第
2 部(モジュール 2)
CTD の概要(サマリー)
2.7 臨床概要
2.7.1 生物薬剤学及び関連する分析法の概要
2.7.2 臨床薬理の概要
2.7.3 臨床的有効性の概要
2.7.4 臨床的安全性の概要
2.7.5 参考文献
2.7.6 個々の試験のまとめ
鳥居薬品株式会社
略号一覧及び用語の定義
略号一覧略号 省略していない表現
英語 日本語
ACT Asthma control test 喘息コントロールテスト AHQ-Japan Asthma health questionnaire-Japan AHQ-Japan QOL 調査票
AU Allergy unit 米国の標準化アレルゲンエキスの活性を表す単位の一つ
Der far Dermatophagoides farinae コナヒョウヒダニ Der pte Dermatophagoides pteronyssinus ヤケヒョウヒダニ FAS Full analysis set 最大の解析対象集団
FEV1 Forced expiratory volume in one second 1 秒間努力呼気容量(又は 1 秒量) %FEV1 % Forced expiratory volume in one second
年齢,性別,身長を基にあらかじめ算出された健常 者の予測1 秒量(FEV1予測値)に対する患者の1 秒 量(FEV1実測値)の比率
HDM House dust mite 室内塵ダニ IgE Immunoglobulin E 免疫グロブリンE IgG Immunoglobulin G 免疫グロブリンG
JRQLQ Japan Rhinitis Quality of Life Questionnaire 日本アレルギー性鼻炎標準QOL 調査票 LLT Lowest level term MedDRA/J の下層語
MedDRA/J Medical dictionary for regulatory activities/J ICH 国際医薬用語集日本語版 PSUR Periodic safety update report 定期的安全性最新報告
PT Preferred term MedDRA/J の基本語 QOL Quality of life 生活の質
RAST Radioallergosorbent test 放射性アレルゲン吸着試験
SCIT Subcutaneous immunotherapy 皮下注射によるアレルゲン免疫療法 SOC System organ class MedDRA/J の器官別大分類
用語の定義
用語 定義
急速法 アレルゲン免疫療法における増量法で,ラッシュ法やクラスター法がある。
従来法 アレルゲン免疫療法における増量法で,50%増量法や 100~200%増量法がある。 ALK 社 デンマークに本社を置く製薬会社(ALK-Abelló 社)
Der f Der far の抽出アレルゲンエキス Der f 1 Der far 糞体由来の主要アレルゲン Der f 2 Der far 虫体由来の主要アレルゲン
Der p Der pte の抽出アレルゲンエキス Der p 1 Der pte 糞体由来の主要アレルゲン Der p 2 Der pte 虫体由来の主要アレルゲン
HDM SCIT ダニアレルゲンを用いた皮下注射によるアレルゲン免疫療法 TO-204 TO-204 皮下注の開発コード
TO-204 皮下注
Der far 及び Der pte の各抽出液を等量混合した注射液で,10,000 AU/mL の活性を有す る。
ALK 社が米国,カナダ等で販売している SCIT 用の Allergenic Extract Standardized Mite と同じものである。
Allergenic Extract Standardized Mite
ALK 社が米国,カナダ等で販売している HDM アレルギー疾患用注射剤の総称で 6 種 類の製剤に分類される(下表参照)。いずれも10,000 AU/mL の活性を有する。
Allergenic Extract Standardized Mite の種類とその構成成分
Allergenic Extract Standardized Mite:製剤 A,B,C,D,E,F の総称 TO-204 皮下注:製剤 F と同じ
構成アレルゲン
Der far エキス Der pte エキス 両者の混合液
皮膚テスト用 製剤A 製剤B 製剤C
2.7.1 生物薬剤学及び関連する分析法の概要 2.7.1.1 背景及び概観 TO-204 皮下注は,皮下注射によるアレルゲン免疫療法(SCIT)のためのアレルゲン製剤であり HDM アレルギー性鼻炎及び HDM アレルギー性喘息の治療薬として製造販売承認申請を行うもの である。 今回の製造販売承認申請の評価資料とする臨床試験としては,HDM アレルギー性鼻炎患者及び HDM アレルギー性喘息患者を対象に,TO-204 皮下注を用いた SCIT を行い,安全性の評価を主目 的としたTO-204 第 III 相臨床試験 長期投与試験を実施した。今回実施した臨床試験はこの TO-204 第III 相臨床試験 1 試験のみである。
今回の臨床試験で使用したTO-204 皮下注は 1 製剤のみで,使用した製剤はガラスバイアルに充 填した液状の注射剤(容量10 mL)であり,1 mL あたり 10,000AU の活性を有する。なお,本剤 はDer far 及び Der pte 由来のアレルゲンエキス(10,000AU/mL)を等量に混合したものである。
TO-204 皮下注の有効成分である主要アレルゲンはタンパク質であることから,皮下投与したア レルゲンは血中に移行した後,組織中及び血液中のタンパク質分解酵素により速やかに分解を受 けると考えられ,TO-204 未変化体の検出は困難である。 以上のことからTO-204 皮下注の生物薬剤学に関連する試験は実施しなかった。 2.7.1.2 個々の試験結果の要約 該当しない。 2.7.1.3 全試験を通しての結果の比較と解析 該当しない。 2.7.1.4 付録 該当する付録はない。
3
2.7.2 臨床薬理の概要
TO-204 皮下注の主要アレルゲンである Der f 1, Der f 2, Der p 1, Der p 2 はいずれもタンパク質で ある。タンパク質は一般に体内で速やかに代謝を受けることから,主要アレルゲンが血中に移行 したとしても速やかに分解,代謝されると考えられ,アレルゲンタンパクそのものが体内循環さ れることは殆ど無いと考えられる。したがって,TO-204 皮下注を用いた臨床薬理学的な検討は必 要ないと考え実施していない。 2.7.2.1 背景及び概観 該当しない。 2.7.2.2 個々の試験結果の要約 該当しない。 2.7.2.3 全試験を通しての結果の比較と解析 該当しない。 2.7.2.4 特別な試験 該当する特別な試験はない。 2.7.2.5 付録 該当する付録はない。
2.7.3 臨床的有効性の概要 2.7.3.1 背景及び概観 HDM アレルギー性鼻炎及び喘息は,本邦ではスギ花粉症と並ぶ重要なアレルギー疾患である。 日本ではこれまで鳥居薬品株式会社が販売する「診断用アレルゲン皮内エキス治療用アレルゲン エキス皮下注「トリイ」ハウスダスト 1),及び治療用アレルゲンエキス皮下注「トリイ」ハウス ダスト2)」がHDM アレルゲンエキスの代用品として HDM 免疫療法に使用されてきたが,これら の「ハウスダスト」はいわゆる家庭の埃であり,主たるアレルゲンとされるダニ成分の活性は低 いという指摘がなされてきた。 鳥居薬品株式会社は,これまで国内における免疫療法のためのアレルゲンエキスを供給してき た唯一の企業として,一般社団法人日本アレルギー学会からアレルゲン免疫療法のための HDM アレルゲンエキスの早期開発の要望を受けてきた。これらの要望に応えるべく,デンマークに本 社を置くALK Abelló 社(以下,ALK 社)からアレルゲンエキス,HDM SCIT 用製剤 TO-204 皮下 注(Allergenic Extract Standardized Mite:以下,TO-204 皮下注)3)の導入を行った。
TO-204 皮下注は,本邦における使用実績はないものの,アメリカ,カナダ等において既に上市 されている。 が,独立行政法人医薬品医療機器総合機構 (以下,PMDA)より示された。これらを踏まえ,鳥居薬品株式会社は, する ことでPMDA との合意に至った( 平成 年 月 日,及び 平成 年 月 日)。 以上の経緯から,TO-204 皮下注の日本人を対象とした臨床試験は,安全性の評価を主目的とす るTO-204 第 III 相臨床試験 長期投与試験(以下,本治験)として計画した。本申請資料に含めた 評価資料としての臨床試験は本治験1 試験のみであり,2.7.3.2 項にその概要を記載した。 2.7.3.2 TO-204 第 III 相臨床試験 長期投与試験の概要 TO-204 第 III 相臨床試験 長期投与試験の概要を表 2.7.3.2-1 に示した。
1
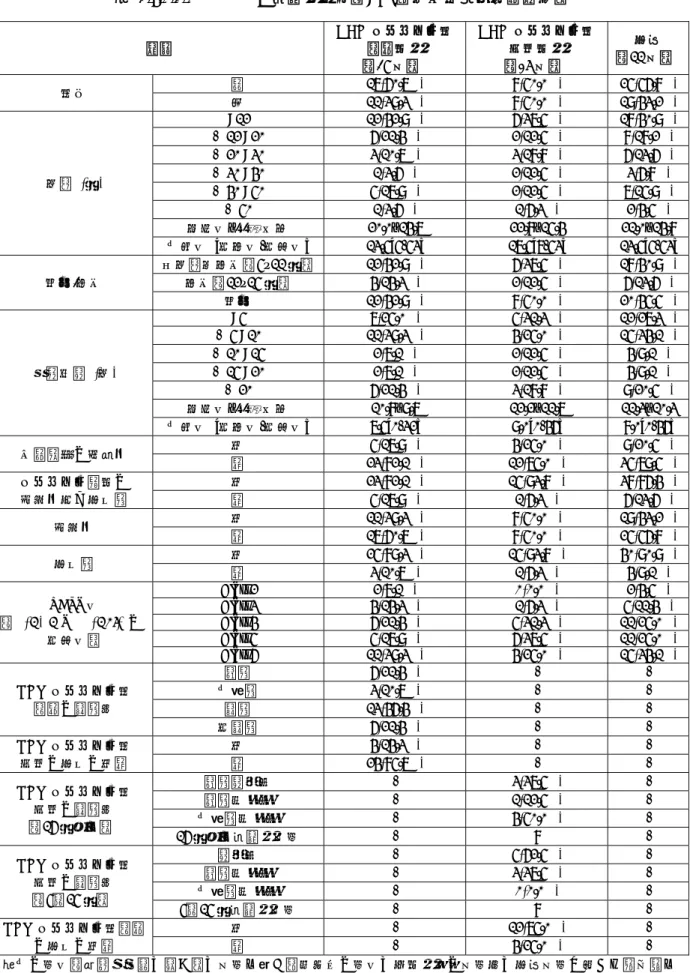
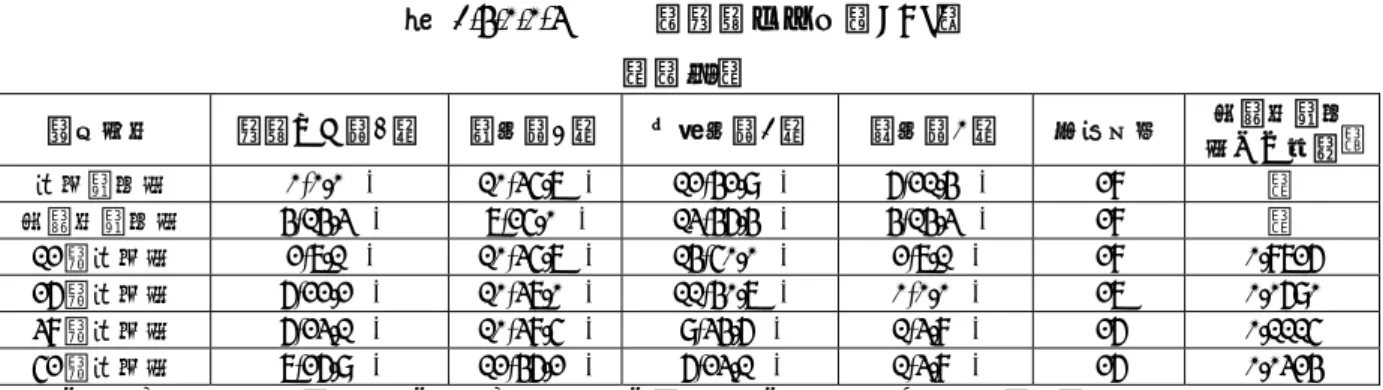
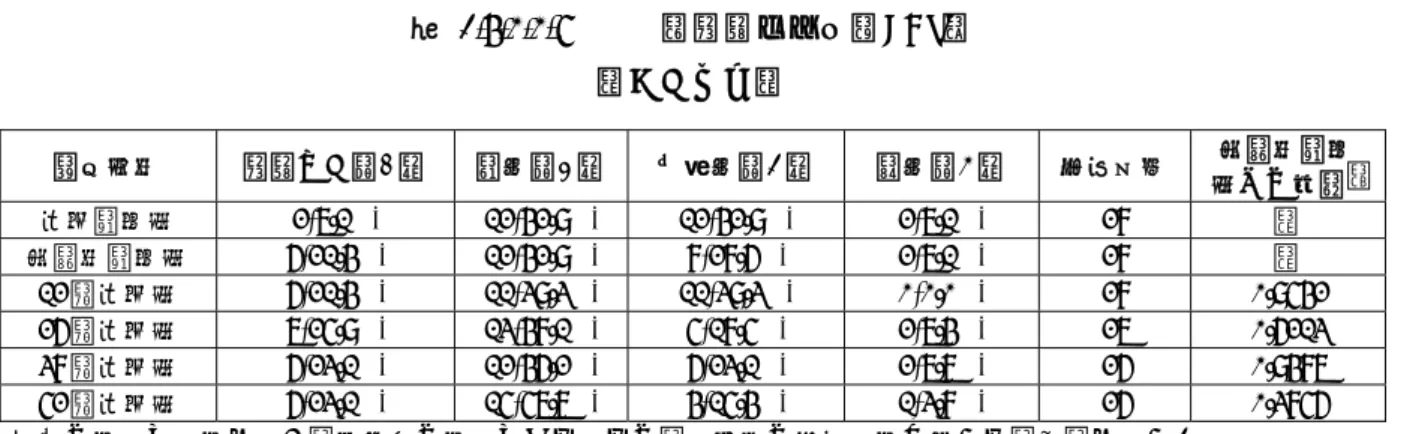
