製造販売承認申請書添付資料
第2部(モジュール2) CTDの概要(サマリー)
2.6. 非臨床試験の概要文および概要表
2.6.1. 緒言
2.6.2. 薬理試験の概要文
2.6.3. 薬理試験概要表
グラクソ・スミスクライン株式会社
項目-頁
2.6.1. 緒言...~xr1i
2.6.2. 薬理試験の概要文...~xr2i
2.6.2.1. まとめ...~xr3i
2.6.2.2. 効力を裏付ける試験...~xr4i
2.6.2.2.1. In vitroにおける作用 ...~xr5i
2.6.2.2.1.1. 作用機序...~xr6i
2.6.2.2.1.1.1. VEGFR、PDGFR、c-KitおよびFGFRに 対する阻害作用...~xr7i
2.6.2.2.1.1.2. 各種キナーゼに対する阻害作用の他剤との 比較...~xr8i
2.6.2.2.1.1.3. 各種細胞におけるキナーゼ阻害作用...~xr9i
2.6.2.2.1.2. 細胞増殖抑制作用 ...~xr10i
2.6.2.2.1.2.1. HUVECの増殖に対する抑制作用 ...~xr11i
2.6.2.2.1.2.2. 各種ヒト腫瘍細胞株の増殖に対する抑制作用 ..~xr12i
2.6.2.2.1.3. 本薬のヒト代謝物の薬理作用 ...~xr13i
2.6.2.2.2. In vivoにおける作用...~xr14i
2.6.2.2.2.1. 作用機序...~xr15i
2.6.2.2.2.1.1. VEGFR-2チロシンリン酸化阻害作用 ...~xr16i
2.6.2.2.2.2. 血管新生阻害作用 ...~xr17i
2.6.2.2.2.2.1. マウスのレーザー誘発脈絡膜血管新生 モデルにおける血管新生阻害作用 ...~xr18i
2.6.2.2.2.2.2. ウサギのVEGF/bFGFペレットモデル における血管新生阻害作用 ...~xr19i
2.6.2.2.2.3. 腫瘍増殖抑制作用 ...~xr20i
2.6.2.2.2.3.1. 悪性軟部腫瘍に対する作用 ...~xr21i
2.6.2.2.2.3.1.1. SW-872ヒト脂肪肉腫細胞株を用いた マウス異種移植モデルにおける 腫瘍増殖抑制作用 ...~xr22i
2.6.2.2.2.3.1.2. SYO-1ヒト滑膜肉腫細胞株を用いた マウス異種移植モデルにおける 腫瘍増殖抑制作用 ...~xr23i
2.6.2.2.2.3.2. 腎細胞癌に対する作用...~xr24i
2.6.2.2.2.3.2.1. CAKI-2ヒト腎細胞癌株を用いた マウス異種移植モデルにおける 腫瘍増殖抑制作用 ...~xr25i
2.6.2.2.2.3.2.2. ACHNヒト腎細胞癌細胞株を用いた マウス異種移植モデルにおける 腫瘍増殖抑制作用 ...~xr26i
2.6.2.2.2.3.2.3. 786-Oヒト腎細胞癌株を用いた マウス異種移植モデルにおける 腫瘍増殖抑制作用 ...~xr27i
2.6.2.2.2.3.3. 卵巣癌に対する作用 ...~xr28i
2.6.1 - p. 1 2.6.2 - p. 1 2.6.2 - p. 1 2.6.2 - p. 2 2.6.2 - p. 4 2.6.2 - p. 4 2.6.2 - p. 4 2.6.2 - p. 5 2.6.2 - p. 6 2.6.2 - p. 7 2.6.2 - p. 7 2.6.2 - p. 8 2.6.2 - p. 9 2.6.2 - p. 10 2.6.2 - p. 10 2.6.2 - p. 10 2.6.2 - p. 12 2.6.2 - p. 12 2.6.2 - p. 14 2.6.2 - p. 15 2.6.2 - p. 15 2.6.2 - p. 15 2.6.2 - p. 16 2.6.2 - p. 18 2.6.2 - p. 18 2.6.2 - p. 19 2.6.2 - p. 20 2.6.2 - p. 21マウス異種移植モデルにおける 腫瘍増殖抑制作用 ...
~xr29i
2.6.2.2.2.3.4. その他のヒト腫瘍細胞株を用いた マウス異種移植モデルにおける増殖抑制作用 ..~xr30i
2.6.2.3. 副次的薬理試験 ...~xr31i
2.6.2.3.1. 各種受容体、チャネルおよびトランスポーターに 対する結合親和性...~xr32i
2.6.2.3.2. 各種キナーゼに対する阻害作用...~xr33i
2.6.2.3.3. ラット摘出心房標本における影響 ...~xr34i
2.6.2.3.4. 骨髄前駆細胞の増殖に及ぼす影響 ...~xr35i
2.6.2.4. 安全性薬理試験 ...~xr36i
2.6.2.4.1. 中枢神経系に及ぼす影響...~xr37i
2.6.2.4.2. 心血管系に及ぼす影響...~xr38i
2.6.2.4.2.1. イヌ摘出プルキンエ線維標本の活動電位に 及ぼす影響 ...~xr39i
2.6.2.4.2.2. hERGテール電流に及ぼす影響...~xr40i
2.6.2.4.2.3. 血圧、心拍数および心電図に及ぼす影響...~xr41i
2.6.2.4.3. 呼吸系に及ぼす影響 ...~xr42i
2.6.2.5. 薬力学的薬物相互作用試験...~xr43i
2.6.2.5.1. 他の抗癌剤の腫瘍組織への送達に及ぼす影響 ...~xr44i
2.6.2.6. 考察および結論 ...~xr45i
2.6.2.6.1. 効力を裏付ける試験 ...~xr46i
2.6.2.6.2. 副次的薬理試験...~xr47i
2.6.2.6.3. 安全性薬理試験...~xr48i
2.6.2.6.4. 薬力学的薬物相互作用試験 ...~xr49i
2.6.2.6.5. 結論...~xr50i
2.6.2.7. 図表 ...~xr51i
2.6.2.8. 参考文献...~xr52i
2.6.3. 薬理試験概要表...~xr53i
2.6.3.1. 薬理試験:一覧表 ...~xr54i
2.6.3.2. 効力を裏付ける試験...~xr55i
2.6.3.3. 副次的薬理試験 ...~xr56i
2.6.3.4. 安全性薬理試験 ...~xr57i
2.6.3.5. 薬力学的薬物相互作用試験...~xr58i
2.6.2 - p. 21 2.6.2 - p. 22 2.6.2 - p. 23 2.6.2 - p. 23 2.6.2 - p. 23 2.6.2 - p. 23 2.6.2 - p. 24 2.6.2 - p. 25 2.6.2 - p. 25 2.6.2 - p. 25 2.6.2 - p. 25 2.6.2 - p. 25 2.6.2 - p. 26 2.6.2 - p. 26 2.6.2 - p. 26 2.6.2 - p. 26 2.6.2 - p. 27 2.6.2 - p. 27 2.6.2 - p. 30 2.6.2 - p. 31 2.6.2 - p. 32 2.6.2 - p. 32 2.6.2 - p. 32 2.6.2 - p. 32 2.6.3 - p. 1 2.6.3 - p. 1 2.6.3 - p. 3 2.6.3 - p. 3 2.6.3 - p. 4 2.6.3 - p. 4略語(略称) 内 容 APD60 60%再分極活動電位持続時間 APD90 90%再分極活動電位持続時間 ATP アデノシン三リン酸 AUC 血漿中濃度-時間曲線下面積 bFGF 塩基性線維芽細胞増殖因子 BrdU ブロモデオキシウリジン C24 投与間隔24 時間での血漿中トラフ濃度 Cmax 最高血漿中濃度 CFU コロニー形成単位 c-Kit 幹細胞因子受容体 DAPI 4’, 6-ジアミジノ-2-フェニルインドール ELISA 酵素結合免疫測定法 FA 蛍光眼底血管造影 FGF 線維芽細胞増殖因子 FGFR 線維芽細胞増殖因子受容体 Flt-3 Fms 様チロシンキナーゼ-3 5-FU フルオロウラシル GIST 消化管間質腫瘍 GM-CSF 顆粒球マクロファージコロニー刺激因子 HEK-293 細胞 ヒト胎児由来腎臓-293 細胞 hERG ヒトether-a-go-go 関連遺伝子 HFF ヒト包皮線維芽細胞株 HPMC ヒドロキシプロピルメチルセルロース 5-HT セロトニン HUVEC ヒト臍帯静脈血管内皮細胞 IC25 25%阻害濃度 IC50 50%阻害濃度 IC75 75%阻害濃度 Ki 阻害定数 Kiapp 見かけ上のKi LC-MS 液体クロマトグラフ/質量分析 LC-MS/MS 液体クロマトグラフ/タンデム質量分析 MEK マイトジェン活性化プロテインキナーゼキナーゼ
MLK1 Mixed lineage kinase 1
MRD 最大脱分極速度 NADH ニコチンアミドアデニンジヌクレオチド PDGF 血小板由来増殖因子 PDGFR 血小板由来増殖因子受容体 PTK5 蛋白質チロシンキナーゼ5 QTc 補正QT RMP 静止膜電位 SCF 幹細胞因子 SCID マウス 重症複合免疫不全マウス SD ラット Sprague Dawley ラット
略語(略称) 内 容
VEGF 血管内皮細胞増殖因子
VEGFR 血管内皮細胞増殖因子受容体
