CTD 第 2 部
2.6 非臨床試験の概要文及び概要表
2.6.6 毒性試験の概要文
用語及び略語一覧
8-MOP 8-methoxypsoralen 8-メトキシソラレン
ALP alkaline phosphatase アルカリホスファターゼ
ALT alanine aminotransferase アラニンアミノトランスフェラーゼ APTT activated partial thromboplastin time 活性化部分トロンボプラスチン時間 AST aspartate aminotransferase アスパラギン酸アミノトランスフェラー
ゼ
ASV asunaprevir, BMS-650032 アスナプレビル AUC area under the plasma concentration-time
curve 血漿中濃度時間曲線下面積
BID twice daily 1 日 2 回
CAC FDA Executive Carcinogenicity Assessment
Committee FDA がん原性評価委員会
CHO Chinese hamster ovary チャイニーズハムスター卵巣 Cmax maximum observed plasma concentration 最高血漿中濃度
CP cyclophosphamide シクロホスファミド
DCV daclatasvir, BMS-790052 ダクラタスビル
DMSO dimethyl sulfoxide ジメチルスルホキシド
E. coli Escherichia coli 大腸菌
F0 founder generation 親世代
F1 first generation 第1 世代
FDA Food and Drug Administration 米国食品医薬品局
GGT gamma glutamyltransferase γ-グルタミルトランスフェラーゼ GLP Good Laboratory Practice 医薬品の安全性に関する非臨床試験の実
施の基準
GSH glutathione グルタチオン
HCV hepatitis C virus C 型肝炎ウイルス
IC50 concentration at 50% inhibition 50%阻害濃度
ICH International Conference on Harmonisation 日米 EU 医薬品規制調和国際会議
LD lactation day 哺育日数
LLOQ lower limit of quantitation 定量下限
MCH mean corpuscular hemoglobin 平均赤血球ヘモグロビン量 MCHC mean corpuscular hemoglobin concentration 平均赤血球ヘモグロビン濃度 MCV mean corpuscular volume 平均赤血球容積
MTD maximum tolerated dose 最大耐量
NA not applicable 該当なし
NMU N-nitrosomethylurea N-ニトロソメチル尿素
NOAEL no-observed-adverse-effect level 無毒性量 NS3 nonstructural protein 3 非構造蛋白3 NS5A nonstructural protein 5A 非構造蛋白5A
PEG polyethylene glycol ポリエチレングリコール
PVP polyvinyl pyrrolidone ポリビニルピロリドン RDW red cell distribution width 赤血球分布幅
S9 liver fraction that contains a high concentration of cytochrome P450 metabolic enzymes
チトクロームP450 代謝酵素を高濃度含 有する肝臓画分
S. typhimurium Salmonella typhimurium ネズミチフス菌
Tg transgenic トランスジェニック
TPGS d-α-tocopheryl polyethylene glycol 1000
succinate d-α-トコフェリルポリエチレングリコール1000 コハク酸エステル
目次
1 まとめ ... 7 2 単回投与毒性試験 ... 14 2.1 マウスにおける単回経口投与毒性試験 ... 14 2.2 ラットにおける単回経口投与毒性試験 ... 15 2.3 イヌにおける単回経口投与トキシコキネティクス及び忍容性試験 ... 15 3 反復投与毒性試験 ... 16 3.1 ラットにおける反復経口投与毒性試験 ... 16 3.2 イヌにおける反復経口投与毒性試験 ... 22 3.3 サルにおける反復経口投与毒性試験 ... 27 3.4 併用投与毒性試験 ... 28 4 遺伝毒性試験 ... 34 4.1 In vitro 試験 ... 34 4.2 In vivo 試験 ... 35 5 がん原性試験 ... 36 5.1 マウスにおけるがん原性試験 ... 37 5.2 ラットにおける2 年間経口投与がん原性試験 ... 40 6 生殖発生毒性試験 ... 42 6.1 受胎能及び着床までの初期胚発生に関する試験 ... 43 6.2 胚・胎児発生に関する試験 ... 44 6.3 出生前及び出生後の発生並びに母体の機能に関する試験 ... 47 7 局所刺激性試験 ... 48 8 その他の毒性試験 ... 48 8.1 光毒性試験 ... 48 8.2 抗原性及び免疫毒性試験 ... 50 8.3 依存性試験 ... 50 8.4 代謝物の毒性試験 ... 50 8.5 不純物の毒性試験 ... 50 8.6 併用投与毒性試験 ... 50 9 考察及び結論 ... 50 10 参考文献 ... 56表一覧
表1-1 アスナプレビルの毒性試験 ... 9 表2.3-1 イヌ単回経口投与試験におけるトキシコキネティクス値 ... 16 表3.1-1 ラット反復経口投与毒性試験におけるAUC 及びヒト AUC との比 ... 17 表3.1.1-1 ラット2 週間経口投与毒性試験におけるトキシコキネティクス値 ... 18 表3.1.1-2 ラット2 週間経口投与毒性試験におけるアスナプレビルの組織中濃度... 18 表3.1.2-1 ラット1 ヵ月間経口投与毒性試験におけるトキシコキネティクス値... 19 表3.1.2-2 ラット1ヵ月間経口投与毒性試験におけるアスナプレビルの肝臓中及び血漿 中濃度 ... 19 表3.1.3-1 ラット6 ヵ月間経口投与毒性試験におけるトキシコキネティクス値... 21 表3.1.3-2 ラット6ヵ月間経口投与毒性試験におけるアスナプレビルの肝臓中及び血漿 中濃度 ... 22 表3.2-1 イヌ反復経口投与毒性試験におけるアスナプレビルのAUC 及びヒト AUC との比 ... 23 表3.2.1-1 イヌ1 ヵ月間経口投与毒性試験におけるトキシコキネティクス値 ... 23 表3.2.1-2 イヌ1ヵ月間経口投与毒性試験におけるアスナプレビルの肝臓中及び血漿中 濃度 ... 24 表3.2.2-1 イヌ9 ヵ月間経口投与毒性試験におけるトキシコキネティクス値 ... 26 表3.2.2-2 イヌ9ヵ月間経口投与毒性試験におけるアスナプレビルの肝臓中及び血漿中 濃度 ... 26 表3.3-1 サル反復経口投与毒性試験におけるアスナプレビルのAUC 及びヒト AUC との比 ... 27 表3.3.1-1 サル1 週間経口投与毒性試験におけるトキシコキネティクス値 ... 28 表3.4.1.1-1 ラット1 ヵ月間併用投与毒性試験におけるトキシコキネティクス値... 29 表3.4.2.1-1 サル1 ヵ月間併用投与毒性試験におけるトキシコキネティクス値 ... 31 表3.4.2.2-1 サル3 ヵ月間併用投与毒性試験におけるトキシコキネティクス値 ... 33 表4.2.1-1 ラット小核試験におけるトキシコキネティクス値 ... 35 表5-1 がん原性試験における定常状態の曝露量及びヒトAUC との比 ... 36 表5.1.1-1 CByB6F1 ハイブリッドマウス 28 日間経口投与毒性試験におけるトキシコキ ネティクス値 ... 37 表5.1.2-1 Tg-rasH2 トランスジェニックマウス 26 週間投与がん原性試験におけるトキ シコキネティクス値 ... 39 表5.1.2-2 Tg-rasH2 トランスジェニックマウス 26 週間投与がん原性試験における生存 率 ... 39 表5.2-1 ラット2 年間投与がん原性試験におけるトキシコキネティクス値 ... 41 表5.2-2 ラット2 年間投与がん原性試験における生存率 ... 41表6-1 ラット受胎能及び着床までの初期胚発生に関する試験におけるアスナプレビ ルのAUC 及びヒト AUC との比 ... 42 表6-2 マウス及びウサギ胚・胎児発生に関する試験におけるアスナプレビルのAUC 及びヒトAUC との比 ... 42 表6-3 ラット出生前及び出生後の発生に関する試験におけるアスナプレビルのAUC 及びヒトAUC との比 ... 43 表6.1-1 ラット受胎能及び着床までの初期胚発生に関する試験におけるトキシコキネ ティクス値 ... 43 表6.2.1-1 妊娠マウス10 日間経口投与用量設定試験におけるトキシコキネティクス値 ... 44 表6.2.2-1 マウス胚・胎児発生に関する試験におけるトキシコキネティクス値... 45 表6.2.3-1 妊娠ウサギ13 日間経口投与用量設定試験におけるトキシコキネティクス値 ... 46 表6.2.4-1 ウサギ胚・胎児発生に関する試験におけるトキシコキネティクス値... 47 表6.3-1 出生前及び出生後の発生並びに母体の機能に関する試験の母動物における トキシコキネティクス値 ... 47 表8.1.2-1 Long-Evans ラット単回経口投与光毒性試験におけるトキシコ キネティクス値 ... 49 表9-1 アスナプレビルの無毒性量及び主な毒性発現用量における曝露量とヒト曝露 量との比 ... 54
