準改訂版が作成され、臨床現場での使用により適したものとな ったが、全般的な概念には変更がなく、他の診断基準で診断された患者集団との差はほとんどな い。外国人 RRMS 患者を対象とした本剤の第 III 相評価試験(試験 C-1801 及び C-1802)では 2001 年の McDonald の診断基準を用い、日本人患者を対象とした試験 101MS203 では McDonald の診断 基準改定版(Polman et al., 2005)を用いた。これらの診断基準は、国内試験を含め、MS を対象と
表 1-1 MSのMcDonaldの診断基準 McDonald の診断基準(2001 年) McDonald の診断基準改訂版(2005 年) 臨床像 MS 診断のために必要とされる追加データ 1 2 回以上の増悪と 2 個以 上の臨床的他覚的病巣 なし a なしb 2 2 回以上の増悪と 1 個の 臨床的他覚的病巣 MRI cによる空間的多発性の証明 又は MS に合致する 2 個以上の MRI 病巣と髄液所見陽性 d 又は 他の病巣に由来する臨床的増悪(を待つ) MRI cによる空間的多発性の証明 又は MS に合致する 2 個以上の MRI 病巣と髄液所見陽性d 又は 他の病巣に由来する臨床的増悪e(を待つ) 3 1 回の増悪と 2 個以上の 臨床的他覚的病巣 MRI fによる時間的多発性の証明 又は 2 回目の臨床的増悪(を待つ) MRI fによる時間的多発性の証明 又は 2 回目の臨床的増悪e(を待つ) 4 1 回の増悪と 1 個の臨床 的他覚的病巣 (単一病巣の CIS) MRI cによる空間的多発性の証明 又は MS に合致する 2 個以上の MRI 病巣と髄液所見陽性d 及び MRI fによる時間的多発性の証明又は 2 回目の臨床的増悪 e (を待つ) MRI cによる空間的多発性の証明 又は MS に合致する 2 個以上の MRI 病巣と髄液所見陽性d 及び MRI fによる時間的多発性の証明 又は 2 回目の臨床的増悪e(を待つ) 5 MS を示唆する進行性の 増悪 (一次性慢性進行型) 髄液所見陽性 d 及び MRI による空間的多発性の証明 ・9 個以上の MRI 病巣又は ・2 個以上の脊髄 MRI 病巣又は ・4~8 個の MRI 病巣と 1 個の脊髄 MRI 病巣又は
・4~8 個の MRI 病巣を伴った VEP 異常g又は 4 個以下の MRI
病巣と 1 個の脊髄 MRI 病巣 及び
MRI fによる時間的多発性の証明又は 1 年間の進行性の増悪
1 年間の進行性の増悪(前向きあるいは後ろ向きに調査決定) そして以下のうち 2 つ
・9 個以上の MRI 病巣又は 4 個以上の MRI 病巣と VEP 異常g
・2 個の脊髄 MRI 病巣
a 追加検査は不要である。しかし、もし MRI や脳脊髄液(CSF)検査が施行され、異常がない場合は、MS と診断するには十分な注意が必要であり、他の疾患を考えなけれ ばならない。MS の診断にはその臨床像には MS 以上に考えられる疾患がないことが必要である。 b 追加検査は不要である。しかし、もし MRI や CSF 検査が施行され、異常がない場合は、MS と診断するには十分な注意が必要であり、他の疾患を考えなければならない。 MS の診断にはその臨床像及びいくつかの客観的エビデンスには MS 以上に考えられる疾患がないことが必要である。 c [M2.7.3 臨床的有効性 8.2 項 表 8-3]の基準を満たすこと。 d 髄液所見陽性とは、確立された方法(望ましいのは等電点電気泳動法)により検出された血清中バンドとは異なるオリゴクローナルバンドあるいは IgG インデックス高 値を指す。 e 発作とは神経障害の発現で、原因の病巣が炎症性で脱髄性である可能性が高い場合と定義する。主観的報告(客観的所見で裏付けられるもの)又は 24 時間以上持続する 事象の客観的所見があること。 f [M2.7.3 臨床的有効性 8.2 項 表 8-4]の基準を満たすこと。 g 視覚誘発電位(VEP)異常は、MS に合致する波形の保たれた潜時の延長をいう。
1.2.3 MSの治療 MS は再発と寛解(突発的な症状悪化と回復)を繰り返すことから、治療は 2 種類に大別され る。急性増悪(再発)に対しては通常、高用量副腎皮質ステロイドの静脈内(IV)投与、経口投 与又はその両方が行われる。再発以外に対しては、再発(急性増悪)予防と MS 進行の抑制を目 的に、主に DMT が行われる。これらの治療法は国内の治療ガイドラインに記載されている(多 発性硬化症治療ガイドライン委員会 2010)。現在、日本、米国(US)及び欧州で承認されている RRMS の治療薬はIFNβ 製剤 2 種類(アボネックス®及びベタフェロン®)及びフィンゴリモド(ジ レニア®/イムセラ® 0.5 mg 1 日 1 回)である。これ以外のインターフェロン(IFN)製剤( 及び )並びに ( )、natalizumab(Tysabri®)及び ( )は本邦未承認である。 日本で最も使用率が高い治療薬は、皮下(SC)注射又は筋肉内(IM)注射用のIFNβ 製剤であ る。IFNβ 製剤は、抗増殖作用、抗ウイルス作用を示すサイトカイン製剤であり、免疫調節作用を 示す MS 治療薬である。いずれのIFNβ 製剤も重要な臨床効果を示すが、その効果は中程度に止ま り、さらに、製剤の特性から使用が制限される。すなわち、IFNβ 製剤は SC 又は IM への自己注 射が必要なため、一部の患者では、針に対する恐怖心やインフルエンザ様症状などのIFNβ 注射に 伴う反応に耐えられないことから、治療の開始を避けるか、これら治療に対する忍容性が得られ ない。 フィンゴリモドは選択的な経口免疫抑制薬であり、リンパ球上の S1P 受容体の機能的アンタゴ ニストである活性代謝物のフィンゴリモドリン酸塩に代謝される。日本では、フィンゴリモド 0.5 mg の 1 日 1 回投与が承認されており、国内治療ガイドラインでは、活動性が極めて高い MS 患者の治療薬として推奨されている(多発性硬化症治療ガイドライン委員会 2010)。一方で、フ ィンゴリモドは、開始時の徐脈、肝障害、黄斑浮腫、感染(特にヘルペス)などの副作用に対す る十分な配慮が必要である。 国内承認薬との比較は、有効性については4.4項で、安全性については5.2項に示す。 本剤はこれまでに得られているデータに基づき、日本人 MS 患者に対して、未充足な医療ニー ズに対して、次の 2 つのニーズを提供することが可能である。その 1 つとしては、本剤の極めて 高い治療効果を求める患者のニーズであり、もう一つは既承認の DMT を使用してもなお、継続 する疾患活動性を示す患者に対する治療ニーズである。このように、本剤は MS 治療の領域に多 大な進歩をもたらすことが期待できる。 1.3 MSの治療におけるナタリズマブ療法 1.3.1 科学的背景 本薬は、マウス骨髄腫細胞(NS/0)内で産生される遺伝子組換え型ヒト化 IgG4κモノクローナ ル抗体であり、選択的接着分子阻害薬である。本薬は、好中球以外の全ての末梢血白血球の表面 に発現しているヘテロ二量体膜貫通タンパク、α4 インテグリン(α4β1 及び α4β7)に特異的に結 合し、これによって、これらのインテグリンと、内皮細胞上の血管細胞接着分子-1、粘膜アドレ シン細胞接着分子-1 などの内因性同族受容体との相互作用を阻害する。これらのインテグリンは、 免疫細胞が CNS などの血管の内皮細胞層に接着する過程において重要な役割を果たしており、こ れに続く免疫細胞の炎症組織への移動を促している。本薬は、この相互作用の阻害を介して自己 反応性免疫細胞の CNS への過剰な動員を抑制し、その結果として、MS の病理検査で特徴的にみ られる炎症性病巣の発生を阻害する。 これらの細胞接着分子を阻害すると、α4 インテグリンを発現する末梢血白血球の増加も生じる。 本剤の投与に影響される白血球分画は、α4 インテグリンの発現レベルに一致する(すなわち、好 中球を除く全ての白血球分画の数が本剤の投与後に増加する)。したがって、α4 インテグリンを
