国際医療福祉大学大学院
医療福祉学研究科博士課程
わが国における臨床研究の現状と改善策の提案
平成
24 年度
保健医療学専攻・創薬育薬医療分野
学籍番号:
10S3040 氏名:鳥越 香織
研究指導教員:中野 重行 教授
副研究指導教員:武藤 正樹 教授
要旨 わが国における臨床研究の現状と改善策の提案 鳥越 香織 医療の質を高め、国民の健康をよりよく守るために、臨床研究によるエビデンスの強化が重要で ある。しかしながら、わが国の臨床研究は制度・実施体制が欧米と比較して遅れていると言われ ている。そこで、その現状を知るために、臨床研究の歴史的背景・制度・実施体制に関して調査 し、米国および英国との比較を行った。また、医師および臨床研究コーディネーター(CRC: Clinical Research Coordinator)を対象とした臨床研究に関する意識調査を実施した。これらの調査研究よ り、臨床研究を育てるための具体的な改善策として、国・政府、臨床研究者、支援スタッフ、患 者を含む一般市民が協働して、臨床研究を推進する必要があることが示唆された。そのためには、 臨床研究に携わる各スタッフが協働して臨床研究の質の向上に努めるとともに、臨床研究の支援 および臨床研究者の育成を国の責務として定めた臨床研究全体を統括した制度的枠組を構築と政 府中央組織の設置が望まれる。 キーワード 臨床研究、治験、臨床研究に関する倫理指針、GCP
ABSTRACT
Present State of and Suggestions for Improving Clinical Research in Japan Kaoru Torigoe
Strengthening of the evidence found in clinical research is important for improving the quality of medical care, and more importantly, protecting national health. However, the system and framework of clinical research in Japan seem to be in a preliminary phase relative to those in the United States and Europe. To investigate the present status of clinical research, we conducted a comparative survey on the historic background, system, and framework of clinical research in Japan, the United States and the United Kingdom. Moreover, I surveyed physicians and clinical research coordinators (CRCs) for awareness of clinical research. The results showed improvement in cooperation between the government, researchers, support staff, and citizens to be concrete ideas for developing clinical research. It is important to establish a controlling system that provides a framework for the duties of the government and central organization toward supporting clinical research and training researchers and clinical research staff to improve the quality of medical care by actively promoting tie-ups and promoting cooperation between the government, researchers, support staff, and citizens.
KEY WORDS Clinical Research, Clinical Trials of Investigational New Drugs, Ethical Guidelines for Clinical Studies, GCP
目次
1.はじめに ... 3 2.研究I:臨床研究の歴史的背景・制度・実施体制に関する国際比較とわが国における改善策 ... 6 2-1.目的 ... 6 2-2.方法 ... 6 2-2-1.調査対象国 ... 6 2-2-2.調査媒体 ... 7 2-2-3.調査項目 ... 7 2-3.調査背景と結果 ... 7 2-3-1.人を対象とした研究に関する歴史と研究倫理ならびに制度の形成 ... 7 2-3-2.臨床研究に関する法体系と臨床研究に関する規制または制度 ... 14 2-3-3.臨床研究に関する組織体制 ... 27 2-3-4.臨床研究の研究予算および資金 ... 30 2-3-5.臨床研究の登録と結果の公表に関する体制 ... 36 2-4.考察 ... 40 2-5.結論 ... 44 3.研究II:わが国における臨床研究の現状と改善策:医師とCRCを対象にした調査研究 ... 46 3-1.目的 ... 46 3-2.方法 ... 46 3-2-1.医師へのアンケート調査 ... 47 3-2-2.CRC へのアンケート調査 ... 48 3-3.倫理上の配慮 ... 49 3-4.結果 ... 49 3-4-1.医師へのアンケート調査 ... 49 3-4-2.CRC へのアンケート調査 ... 57 3-5.考察 ... 68 3-6.結論 ... 78 4.総合考察 ... 81 5.結語 ... 84 6.謝辞 ... 85 7.文献一覧 ... 86 8.資料 ... 92 8-1.略語一覧 ... 92 8-2.参考資料 ... 94 8-2-1.参考資料1 ... 94 8-2-2.参考資料2 ... 95 8-2-3.参考資料3 ... 1078-2-4.参考資料4 ...110
8-2-5.参考資料5 ... 111
8-2-6.参考資料6 ...114
8-2-6.参考資料7 ...115
1.はじめに
わが国では、厚生労働省への医薬品の製造販売の承認申請を目的として実施される治験は法制化さ れている。つまり、薬事法に基づく医薬品の臨床試験の実施の基準に関する省令(以下、GCP 省令 と略す)に則って実施され、独立行政法人医薬品医療機器総合機構(以下、PMDA と略す)が治験 計画の届出を受理調査している1)。一方、人を対象とした治験以外の臨床研究(以下、臨床研究と略 す)は、法制化されていない。遺伝子治療2)とヒト幹細胞治療3)に関しては、各関連指針に基づいて 厚生労働省または文部科学省に届け出る義務があるが、それ以外の臨床研究については、「臨床研究 に関する倫理指針(ガイドライン)」に従って実施され 4)、中央組織によるチェックを受けることな く、研究の倫理性と科学性の審査は、各実施施設の倫理委員会に依存するシステムとなっている。 さらに、各施設の倫理委員会の水準にはバラツキがあり、適正な審議が全ての施設で実施されてい る保証はないといわれている5-6)。その上、GCP 省令では義務化されている被験者への健康被害の補 償制度は、臨床研究では未確立で保険外併用療養費制度は適用されない。そのため、治療費の取り扱 いのし方によっては、混合診療に抵触するという懸念も存在する 7-8)。このように、わが国の臨床研 究システムに改善すべき点があることが指摘されている。 一方、欧米では、治験・臨床研究ともに法制化されている。例えば、欧米では当局にて全ての臨床 試験を管理し規制しており、米国では1974 年に成立した国家研究法(Nationl Research Act、以下、 国家研究法と略す)、英国では欧州連合(EU: European Union、以下 EU と略す)の統一ルールと して2001 年に公布された EU 臨床試験指令(Directive 2001/20/EC: Directive 2001/20/EC of the European Parliament and of the Council of 4 April 2001 on the approximation of the laws, regulations and administrative provisions of the Member States relating to the implementation of good clinical practice in the conduct of clinical trials on medicinal products for human use、以 下EU 臨床試験指令と略す)に従って実施されている9-10)。