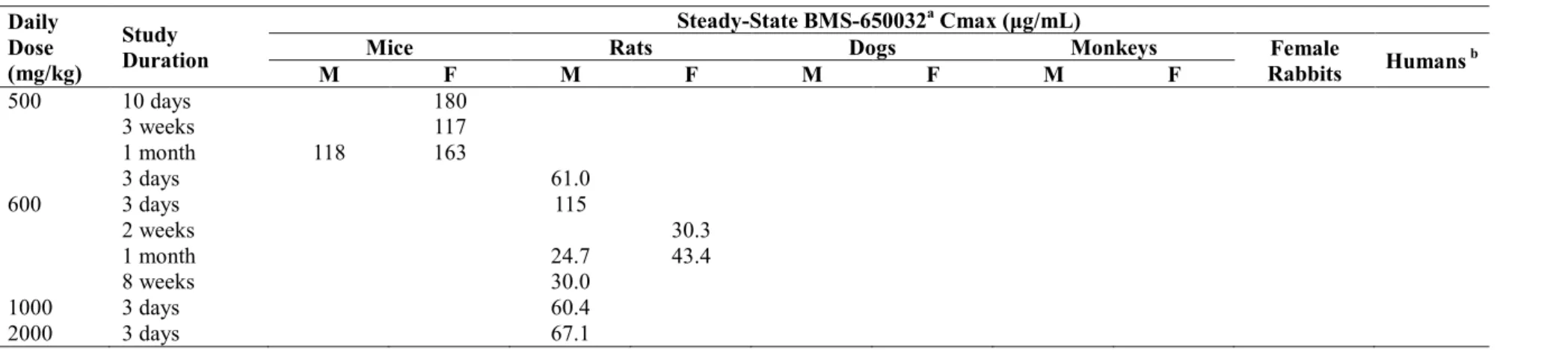
1 まとめ アスナプレビル(BMS-650032)は、C 型肝炎ウイルス(HCV)の非構造蛋白 3(NS3)プロテ アーゼに対する低分子阻害薬(直接作用型抗ウイルス薬)である。本薬はin vitro で広範なジェノ タイプのHCV レプリコンに対し、ナノモル濃度で NS3 阻害作用を示す。また、HCV NS5A 複製 複合体阻害薬であるダクラタスビル塩酸塩[以下、ダクラタスビル(BMS-790052)]など他の直 接作用型抗ウイルス薬との併用で相加的ないし相乗的な阻害作用を示す。これらの特性に基づき、 C型慢性肝炎患者を対象に、ダクラタスビル及びアスナプレビル併用療法(DCV + ASV併用療法) の開発を行った。 アスナプレビルのダクラタスビルとの併用による臨床推奨用量は、100 mg 軟カプセルの 1 日 2 回(BID)投与である。100 mg BID(1 日 200 mg)軟カプセルは予定市販剤形であり、臨床第 2/3 相試験に使用した200 mg 錠剤の BID 投与(1 日 400 mg)と同様の曝露量が得られる。臨床推奨 用量における定常状態でのアスナプレビルの曝露量(以下、ヒト曝露量)は、最高血漿中濃度(Cmax) 0.419 μg/mL、血漿中濃度時間曲線下面積(AUC)3.69 μg•h/mL である。これらのヒト曝露量に基 づき、毒性試験の無毒性量(NOAEL)及び毒性発現量における動物とヒトとの曝露量比(動物の Cmax 又は AUC ÷ ヒトの Cmax 又は AUC)を算出した。In vitro における蛋白結合率は、毒性試 験に用いた動物種間で同程度(マウス、ラット、イヌ及びサル血清中でそれぞれ99.2, 98.8, 98.5 及び97.2%)であり、ヒト血漿中では 99.7%であった(CTD 2.6.4.4)。曝露量の比較においてアス ナプレビル遊離体としての補正は行なわなかった。ヒト血漿中のアスナプレビル遊離体濃度は動 物における遊離体濃度と比較して低い(0.25 倍以下)ため、総濃度に基づく曝露量比は遊離体と して補正した場合より小さいと考えられる。重要な試験における動物とヒトとの曝露量比を表9-1 に示す。 毒性試験に用いた主な動物種であるラット及びイヌにおける血漿中アスナプレビル濃度は、特 に高用量で変動が大きく、特にイヌにおいては嘔吐が血漿中アスナプレビル濃度の変動の要因の 一つと考えられた。ラットにおける反復投与後のアスナプレビルのAUC は、30~200 mg/kg/day で概して用量比を上回って増加したが、より高用量では用量比を下回った。また、30~100 mg/kg/day では性差は概してみられなかったが、100 mg/kg/day を超える用量では雌が雄より高値(約 2 倍) であった。イヌでは、アスナプレビルのAUC は 15~300 mg/kg/day で概して用量比以上に増加し、 雌が雄より高値(約2 倍)であった。反復投与による蓄積はみられなかった。ダクラタスビルと の併用投与では、ラット及びサルにおいてアスナプレビルの AUC の増加傾向がみられたが、個 体間変動が大きいため薬物動態学的相互作用の可能性については明らかでなく、DCV + ASV 併用 療法の臨床試験では薬物相互作用は報告されていない。血漿中濃度の変動が大きく、雌雄の差は 概して2 倍を超えないため、トキシコキネティクス値及び曝露量比は雌雄合計平均値で示した。 アスナプレビルは非臨床試験に用いた動物種及びヒトにおける血清中又は血漿中の主化合物で あった。ヒトに特有の代謝物は検出されなかった。アスナプレビルの消失には複数の経路(胆汁 中排泄及び代謝クリアランスのほか、恐らく直接的腸内分泌)が関与するが、アスナプレビル及 びその代謝物の主な排泄経路は糞中であった。アスナプレビルの代謝には、一酸化、二酸化、 N-脱アルキル化、O-脱メチル化及びアミド加水分解によるイソキノリン環の消失が主に関与してお
り、in vivo で生成される代謝物プロファイルは評価したいずれの動物種でも質的に類似していた (CTD 2.6.5.9)。ヒトでは反復投与により代謝物の曝露量が増加したが、曝露量がアスナプレビル の曝露量の20%を超える、又は未変化体及び代謝物の総曝露量の 10%を超える代謝物はみられな かった。ヒト血漿中に検出されたすべてのアスナプレビル代謝物は試験に用いた動物種(マウス、 ラット、イヌ及びサル)の少なくとも1 種で検出され、ヒトにアスナプレビルの臨床推奨用量を 単回及び反復経口投与したときの代謝物の曝露量は、毒性試験における動物の曝露量より低値で あった。単回投与後の動物(マウス100 mg/kg、ラット 80 mg/kg、イヌ 50 mg/kg)におけるアス ナプレビル代謝物の曝露量は反復投与後のヒト(200 mg BID、10 日間投与)における曝露量を上 回り、動物におけるAUC(0-8)のヒト AUC(0-12)との比は 2.2~1512 倍であった。したがって、代 謝物の毒性試験は実施しなかった。 [14C]アスナプレビルをラット、イヌ、サル及びヒト肝臓ミクロソームとインキュベートした結 果、放射性物質と肝ミクロソーム蛋白との不可逆的結合がみられた。この結合はグルタチオン(GSH) の存在によって減少し、肝ミクロソーム中にGSH 付加体が検出されたことから、反応性中間体の 生成が示唆された。しかし、ラット及びイヌの1 ヵ月間投与毒性試験において、肝毒性を示唆す る変化(両動物種で血清中肝酵素の増加及びイヌでごく軽微~軽微な肝細胞壊死)は高用量(300 mg/kg/day 以上)のみでみられ、これらの用量における AUC は、ラットで 227~371 μg•h/mL、イ ヌで1360~1410 μg•h/mL であった。イヌで肝臓の病理組織学的変化がみられた AUC は、臨床推 奨用量投与時のヒトにおけるAUC(以下、ヒト AUC)の 369~382 倍であった。慢性毒性試験(ラッ ト6 ヵ月間及びイヌ 9 ヵ月間)では肝毒性はみられず、これらの試験における AUC はヒト AUC の87~185 倍(ラット)及び 60~103 倍(イヌ)であった。 重要な毒性試験はいずれも医薬品の安全性に関する非臨床試験の実施の基準(GLP)適合下で 医薬品規制調和国際会議(ICH)ガイドラインに準拠して実施した。用量設定や毒性発現機序解 明のための探索的試験は、一部非GLP 下で実施した。特記する場合を除き、毒性試験の投与経路 は臨床投与経路と同様の経口投与とし、媒体[60% ポリエチレングリコール 400(PEG-400)及 び40% d-α-トコフェリルポリエチレングリコール 1000 コハク酸エステル(TPGS)]に溶解して投 与した。PEG-400・TPGS の媒体は、高用量の投与液にアスナプレビルを溶解させ、高い経口バイ オアベイラビリティを得るために必要であった。媒体のみを投与した対照群に異常はみられず、 マウスがん原性試験において水対照群と媒体対照群における腫瘍の種類及び発生頻度に差は認め られなかった。 アスナプレビル単剤の毒性試験に用いる主要な動物種として、ラット(げっ歯類)及びイヌ(非 げっ歯類)を選択した。ラットは毒性試験に用いる標準的動物種で背景データが豊富であること から選択した。イヌは、経口バイオアベイラビリティ(42%以上)がサル(10%)より高く、高 い全身曝露量が得られることから選択した。 併用投与毒性試験では、単剤の試験で発現した毒性を考慮して動物種を選択した。用量選択に 関しては、明らかな毒性が発現する用量よりも、アスナプレビル及びダクラタスビルそれぞれの 臨床におけるヒト曝露量範囲に関連した AUC が得られる用量を設定した。併用投与毒性試験の 投与期間については、ICH M3(R2)ガイダンス及び FDA の HCV ガイダンス案(Chronic Hepatitis C
