本邦における医薬品副作用報告データベースの評価
方法の検証
著者
土屋 雅美
学位授与機関
Tohoku University
学位授与番号
11301甲第19206号
URL
http://hdl.handle.net/10097/00129261
博士論文
本邦における医薬品副作用報告データベース
(
JADER)の質評価方法の検討
令和元年度
東北大学大学院薬学研究科
医療薬学専攻 病態分子薬学分野
土 屋 雅 美
略語一覧 略語 省略していない表現 CIOMS CT EMA ER FAERS FDA GPSP ICD ICH irAE JADER MedDRA/J MR MRI OECD
Council for International Organization of Medical Sciences: 国際医学団体協 議会
Computed Tomography: コンピューター断層撮影 European Medicines Agency: 欧州医薬品庁 Entity Relation: 実体関連
Food and Drug Administration Adverse Event Reporting System: 米国食品医 薬品局の有害事象報告システム
Food and Drug Administration: 米国食品医薬品局
Good Post-marketing Study Practice: 医薬品の製造販売後の調査及び試験 の実施の基準
International Statistical Classification of Diseases and Related Health Problems: 疾病及び関連保健問題の国際統計分類
International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use: 医薬品規制調和国際会議
Immune- related adverse event: 免疫関連有害事象
Japanese Adverse Drug Event Report database: 医薬品副作用データベース Medical Dictionary for Regulatory Activities/ Japanese version: ICH 国際医薬 用語集日本語版
Medical Representative: 医薬品情報担当者 Magnetic Resonance Imaging: 核磁気共鳴画像法
Organization for Economic Cooperation and Development: 経済協力開発機 構
PMDA PT SNS SOC TTO WHO
Pharmaceuticals and Medical Devices Agency: 独立行政法人医薬品医療機 器総合機構
Preferred Term: 基本語
Social Networking Service: ソーシャル・ネットワーキング・サービス System Organ Class: 器官別大分類
Time-To-Onset: 被疑薬投与から副作用症状出現までの期間 World Health Organization: 世界保健機関
目次
緒論 ... 1
第1 節 WHO documentation grading scheme を用いた JADER の質評価 ... 8
1 方法 ... 8 2 結果 ... 14 3 考察 ... 25 第2 節 vigiGrade を用いた JADER の質評価 ... 34 1. 方法 ... 34 2. 結果 ... 41 3. 考察 ... 52
第3 節 WHO documentation grading scheme と vigiGrade completeness score における質 の高い報告の特性 ... 59 1. 方法 ... 59 2. 結果 ... 60 3. 考察 ... 65 結論 ... 68 謝辞 ... 70 参考文献 ... 71 発表論文リスト ... 78
1
緒論
医薬品を臨床現場で安全・適正に使用するためには、医療従事者による医薬品安全 性情報の創出・収集・活用が必須である。近年、医薬品の市販後安全性を確保するため に、医療従事者や製薬企業による医薬品安全性監視活動(Pharmacovigilance)の重要性 が高まっている。医薬品安全性監視活動とは、世界保健機構(World Health Organization: WHO)の定義によれば、「医薬品の有害な作用または医薬品に関連する諸問題の検出、 評価、理解及び予防に関する科学と活動」とされており1)、医薬品の開発から市販後ま
で、一貫した安全対策を行うことが医薬関係者には求められる。医薬品の開発段階にお いて、その有効性と同時に安全性も評価の対象となるが、実臨床と比較して症例数が少 ないこと、年齢や性別、病態などの患者背景が一定の集団を対象にしていること、投与・ 観察期間が短いことなどの限界(5 TOOS: too few, too simple, too narrow, too median-aged, and too brief)があり2)、医薬品の市販後に、臨床試験とは異なる大規模な集団に医薬品
が使用されて初めて重大な副作用が明らかになる場合も少なくない。さらに近年、医薬 品の世界同時開発・国際共同治験の実施が加速しており、治験における日本人症例数が 少ない状態で医薬品が上市されることや、特に希少疾患などの場合、第2 相試験の結果 に基づいて医薬品の承認申請が行われるなど、本邦において医薬品の市販前に安全性の 充分な評価、検討を行うことは困難となりつつある。例えば、近年開発が進んでいる免 疫チェックポイント阻害薬による免疫関連有害事象(immune-related adverse event: irAE) の場合、その発現頻度は従来の殺細胞性抗がん薬や分子標的治療薬よりも低く、メタア ナリシスの結果によれば重篤な下垂体炎は約0.2%、重篤な甲状腺機能低下症は 0.2%と3)、 治験の症例数では検出が困難である。このような、発現頻度は低いが重篤な有害事象を 早期に検出し、医薬品を安全に使用するためには、医薬品市販後の有害事象個別報告の 収集、評価及び伝達などの安全性監視活動が非常に重要な役割を果たしている。 上述した通り、市販前における医薬品安全性に関連するエビデンスの集積には限界が
2 あり、新薬が上市された時点での医薬品安全性情報は暫定的なものとしかみなすことが できないため、市販後、日常臨床における医薬品の使用によって得られる安全性情報を 収集する必要がある。一般に、副作用の自発報告は市販後の医薬品安全性モニタリング の基盤であり、医薬品による未知で有害な作用の可能性を検出することを目的とし、副 作用が疑われる症例報告を収集・整理する仕組みと定義づけることができる。本邦にお いては医薬品医療機器法第68 条の 10 第 2 項に基づく医薬品・医療機器安全性情報報告 制度として、日常、医療の現場においてみられる医薬品、医療機器又は再生医療等製品 の使用によって発生する健康被害等(副作用、感染症及び不具合)の情報を医療関係者 等が厚生労働大臣に報告する制度であり、副作用自発報告の主要な情報源は医療従事者 である。報告の対象は医薬品、医療機器又は再生医療等製品の使用による副作用、感染 症又は不具合の発生について、保健衛生上の危害の発生又は拡大を防止する観点から報 告の必要があると判断した情報(症例)であり、医薬品等と副作用等の因果関係が必ず しも明確でない場合であっても報告が求められる4)。本邦において集積された副作用報
告の一部は、医薬品副作用データベース(Japanese Adverse Drug Event Report database: JADER database)として一般に公開されており、ウェブサイト上からダウンロードして 利用可能である5)。
医薬品副作用データベースは、WHO の VigiBase や米国食品医薬品局(Food and Drug Administration: FDA)の FAERS(FDA Adverse Event Reporting System)、オランダ医薬監 視センターLareb の Lareb database、欧州医薬品庁(European Medicines Agency: EMA)の Eudravigilance など世界中に存在し、シグナル検出などの薬剤疫学的な手法を用いて市 販後医薬品安全対策に重要な役割を果たしている。医薬品の承認状況や使用に関する法 的規制、保険償還の有無など、医薬品を取り巻く環境の違いにより市販後医薬品の使用 動向は国によって大きく異なる。例えば、抗精神病薬の処方実態調査では、欧米諸国で 単剤または2 剤併用療法が多いのに対し、本邦では 3 剤以上の併用療法が多い傾向が認
