2.7 臨床概要
2.7.5 参考文献
すべて添付せず
1) 厚生省医薬安全局審査管理課.抗菌薬臨床評価のガイドライン.日本化学療法学会雑誌
1998;46:410-437.
2) UTI 研究会 (代表 大越正秋).UTI 薬効評価基準 (第 3 版).Chemotherapy (Tokyo) 1986;
34:408-441.
3) 日本化学療法学会臨床評価法制定委員会呼吸器系委員会報告.呼吸器感染症における新
規抗微生物薬の臨床評価法 (案).日本化学療法学会雑誌 1997;45:762-778.
4) 広津千尋.臨床試験データの統計解析.廣川書店;1992.
5) 日本化学療法学会.「抗菌薬による治験症例における副作用,臨床検査値異常の判定基
準」の一部変更について.日本化学療法学会雑誌 1995;43:巻頭.
6) 日本化学療法学会.抗菌薬による治験症例における副作用,臨床検査値異常の判定基準.
Chemotherapy (Tokyo) 1991;39:687-689.
7) 伊藤康久,武田明久,兼松稔,坂義人,西浦常雄,松田聖士ほか.カルバペネム系抗菌
剤 Imipenem/Cilastatin sodium (MK-0787/MK-0791) のヒト腎・前立腺移行および複雑性
尿路感染症に対する有用性について.Chemotherapy (Tokyo) 1985;33: 825-833.
8) 後藤俊弘,川原元司,牧之瀬信一,山内大司,小濱康彦,大井好忠ほか.尿路感染症に
おける Meropenem の基礎的・臨床的検討.Chemotherapy (Tokyo) 1992; 40:620-630.
9) 佐々木公則,千石一雄,石川睦男,清水哲也,芳賀宏光,長谷川天洙ほか.産婦人科領
域における Meropenem の基礎的・臨床的検討.Chemotherapy (Tokyo) 1992 ; 40:646-650.
10) 張南薫,福永完吾,国井勝昭.産婦人科領域における Imipenem/Cilastatin sodium
(MK-0787/MK-0791) の基礎的,臨床的検討.Chemotherapy (Tokyo) 1985;33:1046-1063.
11) 倉田和男.整形外科領域における Panipenem/Betamipron の血中及び骨組織内濃度の検討.
Jpn J antibiotics 1992;45:155-159.
12) 佐野徳久,桜井実,土肥千里,大山明,室田影久,杉山肇ほか.Meropenem の骨髄血,
骨組織及び関節組織への移行性に関する検討.Jpn J Antibiotics 1993;45:159-163.
13) 馬場駿吉,宮本直哉,山本真一郎,川端五十鈴,田部浩生,河村正三ほか.耳鼻咽喉科
領域感染症における Panipenem/betamipron (CS-976) の基礎的,臨床的検討.耳鼻と臨床
1992;38:37-55.
14) 宮本直哉,山本真一郎,小林武弘,馬場駿吉,伊藤晴夫,伊佐治弘子ほか.Meropenem
の耳鼻咽喉科領域感染症における基礎的・臨床的検討.Chemotherapy (Tokyo) 1992;40:
710-718.
15) 荒田次郎,秋山尚範,神崎寛子,高橋久,田中由比,高浜英人ほか.皮膚科領域におけ
る Panipenem/Betamipron の基礎的・臨床的検討.Jpn J Antibiotics 1992;45:197-207.
16) 神崎寛子,鳥越利加子,山田琢,安部能子,下江敬生,秋山尚範ほか.皮膚科領域にお
ける Meropenem の基礎的・臨床的検討.Chemotherapy (Tokyo) 1992;40:751-754.
17) 大石正夫,坂上富士夫,田沢博,宮尾益也,藤原隆明,石川和男ほか.眼科領域におけ
2.7 臨床概要
672.
18) 大石正夫,坂上富士夫,田沢博,大野重昭,秦野寛,大桃明子ほか.眼科領域における
Meropenem の基礎的・臨床的検討.Chemotherapy (Tokyo) 1992;40:689-700.
19) 金子明寛,佐々木次郎,山根伸夫,岡部孝一,中川清昌,山本悦秀ほか.口腔外科領域
における panipenem/betamipron の基礎的・臨床的検討.Chemotherapy (Tokyo) 1991;39:
647-664.
20) 佐々木次郎.口腔外科領域における Meropenem の基礎的・臨床的検討.Chemotherapy
(Tokyo) 1992;40:732-745.
21) 小野成夫,田中豊治,竹中能文,加藤繁次.外科領域における Meropenem の基礎的,臨
床的検討.Chemotherapy (Tokyo) 1992;40:500-506.
22) 鈴木啓一郎,相川直樹,奥沢星二郎,高橋孝行,石引久弥.Imipenem/Cilastatin sodium
(MK-0787/MK-0791) の腹腔内浸出液移行と外科領域における臨床的検討.Chemotherapy
(Tokyo) 1985;33:942-949.
23) 副島林造,斎藤玲,関根理,勝正孝,斎藤篤,真下啓明ほか.呼吸器感染症に対する
MK-0787/MK-0791 と Piperacillin の薬効比較試験成績.感染症学雑誌 1986;60:345-377.
24) 原耕平,武部和夫,島田馨,柴孝也,小林宏行,松本文夫ほか.慢性気道感染症に対す
る panipenem/betamipron と imipenem/cilastatin の薬効比較試験.Chemotherapy (Tokyo)
1992;40:613-637.
25) 原耕平,武部和夫,島田馨,柴孝也,小林宏行,松本文夫ほか.細菌性肺炎に対する
panipenem/betamipron と imipenem/cilastatin の薬効比較試験.Chemotherapy (Tokyo) 1992;
40:509-531.
26) 原耕平,斎藤玲,武部和夫,斎藤篤,谷本普一,小林宏行ほか.慢性気道感染症に対す
る meropenem と imipenem/cilastatin sodium の薬効比較試験成績.Chemotherapy (Tokyo)
1992;40:1426-1450.
27) 原耕平,斎藤玲,武部和夫,斎藤篤,谷本普一,小林宏行ほか.細菌性肺炎に対する
meropenem と imipenem/cilastatin sodium の薬効比較試験成績.Chemotherapy (Tokyo)
1992;40:1343-1364.
28) 松本文夫,斎藤玲,渡辺彰,島田馨,谷本普一,小林宏行ほか.慢性気道感染症に対す
る biapenem と imipenem/cilastatin の臨床的有用性に関する比較試験.日本化学療法学会
雑誌 1995;43:63-84.
29) 松本文夫,斎藤玲,渡辺彰,島田馨,谷本普一,小林宏行ほか.細菌性肺炎に対する biapenem
と imipenem/cilastatin の臨床的有用性に関する比較試験.日本化学療法学会雑誌 1995;
43:41-62.
30) 松本文夫,渡辺彰,和田光一,小田切繁樹,松島敏春,大泉耕太郎ほか.下部呼吸器感
染症に対する biapenem と imipenem/cilastatin の薬効比較試験.日本化学療法学会雑誌
2000;48:45-67.
2.7 臨床概要
32) 河合伸,小林博之.緑膿菌感染症とバイオフィルム 1) 慢性呼吸器感染症.“緑膿菌の今
日的意味”
.斎藤厚,山口惠三編:医薬ジャーナル社;1996:180-190.
33) UTI 研究会 (代表 大越正秋).UTI 薬効評価基準 (第 3 版) 追補.Chemotherapy (Tokyo)
1991;39:894-933.
34) 馬場駿吉,宮本直哉,山本真一郎,加藤眞二,伊藤晴夫,横田明ほか.化膿性中耳炎に
対する Meropenem の基礎的・臨床的検討.耳鼻と臨床 1992;38:496-508.
35) 大山勝ほか.扁桃炎およびその他の耳鼻咽喉科感染症に対する Meropenem の基礎的・臨
床的検討.耳鼻と臨床 1992;38:524-537.
36) 椎木一雄,大野康亮,山根伸夫,金子明寛,小林寅喆,佐々木次郎.歯科・口腔外科領
域における抗菌薬の臨床評価基準の提案.歯科薬物療法 1998;17:95-122.
37) Knaus WA, Draper EA, Wagner DP, Zimmerman JE.APACHEⅡ:A severity of disease
classification system.Critical Care Medicine 1985;13:818-829.
38) Cometta A, Baumgartner JD, Lew D, Zimmerli W, Pittet D, Chopart P et al.Prospective
randomized comparison of imipenem monotherapy with imipenem plus netilmicin for treatment of
severe infections in nonneutropenic patients.Antimicrob Agents Chemother 1994;June:
1309-1313.
39) Beaucaire G.Clinical activity of cefepime in severe infections.Clin Microbiol Infect 1999;5:
S6-S14.
40) M.P.Fink, D.R.Snydman, M.S.Niderman, K.V.Leeper,Jr., R.H.Johnson, S.O.Heard et al.
Treatment of severe pneumonia in hospitalized patients: Results of a multicenter, randomized,
double-blind trial comparing intravenous ciprofloxacin with imipenem-cilastatin. Antimicrob.