Virus Infection: Developing Direct-Acting Antiviral Agents for Treatment, CDER, September 2010)に基 づき、単剤の高用量による長期投与毒性試験の方が臨床曝露量に関連した AUC が得られる用量 で実施した併用投与毒性試験よりも安全性の評価に有用と考えられることから、最長3 ヵ月間と した。 アスナプレビルの毒性を評価するための非臨床毒性試験として、単回投与毒性試験(マウス、 ラット、イヌ)、反復投与毒性試験(ラット最長6 ヵ月間、イヌ最長 9 ヵ月間)、併用投与(ASV + DCV)毒性試験(ラット、サル)、遺伝毒性試験(in vitro 及び in vivo)、がん原性試験[Tg-rasH2 マウス、ラット]、生殖発生毒性試験(マウス、ラット、ウサギ)、光毒性試験(in vitro 及び in vivo) を実施した。その他、肝臓がヒトにおける治療(HCV 感染)の標的器官であることから、一部の 反復投与毒性試験においてアスナプレビルの肝臓中及び胆汁中濃度を測定した。胆汁中濃度測定 は非GLP 適用下で非臨床試験に用いた動物種及びヒトで実施した。更に、探索的毒性試験として ラット2 週間投与試験及びサル 1 週間投与試験を実施した。 実施したアスナプレビルの毒性試験を表1-1 に示す。これらの試験は、C 型慢性肝炎患者にお けるDCV + ASV 併用療法の安全性を担保するものである。 表1-1 アスナプレビルの毒性試験 試験の種類及び投与期間 投与経路 試験系 単回投与毒性 経口 マウス、ラット、イヌ 反復投与毒性 1 週間投与毒性試験 経口 サル 2 週間投与毒性試験 経口 ラット 1 ヵ月間投与毒性試験 経口 ラット、イヌ 6 ヵ月間投与毒性試験 経口 ラット 9 ヵ月間投与毒性試験 経口 イヌ 併用投与毒性(ASV + DCV) 1 ヵ月間投与毒性試験 経口 ラット、サル 3 ヵ月間投与毒性試験 経口 サル 遺伝毒性
復帰突然変異試験 In vitro S. typhimurium, E. coli
染色体異常試験 In vitro CHO 細胞 小核試験 経口 ラット がん原性 26 週間投与がん原性試験 経口 Tg-rasH2 マウス 2 年間投与がん原性試験 経口 ラット 生殖発生毒性 受胎能及び着床までの初期胚発生に関する試験 経口 ラット 胚・胎児発生に関する試験 経口 マウス、ウサギ 出生前及び出生後の発生並びに母体の機能に関する試験 経口 ラット その他の毒性 光毒性試験 In vitro Balb/c 3T3 マウス線維芽細胞 単回投与光毒性試験 経口 ラット
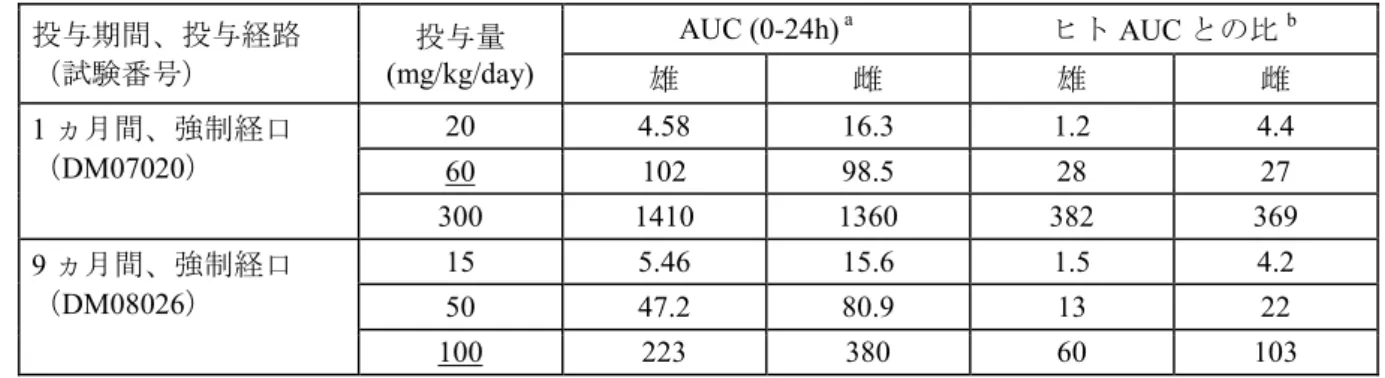
単回投与毒性試験の結果、3 種の動物のいずれでも高用量で胃腸管への影響を示唆する変化が 観察され、マウスでは2000 mg/kg で死亡例がみられた。ラットでは 2000 mg/kg で体重減少など の毒性症状がみられたが、死亡例はみられなかった。イヌでは100 mg/kg まで忍容性は良好であっ たが、最高用量の300 mg/kg で嘔吐が観察された。 反復投与毒性は、重要な試験としてラットでは1 ヵ月間経口投与試験[0(媒体),30, 100 及び 600 mg/kg/day]及び 6 ヵ月間経口投与試験[0(媒体),40, 80 及び 200 mg/kg/day]、イヌでは 1 ヵ 月間経口投与試験[0(媒体),20, 60 及び 300 mg/kg/day)及び 9 ヵ月間経口投与試験[0(媒体), 15, 50 及び 100 mg/kg/day]を実施して評価した。 ラットでは最高用量 200 mg/kg/day(AUC:503 μg•h/mL)を 6 ヵ月間、イヌでは最高用量 100 mg/kg/day(AUC:302 μg•h/mL)を 9 ヵ月間投与し、いずれも忍容性は良好であった。 イヌ1 ヵ月間投与試験では主な変化は肝臓に発現し、300 mg/kg/day(AUC:1385 μg•h/mL)で 肝臓の肝細胞の凝固壊死(ごく軽微~軽微)及びこれに関連したアラニンアミノトランスフェラー ゼ(ALT)増加(対照群の 1.94~4.48 倍)、γ-グルタミルトランスフェラーゼ(GGT)増加(投与 前値の2.7 倍)、総ビリルビン増加(対照群の 1.55~1.66 倍)が認められた。 ラット1 ヵ月間投与試験では主な変化は消化管にみられ、600 mg/kg/day(AUC:299 μg•h/mL) で小腸及び大腸の液体及びガスによる膨満、小腸及び盲腸の腸細胞の肥大(ごく軽微~軽度)並 びに盲腸及び結腸の杯細胞の減少が認められた。また、肝臓に関連した変化として600 mg/kg/day でALT 増加(対照群の 1.79~2.10 倍)、ALP 増加(1.41 倍)、総ビリルビン増加(1.46~1.82 倍) がみられたが、これらに関連した肝臓の病理組織学的所見は認められなかった。 更にラットでは肝臓重量の増加が 1 ヵ月間投与試験では 100 及び 600 mg/kg/day(AUC: 299 μg•h/mL、ヒト AUC の 81 倍)で、6 ヵ月間投与試験では最高用量の 200 mg/kg/day(AUC: 321 μg•h/mL、ヒト AUC の 87 倍)まで用量依存的にみられた。肝臓重量の変化に関連した肝臓の 病理組織学的変化はみられず、肝臓における高濃度のアスナプレビルによる薬物代謝酵素の誘導 に関連した適応性変化と考えられた。 ラット及びイヌのいずれでも肝臓に変化がみられた用量(ラット:1 ヵ月間及び 6 ヵ月間投与 600 mg/kg/day 以下、イヌ:1 ヵ月間投与 300 mg/kg/day)では、肝臓中に高濃度(36~246 μg/g) のアスナプレビルが検出された。 また、ラット及びイヌの1 ヵ月間投与試験では、それぞれ 600 及び 300 mg/kg/day で軽微な血 液学的変化[赤血球分布幅の増加(対照群の1.10~1.20 倍)、平均赤血球容積(MCV)の減少(0.89 ~0.97 倍)]がみられた。これらの変化は赤血球の小型化を反映したものと考えられ、鉄代謝の変 化が示唆された。更に、イヌでは300 mg/kg/day で赤血球に鉄染色陽性の好塩基性細胞封入体(パッ ペンハイマー体)が認められ、ヘモグロビン合成の障害が示唆された 1)。しかし、これらの血液 学的変化の程度は軽微で、骨髄の病理組織学的変化はいずれの試験でも認められなかったことか ら、毒性学的意義の低い変化と考えられた。 その他、アスナプレビルの投与に関連した血液生化学的変化として、ラット及びイヌの1 ヵ月 間投与試験においてぞれぞれ600 及び 300 mg/kg/day(最高投与量)で総蛋白減少(対照群の 0.78
~0.92 倍)、アルブミン減少(0.75~0.96 倍)及びグロブリン減少(0.81~0.87 倍)が認められた。 ラットでは、アスナプレビルによる腸管の変化に関連した消化吸収不全あるいは消化管からの蛋 白損失の促進が血清蛋白の変化に関与している可能性が考えられた。また、ラット1 ヵ月間投与 試験では600 mg/kg/day で尿量の軽度な増加(2.56~3.22 倍)及び尿 pH の上昇(1.10~1.14 倍) が認められた。尿量の増加については、脱水症状がみられず、腎臓の組織学的変化も認められず、 アスナプレビル及びその代謝物は尿中にほとんど排泄されない(アスナプレビル及びその代謝物 による溶質利尿がない)ことから、摂水量の増加(多渇症)による可能性が高く、腎機能の変化 によるものではないと考えられた。ラット6 ヵ月間投与試験及びイヌの反復投与試験では尿の変 化はみられなかった。以上より、ラット及びイヌの1 ヵ月間投与試験における無毒性量は、それ ぞれ100 mg/kg/day(AUC:91 μg•h/mL、ヒト AUC の 25 倍)及び 60 mg/kg/day(AUC:100 μg•h/mL、 ヒトAUC の 27 倍)と考えられた。 ラット6 ヵ月間投与試験及びイヌ 9 ヵ月間投与試験では、1 ヵ月間投与試験と比較して毒性の 進行は認められず、新規の標的器官もみられなかった。それぞれの試験における無毒性量は、 200 mg/kg/day(AUC:503 μg•h/mL)及び 100 mg/kg/day(AUC:302 μg•h/mL)であった。ラット 6 ヵ月間投与試験では 100 mg/kg/day で毒性学的意義の低い軽微な MCV の減少(対照群の 0.93~ 0.94 倍)及び平均赤血球ヘモグロビン量(MCH)の減少(0.91~0.92 倍)、イヌ 9 ヵ月間投与試 験では50 mg/kg/day 以上で回復性の ALP 増加(1.32~2.17 倍)がみられたが、これらの所見以外 にラット及びイヌ1 ヵ月間投与試験でみられたアスナプレビルに関連した所見は認められなかっ た。