このように、米国および英国では、政府 組織にて全ての臨床研究を管理し規制するシステムが整っており、倫理性、科学性が保証された質の 高い臨床研究が多く実施されている。 わが国の臨床研究実施のガイドラインである「臨床研究に関する倫理指針」は、平成15 年 7 月に 制定、平成16 年 12 月および平成 20 年 7 月に全部改正が実施され、平成 21 年 4 月より改正指針が 施行されている4)。平成20 年 7 月の全部改正では、被験者の保護や倫理性の確保について一層の向 上が図られたが、法制化は見送られた 11)。したがって、未だ信頼性が担保された研究実施のハード ルは高く、わが国全体の臨床研究の実施環境が、欧米と比較して低い水準であるといわれている12)。 また、著者らが行った医師の意識調査を実施した先行研究として、改正指針を周知徹底し、臨床研究 の質の向上を目指す過渡期である施行後2 年半を経過した時点(平成 23 年 9 月)の調査報告がある。 この調査結果からは、医師の「臨床研究に関する倫理指針」の理解度は低く、本指針とGCP 省令と の違いを理解している医師は2 割程度と非常に少ない現状にあることが示された13)。 臨床研究の体制整備が遅れていた日本では、1990 年代に医薬品開発の国際調和の動きが加速し、 1997 年に製薬企業が主導で行う治験が GCP 省令によって法制化された。ところが、治験環境の整 備が先行したにも関わらず、臨床研究の方が置き去りにされた。その結果、治験と臨床研究の格差が 大きくなり、臨床研究の活性化が相対的にさらに遅れる状況になってしまった 14)。近年では、臨床 研究の体制整備として、産学官の組織や研究者によって、調査・研究が行われ、様々な方策が立てら れ、少しずつ改善してきている。しかし、今から12 年前の野本らの報告で、治験と比較して臨床研究が軽視され、信頼性の高いエビデンス創出のために臨床研究の基盤整備が必要だと議論されていた ことから15)、治験ではGCP 施行によって 1997 年以来十数年間で大きく前進したのに対し、臨床研 究では、多くの議論は10 年以上前とほとんど変化がないように考えられる。 医療の質を高め、国民の健康をよりよく守ることは、高齢化社会を迎えるわが国にとって、重要な 課題となっている。そのためには、最善であると考えられてきた予防方法、診断方法および治療方法 であっても、その有効性、効率性、利便性および質に関して、臨床研究によって、絶えず再検証され なければならない4)。これらの臨床研究によるエビデンスの強化によって、医療水準の向上、さらに は健康で長寿な社会が実現できると考えられる。したがって、わが国の臨床研究の基盤整備を行うこ とは、国民全体にとって極めて重要な役割を果たすことになるものと考えられる。 これまでのわが国の臨床研究の基盤整備のための調査・研究を取りまとめ、改正指針の周知徹底の 過渡期である現在の状態の評価を十分に行った報告はない。したがって、臨床研究の歴史的背景、制 度および実施体制に関して調査し、日本、米国、英国の比較を行って、わが国における臨床研究の制 度や実施体制について、今後のより良いあり方や改善策を提案することとした。また、これまで著者 が行った先行研究の継続研究となる医師および臨床研究コーディネーター(Clinical Research Coordinator、以下 CRC と略す)を対象とした臨床研究に関する意識調査により、わが国の臨床研 究に関する実際の医療現場での問題点・意識を検証するとともに、具体的に制度や実施体制について 整備していくべき事項や改善策を提案することとした。 なお、本研究では、治験、製造販売後臨床試験、使用成績調査・特定使用成績調査以外の「臨床研 究に関する倫理指針」の適用範囲である臨床研究を対象とした(表1)。 表1 臨床研究分類と本研究の対象(臨床研究推進ガイドライン16)より一部抜粋) Sponsor 資金提供者 Driver・主催者 治験 製薬企業 製薬企業 医師主導治験 国・財団 医師 製造販売後臨床試験 製薬企業 製薬企業 使用成績調査・特定使用成績調査 製薬企業 製薬企業 上記以外の臨床研究(遺伝子治療、 ヒト幹細胞治療は除く) 国・財団・製薬企業 製薬企業・医師 疫学研究 国・財団・製薬企業 製薬企業・研究者(医師) 本論文は二つの調査研究とそれらの総合考察から構成されており、概要は以下のとおり。 研究I:臨床研究の歴史的背景・制度・実施体制に関する国際比較とわが国における改善策 国内外における臨床研究の歴史的背景・制度・実施体制に関して調査し、日本、米国、英国の比較 を行うことで、わが国における臨床研究の制度や実施体制について、今後のより良いあり方や改善策 を提案した。 研究II:わが国における臨床研究の現状と改善策:医師と CRC を対象にした調査研究 医師およびCRC を対象にして、わが国の臨床研究の制度および実施体制における意識調査を実施 本研究 の対象
し、現状を分析した。今後の臨床研究推進のために、医療現場すなわち、臨床研究者および支援スタ ッフの視点から、具体的に制度や実施体制について整備していくべき事項や改善策を提案した。 総合考察 研究I および研究 II の結果を踏まえ、総合的な調査結果を取りまとめて考察するとともに、医療 の質を高め、国民の健康をよりよく守るために必須となる臨床研究を育てるための基盤整備について、 改善策を提案した。
2.研究 I:臨床研究の歴史的背景・制度・実施体制に関する国際比較とわが国におけ
る改善策
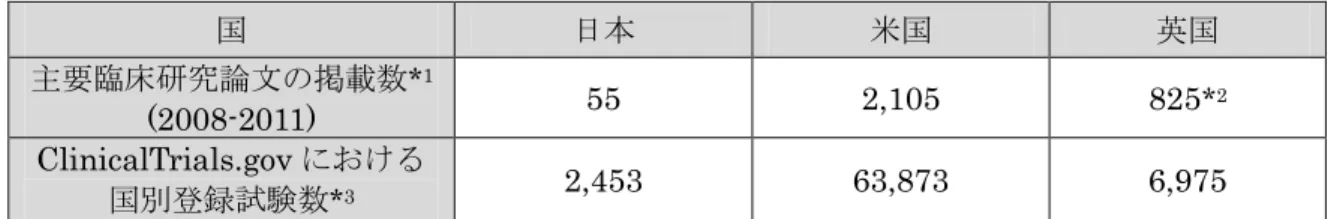
2-1.目的 わが国において、治験は1997 年より法制化され GCP 省令を遵守し実施されている。一方、臨床 研究では「臨床研究に関する倫理指針」というガイドラインに従って実施され、同じ人を対象とする 行為であるにも関わらず、法的な拘束なく行われている。しなしながら、欧米では、治験・臨床研究 ともに法制化されている。 本研究では、このようなわが国における治験と臨床研究のダブルトラックの成立の詳細と、米国お よび英国の諸制度と比較した場合の実施体制上の問題点等を抽出するために、文献調査等により国内 外の臨床研究の歴史的背景と制度の形成、実施体制に関して調査し、各国の比較を行った。さらに、 わが国の臨床研究の制度や実施体制について、今後のより良いあり方や改善策を提案した。 2-2.方法 2-2-1.調査対象国 調査対象国を日本、米国、英国とした。調査対象国(日本以外)の選定理由は、主要臨床研究論文(New Engl J Med、Lancet、JAMA) の掲載数である。米国は世界で圧倒的に多く2,105 報(日本の約 38 倍)、次に多い英国は 825 報(日 本の15 倍)であった17)。また、米国NIH(National Institutes of Health、以下 NIH と略す)の
データベースClinicalTrials.gov.18)を用いて同一のデータベース中の実施国での比較を行ったところ、
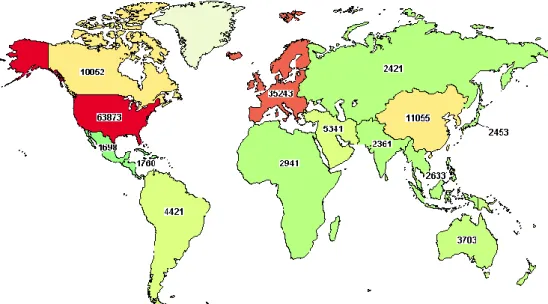
わが国では2,453 試験であったのに対し、米国は 63,873 試験(日本の約 26 倍)、英国は 6,975 試験 (日本の約2.8 倍)であった。
また、英国では、世界初の比較対照試験がジェームス・リンド(James Lind、1716 年 - 1794 年) によって実施されている。リンドは壊血病の予防法としてオレンジとレモンが有効であることを臨床 試験により科学的に証明している。さらに、R.A.フィッシャー(Sir Ronald Aylmer Fisher、1890 年- 1962 年)は生物統計学の基礎を築き、実験計画法を確立しており、医学研究への貢献度は高い。 英国の疫学者であるA.コクラン(Archibald Cochrane、1909 年 - 1988 年)は、世界中の臨床試験 のシステマティック・レビューによる根拠に基づいた医療(Evidence-based medicine、以下 EBM と略す)のための情報インフラストラクチャーを提唱している。このように、英国は臨床研究の発祥 の地であり、臨床研究への貢献度が高い国である。