Agents Chemother. 1994;38:547-557
41) F.A.Lerma, on behalf of the serious infection study group. Efficacy of meropenem as
monotherapy in the treatment of ventilator-associated pneumonia. J. Chemother. 2001 ;
13 :70-81
42) 成人院内肺炎診療の基本的考え方.日本呼吸器学会呼吸器感染症に関するガイドライン
作成委員会.呼吸器感染症に関するガイドライン:日本呼吸器学会;2002
2.7 臨床概要
2.7.6 個々の試験のまとめ
表 2.7.6-1 臨床試験一覧表 (その 1)
試験 区分 試験番号 試験の種類 登録症 例数 用法・用量 投与 期間 試験施設数 場所 治験期間 試験報告書 添付場所 R1411 初 期 忍 容 性 試験 10 例 被験薬:25 mg, 50 mg,125 mg, 250 mg,500 mg 単回点滴静注 単回 1 19 年 月 ∼19 年 月 5.3.3-Report01 R1412 単 回 投 与 試 験 24 例 被験薬:125 mg, 250 mg,500 mg, 1000 mg 単回点滴静注 単回 1 19 年 月 ∼19 年 月 5.3.3-Report02 R1414 反 復 投 与 試 験 8 例 被験薬:500 mg 1 日 2 回点滴静注 プラセボ 6 日間 (11 回) 1 19 年 月 5.3.3-Report03 R1417 高 用 量 反 復 投与試験 8 例 被験薬:1000 mg 1 日 2 回点滴静注 プラセボ 6 日間 (11 回) 1 19 年 月 ∼19 年 月 5.3.3-Report04 健 康 被 験 者 に お け る 薬 物 動 態 試 験 R1419 500 mg×3 回 投与試験 6 例 被験薬:500 mg 1 日 3 回点滴静注 1 日間 (3 回) 1 20 年 月 5.3.3-Report05 R142G 体液・組織内 濃 度 測 定 試 験 ( 泌 尿 器 科領域) 14 例 被験薬: 250 mg,500 mg 単回点滴静注 単回 4 他 19 年 月 ∼19 年 月 5.3.3-Report06 R142H 体液・組織内 濃 度 測 定 試 験 ( 産 婦 人 科領域) 13 例 被験薬:250 mg 単回点滴静注 単回 1 19 年 月 ∼19 年 月 5.3.3-Report07 R143P 体液・組織内 濃 度 測 定 試 験 ( 整 形 外 科領域) 11 例 被験薬:250 mg 単回点滴静注 単回 5 他 20 年 月 ∼20 年 月 5.3.3-Report08 R143Q 体液・組織内 濃 度 測 定 試 験 ( 耳 鼻 咽 喉科領域) 12 例 被験薬:250 mg 単回点滴静注 単回 4 他 20 年 月 ∼20 年 月 5.3.3-Report09 R143S 体液・組織内 濃 度 測 定 試 験 ( 皮 膚 科 領域) 10 例 被験薬:250 mg 単回点滴静注 単回 1 20 年 月 ∼20 年 月 5.3.3-Report10 R143T 体液・組織内 濃 度 測 定 試 験 ( 眼 科 領 域) 5 例 被験薬:250 mg 単回点滴静注 単回 1 20 年 月 ∼20 年 月 5.3.3-Report11 R143V 体液・組織内 濃 度 測 定 試 験 (歯科・口 腔外科領域) 10 例 被験薬:250 mg 単回点滴静注 単回 1 20 年 月 ∼20 年 月 5.3.3-Report12 患 者 に お け る 薬 物 動 態 試 験2.7 臨床概要
表 2.7.6-1 臨床試験一覧表 (その 2)
試験 区分 試験番号 試験の種類 登録症 例数 用法・用量 投与 期間 試験施設数 場所 治験期間 試験報告書 添付場所 R1415 高 齢 者 に お け る 薬 物 動 態試験 6 例 被験薬:250 mg 単回点滴静注 単回 1 20 年 月 5.3.3-Report14 内因性 要因を 検討し た薬物 動態試 験 R1418 腎 機 能 障 害 患 者 に お け る 薬 物 動 態 試験 12 例 被験薬:250 mg 単回点滴静注 単回 1 20 年 月 ∼20 年 月 5.3.3-Report15 外因性 要因を 検討し た薬物 動態試 験 R1416 プ ロ ベ ネ シ ド 併 用 時 の 薬 物 動 態 試 験 8 例 被験薬:250 mg 単回点滴静注 各 1 回 1 20 年 月 ∼20 年 月 5.3.3-Report16 R142R 呼 吸 器 感 染 症 に 対 す る 用 量 検 討 試 験 83 例 被験薬: 250 mg,500 mg 1 日 2 回点滴静 注 最長 14 日間 30 他 19 年 月 ∼19 年 月 5.3.5-Report01 R142U 複 雑 性 尿 路 感 染 症 に 対 す る 用 量 検 討試験 83 例 被験薬: 250 mg,500 mg 1 日 2 回点滴静 注 5 日間 32 他 19 年 月 ∼19 年 月 5.3.5-Report02 R143R 呼 吸 器 感 染 症 に 対 す る 比較試験 219 例 被験薬:250 mg 1 日 2 回点滴静 注 対照薬:(MEPM) 500 mg 1 日 2 回点滴静 注 7 日間 56 他 20 年 月 ∼20 年 月 5.3.5-Report03 比 較 対 照 試 験 R143U 複 雑 性 尿 路 感 染 症 に 対 す る 比 較 試 験 205 例 被験薬:250 mg 1 日 2 回点滴静 注 対照薬:(MEPM) 500 mg 1 日 2 回点滴静 注 5 日間 42 他 20 年 月 ∼20 年 月 5.3.5-Report04 R142A 一 般 臨 床 試 験 ( 内 科 領 域,泌尿器科 領域) 107 例 被験薬: 125 mg 1 日 2 回, 250 mg 1 日 2 回, 250 mg 1 日 3 回, 500 mg 1 日 2 回 点滴静注 3 日間 以上 14 日間 以内 41 他 19 年 月 ∼19 年 月 5.3.5-Report05 R142C 一 般 臨 床 試 験 ( 内 科 領 域) 114 例 被験薬: 250 mg 1 日 2 回, 250 mg 1 日 3 回, 500 mg 1 日 2 回 点滴静注 3 日間 以上 14 日間 以内 55 他 19 年 月 ∼19 年 月 5.3.5-Report06 非 対 照 試 験 R142D 一 般 臨 床 試 験 ( 泌 尿 器 科領域) 45 例 被験薬: 250 mg 1 日 2 回, 250 mg 1 日 3 回, 500 mg 1 日 2 回 点滴静注 3 日間 以上 14 日間 以内 27 他 19 年 月 ∼19 年 月 5.3.5-Report072.7 臨床概要
表 2.7.6-1 臨床試験一覧表 (その 3)
試験 区分 試験番号 試験の種類 登録症 例数 用法・用量 投与 期間 試験施設数 場所 治験期間 試験報告書 添付場所 R142E 一 般 臨 床 試 験 ( 外 科 領 域) 51 例 被験薬: 250 mg 1 日 2 回, 250 mg 1 日 3 回, 500 mg 1 日 2 回 点滴静注 3 日間 以上 14 日間 以内 25 他 19 年 月 ∼19 年 月 5.3.5-Report08 R142F 一 般 臨 床 試 験 ( 産 婦 人 科領域) 59 例 被験薬: 250 mg 1 日 2 回, 250 mg 1 日 3 回, 500 mg 1 日 2 回 点滴静注 3 日間 以上 14 日間 以内 19 他 19 年 月 ∼19 年 月 5.3.5-Report09 R143J 一 般 臨 床 試 験 ( 整 形 外 科領域) 8 例 被験薬: 250 mg 1 日 2 回, 500 mg 1 日 2 回 点滴静注 14 日間 以内 8 他 20 年 月 ∼20 年 月 5.3.5-Report10 R143K 一 般 臨 床 試 験 ( 耳 鼻 咽 喉科領域) 15 例 被験薬: 250 mg 1 日 2 回, 250 mg 1 日 3 回, 500 mg 1 日 2 回 点滴静注 7 日間 9 他 20 年 月 ∼20 年 月 5.3.5-Report11 R143L 一 般 臨 床 試 験 ( 皮 膚 科 領域) 22 例 被験薬: 250 mg 1 日 2 回, 500 mg 1 日 2 回 点滴静注 7 日間 以内 12 他 20 年 月 ∼20 年 月 5.3.5-Report12 R143M 一 般 臨 床 試 験 ( 眼 科 領 域) 15 例 被験薬: 250 mg 1 日 2 回, 250 mg 1 日 3 回, 500 mg 1 日 2 回 点滴静注 14 日間 以内 8 他 20 年 月 ∼20 年 月 5.3.5-Report13 R143N 一 般 臨 床 試 験 (歯科・口 腔外科領域) 24 例 被験薬: 250 mg 1 日 2 回, 250 mg 1 日 3 回, 500 mg 1 日 2 回 点滴静注 7 日間 以内 4 他 20 年 月 ∼20 年 月 5.3.5-Report14 R143E 一 般 臨 床 試 験 ( 外 科 領 域) 15 例 被験薬: 250 mg 1 日 2 回, 250 mg 1 日 3 回, 500 mg 1 日 2 回 点滴静注 14 日間 以内 7 他 20 年 月 ∼20 年 月 5.3.5-Report15 R143A 一 般 臨 床 試 験 [ 敗 血 症 (感染性心内 膜 炎 を 含 む)] 11 例 被験薬: 500 mg 1 日 2 回, 500 mg 1 日 3 回 点滴静注 14 日間 以内 (感染性 心内膜 炎のみ 28 日間 以内) 13 他 20 年 月 ∼20 年 月 5.3.5-Report16 非 対 照 試 験 R1431 一 般 臨 床 試 験 ( 院 内 肺 炎) 18 例 被験薬: 500 mg 1 日 2 回, 500 mg 1 日 3 回, 1000 mg 1 日 2 回 点滴静注 14 日間 以内 14 他 20 年 月 ∼20 年 月 5.3.5-Report172.7 臨床概要
2.7.6.1 健康被験者における薬物動態試験
2.7.6.1.1 第 1 相試験 (初期忍容性)
添付資料 5.3.3-Report01
試験方法の概略を表 2.7.6.1.1-1 に,検査・観察時期を図 2.7.6.1.1-1 に示した.