肝臓の所見がみられない用量(イヌ9 ヵ月試験で 100 mg/kg/day 以下)における肝臓中アス ナプレビル濃度は低値(0.3~4.1 μg/g)であった。慢性毒性試験の無毒性量における AUC とヒト AUC との比は、ラット 6 ヵ月間投与試験で 136 倍、イヌ 9 ヵ月間投与試験で 82 倍であった。 重要な試験以外の反復投与毒性試験として、ラット2 週間経口投与試験及びサル 1 週間経口投 与試験を実施した。これらの試験の成績は、ラット反復投与毒性試験及びサル併用投与毒性試験 の用量設定に用いた。 ラット2 週間投与毒性試験[0(媒体),30, 100 及び 300 mg/kg/day]では、アスナプレビルの 投与に関連した所見として、尿pH の上昇(+0.7)、血清総ビリルビン増加(対照群の 1.9 倍)、腎 臓重量減少(−15%)及び心臓重量減少(−12%)がみられたが、関連した臨床検査値の変化及び 病理学的変化はみられず、生物学的意義の低い変化と考えられた。本試験では最高用量の 300 mg/kg/day(AUC:221 μg•h/mL、ヒト AUC の 60 倍)まで忍容性は良好であり、アスナプレ ビルの投与に関連した肝臓の変化はみられなかった。このことは、肝臓の変化がみられたラット 1 ヵ月間及び 6 ヵ月間投与試験並びにイヌ 1 ヵ月間投与試験と比較して投与期間が短いことによ るものと考えられた。 サル1 週間投与毒性試験[0(媒体),30, 150 及び 300 mg/kg/day]では、アスナプレビルの投 与に関連した所見として、150 mg/kg/day 以上で軽度な総コレステロール減少(対照群の 0.64~0.82 倍)、総蛋白減少(0.93~0.96 倍)及びアルブミン減少(0.84~0.91 倍)、総ビリルビン増加(1.93 ~4.0 倍)がみられ、更に 300 mg/kg/day の雄で胸骨及び肋骨のごく軽微な骨髄細胞数増加(白血 球系細胞の増殖刺激によると考えられる骨髄細胞の増加)がみられた。しかし、これらの所見に
関連した白血球関連値の変化及び他の造血器系組織に病理組織学的変化がみられなかったため、 この骨髄刺激性変化の毒性学的意義は低いと考えられた。本試験では最高用量 300 mg/kg/day (AUC:697 μg•h/mL、ヒト AUC の 189 倍)まで忍容性は良好であった。 複数の試験(ラット試験、イヌ1 ヵ月間試験、サル 1 週間試験)に共通して、総コレステロー ルの減少(対照群の0.55~0.92 倍)がみられたが、高用量(200~600 mg/kg/day、ヒト AUC の 81 ~375 倍)のみでみられた軽度な減少であることから毒性学的及び生物学的意義の低い変化と考 えられた。 アスナプレビルをダクラタスビルと併用投与した場合の毒性学的相互作用について、ラット及 びサルを用いた1 ヵ月間併用投与毒性試験、サルを用いた 3 ヵ月間併用投与毒性試験により評価 した。ラットでは、アスナプレビル及びダクラタスビルを最高用量60 mg/kg/day で 1 ヵ月間併用 投与した。サルでは、アスナプレビル及びダクラタスビルを1 ヵ月間併用投与試験ではそれぞれ 最高用量 129.5 及び 50 mg/kg/day で、3 ヵ月間併用投与試験ではそれぞれ最高用量 80 及び 50 mg/kg/day で投与した。ラット 1 ヵ月間併用投与試験(アスナプレビル AUC:41.4 μg•h/mL、 ヒトAUC の 11倍)、サル 1ヵ月間及び 3ヵ月間併用投与試験(アスナプレビルAUC:67.1 μg•h/mL、 ヒトAUC の 18 倍)のいずれにおいても毒性学的相互作用を示唆する変化は認められなかった。 これらの試験では、ダクラタスビルと併用投与したアスナプレビルの AUC に増加傾向がみられ たが、アスナプレビルの曝露量は個体間変動が大きいためトキシコキネティクスの相互作用の可 能性については明らかでなく、DCV + ASV 併用療法の臨床試験では薬物相互作用は報告されてい ない(AI447009 試験、CTD 2.7.2.2.8)。併用投与試験ではアスナプレビルに関連した肝臓の変化 がみられなかったことと一致して、併用投与試験におけるアスナプレビルの肝臓中濃度(1.1~39 μg/g) は、肝臓の変化が認められたアスナプレビル単剤のラット及びイヌ1 ヵ月間投与毒性試験におけ るアスナプレビルの肝臓中濃度(140~246 μg/g)より低値であった。アスナプレビル単剤のサル 1 週間投与試験ではアスナプレビル肝臓中濃度を測定しなかった。 遺伝毒性に関しては、in vitro 試験(細菌を用いる復帰突然変異試験、CHO 細胞を用いる染色体 異常試験)及びin vivo 試験(ラット小核試験)のいずれも陰性の結果であった。In vitro 試験では、 復帰突然変異及び染色体異常をそれぞれ最高濃度5000 μg/plate 及び 60 μg/mL で評価した。In vivo 試験では、最高用量2000 mg/kg/day(AUC:1190 μg•h/mL、ヒト AUC の 322 倍)をラットに 3 日 間投与した。
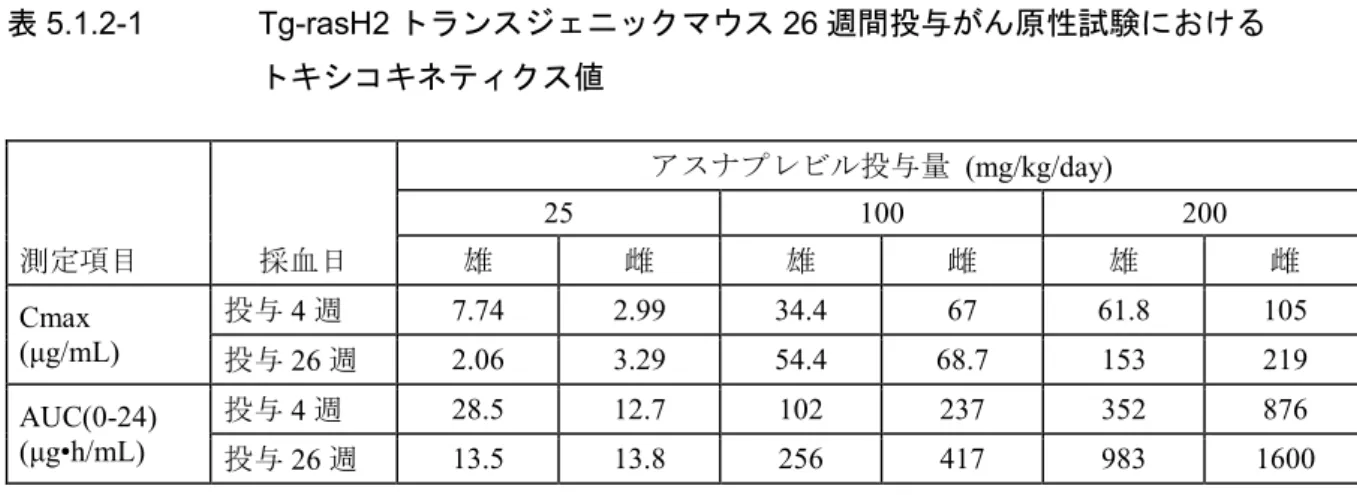
がん原性については、Tg-rasH2 マウス 26 週間投与がん原性試験[0(水),0(媒体),25, 100 及び200 mg/kg/day]の結果、最高用量 200 mg/kg/day(AUC:1292 μg•h/mL、ヒト AUC の 350 倍) までがん原性は認められなかった。アスナプレビル投与群の生存率に対照群との統計学的有意差 は認められなかった。水対照群及び媒体対照群の腫瘍の種類及び発生頻度は同程度であった。陽 性対照[N-ニトロソメチル尿素(NMU)]群では予測されたリンパ腫の発生頻度が媒体対照群と 比較して増加し、導入遺伝子の安定性及びがん原性の検出モデルとしての感度が確認された。ま た、ラット2 年間投与試験[雄: 0(水),0(媒体),50, 75 及び 125 mg/kg/day、雌:0(水),0 (媒体),40, 60 及び 80 mg/kg/day]の結果、雌雄それぞれの最高用量(雌雄合計平均 AUC: 198 μg•h/mL、ヒト AUC の 54 倍)までがん原性は認められなかった。媒体対照群の生存率の低下
により、がん原性を適切に評価するため全群について雄の全生存例を投与84~85 週に、雌の全生 存例を投与92~93 週にそれぞれ剖検した。腫瘍の発生頻度、発生部位及び種類にアスナプレビル の投与に関連した影響は認められなかった。 生殖発生毒性については、ラットの受胎能及び初期胚発生に関する試験[0(媒体),50, 200 及 び600 mg/kg/day]の結果で、最高用量の 600 mg/kg/day[AUC:386(雄)及び 373(雌)μg•h/mL、 ヒトAUC のそれぞれ 105 倍(雄)及び 101 倍(雌)]まで雌雄親動物の生殖能に影響は認められ なかった。 マウス及びウサギの胚・胎児発生に関する試験では、アスナプレビルに選択的な発生毒性は認 められなかった。マウスは、構造が類似した HCV プロテアーゼ阻害薬の試験で胚・胎児毒性に 関してラットより高い感受性を示したため、げっ歯類の動物種として選択した。マウス試験[0(媒 体),10, 50, 250 及び 500 mg/kg/day]における母動物の一般毒性に関する無毒性量は 250 mg/kg/day (AUC:737 μg•h/mL、ヒト AUC の 200 倍)、胚・胎児発生に関する無毒性量は 500 mg/kg/day(AUC: 1740 μg•h/mL、ヒト AUC の 472 倍)と考えられた。また、ウサギ試験[0(媒体),50, 100 及び 200 mg/kg/day]における母動物の一般毒性及び胚・胎児発生に関する無毒性量は、いずれも 200 mg/kg/day(AUC:4.40 μg•h/mL、ヒト AUC の 1.2 倍)と考えられた。 ラット出生前及び出生後の発生並びに母体の機能に関する試験[0(媒体),40, 125 及び 400 mg/kg/day]では、125 mg/kg/day 以上で F0 母動物に副腎重量増加、腹部膨満及び消化管の膨 満がみられ、400 mg/kg/day で F1 出生児の生存率低下、体重及び摂餌量の減少がみられたことに 基づき、F0 母動物の一般毒性に関する無毒性量は 40 mg/kg/day(母動物 AUC:26.8 μg•h/mL、ヒ トAUC の 7.3 倍)、F1 出生児の発達に関する無毒性量は 125 mg/kg/day(母動物 AUC:282 μg•h/mL、 ヒト AUC の 76 倍)、F1 出生児の生殖能に関する無毒性量は 400 mg/kg/day(母動物 AUC: 711 μg•h/mL、ヒト AUC の 193 倍)と考えられた。また、授乳中のラットに[14C]アスナプレビル を80 mg/kgの用量で単回経口投与した結果、アスナプレビルは母動物血漿中に対しCmax比0.174、 AUC 比 0.454(放射能当量)で乳汁中に排泄された(CTD 2.6.4.4)。