発現する白血球分画(例えば、リンパ球)の特異的な増加は、本剤療法の PD マーカーになる。 そのため、本剤の試験ではこの血球数を測定した。さらに、末梢血単核球(PBMC)上のα4 イン テグリン受容体の占有率(飽和度)も本剤療法のマーカーになるため、本薬の開発プログラムを 通じてこの指標を測定した。 1.3.2 日本でナタリズマブのMS治療薬としての開発を行う根拠 本剤は外国人(主に白人)集団において非常に高い有効性を示していることから、日本人 RRMS 患者を対象に試験を行った。現在の治療法に対して、本剤は作用機序が異なることから、新たな 選択肢となる可能性がある。現在の治療法はいずれもまだ不十分であり、治療を継続しても、症 候性又は無症候性の疾患活動性が持続する患者が多い。本剤は、上述したIFNβ とは作用機序が異 なっており、臨床試験では、IFNβ の試験で報告された有効性より最大で 2 倍高い有効性を示し、 MRI 検査により病理学的変化を遅延させることが確認されている(4. 項)。さらに、未治療患者 に投与した場合、又はIFNβ で十分な効果が得られない患者に対して投与した場合のいずれにおい ても、有効性が認められている。すなわち、本剤は満たされていない医療上のニーズを満たすこ とが可能である。 1.3.3 臨床開発プログラムの概要 臨床開発プログラムでは 25 を超える試験を実施しており、主として MS 患者の治療に焦点を当 てているが、他の疾患も対象に含めている。 年代の初期に実施した第 III 相試験では、US、 欧州連合(EU)、カナダ、オーストラリア及びアジアの患者を組み入れた。本剤は、MS を対象と した試験のほか、クローン病(CD)、 及び の患者を対象とした試験も 実施している。MS を対象とした臨床開発プログラムのうち、有効性データについては[M2.7.3 臨 床的有効性]で詳細に論じている。安全性データについては、完全なデータを提示するため、他の プログラムで得られたデータも含めて[M2.7.4 臨床的安全性]に提示している。今回の申請資料中 で論じる評価試験及び参考試験の一覧を表 1-2に示す。詳細は[M5.2 臨床試験一覧表]に示す。
表 1-2 今回の提出資料に含めた試験 試験番号 試験の種類 外国人患者データ 多発性硬化症 C-1801 評価 C-1802 評価 C-1803 評価 AN100226-231 評価 AN100226-200 評価 AN100226-201 評価 AN100226-202 評価 AN100226-221 評価 C-1808 評価 AN100226-101 評価 AN100226-224 評価 C-1804 参考 C-1805 評価 C-1806 評価 101MS001 評価 101MS101 評価 101MS102 評価 101MS201 評価 101MS205 評価 101MS404 参考 101MS406 評価 クローン病 AN100226-CD201 参考 AN100226-CD202 参考 AN100226-CD251 参考 AN100226-CD301 参考 AN100226-CD303 参考 ELN100226-CD305 参考 ELN100226-CD306 参考 ELN100226-CD307 参考 ELN100226-CD351 参考 ELN100226-CD352 参考 ELN100226-CD354 参考 参考 参考 参考 日本人患者データ(多発性硬化症) 101MS203 評価 101MS204 評価
Note: This table does not include the studies contributing only to the anti-JCV antibody
1.3.4 臨床開発の経緯 1.3.4.1 US、EU、その他の国における審査及び承認の状況 本剤は現在、60 を超える国において MS の治療薬として承認されている。US では、RMS 患者 の治療において、身体的機能障害の進行を抑制させ、臨床的増悪(発作)の発現率を減少させる ための単剤療法として承認されている。本剤は、2004 年 11 月 23 日に US で最初に承認された。 2005 年 2 月 日、Biogen Idec 社は、臨床試験において PML の発現が認められたことから、本剤 の販売を自主的に休止した。その後、PML の危険因子に関する詳細な調査を行い、リスク管理計 画を導入した上で、同社は 2006 年 6 月 5 日に本剤の販売を再開した。本剤は EU でも 2006 年 6 月 27 日に承認され、現在、単剤の DMT として以下の高活動性 RRMS 患者に適応されている。 IFNβでの治療にも関わらず、高い疾患活動性を呈する 18 歳以上の成人 上記患者は、完全かつ適切な IFNβによる治療(通常 1 年以上の治療)が無効である患者を 指す。患者は前年の治療中に 1 回以上の再発を認め、頭部 MRI 上で 9 個以上の T2 高信号 病巣又は Gd 造影病巣が認められたものとする。前年と比べて再発率が変わらないか若し くは上昇した患者又は重度の再発中である患者を「無効例」とした。 又は 重度の RRMS の迅速な進行を認めた 18 歳以上の成人患者 上記患者は、1 年のうちに 2 回以上の障害を伴う再発があり、頭部 MRI に 1 個以上の Gd 造影病巣又は前回の MRI に比べて T2 高信号病巣の有意な増加を伴う患者と定義される。 これらの適応の記述は、本剤と PML の関連及びそのリスク分類について詳細な調査を行う前に 定めたものである。Biogen Idec 社は、継続中のデータ収集と世界規模で実施している医薬品安全 性監視活動を通じ、PML の危険因子として、抗 JCV 抗体の陽性、本剤の投与期間(特に、2 年を 超える治療)、及び免疫抑制療法の治療歴の 3 因子を特定した。これらの 3 因子を用い、PML の 発症リスクが高い患者と低い患者の層別並びに、その他の情報に基づく治療方針の決定において 治療医を支援するためのリスク分類アルゴリズムを開発した。 年 月、Biogen Idec 社は、PML 発症リスクが抗 JCV 抗体陰性患者では、抗 JCV 抗体陽 性患者に比べて、非常に低いことを示す 提出し、 。 、 に提出された。 、 、 、 。 1.3.4.2 日本における臨床開発の経緯
Biogen Idec 社は、 年に医薬品医療機器総合機構(PMDA)と本剤の日本での開発方針に関
する相談を開始した。本剤は、 年 月 日に優先対面助言品目に指定され、2008 年 5 月 20 日に希少疾病用医薬品の指定を受けた。PMDA との対面助言は 年 月 日に開催された。 Biogen Idec 社は、 、PMDA と協議した。PMDA との協議の結果に基づき、 、 を追加した。また、 を変更した。 日本人患者を対象に本剤を評価する試験 101MS203 は、 年 月から 年 月にかけて 実施した。12 例がパート A を終了した時点で PK 及び安全性の評価を行い、その結果、パート B では 90 例を対象に 300 mg の固定用量で継続して点滴静注することを決定した。試験 101MS203