以上の背景から、医学論文としての成果が多く、臨床研究実施数が多い米国と、古くから臨床研究 が実施されてきた英国の実態を調査することから、わが国における臨床研究の制度や実施体制につい て、今後のより良いあり方や改善策を検討するヒントを得ることとした。
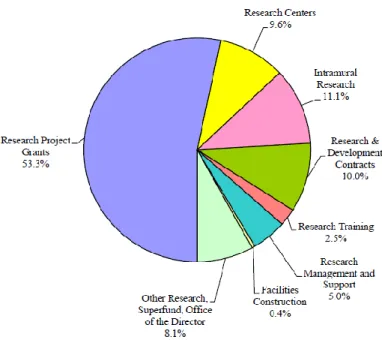
表2 主要臨床研究論文の掲載数と ClinicalTrials.gov における国別登録試験数 国 日本 米国 英国 主要臨床研究論文の掲載数*1 (2008-2011) 55 2,105 825*2 ClinicalTrials.gov における 国別登録試験数*3 2,453 63,873 6,975 *1主要基礎・臨床医学論文掲載数の国際比較17)より引用 *2 イングランド(685)とスコットランド(140)の総計 *3 2012 年 9 月 14 日現在の See Studies on Map19)による試験数
2-2-2.調査媒体
国内外の臨床研究の歴史的背景と各国および国際機関における臨床研究の現状や課題、規制および 制度的システムを把握するために、PubMed、医中誌 Web、NII 論文情報ナビゲータ、NDL-OPAC (国立国会図書館蔵書検索システム)等を用いて文献検索を行った。また、最新の国際機関や各国の 状況を把握するために、世界保健機関(World Health Organization、以下 WHO と略す)、被験者 保護および臨床研究を管轄する各国政府機関(日本、米国、英国)のホームページ等を対象に調査を 行った(参考資料1)。 2-2-3.調査項目 調査項目は、先行研究等で指摘されている問題点や、研究Ⅱにおける医師とCRC を対象にした調 査研究の結果にて抽出された課題を参考として決定した。 本研究の調査項目は、以下のとおりである。 1) 人を対象とした研究に関する歴史と研究倫理ならびに制度の形成 2) 臨床研究に関する法体系と臨床研究に関する規制または制度 3) 臨床研究に関する組織体制 4) 臨床研究の研究予算および資金 5) 臨床研究の登録と結果の公表 2-3.調査背景と結果 2-3-1.人を対象とした研究に関する歴史と研究倫理ならびに制度の形成 2-3-1-1.調査背景(調査項目とした理由) 米国では人体実験の倫理的問題の提起により、生命倫理への関心が高まり、諸規制の整備が行われ てきたのに対し、わが国では第二次世界大戦中に行われた組織的な医学犯罪に対する総括と反省を行 っていないといわれている20)。 同様に、米国では 1960 年代から臨床研究をめぐる倫理的な非行が問題となって、1966 年に非人 道的人体実験がヘンリー・ビーチャーにより告発され21)、1974 年に国家研究法(National Research Act)が制定された。さらに、1979 年には、研究における被験者保護のための倫理原則と指針を定め たベルモント・レポート(The Belmont Report: 生物医学・行動学研究における被験者保護のため の倫理原則およびガイドライン、以下、ベルモント・レポートと略す)が報告されるなど、臨床研究 を実施するための原則が定められた。一方、日本では、大きく遅れて、1990 年代の後半になって医 薬品開発の治験に関してのみ世界標準を採用し、臨床研究の法的整備はなおざりにされてきた、と中
野は報告している14)。 さらに、生命倫理の日本への伝わり方は歴史の前半部分(人を対象とした研究に対する研究倫理史) をスキップして、日常診療の倫理として海外から脈絡なく伝わったとされている22)。より危険度が高 い可能性のある臨床研究を対象とする研究倫理は、それ以前に解決済みでなければならなかったにも かかわらず、後回しにされてきた。したがって、臨床研究を対象とする研究倫理は未熟なままになっ ている。本来ならば国民の手でしっかり決着をつけるべきであった第二次世界大戦中の非人道的人体 実験の責任が、うやむやにされてしまい、ほとんど反省らしい反省がなされなかったと、笹栗は述べ ている22)。 以上より、これらの人を対象とした研究に関する歴史と研究倫理ならびに制度の形成の実態につい て各国の比較調査を行った。 2-3-1-2.日米英の国際比較結果 人を対象とした研究に関する歴史と研究倫理ならびに制度の形成の比較の調査結果を表3 に取り まとめた。
表3 人を対象とした研究に関する歴史と研究倫理ならびに制度の形成の比較 ■人を対象とした研究に関する歴史等 日本 米国 英国 1940 年 代 以前 戦争中 ナチスドイツ、強制収容所内において捕虜・囚人を使った人体実験が広範囲に行われる 731 部隊による人体実験 1945-47 カリフォルニア大、シカゴ大、ロチ ェスター大で、プルトニウムの人体 注射実験 1945 九州大学生体解剖事件 1947 ミッチャーリヒ「人間性なき医学」 ヴァイツゼッカー「安楽死と人体実 験」 1947 ニュルンベルク・コード 1950 年代 1952 新潟精神病院ツツガネムシ 病菌接種事件 1956-72 ニューヨーク州スタッテン島ウィロ ーブルック州立学校(知的障害児施 設)における肝炎感染実験 1952 名古屋市乳児院大腸菌感染 実験 1960 年代 1960 薬事法施行 1960-1 970 人類遺伝諮問機関(HGAC、国家生命倫理委員会の役割も持つ)中 心に研究倫理への対応を行う 1961-2 サリドマイド事件 1963 キセナラミン事件 1962 FD&C 法改正 1964 ヘルシンキ宣言 1966 南光病院事件 1966 ビーチャー「倫理と臨床研究」論文 NIH と FDA で倫理指針を発表 1966 最初の研究審査委員会(REC)が 設立 1967 英国王立内科学会(RCP)の最初 のガイダンスが出される 1969 広島大学原爆放射能研究所 がん治療実験 1967 パップワース「人間モルモット」 バーナードによる心臓移植手術 1968 薬 事 法 (Medicines Act 1968 Part)の施行 1970 年代 1972 タスキーギ研究、マスメディアの批 判により終了 1973 RCP が倫理委員会の運営に関す るガイドラインを発表 1974 国家研究法 1975 「ヘルシンキ宣言」東京改訂 1979 ベルモント・レポート 1975 保健省が各保健当局に倫理委員会 の設置を呼びかけた 1980 年代 1982 治験データ捏造事件 1983 「ヘルシンキ宣言」ベニス改訂 1989 米国保健福祉省(HHS)は科学公正 局(OSI)と科学公正監査局(OSIR) 設立 1984-1 995 ブリストル王立病院での小児心臓 手術の死亡率が異常に高いことが 指摘された 1989 「ヘルシンキ宣言」九龍、香港改訂 1990 年代 1990 旧GCP 施行 1991 コモン・ルール成立 1990-1 993 新生児が親の許可無くベンチレー ターを用いる研究に組み入れられ る 1992 研究公正局(ORI)設立 1991-1 993 小児の死体から摘出した臓器が研 究用に保存される 1991 地域倫理委員会(LREC)制度整 備 1993 CIOMS 人を対象とする生物医学研究の国際倫理指針 1993 ソリブジン薬害事件 1996 ICH「医薬品の臨床試験の実施に関する基準」(ICH-GCP) 1996 「ヘルシンキ宣言」サマーセットウェスト改訂 1997 薬事法改正、新GCP 制定 1999 ゲルシンガー事件 1997 倫理委員会連合会(AREC)発足、 多施設研究倫理委員会(MREC) 制度整備 1998 GCP 施行 2000 年 以 降 2000 「ヘルシンキ宣言」エジンバラ改訂(COI 追加) 2002 「ヘルシンキ宣言」ワシントン改訂 2003 「臨床研究に関する倫理指 針」制定 2002 利益相反に関するAAMC の提言 2000 イングランドのMREC、LREC を 中央で調整する機関である中央倫 理委員会(COREC)の設置 2001 EU 臨床試験指令発令 2004 EU 臨床試験指令の英国での施行 2004 「ヘルシンキ宣言」東京改訂 2006 TGN1412 事件 2007 MHRA より第 I 相試験実施施設認 定基準が提示される 2008 「ヘルシンキ宣言」ソウル改訂 Ⅰ.日本 日本での代表的な非人道的人体実験を行った始まりとなるのは、第二次世界大戦中に関東軍の七三