なお,初期忍容性試験における初回投与量は以下の事項を指標に 1 回 25 mg と設定した.
• 最も感受性の高い動物 (イヌ) の LD
50値 2000 mg/kg 以上の 1/600 以下.
• 最も感受性の高い動物 (イヌ) の 3 ヵ月反復投与毒性試験における無毒性量 100 mg/kg の
1/60 以下.
• 臨床期待用量 1 回 250 mg の 1/10 以下.
表 2.7.6.1.1-1 試験方法
項目 内容 目的 健康成人男性志願者における DRPM の初期忍容性を確認するとともに,併せて体内動態も 検討する. 治験方法 非盲検,非対照,非無作為化試験 単回投与試験 (投与量 25∼500 mg,60 分間点滴静注) 例数 25,50,125,250,500 mg 投与群,各 2 例 対象 健康成人男性 (年齢 24∼49 歳:体重 54∼80 kg) 被験薬,用量及び 投与方法,ロット 番号 被験薬:DRPM 用量及び投与方法:皮内反応検査を実施し,陰性であることを確認した後,DRPM 25∼500 mg を 1 日 1 回輸液ポンプを用いて点滴静注 (60 分間)した. ロット番号: (250 mg), (500 mg) 検査・観察項目 ・臨床所見:自覚症状,他覚所見及び理学所見 (心電図,肺機能,体温,呼吸数,血圧, 脈拍数) ・臨床検査:血液一般 (赤血球数,ヘモグロビン,ヘマトクリット,白血球数,白血球分 画,血小板数),血液生化学 (AST,ALT,Al-P,LAP,LDH,γ-GTP,総蛋白,アルブ ミン,A/G 比,TTT,ZTT,総ビリルビン,総コレステロール,トリグリセライド,BUN, 血清クレアチニン,尿酸,電解質〈Na,K,Cl,Ca,P〉,血糖,β2-ミクログロブリン), 尿 (α1-ミクログロブリン,β2-ミクログロブリン,NAG,尿中クレアチニン,電解質〈Na, K,Cl〉,比重,pH,ビリルビン,蛋白,糖,ケトン体,ウロビリノゲン,沈渣,潜血, クレアチニンクリアランス),その他 (直接クームス試験,プロトロンビン時間,プロト ロンビン活性値,活性化部分トロンボプラスチン時間) ・薬物濃度 (血液,尿) ・代謝物の検索 (血液,尿) 治験総括医師* 治験実施医療機関 治験期間 19 年 月 日 (最初の被験者の同意取得日) ∼19 年 月 日 (最後の被験者の事後診察,検査終了日) *:所属・役職は治験を実施した当時のもの2.7 臨床概要 :治験薬投与 (60 分間点滴静注) a) 500 mg 投与群のみ実施 b) 125 mg,250 mg,500 mg 投与時に実施 c) 2 時間ごと(8∼10hr,10∼12hr) の蓄尿 d) 12 時間 (12∼24hr) の蓄尿
図 2.7.6.1.1-1 検査・観察時期
試験・検査項目 投与 前日 投 与 日 1 日後 3 日後 7 日後 前 0 (hr) 1 1.5 2 3 4 6 8 12 24 宿泊拘束 DRPM 投与 (60 分点滴) 診察 ● ● ● ● ● ● ● ● ● ● 自覚症状確認 ● ● ● ● ● ● ● ● ● ● ● 体温・呼吸数 ● ● ● ● ● 血圧・脈拍 ● ● ● ● ● ● ● ● ● ● 忍 心電図 12 誘導 ● ● モニタリング 肺機能検査 ● ● 容 血液一般検査 ● ● ● ● 血液生化学検査 ● ● ● ● 血糖 ● ● ● ● 性 一般検査 ● ● ● ● 尿 pH ● ● ● ● 潜血反応 ● ● ● ● 直接クームス試験 ● ● ● ● クレアチニン・クリアランス ● ● 血液凝固系試験a) ● ● ● 濃 血中濃度 ● ● ● ● ● ●b) 度 尿中濃度 ● ● 蓄尿 ● 蓄尿 ● 蓄尿 ● 蓄尿 ●c) 蓄尿 ●d) 蓄尿 ● ●2.7 臨床概要
1) 症例構成
スクリーニングにより選定した治験薬投与候補者 11 例における症例構成を図 2.7.6.1.1-2
に示した.11 例全例,皮内反応検査が陰性であることを確認し,治験薬投与前の観察・検査
により最終選定した 10 例を被験者とした.なお,10 例全例治験を完了し,投与後の中止・
脱落例はなかった.
治験薬投与候補者 11 例 (皮内反応検査実施例) 予備被験者 1 例 被験者 (投与症例) 10 例 治験完了被験者 10 例 薬物動態評価対象例 10 例 安全性評価対象例 10 例図 2.7.6.1.1-2 症例構成
2) 安全性
10 例の被験者すべてに,自覚症状,他覚所見及び理学所見の異常は認められなかった.臨
床検査においては,ALT の軽度の上昇が 1 例に認められたが,7 日後には無処置で正常化し
た.被験者の生理的変動内とも考えられたが,DRPM との因果関係も否定し得なかった.そ
の他の異常所見は認められなかった.
3) 薬物動態
DRPM 25 mg,50 mg,125 mg,250 mg,500 mg (各投与量 2 例ずつ) を健康成人男性に 60
分かけて点滴静注した時,実測値から求めた Cmax 及び実測値を用いて台形法により算出し
た投与開始から最終測定時刻までの AUC は投与量に比例した.また,2-コンパートメントモ
デルで解析して得られた t
1/2(
β) は投与量によらず約 1 時間でほぼ一定で,投与開始からの未
変化体の累積尿中排泄率は平均値として約 70%であった.(2.7.2.2.1.1.1 項)
4) 活性代謝物の検索
500 mg 投与時の血漿試料及び尿試料について,DRPM 活性代謝物の検索を TLC-バイオオー
トグラフィーにて行った.血漿中及び尿中には,DRPM に由来する抗菌活性のスポットのみ
2.7 臨床概要
であり,活性代謝物は検出されなかった.
5) 結論
忍容性については,250 mg 投与の 1 例で投与 1 日後,ALT の軽度の上昇が認められ,DRPM
との因果関係が疑われた.7 日後には無処置で正常化した.その他,DRPM に起因する自覚症
状,他覚所見及び理学所見に関して異常所見は認められず,500 mg までの忍容性に問題のな
いことが示唆された.
一方,薬物動態については,Cmax 及び AUC ともおおむね投与量に比例していることが示
唆され,また DRPM の累積尿中排泄率は投与量に関係なく約 70%でほぼ一定であった.血漿
及び尿中には,活性代謝物は検出されなかった.
以上より,DRPM の忍容性及び薬物動態がおおむね確認できたことから,単回投与試験 (用
量相関性) への移行は可能と考えた.
2.7 臨床概要
2.7.6.1.2 第 1 相試験 (単回投与)
添付資料 5.3.3-Report02
試験方法の概略を表 2.7.6.1.2-1 に,検査・観察時期を図 2.7.6.1.2-1 に示した.
表 2.7.6.1.2-1 試験方法
項目 内容 目的 健康成人男性志願者における DRPM 125,250,500,1000 mg 単回投与時における忍容性 と薬物動態を検討し,用量相関性を確認するとともに,血清蛋白結合率,及び代謝物を検 討する. 治験方法 非盲検,非対照,非無作為化試験 単回投与試験 (投与量 125∼1000 mg,30 分間点滴静注) 例数 125,250,500,1000 mg 投与群,各 6 例 対象 健康成人男性 (年齢 26∼45 歳:体重 58.5∼81 kg) 被験薬,用量及び 投与方法,ロット 番号 被験薬:DRPM 用量及び投与方法:皮内反応検査を実施し,陰性であることを確認した後,DRPM 125∼ 1000 mg を 1 日 1 回輸液ポンプを用いて点滴静注 (30 分間)した. ロット番号: (250 mg), (500 mg) 検査・観察項目 ・臨床所見:自覚症状,他覚所見及び理学所見 (心電図,肺機能,体温,呼吸数,血圧, 脈拍数) ・臨床検査:血液一般 (赤血球数,ヘモグロビン,ヘマトクリット,白血球数,白血球分 画,血小板数),血液生化学 (AST,ALT,Al-P,LAP,LDH,γ-GTP,総蛋白,アルブ ミン,A/G 比,TTT,ZTT,総ビリルビン,総コレステロール,トリグリセライド,BUN, 血清クレアチニン,尿酸,電解質〈Na,K,Cl,Ca,P〉,血糖,β2-ミクログロブリン), 尿 (α1-ミクログロブリン,β2-ミクログロブリン,NAG,尿中クレアチニン,電解質〈Na, K,Cl〉,pH,ビリルビン,蛋白,糖,ケトン体,ウロビリノゲン,沈渣,潜血,クレ アチニンクリアランス),その他 (直接クームス試験,プロトロンビン時間,プロトロン ビン活性値,活性化部分トロンボプラスチン時間) ・薬物濃度 (血液,尿) ・代謝物の検索 (血液,尿) 治験総括医師* 治験実施医療機関 治験期間 19 年 月 日 (最初の被験者の同意取得日) ∼19 年 月 日 (最後の被験者の事後診察,検査終了日) *:所属・役職は治験を実施した当時のもの2.7 臨床概要 :DRPM 投与 (30 分間点滴) a) 500 mg,1000 mg 投与時に実施 b) 2 時間ごと (8∼10hr,10∼12hr) の蓄尿 c)12 時間 (12∼24hr)の蓄尿
図 2.7.6.1.2-1 検査・観察時期
試験・検査項目 投与 前日 投 与 日 1 日後 3 日後 7 日後 前 0 (hr) 0.5 0.75 1 1.5 2 3 4 6 8 12 24 宿泊拘束 DRPM 投与 (30 分点滴) 診察 ● ● ● ● ● ● ● ● ● ● ● 自覚症状確認 ● ● ● ● ● ● ● ● ● ● ● ● 体温・呼吸数 ● ● ● ● ● 血圧・脈拍 ● ● ● ● ● ● ● ● ● ● ● 忍 心電図 12 誘導 ● ● モニタリング 肺機能検査 ● ● 容 血液一般検査 ● ● ● ● 血液生化学検査 ● ● ● ● 血糖 ● ● ● ● 性 一般検査 ● ● ● ● 尿 pH ● ● ● ● 潜血反応 ● ● ● ● 直接クームス試験 ● ● クレアチニン・クリアランス ● ● 血液凝固系試験a) ● ● ● 濃 血中濃度 ● ● ● ● ● ● ● ● ● ● 度 尿中濃度 ● 24hr 蓄尿 ● 蓄尿 ● 蓄尿 ● 蓄尿 ● 蓄尿 ●b) 蓄尿 ●c) 蓄尿 血清蛋白結合率 ● ● 血中代謝物 ● ● ● ● ● ●2.7 臨床概要
1) 症例構成
スクリーニングにより選定した治験薬投与候補者 25 例における症例構成を図 2.7.6.1.2-2
に示した.25 例全例,皮内反応検査が陰性であることを確認し,治験薬投与前の観察・検査
により最終選定した 24 例を被験者とした.なお,24 例全例治験を完了し,投与後の中止・
脱落例はなかった.