この多施設共同非盲検長期継続投与試験(試験 101MS204)は、現在進行中である。これまでに 得られている情報を提示するため、今回の申請資料には、投与 1 年目のデータベースロック時点 での長期安全性及び忍容性並びに有効性の評価を含めた。
2. 生物薬剤学に関する概括評価
本項では、MS を適応症とした本剤の使用に際し、臨床試験及び実生産に向けた開発段階にお ける本薬の製造工程の開発経緯を簡単に述べる。表 2-1に、日本での実生産用に計画している製 剤(BG00002-G)に至るまでの臨床試験用及び実生産用の製造工程の概要を示す。 ナタリズマブ原薬(DS)生産用の細胞株は、開発段階を通じて変更していない 社における 2000 L 規模の臨床試験用製剤の製造工程は、基本的に Biogen Idec 社が使用した工程 と同じである。表 2-1の後に記したように、工程を変更するたびに慎重な検討を行い、広範な分 析試験及び生化学的試験、並びにカニクイザル及び/又はヒトにおいて生物学的同等性試験を実 施した。さらに、本薬の活性維持の確認ため、工程を通じて定期的にα4 インテグリン飽和度を測 定した。それぞれの工程変更を支持するために実施した同等性試験については、[M3.2.S.2.6.2 原 薬の同等性/同質性に関する評価結果]に示す。 表 2-1 本剤の製造工程及び開発経緯の概要 用途 培養スケール/製造場所 製造方法名 用途の詳細 臨床試験 用 200 L/ 社 ( , ) AN100226(200 L) 海外の臨床試験 第 I 相:AN100226-101、AN100226-200、 AN100226-221 第 II 相:AN100226-201、AN100226-202 2000 L/ 社( , , ) AN100226(2000 L) 海外の臨床試験 第 I 相:AN100226-224、C-1805 第 II 相:AN100226-231 2000 L/Biogen Idec 社 (Cambridge, MA, US)BG00002-A 海外の臨床試験 第 I 相:C-1806 第 II 相:C-1804 Phase 3:C-1801、C-1802 実生産用 15000 L/Biogen Idec 社 (Research Triangle Park, NC, US) BG00002-B 及び BG00002-B プロセスバリデーション 同等性/同質性試験 海外の臨床試験 第 I 相:C-1805、C-1806 第 II 相:C-1803 第 III 相:C-1808 初期実生産(海外) 15000 L/Biogen Idec 社
(Research Triangle Park, NC, US) BG00002-E プロセスバリデーション 同等性/同質性試験 初期実生産(海外) 海外の臨床試験 第 I 相:101MS101、101MS102 第 II 相:101MS201、101MS205 国内の臨床試験 第 II 相:101MS203、101MS204 15000 L/Biogen Idec 社
(Research Triangle Park, NC, US) BG00002-G プロセスバリデーション 同質性試験 実生産(海外) 社が最初に本薬の製造工程を開発した。第 I 相試験及び初期第 II 相試験用の DS(AN100226[200 L])は、 社( 、 )において 200 L 規模で製造した。
工程を 社( )に移管し、2000 L にスケールアップした(AN100226 [2000 L])。200 L 製造工程による DS と 2000 L 製造工程による DS の同等性を確認するため、広 範な分析試験及び生化学的試験、並びにカニクイザルを用いた前臨床試験を実施した。
その後、臨床試験用製剤の製造工程を 社から Biogen Idec 社(Cambridge、MA、
US)の製造施設に移管した(BG00002-A)。ここでも、 社の製造工程による DS
(AN100226[2000 L])と Biogen Idec 社の製造工程による DS(BG00002-A)の同等性を、広範な 分析試験及び生物分析試験、並びにカニクイザルを用いた前臨床試験により確認した。
Biogen Idec 社において様々な工程の改良を行い、大規模な実生産工程(BG00002-B)に発展さ
せた。BG00002-B では、 も加えた。
BG00002-B 実生産工程は、Biogen Idec 社(Research Triangle Park、NC、US)において 15000 L バ イオリアクターの規模で実施した製造でバリデーションを行っている。生化学的試験、生物物理 学的試験及び生物学的試験により、実生産規模の工程による DS は、特性が完全に明らかにされ ていること、また、 社及び Biogen Idec 社の 2000 L 臨床試験用工程による DS と同 等であることを確認した。カニクイザルを用いた非臨床試験により、実生産規模の工程による DS の PK 及び PD パラメータは臨床試験用 DS(BG00002-A)と同等であることを確認した。さらに、 健康成人被験者を対照に実施した臨床試験 2 試験(試験 C-1805 及び C-1806)において、実生産 規模の工程による DS(BG00002-B)の AN100226(2000 L)及び BG00002-A に対する生物学的同 等性を確認した。両試験とも、最終検体採取時点までの血中濃度時間曲線下面積(AUClast)及び 最高血中濃度(Cmax)の幾何平均比の 90%信頼区間は 80~125%の範囲内にあり、従来の生物学的 同等性の基準に適合した。また、PBMC 上のα4 インテグリン飽和度並びに安全性及び免疫原性プ ロファイルに関して、有意差は認められなかった。 BG00002-B 工程の導入後、 、 、 を加えた( 工程: BG00002-B)。 工程について
は、[M3.2.S.2.6 製造工程の開発の経緯(ナタリズマブ、Biogen Idec, Inc.)]に詳細を示す。最初の
実生産工程(BG00002-B)及び 工程により製造される本剤の製剤プロファイル及び分析に
おける同等性は、[M3.2.S.2.6.2 原薬の同等性/同質性に関する評価結果]に示す。
工程を導入した後、 、ナタリズマブ DS 製造工程を
BG00002-E に発展させた。BG00002-E 工程で行った具体的な変更及び変更の根拠については、 [M3.2.S.2.6 製造工程の開発の経緯(ナタリズマブ、Biogen Idec, Inc..)]に示す。BG00002-E 工程 で製造される DS は、生理化学的及び生化学的方法並びに in vitro 活性により特性が明らかにされ ており、BG00002-B 工程で製造される DS と同等であることが示されている。さらに、BG00002-E と BG00002-B の in vivo 同等性を評価するため、生物学的同等性試験(試験 101MS101)を実施し た。その結果、PK、α4 インテグリン飽和度、安全性、及び免疫原性プロファイルは同等であり、 この試験でも生物学的同等性が証明された。 BG00002-E の導入後、新しい工程(BG00002-G)を開発した。BG00002-G 工程は、 及び するために開発したものである。BG00002-G では、その前の BG00002-E 工程と同じ操作手順を採用している。変更した工程の説明、変更の根拠、及び変更による影響の 評価については、[M3.2.S.2.6 製造工程の開発の経緯(ナタリズマブ、Biogen Idec, Inc..)]に示す。 BG00002-G 工程で製造される DS は、生理化学的及び生化学的方法並びに in vitro 活性により特性 が明らかにされており、BG00002-E 工程で製造される DS と同等であることが示されている。