2.6.1. 緒言 パゾパニブ一塩酸塩(以下、本薬)はおもにVEGFR-1、2 および 3、PDGFR-α および β な らびにc-Kit に対して強力な阻害作用を有する受容体型チロシンキナーゼ阻害薬である(図 2.6.1-1)。本薬は血管内皮細胞の増殖を抑制することにより、血管新生阻害作用および腫瘍 増殖抑制作用を示す。本薬はヒト脂肪肉腫細胞株(SW-872)を移植したマウスにおいて、腫 瘍増殖を抑制することが示されていることから、今回、「進行性悪性軟部腫瘍」を効能・効 果として承認申請を行った。
N
N
H
N
N
H
3C
N
N
CH
3S
CH
3CH
3NH
2O
O
HCl
・
図 2.6.1-1 パゾパニブ一塩酸塩の構造式 進行性悪性軟部腫瘍における申請用法・用量は以下のとおりである。 【用法・用量】 通常、成人にはパゾパニブとして1 日 1 回 800 mg を経口投与する。なお、患者の状態によ り800 mg を超えない範囲で適宜増減する。2.6.2. 薬理試験の概要文 2.6.2.1. まとめ パゾパニブ一塩酸塩(GW786034B;以下、本薬)はおもに VEGFR-1、2 および 3、 PDGFR-α および β ならびに c-Kit に対して強力な阻害作用を有する受容体型チロシンキナー ゼ阻害薬である。各種薬理試験において、本薬は血管新生阻害作用および抗腫瘍作用を有す ることが示されていることから、今回申請する悪性軟部腫瘍をはじめ、腎細胞癌および卵巣 癌などの各種腫瘍に対する治療薬として期待される。 薬理試験では本薬のほかに、一部の副次的薬理試験でパゾパニブ二塩酸塩 (GW786034A)を用いた。用量および濃度は遊離塩基換算量で記載した。 効力を裏付ける試験 本薬は精製キナーゼを用いた in vitro 試験において、VEGFR-1、2 および 3 に対して阻害作 用を示し、IC50はそれぞれ10、30 および 47 nM であった。また、PDGFR-α および β ならび にc-Kit に対しても阻害作用を示し、IC50はそれぞれ71、84 および 80 nM であった。さらに、 VEGFR-1、2 および 3、PDGFR-α および β、c-Kit、Flt-3、B-Raf(野生型)、B-Raf V600E な らびにC-Raf を阻害し、Kiappはそれぞれ15、8、10、30、14、2.4、230、68、160 および 109 nM であった。 本薬は正常ヒト臍帯静脈血管内皮細胞(HUVEC)、ヒト小細胞肺癌細胞株(NCI-H526) および正常ヒト包皮線維芽細胞株(HFF)において、それぞれ VEGFR-2、c-Kit および PDGFR-β のチロシンリン酸化を阻害し、IC50はそれぞれ8、3 および 3 nM であった。 本薬はVEGF および塩基性線維芽細胞増殖因子(bFGF)刺激による HUVEC の増殖をそ れぞれ21 および 721 nM の IC50で抑制した。また、281 種類のヒト腫瘍細胞株のうち、7 種 類の細胞株の増殖に対して抑制作用を示し、そのIC50は50~700 nM であった。
本薬のヒト代謝物であるGSK1268997A は VEGF 刺激による HUVEC の増殖を本薬と同程 度に抑制し、本薬およびGSK1268997A の IC50はそれぞれ39 および 36 nM であった。 マウス in vivo 試験において、本薬は肺における VEGFR-2 キナーゼのチロシンリン酸化を 阻害した。 本薬はマウスのレーザー誘発脈絡膜血管新生モデル(予防的投与:レーザー照射日より 200 mg/kg/日を 13 日間経口投与、治療的投与:レーザー照射 7 日後より 8~200 mg/kg/日を 7 日間経口投与)およびウサギのVEGF/bFGF ペレットモデル(40 mg/kg/日の 26 日間経口投 与)において、いずれのモデルにおいても血管新生阻害作用を示した。 悪性軟部腫瘍の1 種であるヒト脂肪肉腫細胞株(SW-872)を用いたマウス異種移植モデ ルにおいて、本薬は30 および 100 mg/kg を 1 日 1 または 2 回、21 日間経口投与することに より、腫瘍増殖を抑制した。また、ヒト滑膜肉腫細胞株(SYO-1)を用いたマウス異種移植 モデルにおいて、本薬は30 および 100 mg/kg を 1 日 2 回 14 日間経口投与することにより、 腫瘍増殖を抑制した。さらに、10、30 および 100 mg/kg の 1 日 1 または 2 回 21~46 日間投 与により、ヒト腎細胞癌株(CAKI-2、ACHN および 786-O)、卵巣癌細胞株(OVCAR-3)、 結腸癌細胞株(HT29)、頭頸部扁平上皮癌細胞株(HN5)、胃癌細胞株(MKN-45)および 肝細胞癌株(Hep3B)を用いたマウス異種移植モデルにおいて、腫瘍増殖を抑制した。
副次的薬理試験 49 種類の各種受容体、チャネルおよびトランスポーターに対する結合親和性を検討した 結果、パゾパニブ二塩酸塩は10 µM でアデノシン A3、アドレナリンα2およびβ1、ヒスタミ ンH2、ムスカリンM1ならびにセロトニン5-HT1A、5-HT5Aおよび5-HT7に対して50%以上 の結合阻害作用を示したが、これら以外の各種受容体、チャネルおよびトランスポーターに 対して影響は認められなかった。 242 種類のキナーゼ活性に対する阻害作用を検討した結果、本薬は VEGFR-2 阻害作用の IC50の10 倍以内の IC50でAurora-A、c-Raf、MLK1、PTK5 および TAO3 キナーゼに対して阻 害作用を示した。 パゾパニブ二塩酸塩(3、10 および 30 µM)はラットの摘出心房標本において、濃度依存 的に収縮力を増加させ、その作用はプロプラノロールにより抑制されなかった。また、パゾ パニブ二塩酸塩(30 µM)を前処置することにより、イソプロテレノールの最大収縮反応は 減弱した。一方、パゾパニブ二塩酸塩はイソプロテレノールの心房拍動数には影響を及ぼさ なかった。 本薬の骨髄前駆細胞の増殖に及ぼす影響を、GM-CSF、SCF および Flt-3 リガンドの単独ま たは併用の存在下でのCFU 形成能で評価した結果、CFU 形成に対する抑制作用はソラフェ ニブおよびスニチニブと比較して弱かった。 安全性薬理試験 本薬は300 mg/kg までの単回経口投与によりラットの中枢神経系に対して、また、 300 mg/kg までの単回経口投与により呼吸系に影響を及ぼさなかった。hERG テール電流に 対して、溶解限界濃度である4.137 µM(1.810 µg/mL)でわずかな抑制作用を示したが、 80 nM までイヌの摘出プルキンエ線維標本の活動電位パラメータに影響を及ぼさなかった。 覚醒サルの心血管系に対して、本薬は500 mg/kg までの単回経口投与により、一般状態、全 身動脈圧、脈圧、心拍数および心電図パラメータに影響を及ぼさず、また、3.75 mg/kg の単 回静脈内投与により軽度の可逆的な心拍数減少を示したが、心電図に影響を及ぼさなかった。 薬力学的薬物相互作用試験 HT29 ヒト結腸癌および NCI-H460 ヒト肺癌細胞株を用いたマウス異種移植モデルにおい て、本薬は併用した抗癌剤(5-FU、イリノテカン、パクリタキセルおよびカルボプラチン) の腫瘍組織への送達に影響を及ぼさなかった。 2.6.2.2. 効力を裏付ける試験 血管新生は多くの固形癌の増殖および転移において重要な役割を果たしていると考えられ ている。悪性軟部腫瘍においても、細胞増殖および腫瘍悪性度には血管新生が深く関わって おり、良性腫瘍と比較して微小血管密度は有意に高いことが知られている[Baneth, 2005]。 VEGF および PDGF は血管新生および周皮細胞の動員を担う主要な増殖因子であり、それ らの受容体であるVEGFR および PDGFR を介しておもに血管新生を促進し、また、血管透 過性亢進、血管内皮細胞の遊走および周皮細胞の動員などの様々な反応を引き起こすと考え
られている[Holmes, 2007; Ostman, 2007]。おもに、VEGFR-1 は造血および単球の遊走、 VEGFR-2 は血管新生、VEGFR-3 はリンパ管新生、PDGFR-α は腫瘍増殖ならびに PDGFR-β は周皮細胞の動員に関与する受容体であると考えられている(表 2.6.2-1)[Abramsson, 2003; Holmes, 2007; Ostman, 2007]。 腫瘍細胞から放出されるVEGF およびその他の血管内皮細胞増殖因子(bFGF など)はお もにVEGFR を介して血管内皮細胞の増殖および遊走を刺激し、骨髄由来細胞を動員して血 管新生を促進させると考えられている。血管内皮細胞が産生するPDGF は周皮細胞に発現し ているPDGFR に結合して周皮細胞を動員し、微小血管系の成熟を促進する。また、腫瘍細 胞から放出されるPDGF は間質細胞を動員する。間質細胞は線維芽細胞および免疫系細胞 (リンパ球およびマクロファージなど)で構成されており、VEGF を放出して骨髄由来細胞 を動員し、血管新生を促進させると考えられている(図 2.6.2-1)[Ferrara, 2005]。 c-Kit は悪性軟部腫瘍の一部の組織型(脂肪肉腫、肝芽細胞腫、平滑筋肉腫、粘液性脂肪 肉腫、皮膚線維肉腫、癌肉腫および悪性線維性組織球腫)に発現し、SCF による刺激で腫瘍 増殖を促進することが報告されており、c-Kit 阻害作用を有する薬物は腫瘍増殖抑制に関与 する可能性が考えられる[Potti, 2004; Masson, 2009]。 本薬はVEGFR および PDGFR の活性化を阻害することにより悪性軟部腫瘍において細胞 増殖および腫瘍悪性度に関わる血管新生を阻害すること、ならびにc-Kit の阻害を介して直 接的に腫瘍増殖を抑制すると考えられることから、悪性軟部腫瘍をはじめとする種々の悪性 腫瘍に対する治療薬として期待される。 表 2.6.2-1 VEGFR、PDGFR および c-Kit の役割 リガンド 受容体 おもな役割 VEGFR-1 造血および単球の遊走 VEGFR-2 血管新生 VEGF-A など VEGFR-3 リンパ管新生 PDGFR-α 腫瘍増殖 PDGF-A, B, C および D PDGFR-β 周皮細胞の動員 SCF など c-Kit 腫瘍増殖