治験薬投与候補者 25 例 (皮内反応検査実施例) 予備被験者 1 例 被験者 (投与症例) 24 例 治験完了被験者 24 例 薬物動態評価対象例 24 例 安全性評価対象例 24 例図 2.7.6.1.2-2 症例構成
2) 安全性
24 例全例に自覚症状,他覚所見及び理学所見に関して,異常所見は認められなかった.
臨床検査値に関しては,3 例において軽度の変動 (尿潜血陽性,
γ-GTP 上昇,AST 上昇,白
血球増多) がみられたが,DRPM に起因すると考えられる異常変動は認められなかった.
3) 薬物動態
健康成人男性に DRPM 125,250,500,1000 mg を 30 分かけて単回点滴静注したとき,Cmax
及び AUC は投与量に対し比例性を示した.t
1/2(
β) は投与量に依存せず,また,投与開始から
24 時間までの累積尿中排泄率は投与量に依存せずほぼ一定で,DRPM の体内動態は 1000 mg
までの投与量範囲で線形であることが示された.24 時間までの DRPM 未変化体の累積尿中排
泄率は平均値として約 75%で,DRPM が腎排泄型の薬物であることが示された.主代謝物であ
るドリペネムジカルボン酸体 (DRPM-DC) を含めた累積尿中排泄率は,平均値として約 90%
であった.(2.7.2.2.1.1.1 項)
4) 活性代謝物の検索
DRPM 1000 mg 投与時の血漿試料及び尿試料について,DRPM 活性代謝物の検索を TLC-バ
2.7 臨床概要
イオオートグラフィーにて行った.血漿中及び尿中には,DRPM に由来する抗菌活性のスポッ
トのみであり,活性代謝物は検出されなかった.
5) 血漿及び尿中代謝物の検討
DRPM 単回投与後の血漿中及び尿中主要代謝物 DRPM-DC の検討を HPLC 法で行った.
血漿中の代謝物濃度は,投与量にほぼ比例して増加したが,その量はわずかであった.一
方,尿中への 24 時間までの代謝物の累積排泄率 (平均) は,125 mg 投与で 12.7%,250 mg 投
与で 12.1%,500 mg 投与で 15.5%,1000 mg 投与で 17.2%であった (表 2.7.2-2).
測定時間が異なるため正確な比較はできないものの,DRPM-DC の AUC の未変化体 AUC
に対する比は,投与量 250 mg では約 4%,500 mg では約 6%,1000 mg では約 7%であった.な
お,投与量 125 mg では DRPM-DC 濃度のほとんどが定量限界未満であった.DRPM-DC の
Cmax の未変化体 Cmax に対する比は,投与量 250 mg では約 3%,投与量 500 mg では約 3%,
投与量 1000 mg では約 4%であった.また,DRPM 未変化体と主代謝物である DRPM-DC を併
せた 24 時間までの累積尿中排泄率はいずれの投与量においても約 90%であった (表 2.7.2-2∼
3).
6) 血清蛋白結合率
健康成人男性における単回投与試験において,ex vivo で限外ろ過法により血清蛋白結合率
を測定した結果,2.52∼10.8%と被験者間でばらつきが見られたものの,血清蛋白結合率は低
かった.(2.7.2.2.1.2 項)
7) 結論
自覚症状,他覚所見等に DRPM に起因すると考えられる異常所見は認められず,1000 mg
までの忍容性に問題ないことが確認された.
薬物動態については,Cmax と AUC は投与量に比例し,125 mg から 1000 mg の範囲内で線
形であることが示唆された.未変化体の累積尿中排泄率 (平均) は 125 mg から 1000 mg まで
のいずれの投与量でもほぼ一定であり,24 時間までの平均累積尿中排泄率は約 75%であった.
また,DRPM 投与後の代謝物に関し,活性代謝物については血漿及び尿ともに検出されな
かった.一方,DRPM-DC が血漿中及び尿中に検出されたが,その AUC の未変化体 AUC に
対する比は,
投与量 250 mg では約 4%,
500 mg では約 6%,1000 mg では約 7%であった.DRPM-DC
の Cmax の未変化体 Cmax に対する比は,投与量 250 mg では約 3%,投与量 500 mg では約 3%,
投与量 1000 mg では約 4%であった.未変化体 (DRPM) と DRPM-DC を合わせた累積尿中排泄
率はいずれの投与量においても約 90%であった.
血清蛋白結合率は,125∼1000 mg のいずれの投与量,採取時間 (投与開始後 30 分及び 2 時
間) においても低く,各被験者における値は 2.52∼10.8%であった.
以上より,DRPM の 125∼1000 mg 単回投与において,忍容性に問題のないことが確認され,
2.7 臨床概要
2.7.6.1.3 第 1 相試験 (反復投与)
添付資料 5.3.3-Report03
試験方法の概略を表 2.7.6.1.3-1 に,検査・観察時期を図 2.7.6.1.3-1 に示した.
表 2.7.6.1.3-1 試験方法
項目 内容 目的 健康成人男性志願者において DRPM 500 mg を反復投与し,本剤の安全性をプラセボ (生 理食塩液) 投与群と比較するとともに薬物動態の検討を行う.また,併せて腸内細菌叢及 び中咽頭内細菌叢の変動,血清蛋白結合率の検討も行う. 治験方法 単盲検,プラセボ対照,無作為化試験 反復投与試験 (投与量:1 回 500 mg 12 時間ごと計 11 回,30 分間点滴静注) 例数 8 例 (うちプラセボ 2 例) 対象 健康成人男性 (年齢 24∼44 歳:体重 55∼71 kg) 被験薬,用量及び 投与方法,ロット 番号 被験薬:DRPM 用量及び投与方法:皮内反応検査を実施し,陰性であることを確認した後,DRPM 500 mg を 1 日 2 回輸液ポンプを用いて点滴静注 (30 分間)した.なお,投薬は 単盲検にて 6 日間 (11 回) 連続で行った. ロット番号: 検査・観察項目 ・臨床所見:自覚症状,他覚所見及び理学所見 (心電図,脳波,肺機能,体温,呼吸数, 血圧,脈拍数) ・臨床検査:血液一般 (赤血球数,ヘモグロビン,ヘマトクリット,白血球数,白血球分 画,血小板数),血液生化学 (AST,ALT,Al-P,LAP,LDH,γ-GTP,総蛋白,アルブ ミン,A/G 比,TTT,ZTT,総ビリルビン,総コレステロール,トリグリセライド,BUN, 血清クレアチニン,尿酸,電解質〈Na,K,Cl,Ca,P〉,血糖,β2-ミクログロブリン), 尿 (α1-ミクログロブリン,β2-ミクログロブリン,NAG,尿中クレアチニン,電解質〈Na, K,Cl〉,pH,ビリルビン,蛋白,糖,ケトン体,ウロビリノゲン,沈渣,潜血,クレ アチニンクリアランス),その他(直接クームス試験,プロトロンビン時間,プロトロン ビン活性値,活性化部分トロンボプラスチン時間),便 (潜血反応) ・細菌叢 (腸内,中咽頭) ・薬物濃度 (血液,尿,糞便) 治験総括医師* 治験実施医療機関 治験期間 19 年 月 日 (最初の被験者の同意取得日) ∼19 年 月 日 (最後の被験者の事後診察,検査終了日) *:所属・役職は治験を実施した当時のもの:DRPM 投与 (30 分間点滴) *:聴・打診,触診,腱反射は初回投与前及び投与終了 1 日後に実施する. ◎:5 回目投与時に実施
図 2.7.6.1.3-1 検査・観察時期
投与開始日 投与 2∼5 日目 投与 6 日目 投与終了後 試験・検査項目 1日 3日 7日 投 与 前 々 日 投 与 前 日 前 0 0.5 1 1.5 2 3 4 6 8 10 12 0 0.5 前 0 0.5 2 前 0 0.5 前 0 0.5 1 1.5 2 3 4 6 8 10 12 24 36 宿泊拘束 DRPM 投与(30 分点滴) 診察* ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● 自覚症状確認 ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● 体温・呼吸数 ● ● ● ● ● ● ● ● ● 血圧・脈拍 ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● 心電図 12 誘導 ● ● ◎ ● 忍 モニタリング 脳波 ● ◎ 肺機能検査 ● ◎ ● 容 血液一般検査 ● ◎ ● ● ● ● 血液生化学検査 ● ◎ ● ● ● ● 血糖 ● ◎ ● ● ● ● 性 一般検査 ● ◎ ● ● ● ● 尿 pH ● ◎ ● ● ● ● 潜血反応 ● ◎ ● ● ● ● 直接クームス試験 ● ● クレアチニン・クリアランス ● ◎ ● 血液凝固系試験 ● ● ● 便 (潜血反応) ● ● 3∼4 日目 ● 濃 血中濃度 ● ● ● ● ● ● ● ● ● ◎ ◎ ◎ ● ● ● ● ● ● ● ● ● 度 尿中濃度 ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● 蓄尿 蓄尿 蓄尿 蓄尿 蓄尿 蓄尿 蓄尿 蓄尿 蓄尿 蓄尿 血清蛋白結合率 ● ● ● ● 腸内細菌叢 ● ● 3∼4 日目 ● ● 中咽頭内細菌叢 ● ● ◎ ● ● ● 糞便中濃度 ● ● 3∼4 日目 ●1) 症例構成
スクリーニングにより選定した治験薬投与候補者 9 例における症例構成を図 2.7.6.1.3-2 に
示した.9 例全例,皮内反応検査が陰性であることを確認し,治験薬投与前の観察・検査に
より最終選定した 8 例を被験者とした.なお,8 例全例治験を完了し,投与後の中止・脱落
例はなかった.