表 2.7.3.2-1 TO-204 第 III 相臨床試験 長期投与試験の概要 治験の標題 TO-204 第 III 相臨床試験 長期投与試験 -HDM アレルギー性鼻炎患者及び HDM アレルギー性喘息患者を対象とした 皮下免疫療法の検討- 治験実施計画書 識別コード 204-3-1 目的 安全性 実施施設数 13 施設(15 診療科) 治験期間 年 月~医薬品製造販売承認日 開発のフェーズ 治験デザイン 第III 相 非盲検,非対照,多施設共同 対象患者 HDM アレルギー性鼻炎 HDM アレルギー性喘息 安全性解析対象 症例数 44 例 使用製剤 治療薬:TO-204 皮下注(希釈には治療用アレルゲンエキス希釈液「トリイ」 を使用) 皮内閾値検査用薬剤:TO-204 皮下注(希釈には診断用アレルゲン皮内エキス 対照液「トリイ」を使用) 用法・用量 初回投与液の濃度はTO-204 皮下注の皮内閾値検査で求めた閾値又は閾値の 10 倍低い濃度を目安とした。ただし,喘息患者の場合は10 倍低い濃度から開始 した。維持投与は投与可能な最高用量で行った。 増量期では,それぞれの施設が採用している増量方法に従って増量した。 従来法では初回投与液0.02~0.05 mL を皮下に投与し,次回からは 50%ないし 100%ずつ増量し投与液量が 0.5 mL に達した場合は 10 倍高い濃度の投与液を 用いて以下同様に増量することを目安とした。維持期の投与液量は0.1~0.5mL を目安とした。 投与頻度は,従来法では増量期は1 週間に 2 回~2 週間に 1 回の投与を目安と した。ラッシュ法及びクラスター法の場合は1 日 1~5 回を目安とした。維持 期は月1 回投与を目安とした。 治験薬は前腕あるいは上腕の皮下に投与した。 投与期間 52 週間,ただし継続投与を希望する被験者については投与期間を延長できる こととした。 主な選択基準 (1) 同意取得日の満年齢が 5~65 歳の患者
(2) HDM に対する特異的 IgE 抗体が RAST 法で Class 2 以上の患者
(3) 同意取得日から皮内閾値検査日の間又は同意取得日の前 1 年以内に実施さ れたHDM アレルゲン皮膚テストが陽性の患者。ただし,HDM アレルギー 性鼻炎を評価する患者については,同意取得日から皮内閾値検査日の間又 は同意取得日の前1 年以内に実施されたハウスダストアレルゲン鼻誘発試 験が陽性の患者でも可。 <HDM アレルギー性鼻炎患者> (1) 軽症から最重症の HDM アレルギー性鼻炎の症状を有している患者。 (2) HDM アレルギー性喘息を合併している患者については,5~15 歳児は間欠 型,軽症持続型であること。16 歳以上は軽症間欠型であること。 <HDM アレルギー性喘息患者> (1) 5~15 歳児は間欠型,軽症持続型又は中等症持続型であること。16 歳以上 は軽症間欠型,軽症持続型又は中等症持続型であること。なお,HDM ア レルギー性鼻炎の合併の有無は問わない。
また,TO-204 第 III 相臨床試験 長期投与試験の概略を表 2.7.3.2-2 に示した。 なお,試験方法の詳細はCTD 2.7.6 に示した。
表 2.7.3.2-2 TO-204 第 III 相臨床試験 長期投与試験の概略
治験薬 TO-204 皮下注:Allergenic Extract Standardized Mite(ALK-Abelló 社) 投与方法 前腕あるいは上腕の皮下に投与する。 治療方法 それぞれの施設が採用している増量方法に従って増量する。 投与濃度 初回投与液の濃度はTO-204 皮下注の皮内閾値検査で求めた閾値又は閾値 の10 倍低い濃度を目安とする。ただし,喘息患者の場合は 10 倍低い濃度 から開始する。 維持投与は投与可能な最高用量で行う。 投与量(投与 液量) 従来法では初回投与液0.02~0.05 mL を皮下に投与する。次回からは 50% ないしは100%ずつ増量し 0.5 mL に達した場合は 10 倍高い濃度の投与液を 用いて以下同様に増量することを目安とする。維持期の投与液量は0.1~ 0.5mL を目安とする。 投与回数 従来法では増量期は1 週間に 2 回~2 週間に 1 回の投与を目安とする。ラ ッシュ法及びクラスター法の場合は1 日 1~5 回を目安とする。維持期は月 1 回投与を目安とする。 投与期間 52 週間,ただし継続投与を希望する被験者については投与期間を延長でき る。 使用製剤 治療薬:TO-204 皮下注(希釈には治療用アレルゲンエキス希釈液「トリイ」 を使用) 皮内閾値検査用薬剤:TO-204 皮下注(希釈には診断用アレルゲン皮内エキ ス対照液「トリイ」を使用) 目標症例数 30 例(増量期開始症例数として) 安全性の調査 (1) 自覚症状,他覚所見 (2) 生理検査(血圧,脈拍数) (3) 臨床検査(血液学的検査,血液生化学的検査) 有効性に関す る調査 <HDM アレルギー性鼻炎> (1) 医師による総合評価 (2) 鼻の局所所見 (3) 鼻症状スコア (4) 日本アレルギー性鼻炎標準 QOL 調査票(JRQLQ No.1) <HDM アレルギー性喘息> (1) 医師による総合評価 (2) ACT(喘息コントロールテスト,12 歳以上)/ 小児 ACT(5 歳~11 歳) (3) AHQ-Japan(16 歳以上)/ 小児気管支喘息患児と親又は保護者の QOL 調査票 簡易改訂版 2008(5 歳~15 歳) (4) 肺機能検査(FEV1,%FEV1) その他の調査
(1) 免疫学的検査(総 IgE,HDM 特異的 IgE 及び HDM 特異的 IgG4) (2) ヒスタミン遊離試験
(3) 妊娠検査
(4) 治験薬の投薬状況(皮内閾値検査,増量法,及び各投与日における増 量期と維持期の区別を含む)
2.7.3.2.1 TO-204 第 III 相臨床試験 長期投与試験の適切性 1) 非盲検,非対照,多施設共同試験としたこと 本治験は安全性の検討を主目的とした長期投与試験であり,プラセボ対照を置かない非対照, 非盲検試験とした。また,アレルゲン免疫療法に関する知識・経験をもつ医師及び医療機関が, それぞれの経験に従い,投与方法(増量方法)を選択してSCIT を行っていることから,投与方 法の偏りを避けるため,多施設共同とした。 2) それぞれの施設が採用している増量方法に従って増量することとしたこと SCIT は,維持量までの増量の仕方により,従来法,ラッシュ法,クラスター法に分けること ができる。クラスター法はラッシュ法と共に,集中的導入法として位置づけられている。本治 験では,原則として従来法(50%増量法又は 100~200%増量法)に従うことを目安としたが, アレルゲン免疫療法に関する知識・経験をもつ医師及び医療機関においては,それぞれの投与 方法の特長を考慮して投与方法を選択しSCIT を行っているのが実状である。 したがって,本治験では基本的な投与スケジュールは提示しつつも,増量方法及び投与間隔 については,各医療機関の経験に基づいて決定する事が実際的かつ安全な方法と考え,いずれ の方法に対しても一律的な設定は行わないこととした。 3) 目標症例数を 30 例としたこと 本治験での症例数の設定に当たっては,アレルゲン免疫療法に関する知識・経験をもつ専門 医によって治験が安全に実施されて安全性が適切に評価されるべきであるとの考え方に基づき, 症例数の算定を行った。 本件に関し,PMDA と協議を行い,合意に至った( 平成 年 月 日,及び 平成 年 月 日)。また, ことについても,PMDA との合意に至った。 4) SCIT の有効性は公知と考え,試験の主目的を安全性の評価としたこと TO-204 皮下注は,本邦における使用実績はないものの,アメリカ,カナダ等において既に市 販されている。 がPMDA からも示された。 これらを踏まえ, することで PMDA との合意に至った( 平成 年 月 日,及び 平成 年 月 日)。 以上の経緯から,TO-204 皮下注の日本人を対象とした臨床試験は,安全性の検討を主目的と
する長期投与試験として計画した。 5) 治験期間を 52 週間とし,26 週間のデータで申請可能と考えたこと 本治験の投与期間は,「致命的でない疾患に対し長期間の投与が想定される新医薬品の治験 段階において安全性を評価するために必要な症例数と投与期間について(平成7 年 5 月 24 日付, 薬審592 号)」に従い,52 週間とした。52 週間以降の投与に関しては,アレルゲン免疫療法の 主旨を踏まえ,より長期間の投与が可能となるように設定した。 本剤の長期投与時の安全性データについては,「新医薬品の総審査期間短縮に向けた申請に 係る留意事項について(平成22 年 6 月 9 日付け事務連絡)」に従い, を申請資料として添付すること,及び全症例が1 年間の投与を終了したデ ータで最終報告書を作成し, 提 出することとした( ,平成 年 月 日)。 2.7.3.2.2 有効性の評価方法 本治験では,HDM SCIT の有効性は公知であることから対照群は設定せず,病状の把握を目的 とした評価を行い有効性評価とした。 皮内閾値検査で求めた閾値に応じて TO-204 皮下注を初回投与し,安全性を確認後,投与量を 徐々に増量した。従来法(50%増量法又は 100~200%増量法)では,1~2 週間の投与間隔で数ヵ 月を目安に,ラッシュ法では連日投与で数日から数週間を目安に,クラスター法では数日間の連 日投与を 1 セットとして数セット行い,数週間を目安に増量した。また,被験者の安全が確保で きる投与可能な最大量(維持量)まで増量した。投与期間は増量期を含めて52 週間としたが, できることとした。維持期の投与間隔は1 ヵ 月を目安とした。 2.7.3.2.3 有効性の評価項目 <HDM アレルギー性鼻炎> (1) 医師による総合評価 (2) 鼻の局所所見 (3) 鼻症状スコア (4) 日本アレルギー性鼻炎標準 QOL 調査票(JRQLQ No.1) <HDM アレルギー性喘息> (1) 医師による総合評価 (2) ACT(喘息コントロールテスト,12 歳以上)/ 小児 ACT(5 歳~11 歳) (3) AHQ-Japan(16 歳以上)/ 小児気管支喘息患児と親又は保護者の QOL 調査票 簡易改訂版 2008(5 歳~15 歳) (4) 肺機能検査(FEV1,%FEV1) 2.7.3.2.4 有効性解析対象集団
最大の解析対象集団(Full analysis set: FAS)を有効性解析対象集団とした。 FAS 合計(44 例)