図 2.6.2-1 血管新生の機序
Data source: [Ferrara, 2005]の Figure 1
2.6.2.2.1. In vitro における作用 2.6.2.2.1.1. 作用機序 2.6.2.2.1.1.1. VEGFR、PDGFR、c-Kit および FGFR に対する阻害作用 4.2.1.1.1 および 4.2.1.1.2 方法 試験1:ヒト VEGFR-1、2 および 3 にそれぞれの基質ペプチド存在下で本薬を添加して室温 で3 時間反応させたのち、リン酸化された基質量を定量して本薬のキナーゼ阻害活性を算出 した。 試験2:VEGFR-1 および 2 を含む 7 種類のキナーゼのヒト組換え蛋白質に γ-33P-ATP および それぞれの基質ペプチドの存在下で本薬を添加し、30℃で 80 分間反応させた。未反応の γ-33P-ATP を除去したのち、基質ペプチドへ取り込まれた放射能を測定し、各種キナーゼに対 するIC50を算出した。 結果 試験1 において、本薬は VEGFR-1、2 および 3 に対して濃度依存的な阻害作用を示し、そ のIC50はそれぞれ10、30 および 47 nM であった。試験 2 では、本薬の VEGFR-1 および 2 に 対するIC50はそれぞれ13 および 12 nM であった(表 2.6.2-2)。
また、本薬はPDGFR-α および β、c-Kit ならびに FGFR-1 および 3 に対しても阻害作用を 示し、そのIC50はそれぞれ71、84、80、140 および 130 nM であった(表 2.6.2-2)。 表 2.6.2-2 VEGFR、PDGFR、c-Kit および FGFR に対する阻害作用 試験1#1 試験2#2 酵素 IC50 (nM) IC50 (nM) VEGFR-1 10 (n=3) 13 VEGFR-2 30 (n=3) 12 VEGFR-3 47 (n=2)#3 - PDGFR-α - 71 PDGFR-β - 84 c-Kit - 80 FGFR-1 - 140 FGFR-3 - 130 -:実施せず、#1:平均値、#2:n=1 (duplicate)、#3:個別値は 51 および 43 nM Data source: 4.2.1.1.1 の Table 2 および 4.2.1.1.2 の Table A
2.6.2.2.1.1.2. 各種キナーゼに対する阻害作用の他剤との比較 4.2.1.1.3 おもにVEGFR および PDGFR を阻害するマルチキナーゼ阻害薬であるスニチニブおよび ソラフェニブは腎細胞癌などの各種腫瘍に対して抗腫瘍効果を示すことが知られている [Larkin, 2007]。スニチニブまたはソラフェニブの抗腫瘍効果に関わると考えられる各種キナ ーゼに対する本薬の阻害作用をスニチニブおよびソラフェニブの阻害作用と比較検討した。 方法 VEGFR-1、2 および 3、PDGFR-α および β ならびに Flt-3 のヒト組換え蛋白質に、被験物 質を60 分間反応させたのち、基質ペプチドおよび33P-ATP を含む緩衝液を添加して 30 分間 反応させた。未反応の33P-ATP を除去したのち、基質ペプチドに取り込まれた放射能を測定 した。 ヒト c-Kit に被験物質、基質および ATP を含む緩衝液を添加して室温で 50 分間反応させ、 反応停止液を加えたのち、ホモジニアス時間分解蛍光法を用いてリン酸化レベルを測定する ことにより酵素活性を測定した。
ヒトRaf(B-Raf、B-Raf V600E および C-Raf)に被験物質、MEK および ATP を含む緩衝 液を添加して室温で4 時間反応させ、シグナル伝達の下流に位置する MEK ATPase の活性を 反映するNADH の酸化反応を吸光度法を用いて測定することにより酵素活性を算出した。 これらのすべてのキナーゼに対する被験物質の阻害率を算出し、見かけ上のKi(Kiapp)を 求めた。 結果 本薬はVEGFR-1、2 および 3、PDGFR-α および β ならびに c-Kit に対して強い阻害作用を 示し、Kiappはそれぞれ15、8、10、30、14 および 2.4 nM であった(表 2.6.2-3)。また、Flt-3、B-Raf(野生型)、B-Raf V600E および C-Raf に対する阻害作用は弱く、Kiappはそれぞれ 230、68、160 および 109 nM であった。
また、ソラフェニブと比較して、VEGFR-1 の阻害作用は同程度であり、PDGFR-α、Flt-3 お よびRaf の阻害活性は弱かった。さらに、本薬の VEGFR-2 および 3、PDGFR-β ならびに c-Kit に対する阻害活性はおおむねスニチニブおよびソラフェニブと同程度であった(表 2.6.2-3)。 表 2.6.2-3 各種キナーゼに対する阻害作用の他剤との比較 パゾパニブ スニチニブ ソラフェニブ 酵素 Kiapp (nM) n Kiapp (nM) n Ki app (nM) (個別値)#1 n VEGFR-1 15 4 229 3 (6.5, 14) 10 2 VEGFR-2 8 5 51 3 4 4 VEGFR-3 10 3 30 3 6 3 PDGFR-α 30 5 28 5 2 5 PDGFR-β 14 6 7 7 5 7 c-Kit 2.4 6 0.45 6 15 5 Flt-3 230 4 0.6 5 22 4 B-Raf (野生型) 68 3 470 3 (1.90, 2.04) 1.97 2 B-Raf V600E 160 3 3000 3 (4.86, 7.56) 6.1 2 C-Raf 109 3 2000 3 (1.90, 1.82) 1.9 2 平均値
#1:Kiappは、pIC50 (IC50の負の常用対数) を用いて算出しているため、平均値は各個別値の相加平均値とは
異なる場合がある。
Data source: 4.2.1.1.3 の Table 1 および 3
2.6.2.2.1.1.3. 各種細胞におけるキナーゼ阻害作用 4.2.1.1.4 HUVEC ならびに NCI-H526、HFF およびヒト前駆 B 細胞性白血病細胞株(RS4;11)は、 それぞれVEGFR-2、c-Kit、PDGFR-β および Flt-3 を発現していることから、これらの培養細 胞を用いて本薬のチロシンキナーゼ活性に対する作用を検討した。 方法 HUVEC、NCI-H526、HFF および RS4;11 細胞に本薬(0.01 nM~10 µM)を添加して 37℃ で2 時間反応させたのち、それぞれ VEGFR-2、c-Kit、PDGFR-β および Flt-3 のリガンドで 10 分間刺激した。各種細胞を可溶化し、ウエスタンブロット法により VEGFR-2、c-Kit、 PDGFR-β および Flt-3 のチロシンリン酸化を検出した。 結果 本薬はHUVEC、NCI-H526 および HFF 細胞において、それぞれ VEGFR-2、c-Kit および PDGFR-β のチロシンリン酸化を濃度依存的に阻害し(図 2.6.2-2)、その IC50はそれぞれ 8、 3 および 3 nM であった。一方、RS4;11 細胞における Flt-3 のチロシンリン酸化に対する阻害 作用は弱く(図 2.6.2-2)、その IC50は1000 nM 以上であった。
pTyr:リン酸化キナーゼ
図 2.6.2-2 各種細胞におけるキナーゼ阻害作用
Data source: 4.2.1.1.4 の Figure 1
まとめ
本薬はおもにVEGFR、PDGFR および c-Kit に対して阻害作用を示した。本薬の VEGFR-2 および3、PDGFR-β ならびに c-Kit に対する阻害活性は、おおむねスニチニブおよびソラフ ェニブと同程度であり、Flt-3 阻害活性はスチニブおよびソラフェニブよりも弱かった。本 薬のRaf 阻害活性はソラフェニブよりも弱く、スニチニブよりも強かった。 本薬は各種細胞におけるVEGFR-2、PDGFR-β および c-Kit のチロシンリン酸化を強く阻害 した。一方、Flt-3 のチロシンリン酸化に対する阻害作用は弱いことが示された。 2.6.2.2.1.2. 細胞増殖抑制作用 2.6.2.2.1.2.1. HUVEC の増殖に対する抑制作用 4.2.1.1.5 本薬のHUVEC の増殖に対する抑制作用を VEGF または bFGF による刺激下で検討した。 また、コントロール群としてHFF 細胞株の増殖に対する抑制作用についても検討した。 方法 HUVEC に本薬(0.0015 nM~3 µM)を添加し、30 分後に VEGF または bFGF を添加して 48 時間培養したのち、BrdU を添加してさらに 22 時間培養した。また、HFF に本薬(0.0015 ~30 µM)を添加して 72 時間培養したのち、BrdU を添加してさらに 18 時間培養した。細胞 へのBrdU 取込み量を ELISA 法で測定し、それを指標に細胞増殖に及ぼす影響を検討した。