一部隊である。七三一部隊は、旧日本陸軍の、医学者・医者を中心とした部隊として構成され、部隊 の本部は中国ハルビンの郊外平房に置かれていた。その発足から日本の敗戦によってその活動を終え るまでの約10 年間に約 3 千人の人々を生物兵器開発のための人体実験で殺していた23)。さらに、い わゆる九州大学生体解剖事件は、1945 年に福岡県福岡市の九州帝国大学(現在の九州大学)医学部 の敷地内の解剖台の上で米軍捕虜に対する外科手術と死後の解剖が行われた事件である。横浜裁判や その後の調査により事実関係はある程度解明されたが、後に関係者が医学界で活躍したせいもあって か、長い間、表立って生命倫理の話題として議論されることはなかった 24)。その詳細は、実際に医 学生として立ち会うことになった東野利夫著「汚名:九大生体解剖事件の真相」に記載されている 25)。 また、戦後の日本においても、問題性を持った非倫理的事件が起こったが、公的な検討を経た記録 として公表されたものは比較的少なく、法廷で裁かれることもなかった 26)。歴史的文脈の欠如と一 致して、日本の研究倫理は戦後 40 年以上にわたってほとんど放置され、その間、数多くの人々が臨床 試験による健康被害に遭ってきたと考えられる。1952 年名古屋市乳児院大腸菌感染実験、同年の新潟 精神病院ツツガネムシ病菌接種事件、1963 年のキセナラミン事件、1966 年の南光病院事件、1969 年広島大学原爆放射能研究所がん治療実験、等が起きたが、表面化したのは、ごく一部だといわれて いる。日本に倫理指針を作る動きがやっと始まったのは 1980 年代であり、自発的に始めたと言うよ り、医薬品開発競争上の圧力や諸々の不祥事により、取り掛からざるを得なかった、という流れであ った22)。 1990 年から医薬品開発のための治験のルールとして旧 GCP が施行されたが、1993 年にソリブジ ン薬害事件が起こった。ソリブジンは帯状疱疹の治療薬として、当時の厚生省から承認された医薬品 であるが、全国の皮膚科で行われた治験段階ですでに 3 例の死亡例が出ていた。しかし、いずれも がん患者であったことから、ソリブジンとの因果関係の判断が曖昧なままになっていた。実際には、 抗がん剤の5-FU が使用されていた患者にソリブジンが併用された患者で、ソリブジンが 5-FU の代 謝を阻害するという薬物相互作用が生じていたために、5-FU の毒性が強く出たためにがん患者の死 亡を早めてしまった、というのが実情であった。つまり新しいタイプの薬害が生じたものである。し かし、厚生省の承認を得て販売した会社は、ソリブジンの医薬品情報を提供する際に強い警告を発す ることもなかった。そのため、発売後1 か月で 15 名もの死者が出た。そのため、厚生省に「医薬品 安全性確保対策検討会」が設けられ、わが国の治験の実施法、新薬承認審査のあり方、市販後医薬品 の安全性確保のあり方について、議論され、対策が練られた。わが国における治験の実施体制につい ても、根本的な検討が行われた。 その後、薬害エイズ事件等の薬害事件が相次ぎ、1997 年には薬事法が改正され、医薬品の臨床試 験の実施の基準に関する省令(新GCP 省令)が制定された。日米欧三極医薬品規制ハーモニゼイシ ョン国際会議(以下、ICH と略す)の流れに従い、1998 年から新 GCP 省令が施行された22)。丁度、 1990 年代に入り、医薬品開発に要する費用、時間、人的資源を節約するために、医薬品の開発は地 球規模でグローバルに行おうという動きが、日米欧の三極の間で進んでいた。そこで、日米欧の間で 合意された「ICH-GCP」とわが国の旧 GCP の間で調和をさせる必要が生じて、旧 GCP を大改定す る形で、新GCP 省令が誕生した。この新 GCP 省令は薬事法の中に位置づけられており、遵守を義 務付けた法制化が行われたわけである。新GCP 省令の基準では,治験総括医師が治験責任医師へと 変わり、薬剤部が治験薬を管理し、インフォームド・コンセントは文書で得ることを義務付け、当時
の厚生省への報告義務が示された。このようにして被験者保護が篤くなり、治験審査委員会の機能が 強化されて、現在に至っている。一方、臨床研究に関しては、「臨床研究に関する倫理指針」が、2003 年7 月に制定され、2004 年 12 月および 2008 年 7 月に全部改正が実施され、2009 年 4 月より改正 指針が施行されている4)。 しかしながら、GCP 省令も含めて、何か社会問題が起こるたびに、問題の部分だけにつぎを当て るような指針を関係各省が縦割り行政によって作ってきたため、日本の研究倫理規制は、無計画で系 統立っておらず、各指針間に整合性がとれていないと指摘されている。このことが、今日の倫理審査 や研究者等の現場に混乱を招く原因の一つとなっている22)。 以上のような流れより、治験に関しては、欧米の研究倫理の歴史および国際化の動向に合わせて規 制の整備がなされてきた。しかし、日本の生命倫理には、歴史的文脈がなく、米国であったような研 究倫理の歴史(非人道的な人体実験の反省に基づく生命倫理の形成のプロセス)をスキップして、そ の成果だけが伝わり、しかも主として日常診療の倫理として脈絡なく伝わったため、より危険度が高 い可能性のある臨床研究を対象とする研究倫理は、後回しにされ、未だに取り残されたままになって いる20, 22)。 Ⅱ.米国 米国において、人を対象とした研究に関する被験者保護に対する研究倫理の形成は、20 世紀半ば の、第二次世界大戦直後から始まった。1947 年に、国際軍事裁判所はナチスドイツの戦争犯罪行為 である残虐な人体実験への反省から、人類の歴史上、このような悲惨な事件が繰りかえされないよう に、歯止めとして、倫理綱領であるニュルンベルク・コードを発表した。米国が起草したニュルンベ ルク・コードは、人を対象とした研究に関する初の国際的ガイドラインとなった。ニュルンベルク・ コードの影響を受けて、臨床研究にインフォームド・コンセントが取り込まれたが、当初は制度とし ては不十分であった。その後にも、ニューヨーク州スタッテン島ウィローブルック州立学校(知的障 害児施設)における肝炎感染実験が発生した。そして、新薬の臨床試験については、サリドマイド事 件を発端として、1962 年に米国食品医薬品局(FDA: Food and Drug Administration、以下 FDA) では食品・医薬品・化粧品法(Food, Drag, Cosmetic Act)を改正し、治験については規制を強化し た。 一方、臨床研究は、戦後 20 年間程度、法規制がないまま推し進められた。1966 年に非人道的人 体実験がヘンリー・ビーチャーにより告発され、それを受けて同年にFDA は人体実験および臨床試 験に関する新たな指針を、NIH は医療におけるヒトを対象とする臨床的研究への指針(連邦政府か らの助成を受ける場合)を公表するが、法的強制力がないものとなっていた。したがって、黒人男性 約 600 人を対象に米国連邦政府公衆衛生局が行った梅毒研究タスキーギ事件が起こり、非人道的人 体実験が払拭されることはなかった。 これらの背景より、米国内の社会的な論議が活発化し、研究倫理に関して連邦議会で論議されるこ ととなり、1974 年に医学研究全般にわたる規制を目的とした法律である国家研究法が成立した。こ の法のもとに設置された委員会が作成し、1979 年に全米の臨床研究の倫理基準となる、ベルモント・ レポートが報告された。このベルモント・レポートは現在も米国の研究倫理の基礎となり、倫理審査 の基準として用いられている。さらに、ベルモント・レポートの報告を受け、研究倫理に関する行政 令の21CFR50 と 21CFR56(FDA)、および 45CFR46(NIH)を発している。しかし連邦政府全体
を包括する普遍的ルールを作ることが大統領委員会から勧告されたため、1991 年、全ての関係省庁 が参加した共通規則(コモン・ルール)が成立し施行された。これが現在の人を対象とした研究に関 する規制法令となっている20-22, 26-31)。さらに1999 年には、ゲルシンガー事件が勃発した。金銭的な
利益相反(Conflict of Interest:以下 COI と略す)とインフォームド・コンセントに問題があると して、2000 年にはヘルシンキ宣言に COI に関する事項が追記され、2002 年には利益相反に関する AAMC(Association of American Medical Colleges:全米医科大学協会)の提言が発行された32-33)。
Ⅲ.英国
英国における研究倫理の基本は、ニュルンベルク・コードとヘルシンキ宣言にある。遺伝子操作の 技術が発展する1960~1970 年代にかけては、HGAC(Human Genetics Advisory Committee:人 類遺伝諮問機関、国家生命倫理委員会の役割も持つ)が中心となって、研究倫理への対応をしていた と思われる。英国の医療提供体制は、保健省管轄のNHS(National Health Service:国民保健サー
ビス、以下NHS と略す)により行われており、地域ごとに置かれた保健当局が主体となって、倫理
委員会が設立された。1966 年に最初の研究審査委員会(REC)が設立された。その後、1967 年に 英国王立内科学会より査審基準となる最初のガイダンスが出され、その後数回ガイダンスが発行され た。