上記のとおり、工程を変更するたびに、これを支持するための広範な特性評価試験を実施した。 その結果は一貫して、前の工程との同等性を証明するものであった。
3. 臨床薬理に関する概括評価
本薬の PK 及び PD は、外国人患者及び日本人患者の両方で評価した。 検討した用量 本薬の PK 及び PD は、先ず外国人健康成人を対象とした単回投与試験 3 試験において評価し、 次いで外国人 MS 患者を対象とした単回/反復投与試験 16 試験において評価した。本剤は、臨床 開発を通じて 30~60 分間点滴静注した。単回投与試験では、0.03~6 mg/kg の用量又は 300 mg の 固定用量で投与した。第 II 相反復投与試験では、3~6 mg/kg の用量で 4 週間に 1 回、最長 6 ヵ月 間、又は 300 mg の固定用量で 4 週間に 1 回、最長 19 ヵ月間投与した。第 III 相試験では、300 mg を 4 週間に 1 回、6 ヵ月以上投与した。3 試験において、インターフェロン ベータ-1a(IFNβ-1a [AVONEX®]:試験 AN100226-224 及び C-1802)及び GA(Copaxone®:試験 C-1803)を含む他の薬剤に本剤を追加投与したときの本薬の PK 及び PD を評価した。
薬物動態評価項目
各試験において、PK パラメータの推定値は従来の方法を用いて算出した。血中濃度のプロファ イルは非コンパートメント法を用いて解析した。本薬の体内動態の評価に用いたパラメータは、
Cmax、AUClast、0 時間から無限大時間までの濃度-時間曲線下面積(AUC0-inf)、分布容積、クリア
ランス及び消失半減期である。 試験 101MS101 及び C-1801 に組み入れた 641 例のデータを用いて総合的な母集団 PK 解析を実 施した結果、抗ナタリズマブ抗体及び体重を除き、ほとんどの因子が本薬の体内動態に影響を及 ぼさないことが示され、本薬の PK は患者特性が幅広く変化しても一定であると考えられた。 薬力学的評価項目 既に述べたように、本薬はリンパ球上の受容体に影響を及ぼす。α4 インテグリン飽和度及び末 梢血白血球数の増加は、薬物作用のマーカーと考えることができる。本剤 300 mg を 4 週間に 1 回 点滴静注すると、α4 インテグリン飽和度が一貫して高いレベルで維持され、本剤の投与中止に伴 い、飽和度は低下した。また、末梢血白血球数の増加も、開発プログラムを通じて一貫して認め られた。これらの上昇は変動が大きく、投与中止後 16 週間以内に基準値に回復した。 日本人患者における PK 及び PD PK 及び PD は、日本人 MS 患者でも試験 101MS203 のパート A において評価した。日本人患者 集団と外国人患者集団の間で PK を比較した結果、体重の差にもかかわらず、両集団における単 回投与時及び反復投与時の曝露量はほぼ同程度であった。また、日本人患者集団でナタリズマブ 濃度がわずかに高かったものの、これらの差は臨床的に意味のあるものではなかった。α4 インテ グリン飽和度は、外国人患者集団よりは低くかったものの、一貫して高い値を示した。更に、特 徴的な白血球数の増加も認められた。 日本人試験における PK 中間解析の結果は、300 mg の固定用量の使用を支持するものであった。 特別な評価(外国人患者) AVONEX®又は GA への本剤の追加投与について検討した 3 試験において、本薬の PK 及び PD
は認められなかったが、GA への影響については、使用できる生物学的分析法に限界があったため、 評価することができなかった。 実生産品の免疫原性 免疫原性は、評価を行った全ての製剤について低かった。詳細は5.1.2 項に記述する。 3.1 用量の選択の根拠 MS を対象とした第 II 相反復投与用量設定試験(試験 AN100226-231)の PK データが、第 III 相開発における固定用量に対しての初期の妥当性根拠となった。更に、同試験から得られた PD 及び有効性のデータも、投与すべき固定用量の選択根拠となった。その後、300 mg の固定用量を 用いた MS に対する第 III 相試験において有効性及び十分な安全性が示されたが、これは日本以外 で承認されている用量であり、本邦においても本用量を用いた国内試験 101MS203 及び 101MS204 を検討した。 第 II 相試験 AN100226-231:α4 インテグリン飽和度の妥当性の証明 試験 AN100226-231 は、多施設共同無作為化プラセボ対照二重盲検並行群間比較試験であり、 外国人 MS 患者を対象とし、4 週間に 1 回、6 ヵ月間のプラセボ投与、本剤 3 mg/kg 投与又は 6 mg/kg 投与に無作為に割り付け、PK 及び PD(PBMC におけるα4 インテグリン飽和度及び末梢血リンパ 球数の増加)について検討した。 本試験では、血清中ナタリズマブ濃度は用量に比例して上昇し、末梢血リンパ球数の増加及び α4 インテグリン飽和度は、用量及び血清中濃度に関連してプラトーに達すると考えられた。α4 イ ンテグリン飽和度は、血清中ナタリズマブ濃度と並行して、投与直後に最高値に達して、経時的 に低下した。飽和度のプロファイルは 3 mg/kg 群と 6 mg/kg 群で同様であった。投与期間を通じて、 3 mg/kg 群及び 6 mg/kg 群はそれぞれ 70%及び 83%を超えるα4 インテグリン飽和度を維持し、両 用量群間で α4 インテグリン飽和度にかなりの重なりが認められた。これらの結果から、3 mg/kg と 6 mg/kg の両用量群で、投与間隔を通じて非常に高いα4 インテグリン飽和度が維持されること が示された。有効性及び安全性プロファイルは両用量群で同様であった。 固定用量の選択は、本剤の用量に依存しない、一貫した PK 及び有効性プロファイルに基づい て決定した。試験 AN100226-231 では、ベースライン時の平均体重は、3 mg/kg 群及び 6 mg/kg 群 でそれぞれ 72 kg 及び 70 kg であり、その範囲はおよそ 48~100 kg であった。この平均体重は、 それぞれ 216 mg 及び 420 mg の平均投与量に相当する。いずれの用量でも同様の有効性、安全性 及び忍容性プロファイルが得られたことから、第 III 相試験では 300 mg の固定用量を用いること とした。この用量で、3 mg/kg と 6 mg/kg の平均用量の中間の曝露量となる。さらに、300 mg を固 定用量とすると、50 kg 以上の被験者では 6 mg/kg の用量を上回ることがなく、100 kg 以下の被験 者では 3 mg/kg を下回ることがない。 試験 101MS203 パート A:日本人 MS 患者における PK の確認 300 mg を 4 週間に 1 回点滴静注する固定用量の妥当性を日本人の MS 患者を対照とした試験 101MS203 のパート A で確認した。PK 及び PD(α4 インテグリン飽和度)プロファイルは、外国 人 MS 患者を対象として 300 mg を 4 週間に 1 回点滴静注した先行試験と同等であることが明らか になった。そこで、日本人 MS 患者を対象とした試験 101MS203 のパート B でも、300 mg の固定 用量を 4 週間に 1 回点滴静注することとした。