結果
本薬はVEGF および bFGF 刺激による HUVEC の増殖をいずれも抑制し、その IC50はそれ ぞれ約21 および 721 nM であったことから、VEGF 刺激による HUVEC の増殖に対して選択 的な抑制作用を示した。また、HFF の増殖に対する抑制作用は弱く(IC50=1012.3 nM)、 VEGF 刺激による HUVEC の増殖に対する抑制作用と比較して約 1/50 であった(表 2.6.2-4)。 表 2.6.2-4 VEGF または bFGF 刺激による HUVEC の増殖に対する抑制作用 細胞 IC50 (nM) 選択性#1 HUVEC (VEGF 刺激) 21.3±4.5 - HUVEC (bFGF 刺激) 720.9±239.5 33.84 HFF (コントロール) 1012.3±153.3 47.53 平均値±標準誤差 (n=5~15) #1:VEGF 刺激による HUVEC 増殖における IC50に対する各IC50の比
Data source: 4.2.1.1.5 の Table 1
まとめ 本薬は bFGF 刺激と比較して VEGF 刺激による HUVEC の増殖を選択的に阻害した。一方、 HFF の増殖にはほとんど影響を及ぼさなかった。これらのことから、本薬は VEGFR を発現 しているHUVEC に対して選択的に作用を発現することが示唆された。 2.6.2.2.1.2.2. 各種ヒト腫瘍細胞株の増殖に対する抑制作用 4.2.1.1.6 方法 281 種類のヒト腫瘍細胞株のパネルに本薬(0.00032~10 µM)を添加して 72 時間培養した のち、DAPI 染色法を用いて核を蛍光染色し、蛍光強度を指標として細胞数を測定した。な お、培養する細胞株の密度を高および低細胞密度とする2 種類の条件下で検討し、それぞれ 増殖抑制作用のIC50を算出した。 結果 多くの腫瘍細胞に対して、本薬は10 µM まで明らかな増殖抑制作用を示さなかった。一方、 悪性軟部腫瘍細胞株であるA204(横紋筋肉腫)を含む 7 種類の腫瘍細胞に対しては増殖抑 制作用を示し、GDM-1(急性骨髄性白血病)、ARH-77(メラノーマ)、NCI-H716(結腸 癌)、G402(腎平滑筋芽細胞腫)、CGTH-W-1(甲状腺癌)、A204(横紋筋肉腫)および CML-T1(慢性骨髄性白血病)の増殖に対する阻害作用の IC50は、それぞれ高細胞密度の条 件下では0.094、0.037、0.20、0.19、0.21、0.26 および 0.61 µM であり、低細胞密度の条件下 では0.010、0.13、0.18、0.19、0.24、0.27 および 0.79 µM であった。 しかしながら、高細胞密度の培養条件下において、その他の17 種類における各種ヒト悪 性軟部腫瘍細胞株の増殖に対する抑制作用は弱く、IC50は4.8~10 µM 超であった(表 2.6.2-5)。同様に、低細胞密度の培養条件下においても、その他の 17 種類における各種ヒ
ト悪性軟部腫瘍細胞株の増殖に対する抑制作用は弱く、IC50は4.6~10 µM 超であった(表 2.6.2-5)。 表 2.6.2-5 各種ヒト悪性軟部腫瘍細胞株の増殖に対する抑制作用 IC50 (µM) 細胞株名 組織型 高細胞密度 低細胞密度 A204 横紋筋肉腫 0.26 0.27 A673 横紋筋肉腫 9.1 >10 GCT 巨細胞腫 >10 6.4 HOS 骨肉腫 6.2 6.2 HT-1080 線維肉腫 >10 5.1 KHOS-240S 骨肉腫 4.8 4.6 MES-SA 子宮肉腫 >10 7.0 RD 骨肉腫 >10 >10 RD-ES ユーイング肉腫 >10 8.2 Saos-2 骨肉腫 >10 >10 SJRH30 横紋筋肉腫 6.4 9.5 SK-LMS-1 平滑筋肉腫 >10 >10 SK-UT-1 平滑筋肉腫 >10 >10 SW1353 軟骨肉腫 >10 >10 SW684 線維肉腫 >10 >10 SW-872 脂肪肉腫 >10 >10 TE381.T 横紋筋肉腫 5.3 5.2 U-2OS 骨肉腫 5.2 >10 n=1 (triplicate)
Data source: 4.2.1.1.6 の Table 2
まとめ 本薬は検討した281 種類の腫瘍細胞株の多くで増殖を抑制しなかったが(IC50=10 µM 超)、悪性軟部腫瘍細胞株であるA204(横紋筋肉腫)を含む 7 種類の腫瘍細胞株に対して は直接的な増殖抑制作用を示した(IC50=0.05~0.7 µM)。これらの腫瘍細胞に対する増殖 抑制作用のIC50は、VEGF 刺激による HUVEC の増殖に対する抑制作用の IC50(0.0213 µM) と比較して2.4~33 倍の高値であった(表 2.6.2-4)。 2.6.2.2.1.3. 本薬のヒト代謝物の薬理作用 4.2.1.1.7 本薬のヒトでのおもな代謝物であるM24、M26、M27 および M28(2.6.5.2)の薬理活性を 検討するため、それぞれGSK1268992A、GSK1268997A、GSK1071306A および GW700201X を用いてHUVEC の増殖に対する抑制作用を検討した。 方法 HUVEC に本薬、GSK1268992A、GSK1268997A、GSK1071306A および GW700201X(0.01 ~10 µM)を添加し、30 分後に VEGF を添加して 48 時間培養したのち、BrdU を添加して 20 時間培養した。細胞へのBrdU 取込み量を ELISA 法で測定し、それを指標に細胞増殖に及ぼ す影響を検討した。
結果
4 種類のヒト代謝物のうち GSK1268997A は VEGF 刺激による HUVEC の増殖に対して本 薬(IC50=39 nM)と同程度の抑制作用を示し、その IC50は36 nM であった(表 2.6.2-6)。一 方、その他の代謝物であるGSK1268992A、GSK1071306A および GW700201X の増殖抑制作 用は本薬と比較して約1/18~1/10 と弱かった(表 2.6.2-6)。 表 2.6.2-6 ヒト代謝物の VEGF 刺激による HUVEC の増殖に対する抑制作用 IC50 (nM) 化合物 試験1 試験2 平均値 本薬 38 40 39 GSK1268992A 500 254 377 GSK1268997A 20 51 36 GSK1071306A 620 803 712 GW700201X 300 910 605 Data source: 4.2.1.1.7 の Table 2
まとめ
本薬のヒトでのおもな代謝物のうち、GSK1268997A は VEGF 刺激による HUVEC の増殖
に対して本薬と同程度の抑制作用を示した。一方、それ以外の3 種類の代謝物の活性は本薬 の約1/18~1/10 であった。 2.6.2.2.2. In vivo における作用 2.6.2.2.2.1. 作用機序 2.6.2.2.2.1.1. VEGFR-2 チロシンリン酸化阻害作用 4.2.1.1.8 本薬の in vivo における VEGFR-2 のチロシンリン酸化阻害作用を VEGFR のリガンドを投 与したマウスを用いて検討した。 方法 用量依存性の検討では雌Swiss Nude マウスに本薬(3、10、30 および 100 mg/kg)を単回 経口投与し、投与2 時間後に VEGFR に対するリガンドの 1 種である VEGF121(15 µg/匹)を 静脈内投与した。また、作用持続性の検討では、雌Swiss Nude マウスに本薬(30 mg/kg)を 単回経口投与し、投与1、4、8、16 および 24 時間後に VEGF121(15 µg/匹)を静脈内投与し た。それぞれの静脈内投与の 5 分後に血液採取および肺の摘出を行った。肺組織を可溶化し、 抗Flk-1 抗体を用いた免疫沈降法により VEGFR-2 を精製した。ウエスタンブロット解析を 行い、抗チロシンリン酸化抗体を用いてリン酸化VEGFR-2 を検出した。また、血漿中未変 化体濃度をLC-MS/MS 法により測定した。
結果 用量依存性の検討 本薬は10、30 および 100 mg/kg の単回経口投与 2 時間後において、VEGFR-2 のチロシン リン酸化を阻害した(図 2.6.2-3)。3、10、30 および 100 mg/kg 群の単回経口投与 2 時間後 の血漿中未変化体濃度は、それぞれ14.2、38.4、50.3 および 180.1 µM であった。 作用持続性の検討 本薬の30 mg/kg を単回経口投与したとき、VEGFR-2 のチロシンリン酸化阻害作用は投与 8 時間後まで持続し、投与 1、4、8、16 および 24 時間後の血漿中未変化体濃度はそれぞれ 153.9、47.5、41.1、17.3 および 4.3 µM であった(図 2.6.2-4)。 A:ウエスタンブロットの代表例 (用量依存性)、B:ウエスタンブロット解析の定量結果 平均値±標準偏差 (n=3) Vehicle:媒体 (0.5% HPMC-0.1% Tween 80 溶液) 本薬の経口投与2 時間後のデータ 図 2.6.2-3 マウスの肺における VEGF121 誘発 VEGFR-2 チロシンリン酸化 に対する阻害作用