一方、1962 年に明らかになったサリドマイド事件に対応して、保健大臣より 1968 年に薬事法 (Medicines Act 1968 Part)が施行され、医薬品を臨床試験のために供給しようとする者は、承認 申請目的でなくとも保健大臣に市販承認申請と同様の詳細なエビデンスを提出し、臨床試験許可証 (Clinical Trial Certificate、以下 CTC と略す)を取得することが求められた。なお、1968 年の薬 事法の臨床試験の定義は、患者のみを対象とした試験としていた。そのため、健常人試験については、 業界の自主規制に委ねられていた。また、CTC のシステムについては、手続きが煩雑であるため、 安全性上の問題が少ないとされるものや医師が患者に使用する場合には、図1 に示す DDX(Doctors and Dentists Exemption:医師特例許可証)、CTMP(Clinical Trial of a Marketed Product:製造 販売臨床試験)、CTX(Clinical Trial Exemption:臨床試験免除)と称する手続き簡略化のための制 度が導入された。
ところが、1980 年後半から 1990 年前半にかけて数々のスキャンダルが発覚した。主なものとし て、North Starffordshire の病院で新生児が親の許可なくベンチレーターを用いる研究に組み入れら れていたという事件があり、2000 年に調査報告がまとめられた。この事件が研究倫理の枠組みの一 つとなった 34)。これらの背景より、倫理委員会が発展し、1991 年には、地域倫理委員会(Local
Research Ethics Committee、以下 LREC と略す)制度整備が実施された。1997 年には、倫理委員 会連合会(Association of Research Ethics Committee、以下 AREC と略す)が発足し、多施設研究 倫理委員会(Multicentral Research Ethics Committee、以下 MREC と略す)制度整備が実施され た。さらに、英国のMREC、LREC を中央で調整する機関である中央倫理委員会(Central Office for Research Ethics Committee、以下 COREC と略す)が設置された。
2001 年に、EU は、EU 臨床試験指令を発令し、英国では 2004 年 5 月より施行された。これによ り、当局による研究計画の届出・許可制度が、アカデミアの研究や健常人対象の研究にも適用される ようになった。また、倫理委員会による審査のシステムも一律に法規制として整備された10), 35-36)。
全、頭頚部膨張(血管浮腫)、呼吸困難等の全身症状の有害事象が発生し、ICU に搬送されるという 重大な事件が発生した。事件発生を受けて、英国の規制当局である英国医薬品庁(Medicine and Healthcare products Resulatory Agency、以下 MHRA と略す)が外部調査委員会を設置して調査を 実施し、報告書を発表した。これより、MHRA より第 I 相試験実施施設認定基準が提示された。同 様に、欧州医薬品庁、英国製薬工業協会が第I 相試験実施ガイドラインを発行した37-38)。
基本的手続き
CTC (Clinical Trail Certificate:
臨床試験許可制度)
IMP (investigational medical product)を臨床試験のために供給
しようとするものは、申請データを提出し、CTC を取得しなけ
ればならない。MCA(Medical Control Agency:医薬品管理機構) が諮問。
例外
DDX (Doctors and Dentists Exemption:医師特例許可証)
医師・歯科医が、商業的組織のためではなく自分の患者のみに使
う場合は、DDX スキームに基づいて MCA に通知。
CTMP (Clinical Trial of a
Marketed Product) 市販承認をすでに得ている薬剤については、CTMP スキームに基づきMCA に通知
CTX (Clinical Trial Exemption) DDX に該当しない者が CTC を得る場合に提出すべきデータの サマリーを、臨床試験の必要性などについての医師の証明書を添
付しMCA に提出、CTC の手続きを簡略化。
規制対象外
健常人対象の試験 臨床試験に該当せず、企業・アカデミアの自主規制
イギリス臨床試験規制により一本化
CTA (clinical trial
authorization: 臨 床 試 験 許 認可) これまでのCCT、DDX、CTMP、CTX の手続きおよび健常人試験が すべて EU 臨床試験司令に対応したイギリス臨床試験規制による CTA に一本化、GMP/GCP 準拠が求められる。 図 1 臨床試験許可証(CTC)とその他の制度が臨床試験許認可(CTA)へ 1 本化(EU 臨床試験 指令とイギリス臨床試験規則10)より引用) 2-3-1-3.調査結果に基づく提言 先行研究のとおり、臨床研究を対象とする研究倫理は、米国や英国では人体実験の反省より形成さ れ、ニュルンベルク・コードとヘルシンキ宣言に伴って世界的な生命倫理の形成をリードする形で制 度整備が進められてきた。ところが、わが国では、第二次世界大戦中に人体実験が行われ、戦後も数 多くの人々が臨床試験による健康被害に遭ってきたにも関わらず、社会的な生命倫理に対する指摘の 原動力とはならなかった。そのため、研究倫理の形成と制度整備が何もされてこなかったことが明ら かになった。1997 年に治験では、医薬品開発の国際化の流れに従い、薬事法とそれに基づく ICH-GCP に対応した新 GCP 省令が制定された。一方、治験以外の臨床研究においては、未だに米国や欧州の 流れと協調することなく、現在に至っていることが確認された。 臨床研究は、被験者の協力により初めて成立する営みであり、非人道的な行為があってはならない。 被験者が安心して参加できる臨床研究を実施することを目的とし、今後の臨床研究の倫理性、科学性、 品質の向上のために、これまでの人を対象とした研究の健康被害の反省を踏まえ、被験者保護の在り 方について、制度整備を含めた議論することが望ましいと考えらる。
2-3-2.臨床研究に関する法体系と臨床研究に関する規制または制度 2-3-2-1.調査背景(調査項目とした理由) わが国では、厚生労働省への医薬品の製造販売の承認申請を目的として実施される治験は法制化さ れており、薬事法に基づくGCP 省令に則って実施される1)。一方、人を対象とした臨床研究は、法 制化されておらず「臨床研究に関する倫理指針」のガイドラインに従って実施されている4)。 先行研究において、黒川は、わが国では人間を対象とする試験研究を計画・実施してはいけないと する決まりは見あたらず、全体をカバーする法律の不在の下で、治験だけが薬事法に規定され、信頼 性確保などのためGCP 省令などの規制を受けていると述べている。一方、臨床研究を扱う法律がな い中で何らかの目安が必要になった結果、大臣告示などで、それを定めたものと考えることができる、 と述べている39)。 以上より、本調査研究では、これらの臨床研究に関する法体系と規制または制度の実態について詳 細な各国の比較調査を行った。 なお、比較にあたっては、各国の法体系が異なることから、比較検討のために、法体系について述 べた後に、臨床研究および治験の実施基準について詳細を述べた。 2-3-2-2.日米英の国際比較結果 臨床研究および治験の実施基準についての調査結果を、表 4 に取りまとめた。また、臨床研究に 関する規制または制度の各国状況の調査結果を、表5 に取りまとめた。 表4 臨床研究および治験の実施基準 ○:法制化されている、×法制化されていない 国 日本 米国 英国 全体をカバーする法 律の有無 × (なし) ○ 国家研究法 (1974 制定) ○ EU 臨床試験指令 (2004 施行) 臨床研究の実施基準 法制化有無 × 臨床研究に関する倫理 指針(ガイドライン) (2003 制定) ○ コモン・ルール (1991 成立) ○ ICH-GCP *2 (1996 制定) 治験の実施基準 法制化有無 ○ GCP 省令 (1997 制定) ○ 21CFRs*1 ○ ICH-GCP *2, 3 (1996 制定)
*1 21 CFR Part 50: Protection of Human Subjects、21 CFR Part 56: Institutional Review Boards 等を 含み、ICH-GCP に準じた基準となっている。