アスナプレビルは波長290~700 nm の領域の光線を吸収することから光毒性試験を実施した結 果、有色のLong-Evans ラットに最高用量 600 mg/kg/day(AUC:1440 μg•h/m、ヒト AUC の 390 倍)を単回投与した試験において光毒性は認められなかった。 免疫毒性試験及び抗原性試験は、いずれの動物種においてもアスナプレビルの直接的な作用に よる免疫機能への影響を示唆する変化が認められなかったことから実施しなかった。依存性試験 は、アスナプレビルが薬理学的に中枢神経作用を有さず、薬物依存性に関与する受容体等との薬 理学的相互作用は認められず、また、いずれの動物種においても神経系(中枢及び末梢)への影 響を示唆する変化が認められず、ラットにおいて脳への移行が非常に低いことから実施しなかっ た。 不純物の安全性については、安全性確認が必要な閾値(原体0.15%、製剤 0.2%)を超える不純 物が存在しないため、検討していない。 アスナプレビルの毒性を評価した結果、1 ヵ月間反復投与毒性試験では胃腸管及び肝臓が主な
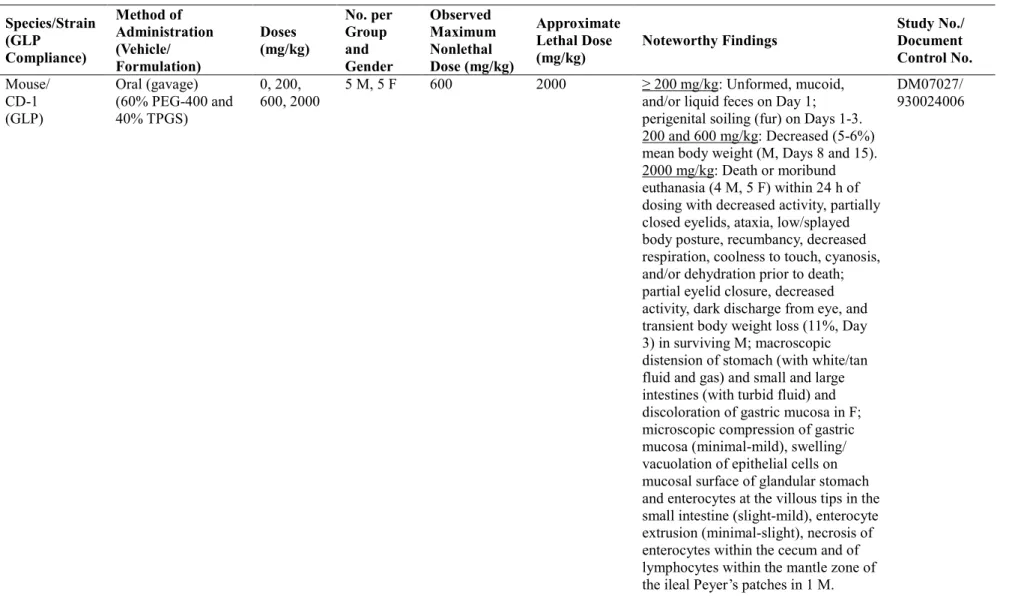
毒性の標的器官と考えられた。しかし、慢性毒性試験(ラット6 ヵ月間、イヌ 9 ヵ月間)におい て毒性の進展を示唆する所見は認められず、1 ヵ月間投与毒性試験と比較して新たな毒性の発現 もみられなかった。慢性毒性試験の無毒性量におけるAUC は、ヒト AUC の 82 倍以上であった。 以上の毒性試験成績は、C 型慢性肝炎患者におけるアスナプレビルのダクラタスビルとの併用 療法の安全性を担保するものと結論した。 2 単回投与毒性試験 マウス及びラットの急性毒性試験、イヌ探索的トキシコキネティクス及び忍容性試験を実施し た。急性毒性試験ではアスナプレビルの曝露量は測定しなかった。イヌでは、アスナプレビル曝 露の増加は用量比を上回り、概して性差はみられなかった。 2.1 マウスにおける単回経口投与毒性試験 (4.2.3.1.1 試験番号 DM07027、概要表 2.6.7.5) CD-1 マウス(1 群雌雄各 5 匹)にアスナプレビルを 0(媒体対照),200, 600 及び 2000 mg/kg の用量で単回経口投与した。投与容量はいずれも20 mL/kg とした。2 週間の観察期間中に生死及 び一般状態観察、体重測定を実施し、観察期間終了後に全例を剖検した。剖検所見に基づき、胃 腸管の組織について病理組織学的検査を実施した。 200 及び 600 mg/kg では試験終了時まで死亡例はみられなかった。2000 mg/kg では雄 4 例及び 雌5 例を投与後約 24 時間以内に死亡発見又は瀕死のため安楽死させた。これらの死亡例では、死 亡前に活動性低下、半眼、運動失調、腹臥位又は四肢伸展、横臥位、呼吸数減少、外皮温低下、 チアノーゼ及び脱水症がみられ、これらは状態悪化を示す症状と考えられた。また、すべての用 量で試験1 日(投与日)に不定形便、粘液便又は水様便、試験 1~3 日に生殖器周囲の皮毛の汚れ が一過性にみられた。更に、2000 mg/kg の雄の生存例でも活動性低下、半眼、暗色の眼脂が認め られた。200 及び 600 mg/kg の雄では、試験 8 日及び 15 日の群平均体重の軽度な減少(−5%~−6%) が認められた。2000 mg/kg の雄の生存例では、試験 3 日の体重が試験 1 日と比較して一過性に減 少した(−11%)。試験 8 日~15 日では、雄の体重増加量はいずれの用量でも対照群と同程度であっ た。雌では体重への影響は認められなかった。 200 及び 600 mg/kg では剖検及び病理組織学的検査で変化は認められなかった。剖検では、 2000 mg/kg の死亡発見例及び瀕死による安楽死例の雌雄で胃腸管の所見(胃の白色又は黄褐色の 液体及びガスによる膨満、小腸及び大腸の混濁液による膨満、雌で胃粘膜面の赤色化)が認めら れた。胃腸管の病理組織学的所見は2000 mg/kg のみで認められた。胃では、胃粘膜面の扁平化(ご く軽微~軽度、液体又はガスによる膨満による変化と考えられる)及び腺胃部表面の粘膜上皮細 胞の腫脹・空胞化(ごく軽微~軽微)が雌雄で認められた。小腸では、腸絨毛先端部の腸細胞剥 脱の減少(ごく軽微~軽微)及び腸絨毛先端部の腸細胞の腫脹・空胞化(軽微~軽度)が雌雄で 認められ、回腸パイエル板外套帯のリンパ球の単細胞壊死(軽微)が雄1 例に認められた。大腸 では、盲腸で腸細胞の単細胞壊死(軽微)が雌雄に認められた。 以上より、マウスにアスナプレビルを単回経口投与した結果、600 mg/kg 以下の用量では軽度
な症状(糞の変化、体重増加抑制)が一過性にみられた一方、2000 mg/kg では死亡及び消化管の 器質的変化が発現した。マウスにおける概略の致死量は2000 mg/kg と考えられた。 2.2 ラットにおける単回経口投与毒性試験 (4.2.3.1.2 試験番号 DM07028、概要表 2.6.7.5) SD ラット(1 群雌雄各 5 匹)にアスナプレビルを 0(媒体対照),200, 600 及び 2000 mg/kg の 用量で単回経口投与した。投与容量はいずれも20 mL/kg とした。2 週間の観察期間中に生死及び 一般状態観察、体重測定を実施し、観察期間終了後に全例を剖検した。 試験終了時まで死亡例はみられなかった。200 mg/kg ではアスナプレビル投与に関連した一般 状態の変化及び体重への影響は認められなかった。600 mg/kg では、試験 1 日~3 日に体幹の汚れ (投与後約 3 時間から発現)がみられたのみであった。2000 mg/kg では、体幹の汚れ(投与後 3 時間から発現し、概して試験 8 日までに消失)、試験 2 日~4 日の糞量減少(低頻度)、雄のみで 活動性低下、半眼及び腹臥位(投与後30 分~1 時間に発現、投与後 2 時間までに消失)が認めら れた。更に2000 mg/kg では雄の群平均体重の減少(−6.7%~−8.5%)が試験 3 日~15 日にみられ、 雌では1 例で試験 3 日に体重減少(−4.3%)が認められた。雄の試験 3 日~15 日の個体別体重増 加量は概して対照群と同程度であった。剖検では、アスナプレビルの投与に関連した所見はいず れの用量でも認められなかった。 以上より、アスナプレビルをラットに単回経口投与した結果、2000 mg/kg で毒性症状が発現し たが死亡はみられなかった。200 mg/kg では本薬に関連した変化はみられなかった。ラットにお ける概略の致死量は2000 mg/kg を超える量と考えられた。 2.3 イヌにおける単回経口投与トキシコキネティクス及び忍容性試験 (4.2.3.1.3 試験番号 DN06076[参考資料]、概要表 2.6.7.5) ビーグル犬(1 群雌雄各 1 匹)にアスナプレビルを 0(媒体対照),30, 100 及び 300 mg/kg の用 量で単回経口投与した。投与容量はいずれも2.5 mL/kg とした。1 週間の観察期間中、一般状態観 察、体重及び摂餌量測定、トキシコキネティクス測定(試験1 日)及び臨床病理学的検査(試験 2 日)を実施した。剖検は実施せず、観察期間終了後に全例を飼育群に戻した。 30~100 mg/kg の間で AUC の増加は用量比を上回り、性差は認められなかった。300 mg/kg で は、投与直後に発現した嘔吐(雌雄で投与後20 分、更に雌では投与後 1 時間)により、正確な曝 露量は評価できなかった可能性が考えられた(表2.3-1)。
表2.3-1 イヌ単回経口投与試験におけるトキシコキネティクス値 アスナプレビル投与量(mg/kg) 30 100 300 測定項目 採血日 雄 雌 雄 雌 雄 雌 Cmax (μg/mL) 投与日 9.83 5.96 85.4 85.9 49.8 97.8 AUC(0-24) (μg•h/mL) 投与日 23.0 15.5 578 486 312 923 アスナプレビルの投与に関連した所見は、300 mg/kg の嘔吐以外にはいずれの用量でも認めら れなかった。 以上より、100 mg/kg(AUC:532 μg•h/mL)以下の用量でアスナプレビルの忍容性は良好であっ た。イヌにおける概略の致死量は300 mg/kg を超える量と考えられた。 3 反復投与毒性試験 重要な試験以外の試験として、ラット2 週間経口投与試験及びサル 1 週間経口投与試験(併用 投与毒性試験の用量設定として利用)を実施した。重要な試験は、ラット(最長6 ヵ月間)及び イヌ(最長9 ヵ月間)を用いて実施した。アスナプレビル単剤の反復投与毒性試験に加え、ダク ラタスビルと併用投与した場合の毒性を評価するため、反復投与による併用投与毒性試験(3.4) を実施した。特記する場合を除き、アスナプレビルは媒体(60% PEG-400 及び 40% TPGS)に溶 解して経口投与した。 3.1 ラットにおける反復経口投与毒性試験 アスナプレビルのラットにおける反復経口投与毒性について、2 週間経口投与毒性試験(GLP 非適用)、1 ヵ月間及び 6 ヵ月間経口投与毒性試験(GLP 適用)を実施して評価した。各試験を通 じてアスナプレビルの血漿中濃度は変動幅が大きかったが、反復投与後のアスナプレビルのAUC は 30~200 mg/kg/day では概して用量比を上回って増加し、より高用量では用量比を下回った。 また、30~100 mg/kg/day では明らかな性差はみられなかったが、100 mg/kg/day を上回る用量で は雌のAUC は雄より高値(約 2 倍)であった。反復経口投与による蓄積は、200 mg/kg/day で投 与13 及び 26 週の AUC が投与 1 日の 2~5 倍であったことを除き、認められなかった。ラットに おける各試験のAUC 及びヒト AUC との比を表 3.1-1 に示す。
