Kyushu University Institutional Repository
公共データの統合・再解析に基づく乾癬発症リスク と関連する機能多型の網羅的探索
久保田, 直人
http://hdl.handle.net/2324/4110434
出版情報:九州大学, 2020, 博士(理学), 課程博士 バージョン:
権利関係:
令和2年度 博⼠論⽂
公共データの統合・再解析に基づく
乾癬発症リスクと関連する機能多型の網羅的探索
九州⼤学⼤学院 システム⽣命科学府
⽬ 次
第1章 緒⾔ 1
1.1. 本論⽂の背景 1
1.2. 本論⽂の⽬的と構成 7
第2章 ⽅法 10
2.1. 機能多型候補同定パイプラインの構築 10
2.1.1. 乾癬感受性SNPSおよびLDvariantsの探索 10
2.1.2. エクソン領域およびスプライス部位の機能多型候補の探索 11
2.1.3. プロモーターおよびエンハンサー領域の機能多型候補の探索 12
2.2. エンリッチメント解析 13
2.3. ゲノム配列の機能解析 14
2.4. プロモーターおよびエンハンサー活性の定量解析 15
2.5. 染⾊体⾼次構造データの可視化 16
2.6. ⽐較ゲノム解析 17
2.7. 転写因⼦結合レベルの⽐較解析 17
2.8. 乾癬患者RNA-seqデータの再解析 19
2.9. 解析環境 19
第3章 結果 20
3.1. 乾癬感受性SNPSと連鎖している多型の網羅的抽出 20
3.2. エクソン領域の機能多型候補 21
3.2.1. rs60542959(COQ10A) 25
3.2.2. rs4600514(TRIM47) 28
3.3. スプライス部位の機能多型候補 30
3.2.1. rs2549797(ERAP2) 32
3.4. 転写制御領域の機能多型候補とターゲット遺伝⼦ 34
3.4.1. エンリッチメント解析 36
3.4.1.1. GO・Pathway 36
3.4.1.2. DNA結合タンパク質制御ネットワーク 38
3.4.1.3. H3K27ac修飾 38
3.4.2. プロモーター領域の機能多型候補 40
3.4.2.1. rs6906175(MICA),rs3132089(HCP5),rs3130923(MICB) 44
3.4.2.2. rs11358218(PAN2) 48
3.4.2.3. rs4889599(SETD1A) 50
3.4.2.4. rs2741432(SYS1) 52
3.4.3. エンハンサー領域の機能多型候補 54
3.4.3.1. rs72635708(ERRFI1) 57
3.4.3.2. rs11231770(PPP1R14B) 69
3.5. 乾癬患者の⽪膚病変部における機能多型候補ターゲット遺伝⼦の発現 74
第4章 考察 75
4.1. 結果のまとめ 75
4.2. エクソン領域およびスプライス部位の機能多型候補 75
4.3. 転写制御領域の機能多型候補 77
第5章 総括および展望 83
5.1. 本論⽂のまとめ 83
5.2. 今後の研究課題 83
謝辞 87
参考⽂献 88
補⾜図表 101
第1章 緒⾔
1.1. 本論⽂の背景
ヒトゲノムの構造と機能
ヒトを含む動物の遺伝情報は,核およびミトコンドリア内に存在するデオキシリボ核酸
(DNA)の配列という形で書き込まれており,遺伝情報の総体はゲノムと呼ばれる.
2001 年に最初のヒトゲノムドラフト配列が報告され 1,2,さらにその後 ENCODE3
(Encyclopedia of DNA Elements, https://www.encodeproject.org/)等の⼤規模国際プロジェ クトによってヒトゲノムの機能的な要素(functional element)が同定されてきた.その 結果,約30 億塩基対の核 DNAにおいてアミノ酸配列をコードする遺伝⼦領域は全体
の2%未満に過ぎず,遺伝⼦をコードしない領域(⾮コード領域)が核 DNA配列の⼤
部分を占めているという事実が明らかになった.当初,⾮コード領域は何の機能も持た ないガラクタ(junk DNA)であると考えられてきたが,研究が進むにつれ細胞の正確な 機能発現に必須であること,そして⾮常に多様な役割を担うことが明らかになってきた.
⾮コード領域には周辺遺伝⼦の転写レベルを調節する領域が存在し,それらは転写制御 領域(regulatory element)と呼ばれる.その中にはプロモーター領域,エンハンサー領 域,サイレンサー領域,インシュレーター領域等が含まれる.プロモーター領域は遺伝
⼦の転写開始点周辺に位置し,基本転写因⼦等から構成される転写開始前複合体の結合 部位として機能するコアプロモーターと,転写アクチベーターが結合し遺伝⼦発現をさ らに制御する近位プロモーターに⼤別される.エンハンサー領域はその多くが遺伝⼦間 領域またはイントロンに存在し,遠位の遺伝⼦発現を活性化するが,その分⼦機構は近 位プロモーターと類似しており,転写因⼦結合配列を含んでいる.サイレンサー領域は,
エンハンサー領域とは反対に遺伝⼦発現を抑制する⽅向に作⽤し,インシュレーター領 域は,転写制御の影響を特定の染⾊体領域に限定する機能を担っている.このように,
⾮コード領域は多様なメカニズムを介して遺伝⼦の転写量を適切に制御することで,正 常な細胞機能の維持に貢献していると考えられる.ゲノム配列は基本的にすべての細胞 において共通であるが,各々の細胞は⾮コード領域による様々な転写調節機構を通して,
細胞種特異的な機能の獲得や分化に伴う表現型の変化等を可能としている.⾮コード領 域の細胞種特異的な機能発現を可能としているメカニズムの実体は,エピゲノムである.
エピゲノムとは「塩基配列の変化を伴わない遺伝情報の可逆的な変化およびその総体」
であり,例えば,シトシンとグアニンが並んだ配列(CpG)にみられるシトシンへのメ
へのメチル基やアセチル基の付加(ヒストン修飾)に代表される4.これらのエピゲノ ム修飾は,転写因⼦のリクルートやヌクレオソームの再配置等のクロマチン構造の変化 を通して,周辺ゲノム領域の転写活性を上⽅もしくは下⽅制御するように働く.エピゲ ノム解析を通して細胞種ごとの⾮コード領域の機能を理解することで,我々は細胞・組 織・器官・個体の機能制御,およびそれらが破綻した結果引き起こされる様々なヒト疾 患について,分⼦メカニズムの詳細を知ることができる.
ゲノム機能エレメントを同定する取り組み
これまで,ヒトゲノムの機能的注釈(functional annotation)を⽬的として,ENCODEや Roadmap Epigenomics5(http://www.roadmapepigenomics.org/),FANTOM6(Functional Annotation of the Mouse/Mammalian Genome, https://fantom.gsc.riken.jp/)等の様々な⼤規模 国際プロジェクトは,主に次世代シーケンシング(Next-Generation Sequencing: NGS)技 術を⽤いてゲノム・エピゲノム・トランスクリプトームデータを⼤量に産⽣してきた(図 1).