A:ウエスタンブロットの代表例 (作用持続性)、B:ウエスタンブロット解析の定量結果 平均値±標準偏差 (n=2)
Vehicle:媒体 (0.5% HPMC-0.1% Tween 80 溶液)
図 2.6.2-4 マウスの肺における VEGF121 誘発 VEGFR-2 チロシンリン酸化 に対する阻害作用の持続性
Data Source: 4.2.1.1.8 の Figure 1
まとめ 本薬は in vivo において VEGFR-2 のリン酸化を持続的に阻害することが示された。また、 血漿中未変化体濃度が約40 µM 以上のときに、十分な VEGFR-2 リン酸化阻害作用を発現す ることが示された。 2.6.2.2.2.2. 血管新生阻害作用 血管新生モデルとして、血管新生を誘発するメカニズムが異なるマウスのレーザー誘発脈 絡膜血管新生モデルおよびウサギのVEGF/bFGF ペレットモデルの 2 種類を用い、本薬の in vivo における血管新生阻害作用を検討した。 2.6.2.2.2.2.1. マウスのレーザー誘発脈絡膜血管新生モデルにおける血管新生阻害作用 4.2.1.1.9 方法 C57BL/6J マウスの網膜にレーザーを照射し(クリプトンレーザー光凝固術)、14 日後に 蛍光ラベルしたデキストランを灌流して、血管新生による脈絡膜の病変面積を測定した。試
験1 では、予防効果を検討するために、レーザー照射の当日から本薬(200 mg/kg/日)を 1 日2 回 13 日間経口投与した。試験 2 では、治療的な病変の縮小効果を検討するために、本 薬(8、40 または 200 mg/kg/日)をレーザー照射 7 日後から 1 日 2 回 7 日間経口投与した。 結果 予防効果の検討(試験 1) 本薬はレーザー照射の当日から200 mg/kg/日を 13 日間経口投与することにより、レーザ ー照射による病変面積の増加を有意(Kruskal-Wallis tests、p<0.0001)に抑制した(媒体群に 対する抑制率は92.6%)(図 2.6.2-5)。 平均値±標準誤差 (n=8~10) Vehicle:媒体 (0.5% HPMC-0.1% Tween 80 溶液) GW786034 の用量:200 mg/kg/日 図 2.6.2-5 マウスのレーザー照射誘発脈絡膜血管新生に対する予防的効果(試験 1)
Data source: 4.2.1.1.9 の Figure 1
治療効果の検討(試験 2)
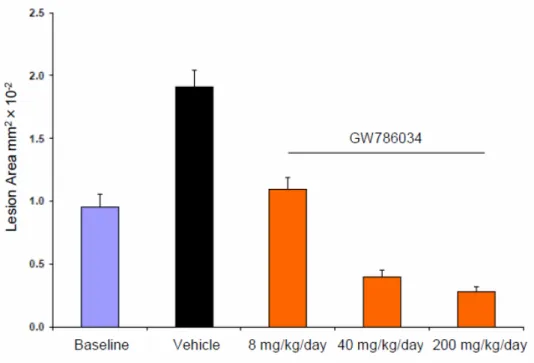
本薬はレーザー照射7 日後から 7 日間経口投与(8、40 および 200 mg/kg/日)することに より、レーザー照射による病変面積を用量依存的かつ有意(Kruskal-Wallis tests、p<0.001) に縮小させ(図 2.6.2-6)、媒体群に対する抑制率はそれぞれ 42.6、79.1 および 85.4%であっ た。
平均値±標準誤差 (n=6~9)
Baseline:レーザー照射 7 日後の病変面積 Vehicle:媒体 (0.5% HPMC-0.1% Tween 80 溶液)
図 2.6.2-6 マウスのレーザー照射誘発脈絡膜血管新生に対する治療効果(試験 2)
Data source: 4.2.1.1.9 の Figure 2
2.6.2.2.2.2.2. ウサギの VEGF/bFGF ペレットモデルにおける血管新生阻害作用 4.2.1.1.10 本試験では、VEGF および bFGF の併用投与により脈絡膜に血管新生を誘発したウサギを 用い、本薬の血管新生阻害作用を検討した。 方法 雄Dutch belted ウサギの脈絡膜上腔(脈絡膜および強膜の間に手術により作製した間隙) にVEGF/bFGF を含有するハイドロンペレットを移植し、その 48 時間後から本薬 (40 mg/kg/日)および類縁物質の 1 種である gsk001* (40 mg/kg/日)を 1 日 2 回 26 日間 経口投与した。ペレットの移植4 週間後まで 1 週間に 1 回、蛍光眼底血管造影(FA)を行い、 血管新生の程度をスコア化して評価した。 結果 本薬および gsk001* は 40 mg/kg/日を 1 日 2 回 26 日間経口投与することにより、血管新 生の指標であるFA スコアの上昇を抑制し、血管新生阻害作用を示した(図 2.6.2-7)。 * 新薬承認情報提供時に置き換え
平均値±標準誤差 (n=8) Control:媒体 (0.5% HPMC-0.1% Tween 80 溶液) FA score:蛍光眼底血管造影スコア 786034:パゾパニブ一塩酸塩、gsk001*:パゾパニブ類縁物質 786034 および gsk001* の用量:40 mg/kg/日の 1 日 2 回 26 日間経口投与 図 2.6.2-7 ウサギの VEGF/bFGF ペレットモデルにおける血管新生阻害作用
Data source: 4.2.1.1.10 の Figure 3
まとめ 本薬は in vivo において血管新生阻害作用を有することが示された。 2.6.2.2.2.3. 腫瘍増殖抑制作用 2.6.2.2.2.3.1. 悪性軟部腫瘍に対する作用 2.6.2.2.2.3.1.1. SW-872 ヒト脂肪肉腫細胞株を用いたマウス異種移植モデルにおける腫 瘍増殖抑制作用 4.2.1.1.11 脂肪肉腫は悪性軟部腫瘍の1 種であり、悪性軟部腫瘍のなかでもっとも多い組織型として 知られている[Lewis, 1998]。本薬の悪性軟部腫瘍に対する抗腫瘍効果を評価する一環として、 SW-872 ヒト脂肪肉腫細胞株を用いたマウス異種移植モデルにおいて腫瘍増殖抑制作用を検 討した。 方法 SW-872 ヒト脂肪肉腫細胞株の懸濁液を雄 SCID マウスの皮下に移植し、その 13 日後に (腫瘍が定着して一定の大きさに増殖したのち)、本薬の100 mg/kg を 1 日 1 回、または 30 および100 mg/kg を 1 日 2 回 21 日間経口投与した。腫瘍の長径と短径を週に 2 回測定し、腫 瘍体積(長径×短径 2×0.5)を算出した。また、週に 2 回体重を測定した。 gsk001*
結果 本薬は100 mg/kg を 1 日 1 回、または 30 および 100 mg/kg を 1 日 2 回 21 日間経口投与す ることにより、腫瘍の増殖を有意に抑制した(図 2.6.2-8)。一方、体重に対して本薬投与に よる有意な影響は認められなかった。 平均値±標準誤差 (n=10) **:p<0.01 vs 媒体群 (Dunnett 多重比較検定) Vehicle:媒体 (0.5% HPMC-0.1% Tween 80 溶液) GW786034:パゾパニブ一塩酸塩、qd:1 日 1 回投与、bid:1 日 2 回投与 図 2.6.2-8 SW-872 ヒト脂肪肉腫細胞株を用いたマウス異種移植モデルにおける 腫瘍増殖抑制作用
Data source: 4.2.1.1.11 の Figure 8.2.2 Tumor Volume
2.6.2.2.2.3.1.2. SYO-1 ヒト滑膜肉腫細胞株を用いたマウス異種移植モデルにおける腫 瘍増殖抑制作用 4.2.1.1.17 滑膜肉腫は悪性軟部腫瘍のなかでも頻度の高い組織型の一つである[日本整形外科学会骨 軟部腫瘍委員会, 2008]。本薬の悪性軟部腫瘍に対する抗腫瘍効果を評価する一環として、 SYO-1 ヒト滑膜肉腫細胞株を用いたマウス異種移植モデルにおいて腫瘍増殖抑制作用を検討 した。 方法
SYO-1 ヒト滑膜肉腫細胞株の懸濁液を雄 BALB/c Slc-nu/nu 免疫不全マウスの皮下に移植し、 その18 日後に(腫瘍が定着して一定の大きさに増殖したのち)、本薬の 10、30 および
100 mg/kg を 1 日 2 回 14 日間経口投与した。腫瘍の長径と短径を週に 2 回測定し、腫瘍体積 (長径×短径2×0.5)を算出した。また、週に 2 回体重を測定した。 結果 本薬は30 および 100 mg/kg を 1 日 2 回 14 日間経口投与することにより、腫瘍の増殖を有 意に抑制した(図 2.6.2-9)。一方、体重に対して本薬投与による有意な影響は認められなか った。 平均値±標準偏差 (n=8) *:p<0.05 vs 媒体群 (Dunnett 多重比較検定) Control:媒体 (0.5% HPMC-0.1% Tween 80 溶液) 図 2.6.2-9 SYO-1 ヒト滑膜肉腫細胞株を用いたマウス異種移植モデルにおける 腫瘍増殖抑制作用(A)および体重への影響(B)
Data source: 4.2.1.1.17 の Figure 4
まとめ
本薬はヒト悪性軟部腫瘍の1 種である SW-872 ヒト脂肪肉腫細胞株および SYO-1 ヒト滑膜 肉腫細胞株を用いたマウス異種移植モデルにおいて、腫瘍増殖を抑制した。本薬投与による 体重への有意な影響は認められなかった。