*2 EU 臨床試験指令に基づき、ICH-GCP を採用している。
表5 臨床研究に関する規制または制度の各国状況 ○:必須、×必須でない、△明示できない 国 日本 米国 英国 規制または制度根拠 臨床研究に関する倫理指 針(ガイドライン) コモン・ルール EU 臨床試験指令*1 対象範囲 疾病の予防・診断・治療方法 の改善、疾病原因および病態 の理解並びに患者の生活の 質の向上を目的として実施 される医学系研究 連邦の補助金を受け ている機関で行われ る臨床研究すべて 販売承認目的に限ら ずあらゆる臨床試験 当局への計画届出 × △*2 ○ IRB 審議 ○ ○ ○ 文書による説明と同意 ○ ○ ○ モニタリング・監査 △*3 ○*4 ○ 有害事象報告 ○ ○ ○ 健康被害の補償措置 ○*5 ×*6 ○*7 臨床試験の事前登録 ○ ○*8 ○ 研究者への教育・研修 ○ ○ ○ IRB 委員への教育 ○ ○ ○ *1 EU 臨床試験指令に基づき、臨床研究実施の基準は ICH-GCP を採用している。
*2 被験者保護局(政府中央組織)(OHRP: Office for Human Research Protections、以下 OHRP と略す) によって米国連邦保証制度(FWA: Federal Wide Assurance、以下 FWA と略す)を得た医療機関の IRB へ計画届出を行う。未承認の新規の医薬品侯補物質については、治験・臨床研究に限定することなく、 IND(IND: Investigational New Drug applications)申請が必要であり、FDA が医薬品の安全性の 審査や認可を行う。 *3 臨床研究機関の長が自己点検し、国の調査に協力する。 *4 被験者の安全性を高めるために研究をモニタリングする過程が用いられていることが必要。また、 国の監査に協力する。 *5 4 社で臨床研究保険を販売。必ずしも金銭的な補償を行う義務が生ずるものではなく、文書同意が必要。 *6 被験者への説明事項。無過失補償の制度的規定はない。 *7 研究実施者とスポンサーが責任を果たすための保険または補償が用意されている。 *8 全ての臨床研究は「臨床研究の登録義務:PUBLIC‐LAW110-85(2007 年 9 月 27 日)」に従い、NIH のデータベースへ登録する。 Ⅰ.日本 日本の法体系は、付録1 のとおり。 わが国の被験者保護に関する法体系は、大きくは、医薬品開発のための治験とその他の臨床研究の 場合に分かれる。厚生労働省への医薬品の承認申請のための臨床試験である治験の場合、薬事法(法 律)に含まれるGCP 省令(省令)によって、厳格な法規の下、被験者保護システムが保たれている。 一方、薬事法・GCP 省令の対象外の医薬品開発を目的としない臨床研究に関する法令は存在しない。 臨床研究に関しては、各局長レベルでの通達・先例等にあたる厚生労働省が定める指針(ガイドライ ン)は、以下の9 つ存在する。さらに研究倫理に関する指針・法律では 9 指針に加えて、利益相反・ 個人情報保護に関する指針・法律も含まれる(表6)41)。
表6 医学研究に関する指針等 医 学 研 究 に 関 す る 指 針 ヒトゲノム・遺伝子解析研究に関する倫理指針 平成20 年 12 月 1 日一部改正 疫学研究に関する倫理指針 平成20 年 12 月 1 日一部改正 遺伝子治療臨床研究に関する指針 平成20 年 12 月 1 日一部改正 臨床研究に関する倫理指針 平成20 年厚生労働省告示第 415 号(平成 21 年 4 月 1 日より施行) 手術等で摘出されたヒト組織を用いた研究開発の在り 方 平成10 年 12 月 6 日 ヒト幹細胞を用いる臨床研究に関する指針 平成22 年 11 月 1 日全部改正 厚生労働省の所管する実施機関における動物実験等の 実施に関する基本指針 平成18 年 6 月 1 日施行 異種移植の実施に伴う公衆衛生上の感染症問題に関す る指針 平成 13 年度厚生科学研究費厚生科学特 別研究事業 ヒト受精胚の作成を行う生殖補助医療研究に関する倫 理指針 平成22 年 12 月 17 日文部科学省 厚生労 働省告示第2 号 厚生労働科学研究における利益相反(COI: Conflict of Interest)の管理に関する指針 平成20 年 3 月 31 日 個人情報の保護に関する法律 平成15 年法律第 57 号 図2 臨床研究からみた各指針の範囲イメージ(「臨床研究に関する倫理指針」の改定について42)よ り引用) わが国の臨床研究実施に係る厚生労働省から発せられているガイドラインである「臨床研究に関す る倫理指針」は、平成15 年 7 月 30 日に最初に示された42)。その後、平成16 年 10 月の世界医師会 総会が東京で開催され、ヘルシンキ宣言の修正に伴う個人情報の保護に関する法律と関連する指針と の整合性をとるために全部改正がなされた。さらに、初めの制定から 5 年毎に見直しを行うことと されており、平成 20 年 7 月の改正は、「ヒトゲノム・遺伝子解析研究に関する倫理指針」(平成 16 年12 月 28 日全部改正、平成 17 年 6 月 29 日一部改正)、および「疫学研究に関する倫理指針」(平 成19 年 8 月 16 日全部改正)との整合性をとるとともに、「臨床研究に関する倫理指針」に対する検
討課題の解決を目的になされ(厚生科学審議会科学技術部会臨床研究の倫理指針に関する専門委員 会・金澤一郎委員長:第1 回平成 19 年 8 月 17 日~第 9 回平成 20 年 7 月 10 日、平成 20 年 7 月 23 日第 46 回厚生科学審議会科学技術部会承認・垣添忠生部会長)、厚労官医政局長より関係機関に通 知され、平成21 年 4 月 1 日から実施された4)。平成20 年の改正では、専門委員会の入念な検討の 末、被験者の生命、健康、プライバシーおよび尊厳を守ることを示した制度が策定された。要点は以 下4 点となる43 -45)。 ① 被験者に対するインフォームド・コンセントの義務付け、説明事項の規定 ② 実施研究機関における被験者の人権保護(情報保護等) ③ 倫理審査委員会の機能(臨床研究実施の適否・継続の審査等) ④ 研究者、臨床研究機関の長等の遵守事項の規定 (1) 適用対象 疾病の予防・診断・治療方法の改善、疾病原因および病態の理解並びに患者の生活の質の向上を 目的として実施される医学系研究を適用対象とする。また、日本国内外において実施される臨床 研究を対象とするが、この指針と比較して当該実施地の法令、指針等の基準が厳格な場合には、 当該基準に従って臨床研究を実施することとする。 なお、次のいずれかに該当するものは、適用対象外とする。 (ア) 診断および治療のみを目的とした医療行為 (イ) 他の法令および指針の適用範囲に含まれる研究 (ウ) 試料等のうち連結不可能匿名化された診療情報(死者に係るものを含む。) 図3 「臨床研究に関する倫理指針」の適用範囲についてのディシジョンツリー(「臨床研究に 関する倫理指針」(改訂)についてのQ&A46)より引用)
(2) 研究者等 研究者等について、要点は以下のとおりとなっておる。 (ア) 被験者または代諾者等によりインフォームド・コンセントを受ける。 (イ) 研究責任者は、臨床研究を実施し、または継続するに当たり、臨床研究機関の長(以下、 機関の長と略す)の許可を受ける。 (ウ) 研究者等は、臨床研究の結果を公表する場合には、被験者を特定できないように行う。 また、利用目的の達成に必要な範囲を超えて、個人情報を取り扱ってはならない。個人 情報の取扱いに関する被験者等からの苦情・問い合わせの適切かつ迅速な対応に努める。 (エ) あらかじめ、登録された臨床研究計画の内容が公開されているデータベース(国立大学 附属病院長会議、財団法人日本医薬情報センターおよび社団法人日本医師会が設置した ものに限る。)に当該研究に係る臨床研究計画を登録する。 (オ) 臨床研究を終了したときは、機関の長にその旨および結果の概要を文書により報告する。 (カ) 研究者等は臨床研究に関する倫理そのほか必要な事項の講習を受けなければならない。 (3) 臨床研究機関と倫理審査委員会 臨床研究機関と倫理審査委員会について、要点は以下のとおりとなっている。 (ア) 機関の長は、臨床研究計画がこの指針に適合しているか否かその他臨床研究の適正な実 施に関し必要な事項について、あらかじめ、倫理審査委員会に審査を行わせなければな らない。 (イ) 機関の長は、外部機関の倫理審査委員会に審査依頼できる(従来から存在する規定であ るが、よりその使用を促すものである)。 (ウ) 機関の長は会議に出席できるが、委員就任・審議・採決は不可。 (エ) 軽微な事項の審査については迅速審査ができる。 (オ) 機関の長は手順書を作成する。 (カ) 倫理審査委員会は、手順書・名簿・会議の概要記録を公表する。 (キ) 機関の長は自己点検・評価を行う。 (ク) 機関の長は毎年 1 回厚生労働大臣等へ委員名簿・開催状況そのほか必要な事項を報告し、 調査に協力する。 (ケ) 倫理審査委員の教育・研修を行う。 (コ) 機関の長は指針と適合していない程度が重大であることを知った場合、速やかに倫理審 査委員会の意見を聴き、必要な対応をしたうえで、その内容を厚労大臣等に報告し、公 表する(過去に実施されたものを含む)。 (サ) 倫理審査委員会の構成員 ① 医学・医療の専門家等自然科学の有識者、法律学の専門家等人文・社会科学の有識 者および一般の立場を代表する者から構成され、かつ、外部委員を構成員として含 む。また、男女両性とする。 ② 審議または採決の際に自然科学分野だけではなく、人文・社会科学分野または一般 の立場を代表する委員が1名以上出席する。