は,DNA結合タンパク質と共沈降するDNA断⽚をNGSで網羅的に読み取る技術であ り,特定の転写因⼦を認識する抗体を⽤いることで,転写因⼦の結合領域をゲノム上の 広範囲にわたって決定することができる.また特定のヒストン修飾を認識する抗体を⽤
いれば,活発に転写が⾏われている領域や,プロモーターやエンハンサー等の転写制御 領域を推定することが可能である.ENCODEやRoadmap Epigenomicsは,ヒトやマウス の様々な細胞や組織における転写因⼦・ヒストン修飾のChIP-seqデータやRNA-seqデ ータ,DNase I-seq8,9およびATAC-seq10データによって検出されるオープンクロマチン 領域データなどを取得し,公共データベース上で公開している.また,FANTOM が率 いるプロジェクトでは,ゲノム上における転写開始点およびその量を精密に計測する CAGE11(Cap Analysis of Gene Expression)を⽤いて,転写産物の発現プロファイルを作 成している.CAGEは,プロモーター毎の正確な転写活性を定量することができ12,ま たエンハンサー領域から双⽅向に転写されている微量で⽐較的短いエンハンサーRNA
(eRNA)も検出・定量することができる13.FANTOMでは,各細胞種におけるプロモ ー タ ー お よ び エ ン ハ ン サ ー 活 性 の 定 量 デ ー タ を FANTOM Human Promoters
(http://slidebase.binf.ku.dk/human_promoters/ ) お よ び FANTOM Human Enhancers
(http://slidebase.binf.ku.dk/human_enhancers/)で公開している.GTEx14(Genotype-Tissue Expression, https://www.gtexportal.org/)では,様々なヒト組織検体からゲノム多型・変異 データとトランスクリプトームデータを同時に取得することで,遺伝⼦発現へ影響を与 える座位(expression Quantitative Trait Locus: eQTL)の網羅的な探索を⾏っている.同定 されたeQTLの詳細はGTEx Portalで公開されている.さらに,GEO15(Gene Expressiom Omnibus, https://www.ncbi.nlm.nih.gov/geo/)には,国際プロジェクトによって産⽣された データのみならず,世界中の研究者が NGSやマイクロアレイ等を⽤いて取得した網羅 的計測データが蓄積され続けている.DeepBlue16(https://deepblue.mpi-inf.mpg.de/)や ChIP-Atlas17(https://chip-atlas.org/),ReMap18(http://remap.univ-amu.fr/)などはChIP-seq データを中⼼とした様々なエピゲノムデータを再解析し独⾃のデータベース上で公開 しており,すべての研究者はそれらのデータを簡単に再利⽤することが可能である.こ のように,複数の国際プロジェクトや個々の研究グループが独⽴して取得した⼤規模デ ータは,公共データベース上で世界中の誰もが利⽤できる形で提供されているため,研 究者はそれぞれの⽬的に合わせてこれらのデータを統合し効率的に使⽤することで,⾃
⾝の研究に役⽴てることが可能となっている.例えば,疾患ゲノミクス研究における公 共データの再活⽤は,低コストで効率的な疾患発症メカニズムの発⾒および創薬ターゲ ット分⼦の探索への展開が期待されるため,⾮常に注⽬されている.
具体的には,以上に列挙したようなプロモーターおよびエンハンサーの位置・活性デ ータとeQTLデータ,さらに染⾊体⾼次構造データを組み合わせることで,転写制御領 域のターゲット遺伝⼦を正確に推定することも可能である.近年,Hi-C19やChIA-PET20,21 など多くの染⾊体⾼次構造解析⼿法が開発されており,とくにHi-C解析によってゲノ ムDNAは全域にわたって約100万塩基(1 Mb)ごとにTAD(Topologically Associating Domain)と呼ばれる空間的機能ドメインを形成しそれらが数珠状に連なった構造をと ることが明らかになった 19,22,23.これまで,疾患の発症リスクと関連する複数の⾮コー ド領域が,数10 kbから数Mb離れた遠位の遺伝⼦領域と物理的に接触し発現制御を⾏
っていることが報告されている24–33.これまで産⽣された染⾊体⾼次構造データの多く は3D Genome Browser34(http://promoter.bx.psu.edu/hi-c/)で閲覧できる
.
多因⼦疾患における遺伝要因
多くのヒト疾患は,遺伝要因と環境要因が複雑に絡み合い発症に⾄る.そのような疾患 を多因⼦疾患(multifactorial disease)あるいは複合性疾患(complex disease)と呼ぶ.多 因⼦疾患の遺伝要因については,単⼀の遺伝⼦が疾患発症に対して強い効果を持つ(メ ンデル型)のではなく,⽐較的弱い効果を持つ複数の遺伝⼦が集積することによって発 症に寄与する(⾮メンデル型).このことから,多遺伝⼦性疾患(polygenic disease)と も称される.個⼈によって保有する多型の組み合わせが異なるうえに,⾷事や喫煙,感 染症等の様々な環境要因が関与するため,その発症機序は⾮常に複雑である.これまで,
多因⼦疾患を引き起こす遺伝要因,すなわち疾患感受性領域の探索を⽬的として,罹患 同胞対解析やその他の連鎖解析が盛んに⾏われてきたが,近年はゲノムワイド関連解析
(Genome-Wide Association Study: GWAS)が最も主要な実験⼿法として⽤いられている.
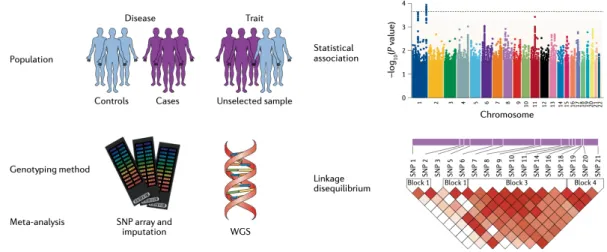
GWASとは,ゲノム中に存在する⼀塩基多型(Single Nucleotide Polymorphism: SNP)の 遺伝型を網羅的に決定し,集団におけるその頻度と疾患発症や量的形質との関連を探索 する⼿法である35(図2).
図2:GWASの概要.Tam et al., 2019より改変・転載.
ヒトゲノム配列が決定され,HapMap project36を筆頭とした国際プロジェクトによっ て多型情報が⼗分に整備されたこと,さらに SNP アレイによるジェノタイピングのコ ストが⼀⼈あたり数千円と⾮常に低コストとなったため,できるだけ⼤きいサンプルサ イズが必要とされるGWAS が現在では広く実施されている.実際には,ヒト集団にお ける連鎖不平衡構造(linkage disequilibrium)を考慮し,他のSNP の遺伝型を効率よく 代理する数万から数百万のSNPs(tag SNPs)が選択されタイピングされている.2002年 に,約10万SNPsを⽤いたGWASによって⼼筋梗塞感受性領域の同定がなされた37こ とを契機に,その後,様々な疾患・形質に対して GWAS が試みられており,新規感受 性領域が報告され続けている.GWASによって報告された感受性領域はGWAS Catalog38
(https://www.ebi.ac.uk/gwas/)に集積され,データベースとして公開されている.しか しながら,GWASで同定された疾患感受性SNPsは単なるマーカーにすぎず,それ⾃体 が近傍遺伝⼦に何らかの影響を与えているケースは⾮常に少ない.なぜなら,前述の通 りGWASでは選択されたtag SNPsのみがタイピングされているため,多くの場合それ らと連鎖不平衡の関係にあるSNPsの中に真の原因領域・原因多型が存在するからであ る.したがって,真に遺伝⼦発現および機能に影響を与えている機能多型(functional variant)の探索が,疾患の詳細な分⼦メカニズムの同定において⾮常に重要である.ま た,これまではコード領域における多型が関与する疾患発症メカニズムが多くの注⽬を 集めてきたが,疾患感受性遺伝⼦の同定率向上が望まれているにもかかわらず,⾮コー ド領域に存在するエンハンサー等の遠位発現制御領域の多型については,ターゲット遺 伝⼦の推定が技術的に困難である等の理由から探索が困難であった.個々の機能多型お よび疾患感受性遺伝⼦を正確に同定しその機能を解析することは,疾患発症メカニズム
の解明,新たな治療薬の開発,予防医学の確⽴のためにも必要であり,喫緊の課題であ る.
乾癬の概観と遺伝要因
代表的な多因⼦疾患の⼀つに,乾癬(psoriasis)が挙げられる.乾癬は慢性炎症性⽪膚 疾患であり,表⽪⾓化細胞の増殖あるいは⾓質の剥離障害による過剰な⾓質肥厚を特徴 とする(図3).
図3:乾癬患者の病変部位.Mantovani et al., 2016より転載.
乾癬の歴史は古く,聖書には leprosy(Zaraath)と記載があり,不快な外観の⽪膚疾 患の総称であったと推測されている.18世紀にWilllanが乾癬を Hansen病と鑑別した が,19世紀になりHebraがpsoriasisを独⽴疾患として報告した39.当時,乾癬に対して すでに遺伝歴および家族歴の存在が指摘されていた39.乾癬患者の⽪膚では,表⽪細胞 のターンオーバーの亢進と,表⽪⾓層内〜直下の⽩⾎球の浸潤(Munro微⼩膿瘍)が認 められる.1970 年代ごろまでは乾癬の病態に関する研究の中⼼は⾓化細胞の過増殖で あり,免疫疾患とは考えられていなかった.しかしながら,病変部位に好中球やT細胞 の浸潤が認められることや,T細胞の機能を抑制する薬剤(Ciclosporin)40,および⽪膚
リックシンドロームを発症すると,脂肪細胞から産⽣・分泌される脂肪組織由来⽣理活 性物質であるアディポサイトカインが作⽤して,様々な病態を引き起こすことが知られ ている.アディポサイトカインは,アディポネクチン,レプチン,レジスチン,TNF-α,
などの総称であり,アディポネクチンは⾼⾎圧や動脈硬化に,レプチン,TNF-αなどは 耐糖能障害・脂質代謝異常に関与している47.実際に,メタボリックシンドロームの肝 臓に お け る 表 現 型 と 考え られ て い る⾮ア ル コ ー ル 性脂 肪 肝 炎(nonalcoholic
steatohepatitis: NASH)患者では乾癬の有病率が有意に⾼いことが報告されている 42–45.
