Clostridium difficile の毒性発現機序に関する研究
日大生産工(院)○強矢 佳織 日大生産工
神野 英毅
1.
緒論
Clostridium difficile
(C. difficile) は偏性嫌気 性有芽胞グラム陽性桿菌で、抗生物質が誘因と なる下痢症あるいは腸炎の主要な原因菌であり、
院内感染症の主原因菌としても知られている。
C.difficile
の病原因子としては
toxin Aおよび
toxin B
があり、本菌を毒素産生性で分類すると
両毒素を産生する
toxin A+B+株、toxin Bのみ産 生する
toxin A-B+株、そして毒素非産生のtoxin A-B-株とに分けられる。臨床的に重要な本菌の毒性発現機序を知ることは非常に意義があると いえる。本研究では両毒素のうち有毒株に共通 の産生毒素である
toxin Bに着目した。
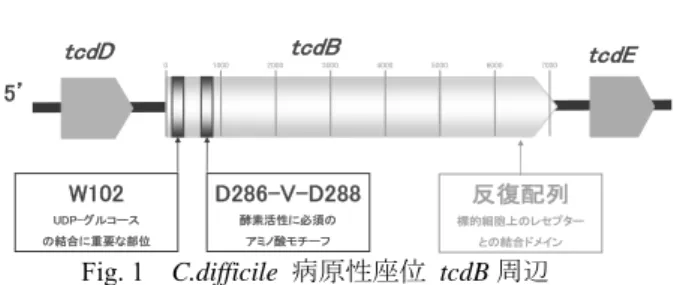
toxin Bは グルコシル転移酵素ドメイン・転移ドメイン・
結合ドメインの
3領域の働きにより細胞毒性を 発現することが明らかにされている。
UDP-グルコースの結合に重要とされる
W102,酵素活性 に必須のアミノ酸モチーフである
D286-V-D288を含む
N末端
546アミノ酸残基にグルコシル転 移酵素ドメイン、C 末端
516アミノ酸残基の反 復配列部位に標的細胞上のレセプターと結合す る結合ドメイン、そして、グルコシル転移酵素 ドメインと結合ドメインの間に毒素がファゴゾ ームから細胞質へ転移する機能を担う転移ドメ インが存在すると推測されている(Fig. 1)。
本研究では
C. difficileの毒性発現機序を解明 すべく、大腸菌の系における遺伝子組換え技術 を用いて、毒素タンパクを構築しているフラグ メントの発現を行なった。
2.
実験方法
2-1.使用菌株
菌体は岐阜大学生命科学総合実験センター嫌 気性菌実験分野より供与された、C.difficile 臨床 分離株
GAI 99093を使用した。本株は
toxin A,toxin B
の両毒素を産生する。
2-2.
遺伝子増幅
フェノール・クロロホルム法により抽出した
DNAを
templateとして
Polymerase Chain Reaction(PCR)法にて遺伝子増幅を行った。UDP-グル コースの結合に重要とされる
W102,酵素活性 に必須のアミノ酸モチーフである
D286-V-D288を含む部位を目的部位とした。primer は
vectorの cloning site に適するように設計した(Table 1)。
forward primer(DK tcdB W102-F)は 5’末端に CACC
配列を置き、直後に開始コドン(ATG)、
続いて導入部位の配列となるようにし、reverse
primer(DK tcdB DVD-R)は導入部位に続き、終 止コドン(TAG)を置き、その相補的な配列と した。これらの
primerを用いて、アニーリング
温度
63℃にて目的部位を増幅した。Fig. 1 C.difficile
病原性座位
tcdB周辺
2-3.
大腸菌への導入
精製した目的部位の増幅産物を
plasmid vector(pET101/D-TOPO)にサブクローニングし、氷 上で大腸菌(TOP10 competent cells)と混合、ヒ ートショックにて導入した。
ampicillinを含む
LB寒天培地に展開し、一晩培養した後に選択し、
コロニーPCR を行い導入の確認をした。コロニ ーPCR には
plasmid vectorの
T7 promoter由来の
primerを用いた(Table 2)。また、これらの
primerを用いて塩基配列解析も行った。
Study on mechanism of toxic manifestation of Clostridium difficile toxins
Kaori SUNEYA and Hideki KOHNO
5’
tcdD 0 1000 2000tcdB3000 4000 5000 6000 7000 tcdE
W102
UDP-グルコース の結合に重要な部位
D286-V-D288
酵素活性に必須の アミノ酸モチーフ
反復配列
標的細胞上のレセプター との結合ドメイン
Table 1
導入部位増幅に用いる
primer塩基配列
primer 塩基配列
DK tcdB W102-F 5'-CACCATGAATAATAATTTAACTCCAG-3' DK tcdB DVD-R 5'-CTACTCTATAGACTCAAATAAGTCTGGTTG-3'
primer 塩基配列
T7 Forward 5'-TAATACGACTCACTATAGGG-3' T7 Reverse 5'-TAGTTATTGCTCAGCGGTGG-3'
Table 2 T7 promoter
由来の
primer塩基配列
2-4. IPTG
による発現誘導
導入が確認されたコロニーから
plasmidを抽 出した。それを氷上で大腸菌(LB21 competent
cells)と混合し、ヒートショックによりに導入した。LB 培地にて一晩培養した後、継代し
Isopropyl-β-D-thiogalactopyranoside(IPTG)によ る発現誘導実験を行なった。IPTG は終濃度
1.0mM
で加え、加えないもの(非誘導)と比較
した。培養液は
1時間毎に採取し、遠心分離
(16,000×g, 30sec)を行い、ペレットをサンプ ルとした。
2-5.