3.2 臨床薬理に関する結論 結論として、PK 及び PD プロファイルは日本人患者と外国人患者の両者で同等であることが示 された。さらに、PK に影響を及ぼす因子はわずかであり、用量は広範囲にわたる患者群で一貫し ていることが示された。300 mg の固定用量で 4 週間に 1 回点滴静注することにより、広範囲にわ たる患者群及び体重群で十分な有効性が認められ、一貫した有効性が示された。α4 インテグリン 飽和度は、日本人 MS 患者を対象とした試験を含めて、上記の試験を通じて一貫しており、用量 調節の必要性はないと考えられる。
4. 有効性の概括評価
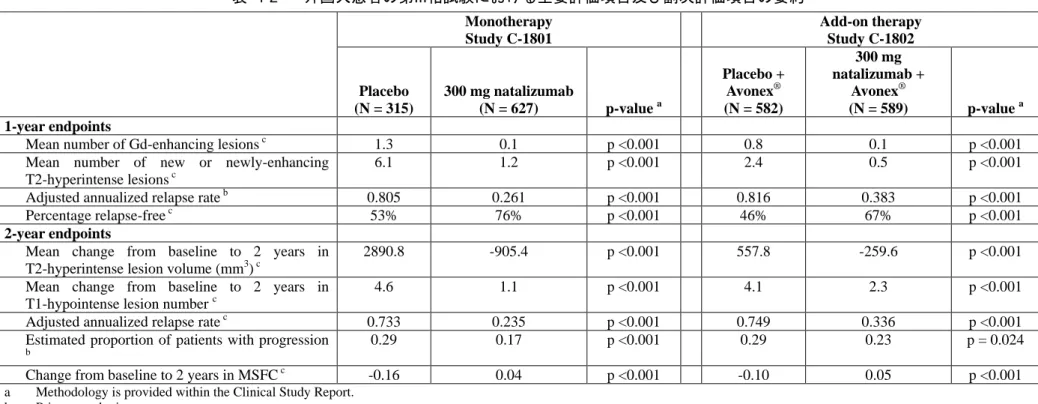
本剤は、世界各国で試験の対象とされた全ての RMS 患者集団において、300 mg を 4 週間に 1 回点滴静注したとき、持続的障害進行開始までの期間の延長、臨床的再発数及び MRI 上の病巣数 の減少において有効性を示した。 4.1 外国人患者における有効性 本剤の有効性は、最初に大規模な多施設共同無作為化プラセボ対照二重盲検並行群間比較試験 3 試験において、RMS を有する外国人患者を対象として評価された。この 3 試験は、4 週間に 1 回、3 mg/kg 又は 6 mg/kg の用量で投与したときの本剤の効果をプラセボと比較した、6 ヵ月間の 第 II 相試験 AN100226-231(表 4-1)と、300 mg の固定用量で 4 週間に 1 回点滴静注した、いず れも 2 年間の第 III 相試験 C-1801 及び C-1802 であった(表 4-2)。これらの試験の重要な評価項 目としては、MRI 上の Gd 造影病巣及び新規又は新規に拡大した T2 高信号病巣の発生で評価した MRI に関する評価項目や、再発の発現率及び総合障害度評価尺度(EDSS)で評価した持続的障害 進行などの臨床的評価項目であった。 第 II 相試験 AN100226-231 第 II 相試験 AN100226-231 では、MRI 指標及び臨床的指標において、本剤 3 mg/kg 及び 6 mg/kg の 2 用量での有効性が示された(表 4-1)。 MRI に関する評価項目 本剤では、プラセボ(9.7 個)と比較して、6 ヵ月間に発生した新規活動性 MRI 病巣の平均累 積数が 92%及び 89%(3 mg/kg[0.8 個]及び 6 mg/kg[1.1 個])減少した。また、Gd 造影病巣の 平均累積数はプラセボ(9.6 個)と比較して 93%及び 89%(3 mg/kg[0.7 個]及び 6 mg/kg[1.1 個])、 新規又は新規に拡大した T2 高信号病巣の平均累積数はプラセボ(2.6 個)と比較して 92%及び 88% (3 mg/kg[0.2 個]及び 6 mg/kg[0.3 個])と有意な減少が認められた。 臨床的評価項目 本剤を 6 ヵ月間投与した後に、81%の被験者が無再発であったのに対し、プラセボの投与を受 けた被験者では 62%のみであった。 PD 及び有効性プロファイルは安定しており、2 種類の用量で同等であったことから、第 III 相 試験 C-1801 及び C-1802 では、4 週間に 1 回、300 mg の固定用量を点滴静注により投与すること を決定した(3.1 項)。 第 III 相試験 C-1801 試験 C-1801 では、主として MS に対して病態修飾薬(DMD)による前治療を受けていない未 治療患者を、単剤療法として、本剤投与又はプラセボ投与のいずれかに無作為に割り付けた(表 4-2)。 MRI に関する評価項目 2 年間の投与後に、本剤では、プラセボと比較して、Gd 造影病巣数が 92%減少し(本剤 0.1 個、プラセボ 1.2 個)、新規又は新規に拡大した T2 高信号病巣数が 83%減少した(本剤 1.9 個、プラセ ボ 11.0 個)。 臨床的評価項目 2 年間の投与後に、本剤では、プラセボと比較して調整した年間再発率が 68%低下し(本剤 0.235、 プラセボ 0.733)、無再発例の割合がプラセボの 41%から本剤の 67%に上昇し、EDSS スコアによ る持続的障害進行開始までの時間をプラセボと比較して 42%延長した(ハザード比 0.58)。2 年目 までに持続的な障害進行が認められた被験者の割合は本剤で 17%、プラセボで 29%と推定された。 第 III 相試験 C-1802 試験 C-1802 は、AVONEX®の投与を継続しているにもかかわらず疾患活動性を有する(治療が 不十分な)患者を対象として、AVONEX®への本剤又はプラセボの追加投与として実施した。その 結果は試験 C-1801(単剤投与)と同様であった(表 4-2)。 MRI に関する評価項目 2 年間の投与後に、プラセボと比較して Gd 造影病巣数が 89%減少し(本剤 + AVONEX® 0.1 個、 プラセボ + AVONEX® 0.9 個)、新規又は新規に拡大した T2 高信号病巣数が 83%減少した(本 剤 + AVONEX® 0.9 個、プラセボ + AVONEX® 5.4 個)。 臨床的評価項目 2 年間の投与後に、プラセボと比較して調整した年間再発率が 55%低下し(本剤 + AVONEX® 0.336、プラセボ + AVONEX® 0.749)、無再発例の割合がプラセボ + AVONEX®の 32%から本 剤 + AVONEX®の 54%に上昇し、EDSS スコアによる持続的障害進行開始までの時間をプラセボと 比較して 24%延長した(ハザード比 0.76)。2 年目までに障害進行が認められた被験者の割合は本 剤 + AVONEX®で 23%、プラセボ + AVONEX®で 29%と推定された。 外国人患者を対象とした全ての試験の結果は統計学的に有意であり、プラセボと比較して、本 剤の効果が MS に対する既承認の DMD より大きいことが示された(表 4-3)。 未治療の被験者や、DMD の投与を継続しているにもかかわらず疾患活動性を有する被験者に対 して、同様に高い有効性が示された。これに加え、これらの試験では、投与前の高い再発率と Gd 造影病巣数が多さから、明らかにべースライン時疾患活動性が高い部分集団を含め、様々な部分 集団において、本剤の有効性が示された。いずれの第 III 相試験においても、再発の活動性の低下 は本剤の投与開始から 6 週間以内で認められ、2 年間にわたって持続した。 第 III 相試験においてみられた本剤の有効性は、現時点で公表されている最大規模の市販後デー タセットでも別途確認された。例えば、本剤投与患者を対象に実施された Swedish National Registry
によって報告された、市販後データにおいても確認されている(Holmén et al., 2011)。
4.2 日本人患者における有効性
日本人患者を対象とした試験 101MS203(表 4-1)は、2 つのパートからなる試験である。パー
ト A では、12 例に本剤 300 mg を 4 週間に 1 回点滴静注して、PK 及び PD を検討した。パート B では、94 例を 6 ヵ月間の本剤投与又はプラセボ投与に無作為に割り付け、MRI 上の新規活動性病
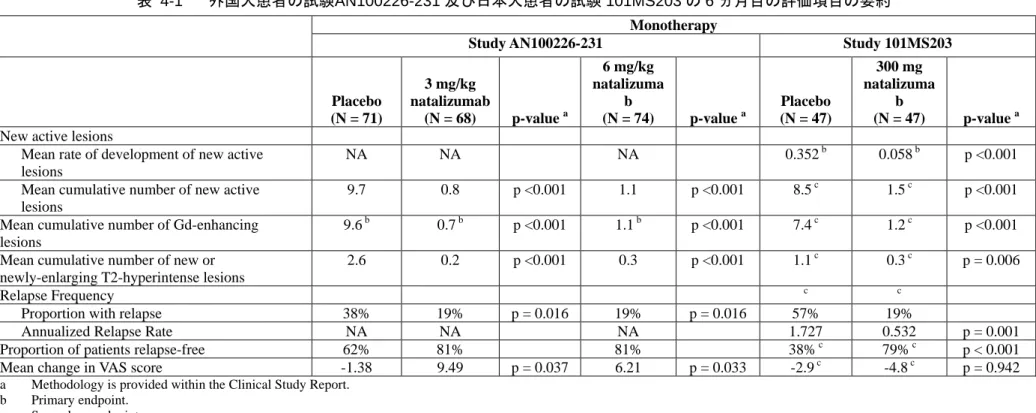