治験薬投与候補者 9 例 (皮内反応検査実施例) 予備被験者 1 例 被験者 (投与症例) 8 例 治験完了被験者 8 例 薬物動態評価対象例 8 例 安全性評価対象例 8 例図 2.7.6.1.3-2 症例構成
2) 安全性
DRPM 500 mg を 1 日 2 回 6 日間 (計 11 回) 反復投与 (30 分間点滴静注) したとき,自覚症
状,他覚所見及び理学所見に関して特に問題となる異常所見は認められなかった.臨床検査
値に関しては,DRPM 群の 1 例において軽度の変動 (赤血球数減少,ヘモグロビン減少) が認
められたが,DRPM に起因すると考えられる異常変動は認められなかった.
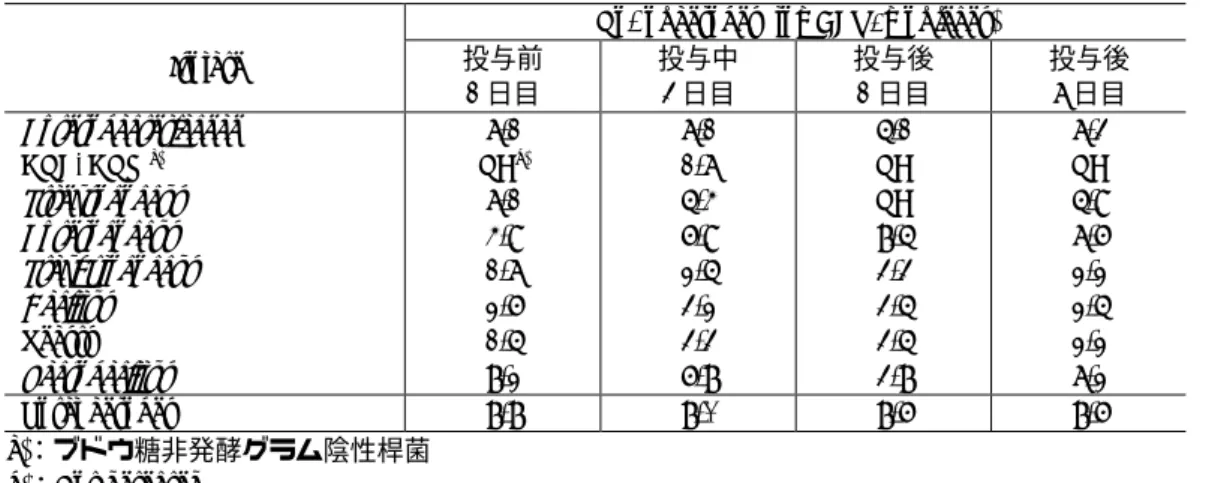
3) 腸内細菌叢の変動
DRPM 投与群の腸内細菌叢の変動を表 2.7.6.1.3-2 及び表 2.7.6.1.3-3 に示した.
腸 内 細 菌 叢 は DRPM の 反 復 投 与 に よ り 一 過 性 に
Enterobacteriaceae
,
Lactobacillus
,
Streptococcus
は減少し,
Enterococcus
は増加したが,投与終了後 7 日目 には投与前の菌数
にほぼ回復した.
C. difficile
は DRPM 投与群のうち投与前の検体から既に検出されていた 1
例を含む合計 4 例から検出された.ただし,全糞便検体共に肉眼的観察において下痢などの
異常所見は認められなかった.
なお,好気性菌及び嫌気性菌の総菌数ではいずれも投与期間中及び投与終了後を通じて大
きな変動は認められなかった.
表 2.7.6.1.3-2 腸内細菌叢の変動 (好気性菌)
No. of aerobes (log CFU/g of feces)
Isolate 投与前 1 日目 投与中 3 日目 投与後 1 日目 投与後 7 日目 Enterobacteriaceae GNF-GNR a) Streptococcus Enterococcus Staphylococcus Bacillus Yeasts Lactobacillus 7.1 NDb) 7.1 4.9 1.7 2.6 1.5 8.2 7.1 1.7 5.4 6.9 2.5 3.2 3.3 6.8 5.1 ND ND 8.5 3.3 3.5 3.5 3.8 7.3 ND 5.9 7.6 2.2 2.5 2.2 7.2 Total aerobes 8.8 8.0 8.6 8.6 a):ブドウ糖非発酵グラム陰性桿菌 b):Not detected
表 2.7.6.1.3-3 腸内細菌叢の変動 (嫌気性菌)
No. of anaerobes (log CFU/g of feces)
Isolate 投与前 1 日目 投与中 3 日目 投与後 1 日目 投与後 7 日目 Bacteroides Fusobacterium Bifidobacterium Eubacterium Lactobacillus Clostridium Peptococcaceae Veillonellaceae 10.1 8.9 10.3 9.6 8.7 8.0 9.0 1.2 9.5 7.3 9.7 9.3 4.1 7.2 8.4 1.1 8.9 7.6 9.5 8.3 NDa) 5.9 7.3 1.1 9.5 8.0 9.8 8.4 6.8 6.5 7.5 2.0 Total anaerobes 10.7 10.2 9.9 10.4 a):Not detected
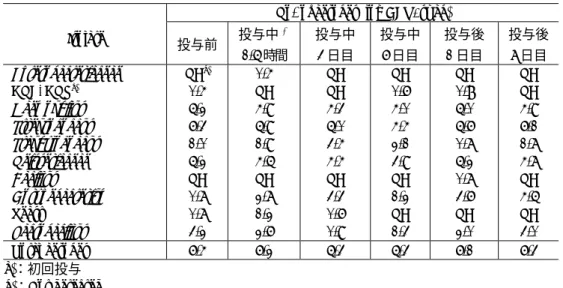
4) 中咽頭内細菌叢の変動
DRPM 投与群の中咽頭内細菌叢の変動を表 2.7.6.1.3-4 及び表 2.7.6.1.3-5 に示した.
中咽頭内細菌叢については DRPM の反復投与により,好気性菌では
Streptococcus
,
Haemophilus
,
Neisseriaceae
の菌数が投与中にわずかに減少し,嫌気性菌では
Bacteroides
,
Fusobacterium
,
Actinomyces
,
Streptococcus
,
Veillonella
の菌数が投与中に減少したが,共に
投与終了後にはほぼ回復した.また,総菌数では好気性菌,嫌気性菌とも投与中 3 日目,6
日目にわずかに減少傾向を示したが,投与終了後にはほぼ回復した.
表 2.7.6.1.3-4 中咽頭内細菌叢の変動 (好気性菌)
No. of aerobes (log CFU/swab) Isolate 投与前 投与中 a) 1.5 時間 投与中 3 日目 投与中 6 日目 投与後 1 日目 投与後 7 日目 Enterobacteriaceae GNF-GNRc) Haemophilus Streptococcus Staphylococcus Neisseriaceae Bacillus Corynebacterium Yeast Lactobacillus NDb) 0.4 5.2 6.3 1.0 5.2 ND 0.7 0.7 3.2 0.4 ND 4.9 5.9 1.9 4.5 ND 2.7 1.2 2.6 ND ND 4.3 5.0 3.4 4.4 ND 3.3 0.6 0.9 ND 0.6 4.0 4.4 2.1 3.9 ND 1.2 ND 1.3 ND 0.8 5.0 5.6 0.7 5.2 0.7 3.6 ND 2.0 ND ND 4.9 6.1 1.7 4.7 ND 4.5 ND 3.0 Total aerobes 6.4 6.2 5.3 5.3 6.1 6.3 a):初回投与 b):Not detected c):ブドウ糖非発酵グラム陰性桿菌
表 2.7.6.1.3-5 中咽頭内細菌叢の変動 (嫌気性菌)
No. of anaerobes (log CFU/swab) Isolate 投与前 投与中a) 1.5 時間 投与中 3 日目 投与中 6 日目 投与後 1 日目 投与後 7 日目 Bacteroides Fusobacterium Actinomyces Bifidobacterium Propionibacterium Eubacterium Peptostreptococcus Streptococcus Veillonella 5.3 3.8 4.2 0.5 NDb) 1.4 3.7 4.8 6.0 5.2 4.8 4.6 0.5 ND 1.8 3.7 5.2 5.4 2.4 2.5 3.1 0.4 ND ND ND 0.4 1.9 1.8 2.5 3.6 1.0 ND ND 0.8 3.1 1.9 3.4 4.5 4.0 0.4 1.0 ND 2.2 3.6 5.2 4.3 3.4 3.8 1.9 ND ND 1.8 3.1 5.4 Total anaerobes 6.5 6.1 4.1 5.1 6.2 5.8 a):初回投与 b):Not detected
5) 薬物動態
健康成人男性に DRPM 500 mg を 1 日 2 回 6 日間 (6 日目は 1 回投与,計 11 回) 30 分かけて
反復点滴静注したとき,DRPM の体内動態は反復投与によりほとんど変化しなかった.