3 められた6)。循環器分野では、心房細動(AF)に対する各種抗凝固薬の薬剤選択に関し て、本邦では直接経口抗凝固薬の処方割合が多いのに対し、国民皆保険制度を採用して いない国や、国民皆保険制度を採用しているが医薬品の保険償還が規制当局により制限 されているような欧米諸国、また直接経口抗凝固薬が未承認の国などではワルファリン の処方割合が多いという知見が得られている7)。医療資源においても、本邦のコンピュ
ー タ ー断 層 撮影 (Computed tomography: CT )装置や核磁気共鳴画像法( Magnetic Resonance Imaging: MRI)装置の人口 100 万人当たりの台数が経済協力開発機構 (Organization for Economic Cooperation and Development: OECD)加盟諸国の平均値と比 してそれぞれ4.1 倍、3.3 倍と多く8)、このような医療環境によって副作用がより早期に
発見される、副作用に関する画像所見などの情報が充実するなどの違いが生じる一因と なっている。独立行政法人医薬品医療機器総合機構(Pharmaceuticals and Medical Devices Agency: PMDA)の副島らは、WHO Collaborating Centre for International Drug Monitoring に収集された副作用のうち、本邦からの副作用報告症例を解析し、間質性肺疾患の副作 用報告が突出して多いことを報告している9)。その理由としてPMDA の岩佐らは、日本 人における遺伝的要因に加え、間質性肺疾患への社会的関心の高さ、漢方製剤の併用、 CT、MRI 保有台数の多さと国民皆保険制度による画像検査へのアクセスのしやすさな どを挙げており 10)、本邦の医薬品を取り巻く環境が副作用報告の内容に影響している ことが示唆される。また、CYP2C19 のような遺伝子多型に起因する薬物動態の差は副 作用の出現状況に影響を与え11)、その多型の頻度が人種間で異なることから、副作用の 発現頻度が人種構成によって異なることが示唆される。JADER と FAERS について、ニュ ーキノロン系抗菌薬による副作用シグナルの検出、比較を行った検討では、共通して検 出されたシグナルと、JADER または FAERS のみで検出されたシグナルとが存在し、薬 剤感受性の人種差や、規制当局による通知の有無などに起因するものと考察されている12)。 自国の安全性情報に基づく医薬品安全対策を行うためには、各国独自の市販後医薬品安
4 全性情報を集積したデータベースが必要であり、JADER も、本邦における医薬品の使 用状況や使用環境を反映した医薬品副作用データベースとして有用であると考えられ る。 これらの医薬品副作用データベースを用いて、より精度の高い副作用シグナルを検出 したり、医薬品と副作用の因果関係評価を適切に行ったりするためには、データベース に含まれる報告の質が高いことが重要であるとFDA の Good Pharmacovigilance Practices and Pharmacoepidemiologic Assessment に記載されている13)。これによれば、「good case
report」は Table 1 に挙げた要素を含むとされており、医薬品と副作用の因果関係につい て 合 理 的 な 可 能 性 を 示 す た め の 基 準 を 示 し た 国 際 医 学 団 体 協 議 会 (Council for International Organization of Medical Sciences: CIOMS) VI ワーキンググループレポート14)
(Table 2)の『A) 個別症例に基づくエビデンス』を用いて評価する上で必要な項目が 含まれている。ここで、医薬品と副作用の因果関係評価を行う上で必要な項目が充分含 まれている副作用報告を「質が高い」と定義したときに、報告の質や情報の最新性、報 告元の地理的な広がり、病歴などの臨床情報の有無などを加味して副作用シグナル検出 を行うことにより、通常のシグナル検出を行うよりも感度、特異度の高いシグナルが検 出可能であるとする研究結果が報告されている15)。また、ソーシャル・ネットワーキン
グ・サービス(Social Networking Service: SNS)から収集した情報を用いて副作用シグナ ル検出を行った場合と、WHO の副作用データベースである vigiBase を用いて副作用シ グナル検出を行った場合とで、vigiBase を用いた方が感度、特異度の高いシグナルが検 出可能であったとの報告もあり、系統的に集積された副作用報告データベースの優位性 が明らかとなった16)。 医薬品と副作用の因果関係評価や副作用シグナル検出に用いるための副作用報告の 質を評価する方法としては、前述したように各個別症例に含まれる項目の充足率 (completeness)を評価するものと、薬剤疫学の専門知識を有する評価者が評価を行う
5
方法とがあり、前者ではWHO の documentation grading scheme17)、VigiBase の質評価の
ために作成されたVigiGrade18)、ATHE Score19)、その他completeness score を算出する方
法20-22)が挙げられる。後者の専門家による評価としては、Rolfes ら23)、Durrieu ら24)、
Tuccori ら25)による研究などがこれまでに報告されている。JADER の医薬品安全性監視
活動に用いるデータベースとしての質や特徴に関する研究は、これまでに、FAERS に 含まれる日本からの症例との比較が行われているのみであり 26)、データベースとして
の特徴の詳細や、含まれる報告の質に関する研究は存在しない。
本研究においては、JADER の質の評価には WHO documentation grading scheme17)と
vigiGrade completeness score18)を用いた。WHO documentation grading scheme は、被疑薬の
投与開始日と副作用症状出現日に関する情報の有無、被疑薬の使用理由、被疑薬再投与 後の副作用症状の再発の有無(リチャレンジ)に関する情報が評価基準に含まれており、 副作用と被疑薬の因果関係の判定における有用性を評価している。一方、vigiGrade completeness score は患者の年齢、性別などの背景情報や、被疑薬の投与開始から副作用 発現日までの期間であるTTO(Time-to-onset)、被疑薬の投与量、被疑薬の使用理由、副 作用の転帰、報告者の職種、報告された国など、副作用報告に含まれる情報量を評価し ている。vigiGrade completeness score の主な用途は、WHO に送られてくる各国の副作用 報告の品質や副作用報告制度の指標であるが、副作用のシグナル検出における有用性に ついても報告されている 27)。本邦における副作用報告データベースである JADER の、
因果関係評価やシグナル検出などの市販後医薬品安全性情報の創出に用いる情報とし ての質を評価することにより、効率的に医薬品のリスクを抽出する方法を見出し、安全 な薬物療法の遂行に必要な情報を創出することが可能であると考える。
以上より、本研究では、WHO documentation grading scheme と VigiGrade を用いた JADER の質評価を行い、報告者や報告の種類等に基づき解析した。
6
Table 1. FDA ガイダンスにおける‘Good case report’に含まれる要素(文献 13 より引用改
変) 1. 有害事象の詳細や経緯、薬の使用から症状の出現までの期間(time-to-onset)など 2. 被疑薬、併用薬とその詳細(i.e., 投与量、ロット番号、投与スケジュール、投与 日、投与間隔など)、(OTC 薬やサプリメント、最近内服していない薬も含む) 3. 患者属性を含む患者背景(e.g., 年齢、人種、性別)、 治療開始前の病状、併存疾 患、併用薬、関連する家族歴、他の危険因子など 4. 有害事象の診断の記載(診断の過程も含む) 5. 有害事象の臨床経過と転帰(e.g., 入院、死亡) 6. ベースライン、治療中、治療後の治療法や検査データ(薬物血中濃度を含む) 7. リチャレンジ、デチャレンジに対する結果の情報 8. 他の関連する情報 (e.g., 有害事象のアセスメントに重要な場合、有害事象に関 する他の詳細、患者が受ける利益に関する情報)