NASHと乾癬を結びつけるメカニズムの詳細は未だ不明であるものの,炎症を起こした 肝臓から放出される種々の炎症性サイトカイン(C 反応性タンパク質,TNF-α,IL-6)
や凝固促進因⼦(フィブリノーゲン,plasminogen activator inhibitor-1),線維化促進性サ イトカイン(TGF-β)などが乾癬の病態⽣理において重要な役割を担っている可能性が 考えられている48–51.炎症性肝臓からのこれらの因⼦の放出は,⽪膚における表⽪細胞 の増殖促進,炎症の誘導,および様々な⾎管細胞接着分⼦の発現上昇に伴う免疫細胞の ホーミングを引き起こすことで,乾癬の発症・重症化に影響を与えるとの仮説が有⼒で ある.また,乾癬の⽪膚病変部位から放出されるIL-6,TNF-α,IL-17などの炎症性サイ トカインが,肝臓の炎症を更に悪化させることも想定される.以上を総合すると,乾癬 は⽪膚疾患でありながら,脂質代謝異常や肝臓における炎症などと深い関連をもつ全⾝
性疾患であるため,その遺伝要因を探索する際には,免疫細胞のみならず肝臓や脂肪を 含めた他の組織についても影響を評価する必要がある.
⽇本における乾癬の罹患率は 0.1~0.3%程度と推定されているが,欧⽶における罹患
率は 2~4%程度である 52,53.この差は⼈種間の遺伝的背景の違いに起因すると考えられ
ている.遺伝要因の関与が⽰唆されているものの,家族性の乾癬は遺伝様式が複雑であ ることから,単⼀遺伝⼦疾患であるとの⾒⽅は否定されている.そのため,発症リスク 上昇に関与する多型の探索を⽬的として,乾癬患者を対象とした⼤規模なGWAS が主 にヨーロッパ⼈集団で⾏われてきた.これまで,GWASによる乾癬感受性SNPsの探索 に関する14報の論⽂が報告されており54–67,同定された感受性SNPsはHLA領域に集 中することや,免疫細胞,とくにT細胞の転写制御領域にエンリッチすることが明らか になっている66,68.しかしながら,実際に遺伝⼦発現や遺伝⼦の機能に影響を与える機 能多型の多くは不明であり,加えて,ほとんどの先⾏研究では⾮コード領域については 免疫細胞に対する影響にのみ着⽬しているため,その他の細胞・組織における影響は⾒
過ごされている可能性が⾼い.このことから,乾癬の分⼦発症メカニズムに対する新知
⾒を得るためには,GWASによって発⾒された疾患感受性SNPs情報を⽤いることによ
って,様々な細胞・組織において影響を与える真の乾癬関連機能多型を探索することが 必要である.
1.2. 本論⽂の⽬的と構成
明らかにすべきこと
2000 年代以降,ヒトの多因⼦疾患の遺伝要因は主に GWAS によって探索されてきた.
とくにヨーロッパ⼈集団における乾癬患者を対象としたGWAS によって,多くの乾癬 感受性領域が同定されてきており,それらは T 細胞をはじめとした免疫細胞の転写制 御領域にエンリッチすることが明らかにされてきた.しかしながら,実際に遺伝⼦の機 能発現制御に影響を与える機能多型については未だその多くが不明なままであり,また 免疫細胞以外の細胞の転写制御領域に存在する有⼒な機能多型の存在もほとんど確認 されていない.⼀般に疾患発症の分⼦メカニズムを理解するためには,GWASで⾒つか った感受性SNPsと強い連鎖不平衡の関係にある多型の中から真の機能多型を網羅的に 探索し,それらが影響を与えている遺伝⼦,パスウェイ,責任細胞・組織を同定する必 要がある.また,複数の疾患感受性領域の情報を元に⼀から実験的に機能多型を探索す るのは⾮常に⾻の折れる作業であるため,可能な限り低コストで,効率的に,かつ網羅 的にそれらを探索するシステムの構築が求められている.疾患発症の分⼦メカニズムに 関与する遺伝⼦およびパスウェイは治療薬のターゲットとなり得るため,それらを同定 するためのストラテジーの確⽴は⾮常に重要である.
以上の問題点を解決するために,本論⽂では以下の2点についてバイオインフォマテ ィクス的アプローチを⽤いて取り組む.
1.遺伝性疾患の発症リスクと関連する機能多型を網羅的に探索する公共データ統合 解析パイプラインを新たに構築する.
フォマティクス的⼿法を提案することにより,低コストで効率的な疾患発症メカニズム の新規発⾒が可能となることが期待される.
本論⽂は多因⼦疾患全般に適⽤できるパイプラインの構築を⽬的としており,乾癬は あくまでモデル疾患である.乾癬をモデルとして選択した理由は次に述べる 3 点であ る.
1.これまで多くのGWAS が実施されており,他の多因⼦疾患と⽐較して⼗分な数の 感受性SNPsが報告されているため,未知の機能多型の存在が期待されること.
2.先⾏研究において,免疫細胞の転写制御領域へのエンリッチなど⾮コード領域の乾 癬感受性SNPsに対する知⾒が蓄積されているため,構築したパイプラインを⽤いて得 た解析結果の妥当性を検証できること.
3.GWASは多く⾏われているものの,とくに⾮コード領域については機能多型そのも のの網羅的探索についての報告は少ないこと.
以上の理由から,本論⽂の解析対象としては乾癬が適当であると判断した.
本論⽂の構成
本論⽂の構成は次の通りである.第2章では,本論⽂における解析の⼿法および⽤いた データセット・ソフトウェアについて述べる.第3章では,構築した解析パイプライン の詳細および抽出された機能多型候補の遺伝⼦機能発現に対する影響について調査し た結果を述べる.また,制御配列の機能多型候補の情報を⽤いて各種エンリッチメント 解析を⾏った結果について記述する.さらに,予測されたターゲット遺伝⼦の乾癬患者 病変部位と正常部位における発現量の変化について解析した結果についても述べる.第 4章では,抽出された機能多型候補およびターゲット遺伝⼦の乾癬発症リスクにおける
⽣物学的意義について考察し,最後に本論⽂で新規に同定した分⼦発症メカニズムとそ の重要性について議論する.第5章では,以上の結果を総合し,今後の展望について述 べる.
第2章 ⽅法
2.1. 機能多型候補同定パイプラインの構築
乾癬の発症リスクと関連する機能多型候補の同定を⽬標として,解析パイプラインの新 規構築を⾏った.データ間の統合にはPythonのPandasライブラリを⽤いた.
2.1.1. 乾癬感受性SNPsおよびLD variantsの探索
最初に,これまでに報告された GWAS データが集積されている公共データベースであ
るGWAS Catalogより形質・疾患感受性SNPsデータをダウンロードした(https://www.
ebi.ac.uk/gwas/docs/file-downloads/gwas_catalog_v1.0.2-associations_e93_r2019-01-11.tsv).
このデータから,乾癬(“Psoriasis”,“Cutaneous psoriasis”,“Psoriasis vulgaris”)の発症 リスクとの関連が報告されているSNPs(乾癬感受性SNPs)のみを抽出し,以降の解析 に使⽤した.GWAS Catalogに登録されている形質・疾患感受性SNPsデータはすべて ゲノムアセンブリhg19(GRCh37)にマップされているため,以降解析パイプライン中 で⽤いるデータセットについてはすべてhg19にマップされたものを選択した.
次に,乾癬発症リスクとの関連が予測される機能多型を探索するために,LDlink69の Application Programming Interface(API)を⽤いて乾癬感受性SNPsと強い連鎖不平衡の 関係にある多型を取得した.API アクセスの際に使⽤したコードは以下の通りである.