(2.7.2.2.1.1.2 項)
6) 血清蛋白結合率
健康成人男性における 500 mg 1 日 2 回 6 日間反復投与試験において,同様に測定した血清
蛋白結合率は,3.55∼12.7%と被験者間でばらつきが見られたものの,小さな値であり,反復
投与による血清蛋白結合率への影響は認められなかった.(2.7.2.2.1.2 項)
7) 糞便中濃度と
β-ラクタマーゼ活性
投与前,投与 3 日目,投与終了 1 日後に糞便中の DRPM 濃度測定を行ったが,いずれの測
定時期においても糞便中の DRPM 濃度は検出限界 (0.15 µg/g) 以下であった (表 2.7.6.1.3.-6).
また,糞便中の
β-ラクタマーゼ活性は個体差が著しく,2 例において投与終了 1 日後に活性
が上昇していたが,その他の例では投与前,投与中,投与終了 1 日後の各測定値に差異がな
かった (表 2.7.6.1.3-7).
表 2.7.6.1.3-6 DRPM 反復投与時における糞便中薬物濃度
被験者 Fecal concentraions (µg/g)№ Before Day 3 Day 7
(投与終了 1 日後) 1 <0.15 <0.15 <0.15 2 <0.15 <0.15 <0.15 3 <0.15 <0.15 <0.15 4 <0.15 <0.15 <0.15 5a) <0.15 <0.15 <0.15 6 <0.15 <0.15 <0.15 7a) <0.15 <0.15 <0.15 8 <0.15 <0.15 <0.15 a):Placebo 群
表 2.7.6.1.3-7 DRPM 反復投与時における糞便中
β−lactamase 活性
被験者 β−lactamase activity№ Before Day 3 Day 7
(投与終了 1 日後) 1 5 3 2 2 1 1 0 3 5 4 5 4 0 0 0 5a) 3 3 5 6 1 2 4 7a) 4 5 5 8 2 2 4
注) β−lactamase 活性は Nitrocefin を変色させる糞便懸濁液の最大希釈倍数 (2E-n)で表した a):Placebo 群
8) 結論
自覚症状,他覚所見,理学所見及び臨床検査値に特に問題となる異常所見は認められず,
腸内細菌叢,中咽頭内細菌叢にもほとんど影響がみられず,反復投与における安全性が確認
された.また,反復投与での薬物動態が確認され,初回投与,最終回投与で Cmax や投与開
始から 12 時間までの累積尿中排泄率に大きな変動はなく,DRPM の反復投与による蓄積傾向
は認められなかった.以上より,第 2 相臨床試験への移行は可能であると考えた.
2.7.6.1.4 第 1 相試験 (高用量反復投与)
添付資料 5.3.3-Report04
試験方法の概略を表 2.7.6.1.4-1 に,検査・観察時期を図 2.7.6.1.4-1 に示した.
表 2.7.6.1.4-1 試験方法
項目 内容 目的 健康成人男性志願者において DRPM 1000 mg を反復投与し,安全性を確認し,併せて薬 物動態の検討を行う. 治験方法 単盲検,プラセボ対照,無作為化試験 反復投与試験 (投与量:1 回 1000 mg 12 時間ごと計 11 回,30 分間点滴静注) 例数 8 例 (うちプラセボ 2 例) 対象 健康成人男性 (年齢 25∼41 歳:体重 57∼67.8 kg) 被験薬,用量及び 投与方法,ロット 番号 被験薬:DRPM 用量及び投与方法:皮内反応検査を実施し,陰性であることを確認した後,DRPM 1000 mg を 1 日 2 回輸液ポンプを用いて点滴静注 (30 分間)した.なお,投薬は 単盲検にて 6 日間 (11 回) 連続で行った. ロット番号: (500 mg), (皮内反応検査薬) 検査・観察項目 ・臨床所見:自覚症状,他覚所見及び理学所見 (心電図,脳波,肺機能,体温,呼吸数, 血圧,脈拍数) ・臨床検査:血液一般 (赤血球数,ヘモグロビン,ヘマトクリット,白血球数,白血球分 画,血小板数),血液生化学 (AST,ALT,Al-P,LAP,LDH,γ-GTP,総蛋白,アルブ ミン,A/G 比,TTT,ZTT,総ビリルビン,総コレステロール,トリグリセライド,BUN, 血清クレアチニン,尿酸,電解質〈Na,K,Cl,Ca,P〉,血糖,β2-ミクログロブリン), 尿 (α1-ミクログロブリン,β2-ミクログロブリン,NAG,尿中クレアチニン,電解質〈Na, K,Cl〉,pH,ビリルビン,蛋白,糖,ケトン体,ウロビリノゲン,沈渣,潜血,クレ アチニンクリアランス),その他 (CRP 直接クームス試験,プロトロンビン時間,プロト ロンビン活性値,活性化部分トロンボプラスチン時間,便潜血) ・腸内細菌叢 ・薬物濃度 (血液,尿,糞便) 治験総括医師* 治験実施医療機関 治験期間 19 年 月 日 (最初の被験者の同意取得日) ∼19 年 月 日 (最後の被験者の事後診察,検査終了日) *:所属・役職は治験を実施した当時のもの観察日 前々 日 前日 1 日目 2 日目 3 日目 4 日目 5 日目 6 日目 7 日 目 12 日 目 13 日 目 DRPM 投与 1 2 3 4 5 6 7 8 9 10 11 回目 宿泊拘束 入院 退院 入院 退院 皮内反応 ○ 自覚症状 診察 ○ 体温,呼吸数 ○ 血圧,脈拍数 ○ 心電図 ○ ○ ○ 脳波 ○ ○ ○ 肺機能検査 ○ ○ ○ 血液 血液一般検査 ○ ○ ○ ○ ○ 血液生化学検査 ○ ○ ○ ○ ○ CRP ○ ○ ○ ○ 直接クームス試験 ○ ○ 血液凝固系試験 ○ ○ 血中薬物濃度 ○ ○ ○ 血清蛋白結合率 ○ ○ 尿 一般検査 ○ ○ ○ ○ pH ○ ○ ○ ○ 潜血反応 ○ ○ ○ ○ 尿ウロビリノゲン ○ 薬物尿中濃度 (蓄尿) 糞便 便(潜血反応) ○ ○ ○ 腸内細菌叢 ○ ○ ○ ○ 糞便中濃度,β−ラ クタマーゼ活性 ○ ○ ○ :期間内実施, :期間内の規定の時間に実施, ○ :期間内に 1 回採取
図 2.7.6.1.4-1 検査・検査時期
1) 症例構成
スクリーニングにより選定した治験薬投与候補者 10 例における症例構成を図 2.7.6.1.4-2
に示した.10 例全例,皮内反応検査は陰性であった.治験薬投与前の観察・検査にて,1 例
は胃あるいは十二指腸潰瘍が疑われたため,もう 1 例は入院後咳・痰がみられ他の被験者を
選択したため除外とした.被験者として選定された 8 例は全例治験を完了し,投与後の中止・
脱落例はなかった.
治験薬投与候補者 10 例 (皮内反応検査実施例) 除外 2 例 被験者 (投与症例) 8 例 治験完了被験者 8 例 薬物動態評価対象例 8 例 安全性評価対象例 8 例図 2.7.6.1.4-2 症例構成
2) 安全性
DRPM 投与群で認められた有害事象のうち自他覚所見は,口内炎及び軟便各 1 例であり,
いずれも軽度で,DRPM との因果関係は可能性小であり,DRPM の副作用とされた.対照群
では動悸,めまいの自他覚所見が認められた.
臨床検査については,AST 上昇,ALT 上昇を示した 1 例,ALT 上昇を示した 1 例,ALT 上
昇,LDH 上昇を示した 1 例の計 3 例を有害事象として取り上げ,因果関係は可能性大であり,
DRPM の副作用とされたが,いずれの検査項目についても速やかに正常域に復した.その他
の異常値を示した臨床検査値は生理学的変動範囲内の変化であり,臨床的に問題となるもの
はなかった.
血圧,脈拍数,体温,呼吸数のバイタルサイン及び体重においては DRPM に起因する異常
変動は認められなかった.また,心電図においても DRPM による影響は認められなかった.
3) 腸内細菌叢
DRPM 投与による腸内細菌叢への影響について,各菌種の生菌数変化を調べ検討した.
好気性菌では,投与により
Enterobacteriaceae
は減少し
Enterococcus
は増加したが,投与終
了 7 日目には各々投与前の菌数にほぼ回復した.総好気性菌数の変動はほとんど認められな
かった.