7 Table 2. CIOMS VI ワーキンググループが提案した因果関係の判断材料(文献 14 よ り引用改変) A) 個別症例に基づくエビデンス 1. リチャレンジ陽性(訳注;再投与による再発) 2. 疑いの余地がない(明確に特徴づけられ,文書化された症例の病歴が存在する) 3. 事象発現までの時間に説得力がある 4. デチャレンジ陽性(訳注;投与中止で消失) 5. 交絡するリスク因子がない 6. 曝露量や曝露期間から原因と効果の一貫した説得力のある説明が可能 7. 正確な病歴が裏付けられている 8. その症例の場合疑いの余地がなく容易に評価できる 9. 併用治療が原因である可能性が低い 10. その他の治験責任(分担)医師による判断 11. 他に説明できる原因がない B) 複数の症例に基づくエビデンス 1. 安全性に的を絞った試験でのポジティブな結果 2. 発現割合がプラセボや対照薬に対して一貫して高い (統計的に有意であるかは問わない) 3. 用量反応関係が認められる(固定用量あるいは漸増法の試験) 4. その事象による中止症例の割合が対照群より高い 5. 対照群に比較して,より早期に発現している,あるいは重症度が高い 6. 関連する症状のパターンに一貫性がある 7. 発現までの時間に一貫性がある 8. 異なる試験間で一貫した傾向が観察される 9. 臨床的状態や潜伏のパターンが一貫している C) 有害事象,薬剤(薬効群)についての既知の知見 1. 過量投与の結果として知られている 2. 対象となる患者集団では(その薬なしで)起こることは稀な事象である 3. 歴史的に,薬剤性の事象であることが知られている(スティーヴンズ・ジョン ソン症候群,好中球減少症など) 4. 薬物相互作用などの臨床薬理学的エビデンス 5. 既知の作用機序 6. 既知の同種同効薬効果 7. 動物モデル,in vitro モデルでの同様な所見 8. その事象を引き起こす他の薬剤との特性の類似性
8
第1 節 WHO documentation grading scheme を用いた JADER の質評価
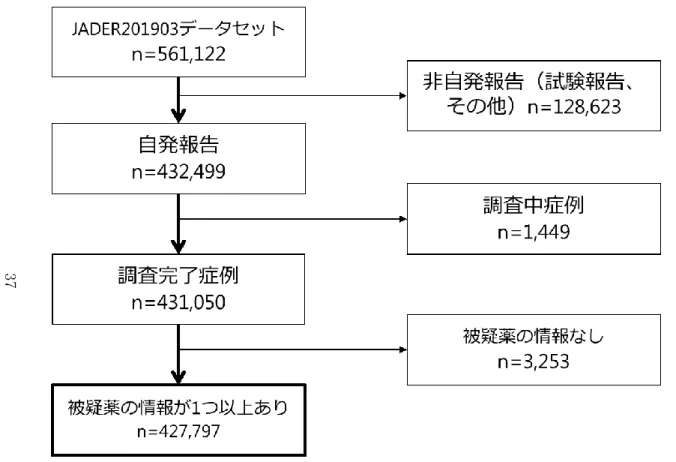
1 方法 1.1 データソース JADER は、PMDA が受理した副作用報告の一部をデータベース化したものであり、 国内製薬企業からの副作用報告、医療機関からの直接報告、臨床試験等で得られた副 作用情報などが含まれている26)。JADER は PMDA のウェブサイトよりダウンロード 可能であり5)、本研究では2004 年 4 月から 2017 年 7 月までの副作用報告を含む JADER201711 データセットを使用した。JADER は医薬品規制調和国際会議
(International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use: ICH)E2B-M2(R3)28)に基づいた報告様式となっており、ダウンロード可
能なデータベースは以下の4 つのテーブルから構成されている。①症例一覧テーブル (demo)(478,508 件); ②医薬品情報テーブル(drug) (3,021,893 件); ③副作用情 報テーブル(reac)(757,109 件); ④原疾患テーブル(hist)(960,311 件)(Figure 1) ①症例一覧テーブルには、患者の年齢、性別、身長、体重などの詳細情報のほか、報 告ごとに付与される固有の識別番号、報告回数、報告者の資格(医師、薬剤師、医療 関係者、弁護士、消費者等)、報告の種類(自発報告、試験、その他)、報告年度(四 半期)、症例の調査状況(調査中、調査完了)などの副作用報告の管理上の情報が記載 されている。識別番号には、医療機関報告と企業報告を分類するアルファベットが含 まれている。JADER においては、医療機関から PMDA に直接報告され、かつ PMDA により詳細調査が行われた症例が「医療機関報告」、医療機関から直接、または製薬企 業を介してPMDA に報告された症例のうち、製薬企業によって詳細調査が行われた症 例が「企業報告」として格納されている。報告者の資格について、医療機関からの自 発報告については、2017 年より 1 件の副作用報告には報告者 1 職種分のみが記載され るように取り扱われているが、その他の報告に関しては、1件の副作用報告に対して
9 複数の職種が報告者として記載されることもあるため(例:医師と薬剤師、医師と医 療関係者など)、本研究では、報告者の職種の項の1 番目に記載されている職種を「報告 者」として取り扱った。報告の種類は、ICH E2B(R3)28)で規定されるように、医薬関係 者等からの自発的な報告(文献や学会発表等も含む場合がある)に由来する「自発報 告」、市販後臨床試験やレジストリ、使用成績調査、特定使用成績調査など、医薬品の 製造販売後の調査及び試験の実施の基準に関する省令(Good Post-marketing Study Practice: GPSP 省令)に規定されるような、試験や系統的なデータ収集スキームで得ら れた「試験報告」、「自発報告」「試験報告」いずれにも該当しない「その他」、報告の 種類に関する情報がない「不明」と定義されている。②医薬品情報テーブルには、被 疑薬または併用薬の情報、医薬品の商品名、一般名、投与量、投与経路、投与開始 日・終了日、医薬品の適用理由、再投与再発有無などの情報が含まれている。再投与 再発有無、すなわち、副作用が出現したあとに被疑薬を再投与したか、また再投与に よって副作用症状が再度出現したか(リチャレンジ陽性)、出現しなかったか(リチャ レンジ陰性)の情報は、被疑薬と副作用の因果関係を推定するのに有力な情報であ る。③副作用情報テーブルには、有害事象名、有害事象の発現日、転帰が記載されて いる。④原疾患テーブルには、患者の原疾患名(既往歴)が記載されている。②医薬 品情報テーブルの医薬品の適用理由(疾患名)、③副作用情報テーブルの有害事象名、 ④原疾患テーブルの原疾患名は、ICH 国際医薬用語集日本語版(Medical Dictionary for Regulatory Activities/ Japanese version: MedDRA/J)(Version 20.1)の 基本語(preferred term: PT)を用いて記載されている。
10
Figure 1. JADER の各テーブルの ER 図(Entity Relation Diagram)
(https://www.pmda.go.jp/safety/info-services/drugs/adr-info/suspected-adr/0004.html から引 用)
11 1.2 解析方法 各副作用報告は報告者(医師、薬剤師、その他)、報告の種類(自発報告、試験報 告、その他または不明)、報告元(医療機関、製薬企業)で分類し、解析を行った。本 研究における解析対象は、自発報告または試験報告のうち、症例の調査状況が「調査 完了」、すなわちPMDA または企業による詳細調査が完了しており、「報告者」に医師 または薬剤師を含む報告とした(医師と医療関係者、医師と弁護士、医師と消費者、 薬剤師と医療関係者、薬剤師と弁護士、薬剤師と消費者など)。報告者の職種として医 師と薬剤師の両方が含まれている報告は解析対象から除外した(Figure 2)。副作用報 告の質評価には、前述したWHO の documentation grading scheme(Figure 3)17)を用い
た。医療機関からの副作用報告件数について、報告者(医師、薬剤師)別に年次推移 を算出した。医療機関からの副作用報告件数に対する病棟薬剤師業務の影響を検証す るため、医療機関報告の件数と病棟薬剤業務実施加算届出施設件数を算出、比較し た。さらに、有害事象毎の副作用報告の件数等を解析するため、有害事象連番1 番目 の有害事象名を集計し、報告の種類別に比較した。報告者別、報告元別に分類した副 作用報告のgrade 毎の分布の比較にはカイ二乗検定を用いた。医療機関報告の件数と 病棟薬剤業務実施加算届出施設の件数の相関関係についてSpearman の順位相関件数 を、またこれらの件数年次推移の傾向性をCochran-Armitage 検定を用いて評価した。 ここで用いた病棟薬剤業務実施加算届出施設の件数データは厚生労働省の公開データ から引用した29)。解析対象における有害事象の分布について、各PT を器官別大分類
(System Organ Class: SOC)に集約して集計を行った。いずれの統計検定も、p<0.05 を 有意とし、すべての解析にはSAS version 9.4(SAS Institute Inc.,Cary,NC,USA)を 用いた。
12 Figure 2. 解析対象
13