$ wget https://ldlink.nci.nih.gov/LDlinkRest/ldproxy?var=rs???&p op=EUR&r2_d=r2&token=mytoken
1000 Genomes Project(Phase 3)70によって収集されたヨーロッパ⼈集団503⼈分(Utah Residents from North and West Europe, Toscani in Italia, Finnish in Finland, British in England and Scotland, Iberian population in Spain)のゲノム多型データに基づき計算された r-
!" = (&'(&)*− &'*&)()"
&'&)&(&*
2.1.2. エクソン領域およびスプライス部位の機能多型候補の探索
エクソン領域およびスプライス部位に位置する多型の抽出にはVariant Effect Predictor74
を⽤い,それらをexonic variantsと定義した.エクソン領域の多型に関しては,PolyPhen-
275によるHDIV prediction scoreが“probably damaging”もしくは“possibly damaging”であ るmissense多型,frame shift多型,start lost多型およびstop gained多型を機能多型候補 と定義した.PolyPhen-2は塩基配列情報,進化的保存性,タンパク質⽴体構造情報に基
づいてnonsynonymous多型の有害性をin silicoで総合的に評価できるソフトウェアであ
る.スプライス部位の多型に関しては,リスクアリル・⾮リスクアリル間のMaxEntScan
score76の差をスプライス部位選択性の変化の評価に⽤いた.MaxEntScanは,5´末端スプ
ライス部位の9塩基(エクソン側3塩基 + イントロン側6塩基)および3´末端スプラ イス部位の23塩基(エクソン側3塩基 + イントロン側20塩基)の配列情報のみを⽤
いて,それぞれのスプライス部位のスプライシング強度,すなわちスプライス部位の選
択性をin silicoで評価するソフトウェアである.個⼈間で使⽤頻度が異なるエクソンの
スプライス部位が SNP によってスプライシング強度を変化させる場合,MaxEntScan
score の差が 5.0 以上であればエクソン使⽤頻度が異なる場合が多いという結果を所属
研究室の解析によってすでに得ているため(2020年5⽉30⽇現在,論⽂投稿中),本
論⽂ではMaxEntScan scoreの差が5.0以上である場合にみられた多型をスプライス部位
の機能多型候補と定義した.本論⽂では,エクソン領域およびスプライス部位における 機能多型候補をまとめてexonic functional variantsと定義した.Exonic functional variants の遺伝⼦発現量に対する影響については,GTEx Analysis V7 project77によって収集され たゲノムデータおよびトランスクリプトームデータから計算された eQTL データを⽤
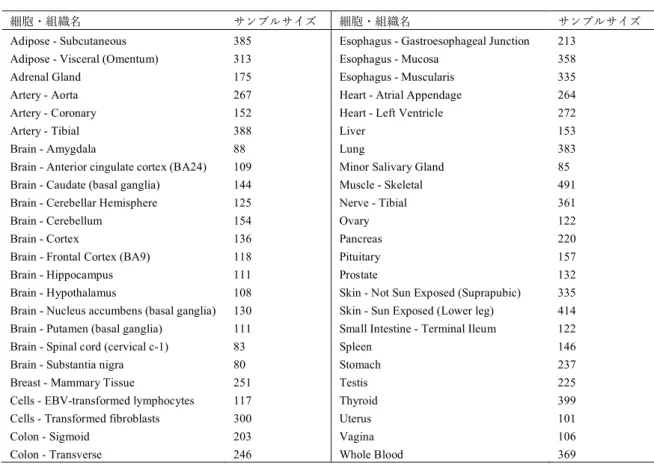
いて解析を⾏った(表1,dbGaP accession: phs000424.v7.p2).
表1:eQTL解析に⽤いた細胞・組織とそのサンプルサイズ
細胞・組織名 サンプルサイズ 細胞・組織名 サンプルサイズ
Adipose - Subcutaneous 385 Esophagus - Gastroesophageal Junction 213
Adipose - Visceral (Omentum) 313 Esophagus - Mucosa 358
Adrenal Gland 175 Esophagus - Muscularis 335
Artery - Aorta 267 Heart - Atrial Appendage 264
Artery - Coronary 152 Heart - Left Ventricle 272
Artery - Tibial 388 Liver 153
Brain - Amygdala 88 Lung 383
Brain - Anterior cingulate cortex (BA24) 109 Minor Salivary Gland 85
Brain - Caudate (basal ganglia) 144 Muscle - Skeletal 491
Brain - Cerebellar Hemisphere 125 Nerve - Tibial 361
Brain - Cerebellum 154 Ovary 122
Brain - Cortex 136 Pancreas 220
Brain - Frontal Cortex (BA9) 118 Pituitary 157
Brain - Hippocampus 111 Prostate 132
Brain - Hypothalamus 108 Skin - Not Sun Exposed (Suprapubic) 335
Brain - Nucleus accumbens (basal ganglia) 130 Skin - Sun Exposed (Lower leg) 414 Brain - Putamen (basal ganglia) 111 Small Intestine - Terminal Ileum 122
Brain - Spinal cord (cervical c-1) 83 Spleen 146
Brain - Substantia nigra 80 Stomach 237
Breast - Mammary Tissue 251 Testis 225
Cells - EBV-transformed lymphocytes 117 Thyroid 399
Cells - Transformed fibroblasts 300 Uterus 101
Colon - Sigmoid 203 Vagina 106
Colon - Transverse 246 Whole Blood 369
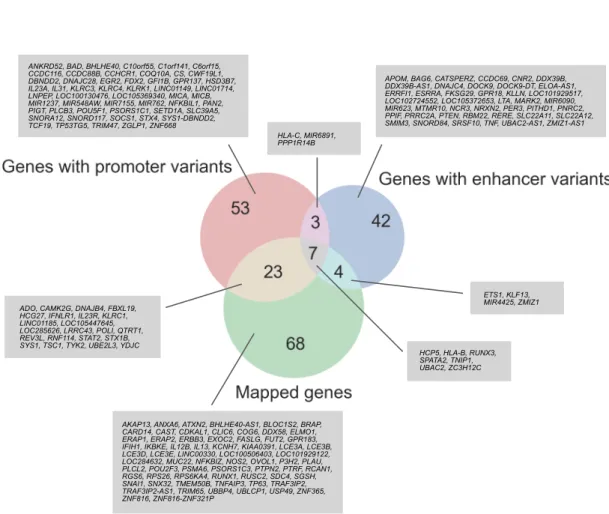
2.1.3. プロモーターおよびエンハンサー領域の機能多型候補の探索
本論⽂では,転写開始点から上流2 kbの領域をプロモーター領域と定義し,機能多型 候補の探索を⾏った.UCSC table Browser からプロモーター領域の BEDファイル(N CBI RefSeq Curated Genes)を取得後,bedtools78(version 2.27.1)を⽤いてプロモータ ー領域に存在するLD variants を抽出し,それらをpromoter variantsと定義した.エン ハンサー領域の多型およびそのターゲット遺伝⼦に関しては,FANTOM によって提供
与える多型は,それぞれ promoter eQTL variants,enhancer eQTL variants と定義した.
次に,SNP2TFBS79(https://ccg.epfl.ch//snp2tfbs/)を⽤いて,転写因⼦の結合レベルに影 響を与える可能性がある機能多型候補の探索を⾏った.SNP2TFBSは,転写因⼦結合配 列(Transcription Factor Binding Site: TFBS)の位置特異的重み⾏列(Position Weight Matrix: PWM)スコアを計算し,多型(SNP/indel)を含む際のスコアの変化量に基づき,
転写因⼦の結合に対する多型の影響をin silicoで評価するツールである.SNP2TFBSに よる解析を通して,promoter eQTL variants および enhancer eQTL variants の中で転写 因⼦結合配列に位置しているものを抽出し,それぞれpromoter TFBS variants,enhance
r TFBS variants と定義した.結合配列中に存在するものの PWM スコアを全く変化さ
せない多型は,転写因⼦の結合に影響を及ぼさないことが予想されたことからTFBS v
ariantsには含めなかった.さらに,その転写因⼦結合配列が機能的であることを確かめ
るために,対応する転写因⼦のChIP-seqデータを⽤いて,何らかの細胞・組織において 実際に転写因⼦の結合が認められるかどうか検証した.ChIP-seqのピークコールデータ は,公共エピゲノムデータベースであるDeepBlueのAPIアクセスを⽤いて取得した.