HDM アレルギー性鼻炎患者:28 例 HDM アレルギー性喘息患者:16 例 2.7.3.2.5 被験者の内訳 被験者の内訳を図 2.7.3.2-1 に示した。 図 2.7.3.2-1 被験者の内訳 引用元:CTD 5.3.5.1.2-1 の図 10.1-1 49 例 44 例 5 例 HDMアレルギー性鼻炎患者 0 例* HDMアレルギー性喘息患者 0 例* HDMアレルギー性鼻炎患者 1 例* HDMアレルギー性喘息患者 0 例* HDMアレルギー性鼻炎患者 2 例* HDMアレルギー性喘息患者 1 例* HDMアレルギー性鼻炎患者 26 例 HDMアレルギー性鼻炎患者 2 例* HDMアレルギー性喘息患者 15 例 HDMアレルギー性喘息患者 1 例* 例数 例数 被験者の申し出 1 有害事象の発現 1 選択基準不適 0 原疾患の悪化 0 除外基準抵触 4 被験者から中止の申し出 1 その他 0 対象として不適切 0 被験者の都合 1 妊娠 0 その他 0 * 累積の中止例数を表示した。 観察期に脱落した理由 投与を中止した被験者の内訳 同意を取得した被験者 投与を開始した被験者 観察期に脱落した被験者 12週観察日までに投与を中止した被験者 52週観察日まで投与を完了した被験者 52週観察日までに投与を中止した被験者 26週観察日までに投与を中止した被験者 38週観察日までに投与を中止した被験者
2.7.3.2.6 人口統計学的特性及び他の基準値の特性
主な被験者背景(FAS,安全性解析対象集団)を表 2.7.3.2-3 に示した。
表 2.7.3.2-3 主な被験者背景(FAS,安全性解析対象集団) 項目 HDM アレルギー性 鼻炎患者 (28 例) HDM アレルギー性 喘息患者 (16 例) 合計 (44 例) 性別 男 17(60.7%) 8(50.0%) 25(56.8%) 女 11(39.3%) 8(50.0%) 19(43.2%) 年齢 (歳) <12 12(42.9%) 6(37.5%) 18(40.9%) ≧12 <20 6(21.4%) 2(12.5%) 8(18.2%) ≧20 <30 3(10.7%) 3(18.8%) 6(13.6%) ≧30 <40 1(3.6%) 2(12.5%) 3(6.8%) ≧40 <50 5(17.9%) 2(12.5%) 7(15.9%) ≧50 1(3.6%) 1(6.3%) 2(4.5%) 平均値±標準偏差 20.0±14.7 22.8±15.4 21.0±14.8 中央値 [最小値-最大値] 13.5[5-53] 17.5[7-53] 13.5[5-53] 成人/小児 低年齢層小児(5~11 歳) 12(42.9%) 6(37.5%) 18(40.9%) 小児(12~15 歳) 4(14.3%) 2(12.5%) 6(13.6%) 成人 12(42.9%) 8(50.0%) 20(45.5%) 罹病期間 (年) <5 7(25.0%) 5(31.3%) 12(27.3%) ≧5 <10 11(39.3%) 4(25.0%) 15(34.1%) ≧10 <15 2(7.1%) 2(12.5%) 4(9.1%) ≧15 <20 2(7.1%) 2(12.5%) 4(9.1%) ≧20 6(21.4%) 3(18.8%) 9(20.5%) 平均値±標準偏差 10.8±9.7 12.2±11.7 11.3±10.3 中央値 [最小値-最大値] 7.5[0-32] 9.0[0-46] 8.0[0-46] 免疫療法の施行歴 有 5(17.9%) 4(25.0%) 9(20.5%) 無 23(82.1%) 12(75.0%) 35(79.5%) アレルギー疾患の 既往歴及び合併症 有 23(82.1%) 15(93.8%) 38(86.4%) 無 5(17.9%) 1(6.3%) 6(13.6%) 既往歴 有 11(39.3%) 8(50.0%) 19(43.2%) 無 17(60.7%) 8(50.0%) 25(56.8%) 合併症 有 25(89.3%) 15(93.8%) 40(90.9%) 無 3(10.7%) 1(6.3%) 4(9.1%) RAST 値 (Der far と Der pte の
最大値) Class2 2(7.1%) 0(0.0%) 2(4.5%) Class3 4(14.3%) 1(6.3%) 5(11.4%) Class4 6(21.4%) 5(31.3%) 11(25.0%) Class5 5(17.9%) 6(37.5%) 11(25.0%) Class6 11(39.3%) 4(25.0%) 15(34.1%) HDM アレルギー性 鼻炎の重症度 軽症 6(21.4%) - - 中等症 3(10.7%) - - 重症 13(46.4%) - - 最重症 6(21.4%) - - HDM アレルギー性 喘息の合併の有無 有 4(14.3%) - - 無 24(85.7%) - - HDM アレルギー性 喘息の重症度 (16 歳以上) 軽症間欠型 - 3(37.5%) - 軽症持続型 - 1(12.5%) - 中等症持続型 - 4(50.0%) - 16 歳以上被験者例数 - 8 - HDM アレルギー性 喘息の重症度 (5~15 歳) 間欠型 - 5(62.5%) - 軽症持続型 - 3(37.5%) - 中等症持続型 - 0(0.0%) - 5~15 歳被験者例数 - 8 - HDM アレルギー性鼻炎 の合併の有無 有 - 12(75.0%) - 無 - 4(25.0%) - 表中の数値(記述統計量は除く)は例数を示し,括弧内の数値は各患者群の例数又は合計例数に対する百分率を 示す。引用元:CTD 5.3.5.1.2-1 の表 11.2-1
2.7.3.2.7 投与法(増量法)の内訳 投与法(増量法)の内訳を表 2.7.3.2-4 に示した。 表 2.7.3.2-4 投与法(増量法)の内訳(FAS,安全性解析対象集団) 投与法(増量法) HDM アレルギー性 鼻炎患者(28 例) HDM アレルギー性 喘息患者(16 例) 合計(44 例) 従来法 50%増量法 4(14.3%) 2(12.5%) 6(13.6%) 21(47.7%) 100~200%増量法 9(32.1%) 6(37.5%) 15(34.1%) 急速法 ラッシュ法 13(46.4%) 6(37.5%) 19(43.2%) 23(52.3%) クラスター法 2(7.1%) 2(12.5%) 4(9.1%) 表中の数値は例数を示し,括弧内の数値はそれぞれの患者群の例数又は合計例数に対する百分率を示す。 引用元:CTD 5.3.5.1.2-1 の表 11.4-3 2.7.3.2.8 有効性の評価期間 本治験の試験期間は52 週間,さらに継続投与を希望する場合は承認日までとした。本資料では 52 週観察日までに得られたデータを集計,解析した。 2.7.3.2.9 被験者ごとの増量法,閾値,初回投与量,増量期間,維持量,投与期間,累積投与量 被験者ごとの増量法,閾値,初回投与量,増量期間,維持量,投与期間,累積投与量を表 2.7.3.2-5 に示した。
9
表 2.7.3.2-5 被験者ごとの増量法,閾値,初回投与量,増量期間,維持量,投与期間,累積投与量 (FAS,安全性解析対象集団) 原疾患 増量法 被験者識 別コード 年齢 性別 閾値 (AU/mL) 初回投与量 (AU) 増量 期間 (週) 増量期の 投与回数 (回) 維持量 (AU) 投与 期間 (週) 累積 投与量 (AU) HDM アレルギー性鼻炎 50%増量法 3 男性 0.01 0.00002 23 19 0.02* 23 0.05 1 男性 0.1 0.0002 34 32 0.2 52 2.75 男性 0.1 0.0002 30 27 0.2 52 2.77 4 女性 0.0001 0.0000005 52 50 10* 52 46.98 100~200%増量法 4 男性 0.1 0.005 9 8 1 52 12.50 4 男性 0.1 0.005 20 14 10 51 165.00 2 男性 1 0.05 15 11 100 51 1499.95 男性 0.0000001 0.000000005 24 20 20 54 173.66 5 女性 0.01 0.0005 18 17 30 55 336.67 4 女性 1 0.05 10 5 1 34 11.45 4 女性 0.1 0.005 19 17 20 53 245.33 1 女性 0.1 0.005 21 17 20 53 225.33 女性 ― 0.005 19 17 20 56 265.33 ラッシュ法 1 男性 0.01 0.00005 4 31 4 55 63.62 1 男性 0.1 0.005 7 15 20 55 323.89 1 男性 0.1 0.005 6 15 20 53 348.89 1 男性 0.00001 0.00000001 2 24 100 53 2044.44 1 男性 0.01 0.00005 4 30 10 52 190.58 1 男性 0.001 0.000005 36 29 50 53 373.33 男性 0.01 0.0001 5 17 50 53 759.44 男性 0.0000001 0.00000001 8 27 100 53 1484.44 2 女性 0.0001 0.000005 13 21 20 52 244.45 1 女性 0.0001 0.000002 7 22 100 52 2009.44 1 女性 0.0001 0.0000005 28 33 50 51 373.33 1 女性 0.1 0.005 23 20 200 53 2638.89 女性 0.0000001 0.000000001 8 28 100 53 1364.44 クラスター法 2 男性 0.0001 0.0000005 27 36 50 53 573.33 男性 0.001 0.0001 25 22 100 54 1309.94 HDM アレルギー性喘息 50%増量法 4 男性 0.001 0.000005 49 45 100 52 839.44 3 女性 0.01 0.00005 53 52 100* 53 1577.56 100~200%増量法 男性 0.1 0.0005 17 14 10 52 110.00 5 女性 0.0001 0.000001 23 29 50 54 672.22 4 女性 1 0.005 5 4 0.02 33 0.22 3 女性 1 0.005 12 7 1 52 13.45 2 女性 0.000001 0.000000005 53 47 0.5* 53 1.65 2 女性 0.1 0.0001 28 26 50 53 622.23 ラッシュ法 1 男性 0.001 0.00001 7 20 50 53 810.44 1 男性 0.01 0.0001 7 17 50 53 809.44 1 男性 0.000001 0.00000002 6 24 100 52 2109.44 男性 10 0.1 5 14 200 53 2909.40 男性 0.00001 0.0000002 7 21 50 53 909.44 2 女性 0.001 0.000005 17 31 5 55 493.05 クラスター法 1 男性 0.001 0.00001 25 24 100 54 1209.94 1 女性 0.001 0.00001 25 24 100 54 1309.94 年齢は同意取得時 * 維持期未達のため,最終投与時のデータを補完 被験者識別コード , , :中止例 被験者識別コード :閾値欠値 引用元:CTD 5.3.5.1.2-1 の表 11.4-5 1-9* 1-9* 1-9* 1-9* 1-9* 1-9* 1-9* 1-9* 1-9* 1-9*