14 2 結果 2.1 解析対象 解析に使用したJADER データセット(JADER201711)には 2004 年 4 月から 2017 年 7 月の間に PMDA が受理した副作用報告の一部である 478,508 件が含まれており、その うちの365,590 件(76.4%)が自発報告、92,511 件(19.3%)が試験報告、20,395 件(4.3%) がその他、12 件(0.1%以下)が不明であった。自発報告 365,590 件の内訳は、6,051 件 (1.7%)が医療機関報告、359,539 件(98.3%)が企業報告であった。報告者の職種とし ては、医師、薬剤師、医療関係者(看護師など)、弁護士、消費者が含まれていた。報告 者が不明またはデータが欠損している報告は21,523 件(4.5%)であった。PMDA また は製薬企業による詳細調査が完了している「調査完了」の報告は 476,500 件(99.6%)、 詳細調査が途中である「調査中」の報告は2,008 件(0.4%)であった。 本研究の解析対象は1.2 で述べた通り、JADER に含まれる全報告 478,508 件のうち、 報告の種類が自発報告または試験報告のうち詳細調査が完了しているもので、報告者に 医師または薬剤師を含む(医師と薬剤師の両方を含むものを除く)報告395,091 件であ り、JADER に含まれる全報告の 82.6%に相当する。 解析対象 395,091 件について、報告の種類別では、308,926 件(78.2%)が自発報告、 86,165 件(21.8%)が試験報告であった。自発報告の報告元については、5,989 件(1.9%) が医療機関からの報告、302,937 件(98.1%)が企業からの報告であった。報告者の職種 について、356,713 件(90.3%)が報告者に医師を含む報告であり、38,378 件(9.7%)が 報告者に薬剤師を含む報告であった。 2.2 欠損項目 JADER 全報告と解析対象について、各項目の欠損率を Table 3 に示した。患者の基本 的な情報となる年齢は全報告の5.9%、解析対象の 5.0%で、また性別はそれぞれ 3.8%、
15 3.9%で欠損していた。医薬品の一般名は全報告、解析対象ともにすべての報告で記載さ れている一方で、医薬品の商品名はそれぞれ 4.5%、3.5%の報告で得られなかった。被 疑薬と有害事象の因果関係判定に重要な要素である被疑薬のリチャレンジに関する情 報については、全報告、解析対象ともに90%以上の報告で欠損していた。 2.3 MedDRA による解析
Figure 4, 5 は、MedDRA の SOC 別に集計した各有害事象の割合について、医療機関 報告、企業報告で分類し、報告者の職種で比較したものである。SOC 分類「感染症およ び寄生虫症(infections and infestations)」では、医師で報告割合が多く、一方、「臨床検 査(investigations)」は薬剤師で報告割合が多かった。「妊娠、産褥および周産期の状態 (pregnancy, puerperium and perinatal conditions)」と「生殖系および乳房障害(reproductive system and breast disorders)」に関する報告について、医療機関報告では医師、薬剤師と もにほとんど認められなかった。
16 Table 3. JADER の副作用報告における各項目の欠損率 JADER の項目 全報告 [%] 解析対象 [%] (n=478,508) (n=395,091) 患者情報 年齢 5.9 5.0 性別 3.8 3.9 体重 53.7 50.9 身長 59.2 56.6 原疾患 26.6 23.7 被疑薬の情報 商品名 4.5 3.5 一般名 0.0 0.0 投与量 26.8 24.1 投与開始日 27.1 25.7 有害事象の情報 発現日 25.8 23.6 転帰 19.6 17.7 リチャレンジ 94.5 94.2 JADER: Japanese Adverse Drug Event Report database
17 Figure 4. 自発報告(医療機関報告)における有害事象の PT 別分布 0 5 10 15 20 製品の問題 社会環境 外科および内科処置 傷害、中毒および処置合併症 臨床検査 一般・全身障害および投与部位の状態 先天性、家族性および遺伝性障害 生殖系および乳房障害 妊娠、産褥および周産期の状態 腎および尿路障害 筋骨格系および結合組織障害 皮膚および皮下組織障害 肝胆道系障害 胃腸障害 呼吸器、胸郭および縦隔障害 血管障害 心臓障害 耳および迷路障害 眼障害 神経系障害 精神障害 代謝および栄養障害 内分泌障害 免疫系障害 血液およびリンパ系障害 良性、悪性および詳細不明の新生物 (嚢胞およびポリープを含む) 感染症および寄生虫症
自発報告 - 医療機関報告 (n=5,989)
医師 (n=2,056) 薬剤師 (n=3,933)18 Figure 5. 自発報告(企業報告)における有害事象の PT 別分布 0 5 10 15 20 製品の問題 社会環境 外科および内科処置 傷害、中毒および処置合併症 臨床検査 一般・全身障害および投与部位の状態 先天性、家族性および遺伝性障害 生殖系および乳房障害 妊娠、産褥および周産期の状態 腎および尿路障害 筋骨格系および結合組織障害 皮膚および皮下組織障害 肝胆道系障害 胃腸障害 呼吸器、胸郭および縦隔障害 血管障害 心臓障害 耳および迷路障害 眼障害 神経系障害 精神障害 代謝および栄養障害 内分泌障害 免疫系障害 血液およびリンパ系障害 良性、悪性および詳細不明の新生物 (嚢胞およびポリープを含む) 感染症および寄生虫症
自発報告 - 企業報告 (n=302,937)
医師 (n=268,849) 薬剤師 (n=34,088)19
2.4 WHO documentation grading scheme によるグレード分類
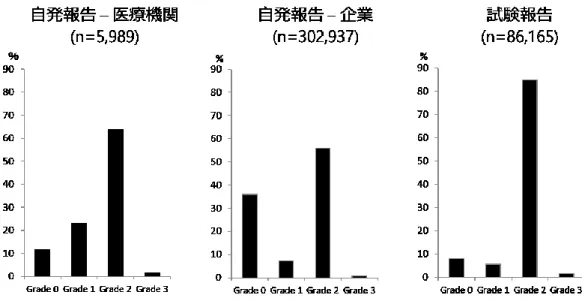
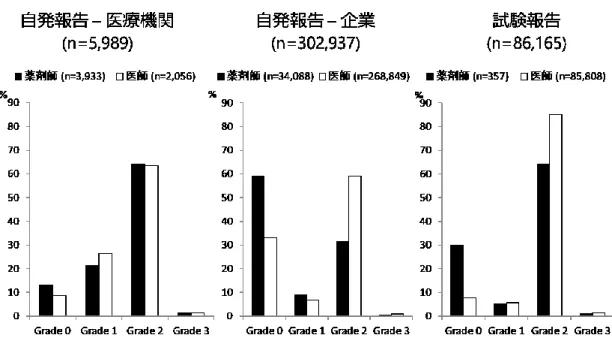
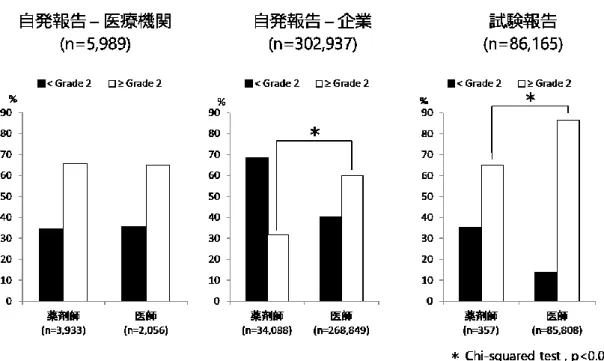
解析対象395,091 件について、WHO documentation grading を用いてグレード評価を行 った。解析対象を報告の種類と報告元で分類したとき、医療機関からの自発報告では Grade 2 の割合が最も高く、次いで Grade 1、Grade 0、Grade 3 の順番であった。企業か らの自発報告と試験報告では、Grade 2 の割合が最も高く、次いで Grade 0、Grade 1、 Grade 3 の順番であった(Figure 6)。 Figure 7 に、報告の種類と報告元、報告者の職種で分類した各グレードの分布を示し た。医療機関からの自発報告について、報告者の職種で比較した場合、報告者に医師を 含む報告と薬剤師を含む報告いずれにおいてもGrade 2 の割合が最も高く、職種間での グレードの分布に差はなかった。報告者に薬剤師を含む報告の件数は、報告者に医師を 含む報告の件数の約2 倍であった(n=3,933 vs n=2,056)。企業からの自発報告について、 報告者に薬剤師を含む報告ではGrade 0 の割合が最も高いのに対し、報告者に医師を含 む報告ではGrade 2 の割合が最も高かった。報告者に医師を含む報告の件数は、報告者 に薬剤師を含む件数の約8 倍であった(n=268,849 vs n=34,088)。企業からの試験報告に ついて、いずれの職種でもGrade 2 の割合が最も高いが、特に報告者に医師を含む報告 でGrade 2 の割合が高かった。試験報告については、報告者に医師を含む報告が 90%以 上を占めており、報告者に薬剤師を含む報告はごく少数であった。 Grade 2 以上を「質が高い報告」と定義した時に、報告元、報告の種類、報告者の職 種で質が高い報告の割合を比較した結果をFigure 8 に示した。医療機関からの自発報告 について、質が高い報告の割合は職種間で差がないのに対し、企業からの自発報告、試 験報告においては、報告者に医師を含む報告で、質の高い報告の割合が有意に高かった (いずれもp<0.0001)。
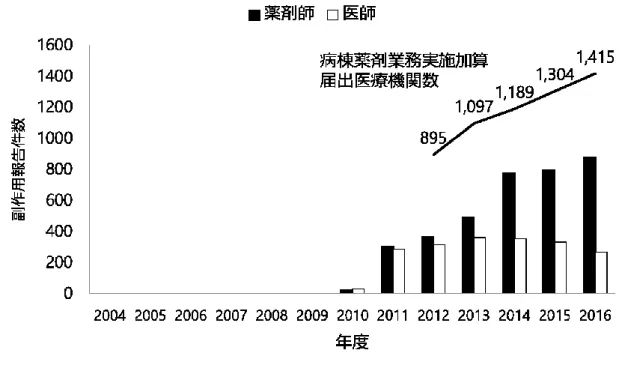
20 2.5 副作用報告件数の年次推移 Figure 9 に医療機関からの自発報告件数と、病棟薬剤業務実施加算届出施設件数の年次 推移を示した。薬剤師による副作用報告件数は期間全体を通して増加傾向を示している が、特に、診療報酬改定により病棟薬剤業務実施加算が新設された2012 年以降、薬剤 師からの報告は有意に増加しており、医師からの報告は有意に減少傾向であった (Cochran-Armitage 検定, ともに p<0.0001)。さらに、病棟薬剤業務実施加算届出施設件 数の増加に同調して、薬剤師からの報告件数は有意に増加していた(Spearman の順位相 関係数 r=1.00, p<0.0001)。