DeepBlue は,主要な国際プロジェクト(ENCODE,Roadmap Epigenomics,DEEP(htt
p://www.deutsches-epigenom-programm.de/),BLUEPRINT Epigenome(http://www.bluepr int-epigenome.eu/),CEEHRC(http://www.epigenomes.ca/),CREST(http://crest-ihec.jp
/))によって産⽣された ChIP-seq を含むエピゲノムデータと,ChIP-Atlas によって再
解析された⾮国際プロジェクトのエピゲノムデータを⼀括して検索できるデータベー スであり,ウェブインターフェースやAPIアクセスを介して興味のあるゲノム領域のC
hIP-seq ピークコールデータ等を⼀挙に取得することが可能であるため,本論⽂で採⽤
した.
2.2. エンリッチメント解析
本解析パイプラインで抽出された機能多型候補のターゲット遺伝⼦群のエンリッチメ ン ト 解 析 に は Metascape80(http://metascape.org/gp/index.html) お よ び ChEA381
(https://amp.pharm.mssm.edu/chea3/)を⽤いた.MetascapeはGOなどのオントロジーデ ータに基づき任意の遺伝⼦群における機能の偏りを検出するエンリッチメント解析ツ ールであり,ChEA3 は ENCODE や ReMap 等の公共 ChIP-seq データや,GTEx や
ARCHS482 等の公共 RNA-seq データに基づく共発現プロファイルによって推定された
DNA結合タンパク質–ターゲット遺伝⼦相互作⽤の情報を⽤いて,任意の遺伝⼦群の発 現制御に関与するDNA結合タンパク質を同定するためのエンリッチメント解析ツール
である.Metascapeにおいて設定したパラメータは次の通りである;“Input as Species” –
“H. sapiens”,“Analysis as Species” – “H. sapiens”,“Ontology source” – “KEGG Pathway, GO Biological Processes, Reactome Gene Sets, Canonical Pathways and CORUM”.また,機 能多型候補がターゲットとする遺伝⼦のプロモーター領域におけるヒストン修飾のエ ン リ ッ チ メ ン ト 解 析 に は ChIP-Atlas の “Enrichment Analysis ” (http://chip- atlas.org/enrichment_analysis)機能を⽤いた.NCBI RefSeq Curated Genesの転写開始点か
ら上流 2 kb の領域を対照データとして⽤いた.本論⽂において設定したパラメータは
次の通りである;“Antigen class” – “Histone”,“Cell type Class” – “All cell types”,“Threshold for Significance” – “50”.算出されたP値はBenjamini-Hochberg法83により多重⽐較のた めの補正を⾏い,adjP < 0.1を有意とみなした.
2.3. ゲノム配列の機能解析
機能多型候補が存在しているゲノム領域のエピゲノム修飾状態の可視化には UCSC Genome Browser84(https://genome.ucsc.edu)を⽤いた.7つの細胞株(GM12878, H1-hESC, HSMM, HUVEC, K562, NHEK, NHLF)のヒストン修飾のChIP-seqシグナル(H3K4me3, H3K4me1, H3K27ac)はtransparent overlay methodで可視化した.また,様々な細胞・組 織におけるクロマチン状態の解析にはChromHMM85–87によって定義されたimputed 25- STATE MODEL を⽤いた.解析に⽤いたヒト細胞・組織(Liver, Foreskin Fibroblasts, Foreskin Keratinocytes, Foreskin Melanocytes, Adipose Nuclei, Lung, Ovary, Skeletal Muscle Female, Skeletal Muscle Male, Thymus, Pancreatic Islets, Spleen, Stomach Mucosa, Small Intestine, Sigmoid Colon, Hematopoietic stem cells, T helper memory cells from peripheral blood, T CD8+ naive cells from peripheral blood, T cells from cord blood, T cells from peripheral blood, Hematopoietic stem cells G-CSF-mobilized Female, Hematopoietic stem cells G-CSF-mobilized Male, T helper naive cells from peripheral blood, B cells from cord blood, B cells from peripheral blood, Neutrophils from peripheral blood, T helper cells from peripheral blood, Monocytes from
したPWMスコア(REST:MA0138.2,Arnt:MA0004.1,BHLHE40:MA0464.2,Tcf3:
MA0464.2,Spi1:MA0080.3,EGR1:MA0162.2,CEBPB:MA0466.1,Bach1::Mafk:MA0591.1,
FOS:MA0476.1,JUND:MA0491.1,SP1:MA0079.3,SP2:MA0516.1,ZNF263:MA0528.1)
に基づき,WebLogo89(version 3.7.1)を⽤いて作成した.
2.4. プロモーターおよびエンハンサー活性の定量解析
プロモーターおよびエンハンサー活性の定量には,FANTOM によって産⽣・解析され た CAGE データを使⽤した.機能多型候補が存在するプロモーターもしくはエンハン サーのターゲット遺伝⼦の活性の定量データはFANTOM Human Promotersから取得し た.機能多型候補が存在するエンハンサーの活性の定量データは FANTOM Human En hancersから取得した.
2.5. 染⾊体⾼次構造データの可視化
ヒト肝臓および IMR90 細胞株における染⾊体のコンタクトマップは 3D Genome
Browser を⽤いて Hi-C データから作成し,それぞれを CTCF の ChIP-seq シグナル
(ENCODE accession number of Liver CTCF ChIP-seq: ENCFF555SBI,table name of IMR90 CTCF ChIP-seq in the UCSC Genome Browser: wgEncodeSydhTfbsImr90CtcfbIggrabSig)と ともに可視化した.コンタクトマップの解像度は10 kbとした.Hi-C データの4C-like plotの作成には3DIV90(http://kobic.kr/3div/)を⽤いた.K562細胞株およびMCF-7細胞 株におけるRNAポリメラーゼIIのChIA-PETデータから⽣成されたクロマチン相互作
⽤データは,UCSC Genome Browserを⽤いてFANTOMのenhancer–promoter correlation データとともに可視化した.CTCF 結合モチーフの位置および⽅向は JASPAR database を⽤いて探索した.
2.6. ⽐較ゲノム解析
複数の⽣物種のゲノム配列⽐較解析を⽬的として,UCSC Genome Browser を⽤いてゲ ノムアラインメントを⾏った.解析に⽤いた⽣物種およびゲノムアセンブリバージョン を表2に⽰す.
表2:ゲノムアラインメントに使⽤した⽣物種およびゲノムアセンブリバージョン
⽣物種 ゲノムアセンブリバージョン
Human GRCh37/hg19
Chimp WUGSC Pan_troglodytes-2.1.4/panTro4
Mouse GRCm38/mm10
Rabbit Broad/oryCun2
Cow Baylor Btau_4.6.1/bosTau7
Cat ICGSC Felis_catus 6.2/felCat5
Dog Broad/canFam3
Elephant Broad/loxAfr3
Chicken ICGSC Gallus_gallus-4.0/galGal4 X. tropicalis JGI 7.0/xenTro7
Zebrafish Zv9/danRer7
Lamprey WUGSC 7.0/petMar2
2.7. 転写因⼦結合レベルの⽐較解析
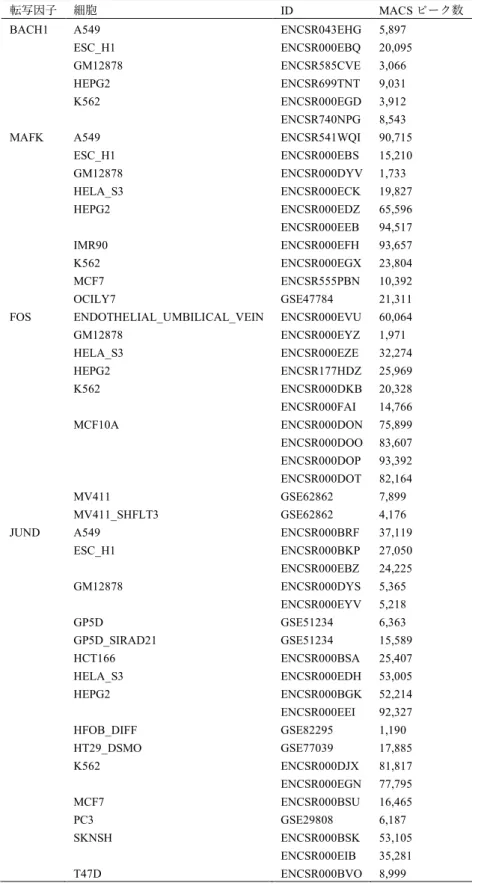
機能多型候補の転写因⼦結合レベルに対する影響を評価するために,様々なヒト細胞に
の Scipy ライブラリを⽤いて Mann–Whitney の U 検定を⾏った.算出した P 値は Benjamini-Hochberg 法により多重⽐較のための補正を⾏い,adjP < 0.1 を有意とみなし た(adjP < 0.1の場合には*を,adjP < 0.01の場合には**を,adjP < 0.001の場合には***
を付した).解析に使⽤したデータセットは表3に⽰す.