2.7.3.2.10 解析計画 本治験では,有効性は公知であるとの判断から,通常の検証試験で要求される厳格な有効性評 価指標は設定せず,適切な検出力で有意差を検出するための症例数設定を行なわなかった。した がって,有効性の評価で得られた p 値については統計学的有意差の参考データとはしたが,有意 水準を設定せず,検証的な判断は行わなかった。 1) 解析対象集団 (1) 有効性解析対象集団
最大の解析集団(Full analysis set: FAS)を主たる解析対象集団とした。 (2) 安全性解析対象集団 治験薬が投与され,安全性評価項目の調査が実施された症例とした。安全性解析対象集団 はFAS と同じ集団となった。 2) 外れ値,欠測値の取り扱い 適正に測定が実施され測定値に誤りがないと考えられる外れ値については,そのまま集計に 含めた。中止又は不測の事態により測定値が欠測した場合は,補完を実施しなかった。 3) 集計データの取り扱い 有効性に関する調査項目に関しては,治験薬投与開始後の初回規定観察日以降の測定値が存 在する症例を対象に,当該症例の各調査項目の最終の観察日の測定値(中止時観察日の測定値 を含む)を用いて投与終了時の集計を行った。 最終症例が52 週観察日までの測定値(中止時観察日の測定値を含む)を用いて集計を行った。 4) 終了例及び中止例の分類 終了例については,52 週での投与終了例,承認日での投与終了例,製造販売後臨床試験投与 終了例に分類する。 中止例については,52 週までの投与中止例,52 週から承認日までの投与中止例,製造販売後 臨床試験投与中止例に分類する。 5) 有意水準及び多重性 検定手法を用いる場合,得られた p 値については統計学的有意差の参考データとはしたが, 有意水準を設定せず,検証的な判断は行わなかった。 多重性の調整は行わなかった。 6) 有効性の解析 <HDM アレルギー性鼻炎> (1) 医師による総合評価 (2) 鼻の局所所見 (3) 鼻症状スコア (4) 日本アレルギー性鼻炎標準 QOL 調査票(JRQLQ No.1)
11
<HDM アレルギー性喘息> (1) 医師による総合評価 (2) ACT(12 歳以上) (3) 小児 ACT(5 歳~11 歳) (4) AHQ-Japan(16 歳以上) (5) 小児気管支喘息患児と親又は保護者の QOL 調査票 簡易改訂版 2008(5 歳~15 歳) (6) 肺機能検査(FEV1,%FEV1) 7) 安全性の解析 安全性評価項目について統計学的解析は実施しなかった。 (1) 自覚症状・他覚所見 自覚症状を被験者から聴取した。他覚所見は医師の診察(問診,聴診,打診,視診,触診 等)により調査した。有害事象が認められた場合には以下の有害事象の集計に従って検討を 行った。 (2) 臨床検査及び生理検査 臨床検査及び生理検査における安全性の評価は,治験薬投与前及び投与後,それぞれ1 回 以上の検査が実施された被験者を対象に行った。臨床検査及び生理検査結果は,それぞれの 項目について集計し,本剤の安全性について評価した。 (3) 有害事象の集計
症例報告書に記載された有害事象名は,MedDRA/J V.15.0 の下層語(Lowest level term:LLT) に読み替えて表記した。集計・分析に際しては,基本語(Preferred term:PT)又は器官別大 分類(System organ class:SOC)を用いた。集計に関しては,疾患別,増量法別,年齢別,投 与期別の集計も行った。 投与期(増量期開始前,増量期,維持期)別有害事象の発現時期は,報告された有害事象 発現日時に基づき判断した。有害事象発現日が増量期開始日,若しくは維持期開始日と同一 で,かつ日時が欠測の場合には,症例検討会の判定結果に基づき,有害事象の発現時期を決 定した。 有害事象の重篤度は,薬事法施行規則第273 条第 1 項,及び平成 22 年 7 月 29 日発出の「副 作用等報告に関する Q&A についての改訂について」(厚生労働省医薬食品局審査管理課, 同安全対策課)の基準に従い,重篤又は非重篤に分類した。また,有害事象の重症度を,軽 度,中等度,高度の3 段階に分類し,重症度別に有害事象を集計し評価した。 有害事象及び重篤な有害事象は,被験者ごとに年齢,性別,有害事象名,因果関係,転帰 などを一覧に示した。 同一被験者に同一事象が複数回発現した場合には,発現被験者数は 1 例と数えるものとし た。 有害事象発現までの日数は,有害事象が増量期開始日以降の場合には,「有害事象発現日」 -「増量期開始日」+1 とし,増量期開始日より前の場合には,「有害事象発現日」-「増 量期開始日」とした。有害事象の持続期間は「有害事象消失日(転帰日)」-「有害事象発
現日」+1 とした。 有害事象及び因果関係の否定できない有害事象(副作用)を集計し,TO-204 皮下注の安全 性を評価した。なお,安全性データについては統計学的な解析は実施しなかった。 2.7.3.3 有効性の結果 有効性の結果を以下に示す。 なお,2.7.3.2.10 項で述べたように,本治験では,統計学的有意差の参考データとして p 値は求 めたが,有意水準を設定せず,検証的な判断は行わなかった。 2.7.3.3.1 HDM アレルギー性鼻炎患者 2.7.3.3.1.1 医師による総合評価 結果を表 2.7.3.3-1 に示した。 26 週観察日は観察開始日に比べ,「良い」が増加し,「少し悪い」,「悪い」が減少した(p=0.0002)。 52 週観察日も観察開始日に比べ,「良い」,「少し良い」が増加し,「普通」,「少し悪い」, 「悪い」が減少した(p<0.0001)。 表 2.7.3.3-1 医師による総合評価(FAS) 評価時期 良い 少し良い 普通 少し悪い 悪い 合計 例数 観察開始日 との比較* 観察開始日 3(10.7%) 3(10.7%) 6(21.4%) 7(25.0%) 9(32.1%) 28 - 26 週観察日 14(51.9%) 3(11.1%) 7(25.9%) 3(11.1%) 0(0.0%) 27 0.0002 52 週観察日 18(69.2%) 4(15.4%) 2(7.7%) 2(7.7%) 0(0.0%) 26 <.0001 表中の数値は例数を示し,括弧内の数値はそれぞれの評価時期の合計例数に対する百分率を示す。 * Wilcoxon 符号付順位検定に基づく p 値 引用元:CTD 5.3.5.1.2-1 の表 11.4-6 2.7.3.3.1.2 鼻の局所所見 1) 下鼻甲介粘膜の腫脹 結果を表 2.7.3.3-2 に示した。 26 週観察日と観察開始日との間に,大きな変化はなかった(p=0.1125)。 52 週観察日は観察開始日に比べ,症状の軽い被験者(-,1+)が増加し,症状の重い被験 者(2+,3+)が減少した(p=0.0024)。
13
表 2.7.3.3-2 鼻の局所所見(FAS) -下鼻甲介粘膜の腫脹- 評価時期 - (なし) 1+ (中鼻甲介中央 までみえる) 2+ (3+と 1+ の中間) 3+ (中鼻甲介 みえず) 合計 例数 観察開始日 との比較* 観察開始日 1(3.6%) 9(32.1%) 15(53.6%) 3(10.7%) 28 - 12 週観察日 3(10.7%) 13(46.4%) 9(32.1%) 3(10.7%) 28 0.1865 26 週観察日 0(0.0%) 17(63.0%) 9(33.3%) 1(3.7%) 27 0.1125 38 週観察日 3(11.5%) 16(61.5%) 6(23.1%) 1(3.8%) 26 0.0092 52 週観察日 4(15.4%) 16(61.5%) 6(23.1%) 0(0.0%) 26 0.0024 表中の数値は例数を示し,括弧内の数値はそれぞれの評価時期の合計例数に対する百分率を示す。 * Wilcoxon 符号付順位検定に基づく p 値 引用元:CTD 5.3.5.1.2-1 の表 11.4-7 2) 下鼻甲介粘膜の色調 結果を表 2.7.3.3-3 に示した。 26 週観察日と観察開始日との間に大きな変化はなかった(p=0.2254)。 52 週観察日は観察開始日に比べ,症状の軽い被験者(-,1+)が増加し,症状の重い被験 者(2+,3+)が減少した(p=0.0060)。 表 2.7.3.3-3 鼻の局所所見(FAS) -下鼻甲介粘膜の色調- 評価時期 - (なし) 1+ (薄赤) 2+ (赤) 3+ (蒼白) 合計例数 観察開始日と の比較* 観察開始日 1(3.6%) 13(46.4%) 5(17.9%) 9(32.1%) 28 - 12 週観察日 3(10.7%) 10(35.7%) 8(28.6%) 7(25.0%) 28 0.5745 26 週観察日 2(7.4%) 12(44.4%) 9(33.3%) 4(14.8%) 27 0.2254 38 週観察日 5(19.2%) 15(57.7%) 4(15.4%) 2(7.7%) 26 0.0028 52 週観察日 3(11.5%) 18(69.2%) 3(11.5%) 2(7.7%) 26 0.0060 表中の数値は例数を示し,括弧内の数値はそれぞれの評価時期の合計例数に対する百分率を示す。 * Wilcoxon 符号付順位検定に基づく p 値 引用元:CTD 5.3.5.1.2-1 の表 11.4-8 3) 水性分泌量 結果を表 2.7.3.3-4 に示した。 26 週観察日は観察開始日に比べ,症状の軽い被験者(-)が増加し,症状の重い被験者(2 +,3+)が減少した(p=0.0024)。 52 週観察日も観察開始日に比べ,症状の軽い被験者(-)が増加し,症状の重い被験者(2 +,3+)が減少した(p=0.0002)。