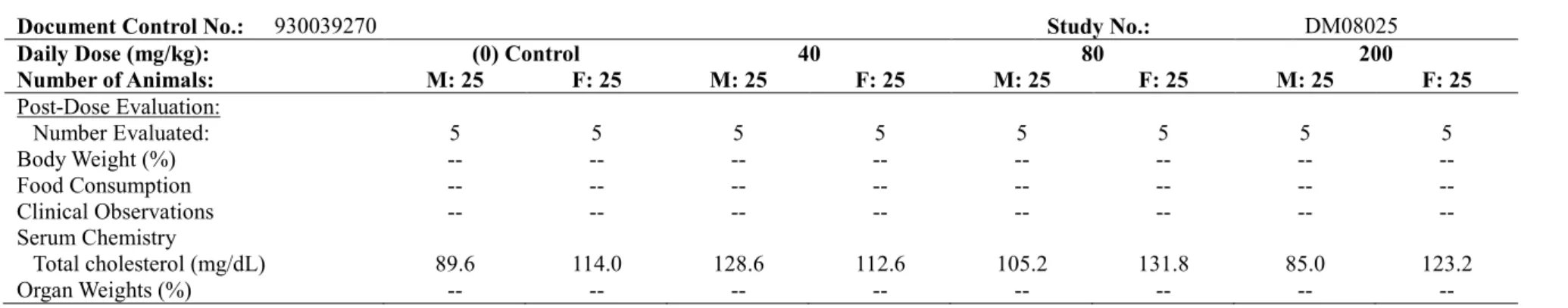
表3.1-1 ラット反復経口投与毒性試験におけるAUC及びヒトAUCとの比 投与期間、投与経路 (試験番号) 投与量 (mg/kg/day) AUC(0-24h) (μg•h/mL) a ヒトAUC との比b 雄 雌 雄 雌 2 週間、強制経口 (DN07002) 30 4.09 4.20 1.1 1.1 100 135 155 37 42 300 147 294 40 80 1 ヵ月間、強制経口 (DM07024) 30 1.88 1.34c 0.5 0.4 100 83.2 98.2 23 27 600 227 371 62 101 6 ヵ月間、強制経口 (DM08025) 40 3.99 11.6 1.1 3.1 80 39.3 144 11 39 200 321 684 87 185 下線を施した投与量は無毒性量を示す。 a 試験終了時の測定値
b 臨床推奨用量におけるAUC(3.69 μg•h/mL)に基づき算出(動物 AUC ÷ ヒト AUC)
c 投与後24 時間の AUC が LLOQ(5 ng/mL)未満であったため、AUC(0-12h)として算出
3.1.1 ラット 2 週間経口投与毒性試験 (4.2.3.2.1 試験番号 DN07002[参考資料]、概要表 2.6.7.6) SD ラット(1 群雌雄各 6 匹)にアスナプレビルを 0(媒体対照),30, 100 及び 300 mg/kg/day の 用量で1 日 1 回、2 週間経口投与した。媒体には 80% PEG-400 及び 20% TPGS を用い、投与容量 はいずれも5 mL/kg とした。生死及び一般状態観察、体重及び摂餌量測定、理学的検査、臨床病 理学的検査、器官重量測定、剖検及び病理組織学的検査(心臓、腎臓、肝臓、肺、大腸、小腸、 骨格筋及び脾臓)を実施した。また、投与1 日及び 14 日のトキシコキネティクスを測定し、剖検 時に採取した心臓、肝臓及び脾臓中のアスナプレビル濃度(1 群雌雄各 3 匹又は 6 匹)を測定し た。 投与1 日及び 14 日におけるアスナプレビルの全身曝露量は、30~100 mg/kg/day では用量比を 上回って増加したが(Cmax:約 10~12 倍、AUC:約 29~37 倍)、100~300 mg/kg では投与 1 日 で増加せず、投与14 日で約 2 倍に増加した。30 及び 100 mg/kg/day では Cmax 及び AUC に性差 はみられなかったが、300 mg/kg/day では雄の AUC が雌と比較してわずかに低値であった。投与 14 日の曝露量は投与 1 日と比較して、30 及び 100 mg/kg/day では AUC が約 50%減少したが、 300 mg/kg では雄で同等、雌で約 20%~36%増加し、2 週間の反復投与による明らかな蓄積は認め られなかった(表3.1.1-1)。
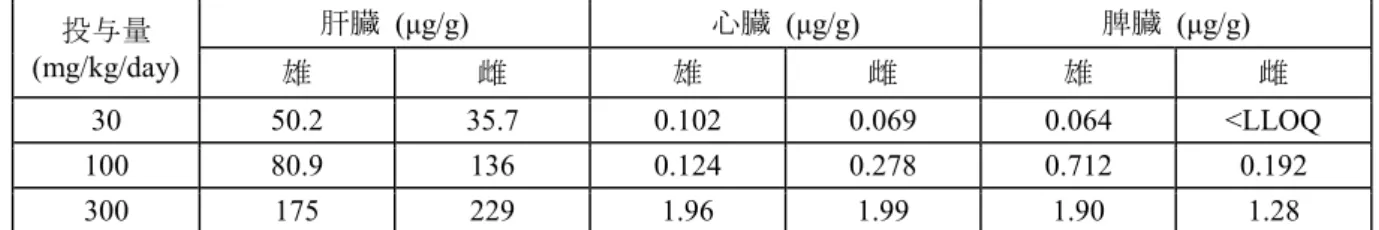
表3.1.1-1 ラット2 週間経口投与毒性試験におけるトキシコキネティクス値 アスナプレビル投与量 (mg/kg/day) 30 100 300 測定項目 採血日 雄 雌 雄 雌 雄 雌 Cmax (μg/mL) 投与1 日 1.67 2.00 19.4 21.0 17.5 16.6 投与14 日 1.01 1.19 9.73 12.3 19.6 22.6 AUC(0-24) (μg•h/mL) 投与1 日 8.45 9.28 246 275 164 244 投与14 日 4.09 4.20 135 155 147 294 肝臓、心臓及び脾臓の組織中アスナプレビル濃度はいずれも用量に比例して増加し、概して性 差はみられなかった。すべての用量において、アスナプレビルの心臓及び脾臓(抗ウイルス作用 の非標的器官)中濃度は、肝臓(治療器官)の約0.1%~1%であった(表 3.1.1-2)。 表3.1.1-2 ラット2 週間経口投与毒性試験におけるアスナプレビルの組織中濃度 投与量 (mg/kg/day) 肝臓 (μg/g) 心臓 (μg/g) 脾臓 (μg/g) 雄 雌 雄 雌 雄 雌 30 50.2 35.7 0.102 0.069 0.064 <LLOQ 100 80.9 136 0.124 0.278 0.712 0.192 300 175 229 1.96 1.99 1.90 1.28 試験期間中の死亡例はみられず、最高用量の 300 mg/kg/day まで忍容性は良好であった。アス ナプレビルの投与に関連した一般症状、剖検及び病理組織学的所見はいずれの用量でも認められ なかった。 300 mg/kg/day では、アスナプレビルに関連した変化として尿 pH の軽微な上昇(+0.7 pH)、血 清ビリルビンの軽度な増加(雌、対照群の1.9 倍)、雄における腎臓重量減少(15%)及び心臓重 量減少(−12%)が認められたが、血液生化学的検査及び剖検で関連した変化は認められなかった ため、これらの変化は生物学的意義の低い変化と考えられた。 以上より、アスナプレビルを 2 週間経口投与したラットにおいて、最高用量の 300 mg/kg/day (AUC:221 μg•h/mL)まで忍容性は良好であり、本薬の投与に関連した毒性学的意義のある変化 は認められなかったことから、本試験における無毒性量は300 mg/kg/day と考えられた。 3.1.2 ラット 1 ヵ月間経口投与毒性試験 (4.2.3.2.2 試験番号 DM07024、概要表 2.6.7.7B) SD ラット(1 群雌雄各 15 匹)にアスナプレビルを 0(媒体対照),30, 100 及び 600 mg/kg/day の用量で1 日 1 回、1 ヵ月間経口投与した。投与容量はいずれも 5 mL/kg とした。別途トキシコ キネティクス測定用サテライト群(1 群雌雄各 9 匹)を設け、同様に投与した。生死及び一般状 態観察、体重及び摂餌量測定、理学的検査及び眼科学的検査、臨床病理学的検査、器官重量測定、
剖検及び病理組織学的検査を実施した。また、投与1 日及び 28 日のトキシコキネティクス測定及 び剖検時の肝臓中及び血漿中アスナプレビル濃度測定を実施した。投与期間終了時に各群雌雄各 10 匹を剖検し、2 週間の休薬期間後に各群雌雄各 5 匹を剖検した。
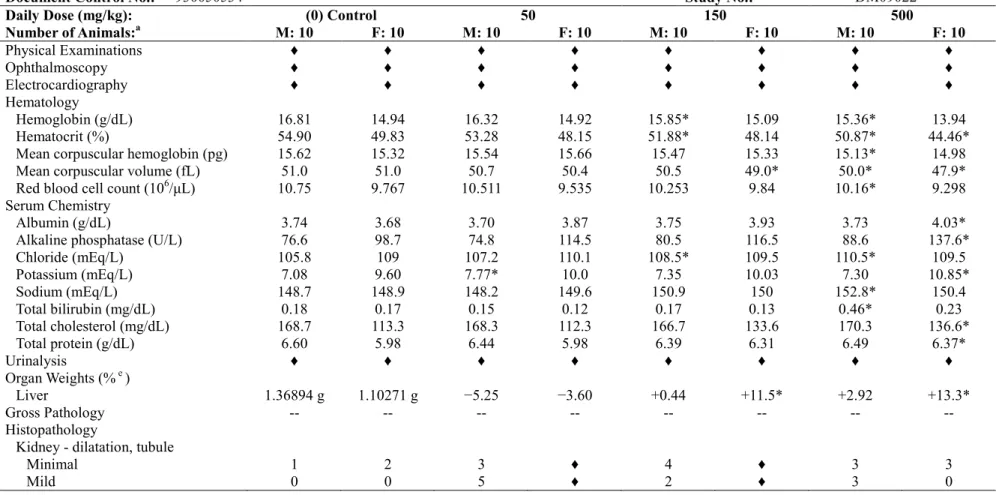
反復投与後のアスナプレビルのCmax及びAUCは30~100 mg/kg/dayでは概して用量比を上回っ て増加し(23~73 倍)、100~600 mg/kg/day では用量比を下回った(2.3~3.8 倍)。いずれの用量 でも性差はみられなかった。投与28 日における雌雄の Cmax 及び AUC は、30 mg/kg/day では投 与1 日と比較して 29%~72%低値であったが 100 mg/kg/day 以上では同程度であり、蓄積はないと 考えられた(表3.1.2-1)。 表3.1.2-1 ラット1 ヵ月間経口投与毒性試験におけるトキシコキネティクス値 アスナプレビル投与量 (mg/kg/day) 30 100 600 測定項目 採血日 雄 雌 雄 雌 雄 雌 Cmax (μg/mL) 投与1 日 0.658 0.785 14.9 14.5 29.2 20.1 投与28 日 0.290 0.306 11.1 13.7 24.7 43.4 AUC(0-24) (μg•h/mL) 投与1 日 2.65a 4.73 129 108 291 262 投与28 日 1.88 1.34a 83.2 98.2 227 371 a 投与後24 時間のアスナプレビル濃度が LLOQ (5 ng/mL)を下回ったため、AUC(0-12h)を算出した。 剖検時(最終投与後の約24 時間)に採取した肝臓中のアスナプレビル濃度は、用量比を下回っ て増加した。アスナプレビルの肝臓中濃度は血漿中濃度より高値であり、性差はみられなかった (表3.1.2-2)。 表3.1.2-2 ラット1 ヵ月間経口投与毒性試験におけるアスナプレビルの肝臓中及び 血漿中濃度 投与量 (mg/kg/day) 肝臓 (μg/g) a 血漿 (μg/mL) a 肝臓中/血漿中濃度比 雄 雌 雄 雌 雄 雌 30 49.0 31.5 0.0064 <LLOQ 7656 NA 100 96.9 93.1 0.0062 0.0188 15680 4952 600 228 264 0.371 3.74 615 71 a 肝臓及び血漿中アスナプレビル濃度は投与期間終了時の剖検(最終投与後約24 時間)において測定 アスナプレビルに関連した死亡例及び一般症状はみられず、30 mg/kg/day では本薬投与に関連 した変化は認められなかった。100 mg/kg/day では、血清クレアチニンの減少(対照群の 0.90 倍) 及び肝臓重量増加(+10%)が認められたのみであった。 600 mg/kg/day では、アスナプレビルに関連した影響として、投与 1 週に雄で体重減少(−6.7%) 及び摂餌量減少(−26%)、雌で摂餌量減少(−15%)が認められた。また、雌において投与 3 週及