表3:ReMapからダウンロードした転写因⼦ChIP-seqデータセット
転写因⼦ 細胞 ID MACSピーク数
BACH1 A549 ENCSR043EHG 5,897
ESC_H1 ENCSR000EBQ 20,095
GM12878 ENCSR585CVE 3,066
HEPG2 ENCSR699TNT 9,031
K562 ENCSR000EGD 3,912
ENCSR740NPG 8,543
MAFK A549 ENCSR541WQI 90,715
ESC_H1 ENCSR000EBS 15,210
GM12878 ENCSR000DYV 1,733
HELA_S3 ENCSR000ECK 19,827
HEPG2 ENCSR000EDZ 65,596
ENCSR000EEB 94,517
IMR90 ENCSR000EFH 93,657
K562 ENCSR000EGX 23,804
MCF7 ENCSR555PBN 10,392
OCILY7 GSE47784 21,311
FOS ENDOTHELIAL_UMBILICAL_VEIN ENCSR000EVU 60,064
GM12878 ENCSR000EYZ 1,971
HELA_S3 ENCSR000EZE 32,274
HEPG2 ENCSR177HDZ 25,969
K562 ENCSR000DKB 20,328
ENCSR000FAI 14,766
MCF10A ENCSR000DON 75,899
ENCSR000DOO 83,607 ENCSR000DOP 93,392 ENCSR000DOT 82,164
MV411 GSE62862 7,899
MV411_SHFLT3 GSE62862 4,176
JUND A549 ENCSR000BRF 37,119
ESC_H1 ENCSR000BKP 27,050
ENCSR000EBZ 24,225
GM12878 ENCSR000DYS 5,365
ENCSR000EYV 5,218
GP5D GSE51234 6,363
GP5D_SIRAD21 GSE51234 15,589
HCT166 ENCSR000BSA 25,407
HELA_S3 ENCSR000EDH 53,005
HEPG2 ENCSR000BGK 52,214
2.8. 乾癬患者 RNA-seq データの再解析
公共データベースに登録されている乾癬患者⽪膚病変部の RNA-seq データを⽤いた発 現変動遺伝⼦の抽出には,BioJupies92(https://biojupies.cloud/)を⽤いた.BioJupiesでは,
公共データベースに登録されている RNA-seq データから kallisto93を⽤いて転写産物の 発現量を定量し,The characteristic direction94を⽤いて発現変動遺伝⼦を検出している.
発現変動についてはadjP < 0.1を有意とみなした.使⽤したデータセットの詳細および 解析結果のURLを表4に⽰す.
表4:BioJupiesで解析した乾癬患者⽪膚病変部のRNA-seqデータセット
GEO ID サンプルサイズ
(疾患群 vs 対照群)
PubMed
ID 解析結果のURL
GSE47944 8 vs 5 24909886 https://biojupies.cloud/notebook/BbDYIahv7 GSE74697 36 vs 16 27015450 https://biojupies.cloud/notebook/MU7vHmPf5
2.9. 解析環境
本論⽂におけるすべての解析はPython(version 3.7.3)およびGNU Bash(version 3.2)
環境下で⾏った.Python環境下で⽤いたライブラリを表5に⽰す.
表5:使⽤したPythonライブラリ
ライブラリ バージョン 解析
Matplotlib 3.1.1 図表作成
Seaborn 0.9.0 図表作成
Pandas 0.24.2 データ統合
Numpy 1.16.4 数値計算
Scipy 1.2.1 統計検定
第3章 結果
3.1. 乾癬感受性 SNPs と連鎖している多型の網羅的抽出
本論⽂では,乾癬の発症リスクに関与する機能多型の網羅的同定を⽬的として,複数の 公共データセットを統合した解析パイプラインを新たに構築した.その概要を図4に⽰
す.
"Psoriasis", "Psoriasis vulgaris"
"Cutaneous psoriasis"
2,894 LD variants r2 > 0.8 in European population 1000 Genomes Project, LDlink
Enhancer-Promoter correlations FANTOM5 2 kb upstream regions of TSS
NCBI RefSeq
210 exonic variants 93 genes Data
Database
Analysis Tools
eQTL GTEx v7
94 promoter eQTL variants 45 genes
11 enhancer eQTL variants 5 genes
21 promoter TFBS variants 18 genes
2 enhancer TFBS variants 2 genes Variants on TFBS
SNP2TFBS
Exonic variant annotation Variant Effect Predictor Intersection
bedtools
Intersection bedtools
26 enhancer variants 56 genes 186 promoter variants
86 genes
147 psoriasis risk SNPs
Deleterious variant in exons or splice sites 70,459 GWAS SNPs
GWAS Catalog
̲
3.1.
GWAS Catalogに蓄積されている形質・疾患関連座位の総数は70,459個であり,この 内,乾癬(“Psoriasis”,“Cutaneous psoriasis”,“Psoriasis vulgaris”)の発症リスクとの関連 が報告されている多型の総数は147個であった.次に,ヨーロッパ⼈集団において乾癬 感受性SNPsと強い連鎖不平衡にある多型(LD variants)を探索した結果,総数は2,894 個であった.それらを機能多型の候補とし,以降の解析に使⽤した.本論⽂において探 索対象とした機能多型を図5に⽰す.
図5:本論⽂で探索対象とした機能多型
3.2. エクソン領域の機能多型候補
エクソン領域においては,タンパク質の構造や機能,発現量に重⼤な影響を与えること が予想される多型を 2,894個のLD variantsの中から探索した.解析の結果,12個の機 能多型(exonic functional variants)候補を同定した.そのうちの10個はすでに乾癬や他 の免疫疾患との関連が報告されているものであった(表 6)が,2 個は本論⽂によって 新規に検出された乾癬関連機能多型であった(表7).また,エクソン領域中の多型は mRNAの⾼次構造を変化させたり,small RNAの標的配列を変化させたりすることで遺 伝⼦の発現量に影響を与えることが知られているため,多型と遺伝⼦発現量との関連を 探索した.その結果,5個の機能多型候補はその遺伝型に依存して⾃⾝の遺伝⼦発現が 変化する多型(eQTL)であった(図6).