21
22
Figure 7. WHO documentation grading scheme による分類(報告の種類、報告元、職種
23
24
25
3 考察
3.1 解析対象
本研究では、日本の副作用等報告データベースであるJADER の特性を明らかにし、 WHO documentation grading scheme を用いて副作用報告の質評価を行った。本研究は、 JADER の特性とそこに含まれる報告の質を明らかにしたはじめての研究である。 本研究において、JADER に含まれる副作用報告の約 76%が自発報告、約 19%が試験 報告、約5%がその他または不明であることが示された。自発報告の内訳は、1.7%が医 療機関からの報告、98.3%が企業からの報告であり、企業からの副作用報告が多数を占 めていることが明らかとなった。製薬企業は医療従事者からのヒアリングや学会発表、 論文等から入手した副作用情報や、市販直後調査、特定使用成績調査などによって得た 副作用情報について、死亡または重篤で未知の副作用の場合は15 日以内、重篤で既知 の副作用の場合は30 日以内に規制当局へ報告しなければならない義務があることから、 このことが副作用報告件数の多さに寄与したと考えられる。また、医療機関から直接 PMDA に副作用情報が報告された場合においても、企業が詳細調査を行った場合は、企 業からの自発報告扱いとなるため、JADER を解析して得られた医療機関からの自発報 告の件数は、実際の医療機関報告からの直接報告の件数とは乖離している可能性がある。 それでも医療機関からの副作用報告件数が少ないことは事実であり、総務省からの2 度 の勧告30, 31)が発出され、2017 年には医薬関係者の副作用報告ガイダンス骨子32)が発出、 2019 年には副作用報告の基準案が作成されるなど、医療機関からの直接報告の推進が 掲げられている。 3.2 欠損項目 Table 3 に示すように、患者の年齢は解析対象の 5.0%で、患者の性別は 3.9%で欠損し ているのみであった。先行研究においても同様の結果であり33)、年齢や性別など患者の
26
基本情報は医療現場からの報告だけでなく、学会発表や論文などの情報ソースにおいて も容易に入手可能であることに起因すると考えられる。患者の身長、体重などの身体情 報は解析対象の約50%以上で欠損しており、正確な身体情報の記載なしに約半数の報告 が行われていることが示唆される。一方で、身長や体重、年齢などの情報は被疑薬と有 害事象の因果関係評価に直接用いられることは少なく、WHO documentation grading scheme によるグレード評価に及ぼす影響は少ないと考えられる。医薬品の一般名は全 報告、解析対象ともにすべての報告で記載されていたが、英語名称や治験時のコード名 で記載しているものや、医薬品名に誤記があるものなどがあり、表記の正確性は今回の 評価対象とはしていない。また、解析対象において、リチャレンジに関する情報は解析 対象の 90%以上で欠損していた。被疑薬との関連が疑われる有害事象が発現した場合 に、被疑薬を再投与することは有害事象を再発させる恐れがあり、倫理的側面からも回 避されることが多い。そのため、実臨床においてリチャレンジに関する情報が得られる ことは稀であり、多くの副作用報告で情報が欠損したものと考えられる。 3.3 MedDRA による解析
MedDRA の SOC 別の分類では、SOC 分類「感染症および寄生虫症(infections and infestations)」において、報告者に医師を含む報告と薬剤師を含む報告とで、報告件数全 体に占める割合が異なっていた。この違いの要因として、医薬品使用に続発して発生し た感染症や寄生虫症は、細菌培養検査やX 線、CT スキャンなどによる診断、診療行為 を必要とするため、薬剤師が発見・報告者である割合が低くなったものと考えられる。 一方、SOC 分類「臨床検査(investigations)」に関する報告の割合は、報告者に薬剤師を 含む報告で多かった。薬剤師による薬学的管理には臨床検査値等の確認も含まれるため、 医薬品との関連性が疑われる臨床検査値異常を薬剤師がより発見しやすかったことが 考えられる。Gedde-Dahl らによる先行研究では34)、薬剤師による報告は「一般・全身障
27
害および投与部位の状態(general disorders and administration site conditions)」に分類され る項目が多く、これらは臨床検査値などの客観的指標に基づかないことから、副作用の シグナル検出に用いることが困難であるとされている。また、Gedde-Dahl34)ら、van Gootheest ら35)らの先行研究では、薬剤師による副作用報告は非重篤で自覚症状を伴い、 患者自身が発見可能な症状に関するものであることが多いとされており、その理由とし ては、症状が非重篤であることから医師ではなく薬剤師に容易に相談することが可能で あると説明している。これらの先行研究の「薬剤師」はその大半がcommunity pharmacist またはdispensing pharmacist(開局薬剤師)であり、報告者「薬剤師」の大半を病院薬剤 師が占めていると想定されるJADER とは異なっており、副作用の SOC 分類の傾向の違 いはここに起因していると考えられる。本邦では近年、院外処方箋への臨床検査値の印 字が各医療機関で行われるようになり、患者の診療記録に直接アクセス可能な病院薬剤 師だけでなく、調剤薬局薬剤師も患者の臨床検査値に基づく薬学的管理を行うことが可 能となった。また、平成30 年度診療報酬改定では、調剤薬局に対する診療報酬上の加 算である地域支援体制加算において「副作用報告に係る手順書を作成し、報告を実施す る体制を有していること。」という要件が追加されており、調剤薬局薬剤師による副作 用報告の件数は少ないものの増加傾向である。本邦では今後、調剤薬局・病院薬剤師の いずれもが、客観的指標に基づいて副作用を発見・報告し、副作用のシグナル検出など の医薬品安全対策に寄与することが可能であると考えられる。 3.4 報告元の解析 医療機関報告において、Grade 2 の報告が最も多く、次いで Grade 1 の報告が多かっ た。Grade 3 は最も質が高いが、医療機関報告全体に占める割合は 1.5%と少なかった。 先行研究においても、ノルウェーの副作用報告ではGrade 3 は 5%以下34)、オランダで も10%以下35)と同様の傾向が認められた。Grade 3 は患者情報や副作用発現日、被疑薬
28 投与開始日などの情報の他に、因果関係評価において重要な要素である被疑薬の再投与 (リチャレンジ)に関する情報があるものと定義されているが、前述の通り副作用、特 に重篤なものが出現した場合に再投与を行うことは安全、倫理的観点から行われること はまれであり、リチャレンジに関する情報が不足していることが、Grade 3 の報告が少 ない理由であると考えられる。JADER における医療機関報告は、PMDA に報告された 重篤な副作用のうち、PMDA が詳細調査を行ったものと定義される。この医療機関報告 は、企業報告と比較して質の高い報告の割合が高く認められた。先行研究でも同様に、 医療機関報告の質が高いという結果が得られており18)、その要因としては、規制当局に よる報告者への詳細調査の影響が挙げられている。 企業報告においては、医療機関報告と同様Grade 2 の報告が最も多く、次いで Grade 0 の報告が多かった。JADER における企業報告は、医療機関からの自発報告、市販直後 調査、学会発表や論文、インターネットなどの様々な情報源から成り立っており、副作 用症例についての詳細な情報が得られない場合も多く、これが質の低い報告の割合が高 く認められた理由の一つであることが示唆される。JADER において、個々の副作用報 告の詳細な情報源は明らかにされておらず、Grade 0 の報告の情報源についてもその詳 細は不明であるが、学会発表や論文、インターネットなど、詳細調査が困難な情報源に 起因する可能性がある36)。 試験報告では、医療機関報告、企業報告と比較してGrade 2 の報告の割合が明らかに 高かった。JADER における試験報告は、製造販売後臨床試験、使用成績調査、特定使用 成績調査などの「依頼に基づく非自発的な情報源(solicited sources)」から得られる情報 に基づく報告であり、医療機関と製薬企業との契約に基づき収集されるものである。報 告の際には症例の詳細について記載可能な様式が用いられ、企業の医薬品情報担当者 (medical representative: MR)が医師を直接訪問して情報を収集することから、質が高い 報告の割合が高いことが考えられる。