表 2.7.3.3-4 鼻の局所所見(FAS) -水性分泌量- 評価時期 - (なし) 1+ (付着程度) 2+ (3+と 1+の 中間) 3+ (充満) 合計例数 観察開始日 との比較* 観察開始日 3(10.7%) 14(50.0%) 10(35.7%) 1(3.6%) 28 - 12 週観察日 7(25.0%) 9(32.1%) 10(35.7%) 2(7.1%) 28 0.5930 26 週観察日 11(40.7%) 12(44.4%) 4(14.8%) 0(0.0%) 27 0.0024 38 週観察日 9(34.6%) 14(53.8%) 3(11.5%) 0(0.0%) 26 0.0015 52 週観察日 10(38.5%) 13(50.0%) 3(11.5%) 0(0.0%) 26 0.0002 表中の数値は例数を示し,括弧内の数値はそれぞれの評価時期の合計例数に対する百分率を示す。 * Wilcoxon 符号付順位検定に基づく p 値 引用元:CTD 5.3.5.1.2-1 の表 11.4-9 4) 鼻汁の性状 結果を表 2.7.3.3-5 に示した。 26 週観察日は観察開始日に比べ,症状の軽い被験者(-,1+)が増加し,症状の重い被験 者(2+,3+)が減少した(p=0.0144)。 52 週観察日も観察開始日に比べ,症状の軽い被験者(-,1+)が増加し,症状の重い被験 者(3+)が減少した(p=0.0010)。 表 2.7.3.3-5 鼻の局所所見(FAS) -鼻汁の性状- 評価時期 - (なし) 1+ (膿性) 2+ (粘性) 3+ (水性) 合計例数 観察開始日 との比較* 観察開始日 4(14.3%) 1(3.6%) 7(25.0%) 16(57.1%) 28 - 12 週観察日 9(32.1%) 1(3.6%) 9(32.1%) 9(32.1%) 28 0.0586 26 週観察日 12(44.4%) 2(7.4%) 4(14.8%) 9(33.3%) 27 0.0144 38 週観察日 11(42.3%) 2(7.7%) 5(19.2%) 8(30.8%) 26 0.0043 52 週観察日 10(38.5%) 3(11.5%) 6(23.1%) 7(26.9%) 26 0.0010 表中の数値は例数を示し,括弧内の数値はそれぞれの評価時期の合計例数に対する百分率を示す。 * Wilcoxon 符号付順位検定に基づく p 値 引用元:CTD 5.3.5.1.2-1 の表 11.4-10 2.7.3.3.1.3 鼻症状スコア 1) 鼻汁 結果を表 2.7.3.3-6 に示した。 26 週観察日は増量期開始日に比べ,症状の軽い被験者(0 点,1 点)が増加し,症状の重い 被験者(2 点,3 点)が減少したが,大きな変化はなかった(p=0.0690)。 52 週観察日は増量期開始日に比べ,症状の軽い被験者(0 点,1 点)が増加し,症状の重い 被験者(2 点,3 点)が減少した(p=0.0324)。
15
表 2.7.3.3-6 鼻症状スコア(FAS) -鼻汁- 評価時期 症状なし:0 点 軽度:1 点 中等度:2 点 重度:3 点 合計例数 増量期開始 日との比較* 観察開始日 0(0.0%) 10(35.7%) 12(42.9%) 6(21.4%) 28 - 増量期開始日 4(14.3%) 7(25.0%) 13(46.4%) 4(14.3%) 28 - 12 週観察日 2(7.1%) 10(35.7%) 14(50.0%) 2(7.1%) 28 0.8726 26 週観察日 6(22.2%) 10(37.0%) 11(40.7%) 0(0.0%) 27 0.0690 38 週観察日 6(23.1%) 10(38.5%) 9(34.6%) 1(3.8%) 26 0.1115 52 週観察日 7(26.9%) 12(46.2%) 6(23.1%) 1(3.8%) 26 0.0324 表中の数値は例数を示し,括弧内の数値はそれぞれの評価時期の合計例数に対する百分率を示す。 * Wilcoxon 符号付順位検定に基づく p 値 引用元:CTD 5.3.5.1.2-1 の表 11.4-11 2) 鼻閉 結果を表 2.7.3.3-7 に示した。 26 週観察日は増量期開始日に比べ,症状のない被験者(0 点)が増加し,症状の重い被験者 (2 点,3 点)が減少した(p=0.0023)。 52 週観察日も増量期開始日に比べ,症状の軽い被験者(0 点)が増加し,症状の重い被験者 (2 点,3 点)が減少した(p=0.0156)。 表 2.7.3.3-7 鼻症状スコア(FAS) -鼻閉- 評価時期 症状なし:0 点 軽度:1 点 中等度:2 点 重度:3 点 合計例数 増量期開始 日との比較* 観察開始日 8(28.6%) 12(42.9%) 5(17.9%) 3(10.7%) 28 - 増量期開始日 9(32.1%) 11(39.3%) 4(14.3%) 4(14.3%) 28 - 12 週観察日 13(46.4%) 11(39.3%) 3(10.7%) 1(3.6%) 28 0.0582 26 週観察日 16(59.3%) 11(40.7%) 0(0.0%) 0(0.0%) 27 0.0023 38 週観察日 16(61.5%) 9(34.6%) 1(3.8%) 0(0.0%) 26 0.0123 52 週観察日 16(61.5%) 8(30.8%) 1(3.8%) 1(3.8%) 26 0.0156 表中の数値は例数を示し,括弧内の数値はそれぞれの評価時期の合計例数に対する百分率を示す。 * Wilcoxon 符号付順位検定に基づく p 値 引用元:CTD 5.3.5.1.2-1 の表 11.4-12 3) くしゃみ 結果を表 2.7.3.3-8 に示した。 26 週観察日,52 週観察日と増量期開始日との間に,いずれも大きな変化はなかった(p=0.6213, p=0.3856)。
表 2.7.3.3-8 鼻症状スコア(FAS) -くしゃみ- 評価時期 症状なし:0 点 軽度:1 点 中等度:2 点 重度:3 点 合計例数 増量期開始 日との比較* 観察開始日 2(7.1%) 12(42.9%) 12(42.9%) 2(7.1%) 28 - 増量期開始日 6(21.4%) 12(42.9%) 8(28.6%) 2(7.1%) 28 - 12 週観察日 6(21.4%) 11(39.3%) 11(39.3%) 0(0.0%) 28 0.9542 26 週観察日 7(25.9%) 13(48.1%) 5(18.5%) 2(7.4%) 27 0.6213 38 週観察日 6(23.1%) 12(46.2%) 6(23.1%) 2(7.7%) 26 0.9487 52 週観察日 6(23.1%) 15(57.7%) 4(15.4%) 1(3.8%) 26 0.3856 表中の数値は例数を示し,括弧内の数値はそれぞれの評価時期の合計例数に対する百分率を示す。 * Wilcoxon 符号付順位検定に基づく p 値 引用元:CTD 5.3.5.1.2-1 の表 11.4-13 4) そう痒感 結果を表 2.7.3.3-9 に示した。 26 週観察日は増量期開始日に比べ,症状のない被験者(0 点)が増加し,症状の重い被験者 (2 点,3 点)が減少した(p=0.0005)。 52 週観察日も増量期開始日に比べ,症状のない被験者(0 点)が増加し,症状の重い被験者 (2 点,3 点)が減少した(p=0.0023)。 表 2.7.3.3-9 鼻症状スコア(FAS) -そう痒感- 評価時期 症状なし:0 点 軽度:1 点 中等度:2 点 重度:3 点 合計例数 増量期開始 日との比較* 観察開始日 1(3.6%) 15(53.6%) 9(32.1%) 3(10.7%) 28 - 増量期開始日 2(7.1%) 13(46.4%) 11(39.3%) 2(7.1%) 28 - 12 週観察日 9(32.1%) 11(39.3%) 7(25.0%) 1(3.6%) 28 0.0321 26 週観察日 10(37.0%) 13(48.1%) 4(14.8%) 0(0.0%) 27 0.0005 38 週観察日 13(50.0%) 10(38.5%) 3(11.5%) 0(0.0%) 26 0.0003 52 週観察日 7(26.9%) 16(61.5%) 3(11.5%) 0(0.0%) 26 0.0023 表中の数値は例数を示し,括弧内の数値はそれぞれの評価時期の合計例数に対する百分率を示す。 * Wilcoxon 符号付順位検定に基づく p 値 引用元:CTD 5.3.5.1.2-1 の表 11.4-14 2.7.3.3.1.4 日本アレルギー性鼻炎標準 QOL 調査票(JRQLQ No.1) 1) 鼻・眼の症状 (1) 水っぱな 治験期間中の水っぱなの程度を表 2.7.3.3-10 に示した。 26 週観察日は観察開始日に比べ,症状のない被験者(0 点)が増加し,症状の軽い被験者 (1 点),症状のやや重い被験者(2 点),症状の重い被験者(3 点,4 点)が減少した(p=0.0072)。 52 週観察日も観察開始日に比べ,症状のない被験者(0 点)が増加し,症状のやや重い被 験者(2 点),症状の重い被験者(3 点,4 点)が減少した(p=0.0300)。
17