2.6.2.2.2.3.2. 腎細胞癌に対する作用 今回申請する適応癌種である悪性軟部腫瘍のほかに、開発中であるヒト腎細胞癌の細胞株 を用いたマウス異種移植モデル試験を実施し、本薬の腫瘍増殖抑制作用を検討した。 2.6.2.2.2.3.2.1. CAKI-2 ヒト腎細胞癌株を用いたマウス異種移植モデルにおける腫瘍増 殖抑制作用 4.2.1.1.12 方法 CAKI-2 ヒト腎細胞癌株の懸濁液を雌 CB-17 SCID マウスの皮下に移植し、腫瘍が定着して 一定の大きさに増殖したのち、本薬(10、30 および 100 mg/kg)を 1 日 1 回 24 日間経口投与 した。2.6.2.2.2.3.1.1 と同様の方法で腫瘍体積および体重を週に 2 回測定した。 結果 本薬は10、30 および 100 mg/kg を 1 日 1 回 24 日間経口投与することにより、腫瘍の増殖 を抑制した(図 2.6.2-10)。10、30 および 100 mg/kg における最終投与後の抑制率は、それ ぞれ90、77 および 99%であった。一方、本薬投与による体重への有意な影響は認められな かった。 平均値±標準誤差 (n=8) Vehicle:媒体 (0.5% HPMC-0.1% Tween 80 溶液) GW786034B:パゾパニブ一塩酸塩、QD:1 日 1 回投与 図 2.6.2-10 CAKI-2 ヒト腎細胞癌株を用いたマウス異種移植モデルにおける 腫瘍増殖抑制作用
2.6.2.2.2.3.2.2. ACHN ヒト腎細胞癌細胞株を用いたマウス異種移植モデルにおける腫 瘍増殖抑制作用 4.2.1.1.12 方法 ACHN ヒト腎細胞癌株の懸濁液を雌 Swiss ヌードマウスの皮下に移植し、腫瘍が定着して 一定の大きさに増殖したのち、本薬(10、30 および 100 mg/kg)を 1 日 2 回 46 日間経口投与 した。2.6.2.2.2.3.1.1 と同様の方法で腫瘍体積および体重を週に 2 回測定した。 結果 本薬は10、30 および 100 mg/kg を 1 日 2 回 46 日間経口投与することにより、腫瘍の増殖 を抑制した(図 2.6.2-11)。10、30 および 100 mg/kg における最終投与後の抑制率は、それ ぞれ38、28 および 46%であった。一方、本薬投与による体重への有意な影響は認められな かった。 平均値±標準誤差 (n=8) Vehicle:媒体 (0.5% HPMC-0.1% Tween 80 溶液) GW786034B:パゾパニブ一塩酸塩、BID:1 日 2 回投与 図 2.6.2-11 ACHN ヒト腎細胞癌株を用いたマウス異種移植モデルにおける 腫瘍増殖抑制作用
2.6.2.2.2.3.2.3. 786-O ヒト腎細胞癌株を用いたマウス異種移植モデルにおける腫瘍増 殖抑制作用 4.2.1.1.12 方法 786-O ヒト腎細胞癌株の懸濁液を雌 Swiss ヌードマウスの皮下に移植し、腫瘍が定着して 一定の大きさに増殖したのち、本薬(10、30 および 100 mg/kg)を 1 日 2 回 31 日間経口投与 した。2.6.2.2.2.3.1.1 と同様の方法で腫瘍体積および体重を週に 2 回測定した。 結果 本薬は30 および 100 mg/kg を 1 日 2 回 31 日間経口投与することにより、腫瘍の増殖を抑 制した(図 2.6.2-12)。30 および 100 mg/kg における最終投与後の抑制率は、それぞれ 37 お よび30%であった。一方、本薬投与による体重減少は認められなかった。 平均値±標準誤差 (n=8) Vehicle:媒体 (0.5% HPMC-0.1% Tween 80 溶液) GW786034B:パゾパニブ一塩酸塩、BID:1 日 2 回投与 図 2.6.2-12 786-O ヒト腎細胞癌株を用いたマウス異種移植モデルにおける 腫瘍増殖抑制作用
2.6.2.2.2.3.3. 卵巣癌に対する作用 ヒト腎細胞癌と同様に開発中であるヒト卵巣癌の細胞株を用いたマウス異種移植モデル試 験も実施し、本薬の腫瘍増殖抑制作用を検討した。 2.6.2.2.2.3.3.1. OVCAR-3 ヒト卵巣癌細胞株を用いたマウス異種移植モデルにおける腫 瘍増殖抑制作用 4.2.1.1.13 方法 OVCAR-3 ヒト卵巣癌細胞株の懸濁液を雌 CB-17 SCID マウスの皮下に移植し、腫瘍が定 着して一定の大きさに増殖したのち、本薬(10、30 および 100 mg/kg)を 1 日 2 回 22 日間経 口投与した。2.6.2.2.2.3.1.1 と同様の方法で投与期間中、腫瘍体積および体重を測定した。 結果 本薬は10、30 および 100 mg/kg を 1 日 2 回 22 日間経口投与することにより、腫瘍の増殖 を抑制した(図 2.6.2-13)。10、30 および 100 mg/kg における最終投与後の抑制率は、それ ぞれ40、59 および 81%であった。一方、本薬投与による体重減少は認められなかった。 平均値±標準誤差 (n=4) Vehicle:媒体 (0.5% HPMC-0.1% Tween 80 溶液)、GW786034B:パゾパニブ一塩酸塩、 BID:1 日 2 回投与 図 2.6.2-13 OVCAR-3 ヒト卵巣癌細胞株を用いたマウス異種移植モデルにおける 腫瘍増殖抑制作用
2.6.2.2.2.3.4. その他のヒト腫瘍細胞株を用いたマウス異種移植モデルにおける増殖抑 制作用 4.2.1.1.14、4.2.1.1.15 および 4.2.1.1.16 今回申請する適応癌種である悪性軟部腫瘍ならびに開発中である腎細胞癌および卵巣癌の ほかに、ヒト結腸癌、頭頸部扁平上皮癌、胃癌および肝細胞癌の細胞株を用いたマウス異種 移植モデル試験を実施し、本薬の腫瘍増殖抑制作用を検討した。 方法 各種ヒト腫瘍細胞株の懸濁液を各種ヌードマウスの皮下に移植し、腫瘍が定着して一定の 大きさに増殖したのち、本薬(10、30 および 100 mg/kg)を 1 日 1 回または 2 回 21 日間経口 投与した。2.6.2.2.2.3.1.1 と同様の方法で腫瘍体積および体重を週 2 回または連日測定した。 結果 いずれの癌細胞異種移植マウスにおいても、本薬は10~100 mg/kg の範囲で腫瘍増殖抑制 作用を示したが(表 2.6.2-7)、体重の減少および死亡例は認められず、十分な忍容性が示さ れた。 表 2.6.2-7 その他のヒト腫瘍細胞株を用いたマウス異種移植モデルにおける 腫瘍増殖抑制作用 癌種 細胞株 使用動物 (例数) 投与量 (mg/kg) 投与 日数 腫瘍増殖抑制作用 資料番号 10、30、100 1 日 1 回 21 10 mg/kg で 39%、30 mg/kg で20%、100 mg/kg で 65% 抑制 結腸癌 HT29 雌Swiss ヌー ドマウス (n=8) 10、30、100 1 日 2 回 21 10 mg/kg で 15%、30 mg/kg で54%、100 mg/kg で 66% 抑制 4.2.1.1.14 10、30、100 1 日 1 回 21 10 mg/kg で 53%、30 mg/kg で13%、100 mg/kg で 90% 抑制 頭頸部扁 平上皮癌 HN5 雌CD-1 ヌー ドマウス (n=8) 10、30、100 1 日 2 回 21 10 mg/kg で 40%、30 mg/kg で33%、100 mg/kg で 90% 抑制 4.2.1.1.14 30、100 1 日 1 回 21 30 mg/kg で腫瘍増殖を有意 に抑制、100 mg/kg で抑制 傾向 胃癌 MKN-45 雌無胸腺ヌ ードマウス (n=9~10) 100 1 日 2 回 21 100 mg/kg で腫瘍増殖を有 意に抑制 4.2.1.1.15 肝細胞癌 Hep3B 雌無胸腺ヌ ードマウス (n=9) 100 1 日 1 回 21 100 mg/kg で腫瘍増殖を有 意に抑制 4.2.1.1.16 Data source: 4.2.1.1.14 の Table 1、4.2.1.1.15 の Table 2 および 4.2.1.1.16 の Table 3
まとめ 本薬は腎細胞癌、卵巣癌、結腸癌、頭頸部扁平上皮癌、胃癌および肝細胞癌に対しても抗 腫瘍効果を有することが示された。 2.6.2.3. 副次的薬理試験 2.6.2.3.1. 各種受容体、チャネルおよびトランスポーターに対する結合親和性 4.2.1.2.1 方法 哺乳類由来またはヒト遺伝子組換え体の49 種類の各種受容体、チャネルおよびトランス ポーターに対するパゾパニブ二塩酸塩の結合親和性を放射性標識リガンド結合試験で検討し た。 結果 パゾパニブ二塩酸塩は10 µM でアデノシン A3、アドレナリンα2およびβ1、ヒスタミンH2、 ムスカリンM1ならびにセロトニン5-HT1A、5-HT5Aおよび5-HT7に対して50%以上の結合阻 害作用を示したが、これら以外の各種受容体、チャネルおよびトランスポーターに対して影 響は認められなかった。 2.6.2.3.2. 各種キナーゼに対する阻害作用 4.2.1.2.2 本薬の242 種類のキナーゼ活性に対する阻害作用を検討した。 方法 242 種のキナーゼ溶液に γ-33P-ATP およびそれぞれの基質ペプチドの存在下で本薬(0.3 お よび10 µM)を添加し、室温で 40 分間反応させた。リン酸水溶液を添加して反応を終了さ せたのち、リン酸化された基質ペプチドをフィルターマットに吸着させ、放射能を測定する ことによりキナーゼ活性を測定した。 結果 本薬は0.3 µM の濃度で 29 種のキナーゼに対して、10 µM で 94 種のキナーゼに対して 50%超の結合阻害作用を示した。そのうち、腫瘍血管新生に直接関わらない 13 種のキナー ゼに対するIC50が1 µM 未満であり、その中でも Aurora-A、c-Raf、MLK1、PTK5 および TAO3 に対する IC50はVEGFR-2 に対する IC50(12 および 30 nM:表 2.6.2-2)の 10 倍以内で あった。 2.6.2.3.3. ラット摘出心房標本における影響 4.2.1.2.3 本薬のβ アドレナリン受容体に対する作用を検討するために、ラットの摘出心房標本にお ける、プロプラノロール(β アドレナリン受容体アンタゴニスト)およびイソプロテレノー ル(β アドレナリン受容体アゴニスト)の作用に及ぼすパゾパニブ二塩酸塩の影響を検討し た。