29 3.5 報告者職種の解析 Figure 7 に示す通り、医療機関報告において、医師による報告と薬剤師による報告と の間で、報告のグレードの分布に差はなかった。医療機関報告のうち、薬剤師による報 告件数は3,933 件と、医師による報告 2,056 件よりも多く、医療機関報告件数において、 薬剤師の貢献が大きいことが示唆された。報告の質に差がなかった理由としては、前述 した通り規制当局による詳細調査の影響で報告が均質化されたことが考えられる。企業 報告について、Grade 2 +Grade 3 の報告の割合は、薬剤師による報告と比較して医師に よる報告で有意に高く(Figure 8, p<0.0001)、報告件数については医師による報告が 268,849 件と、薬剤師による報告件数 34,088 件の約 8 倍であった。企業報告の場合、医 療機関報告のように全例が詳細調査の対象となっているわけではなく、報告者の医学的 知識や臨床経験、患者情報へのアクセスの状況により報告の質に差が出た可能性が考え られる。とりわけ医師については、患者情報へのアクセスが薬剤師よりも容易であるこ と、臨床経験における優位性などから、薬剤師と比較して質の高い報告を数多く行った ことが示唆される。試験報告については、Grade 2+ Grade 3 の報告の割合は、薬剤師に よる報告と比較して医師による報告で有意に高く(Figure 8, p<0.0001)、報告件数につい ては医師による報告が85,808 件、薬剤師による報告が 357 件と、医師による報告が 90% 以上を占めていた。試験報告の対象となる「依頼に基づく非自発的な報告(solicited report)」は企業と医療機関との契約に基づいて報告されるが、その際、患者情報の収集 は主に医師の業務となっている37)。このことから、試験報告における報告の質の高さに 関しては医師の貢献が大きいことが示唆される。
Gedde-Dahl らの先行研究では、薬剤師からの報告の約 70%が Grade 0 であり34)、van
Gootheest らの先行研究では薬剤師からの報告の約 5%が Grade 0、約 50%以上が Grade 1 であった35)。本研究の解析対象においては、薬剤師からの報告の約54%が Grade 0 の報
30 であった。本邦においては、製薬企業は副作用情報を臨床現場から収集し、規制当局へ 報告することが法律上求められている。臨床現場において、医療従事者により発見・収 集された副作用報告は、医療機関から製薬企業に送られ、製薬企業から規制当局へと報 告されることがしばしば行われる。一方で、諸外国においては、医療機関または製薬企 業から直接規制当局へ副作用が報告されることが多い38)。また、副作用報告様式や項目 も、ICH-E2B に基づき標準化が進められているものの、その詳細やデータの粒度は各 規制当局により異なる39)。このように、市販後医薬品安全対策に関わる体制の違いや、 副作用報告の際に求められる項目の違いなどから、諸外国における副作用報告の実態や その質と、本邦における副作用報告の実態や質を直接比較することは困難であるが、 JADER の医療機関報告における薬剤師からの副作用報告の質は、先行研究にあるよう な他国のそれと比較して高いことが示唆された。 3.6 副作用報告件数の年次推移 医療機関報告のうち、薬剤師からの副作用報告件数は2012 年度以降、有意に増加傾 向を示した(Figure 9)。2012 年は、同年 4 月の診療報酬改定により病棟薬剤業務実施加 算が新設され、薬剤師による病棟業務に診療報酬上の点数が付与された年であり、病棟 における薬剤師業務の普及により、薬剤師がベッドサイドで患者の副作用を発見する機 会が増加したと考えられる。さらには病棟薬剤業務実施加算の要件である病棟担当薬剤 師と医薬品情報担当薬剤師との連携により、薬剤師がより多くの副作用情報を収集、報 告することが可能になった。JADER において、2010 年度以前は医療機関報告に分類さ れる症例が存在しなかったことが本研究で明らかになった(Figure 9)。前項で述べたと おり、JADER における医療機関報告の定義は、医療機関から PMDA に報告された副作 用報告のうち、PMDA が詳細調査を行ったものとされている。2010 年に本邦における 副作用自発報告の実施要項の改訂が行われ 40)、副作用等報告を行った医療機関等に対
31 し、詳細な調査が必要な場合にPMDA による調査が行われることとなった。2010 年度 以前、医療機関から PMDA に報告された副作用報告は製薬企業に送られたのちに企業 による詳細調査が行われていたものと考えられるが、このようなデータの管理方法の変 更に関する詳細は公表されておらず不明である。このような副作用報告に関連する制度 の変更が、JADER において 2010 年度以前に医療機関報告が存在しない理由の一つだと 考えられる。 本研究において、我々は医療機関報告件数の年次推移と、病棟薬剤業務実施加算の届 出施設数との関連について注目した。医療機関報告件数の増加と、病棟薬剤業務実施加 算の届出施設数の増加は同調しており(r=1.00, p<0.0001)、上記で述べた薬剤師による 病棟業務の推進が、医療機関報告の増加に寄与していることが示唆される。 日本と諸外国の間のドラッグラグが解消されつつあることにより、日本において、世 界に先駆けて承認される新規医薬品の数も増加傾向である 41)。本邦の医薬関係者につ いては、新規医薬品の市販後安全性情報を収集、報告することが以前に増して求められ ている。一方で、医薬品医療機器法に規定されているにも関わらず、製薬企業による副 作用報告漏れは後を絶たず、副作用報告義務違反で初めて業務改善命令が発出された 2014 年 7 月以降、2017 年までの間に業務停止処分や業務改善命令など 7 件の行政処分 が行われている。企業を介さない、医療機関から PMDA への直接報告は、今後の市販 後医薬品安全対策に大きく貢献することが予想され、とりわけ医療機関におけるタス ク・シフティングが推進される昨今においては、薬剤師による医薬品安全性情報の創出 はより重要な役割を果たすことが考えられる。 3.7 本研究の強みと限界 本研究は、本邦の副作用報告データベースであるJADER の質評価を行った最初の研 究であり、これまで知られていなかったJADER の副作用データベースとしての質を評
32
価し、他国の副作用データベースの特徴や質と比較したものである。本研究結果は、 JADER を市販後医薬品安全対策活動や医薬品安全性シグナルの検出などに用いる際の 一助となると考えられる。
一般的に、副作用報告データベースを用いた副作用シグナル検出などの研究において は、過少報告、Notoriety bias、Ripple effect42)、Weber effect43)などの報告バイアスの影響
が存在することが知られている。また、副作用報告データベース単体では医薬品の使用 患者数を把握することができないために副作用の発現頻度が求められないことから、対 照群を置いたリスクの定量的かつ適切な評価等が行えず、安全対策上の判断が困難な場 合も存在する。これらは副作用報告データベースを用いた研究を実施する上で、つねに 研究の限界となりうる44, 45)。さらに、PMDA に報告された副作用の一部のみが JADER に格納されていることから、JADER を副作用シグナル検出や副作用の発現好発時期の 推定などに用いる際などには、JADER が必ずしも本邦の副作用報告の全てを正確に反 映しているわけではないことに留意が必要である。本研究で行われたJADER の質評価 についても同様であり、本邦の副作用報告全体の質とは異なる可能性があることに注意 しなければならない。また、JADER における被疑薬、併用薬などの医薬品名は報告者 による自由記載であり、英語での記載やスペルミス、開発コードでの記載など、不正確 なものも散見される46)。後発医薬品の場合、一般名のみで屋号まで記載されていないケ ースが多く、商品名が欠損している場合、先発品と後発品との区別は困難であり、この 点もJADER を市販後医薬品の安全性評価に用いる上での課題である。
本研究で用いたWHO documentation grading scheme は、副作用報告に含まれる情報の 内容について評価したものではなく、あくまで評価対象項目の情報の有無に基づき評価 を行ったものである。副作用報告の内容に関する評価を行うためには、緒論で述べたよ うに薬剤疫学の専門知識を有する評価者による、統一した評価基準を用いた評価が必要 となり、大規模なデータベース全体の質評価を行うのには人的、時間的資源の面から不