評価項目によって、本剤の有効性を評価した。試験 101MS204 は、日本人患者における有効性及 び長期安全性を検討するために試験 101MS203 で本剤 300 mg を 4 週間に 1 回、点滴静注した 97 例を対象とした多施設共同非盲検長期継続投与試験である。 試験 101MS203 パート A:PK 及び PD パート A では、12 例を対象として試験を行ったところ、本剤は外国人患者集団の場合と同等の PK(3. 項)を示した。α4 インテグリン飽和度は他の患者集団より低かったものの、一貫して高 く、臨床的意義が認められた。300 mg の用量で 4 週間に 1 回点滴静注することの妥当性が同試験 のパート B で確認された。 試験 101MS203 パート B パート B では、MS 患者 94 例を対象として、6 ヵ月間の本剤 300 mg の点滴静注による投与に ついて、プラセボと比較検討した。 パート B:MRI に関する評価項目 パート B では、本剤 300 mg を 4 週間に 1 回点滴静注したところ、プラセボと比較して 84%と 有意な MRI 上の新規活動性病巣の発生率の低下(本剤 0.058、プラセボ 0.352)や、新規活動性病 巣の平均累積数(本剤 1.5 個、プラセボ 8.5 個、82%減少)、新規 Gd 造影病巣の平均累積数(本剤 1.2 個、プラセボ 7.4 個、84%減少)及び新規又は新規に拡大した T2 高信号病巣の平均累積数(本 剤 0.3 個、プラセボ 1.1 個、73%減少)の有意な減少が認められた。 パート B:臨床的評価項目 本剤では、臨床的評価項目に関してプラセボを上回る有意な改善が認められ、調整した年間再 発率は 69%低下し(本剤 0.532、プラセボ 1.727)、無再発例の割合はプラセボの 38%から本剤の 79%に上昇した。 試験 101MS204:有効性 有効性評価指標は、投与 48 週目の EDSS スコア、投与 52 週目(投与 1 年時点での中間報告) の年間再発率及び無再発例の割合である。ベースラインから投与 48 週目までの EDSS スコアの平 均変化量は、旧本剤群、旧プラセボ群、及び全体でそれぞれ−0.02、−0.19、及び−0.09 であった。 また、投与 52 週目の調整した年間再発率は、旧プラセボ群(0.451)と比較して旧本剤群(0.302) で低かった。予測どおり、旧プラセボ群の被験者は試験 101MS204 の投与 1 年目に本剤の投与を 開始し、既に本剤の投与を受けていた旧本剤群の被験者と比較して、MS 再発が若干多かった。 投与 52 週目の無再発例の割合は、旧本剤群で 72%、旧プラセボで 60%であった。 4.3 日本人患者のデータの外国人患者のデータとの比較 日本人患者を対象として実施した試験 101MS203 及び外国人患者を対象として実施した試験 AN100226-231 は同様のデザインであった。これらはいずれも 6 ヵ月間の試験であり、同様の MRI に関する評価項目及び臨床的評価項目を用いた。これら 2 試験を比較したところ、各試験の開始 時期には 10 年以上の隔たりがあり、ベースライン時の疾患特性が一致していない点もあったが、 本剤の効果の程度は 2 つの患者集団で極めて類似していることが示された(表 4-1)。
MRI に関する評価項目 MRI 上の新規活動性病巣の平均累積数は、日本人患者(試験 101MS203)ではプラセボ(8.5 個) と比較して本剤(1.5 個)により 82%減少し、試験 AN100226-231 ではプラセボ(9.7 個)と比較 して本剤 3 mg/kg(0.8 個)及び 6 mg/kg(1.1 個)で 89%以上の減少であった。同様に、Gd 造影病 巣の平均累積数は日本人患者でプラセボ(7.4 個)と比較して本剤(1.2 個)により 84%減少し、 外国人患者ではプラセボ(9.6 個)と比較して本剤 3 mg/kg(0.7 個)及び 6 mg/kg(1.1 個)で 89% 以上の減少であった。新規又は新規に拡大した T2 高信号病巣の平均累積数は、日本人患者ではプ ラセボ(1.1 個)と比較して本剤(0.3 個)により 73%減少し、外国人患者ではプラセボ(2.6 個) と比較して本剤 3 mg/kg(0.2 個)及び 6 mg/kg(0.3 個)で 88%以上の減少であった。また、上記 のとおり、日本人患者におけるこのような無症候性疾患活動性の低下は、外国人患者を対象とし た大規模な第 III 相試験でも一致して認められた。 臨床的評価項目 臨床的評価項目に対する効果は本試験でも認められ、MRI によって評価された無症候性疾患活 動性に対する本剤の効果同様に、日本人患者と外国人患者で一致した。調整した年間再発率は、 試験 101MS203 における日本人患者ではプラセボ(1.727)と比較して本剤(0.532)で 69%低下し、 試験 C-1801 において 2 年間で認められたプラセボ(0.733)と比較して本剤(0.235)で 68%の低 下と同様であった。無再発例の割合は、日本人患者では 6 ヵ月間でプラセボの 38%から本剤の 79% に上昇し、試験 AN100226-231 の外国人患者では 62%から 81%に上昇した。試験 101MS204 の投 与 1 年目で得られた有効性評価指標(年間再発率及び EDSS スコア)の所見は、試験 101MS203 の有効性プロファイルと一致した。 以上のことから、4 週間に 1 回の本剤 300 mg の点滴静注の有効性は、日本人 RRMS 患者と外国 人患者で同様であった。
表 4-1 外国人患者の試験AN100226-231 及び日本人患者の試験 101MS203 の 6 ヵ月目の評価項目の要約 Monotherapy
Study AN100226-231 Study 101MS203
Placebo (N = 71) 3 mg/kg natalizumab (N = 68) p-value a 6 mg/kg natalizuma b (N = 74) p-value a Placebo (N = 47) 300 mg natalizuma b (N = 47) p-value a
New active lesions
Mean rate of development of new active lesions
NA NA NA 0.352 b 0.058 b p <0.001
Mean cumulative number of new active lesions
9.7 0.8 p <0.001 1.1 p <0.001 8.5 c 1.5 c p <0.001 Mean cumulative number of Gd-enhancing
lesions
9.6 b 0.7 b p <0.001 1.1 b p <0.001 7.4 c 1.2 c p <0.001 Mean cumulative number of new or
newly-enlarging T2-hyperintense lesions
2.6 0.2 p <0.001 0.3 p <0.001 1.1 c 0.3 c p = 0.006
Relapse Frequency c c
Proportion with relapse 38% 19% p = 0.016 19% p = 0.016 57% 19%
Annualized Relapse Rate NA NA NA 1.727 0.532 p = 0.001
Proportion of patients relapse-free 62% 81% 81% 38% c 79% c p < 0.001
Mean change in VAS score -1.38 9.49 p = 0.037 6.21 p = 0.033 -2.9 c -4.8 c p = 0.942
a Methodology is provided within the Clinical Study Report.
b Primary endpoint.
c Secondary endpoint.