③ 機関の長および臨床研究に携わる者は、審議または採決に参加してはならない。 (4) 健康被害・不具合 健康被害・不具合についての要点は以下のとおりとなっている。 (ア) 臨床研究に伴い被験者に生じた健康被害の補償のための保険等必要な措置をとる(金銭 の支払い以外に医療の提供・物またはサービスの提供を含む)(図4)。 (イ) 機関の長は,重篤な有害事象・不具合などが発生した場合は速やかに必要な対応をした うえで、倫理審査委員会の意見を聴き、措置をとる。 (ウ) 予期しない重篤な有害事象・不具合などが発生した場合は、(イ)の対応の状況・結果を 公表し、厚労大臣に逐次報告する。 (エ) 研究責任者は、毎年一回、臨床研究の進捗状況並びに有害事象および不具合等の発生状 況を臨床研究機関の長に報告する。 図4 健康被害に伴う補償措置を求められる範囲(「臨床研究に関する倫理指針」の改定について43) より引用) (5) 匿名化 連結可能匿名化、連結不可能匿名化を明記した。他の指針において規定され、概念は周知された 状況にあり、他の指針との整合性をとるために明記された。 (6) 指針の運用 国・関係機関からの研究費について指針の遵守が交付要件となることから、指針違反があった場 合は交付規則などにより研究者等に罰則などが課せられることがある。機関の長は指針の遵守を 徹底し、それに従わない研究者等に対して適切に是正措置などの対応を行う。 Ⅱ.米国 米国の法体系は、付録 2 のとおりとなっている。米国は、連邦制を採用しているため、連邦法(federal law)と州法(state law)との関係が問題になるが、両者は別個独自のものとされ、この二元性がアメ
リカ法の特徴となっている。
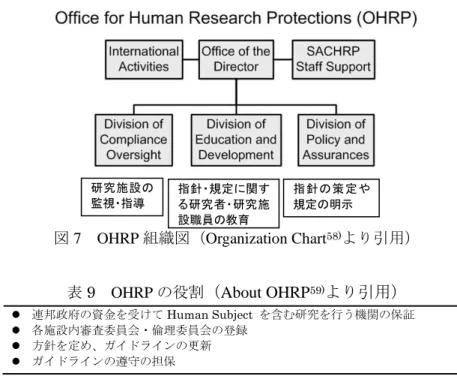
米国の被験者保護に関する法体系は、新薬の臨床試験の実施に適用されるFDAの規則(21 CFR Part 50: Protection of Human Subjects、21 CFR Part 56: Institutional Review Boardsを含む)と、 人を対象とする研究に適用される連邦の省庁の規則(いわゆるコモン・ルール)の二つに分けられる。 前者は、州際通商規制権限(interstate commerce clause)に基づいて制定された食品・医薬品・化 粧品法を根拠に制定されたものであり、後者は、歳出権限などに基づいて制定された法律を根拠に制 定されたものである9, 48)。
米国の人を対象とする研究に適用される連邦の省庁の規則の最上位の法は、国家研究法となり、倫 理委員会(IRB: Istitutional Review Board、以下IRBと略す)の法的位置付けと被験者保護に関する国 家委員会設置を示したものである。その実施の基準を具体化したものとしてコモン・ルール、研究倫 理に関する考え方に関してベルモント・レポートがある。両者は現在でも研究倫理審査等の重要な基 準となっている35)。
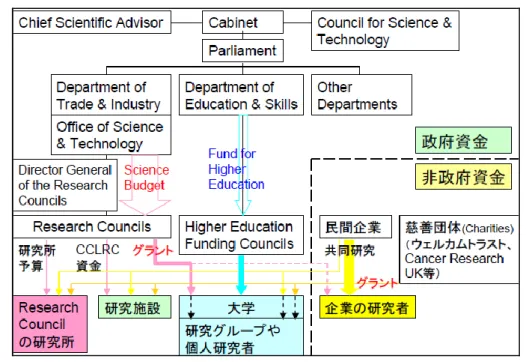
図5 人を対象とした研究における米国連邦政府の被験者保護制度(MORAL SCIENCE Protecting Participants in Human Subjects Research46)より引用)
1981 年に被験者の保護のための規制として、連邦行政命令 45part46 が施行され、1991 年に連邦 政府規則集45CFR46 A 項が、17 の連邦政府機関によって採択されるコモン・ルールとして成立し ている。農務省、エネルギー省、商務省、住宅・都市開発省、司法省、国防総省、教育省、復員軍人 援護局、運輸省、保健福祉省(Department of Health and Human Services、以下 HHS と略す)、 国立科学財団、航空宇宙局、環境保護庁、国際開発庁、社会保障庁、中央情報局、消費者製品安全委 員会の各省庁・関係機関が支援する連邦政府資金による研究の実施を規制するものである31)。なお、
2011 年 7 月に、コモン・ルール改定のための一般意見を集めるための事前通知(ANPRM: Advanced Notice of Proposed Rule Making)が出版されている49)。
コモン・ルールに示される合衆国の規制の主な特徴は、以下のとおりである50)。
に対して、被験者を保護すること(現実には、コモン・ルールが定める要件を遵守するこ と)を確約する書面(Written Assurance)の提出を求める ② 被験者保護の責任を担う機関として、各研究施設に対して、その施設で実施されるすべての 研究について、その内容を事前に審査し、かつ実施の監視に当たる施設内IRB を設置する ことを求めるとともに、その構成・職務について要件を定める ③ 個々の被験者の保護のかなめになるものとして、インフォームド・コンセント(Informed Consent、以下 IC と略す)の要件について、詳細な規定をおく (1) 適用対象 連邦の補助金を受けている機関で行われる臨床研究を適用対象とする31)。 (2) 米国連邦保証制度:FWA 連邦の省庁が実施・補助する研究に参加する研究施設は、特に被験者の保護に関して、適切な手 段、方法、手順を通じて、倫理原則、適用法、規制要件などを遵守することを保証する文書(施 設保証)を、被験者保護局に提出し、その承認を申請する。OHRP への IRB 登録には付録 3 に 示す事項が必須である。 (3) IRB 研究者はIRB へプロトコールを提出し、審議の上、承認を得た後に、初めて実施することがで きる。IRB の構成員についても規定されており、承認の基準についても定めている。IRB につ いての要点は以下のとおりである31)。 (ア) IRB の構成員 ① 少なくとも 5 人の委員とする。 ② 科学的・非科学的な領域について専門的な知識をもつ者である必要があり、次に挙 げる要件を満たす委員を1 人ずつ含む必要がある。 ・ 科学的な領域に主に関心のある人物 ・ 非科学的な領域に主に関心がある人物 ・ 研究組織に所属していない人物、そして審査においてIRB 委員の専門性を超え る、もしくはさらに専門家を追加する必要がある場合には審査を行う助けとな る特別な領域の専門家を招いても良い。 (イ) IRB の研究審査の基準 研究が承認されるためには、IRB によって、その研究が最低限の要件を満たすことを 判定されなければならない。本規則で示されているIRB の審査と承認の最低限の基準 を付録4 に示した。 (ウ) 危険の程度に応じた適切な頻度(ただし、年 1 回以上)で継続的な審議を行う。 (エ) 最小限の危険しか想定されない一定の種類の研究および承認された研究における軽微 な変更に関しては迅速審査を可能とする。 (オ) 以下を含む IRB の活動記録を作成し、少なくとも 3 年間は保存する。
・ 審議資料、経過報告 ・ 健康被害の報告 ・ IRB 会議の議事録 ・ 委員名簿 (4) インフォームド・コンセント(IC) 研究者は、被験者の自己決定権を尊重して、丁寧で十分なIC を行わなければならない。IRB は、 IC の際に使用する同意説明文書に必要な事項が漏れなく含まれているかを審査し、これを保証 する役割を持っている。また、被験者の読解力のレベルに応じて書かれているか、体裁は整って いるか、これから被験者となる人に理解可能であるかなども同様にIRB によって審査される。 本規則で定めるIC の一般要件は付録 5 のとおりである31)。 Ⅲ.英国 英国では、EU での意思決定制度により地域法を制定する。また、EU の諸法は批准手続きや拘束 性により次の付録6 のとおり分類される。EU における「指令」とは、加盟各国がそれぞれの国で達 成すべき結果について各国を拘束するものであり、達成のための形式・手段は各国に委ねられる52)。 英国の被験者保護に関する法体系は、EU 統一のルールを導入している。2001 年に EU は、人を 用いる医薬品の臨床試験においてGCP を履行させる EU 臨床試験指令を発令した。それに伴い、英 国では2004 年 5 月より施行された。本指令は販売承認目的に限らず、すべての臨床試験に適用され る。これにより、当局による研究計画の届出・許可制度が、アカデミアの研究や健常人対象の研究に も適用されるようになった。また、倫理委員会による審査のシステムも一律に法規制として整備され た。