び4 週に摂餌量の増加(+9.0%及び+9.8%)が認められたが、比較的軽度な変化であり、関連した 体重増加はみられなかった。 また、血液学検査及び凝固検査では、600 mg/kg/day の雄及び雌でヘモグロビン減少(対照群の 0.95 及び 0.93 倍)、ヘマトクリット減少(0.95 倍)、MCV 減少(0.95 及び 0.97 倍)、MCH 減少(0.94 及び0.95 倍)、フィブリノゲン減少(0.76 及び 0.86 倍)が認められた。更に雌では、赤血球分布 幅(RDW)の増加(1.10 倍)、平均赤血球ヘモグロビン濃度(MCHC)の減少(0.98 倍)が認め られた。アスナプレビルに関連したこれらの血液学的変化から、赤血球の小型化が示唆された。 血液生化学的検査では、雄及び雌でALT 増加(対照群の 1.79 及び 2.10 倍)、総ビリルビン増加(1.82 及び1.46 倍)、塩素増加(1.02 倍)及びアルブミン/グロブリン比上昇(1.12 及び 1.11 倍)、総蛋 白減少(0.90 及び 0.92 倍)、グロブリン減少(0.84 及び 0.87 倍)、グルコース減少(0.84 及び 0.86 倍)、尿素窒素減少(0.69 及び 0.79 倍)、クレアチニン減少(0.87 倍)及び総コレステロール減少 (0.71 及び 0.70 倍)が認められた。また、雄でトリグリセリド減少(対照群の 0.43 倍)及びカル シウム減少(0.97 倍)、アルブミン減少(0.96 倍)、雌で ALP 増加(1.41 倍)が認められた。これ らの臨床検査値の変化は、ALT 及び総ビリルビンの変化を除き、比較的軽度な変化でほとんどの 平均値及び個別値は試験施設背景値2) の範囲内あるいは生物学的意義のない変動であったことか ら、毒性学的意義は低いと考えられた。赤血球関連値、ALT、総ビリルビン及び血清蛋白の変化 は、イヌ1 ヵ月間投与毒性試験でも 300 mg/kg/day で認められた。尿検査では、600 mg/kg/day で 尿量の増加(対照群の2.56~3.22 倍)並びにこれに関連して尿比重低下(0.60 倍)及び尿 pH 上 昇(1.10~1.14 倍)が認められた。尿量の増加については、脱水症状が認められずアスナプレビ ル及び代謝物の尿排泄はほとんどないことから、摂水量の増加(多飲)によるものであり、腎障 害を示唆する所見ではないと考えられた。腎臓にアスナプレビルの投与に関連した病理組織学的 変化は認められなかった。 器官重量測定では、600 mg/kg/day の雄で甲状腺重量増加(+27%)、雌で副腎及び肝臓重量増加 (+17%及び+21%)が認められた。これらの器官重量の変化に関連した病理組織学的所見は認め られなかった。雌における肝臓重量の増加は、肝臓における高濃度のアスナプレビルによる肝薬 物代謝酵素の誘導に関連した適応性変化である可能性が考えられた。剖検では、雌雄で透明~黄 色の液体及びガスによる小腸及び大腸の膨満(小腸で顕著)がみられ、雄1 例で小腸壁の肥厚が 認められた。病理組織学的検査では、雌雄で腸細胞の肥大(ごく軽微~軽度)が小腸(十二指腸 及び空腸)で高頻度に認められ、大腸(盲腸)でも低頻度に認められた。また、雄のみで盲腸及 び結腸の杯細胞の減少が認められた。 更に 600 mg/kg/day の雌において、副腎皮質索状帯に多巣性の細胞変性(ごく軽微)の発現頻 度増加が認められ、ラットにおける自然発生性の変化がアスナプレビルの投与により亢進したも のと考えられた。 100 mg/kg/day 以上で用量依存性に認められた変化は、血清クレアチニン減少(対照群の 0.87 ~0.90 倍)及び肝臓重量増加(+10%~+17%)のみであった。2 週間の休薬期間により、上記の 変化はいずれも回復した。なお、600 mg/kg/day の雌で休薬期間中の摂餌量増加(22%及び 15%) 及び600 mg/kg/day の雄でグルコース減少(0.79 倍)が認められたが、毒性学的意義の低い変化
と考えられた。 以上より、アスナプレビルを1 ヵ月間経口投与したラットにおいて、100 mg/kg/day まで忍容性 は良好であった。主要な毒性の標的器官は、600 mg/kg/day(AUC:299 μg•h/mL)腸細胞の肥大が 認められたことから、小腸と考えられた。2 週間の休薬により、アスナプレビルに関連した毒性 学的意義のある所見は完全に回復した。本試験における無毒性量は100 mg/kg/day(AUC:91 μg•h/mL、 ヒトAUC の 25 倍)と考えられた。 3.1.3 ラット 6 ヵ月間経口投与毒性試験 (4.2.3.2.4 試験番号 DM08025、概要表 2.6.7.7C) SD ラット(1 群雌雄各 25 匹)にアスナプレビルを 0(媒体対照),40, 80 及び 200 mg/kg/day の 用量で1 日 1 回、6 ヵ月間経口投与した。最高用量の 200 mg/kg/day は、1 ヵ月間投与毒性試験の 所見に基づき、投与期間の延長による毒性の亢進の可能性を考慮し、十分な曝露量(AUC)が得 られる用量として選択した。投与容量はいずれも5 mL/kg とした。生死及び一般状態観察、体重 及び摂餌量測定、理学的検査、眼科学的検査、臨床病理学的検査、器官重量測定、剖検及び病理 組織学検査を実施した。また、投与1 日、投与 13 週及び 26 週のトキシコキネティクス測定、剖 検時の肝臓中及び血漿中アスナプレビル濃度測定を実施した。6 ヵ月間の投与期間終了時に各群 雌雄各20 匹を剖検し、1 ヵ月間の休薬期間後に各群雌雄各 5 匹を剖検した。 反復投与後のアスナプレビルの AUC は、いずれの用量及び測定時期においても概して用量比 を上回って増加したが、雄の平均AUC は雌より低値(0.3~0.8 倍)であった。40 及び 80 mg/kg/day では蓄積あるいは酵素誘導による曝露量の減少は認められなかったが、投与13 週及び 26 週にお ける200 mg/kg/day の平均 AUC は投与 1 日と比較して 2~5 倍高値であった(表 3.1.3-1)。 表3.1.3-1 ラット6 ヵ月間経口投与毒性試験におけるトキシコキネティクス値 アスナプレビル投与量 (mg/kg/day) 40 80 200 測定項目 採血日 雄 雌 雄 雌 雄 雌 Cmax (μg/mL) 投与1 日 2.01 2.51 17.4 17.9 22.3 12.0 投与13 週 1.33 3.39 15.1 16.3 62.3 89.2 投与26 週 1.16 2.65 8.67 22.7 38.7 124 AUC(0-24) (μg•h/mL) 投与1 日 8.80 14.7 80.9 119 193 133 投与13 週 6.24 13.6 63.2 83.4 442 650 投与26 週 3.99 11.6 39.3 144 321 684 投与期間終了後の剖検時(最終投与後約24~30 時間)に採取した肝臓中のアスナプレビル濃度 は血漿中濃度より高値であり、用量に伴う増加は概して用量比を下回った(表3.1.3-2)。
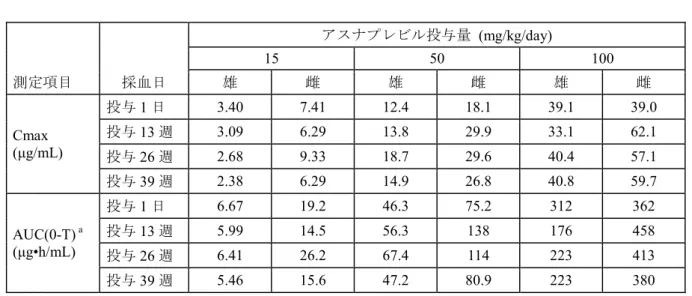