方法 雄SD ラットから右心房を摘出し、Krebs 緩衝液を満たしたオルガンバス中に心房標本を 懸垂し、圧トランスデューサーを接続したポリグラフを用いて等尺性張力および心房拍動数 を測定した。 試験1:パゾパニブ二塩酸塩(3、10 および 30 µM)を累積的に添加し、つづいてプロプラ ノロール(1 µM)を添加して 45 分間インキュベートしたのち、パゾパニブ二塩酸塩(3、10 および30 µM)を累積的に添加して張力を測定した。 試験2:パゾパニブ二塩酸塩(30 µM)存在下でプレインキュベートしたのち、イソプロテ レノール(1、3 および 10 nM)を累積的に添加して張力および心房拍動数を測定した。 結果 試験1:パゾパニブ二塩酸塩(3、10 および 30 µM)は濃度依存的に収縮力を増加させ、そ の作用はプロプラノロールにより抑制されなかった。 試験2:パゾパニブ二塩酸塩(30 µM)を前処置することにより、イソプロテレノールの最 大収縮反応は減弱した。一方、パゾパニブ二塩酸塩はイソプロテレノールの心房拍動数には 影響を及ぼさなかった。 まとめ パゾパニブ二塩酸塩はβ アドレナリン受容体を介さない陽性変力作用を有することが示唆 された。また、イソプロテレノールの最大収縮反応を抑制したことから、高濃度では心房の β アドレナリン受容体を阻害する可能性が示されたが、心房拍動数には影響を示さなかった ことから、本薬のβ アドレナリン受容体に対する作用は弱いと考えられる。 2.6.2.3.4. 骨髄前駆細胞の増殖に及ぼす影響 4.2.1.2.4 Flt-3 および c-Kit は骨髄前駆細胞に発現しており、細胞の増殖および分化などに関与して いることから[Kumar, 2009]、これらの受容体を阻害することは骨髄抑制を引き起こすと考え られる。そこで、骨髄抑制を評価する標準的なGM-CSF を用いたコロニー形成細胞アッセ イ[Pessina, 2001]に加えて、本薬が阻害作用を示す可能性のある Flt-3 のリガンドおよび c-Kit のリガンドであるSCF を組み合わせた試験により本薬の骨髄抑制作用を評価し、ソラフェ ニブおよびスニチニブの作用と比較した。 方法 ヒト骨髄前駆細胞の懸濁液にヒト組換え体の増殖因子(GM-CSF、SCF および Flt-3 リガン ド)の単独または併用の存在下で本薬(0.1~10000 nM)、ソラフェニブ(0.1~10000 nM) またはスニチニブ(0.01~10000 nM)を添加し、10 日間培養したのち、顕微鏡下で CFU (細胞数50 個超のコロニー)をカウントした。 結果 本薬は濃度依存的なCFU 形成抑制作用を示したが、GM-CSF 単独による CFU 形成に対す る阻害作用のIC50は1079 nM であり、ソラフェニブおよびスニチニブと比較して同程度また
は弱い作用であった(表 2.6.2-8)。また、Flt-3 リガンド単独による CFU 形成に対する本薬 の抑制作用のIC50は8984 nM であり、ソラフェニブおよびスニチニブと比較して弱かった。 SCF 存在下では、本薬の GM-CSF または Flt-3 リガンドによる CFU 形成に対する抑制作用 はSCF 非存在下と比較して増強されたが、その程度はソラフェニブおよびスニチニブより も弱かった(表 2.6.2-8)。 表 2.6.2-8 各種増殖因子によるコロニー形成作用に対する抑制作用(類薬との比較) IC50 (nM) 増殖因子 パゾパニブ ソラフェニブ スニチニブ GM-CSF 1079±405 1671±1059 32.7±10.0 GM-CSF+SCF 173±45 86±18 1.1±0.7 GM-CSF+SCF+Flt-3 604±299 562±268 1.4±0.7 Flt-3 8984±3071 795±278 14.8±7.8 Flt-3+SCF 527±462 54±55 0.6±0.3 平均値±標準偏差 (n=3)
Data source: 4.2.1.2.4 の Table 1、2 および 3
まとめ 本薬の骨髄抑制作用はソラフェニブおよびスニチニブと比較して弱い可能性が示された。 2.6.2.4. 安全性薬理試験 2.6.2.4.1. 中枢神経系に及ぼす影響 4.2.1.3.1 雌SD ラットにおいて、本薬(3、10、100 および 300 mg/kg)は単回経口投与により、一 般状態および神経学的行動に影響を及ぼさず、死亡例も認められなかった。 2.6.2.4.2. 心血管系に及ぼす影響 2.6.2.4.2.1. イヌ摘出プルキンエ線維標本の活動電位に及ぼす影響 4.2.1.3.2 雌雄ビーグル犬から摘出したプルキンエ線維標本において、本薬は40 および 80 nM の濃 度で、1 または 0.5 Hz での刺激条件下で APD60、APD90、MRD、RMP および UA などの活動 電位パラメータに影響を及ぼさなかった。 2.6.2.4.2.2. hERG テール電流に及ぼす影響 4.2.1.3.3 hERG cDNA を安定導入した HEK-293 細胞において、本薬は 1.241 µM(0.543 µg/mL)お よび4.137 µM(1.810 µg/mL;溶解限界濃度)で hERG テール電流をそれぞれ 19.5 および 18.5%抑制したが、濃度依存性は認められなかった。抑制作用が弱かったため、IC25、IC50お よびIC75は算出できなかった。
2.6.2.4.2.3. 血圧、心拍数および心電図に及ぼす影響 4.2.1.3.4 および 4.2.1.3.5 覚醒雄カニクイザルにおいて、本薬(5、50 および 500 mg/kg)は単回経口投与により、 一般状態、全身動脈圧、脈圧、心拍数、心電図パラメータおよび体温に影響を及ぼさなかっ た。 覚醒雄カニクイザルにおいて、本薬(3.75 mg/kg)は単回静脈内投与により、可逆的で軽 度の心拍数減少を示したが、動脈圧および心電図パラメータに影響を及ぼさなかった。また、 心電図の波形異常および不整脈は認められず、一般状態および体温への影響も認められなか った。 2.6.2.4.3. 呼吸系に及ぼす影響 4.2.1.3.6 雄SD ラットにおいて、本薬(3、10、100 および 300 mg/kg)は単回経口投与により、呼 吸数、1 回換気量および分時換気量などの呼吸系パラメータに影響を示さなかった。また、 一般状態の異常および死亡例も認められなかった。 2.6.2.5. 薬力学的薬物相互作用試験 2.6.2.5.1. 他の抗癌剤の腫瘍組織への送達に及ぼす影響 4.2.1.4.1 血管新生阻害薬は他の抗癌剤と併用したときに、他の抗癌剤の腫瘍組織への送達を高める という仮説が提唱されている[Jain, 2005]。そこで、本薬および他の抗癌剤(5-FU、イリノテ カン、パクリタキセルおよびカルボプラチン)を併用したときの、本薬が併用した抗癌剤の 腫瘍組織への送達に及ぼす影響を検討した。また、比較対照薬としてVEGF に対するモノク ローナル抗体であり、血管新生阻害薬であるベバシズマブ[Van Meter, 2010]の影響をあわせ て検討した。 方法 HT29(ヒト結腸癌細胞株)または NCI-H460(ヒト非小細胞肺癌細胞株)の懸濁液を雌 CD-1 ヌードマウスの皮下に移植し、腫瘍が定着して一定の大きさに増殖したのち、本薬 (100 mg/kg)を 1 日 2 回 7 日間経口投与またはベバシズマブ(10 mg/kg)を 7 日間に 3 回 (Day 1、3 および 6)腹腔内投与した。 HT29 を用いたマウス異種移植モデルでは、本薬投与 7 日目に 5-FU(100 mg/kg)または イリノテカン(100 mg/kg)を単回腹腔内投与した。また、NCI-H460 を用いたマウス異種移 植モデルでは、本薬投与7 日目にパクリタキセル(20 mg/kg)またはカルボプラチン (50 mg/kg)を単回腹腔内投与した。 抗癌剤投与2 および 8 時間後に血液および腫瘍組織を採取し、LC-MS 法により血中およ び腫瘍組織中薬物濃度を測定した。 結果 HT29 を用いたマウス異種移植モデルにおいて、本薬(100 mg/kg、1 日 2 回 7 日間経口投 与)およびベバシズマブ(10 mg/kg、7 日間に 3 回腹腔内投与:Day 1、3 および 6)は、腫