NOTE: The numbers presented in this table reflect those shown in corresponding Clinical Study Reports (included in Module 5).
Source: M5.3.5.1-3 AN100226-231 Table 11.3-1, Table 11.3-13, Table 11.3-21, Table 11.4-5, Table 11.4-10, M5.3.5.1-4 101MS203 Table 16, Table 18, Table 19, Table 20, Table 76, Table 79, Table 81, Table 84, Table 86
表 4-2 外国人患者の第III相試験における主要評価項目及び副次評価項目の要約 Monotherapy Study C-1801 Add-on therapy Study C-1802 Placebo (N = 315) 300 mg natalizumab (N = 627) p-value a Placebo + Avonex® (N = 582) 300 mg natalizumab + Avonex® (N = 589) p-value a 1-year endpoints
Mean number of Gd-enhancing lesions c 1.3 0.1 p <0.001 0.8 0.1 p <0.001
Mean number of new or newly-enhancing T2-hyperintense lesions c
6.1 1.2 p <0.001 2.4 0.5 p <0.001
Adjusted annualized relapse rate b 0.805 0.261 p <0.001 0.816 0.383 p <0.001
Percentage relapse-free c 53% 76% p <0.001 46% 67% p <0.001
2-year endpoints
Mean change from baseline to 2 years in T2-hyperintense lesion volume (mm3) c
2890.8 -905.4 p <0.001 557.8 -259.6 p <0.001
Mean change from baseline to 2 years in T1-hypointense lesion number c
4.6 1.1 p <0.001 4.1 2.3 p <0.001
Adjusted annualized relapse rate c 0.733 0.235 p <0.001 0.749 0.336 p <0.001
Estimated proportion of patients with progression
b 0.29 0.17 p <0.001 0.29 0.23 p = 0.024
Change from baseline to 2 years in MSFC c -0.16 0.04 p <0.001 -0.10 0.05 p <0.001 a Methodology is provided within the Clinical Study Report.
b Primary endpoint.
c Secondary endpoint.
NOTE: The numbers presented in this table reflect those shown in corresponding Clinical Study Reports (included in Module 5).
Source: M5.3.5.1-5 C-1801 Table 11.4-1, Table 11.4-6, Table 11.4-8, Table 11.4-12, Table 11.4-14, Table 11.4-15, Table 11.4-16, Table 11.4-18, Table 11.4-21, M5.3.5.1-6 C-1802 Table 11.4-1, Table 11.4-6, Table 11.4-8, Table 11.4-12, Table 11.4-14, Table 11.4-15, Table 11.4-16, Table 11.4-18, Table 11.4-21
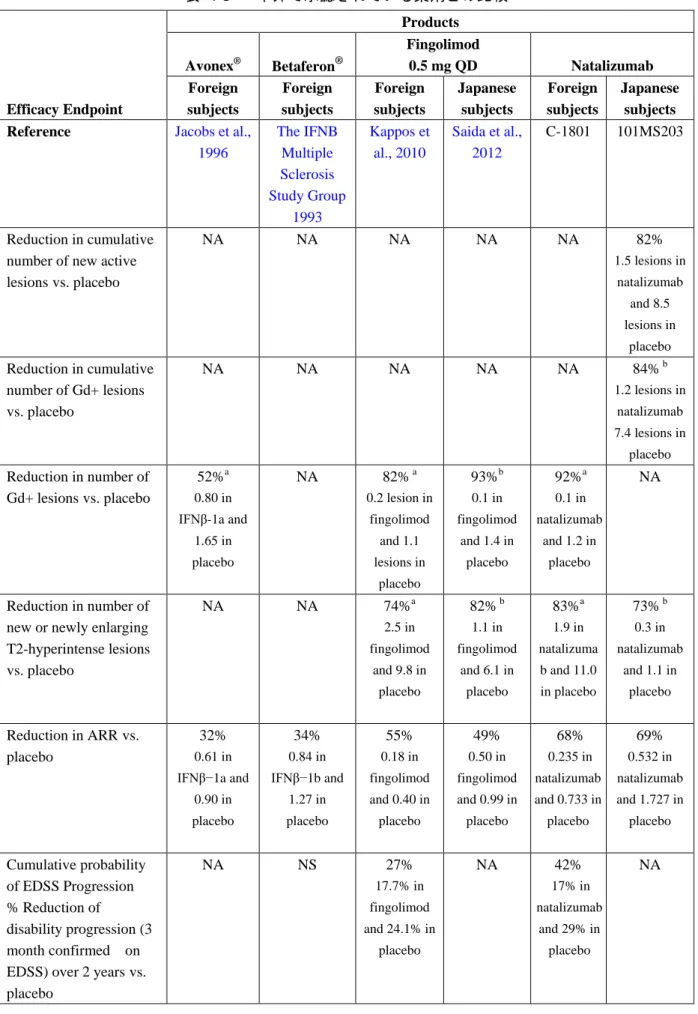
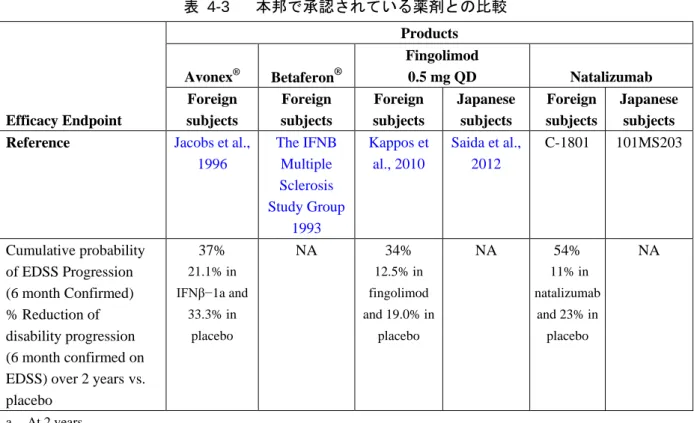
4.4 本邦で承認されている他のMS治療薬とのナタリズマブの有効性の比較 現在本邦で承認されている薬剤は 2 種類のIFNβ(アボネックス® 及びベタフェロン® )及びフィ ンゴリモド(ジレニア®/イムセラ®)である。 これらの薬剤の有効性も外国人 MS 患者を対象として評価されている。評価試験ごとに評価項 目の評価方法に相違はあるものの、それぞれの評価試験でみられたプラセボを上回る効果により、 重要度の高い臨床的評価及び MRI に関する評価項目に対して、治療薬の有効性の比較を試みるこ とが可能である。 以下の表 4-3に本邦で承認されている他の薬剤と本剤の臨床試験から得られた主な結果を示す。 それぞれの試験について全ての評価項目が示されているわけではなく、また試験期間が異なる場 合もある。本剤との直接比較試験ではないため、比較は困難である一方、明確な傾向をみること はできる。 MRI に関する評価項目 MRI 上の病巣の減少は本剤の方が IFNβ より大きく、フィンゴリモドと同程度であった。新規 活動性病巣の平均累積数は、本剤(1.5 個)ではプラセボ(8.5 個)と比較して 82%の減少が認め られたが(試験 101MS203)、この評価項目は他の薬剤では報告されていない。新規 Gd 造影病巣 の平均累積数は、本剤(1.2 個)ではプラセボ(7.4 個)と比較して 84%の減少が認められた(試 験 101MS203)。Gd 造影病巣数は、本剤(0.1 個)ではプラセボ(1.2 個)と比較して 92%の減少 が認められた(試験 C-1801)のに対し、Kappos らの報告ではフィンゴリモド 1 日 1 回 0.5 mg(0.2 個)ではプラセボ(1.1 個)と比較して 82%の減少(Kappos et al., 2010)、Saida らの報告ではフィ
ンゴリモド 1 日 1 回 0.5 mg(0.1 個)ではプラセボ(1.4 個)と比較して 93%の減少(Saida et al., 2012)、
IFNβ-1a(0.80 個)ではプラセボ(1.65 個)と比較して 52%の減少であった(Jacobs et al., 1996)。
新規又は新規に拡大した T2 高信号病巣の平均累積数は、本剤(0.3 個)では日本人患者でプラセ ボ(1.1 個)と比較して 73%(試験 101MS203)の減少、本剤の外国人患者(1.9 個)でプラセボ (11.0 個)と比較して 83%(試験 C-1801)減少したのに対し、フィンゴリモド 1 日 1 回 0.5 mg では、日本人患者でプラセボ(6.1 個)と比較してフィンゴリモド 1 日 1 回 0.5 mg(1.1 個)で 82% 減少(Saida et al., 2012)、外国人患者でプラセボ(9.8 個)と比較してフィンゴリモド 1 日 1 回 0.5 mg (2.5 個)で 74%減少であった(Kappos et al., 2010)。 臨床的評価項目 年間再発率に対しては、本剤では日本人患者(プラセボ[1.727]と比較して本剤[0.532]で 69%低下、試験 101MS203)及び外国人患者(プラセボ[0.733]と比較して本剤[0.235]で 68% 低下、試験 C-1801)の両者とも、IFNβ(外国人患者:プラセボ[0.90]と比較して IFNβ-1a[0.61]