嫌気性菌については,
Bacteroides
,
Bifidobacterium
等の大半の菌種の生菌数及び総嫌気性菌
数の変動がほとんど認められなかった.
C. difficile
については,いずれの検体からも検出され
ず,また全糞便検体とも下痢のような異常は認められなかった.
以上の結果より,DRPM 1 回 1000 mg 1 日 2 回反復投与による腸内細菌叢に及ぼす大きな影
響はないものと判断された.
4) 薬物動態
健康成人男性に DRPM 1000 mg を 1 日 2 回 6 日間 (6 日目は 1 回投与,計 11 回) 30 分かけ
て反復点滴静注したとき,DRPM の体内動態は反復投与によりほとんど変化しなかった.
(2.7.2.2.1.1.2 項)
5) 血清蛋白結合率
1000 mg 1 日 2 回 6 日間反復投与試験における血清蛋白結合率は,1.5∼23.8%と被験者間で
ばらつきが見られたものの,小さな値であり,血漿中濃度推移に影響を示すような差は認め
られなかった.(2.7.2.2.1.2 項)
6) 糞便中薬物濃度と
β-ラクタマーゼ活性
糞便中の DRPM 濃度は,投薬前,投薬中 (投与 3∼4 日目),投薬後 (投与 6∼7 日目) のい
ずれにおいても,検出限界 (0.15 µg/g) 以下であった.
糞便中の
β-ラクタマーゼ活性は個人差がみられ,被験薬群,対照群に関係なく,投薬前,
投薬中あるいは投薬後に上昇あるいは低下する場合がみられた.
7) 結論
自覚症状,他覚所見,理学所見及び臨床検査値に特に問題となる異常所見は認められず,
腸内細菌叢に対する影響は少なかったことから,DRPM 高用量の反復投与時 (1 回 1000 mg 1
日 2 回) における安全性が確認された.また,体内動態に変化はみられず,また蓄積性はな
いものと考えられた.
2.7.6.1.5 第 1 相試験 (500 mg×3 回投与)
添付資料 5.3.3-Report05
試験方法の概略を表 2.7.6.1.5-1 に,検査・観察時期を図 2.7.6.1.5-1 に示した.
表 2.7.6.1.5-1 試験方法
項目 内容 目的 健康成人男性志願者において DRPM を 1 回 500 mg,1 日 3 回投与した時の朝投与時と夕投 与時の薬物動態を比較検討し,1 日 3 回投与時における DRPM の薬物動態を確認する. 治験方法 非盲検,非対照,非無作為化試験 反復投与試験 (投与量:1 回 500 mg 朝・昼・夕に 6 時間間隔で計 3 回,30 分間点滴静注) 例数 6 例 対象 健康成人男性 (年齢 21∼25 歳:体重 53.2∼70.0 kg) 被験薬,用量及び 投与方法,ロット 番号 被験薬:DRPM 用量及び投与方法:皮内反応検査を実施し,陰性であることを確認した後,DRPM 500 mg を 1 日 3 回輸液ポンプを用いて点滴静注 (30 分間)した.なお,投薬は 3 回連続で行った. ロット番号: (500 mg), (皮内反応検査薬) 検査・観察項目 ・臨床所見:自覚症状,他覚所見及び理学所見 (体温,呼吸数,血圧,脈拍数) ・臨床検査:血液一般 (赤血球数,ヘモグロビン,ヘマトクリット,白血球数,白血球分 画,血小板数),血液生化学 (AST,ALT,Al-P,LAP,LDH,γ-GTP,総ビリルビン, BUN,血清クレアチニン,電解質〈Na,K,Cl〉),尿 (蛋白,糖,ウロビリノゲン,沈 渣※),その他(CRP) ※:尿蛋白が陽性の場合にのみ実施. ・薬物濃度 (血液,尿) 医学専門家* 治験実施医療機関 治験期間 20 年 月 日 (最初の被験者の同意取得日) ∼20 年 月 日 (最後の被験者の事後診察,検査終了日) * :所属・役職は治験を実施した当時のもの被験者選択期 治験薬投与期 終了期 事後 診察期 項 目 投与 2 週前∼ 投与 1 週前 投与 1 日前 投与前 (投与日) 朝 投与時 昼 投与時 夕 投与時 投与 1 日後 投与 7 日後 同意取得 入院 投与被験者の決定 退院 通院 被験者特性の調査 ● 心電図検査 ● 皮内反応検査 ● 治験薬投与 ● ● ● 理学的検査 ● ● ● ● 臨床検査 (採血・採尿) ● ● ● ● 血漿中濃度 ● ● ● 薬 物 動 態 尿中濃度 (蓄尿) ● ● ● ● 自覚症状・他覚所見 ● ● ● ● ● ● ●
図 2.7.6.1.5-1 検査・観察時期
1) 症例構成
スクリーニングにより選定した治験薬投与候補者 10 例における症例構成を図 2.7.6.1.5-2
に示した.10 例のうち,1 例は入院前に治験参加辞退の申し出があった.また他の 1 例は入
院当日感冒のため来院しなかった.このため,残り 8 例が入院し,皮内反応検査を実施し,8
例全員陰性であることを確認した.この 8 例の中から,肘皮静脈が細かった 1 例と,治験薬
投与前の検査にて,血中カリウムが基準値上限を超えていた 1 例を除外とした.被験者とし
て選定された 6 例は全例治験を完了し,投与後の中止・脱落例はなかった.
治験薬投与候補者 10 例 脱落 2 例 皮内反応検査実施例 8 例 除外 2 例 被験者 (投与症例) 6 例 治験完了被験者 6 例 薬物動態評価対象例 6 例 安全性評価対象例 6 例
図 2.7.6.1.5-2 症例構成
2) 薬物動態
健康成人男性に DRPM 500 mg を 1 日 3 回 (6 時間間隔,計 3 回) 30 分かけて反復点滴静注
したとき,朝投与 (初回投与) と夕投与 (最終回投与) とで DRPM の体内動態にほとんど差は
なかった.(2.7.2.2.1.1.3 項)
3) 安全性
認められた有害症状は 2 例 ( 4 件) で,いずれも軽度であり,処置なく回復した.このうち,
DRPM との因果関係が否定できない有害症状は 1 件 [頭重 (感)] であった.臨床検査値の異常
変動は認められなかった.
4) 結論
1 日 3 回反復投与における朝投与時と夕投与時とで DRPM の体内動態に差はないことが示
された.安全性については,特記すべき異常は認められず,忍容性に問題はなかった.
2.7 臨床概要
2.7.6.2 患者における薬物動態試験
2.7.6.2.1 体液・組織中濃度測定試験 (泌尿器科領域)
添付資料 5.3.3-Report06
試験方法の概略を表 2.7.6.2.1-1 に示した.
表 2.7.6.2.1-1 試験方法
項目 内容 目的 前立腺組織への移行を検討し,DRPM の後期第 2 相試験で得られた前立腺炎に対する効果 を基礎的に確認する. 治験方法 多施設共同による非盲検,非対照,非無作為化試験 単回投与試験 (投与量 250 mg 又は 500 mg,30∼60 分間点滴静注) 症例数 目標症例数:20 例程度 集積症例数:14 例 投与症例数〔登録症例数〕:14 例 薬物動態評価対象例数:13 例 副作用 (症状) 評価対象例数:13 例 副作用 (臨床検査値) 評価対象例数:12 例 対象及び主要な組 み入れ基準 対象: 前立腺摘出術等の手術患者で,抗菌薬の投与を必要とする患者 選択基準: 1) 20 歳以上 79 歳以下の患者 除外基準: 1) 重篤な心,肝又は腎機能障害を有する患者 2) てんかんの既往歴あるいは中枢神経系障害を有する患者 3) β-ラクタム系抗菌薬に薬剤アレルギーの既往のある患者 4) 併用禁止薬の併用を必要とする患者 5) DRPM の治験に一度組み入れられたことのある患者 6) 過去 6 ヵ月以内に他の治験薬が投与された患者 7) その他,治験担当医師が本治験の対象として不適当と判断した患者 被験薬,用量及び 投与方法,ロット 番号 被験薬:DRPM 用量及び投与方法:DRPM の皮内反応が陰性であることを確認した後,1 回 250 mg (力価) 又は 500 mg (力価) 点滴静注 (30∼60 分間) した. ロット番号:250 mg バイアル ,500 mg バイアル ,皮内反応検査薬 投与期間 単回投与 併用禁止薬 1) 他の抗菌薬,2) ループ利尿薬,3) バルプロ酸ナトリウム,4) 他の治験薬 検体採取時期 DRPM 点滴開始後手術を施行し,点滴終了 1 時間後又は 2 時間後に血液及び前立腺組織を 同時に採取することとし,対象に応じて,症例個々の検体採取時期は治験担当医師が決定 した. 評価基準 薬物動態: Bioassay 法で測定した. 安全性: 1) 有害事象 ①有害症状 有害症状の程度は,日本化学療法学会『「抗菌薬による治験症例における副作用,臨 床検査値異常の判定基準」の一部変更について』5) に準じて軽度,中等度,重度の 3 段階で判定した.発現した有害症状と本剤との因果関係は 5 段階で判定した. 1.関係がある 2.多分関係がある 3.関係があるかもしれない 4.多分関係がない 5.関係がない 因果関係が上記の 1,2 あるいは 3 と判定されたものを副作用 (症状) として取り扱っ2.7 臨床概要
表 2.7.6.2.1-1 試験方法 (続き)
項目 内容 評価基準 (続き) ②臨床検査値異常変動 異常変動の有無は,日本化学療法学会「抗菌薬による治験症例における副作用,臨 床検査値異常の判定基準」6) に準じて判定した.程度については有害症状と同様に判 定した.発現した臨床検査値異常変動と本剤との因果関係は 5 段階で判定した. 1.関係がある 2.多分関係がある 3.関係があるかもしれない 4.多分関係がない 5.関係がない 因果関係が上記の 1,2 あるいは 3 と判定されたものを副作用 (臨床検査値) として 取り扱った. 治験総括医師* 治験実施医療機関 他 計 4 施設 治験期間 19 年 月 日 (最初の被験者の投与開始日) ∼19 年 月 日 (最後の被験者の投与終了日) * :所属・役職は治験を実施した当時のもの1) 症例構成
集積症例 14 例に対し,薬物動態評価対象例は 13 例であった (図 2.7.6.2.1-1).薬物動態評
価対象外例 1 例の解析除外理由は,投与方法違反 (点滴時間が規定の範囲を超えていた) で
あった.なお,投与中止例はなかった.