33
向きである。従って、大規模な副作用報告データベースに含まれる報告の質評価を包括 的に行う方法としては、本研究で用いたdocumentation grading scheme など、項目の充足 率を機械的に評価する方法が適していると考えられる。
34
第2 節 vigiGrade を用いた JADER の質評価
1. 方法
1.1 データソース
本研究では2004 年 4 月から 2018 年 11 月までの副作用報告を含む JADER201903 デ ータセットを使用した。JADER は前項で述べた通り ICH E2B-M2(R3)に基づいた報告 様式となっており、①症例一覧テーブル(demo)(561,122 件); ②医薬品情報テーブル (drug) (3,390,826 件); ③副作用情報テーブル(reac)(887,636 件); ④原疾患テー ブル(hist)(1,156,673 件)の 4 つのテーブルから構成されている。 1.2 除外基準 本研究において、(1)自発報告ではない報告(例: 試験報告など非自発的な情報源に 基づく報告、その他、不明)、(2)PMDA または製薬企業による詳細調査が完了してい ない(報告の状態が「調査中」の)報告、(3)被疑薬が 1 つも含まれない報告(薬物間 相互作用のみ)は解析対象から除外した。詳細調査が完了していない報告は、医薬品と 有害事象の因果関係評価に必要な項目や副作用好発時期の解析に必要な項目などが今 後追加される可能性があるため、(2)を除外基準に加えた(Figure 10)。
1.3 vigiGrade completeness score
JADER に含まれる副作用報告の質を評価するために、本研究では WHO Uppsala Monitoring Centre により提唱された vigiGrade completeness score18) を使用した。vigiGrade
completeness score は副作用報告に含まれる複数の項目の充足度を、後述する計算式に基 づき計算したものであり、評価の対象となる項目は①time-to-onset(被疑薬開始から副 作用発現までの期間)、②被疑薬の使用理由、③副作用の転帰、④患者の性別、⑤患者 の年齢、⑥被疑薬の投与量、⑦報告元の国、⑧第一報告者、⑨報告の種類、⑩報告者意
35 見、の10 項目である。これらの項目に Table 4 に示すような独自の重みづけとペナルテ ィの設定が行われており、各項目のスコアは情報が欠損していれば0、粒度の高い情報 が存在すれば1 として、0~1 の間の値を取る。例えば、ある項目の情報が過不足なく存 在すればペナルティは0 で、その項目のスコアは 1 となる。情報が欠損していた場合の ペナルティの設定は、因果関係評価を行うために必要不可欠なtime-to-onset は最大 50%、 因果関係評価を行うために重要な被疑薬の使用理由、副作用の転帰、性別、年齢などの 項目は30%、被疑薬の投与量や報告元の国、報告者、報告の種類、報告者意見などの補 助的な情報を含む項目は 10%とされており、本研究もそれに従った。被疑薬-副作用の 組み合わせのvigiGrade completeness score を算出する計算式は以下の通りであり、0.07 から1.0 の間の値をとる。
Completeness score =∏10𝑖=1(1 − 𝑃𝑖) = (1 − 𝑃1) … (1 − 𝑃10) 18),
注:Pi は Table 4 に示すように、項目 i に対するペナルティの数値である。
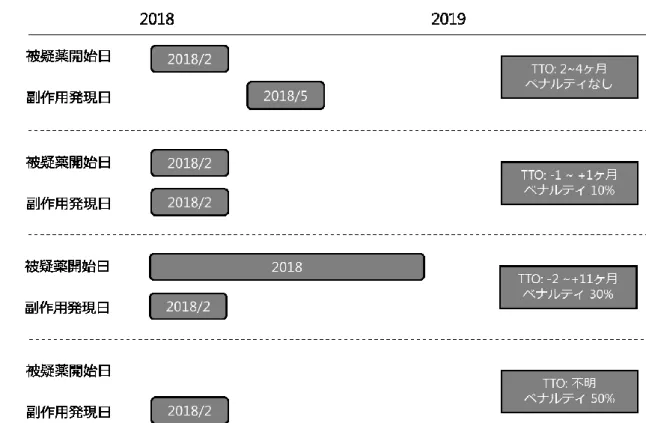
vigiGrade completeness score の算出に必要な項目は全て JADER に含まれているものを使 用した。Time-to-onset は、被疑薬の使用開始日と副作用の発症日から算出し、vigiGrade completeness score の算出に用いた(Figure 11)。
本 研 究 に お い て は 、 評 価 項 目 や ペ ナ ル テ ィ の 一 部 を オ リ ジ ナ ル の vigiGrade completeness score から改変して用いた(Table 5)。JADER では、個人情報保護の観点か ら患者の年齢は年齢層(10 代、20 代、30 代など)または世代(高齢者、新生児、小児 など)を用いて記載されており、患者年齢の項目の評価にあたっては、年齢が世代を用 いて記載されている場合は10%のペナルティ(0.9 を乗算)を、年齢層を用いて記載さ れている場合はペナルティなし(1.0 を乗算)と設定した。また、本研究の解析対象で ある自発報告はすべて日本国内からの報告であり、報告元の国については評価の対象外 とした。また、医療機関から PMDA に副作用報告を行う際のフォーマットには報告者 の意見を自由記載する欄があるが、その内容はJADER には反映されていない。そのた
36
め、報告者意見についても評価の対象外とした。従って、本研究における各副作用報告 の vigiGrade completeness score は最大で 1.0、最小で 1×0.5×0.74×0.93 = 0.088 となる。
WHO Uppsala Monitoring Centre による先行研究では、vigiGrade completeness score>0.8 を ‘well-documented report’と定義している 18)。本研究においては、報告元の国と報告者意
見について評価の対象外としているが、いずれも補助的な情報(10%ペナルティ)とい う扱いであり、先行研究と同様に、‘well-documented report’は vigiGrade completeness score>0.8 であると定義した。また、‘well-documented report’の割合の年次推移を、報告 者の職種と報告の種類で分類し、解析を行った。いずれの統計検定も、p<0.05 を有意と し、すべての解析にはSAS version 9.4(SAS Institute Inc.,Cary,NC,USA)を用いた。
37
38
Table 4. vigiGrade completeness score の評価項目とペナルティ(文献 18 より作成)
項目 説明 検討事項 ペナルティ[%] Time-to-onset 被疑薬使用から副作用発現までの期間 被疑薬の使用開始と副作用発現との時間的前後関 係が不明な場合はペナルティ50% 被疑薬の使用開始と副作用発現との間隔が±1 ヶ月 以上の場合はペナルティ30% それ以外は10%(Figure 9 参照) 50 30 10 被疑薬の使用理由 被疑薬を何の治療のために用いていたか 情報が欠損、または標準化された病名(ICD や MedDRA など)で記載されていない場合 30 転帰 副作用症状の転帰 30 性別 患者の性別 「不明」は欠損として扱う 30 年齢 副作用発現時の患者の年齢 世代(高齢者、小児など)のみの場合はペナルテ ィ10% 30 10 投与量 医薬品の投与量 10 報告元の国 報告元の国名 診療行為の内容や、副作用報告制度は国によって 異なるため評価対象として設定 10 第一報告者 報告者の職種 副作用報告の内容や解釈は報告者の職種によって 異なるため評価対象として設定 10 報告の種類 報告の種類(自発報告、試験報告、他) 「不明」は欠損として扱うが、「その他」はペナル ティを課さない 10 報告者意見 自由記載による報告者の意見 情報に乏しい断片的なテキストなどは除外する 10 *ICD: International Statistical Classification of Diseases and Related Health Problems
39
Figure 11. Time-to-onset の算出方法とペナルティ(文献 13 より作成)
40
Table 5. 本研究で用いた vigiGrade completeness score の評価項目とペナルティ(文献 18 より改変)