表 2.7.3.3-10 JRQLQ No.1(FAS) -水っぱな- 評価時期 症状なし 0 軽い 1 やや重い 2 重い 3 非常に重い 4 合計例数 観察開始日 との比較* 観察開始日 5(17.9%) 11(39.3%) 8(28.6%) 3(10.7%) 1(3.6%) 28 - 26 週観察日 12(44.4%) 10(37.0%) 5(18.5%) 0(0.0%) 0(0.0%) 27 0.0072 52 週観察日 10(38.5%) 10(38.5%) 5(19.2%) 0(0.0%) 1(3.8%) 26 0.0300 表中の数値は例数を示し,括弧内の数値はそれぞれの評価時期の合計例数に対する百分率を示す。 * Wilcoxon 符号付順位検定に基づく p 値 引用元:CTD 5.3.5.1.2-1 の表 11.4-15 (2) くしゃみ 治験期間中のくしゃみの程度を表 2.7.3.3-11 に示した。 26 週観察日は観察開始日に比べ,症状のない被験者(0 点)が増加し,症状の軽い被験者 (1 点),症状の重い被験者(3 点)が減少した。しかし,症状のやや重い被験者(2 点)が 増加し,全体として大きな変化はなかった(p=0.0816)。 52 週観察日は観察開始日に比べ,症状のない被験者(0 点)が増加し,症状の軽い被験者 (1 点),症状の重い被験者(3 点)が減少した(p=0.0394)。 表 2.7.3.3-11 JRQLQ No.1(FAS) -くしゃみ- 評価時期 症状なし 0 軽い 1 やや重い 2 重い 3 非常に重い 4 合計例数 観察開始日 との比較* 観察開始日 0(0.0%) 18(64.3%) 6(21.4%) 3(10.7%) 1(3.6%) 28 - 26 週観察日 8(29.6%) 10(37.0%) 8(29.6%) 0(0.0%) 1(3.7%) 27 0.0816 52 週観察日 8(30.8%) 10(38.5%) 7(26.9%) 0(0.0%) 1(3.8%) 26 0.0394 表中の数値は例数を示し,括弧内の数値はそれぞれの評価時期の合計例数に対する百分率を示す。 * Wilcoxon 符号付順位検定に基づく p 値 引用元:CTD 5.3.5.1.2-1 の表 11.4-16 (3) 鼻づまり 治験期間中の鼻づまりの程度を表 2.7.3.3-12 に示した。 26 週観察日は観察開始日に比べ,症状のない被験者(0 点),症状の軽い被験者(1 点) が増加し,症状のやや重い被験者(2 点),症状の重い被験者(3 点,4 点)が減少した(p<0.0001)。 52 週観察日も観察開始日に比べ,症状のない被験者(0 点),症状の軽い被験者(1 点) が増加し,症状のやや重い被験者(2 点),症状の重い被験者(3 点,4 点)が減少した(p=0.0014)。
表 2.7.3.3-12 JRQLQ No.1(FAS) -鼻づまり- 評価時期 症状なし 0 軽い 1 やや重い 2 重い 3 非常に重い 4 合計例数 観察開始日 との比較* 観察開始日 4(14.3%) 5(17.9%) 9(32.1%) 7(25.0%) 3(10.7%) 28 - 26 週観察日 11(40.7%) 13(48.1%) 3(11.1%) 0(0.0%) 0(0.0%) 27 <.0001 52 週観察日 8(30.8%) 12(46.2%) 5(19.2%) 0(0.0%) 1(3.8%) 26 0.0014 表中の数値は例数を示し,括弧内の数値はそれぞれの評価時期の合計例数に対する百分率を示す。 * Wilcoxon 符号付順位検定に基づく p 値 引用元:CTD 5.3.5.1.2-1 の表 11.4-17 (4) 鼻のかゆみ 治験期間中の鼻のかゆみの程度を表 2.7.3.3-13 に示した。 26 週観察日は観察開始日に比べ,症状のない被験者(0 点),症状の軽い被験者(1 点) が増加し,症状のやや重い被験者(2 点),症状の重い被験者(3 点,4 点)が減少した(p=0.0009)。 52 週観察日も観察開始日に比べ,症状のない被験者(0 点),症状の軽い被験者(1 点) が増加し,症状のやや重い被験者(2 点),症状の重い被験者(3 点,4 点)が減少した(p=0.0018)。 表 2.7.3.3-13 JRQLQ No.1(FAS) -鼻のかゆみ- 評価時期 症状なし 0 軽い 1 やや重い 2 重い 3 非常に重い 4 合計例数 観察開始日 との比較* 観察開始日 3(10.7%) 10(35.7%) 11(39.3%) 3(10.7%) 1(3.6%) 28 - 26 週観察日 10(37.0%) 12(44.4%) 3(11.1%) 2(7.4%) 0(0.0%) 27 0.0009 52 週観察日 10(38.5%) 12(46.2%) 3(11.5%) 1(3.8%) 0(0.0%) 26 0.0018 表中の数値は例数を示し,括弧内の数値はそれぞれの評価時期の合計例数に対する百分率を示す。 * Wilcoxon 符号付順位検定に基づく p 値 引用元:CTD 5.3.5.1.2-1 の表 11.4-18 (5) 眼のかゆみ 治験期間中の眼のかゆみの程度を表 2.7.3.3-14 に示した。 26 週観察日,52 週観察日と観察開始日との間に,いずれも大きな変化はなかった(p=0.5112, p=0.1430)。 表 2.7.3.3-14 JRQLQ No.1(FAS) -眼のかゆみ- 評価時期 症状なし 0 軽い 1 やや重い 2 重い 3 非常に重い 4 合計例数 観察開始日 との比較* 観察開始日 7(25.0%) 11(39.3%) 4(14.3%) 3(10.7%) 3(10.7%) 28 - 26 週観察日 9(33.3%) 9(33.3%) 4(14.8%) 3(11.1%) 2(7.4%) 27 0.5112 52 週観察日 9(34.6%) 11(42.3%) 2(7.7%) 3(11.5%) 1(3.8%) 26 0.1430 表中の数値は例数を示し,括弧内の数値はそれぞれの評価時期の合計例数に対する百分率を示す。 * Wilcoxon 符号付順位検定に基づく p 値 引用元:CTD 5.3.5.1.2-1 の表 11.4-19
19
(6) 涙眼 治験期間中の涙眼の程度を表 2.7.3.3-15 に示した。 26 週観察日,52 週観察日と観察開始日との間に,いずれも大きな変化はなかった(p=0.1146, p=0.1836)。 表 2.7.3.3-15 JRQLQ No.1(FAS) -涙眼- 評価時期 症状なし 0 軽い 1 やや重い 2 重い 3 非常に重い 4 合計例数 観察開始日 との比較* 観察開始日 14(50.0%) 7(25.0%) 2(7.1%) 3(10.7%) 2(7.1%) 28 - 26 週観察日 17(63.0%) 6(22.2%) 3(11.1%) 0(0.0%) 1(3.7%) 27 0.1146 52 週観察日 14(53.8%) 9(34.6%) 2(7.7%) 0(0.0%) 1(3.8%) 26 0.1836 表中の数値は例数を示し,括弧内の数値はそれぞれの評価時期の合計例数に対する百分率を示す。 * Wilcoxon 符号付順位検定に基づく p 値 引用元:CTD 5.3.5.1.2-1 の表 11.4-20 1) QOL 質問項目 結果を図 2.7.3.3-1,表 2.7.3.3-16 に示した。 26 週観察日は観察開始日に比べ,6 つの領域のうち「日常生活」,「社会生活」,「睡眠」, 「身体」,「精神生活」の領域別スコアが低下した(QOL が良くなった)(それぞれ,p=0.0016, p=0.0281,p=0.0381,p=0.0248,p=0.0220)。一方,「戸外行動」には大きな変化はなかった(p=0.0961)。 52 週観察日は観察開始日に比べ,6 つの領域のうち「日常生活」,「睡眠」,「身体」,「精 神生活」の領域別スコア低下した(QOL が良くなった)(それぞれ,p=0.0009,p=0.0209,p=0.0218, p=0.0246)。一方,「戸外行動」,「社会生活」には大きな変化はなかった(p=0.4281,p=0.5315)。 図 2.7.3.3-1 JRQLQ No.1(FAS) -領域別スコア(平均値)の比較- 引用元:CTD 5.3.5.1.2-1 の図 11.4-1
表 2.7.3.3-16 JRQLQ No.1(FAS) -総括的状態- 評価時期 総括的状態 合計 例数 観察開始日 との比較* 0 晴れ晴れ 1 2 3 4 泣きたい 観察開始日 3(10.7%) 4(14.3%) 14(50.0%) 6(21.4%) 1(3.6%) 28 - 26 週観察日 6(22.2%) 13(48.1%) 5(18.5%) 3(11.1%) 0(0.0%) 27 0.0005 52 週観察日 6(23.1%) 10(38.5%) 8(30.8%) 1(3.8%) 1(3.8%) 26 0.0202 表中の数値は例数を示し,括弧内の数値はそれぞれの評価時期の合計例数に対する百分率を示す。 * Wilcoxon 符号付順位検定に基づく p 値 引用元:CTD 5.3.5.1.2-1 の表 11.4-22 2.7.3.3.2 HDM アレルギー性喘息患者 2.7.3.3.2.1 医師による総合評価 結果を表 2.7.3.3-17 に示した。 26 週観察日は観察開始日に比べ,「良い」が増加し,「普通」,「悪い」が減少したが,大き な変化はなかった(p=0.1875)。 52 週観察日は観察開始日に比べ,「良い」,「少し良い」が増加し,「普通」,「悪い」が減 少したが,大きな変化はなかった(p=0.1250)。 表 2.7.3.3-17 医師による総合評価(FAS) 評価時期 良い 少し良い 普通 少し悪い 悪い 合計 例数 観察開始日 との比較* 観察開始日 10(62.5%) 1(6.3%) 3(18.8%) 0(0.0%) 2(12.5%) 16 - 26 週観察日 13(81.3%) 1(6.3%) 2(12.5%) 0(0.0%) 0(0.0%) 16 0.1875 52 週観察日 12(80.0%) 2(13.3%) 1(6.7%) 0(0.0%) 0(0.0%) 15 0.1250 表中の数値は例数を示し,括弧内の数値はそれぞれの評価時期の合計例数に対する百分率を示す。 * Wilcoxon 符号付順位検定に基づく p 値 引用元:CTD 5.3.5.1.2-1 の表 11.4-23 2.7.3.3.2.2 ACT(喘息コントロールテスト,12 歳以上) 結果を表 2.7.3.3-18 に示した。 26 週観察日は観察開始日に比べ,ACT スコア(合計点)が上昇した(喘息コントロールの状態 が良くなった)が,大きな変化はなかった(p=0.0528)。 52 週観察日は観察開始日に比べ,ACT スコア(合計点)が更に上昇した(p=0.0184)。
21