Gene Gene
Psoriasis Marker Variant Enhancer Functional Variant Promoter Functional Variant Exonic Functional Variant
< 2 kb Enhancer-Promoter Correlations
High LD
High LD
High LD
√ eQTL
√ TFBS disruption
√ TF ChIP-seq peak
√ eQTL
√ TFBS disruption
√ TF ChIP-seq peak
√ damaging missense
√ frameshift
√ stop gain
√ start lost
√ splice site
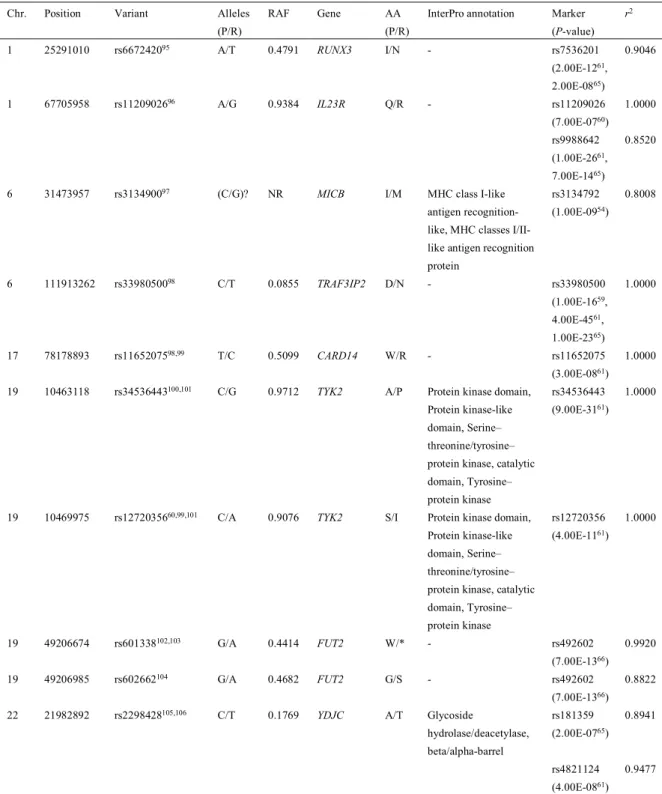
表6:エクソン領域における既知の機能多型
Chr. Position Variant Alleles (P/R)
RAF Gene AA
(P/R)
InterPro annotation Marker (P-value)
r2
1 25291010 rs667242095 A/T 0.4791 RUNX3 I/N - rs7536201
(2.00E-1261, 2.00E-0865)
0.9046
1 67705958 rs1120902696 A/G 0.9384 IL23R Q/R - rs11209026
(7.00E-0760) 1.0000
rs9988642 (1.00E-2661, 7.00E-1465)
0.8520
6 31473957 rs313490097 (C/G)? NR MICB I/M MHC class I-like antigen recognition- like, MHC classes I/II- like antigen recognition protein
rs3134792 (1.00E-0954)
0.8008
6 111913262 rs3398050098 C/T 0.0855 TRAF3IP2 D/N - rs33980500
(1.00E-1659, 4.00E-4561, 1.00E-2365)
1.0000
17 78178893 rs1165207598,99 T/C 0.5099 CARD14 W/R - rs11652075
(3.00E-0861) 1.0000
19 10463118 rs34536443100,101 C/G 0.9712 TYK2 A/P Protein kinase domain, Protein kinase-like domain, Serine–
threonine/tyrosine–
protein kinase, catalytic domain, Tyrosine–
protein kinase
rs34536443 (9.00E-3161)
1.0000
19 10469975 rs1272035660,99,101 C/A 0.9076 TYK2 S/I Protein kinase domain, Protein kinase-like domain, Serine–
threonine/tyrosine–
protein kinase, catalytic domain, Tyrosine–
protein kinase
rs12720356 (4.00E-1161)
1.0000
19 49206674 rs601338102,103 G/A 0.4414 FUT2 W/* - rs492602
(7.00E-1366) 0.9920
19 49206985 rs602662104 G/A 0.4682 FUT2 G/S - rs492602
(7.00E-1366) 0.8822
表7:エクソン領域における新規の機能多型候補
Chr. Position Variant Alleles (P/R)
RAF Gene AA
(P/R)
InterPro annotation Marker (P-value)
r2
12 5660905 rs60542959 T/G 0.9344 COQ10A I/M - rs2066807
(5.00E-1263) 0.9523
rs2066808 (6.00E-1062)
0.9388
rs2066819 (5.00E-1761)
0.9517
17 73874071 rs4600514 G/A 0.1650 TRIM47 R/W B-box-type zinc finger, Zinc finger
RING/FYVE/PHD- type
rs55823223 (1.00E-0866)
0.9075
P/R, protective/risk; RAF, risk allele frequency; AA, amino acids; r2, 乾癬感受性SNPと機能多型間の r-squared value
3.2.1. rs60542959(COQ10A)
機能多型候補rs60542959は,COQ10A遺伝⼦(NM_144576.3)の第1エクソン領域に位 置していた.乾癬発症に対する⾮リスクアリルであるrs60542959-Tは,1番⽬のアミノ 酸残基メチオニン(ATG)をイソロイシン(ATT)に変化させるstart lost多型であった.
この機能多型候補は,STAT2遺伝⼦領域内に位置する3個の乾癬感受性SNPsと強い連 鎖不平衡の関係にあった(rs2066807, [r2 = 0.9523]; rs2066808, [r2 = 0.9388]; rs2066819, [r2
= 0.9517])(図7a).COQ10A遺伝⼦は2種類の転写産物を有しており(NM_144576.3,
NM_001099337.1)(図7b),CAGEデータに基づくと,rs60542959-Tによってstart lost が引き起こされるNM_144576.3(CAGE_peak_1_at_COQ10A_5end)は,natural killer cells,
T cells,およびlymphocytes of B cell lineageにおいて⾼発現しており(図7c),start lost が引き起こされない NM_001099337.1(CAGE_peak_3_at_COQ10A_5end)は,intestinal epithelial cells,hepatocyte,およびpericyte cellにおいて⾼発現していた(図7d).転写
産物NM_144576.3の開始コドンは,哺乳類において広く保存されていた(図7e).GTEx
eQTL データを参照すると,esophagus mucosaにおいてのみ,マイナーアリル(⾮リス クアリル)rs60542959-Tの本数はCOQ10A遺伝⼦の発現量と負の相関を⽰していた(P
= 2.51e-05)(図6, 7f;補⾜図1).
Window Position Scale chr12:
Human Feb. 2009 (GRCh37/hg19) chr12:56,658,001-56,757,000 (99,000 bp)
20 kb hg19
56,670,000 56,680,000 56,690,000 56,700,000 56,710,000 56,720,000 56,730,000 56,740,000 56,750,000 COQ10A
COQ10A CS
CNPY2 CNPY2
PAN2PAN2 PAN2
IL23A STAT2 STAT2
APOF
Layered H3K4Me3150 - 0 _ Layered H3K4Me150 - 0 _ Layered H3K27Ac100 - 0 _ 100 Vert. Cons
1 - 0 _
rs11358218 rs2066808 rs2066807 rs2066819
rs60542959
0.939 0.939
0.952 0.952
1.0 0.954
0.893
0.954 0.893
0.936
a
b
Window Position Scale chr12:
Human Feb. 2009 (GRCh37/hg19) chr12:56,660,001-56,665,000 (5,000 bp)
2 kb hg19
56,661,000 56,662,000 56,663,000 56,664,000 COQ10A
COQ10A
NM_144576.3 NM_001099337.1 rs60542959
c d
COQ10A
e f
5.903 3.556
3.305 2.487 1.946 1.634 1.609 1.588 1.584 1.545 8.286
8.120 7.336 6.777 5.964 5.491 5.192 5.011 4.723 4.421
(continued from previous page)
図7:COQ10A遺伝⼦座の機能多型候補rs60542959
a. 12番染⾊体の乾癬感受性SNPs・機能多型候補の座位およびヨーロッパ⼈集団におけ
るr-squared value.⻘縦線は乾癬感受性SNPs,⾚縦線は機能多型候補の座位を⽰す.
b. COQ10A遺伝⼦座と機能多型候補rs60542959.COQ10A遺伝⼦は2種類の転写産物 を有する(NM_144576.3, NM_001099337.1).
c. CAGEによって測定された転写産物NM_144576.3(CAGE_peak_1_at_COQ10A_5end)
の発現量.縦軸に⾼発現する細胞10種を,横軸に“Percentage of Expression”を⽰す.
各細胞種における“Percentage of Expression”とは,解析したすべての細胞において測 定された全発現量(normalized CAGE counts from all cells)に対する発現量の⽐を表し ている.グラフ中の数値はそれぞれの細胞種におけるTPM(Tags Per Million)を⽰
す.
d. CAGE に よ っ て 測 定 さ れ た 転 写 産 物 NM_001099337.1
(CAGE_peak_3_at_COQ10A_5end)の発現量.縦軸に⾼発現する細胞10種を,横軸 に“Percentage of Expression”を⽰す.
e. NM_144576.3 スタートコドン付近の脊椎動物におけるゲノムアラインメント.機能
多型候補rs60542959の位置を⾚箱で⽰す.
f. Esophagus mucosaにおける機能多型候補rs60542959の遺伝型とCOQ10A遺伝⼦の発 現.横軸にrs60542959の遺伝型とそれぞれのサンプル数を⽰す.アスタリスク(*)
は統計的有意性を⽰す.