表3.1.3-2 ラット6 ヵ月間経口投与毒性試験におけるアスナプレビルの肝臓中及び血漿中 濃度 投与量 (mg/kg/day) 肝臓 (ng/g) a 血漿 (ng/mL) a 肝臓中/血漿中濃度比b 雄 雌 雄 雌 雄 雌 40 41.9 31.0 0.018 0.016 3093 2724 80 53.8 46.1 0.025 0.052 2713 1362 200 83.8 54.1 0.117 0.090 1512 1144 a 肝臓及び血漿中アスナプレビル濃度は投与期間終了時の剖検(最終投与後約24 時間)で測定 b 個体別の肝臓/血漿中濃度比より算出 アスナプレビルの投与に関連した死亡例はみられなかった。本薬の投与に関連した一般症状は、 200 mg/kg/day の雌雄で投与期間中に散発的にみられた軟便及び被毛の濡れ(鼻口部、胸部、頸部 又は腹部)の発現頻度増加のみであった。また、200 mg/kg/day の雄で軽微な体重増加(対照群の +9.7%~+11.3%)及び摂餌量増加(+8.8%~+13.7%)が認められた。これら以外に体重及び摂餌 量の変化はみられず、アスナプレビルの投与に関連した眼科学的検査、理学的検査、血液学的検 査、凝固検査及び尿検査所見は最高用量の200 mg/kg/day まで認められなかった。 血液生化学的検査において、200 mg/kg/day の雌雄で総コレステロール減少(対照群の 0.79~0.92 倍)がみられたが、休薬により回復した。 器官重量測定では、雄の全用量で肝臓重量の用量依存性の増加(+8%~+23%)が認められた。 これらの肝臓重量増加に関連した病理組織学的所見は認められず、肝臓における高濃度のアスナ プレビルによる肝薬物代謝酵素の誘導に関連した適応性変化である可能性が考えられた。剖検及 び病理組織学的検査では、本薬投与に関連した所見はいずれの用量でも認められなかった。 以上より、アスナプレビルを最高用量200 mg/kg/day で 6 ヵ月間経口投与したラットにおける 忍容性は良好であった。アスナプレビルの投与に関連した所見はいずれも有害性の低いものと考 えられ、特定の器官への毒性はみられなかった。本試験における無毒性量は200 mg/kg/day(AUC: 503 μg•h/mL、ヒト AUC の 136 倍)と考えられた。 3.2 イヌにおける反復経口投与毒性試験 アスナプレビルのイヌにおける反復経口投与毒性について、1 ヵ月間及び 9 ヵ月間経口投与毒 性試験(GLP 適用)を実施して評価した。イヌでは経口投与時のバイオアベイラビリティ(42% 以上)がサル(10%)と比較して高く、より高いアスナプレビルの全身曝露が得られたため、非 げっ歯類の動物種としてイヌを選択した。イヌにおける代謝プロファイルはヒトと質的に類似し た。いずれの試験でもアスナプレビルのAUC は 15~300 mg/kg/day では概して用量比を上回って 増加した。雌のAUC は雄より高く(約 2 倍)、反復投与による蓄積はみられなかった。(表3.2-1)。
表3.2-1 イヌ反復経口投与毒性試験におけるアスナプレビルのAUC及び ヒトAUCとの比
下線を施した投与量は無毒性量を示す。
a 試験終了時の測定値
b 臨床推奨用量におけるAUC(3.69 μg•h/mL)に基づき算出(動物 AUC ÷ ヒト AUC)
3.2.1 イヌ 1 ヵ月間経口毒性試験 (4.2.3.2.5 試験番号 DM07020、概要表 2.6.7.7D) ビーグル犬(1 群雌雄各 3 匹)にアスナプレビルを 0(媒体対照),20, 60 及び 300 mg/kg/day の 用量で1 日 1 回、1 ヵ月間経口投与した。投与容量はいずれも 2.5 mL/kg とした。生死及び一般状 態、体重測定、摂餌量観察、理学的検査(神経学的検査、眼科学的検査及び呼吸系検査を含む)、 心電図検査、動脈血酸素飽和度測定、臨床病理学的検査、器官重量測定、剖検及び病理組織学的 検査を実施した。また、投与1 日及び 28 日のトキシコキネティクス測定及び剖検時の肝臓中及び 血漿中アスナプレビル濃度測定を実施した。 反復投与後のアスナプレビルのAUC は、概して用量比を上回って増加した。20 mg/kg/day では 雌のAUC が雄より高値(約 1.8~3.6 倍)であったが、60 mg/kg/day 以上では同程度であった。個 体間変動が大きいため、蓄積は雌雄のいずれの用量でも明らかでなかった(表3.2.1-1)。 表3.2.1-1 イヌ1 ヵ月間経口投与毒性試験におけるトキシコキネティクス値 アスナプレビル投与量 (mg/kg/day) 20 60 300 測定項目 採血日 雄 雌 雄 雌 雄 雌 Cmax (μg/mL) 投与1 日 1.96 4.47 19.6 21.9 60.5 97.5a 投与28 日 1.69 6.12 28.9 26.3 114 106a AUC(0-24h) (μg•h/mL) 投与1 日 6.10 11.1 78.3 55.6 672 952a 投与28 日 4.58 16.3 102 98.5 1410 1360a a 2 例の平均値(1 例で投与 1 日及び 28 日の投与後 1 時間に一部溶解した投与カプセルの嘔吐がみられたため、 平均値の算出から本例を除外した) 肝臓中のアスナプレビル濃度の用量に伴う増加は、20~60 mg/kg/dayでは用量比を下回ったが、 投与期間、投与経路 (試験番号) 投与量 (mg/kg/day) AUC (0-24h) a ヒトAUC との比b 雄 雌 雄 雌 1 ヵ月間、強制経口 (DM07020) 20 4.58 16.3 1.2 4.4 60 102 98.5 28 27 300 1410 1360 382 369 9 ヵ月間、強制経口 (DM08026) 15 5.46 15.6 1.5 4.2 50 47.2 80.9 13 22 100 223 380 60 103
60~300 mg/kg/day では用量比を上回った。肝臓中/血漿中濃度比はいずれの用量でも概して同程 度であり、肝臓への取り込みが飽和しておらず、肝臓及び血漿からのアスナプレビルの消失は同 程度であることが示唆された。肝臓中濃度及び肝臓中/血漿中濃度比に性差は認められなかった (表3.2.1-2)。 表3.2.1-2 イヌ1 ヵ月間経口投与毒性試験におけるアスナプレビルの肝臓中及び 血漿中濃度 投与量 (mg/kg/day) 肝臓 (μg/g) a 血漿 (μg/mL) a 肝臓/血漿濃度比b 雄 雌 雄 雌 雄 雌 20 1.01 1.17 0.031 0.032 47 44 60 2.10 1.72 0.030 0.040 89 43 300 146 133 13.0 7.72 54 17 a 肝臓及び血漿中アスナプレビル濃度は投与期間終了時の剖検(最終投与後約24 時間)において測定 b 個体別の肝臓中/血漿中濃度比より算出 試験期間中の死亡例はみられなかった。60 mg/kg/day 以下の用量ではアスナプレビルの投与に 関連した所見は認められず、300 mg/kg/day では呼吸及び体温、神経学的検査、眼科学的検査、心 電図検査及び尿検査に変化は認められなかった。アスナプレビルに関連した一般症状として、 300 mg/kg/day の雌雄で嘔吐及び不定形・水様便の発現頻度が投与期間を通して増加し、体重減少 (−6.9%~−9.1%)及び摂餌量減少が認められた。 血液学的検査では、300 mg/kg/day の雌雄で MCV 減少(対照群の 0.90 及び 0.89 倍)、MCH 減 少(0.89 及び 0.86 倍)、RDW 増加(1.20 及び 1.16 倍)、雌でヘモグロビン減少(0.85 倍)が認め られ、赤血球の小型化が示唆された。また、雌雄の数例で有核赤血球及び鉄染色陽性の微小な好 塩基性細胞封入体(パッペンハイマー体)を有する赤血球が少数認められた。パッペンハイマー 体はヘモグロビン合成の障害との関連性が知られている1)。血液生化学的検査では、300 mg/kg/day の雄及び雌でALT 増加(対照群の 4.48 倍及び 1.94 倍)、総ビリルビン増加(1.66 倍及び 1.55 倍)、 総蛋白減少(0.90 倍及び 0.78 倍)、アルブミン減少(0.87 倍及び 0.75 倍)、総コレステロール減少 (0.61 倍及び 0.55 倍)が認められた。更に雄 1 例で GGT 増加(投与前値の 2.7 倍)、雌で塩素増 加(対照群の1.03 倍)、グロブリン減少(0.81 倍)、カルシウム減少(0.91 倍)が認められた。こ れらの臨床検査値の変化は、ALT、GGT 及び総ビリルビンの増加を除き、比較的軽度でほとんど の平均値及び個別値は概して試験施設背景値3) の範囲内あるいは生物学的意義のない変化であっ たことから、毒性学的意義は低いと考えられた。ALT、GGT 及び総ビリルビンの増加は、病理組 織学的検査で観察された肝細胞凝固壊死(後述)に関連した変化と考えられた。赤血球関連値、 ALT、総ビリルビン及び血清蛋白の変化は、ラット 1 ヵ月間投与毒性試験でも 600 mg/kg/day で認 められた。また、血液凝固検査において、雄及び雌で活性化部分トロンボプラスチン時間(APTT) の軽度な短縮(0.82 及び 0.83 倍)がみられた。尿検査では、アスナプレビルに関連した変化はい ずれの用量でも認められなかった。 器官重量測定では、300 mg/kg/day の雌で副腎重量増加(+39%~+42%)及び脾臓重量増加(+45%