さらに、医薬品の許認可などに関しては、欧州医薬品庁(European Medicines Agency、以下 EMA
と略す)が統括している。実施の基準は EU 臨床試験指令が適用され、その他、EMA が管理する
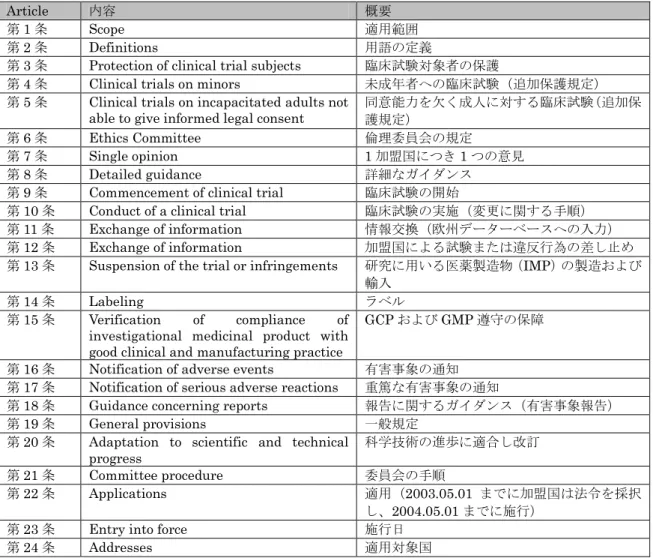
EudraLex というデータベースに収載している医薬品に関する法律である DIRECTIVE 2001/83/EC OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL OF 6 NOVEMBER 2001 ON THE COMMUNITY CODE RELATING TO MEDICINAL PRODUCTS FOR HUMAN USE(医薬 品の中央販売承認における申請の手順、申請に必要な要件、GMP 製造、承認後のフォローアップな どを規定する指令)等も適用される47)。 EU 臨床試験指令の目的は Article 1 に記載されているように、GCP の履行および GMP、その他 関連する規定を確立するためのものである。主な要点は以下のとおりとなっている。構成は表 7 に 示した10, 47)。 ① 被験者を保護する観点でのインフォームドコンセント(IC)に関する考え方 ② 倫理委員会の機能業務、責任
③ Investigational Medicinal Product(以下、IMP と略す)について臨床試験を開始するため には、各国の当局および倫理委員会に対する手続きを行う。
表7 EU 臨床試験指令の構成(EU 臨床試験指令とイギリス臨床試験規則10)より引用)
Article 内容 概要
第1 条 Scope 適用範囲
第2 条 Definitions 用語の定義
第3 条 Protection of clinical trial subjects 臨床試験対象者の保護
第4 条 Clinical trials on minors 未成年者への臨床試験(追加保護規定)
第5 条 Clinical trials on incapacitated adults not able to give informed legal consent
同意能力を欠く成人に対する臨床試験(追加保 護規定)
第6 条 Ethics Committee 倫理委員会の規定
第7 条 Single opinion 1 加盟国につき 1 つの意見
第8 条 Detailed guidance 詳細なガイダンス
第9 条 Commencement of clinical trial 臨床試験の開始
第10 条 Conduct of a clinical trial 臨床試験の実施(変更に関する手順)
第11 条 Exchange of information 情報交換(欧州データーベースへの入力)
第12 条 Exchange of information 加盟国による試験または違反行為の差し止め
第13 条 Suspension of the trial or infringements 研究に用いる医薬製造物(IMP)の製造および
輸入
第14 条 Labeling ラベル
第15 条 Verification of compliance of
investigational medicinal product with good clinical and manufacturing practice
GCP および GMP 遵守の保障
第16 条 Notification of adverse events 有害事象の通知
第17 条 Notification of serious adverse reactions 重篤な有害事象の通知
第18 条 Guidance concerning reports 報告に関するガイダンス(有害事象報告)
第19 条 General provisions 一般規定
第20 条 Adaptation to scientific and technical
progress 科学技術の進歩に適合し改訂
第21 条 Committee procedure 委員会の手順
第22 条 Applications 適用(2003.05.01 までに加盟国は法令を採択
し、2004.05.01 までに施行)
第23 条 Entry into force 施行日
第24 条 Addresses 適用対象国 (1) 適用対象 適用対象となる臨床試験については、第 2 条(a)で定義されているように、非介入的臨床試験を 除くすべての臨床試験が対象になる。また、その際、研究に用いるIMP(第 2 条(d))は、未承 認薬の場合は当然のことながら、販売承認を既に得ている薬剤でも、剤型、用量が承認を得てい るものと異なる場合はIMP とみなされる。 スポンサー(治験の開始、管理、資金の責任者)としては、個人、企業、機関もしくは組織がす べて含まれる。なお、医師が個人的に行う臨床試験の場合でも本指令に準拠して実行されなけれ ばならない。 (2) 被験者の保護 被験者の保護についての要点は以下のとおりとなっている。 (ア) 臨床試験への参加を自由意思で決定すること。インフォームド・コンセントは日付と 署名を記載した文書で取得されなければならない。 (イ) 指令 95/46/EC に基づいて、被験者の個人情報が保護される。 (ウ) 研究実施者とスポンサーが責任を果たすための保険または補償が用意されている。
(3) 倫理委員会について 英国の研究倫理委員会は、米国のように施設ごとでなく、地域ごとに、保健当局が責任主体とな る諮問機関として、NHS の自発的な活動をとおして発展してきた。法的拘束力のないガイドラ インによる諮問委員会である一方、医薬品臨床試験に限らず、社会学的研究や記録調査なども、 NHS のスタッフ・施設・患者が関与するものはすべて審査を受けるべきものとされてきた。2004 年3 月からは、少数の地域研究倫理委員会(LREC)や多施設研究倫理委員会(MREC)に審査 が集中しないよう、EU 臨床試験指令に該当する医薬品の治験と、複数の地域研究倫理委員会に またがる研究は、主たる審査委員会を中央局(COREC)が決めるようになった。 (ア) 倫理委員会は、臨床試験が開始される前に、その内容について検討し、意見を述べな ければならない。特に以下を考慮する[第 3 条.3]。 ① 臨床試験の適切性とデザイン ② 予想される不利益、危険の評価が十分なもので結論が正当化できるか ③ 研究計画書 ④ 研究実施者の適格性 ⑤ 設備の適格性 ⑥ インフォームド・コンセントの手順と適切性、完全性 ⑦ 予想される障害、死亡に対する補償または損失補填 ⑧ スポンサーと実施施設の間の契約 ⑨ 対象者募集の方法などを評価してその臨床試験の妥当性について肯定的か否定的 かの意見をいうことで、詳細な機能はArticle 3.3 に記述されている。 (イ) 倫理委員会の構成員[第 2 条(k)] ① ヘルスケアの専門家 ② 医学専門家ではない委員 (ウ) 倫理委員会がスポンサーから意見を求める申請書を受理してから申請者および当該当 局に意見を提出するまでの期間が次のように厳格に決められている。 ① 通常の薬剤の場合は最大 60 日以内 ② 遺伝子治療、体細胞治療、遺伝子改変生物治療の場合プラス 30 日の延長 ③ 異種動物細胞の場合期限なし (4) 当該当局への臨床研究(治験)開始の許可申請 スポンサーが臨床研究(治験)を開始するための手順については第 9 条に記述されている。 スポンサーは、倫理委員会に意見を求める申請書を提出するとともに、各国の当該当局に臨床研 究(治験)開始の申請書を提出しなければならない。スポンサーは、臨床研究(治験)を開始す るためには、倫理委員会より肯定的な意見を得ると同時に、当局から書面による承認の通知を受 領しているか、あるいは期間内に拒否の通知を受け取っていない事が条件になる。当局の検討期 間および開始の可否の判断の通知の仕方はIMP の性格によって異なり、次のように規定されて いる。 最初の申請が不許可の場合スポンサーは 1 回限り計画変更できるが、再度不許可になった場