COQ10A遺伝⼦(NM_144576.3)がコードするタンパク質のアミノ酸配列を参照する
と,rs60542959-Gを含むアリルでは1番⽬と44番⽬のアミノ酸残基がメチオニンであ る⼀⽅で,rs60542959-T を含むアリルでは 1 番⽬のアミノ酸残基はイソロイシンで 44 番⽬のアミノ酸残基がメチオニンであった.このことから,rs60542959-Tを含むアリル では44番⽬のメチオニンから翻訳が開始することが予想された.タンパク質のドメイ ン検索を⾏うと,24番⽬から43番⽬のアミノ酸残基(SLSPGAQPAPPPGPLPPPRP)は,
低複雑度領域(Low Complexity Region: LCR)であった(図8).
図8:機能多型候補rs60542959を含む転写産物から翻訳される COQ10A遺伝⼦のアミノ酸配列
Rs60542959-Gおよびrs60542959-Tを含むアリルから転写・翻訳されるCOQ10A遺伝⼦
(NM_144576.3)のアミノ酸配列.メチオニン残基は⾚⾊の“M”で⽰す.低複雑度領域を
⿊箱で⽰す.
rs60542959-G: MAWAGSRRVPAGTRAAAERCCRLSLSPGAQPAPPPGPLPPPRPMRFLTSC...
rs60542959-T: IAWAGSRRVPAGTRAAAERCCRLSLSPGAQPAPPPGPLPPPRPMRFLTSC...
COQ10A (NM_144576.3) protein sequence
low complexity region (aa 24~43)
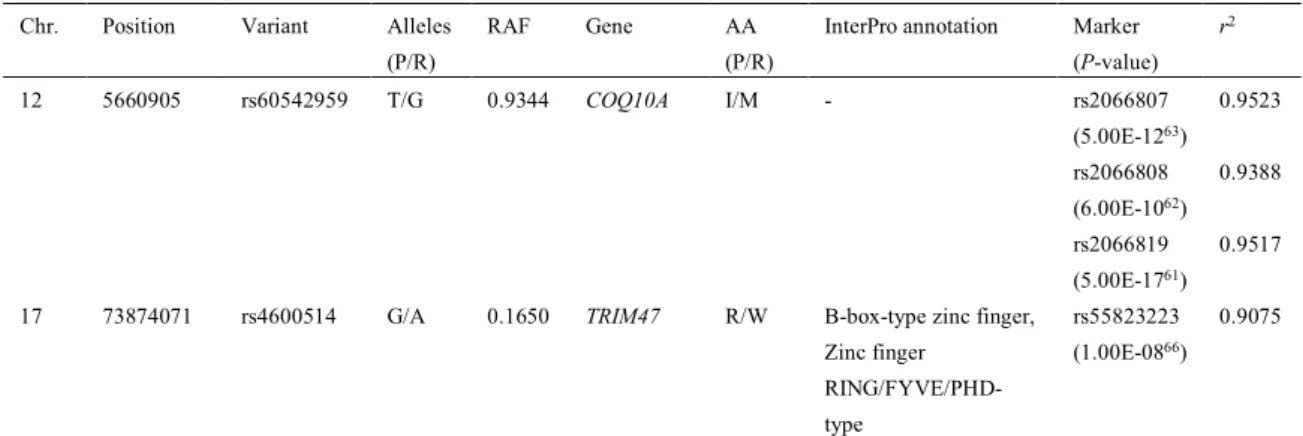
3.2.2. rs4600514(TRIM47)
機能多型候補rs4600514は,TRIM47遺伝⼦(NM_033452.2)の第1エクソン領域に位置
していた(図9a).この機能多型候補は,TRIM65遺伝⼦領域内に位置する乾癬感受性
SNP rs55823223と強い連鎖不平衡の関係にあった(r2 = 0.9075)(図9a).乾癬発症に
対するリスクアリルであるrs4600514-Aは,187番⽬のアミノ酸残基アルギニン(CGG)
をトリプトファン(TGG)に変化させるmissense多型であった.TRIM47タンパク質の 187番⽬のアミノ酸残基はB-box type zinc finger domain内に位置していた(図 9b).
CAGEデータに基づくと,NM_033452.2(CAGE_peak_1_at_TRIM47_5end)は,mast cell
やmacrophage等のリンパ球,および様々なepithelial cellにおいて⾼発現していた(図
9c).GTEx eQTLデータを参照すると,この機能多型候補の遺伝型に依存したTRIM47
遺伝⼦の発現量の変化に統計的有意差は確認されなかった(図6;補⾜図1).
図9:TRIM47遺伝⼦座の機能多型候補rs4600514
a. 17番染⾊体の乾癬感受性SNP rs55823223と機能多型候補rs4600514の座位およびヨ ーロッパ⼈集団におけるr-squared value.⻘縦線は乾癬感受性SNP rs55823223,⾚縦 線は機能多型候補rs4600514の座位を⽰す.
b. TRIM47タンパク質のアミノ酸配列の⼀部.187番アミノ酸残基の位置をピンク⾊の
縦線(*W*)および⽔⾊の「R」で⽰す.B-box type zinc finger domainを⾚箱で,亜 鉛の結合部位を⻘丸で⽰す.
c. CAGEによって測定された転写産物NM_033452.2(CAGE_peak_1_at_TRIM47_5end)
の発現量.縦軸に⾼発現する細胞10種を,横軸に“Percentage of Expression”を⽰す.
各細胞種における“Percentage of Expression”とは,解析したすべての細胞において測 定された全発現量(normalized CAGE counts from all cells)に対する発現量の⽐を表し ている.グラフ中の数値はそれぞれの細胞種におけるTPM(Tags Per Million)を⽰
す.
Window Position Scale chr17:
Human Feb. 2009 (GRCh37/hg19) chr17:73,869,001-73,894,000 (25,000 bp)
10 kb hg19
73,875,000 73,880,000 73,885,000 73,890,000
TRIM47 TRIM65
TRIM65 Layered H3K4Me3150 -
0 _ Layered H3K4Me150 - 0 _ Layered H3K27Ac100 - 0 _ 100 Vert. Cons
1 -
0 _
rs55823223 rs4600514
r2 = 0.9075
a
b
c
41.436 32.995 26.283 20.714 20.347 18.587 17.802 15.575 15.402 15.174
3.3. スプライス部位の機能多型候補
スプライス部位においては,スプライシング活性測定ソフトウェアである MaxEntScan によって算出されたスコアに基づき,スプライス部位の選択に影響を与えることが予想 される多型を探索した.解析の結果,2個の機能多型候補を同定した.そのうちの1個 はすでに乾癬や他の免疫疾患との関連が報告されているものであったが(表8),1個 は本論⽂によって新規に同定された乾癬関連機能多型であった(表9).また,その新 規機能多型候補は,その遺伝型に依存して⾃⾝の遺伝⼦発現が変化する多型(eQTL)
であった(図10)
表8:スプライス部位における既知の機能多型
Chr. Position Variant Alleles (P/R)
RAF Gene ss InterPro
annotation
Marker (P-value)
r2
6 31380000 rs199503730107 G/- 0.0447 MICA 5´ ss - rs2395029 (2.00E-2655)
0.8860
P/R, protective/risk; RAF, risk allele frequency; ss, splilce site; r2, 乾癬感受性SNPと機能多型間のr- squared value
表9:スプライス部位における新規の機能多型候補
Chr. Position Variant Alleles (P/R)
RAF Gene ss InterPro
annotation
Marker (P-value)
r2
5 96245518 rs2549797 G/A 0.5199 ERAP2 5´ ss - rs2910686
(2.00E-0861)
0.8051
P/R, protective/risk; RAF, risk allele frequency; ss, splilce site; r2, 乾癬感受性SNPと機能多型間のr- squared value
図10:スプライス部位における機能多型とeQTL normalized effect size 縦軸は解析した細胞・組織,横軸は機能多型とそれらを有する遺伝⼦を⽰す.セル内 の数値は各多型のマイナーアリルが遺伝⼦発現に与える影響のスコア(normalized effect size)であり,マイナーアリルの本数と遺伝⼦発現量が正の相関を⽰す場合は
⾚⾊,負の相関を⽰す場合は⻘⾊で⽰している.灰⾊で⽰す“n.s.”は,統計学的に有 意な相関がみられなかったことを⽰す.