表 2.7.3.3-18 ACT(12 歳以上)(FAS) -合計点- 評価時期 例数 評価点数(合計点) 平均値 標準偏差 中央値 最小値 最大値 観察開始日との比較 差の平均値 p 値* 観察開始日 10 21.7 2.1 22.0 18 25 - - 12 週観察日 10 21.6 3.9 22.0 12 25 -0.1 0.9140 26 週観察日 10 23.3 1.4 23.0 21 25 1.6 0.0528 38 週観察日 9 22.4 2.9 23.0 16 25 0.7 0.4186 52 週観察日 9 23.9 1.2 24.0 22 25 2.1 0.0184 * 対応のある t 検定に基づく p 値 引用元:CTD 5.3.5.1.2-1 の表 11.4-24 2.7.3.3.2.3 小児 ACT(小児喘息コントロールテスト,5 歳~11 歳) 結果を表 2.7.3.3-19 に示した。 26 週観察日は観察開始日に比べ,小児 ACT スコア(合計点)が上昇した(喘息コントロール の状態が良くなった)が,大きな変化ではなかった(p=0.2229)。 52 週観察日は観察開始日に比べ,小児 ACT スコア(合計点)が更に上昇した(喘息コントロ ールの状態が良くなった)(p=0.0264)。 表 2.7.3.3-19 小児ACT(11 歳以下)(FAS) -合計点- 評価時期 例数 評価点数(合計点) 平均値 標準偏差 中央値 最小値 最大値 観察開始日との比較 差の平均値 p 値* 観察開始日 6 23.3 3.4 24.0 18 27 - - 12 週観察日 6 22.3 5.4 23.5 12 27 -1.0 0.6238 26 週観察日 6 25.3 2.3 26.5 22 27 2.0 0.2229 38 週観察日 6 24.7 3.0 25.5 19 27 1.3 0.3075 52 週観察日 6 26.2 2.0 27.0 22 27 2.8 0.0264 * 対応のある t 検定に基づく p 値 引用元:CTD 5.3.5.1.2-1 の表 11.4-25 2.7.3.3.2.4 AHQ-Japan(16 歳以上) 1) AHQ-Japan の質問項目 26 週観察日は観察開始日に比べ,「喘息症状」,「感情面」,「活動の制限や困難」,「喘 息症状の増悪因子」,「社会活動の制限」,「経済的側面」の 6 つの領域別スコアがいずれも 低下した(QOL が良くなった)が,大きな変化ではなかった(それぞれ,p=0.6253,p=0.2679, p=0.0796,p=0.7086,p=0.0729,p=0.2849)。 52 週観察日も観察開始日に比べ,6 つの領域別スコアがいずれも低下し,特に「感情面」, 「社会活動の制限」,「経済的側面」で低下した(QOL が良くなった)が,大きな変化ではな
かった(それぞれ,p=0.2621,p=0.2953,p=0.6891,p=0.5347,p=0.1689,p=0.3208)(図 2.7.3.3-2)。 図 2.7.3.3-2 AHQ-Japan(FAS) -領域別スコア(平均値)の比較- 引用元:CTD 5.3.5.1.2-1 の図 11.4-2 2) 過去 7 日間の全体的な生活の質(QOL) 26 週観察日は観察開始日に比べ,「0」が増加し,「4」が減少した(QOL が良くなった)が, 全体として大きな変化はなかった(p=0.5313)。 52 週観察日は観察開始日に比べ,「4」が減少した(QOL が良くなった)が,全体として大 きな変化はなかった(p=0.7500)(表 2.7.3.3-20)。 表 2.7.3.3-20 AHQ-Japan(FAS) -この7 日間,全体的な生活の質(QOL)はどのようでしたか?- 評価時期 0 1 2 3 4 合計 例数 観察開始日 との比較* 観察開始日 3(37.5%) 3(37.5%) 1(12.5%) 0(0.0%) 1(12.5%) 8 - 26 週観察日 5(62.5%) 1(12.5%) 1(12.5%) 1(12.5%) 0(0.0%) 8 0.5313 52 週観察日 3(42.9%) 3(42.9%) 0(0.0%) 1(14.3%) 0(0.0%) 7 0.7500 表中の数値は例数を示し,括弧内の数値はそれぞれの評価時期の合計例数に対する百分率を示す。 * Wilcoxon 符号付順位検定に基づく p 値 引用元:CTD 5.3.5.1.2-1 の表 11.4-27
23
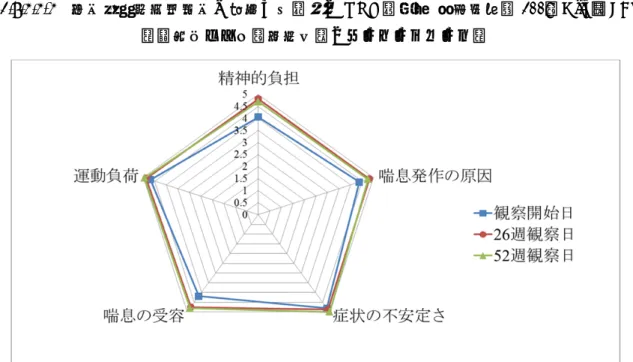
2.7.3.3.2.5 小児気管支喘息患児と親又は保護者の QOL 調査票 簡易改訂版 2008(5 歳~15 歳) 26 週観察日は観察開始日に比べ,5 つの領域のうち「精神的負担」,「喘息の受容」の領域別 スコアが上昇した(QOL が良くなった)(p=0.0263,p=0.0256)。一方,「喘息発作の原因」, 「症状の不安定さ」,「運動負荷」の領域別スコアも上昇したが,大きな変化はなかった(それぞ れ,p=0.1949,p=0.3506,p=0.1970)。 52 週観察日は観察開始日に比べ,5 つの領域のうち「喘息の受容」が上昇した(QOL が良くな った)(p=0.0053)。一方,「精神的負担」,「喘息発作の原因」,「症状の不安定さ」,「運 動負荷」の領域別スコアも上昇したが,大きな変化はなかった(それぞれ,p=0.0950,p=0.2654, p=0.1970,p=0.1705)(図 2.7.3.3-3)。
図 2.7.3.3-3 小児気管支喘息患児と親又は保護者の QOL 調査票 簡易改訂版 2008(Gifu)(FAS)
-領域別スコア(平均値)のレーダーチャート- 引用元:CTD 5.3.5.1.2-1 の図 11.4-3 2.7.3.3.2.6 肺機能検査(FEV1,%FEV1) 1) 肺機能検査(FEV1)の記述統計量 26 週観察日,52 週観察日は観察開始日に比べ,いずれも FEV1が増大した(p=0.0086,p=0.0058)。 2) 肺機能検査(%FEV1)の記述統計量 26 週観察日,52 週観察日は観察開始日に比べ,いずれも%FEV1が増大したが,大きな変化 ではなかった(p=0.1031,p=0.2619)。
2.7.3.4 有効性の結論 HDM アレルギー性鼻炎患者及び HDM アレルギー性喘息患者を対象として,TO-204 皮下注に よる免疫療法を52 週間行った結果,以下の結論を得た。なお,本治験の有効性評価では検証的な 判断は行わなかった。 1) HDM アレルギー性鼻炎患者 観察開始日又は増量期開始日に比べ,52 週観察日において, ・医師による総合評価は,「良い」,「少し良い」が増加し,「普通」,「少し悪い」,「悪 い」が減少した。 ・鼻の局所所見は,下鼻甲介粘膜の腫脹,下鼻甲介粘膜の色調,水性分泌量,鼻汁の性状の いずれも症状の軽い被験者が増加し,症状の重い被験者が減少した。 ・鼻症状スコアは,鼻汁,鼻閉,そう痒感は,症状のない被験者が増加し,症状の重い被験 者が減少した。一方,くしゃみには大きな変化はなかった。 ・JRQLQ No.1(QOL 調査)の鼻・眼の症状は,水っぱな,くしゃみ,鼻づまり,鼻のかゆみ は,症状のない被験者が増加し,症状の重い被験者が減少した。一方,眼のかゆみ,涙眼 には大きな変化はなかった。 ・JRQLQ No.1(QOL 調査)の QOL 質問項目を 6 つの領域に分類して検討した結果,日常生 活,睡眠,身体,精神生活は,評価点数が良くなった。一方,戸外行動,社会生活には大 きな変化はなかった。 ・JRQLQ No.1(QOL 調査)の総括的状態は,0(晴れ晴れ),1 が増加し,2,3 が減少した。 2) HDM アレルギー性喘息患者 観察開始日に比べ,52 週観察日において, • 医師による総合評価は,「良い」,「少し良い」が増加し,「普通」,「悪い」が減少した が,大きな変化はなかった。 • 12 歳以上の被験者の ACT スコア(合計点)は上昇した(喘息コントロールの状態が良くな った)。 • 5~11 歳の被験者の小児 ACT スコア(合計点)は上昇した(喘息コントロールの状態が良 くなった)。 • AHQ-Japan(16 歳以上の QOL 調査)の調査項目を 6 つの領域に分類して検討した結果,喘 息症状,感情面,活動の制限や困難,喘息症状の増悪因子,社会活動の制限,経済的側面の いずれも領域別スコアが低下した(QOL が良くなった)が,大きな変化はなかった。 • AHQ-Japan(16 歳以上の QOL 調査)の過去 7 日間の全体的な生活の質(QOL)は,「4」 が減少した(QOL が良くなった)が,大きな変化はなかった。 • 小児気管支喘息と親又は保護者の QOL 調査(5~15 歳の QOL 調査)の調査項目を 5 つの領 域に分類して検討した結果,喘息の受容の領域別スコアが上昇した(QOL が良くなった)。 一方,精神的負担,喘息発作の原因,症状の不安定さ,運動負荷の領域別スコアも上昇した が,大きな変化はなかった。
25
• FEV1が増大した。%FEV1も増大したが,大きな変化ではなかった。 3) まとめ
TO-204 を 52 週間投与した結果,検証的判断は実施していないものの,HDM アレルギー性鼻 炎患者の鼻症状及びQOL に改善が認められ,HDM アレルギー性喘息患者の喘息コントロール 状態及び呼吸機能の一部に改善が認められた。