発現の確認
IPTG
による発現誘導で得られたペレットを
Lysis buffer
にて溶解し、凍結融解を行った。遠
心分離(16,000×g, 1min)にて、上清とペレッ トに分け、それぞれを
SDS-PAGEにて確認した。
3.
結果および考察
TOP10
への形質転換におけるコロニーPCR で
は plasmid vector の
T7 promoter由来の
primerを 用いたため、T7 promoter 周辺の塩基(282bp)
も含めて増幅されている。よって、目的の
W102および
D286-V-D288を含む断片(655bp)が組込ま れた場合は
937bpに、組込まれない場合は
T7promoter
周辺のみの
282bpにバンドが検出でき
る。コロニーPCR による増幅産物をアガロース ゲル電気泳動した結果を
Fig. 2に示す。
Lane1, 2では増幅が行われなかった。Lane3, 5~7 では
T7promoter
周辺のみのバンドが確認できた。そし
て、Lane4 では目的のバンドが検出されたことが 確認できる。
また、colony D の塩基配列を解析した結果を
Fig. 3に示す。
vector配列に続いて
CACC配列か ら始まる目的導入部位の配列が確認できた。こ のことからも、目的部位が正しく導入されたこ とが確認できた。
次に、colony D 由来のプラスミドDNA を導入し た大腸菌(BL21)で
IPTGによる発現誘導を行 った。Lysis buffer を加えた凍結融解の後、遠心 分離によってペレットと上清に分け
SDS-PAGEを行った。
216アミノ酸残基からなる目的の毒素 タンパクのフラグメントサイズは約
24kDaであ る。ペレットの
SDS-PAGEの結果、24kDa 付近 に時間経過とともに濃くなるバンドが確認され た(Fig. 4)。このバンドは非誘導では薄く、ほ とんど確認できない。誘導によって目的のタンパ クが過剰に発現されたものと考えられる。
4.
結論
大腸菌の系における遺伝子組換え技術を用い
て、毒素タンパクを構築しているフラグメント の発現に成功したといえる。そして、同様にし て他部位のフラグメントを発現させ、それらを 比較することにより毒性発現機序の解明が行え ることも分かった。
【参考文献】
1)F.Hofmann et al. , Localization of the glucosyl transferase activity of Clostridium difficile toxin B to the N-terminal part of the holotoxin, Journal of Biological Chemistry, 272, 11074-11078, 1997 2
)
Maja Rupnik et al. , New Types of Toxin A-Negative,Toxin B-Positive Strains among Clostridium difficile Isolates from Asia, JOURNAL OF CLINICAL MICROBIOLOGY, 41(3), p. 1118–1125, 2003
Vector sequence Inserted gene
(kDa)
25 20
M 1 2 3 4 5 6 7 8 9 10 11
Fig. 4 IPTG
による誘導実験
(Lysis ペレット)
1
:誘導
0h 2:誘導
1h 3:誘導
2h 4:誘導
3h 5:誘導
4h 6:誘導
5h 7:非誘導
1h 8:非誘導
2h 9:非誘導
3h 10:非誘導
4h 11:非誘導
5h M:WIDE-VIEWTMPrestained Protain Size MarkerⅡ(Wako)
1 2 3 4 5 6 7
M4
603 bp 310 bp 872 bp 1078 bp 1353 bp
937 bp
M4:Marker4
(φX174/Hae Ⅲ digest) 1: colony A 2: colony B 3: colony C 4: colony D 5: colony E 6: colony F 7: colony G
Fig. 2
コロニーPCR による増幅産物のゲル電気泳動結果
Fig. 3 Cloning site