項目 説明 検討事項 ペナルティ[%] Time-to-onset 被疑薬使用から副作用発現までの期間 被疑薬の使用開始と副作用発現との時間的前後関 係が不明な場合はペナルティ50% 被疑薬の使用開始と副作用発現との間隔が±1 ヶ月 以上の場合はペナルティ30% それ以外は10%(Figure 9 参照) 50 30 10 被疑薬の使用理由 被疑薬を何の治療のために用いていたか 情報が欠損、または標準化された病名(ICD や MedDRA など)で記載されていない場合 30 転帰 副作用症状の転帰 30 性別 患者の性別 「不明」は欠損として扱う 30 年齢 副作用発現時の患者の年齢 世代(高齢者、小児など)による記載の場合はペ ナルティ10% 年齢層(10 代、20 代、30 代など)を用いた記載に はペナルティなし 30 10 投与量 医薬品の投与量 10 報告元の国 報告元の国名 JADER はすべて国内報告であり、除外 0 第一報告者 報告者の職種 副作用報告の内容や解釈は報告者の職種によって 異なるため評価対象として設定 10 報告の種類 報告の種類(自発報告、試験報告、他) 「不明」は欠損として扱うが、「その他」はペナル ティを課さない 10 報告者意見 自由記載による報告者の意見 JADER には自由記載内容は含まれておらず、除外 0
41 2. 結果 2.1 解析対象 本研究で用いたJADER201903 データベースの副作用報告 561,122 件のうち、自発報 告は432,499 件(77.1%)、試験報告は 106,822 件(19.0%)、その他・不明な報告は 21,801 件(0.1%)であった。561,122 件の副作用報告症例に対し、887,636 件の有害事象、1,353,348 件の被疑薬または併用薬とそのうち557,507 件の第一被疑薬の情報が含まれていた。本 研究ではこれらの副作用報告のうち、Figure 10 の抽出方法に従い、427,797 件について 解析を行った。解析対象427,797 件のうち、419,540 件(98.1%)が製薬企業からの報告、 8,257 件(1.9%)が医療機関からの直接報告であった。報告者職種別では、314,077 件 (73.4%)が報告者に医師を含む報告、48,192 件(11.3%)が報告者に薬剤師を含む報告、 23,638 件(5.5%)が報告者に医師と薬剤師両方を含む報告、41,890 件(9.8%)がその他 の報告者(医療従事者、弁護士、消費者など)による報告であった。
2.2 vigiGrade completeness score
Figure 12 に解析対象 427,797 件における vigiGrade completeness score の分布を示した。 本研究の解析対象のうち、211,774 件(49.5%)が vigiGrade completeness score>0.8 の‘well-documented report’であった。報告元で分類すると、医療機関報告 8,257 件のうち 6,240 件(75.6%)が、また、企業報告 419,540 件のうち 205,534 件(49.0%)が、vigiGrade completeness score>0.8 の‘well-documented report’であった。カイ二乗検定の結果、医療機 関報告における‘well-documented report’の割合は、企業報告と比較して有意に高かった (p<0.0001)(Figure 13)。報告者の職種で分類すると、報告者に薬剤師を含む報告 48,192 件のうち15,799 件(32.8%)が、また、報告者に医師を含む報告 314,077 件のうち 170,933 件(54.4%)が‘well-documented report’であった(Figure 14)。
42 2.3 項目欠損の状況 本研究の解析対象である副作用報告427,797 件のうち、33,580 件(7.9%)で年齢に 関する情報が欠損しており、11,510 件(2.7%)が年齢層ではなく世代による記載のみ であった。また、21,247 件(5.0%)で患者性別に関する情報が欠損しており(“不明” も含む)、被疑薬の投与量、副作用の転帰、被疑薬の使用理由はそれぞれ133,299 件 (31.2%)、98,136 件(22.9%)、57,090 件(13.4%)で欠損していた。報告者の職種の 情報は17,541 件(4.1%)で欠損していたが、報告の種類に関する情報は解析対象のう ち全ての報告に含まれていた。被疑薬の使用開始から副作用発現までの期間(“time-to-onset”)について、174,255 件(40.7%)が被疑薬の使用開始日(154,110 件, 36.0%) または副作用発現日(137,140 件, 32.1%)の情報がないために算出不可能であり、 10,385 件(2.4%)で被疑薬の使用開始と副作用発現との間隔が±1 ヶ月以上であった。 Table 6 に報告元で分類した項目毎の情報の欠損について示した。医療機関報告と比較 し、企業報告では各項目における欠損情報の割合が有意に高かった。特に、“time-to-onset”が算出不可能であった報告の割合は、医療機関報告では 1,228 件(14.9%)であ ったのに対し、企業報告では173,027 件(41.2%)と、企業報告で有意に高かった(カ イ二乗検定、p<0.0001)。 2.4 ‘well-documented report’の割合の年次推移 JADER において、副作用報告件数は年々増加傾向である一方で、‘well-documented report’の割合は減少傾向を示した(Figure 15)。Figure 16 は報告者の職種別に‘well-documented report’の割合の年次推移を示したものである。報告者に医師を含む報告、そ の他の職種による報告では年々減少傾向であったのに対し、報告者に薬剤師を含む報告、 報告者に医師と薬剤師の両方を含む報告ではわずかに減少傾向を示したのみであった。 Figure 17 は報告元別で分類した‘well-documented report’の年次推移を示したものである。
43
医療機関報告における‘well-documented report’の割合は調査期間中約 70-80%を維持して いたのに対し、企業報告では時間経過とともに約75%から約 30%へと低下していた。
2.5 ‘well-documented report’ の特徴
JADER201903 データベースの副作用報告 561,122 件のうち、第一被疑薬が特定可能で あった557,707 件について、‘well-documented report’と‘not well-documented report’の基 礎属性を比較し、Table 7 に示した。‘well-documented report’は 307,450 件(55.2%)と、 上記で解析対象にした自発報告、調査完了のみのデータセットよりも高い割合で認められ た。‘Well-documented report’と‘not well-documented report’を比較した時に、‘ well-documented report’における医療機関報告の割合が高く(2.0% vs 0.8%, p<0.0001)、調査 中の報告の割合は低かった(0.2% vs 0.5%, p<0.0001)。報告者別に見た場合、’well-documented report’はその他の報告者(看護師、弁護士、消費者など)からの報告の割合が 低く(4.8% vs 16.1%)、報告の種類は試験報告の割合が高かった(28.2% vs 7.9%)。
44
45
Figure 13. 企業報告または医療機関報告における‘well-documented report’の割合
(n=427,797) *カイ二乗検定
46
47 Table 6. 各項目の情報欠損割合 (n=427,797) 項目 医療機関報告 (n=8,257) 企業報告 (n=419,540) p 値* Time-to-onset (n, %) 1,228 (14.9) 173,027 (41.2) <0.0001 副作用の発現日 (n, %) 76 (0.9) 137,064 (32.7) <0.0001 被疑薬の開始日 (n, %) 1,183 (14.3) 152,927 (36.5) <0.0001 被疑薬の中止日 (n, %) 1,270 (0.7) 186,486 (44.5) <0.0001 被疑薬の使用理由(n, %) 650 (7.9) 56,440 (13.5) <0.0001 転帰 (n, %) 297 (3.6) 97,839 (23.3) <0.0001 性別 (n, %) 27 (0.3) 21,220 (5.1) <0.0001 年齢 (n, %) 36 (0.4) 33,544 (8.0) <0.0001 投与量 (n, %) 902 (10.9) 132,397 (31.6) <0.0001 第一報告者(n, %) 13 (0.2) 17,528 (4.2) <0.0001 報告の種類(n, %) 0 (0) 0 (0) *カイ二乗検定
48
49
50