2.7.4 臨床的安全性の概要 2.7.4.1 安全性評価に用いた試験 2.7.3.1 項で述べたように,これまでの長い歴史の中で,アレルギー性鼻炎及び喘息に対する SCIT の有効性が確立し,各種ガイドライン等においても SCIT の有用性が記されている。また, TO-204 皮下注は,本邦における使用実績はないものの,アメリカ,カナダ等では既に上市されて いる。 これらを踏まえ, することでPMDA との合意に至った。本 申請に当たっては,TO-204 第 III 相臨床試験 長期投与試験 1 試験のみを評価資料として申請資料 に含めた。本治験の概要を表 2.7.3.2-1 に示した。 なお,国内外の公表文献で示された標準化HDM SCIT の臨床試験における安全性を 2.7.4.4 項 に,SCIT におけるアナフィラキシー等の全身性反応の発現状況を 2.7.4.5 項に記載した。 2.7.4.2 安全性の評価方法 1) 安全性解析対象集団 治験薬が投与され,安全性評価項目の調査が実施された症例を安全性解析対象集団とした。 本治験では,44 例の被験者に対して TO-204 皮下注を用いた皮内閾値検査を実施して以降, すべての被験者(44 例)に対して少なくとも一回,安全性の評価が行われた。したがって,本 治験での安全性解析対象集団はFAS と同様に 44 例となった。 2) 安全性の評価項目 (1) 有害事象及び副作用 (2) 臨床検査 (3) 生理検査 3) 有害事象及び副作用 皮内閾値検査後から52 週観察日まで,又は中止時観察日までに発現した有害事象について, 有害事象の名称,発現日,発現時刻,治療の有無,治験薬の処方変更,重篤度,重症度,因果 関係,転帰を調査した。 安全性の解析については,2.7.3.2.10 項の解析計画に記した。 本治験では,被験者数が少ないため被験者全体(44 例)における有害事象(又は副作用)発 現率は,1 例の発現でも 2.3%となり,比較的よく見られる基準として通常設定される 2%を超え ることから,本治験における比較的よく見られる基準は被験者全体の5%とした。 また,有害事象(又は副作用)発現率を部分集団(疾患別,増量法別,年齢別)で比較する 場合には,各集団の被験者数が更に少なくなることから(1 例の発現で発現率は 5%前後),発 現率又は発現率の集団間差は 10%を基準として評価とした。なお,投与期別(増量期開始前, 増量期,維持期)に比較する場合には,各期の被験者数は44 例又は 40 例であったことから, 発現率又は発現率の差は5%を基準として評価した。本申請資料における有害事象の調査期間は,
1
皮内閾値検査後から,52 週観察日まで(中止時観察日の測定値を含む)とした。 副作用の集計及び評価は有害事象と同様に行った。なお,安全性データについては統計学的 な解析は実施しなかった。 2.7.4.2.1 全般的な曝露状況 本治験の安全性に関しては,利用可能なすべてのデータを評価するため,52 週観察日までに得 られたすべてのデータを集計した。 被験者ごとの増量法,閾値,初回投与量,増量期間,維持量,投与期間,累積投与量を表 2.7.3.2-5 に示した。また,維持量に到達した被験者は44 例中 40 例だった。 2.7.4.2.2 治験対象集団の人口統計学的特性及びその他の特性 主な被験者背景(FAS,安全性解析対象集団)を表 2.7.3.2-3 に示した。 2.7.4.3 安全性の結果 2.7.4.3.1 有害事象 2.7.4.3.1.1 比較的よく見られる有害事象 比較的よく見られる(被験者全体における発現率が5%以上)有害事象を,被験者全体における 発現率順に表 2.7.4.3-1 に示した。
表 2.7.4.3-1 比較的よく見られる有害事象(被験者全体における発現率が5%以上) (安全性解析対象集団) 有害事象名 PT (MedDRA/J V15.0) HDM アレルギー性 鼻炎患者(28 例) HDM アレルギー性 喘息患者(16 例) 合計(44 例) 件数 例数 発現率 (%)* 件数 例数 発現率 (%)* 件数 例数 発現率 (%)* 鼻咽頭炎 34 17 60.7 13 7 43.8 47 24 54.5 頭痛 13 9 32.1 8 5 31.3 21 14 31.8 上気道の炎症 10 7 25.0 4 4 25.0 14 11 25.0 咳嗽 8 8 28.6 1 1 6.3 9 9 20.5 蕁麻疹 11 4 14.3 8 4 25.0 19 8 18.2 注射部位疼痛 7 4 14.3 4 3 18.8 11 7 15.9 嘔吐 5 4 14.3 3 3 18.8 8 7 15.9 胃腸炎 4 4 14.3 3 3 18.8 7 7 15.9 注射部位そう痒感 10 6 21.4 0 0 0.0 10 6 13.6 下痢 6 4 14.3 2 2 12.5 8 6 13.6 インフルエンザ 3 3 10.7 3 3 18.8 6 6 13.6 注射部位腫脹 16 4 14.3 1 1 6.3 17 5 11.4 アナフィラキシー反応 4 4 14.3 1 1 6.3 5 5 11.4 注射部位紅斑 5 3 10.7 3 1 6.3 8 4 9.1 悪心 2 2 7.1 4 2 12.5 6 4 9.1 発熱 2 1 3.6 4 3 18.8 6 4 9.1 注射部位反応 2 2 7.1 3 2 12.5 5 4 9.1 扁桃炎 3 3 10.7 1 1 6.3 4 4 9.1 膿痂疹 5 2 7.1 1 1 6.3 6 3 6.8 注射部位熱感 5 3 10.7 0 0 0.0 5 3 6.8 気管支炎 1 1 3.6 2 2 12.5 3 3 6.8 齲歯 1 1 3.6 2 2 12.5 3 3 6.8 湿疹 1 1 3.6 2 2 12.5 3 3 6.8 潮紅 3 3 10.7 0 0 0.0 3 3 6.8 麦粒腫 2 2 7.1 1 1 6.3 3 3 6.8 中耳炎 3 3 10.7 0 0 0.0 3 3 6.8 口腔咽頭不快感 3 3 10.7 0 0 0.0 3 3 6.8 *それぞれ該当する例数に対する百分率を示す。 引用元:CTD 5.3.5.1.2-1 の表 12.2-7
3
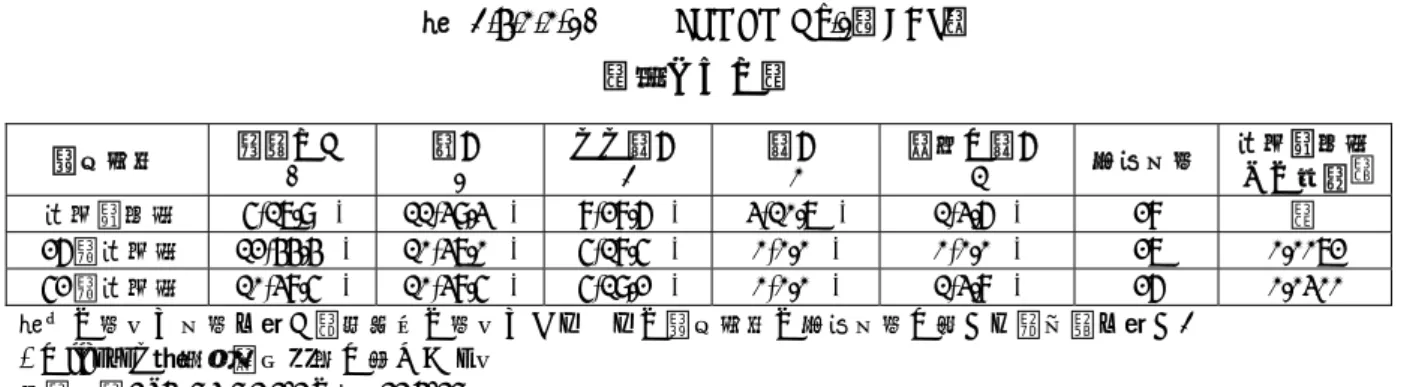
2.7.4.3.1.2 比較的よく見られる副作用 本治験で発現した比較的よく見られる(被験者全体における発現率が5%以上)副作用を,被験 者全体における発現率順に表 2.7.4.3-2 に示した。 表 2.7.4.3-2 比較的よく見られる副作用(被験者全体における発現率が5%以上) (安全性解析対象集団) 副作用名 PT (MedDRA/J V15.0) HDM アレルギー性 鼻炎患者(28 例) HDM アレルギー性 喘息患者(16 例) 合計(44 例) 件数 例数 発現率 (%)* 件数 例数 発現率 (%)* 件数 例数 発現率 (%)* 注射部位疼痛 7 4 14.3 4 3 18.8 11 7 15.9 蕁麻疹 11 4 14.3 3 2 12.5 14 6 13.6 注射部位そう痒感 10 6 21.4 0 0 0.0 10 6 13.6 注射部位腫脹 16 4 14.3 1 1 6.3 17 5 11.4 咳嗽 4 4 14.3 1 1 6.3 5 5 11.4 注射部位反応 2 2 7.1 3 2 12.5 5 4 9.1 アナフィラキシー反応 3 3 10.7 1 1 6.3 4 4 9.1 注射部位紅斑 4 2 7.1 3 1 6.3 7 3 6.8 注射部位熱感 5 3 10.7 0 0 0.0 5 3 6.8 *それぞれ該当する例数に対する百分率を示す。 引用元:CTD 5.3.5.1.2-1 の表 12.2-8 1) 比較的よく見られる副作用の初回発現時の投与後発現時間 本剤投与によって発現した比較的よく見られる副作用(表 2.7.4.3-2 参照)に対し,初回発現 時の投与後発現時間を検討した。結果を表 2.7.4.3-3,表 2.7.4.3-4,表 2.7.4.3-5,表 2.7.4.3-6, 表 2.7.4.3-7,表 2.7.4.3-8 に示した。 副作用全体の累積発現件数は,投与後30 分以内に 43 件中 27 件(62.8%),投与後 1 時間以 内に43 件中 33 件(76.7%),投与後 2 時間以内では 43 件中 40 件(93.0%)であった。さらに, 投与2 時間を超えてから発現した副作用は 43 件中 3 件(7.0%)のみであった。このことから, 本治験では,投与後2 時間を超えて新たな副作用が発現する割合は低かったことが確認された。
表 2.7.4.3-3 比較的よく見られる副作用(被験者全体における発現率が5%以上) の初回発現時の投与後発現時間 件数(その 1) 副作用名 (MedDRA/J V.15.0) 全体(44 例) 30 分 以内 30 分< ≦1 時間 1 時間< ≦2 時間 PT 件数 例数 件数の 割合(%) 件数 例数 件数の 割合(%) 件数 例数 件数の 割合(%) 全体 27 15 62.8 6 5 14.0 7 6 16.3 アナフィラキシー反応 1 1 25.0 1 1 25.0 2 2 50.0 咳嗽 2 2 40.0 2 2 40.0 1 1 20.0 蕁麻疹 2 2 33.3 1 1 16.7 2 2 33.3 注射部位紅斑 3 3 100.0 0 0 0.0 0 0 0.0 注射部位疼痛 7 7 100.0 0 0 0.0 0 0 0.0 注射部位そう痒感 3 3 50.0 2 2 33.3 0 0 0.0 注射部位反応 4 4 100.0 0 0 0.0 0 0 0.0 注射部位熱感 2 2 66.7 0 0 0.0 1 1 33.3 注射部位腫脹 3 3 60.0 0 0 0.0 1 1 20.0 引用元:CTD 5.3.5.1.2-1 の表 12.2-22 表 2.7.4.3-4 比較的よく見られる副作用(被験者全体における発現率が5%以上) の初回発現時の投与後発現時間 件数(その 2) 副作用名 (MedDRA/J V.15.0) 全体(44 例) 2 時間< ≦4 時間 4 時間< ≦8 時間 8 時間< ≦12 時間 PT 件数 例数 件数の 割合(%) 件数 例数 件数の 割合(%) 件数 例数 件数の 割合(%) 全体 1 1 2.3 1 1 2.3 0 0 0.0 アナフィラキシー反応 0 0 0.0 0 0 0.0 0 0 0.0 咳嗽 0 0 0.0 0 0 0.0 0 0 0.0 蕁麻疹 0 0 0.0 1 1 16.7 0 0 0.0 注射部位紅斑 0 0 0.0 0 0 0.0 0 0 0.0 注射部位疼痛 0 0 0.0 0 0 0.0 0 0 0.0 注射部位そう痒感 0 0 0.0 0 0 0.0 0 0 0.0 注射部位反応 0 0 0.0 0 0 0.0 0 0 0.0 注射部位熱感 0 0 0.0 0 0 0.0 0 0 0.0 注射部位腫脹 1 1 20.0 0 0 0.0 0 0 0.0 引用元:CTD 5.3.5.1.2-1 の表 12.2-23