集積症例 14 例 投与症例〔登録症例〕 14 例 薬物動態評価対象例 13 例 不採用症例 1 例 処置違反 1 例 (点滴時間超過 1 例) 副作用 (症状) 評価対象集団 13 例 不採用症例 1 例 処置違反 1 例 (点滴時間超過 1 例) 副作用 (臨床検査値) 評価対象集団 12 例 不採用症例 2 例 処置違反 2 例 (点滴時間超過 1 例,検査日のずれ 1 例)図 2.7.6.2.1-1 症例構成
2.7 臨床概要
2) 人口統計学的及び他の基準値の特性
薬物動態評価対象例における背景因子の分布を表 2.7.6.2.1-2 に示した.
表 2.7.6.2.1-2 薬物動態評価対象例の背景因子の分布
項目 カテゴリー 例数 性 男 13 60 歳以上 70 歳未満 5 70 歳以上 80 歳未満 8 平均値 (歳) 69.9 標準偏差 (歳) 4.9 最小値 (歳) 61.0 中央値 (歳) 72.0 年齢 最大値 (歳) 75.0 40 kg 以上 50 kg 未満 1 50 kg 以上 60 kg 未満 6 60 kg 以上 70 kg 未満 6 平均値 (kg) 59.2 標準偏差 (kg) 6.3 最小値 (kg) 46.0 中央値 (kg) 59.0 体重 最大値 (kg) 69.5 性 男 13 入院 13 入院・外来の区分 外来 0 無 11 基礎疾患の有無 有 2 無 13 合併症の有無 有 0 無 13 治験薬投与直前の化学療法の有無 有 03) 薬物動態
DRPM 250 mg を 30 分もしくは 60 分かけて単回点滴静注したとき,点滴静注開始後 60∼160
分の前立腺組織中濃度は 0.76∼10.3 µg/g (8 例),同時期に測定された血漿中濃度は 2.42∼10.2
µg/mL (8 例) で,前立腺組織中/血漿中濃度比は 1 例について 425.6%と非常に大きな値を示
したものの,その他の症例については 15.0∼72.4% (7 例) であった.また,DRPM 500 mg を
30 分もしくは 60 分かけて単回点滴静注したとき,点滴静注開始後 90∼130 分の前立腺組織
中濃度は 1.04∼4.51 µg/g (5 例),
同時期に測定された血漿中濃度は 5.55∼15.6 µg/mL (5 例) で,
前立腺組織中/血漿中濃度比は 18.7∼49.9% (5 例) であった.(2.7.2.2.5.4 項)
4) 安全性
A) 副作用 (症状) 及び有害症状
2.7 臨床概要
mg 投与が 5 例であった.
有害症状は 13 例全例に認められなかった.
B) 副作用 (臨床検査値) 及び臨床検査値異常変動
副作用 (臨床検査値) の評価対象例数は 12 例であり,評価から除外された 2 例は,投与方
法違反 (点滴時間が規定の範囲を超えていた) 症例 1 例と投与前検査日がずれた症例 1 例で
あった.投与量別内訳は 250 mg 投与が 7 例,500 mg 投与が 5 例であった.
臨床検査値異常変動は評価症例 12 例中 3 例 (発現率 25.0%),10 件みられ,そのうち本剤に
よる副作用 (臨床検査値) と判定されたのは本剤 250 mg 投与における 1 例 (発現率 8.3%),4
件であった.副作用 (臨床検査値) の内訳は AST 上昇,ALT 上昇,
γ-GTP 上昇及び血清β
2ミ
クログロブリン上昇であり,程度はいずれも軽度であった.追跡調査は実施されなかった.
5) 結論
DRPM 250 mg 又は 500 mg,30∼60 分間点滴静注により,前立腺組織へ既存のカルバペネム
系抗生物質
7-8)と同程度の満足な移行性が確認されたことから,前立腺炎に対して良好な臨床
成績を示すものと考えられた.
2.7 臨床概要
2.7.6.2.2 体液・組織中濃度測定試験 (産婦人科領域)
添付資料 5.3.3-Report07
試験方法の概略を表 2.7.6.2.2-1 に示した.
表 2.7.6.2.2-1 試験方法
項目 内容 目的 DRPM の子宮・子宮付属器の各組織内濃度又は骨盤死腔液中濃度を測定し,産婦人科領域 感染症に対する投与量の確認のための資料とする. 治験方法 非盲検,非対照,非無作為化試験 単回投与試験 (投与量 250 mg,30 分間点滴静注) 症例数 目標症例数:13 例 (子宮・子宮付属器の各組織内濃度:10 例,骨盤死腔液中濃度 3 例) 集積症例数:13 例 投与症例数〔登録症例数〕:13 例 薬物動態評価対象症例数:13 例 副作用 (症状) 評価対象症例数:13 例 副作用 (臨床検査値) 評価対象症例数:13 例 対象及び主要な組 み入れ基準 対象: 20 歳以上 70 歳未満で抗菌薬の投与が必要と考えられる下記患者 1) 子宮・子宮付属器の各組織移行 産婦人科領域の疾患のため,子宮全摘手術予定の患者 2) 体液 (骨盤死腔液) 移行 産婦人科領域疾患のため子宮全摘出を施行し,骨盤死腔液の採取が可能な患者 除外基準: 1) 本治験の対象として不適当と考えられる重篤な基礎疾患又は合併症を有する患者 2) β-ラクタム系抗生物質に起因する重篤な副作用又は薬剤アレルギーの既往のある患者 3) てんかんの既往歴あるいは中枢神経系障害を有する患者 4) 妊婦,授乳中及び妊娠している可能性のある患者 5) 併用禁止薬の併用を必要とする患者 6) 過去 6 ヵ月以内に治験薬 (DRPM を含む) の投与を受けた患者 7) ウイルス性疾患 (肝炎,HIV) の既往歴を有する患者 8) その他,治験責任 (分担) 医師が本治験の対象として不適当と判断した患者 被験薬,用量及び 投与方法,ロット 番号 被験薬:DRPM 用量及び投与方法:DRPM の皮内反応が陰性であることを確認した後,1 回 250 mg (力価) 点滴静注 (30 分間) した. ロット番号:250 mg バイアル ,皮内反応検査薬 投与期間 単回投与 併用禁止薬 1) 他の抗菌薬,2) ループ利尿薬,3) バルプロ酸ナトリウム,4) 他の治験薬 検体採取時期 1) 子宮・子宮付属器の各組織移行 投与終了の約 1∼3 時間後に手術を施行し,血液及び組織を採取した.なお,手術の都 合により,投与終了から検体採取の時間を完全に規定することは困難と考えられるが, 投与終了 1 時間後に 2 例,2 時間後に 4 例,3 時間後に 4 例の集積を目安とした. 2) 体液 (骨盤死腔液) 移行 投与終了 15 分前,投与終了時,30 分後,1 時間後,2 時間後,4 時間後,6 時間後に血 液及び骨盤死腔液を経時的に採取した. 評価基準 薬物動態: Bioassay 法で測定した.2.7 臨床概要
表 2.7.6.2.2-1 試験方法 (続き)
項目 内容 評価基準 (続き) 安全性: 1) 有害事象 ①有害症状 有害症状の程度は,日本化学療法学会『「抗菌薬による治験症例における副作用,臨 床検査値異常の判定基準」の一部変更について』5) に準じて軽度,中等度,重度の 3 段階で判定した.発現した有害症状と本剤との因果関係は 5 段階で判定した. 1.関係がある 2.多分関係がある 3.関係があるかもしれない 4.多分関係がない 5.関係がない 因果関係が上記の 1,2 あるいは 3 と判定されたものを副作用 (症状) として取り扱っ た. ②臨床検査値異常変動 異常変動の有無は,日本化学療法学会「抗菌薬による治験症例における副作用,臨 床検査値異常の判定基準」6) に準じて判定した.程度については有害症状と同様に判 定した.発現した臨床検査値異常変動と本剤との因果関係は 5 段階で判定した. 1.関係がある 2.多分関係がある 3.関係があるかもしれない 4.多分関係がない 5.関係がない 因果関係が上記の 1,2 あるいは 3 と判定されたものを副作用 (臨床検査値) として 取り扱った. 医学専門家* 治験実施医療機関 治験期間 19 年 月 日 (最初の被験者の投与開始日) ∼19 年 月 日 (最後の被験者の投与終了日) * :所属・役職は治験を実施した当時のもの1) 症例構成
集積症例 13 例に対し,全例薬物動態評価対象例であった (図 2.7.6.2.2-1).なお,投与中止
例はなかった.
集積症例 13 例 投与症例〔登録症例〕 13 例 薬物動態評価対象例 13 例 副作用 (症状) 評価対象集団 13 例 副作用 (臨床検査値) 評価対象集団 13 例2.7 臨床概要