で 32%低下[Jacobs et al., 1996]、プラセボ[1.27]と比較してIFNβ-1b 8 MIU[0.84]で 34%低下
[The IFNB Multiple Sclerosis Study Group 1993])又はフィンゴリモド 1 日 1 回 0.5 mg(日本人患
者:プラセボ[0.99]と比較してフィンゴリモド[0.50]で 49%低下[Saida et al., 2012]、外国人
患者:プラセボ[0.40]と比較してフィンゴリモド[0.18]で 55%低下[Kappos et al., 2010])に 比べて、大幅な低下が認められた。外国で実施した試験 C-1801 において、本剤では持続的障害進 行までの時間をプラセボと比較して 42%(本剤 17%、プラセボ 29%)~54%(本剤 11%、プラセ ボ 23%)延長したのに対し、フィンゴリモドではプラセボと比較して 27%(フィンゴリモド 17.7%、 プラセボ 24.1%)~34%(フィンゴリモド 12.5%、プラセボ 19.0%)延長(Kappos et al., 2010)、IFNβ-1a
ではプラセボと比較して 37%(IFNβ-1a 21.1%、プラセボ 33.3%)延長(Jacobs et al., 1996)であっ
表 4-3 本邦で承認されている薬剤との比較 Efficacy Endpoint Products Avonex® Betaferon® Fingolimod 0.5 mg QD Natalizumab Foreign subjects Foreign subjects Foreign subjects Japanese subjects Foreign subjects Japanese subjects
Reference Jacobs et al.,
1996 The IFNB Multiple Sclerosis Study Group 1993 Kappos et al., 2010 Saida et al., 2012 C-1801 101MS203 Reduction in cumulative number of new active lesionsvs. placebo NA NA NA NA NA 82% 1.5 lesions in natalizumab and 8.5 lesions in placebo Reduction in cumulative number of Gd+ lesions vs. placebo NA NA NA NA NA 84% b 1.2 lesions in natalizumab 7.4 lesions in placebo Reduction in number of Gd+ lesionsvs. placebo 52%a 0.80 in IFNβ-1a and 1.65 in placebo NA 82% a 0.2 lesion in fingolimod and 1.1 lesions in placebo 93%b 0.1 in fingolimod and 1.4 in placebo 92%a 0.1 in natalizumab and 1.2 in placebo NA Reduction in number of new or newly enlarging T2-hyperintense lesions vs. placebo NA NA 74%a 2.5 in fingolimod and 9.8 in placebo 82% b 1.1 in fingolimod and 6.1 in placebo 83%a 1.9 in natalizuma b and 11.0 in placebo 73% b 0.3 in natalizumab and 1.1 in placebo Reduction in ARRvs. placebo 32% 0.61 in IFNβ−1a and 0.90 in placebo 34% 0.84 in IFNβ−1b and 1.27 in placebo 55% 0.18 in fingolimod and 0.40 in placebo 49% 0.50 in fingolimod and 0.99 in placebo 68% 0.235 in natalizumab and 0.733 in placebo 69% 0.532 in natalizumab and 1.727 in placebo Cumulative probability of EDSS Progression % Reduction of disability progression (3 month confirmed on NA NS 27% 17.7% in fingolimod and 24.1% in placebo NA 42% 17% in natalizumab and 29% in placebo NA
表 4-3 本邦で承認されている薬剤との比較 Efficacy Endpoint Products Avonex® Betaferon® Fingolimod 0.5 mg QD Natalizumab Foreign subjects Foreign subjects Foreign subjects Japanese subjects Foreign subjects Japanese subjects
Reference Jacobs et al.,
1996 The IFNB Multiple Sclerosis Study Group 1993 Kappos et al., 2010 Saida et al., 2012 C-1801 101MS203 Cumulative probability of EDSS Progression (6 month Confirmed) % Reduction of disability progression (6 month confirmed on EDSS) over 2 yearsvs. placebo 37% 21.1% in IFNβ−1a and 33.3% in placebo NA 34% 12.5% in fingolimod and 19.0% in placebo NA 54% 11% in natalizumab and 23% in placebo NA a At 2 years b At 6 months 4.5 要約及び結論 本剤は様々な患者集団に対して、様々な疾患活動性に関する MRI に関する評価項目及び臨床的 評価項目において一貫して高い有効性を示した。この患者集団には、試験 C-1801 に代表される MS に対する治療歴のない患者や、試験 C-1802 に代表される DMD の投与を継続しているにもか かわらず疾患活動性を有する患者が含まれていた。本剤の本邦及び海外における評価試験で認め られた有効性の結果は、既に利用可能な他の DMD の臨床試験で報告されている有効性を上回る ものと考えられた。 本剤の有効性について、MS を有する日本人患者と外国人患者で直接比較を行ったところ、関 連する全ての臨床的評価項目及び MRI 評価項目において極めて類似した効果が示されたことから、 この本剤の用法・用量(ナタリズマブとして 1 回 300 mg を 4 週に 1 回 1 時間かけて点滴静注す る)を、MS を有する日本人患者の治療に用いる妥当性が裏付けられた。IFNβ 及びフィンゴリモ ドと比較しても本剤の有効性が優れていることや、前治療が無効であった MS 患者に対しても有 効性が示されていることから、より効果的な MS の治療法として実質的に満たされていない医療 上のニーズに貢献すること考えられる。