瘍組織中の5-FU およびイリノテカン濃度に影響を及ぼさなかった。また、本薬およびベバ シズマブは5-FU およびイリノテカンの血中濃度にも影響を及ぼさなかった。 同様に、NCI-H460 を用いたマウス異種移植モデルにおいても、本薬およびベバシズマブ はパクリタキセルおよびカルボプラチンの血中および腫瘍組織中濃度に影響を及ぼさなかっ た。 まとめ マウス異種移植モデルにおいて、本薬は他の抗癌剤と併用したときに、腫瘍組織中の併用 薬の濃度に影響を及ぼさなかった。 2.6.2.6. 考察および結論 2.6.2.6.1. 効力を裏付ける試験 作用機序および細胞増殖抑制作用 悪性軟部腫瘍において、血管内皮増殖因子であるVEGF の発現レベルおよび腫瘍悪性度、 ならびに腫瘍内の微小血管密度および腫瘍悪性度の間に有意な相関が認められている[Pakos, 2005]。また、各種悪性軟部腫瘍において PDGF-B の発現レベルは病態の悪性度および増殖 マーカーと相関することが示されており[Wang, 1994]、悪性軟部腫瘍の 1 種である血管肉腫 では、VEGFR-2 の発現も確認されている[Itakura, 2008]。これらのことから、悪性軟部腫瘍 の病態には、VEGFR および PDGFR を介した血管新生が深く関与していると考えられる。 さらに、c-Kit 阻害作用を有するイマチニブは GIST、白血病および一部の固形癌の増殖を直 接的に(血管新生阻害作用を介さずに)抑制することが示唆されており[Mukherjee, 2009]、 c-Kit は一部の悪性軟部腫瘍の細胞膜上にも発現していることから、腫瘍増殖に関与してい る可能性が考えられている[Potti, 2004]。 In vitro における各種キナーゼ阻害作用を検討した結果、本薬は遺伝子組換えヒト VEGFR-1、2 および 3、PDGFR-α および β ならびに c-Kit に対してキナーゼ阻害作用(IC50=10~ 84 nM、Kiapp=2.4~30 nM)を示した。また、本薬は HUVEC の VEGFR-2、HFF の PDGFR-β および腫瘍細胞である NCI-H526 の c-Kit のリン酸化を阻害(IC50=3~8 nM)した。さらに、 本薬はbFGF(IC50=721 nM)よりも VEGF(IC50=21 nM)刺激により誘導される HUVEC 増殖作用を強力に抑制した。 本薬は in vitro において、281 種類のうち、ほとんどの腫瘍細胞株の増殖には影響を示さな かったが、A204 ヒト悪性軟部腫瘍(横紋筋肉腫)細胞株を含む 7 種類の細胞株に対しては 増殖抑制作用を示したことから(IC50=0.05~0.7 µM)、これらの腫瘍細胞に対しては血管 新生阻害作用を介さない直接的な増殖抑制作用を示す可能性が示唆された。 また、本薬のA204 ヒト横紋筋肉腫細胞株の増殖に対する抑制作用(IC50=0.26 µM)は、 VEGF 刺激による HUVEC の増殖に対する抑制作用(IC50=0.0213 µM)と比較して約 1/10 と 弱かったことから、本薬の in vitro における各種ヒト悪性軟部腫瘍細胞株の増殖に対する抑 制作用は弱いことが示された。なお、A204 細胞株は VEGFR-1 を発現しており[Onisto, 2005]、 VEGFR-1 が A204 細胞株の増殖に関与している可能性が考えられる。本薬はおもに VEGFR、
PDGFR および c-Kit に対する阻害作用を有することから、VEGFR-1 阻害作用により A204 細 胞株に対して直接的な増殖抑制作用を発現したと推察される。 一方、検討したほとんどの腫瘍細胞に対する in vitro での腫瘍増殖抑制作用も弱かったこ とから、本薬の腫瘍増殖抑制作用はおもに血管新生阻害作用を介しており、一部、腫瘍細胞 に対する直接的な増殖抑制作用が関与していると考えられる。 本薬は in vivo において、約 40 µM(17.5 µg/mL)以上の血漿中濃度でマウス肺組織の VEGFR-2 リン酸化に対して阻害作用を示した。この血漿中濃度は予定臨床用量である 800 mg を日本人固形癌患者に 1 日 1 回 22 日間経口投与したときの C24(血漿中未変化体の トラフ濃度、22.0 µg/mL、約 50 µM)と同程度であったことから(2.7.2.2.2.2.1)、本薬はヒ トに予定臨床用量(800 mg/日)を投与したときに、VEGFR のリン酸化に対して阻害作用を 示すと考えられる。 以上のことから、本薬はVEGFR-1、2 および 3、PDGFR-α および β ならびに c-Kit に対し て阻害作用を有するマルチキナーゼ阻害薬であることが示された。また、本薬は in vitro に おいてVEGF 刺激による HUVEC の増殖を強く抑制するものの、腫瘍細胞に対する直接的な 増殖抑制作用は弱いと考えられる。 血管新生阻害作用 In vivo において、本薬が血管新生阻害作用を有するか否かを血管新生のメカニズムがそれ ぞれ異なるマウスのレーザー誘発脈絡膜血管新生モデルおよびウサギのVEGF/bFGF ペレッ トモデルで検討した結果、本薬はマウスのレーザー誘発脈絡膜血管新生モデル(予防的投 与:レーザー照射日より200 mg/kg/日を 13 日間投与、治療的投与:レーザー照射 7 日後よ り8~200 mg/kg/日を 7 日間投与)およびウサギの VEGF/bFGF ペレットモデル(40 mg/kg/日 の26 日間投与)において、いずれのモデルにおいても血管新生阻害作用を示した。本薬は
in vitro において VEGF および bFGF による HUVEC の増殖に対して抑制作用を示したことか ら(2.6.2.2.1.2.1)、メカニズムの異なる様々なタイプの血管新生に対して効果を示すと考え られる。また、血管内皮増殖因子としてVEGF が血管新生において中心的な役割を果たして いると考えられているが、bFGF も血管内皮細胞増殖作用を有していることが示唆されてい ることから[Ellenberg, 2010]、病態時には VEGF および bFGF が協調的に働いて血管新生を引 き起こしていると考えられる。ウサギのVEGF/bFGF ペレットモデルでは血管内皮細胞増殖 因子としてVEGF および bFGF を併用しているが、本薬はこれらによる血管新生を完全に抑 制することから、複数の増殖因子が関与する病態時においても強力な血管新生阻害作用を示 すと考えられる。また、本薬はVEGFR-2 と比較すると弱いものの、FGFR-1 および 3 に対し ても阻害作用を有することが示されていることから(表 2.6.2-2)、bFGF による血管新生に 対する阻害作用には、本薬のFGFR-1 および 3 阻害作用が関与していると推察される。 抗腫瘍効果 脂肪肉腫は悪性軟部腫瘍のなかでもっとも頻度が高く、良性腫瘍と比較して血管密度が高 いことも示されている[Lewis, 1998, Baneth, 2005]。In vitro において、本薬の SW-872 ヒト脂 肪肉腫細胞株の増殖に対する抑制作用は弱く、IC50は10 µM 超であったが、in vivo では本薬
(100 mg/kg を 1 日 1 回、または 30 および 100 mg/kg を 1 日 2 回 21 日間経口投与)は SW-872 細胞株を用いたマウス異種移植モデルにおいて増殖抑制作用を示した。これらの結果か ら、本薬の脂肪肉腫細胞株に対する増殖抑制作用はおもに血管新生阻害作用を介して発現す ると考えられる。 本薬は悪性軟部腫瘍のなかでも頻度の高い組織型である脂肪肉腫および滑膜肉腫[日本整 形外科学会骨軟部腫瘍委員会, 2008]に対する増殖抑制作用を有する可能性が示唆されており、 様々な組織型の悪性軟部腫瘍に効果を示す可能性が考えられる。 SW-872 ヒト脂肪肉腫細胞株を用いたマウス異種移植モデルにおいて、本薬は 100 mg/kg を1 日 1 回、または 30 および 100 mg/kg を 1 日 2 回 21 日間経口投与により腫瘍増殖を抑制 した。また、SYO-1 ヒト滑膜肉腫細胞株を用いたマウス異種移植モデルにおいて、本薬は 30 および 100 mg/kg を 1 日 2 回 14 日間経口投与により腫瘍増殖を抑制した。マウスに本薬 の100 mg/kg を 1 日 1 回経口投与したときの投与 7 日目における血漿中未変化体濃度の投与 前値は約3 µg/mL であり(4.2.2.2.1)、予定臨床用量である 800 mg を日本人固形癌患者に 1 日1 回 22 日間経口投与したときの C24である22.0 µg/mL(2.7.2.2.2.2.1)よりも低かった。こ のように、マウスではヒトよりも低い血漿中濃度で抗腫瘍効果が認められていることから、 本薬は悪性軟部腫瘍に対して治療効果を示す可能性が考えられる。 なお、SYO-1 ヒト滑膜肉腫細胞株では PDGFR の発現が確認されており(4.2.1.1.17)、本 薬はPDGFR に対する阻害作用を有することから、SYO-1 細胞株を用いたマウス異種移植モ デルにおいて認められた腫瘍増殖抑制作用には、血管新生阻害作用のほかに、PDGFR 阻害 作用を介した直接的な細胞増殖抑制作用が関与している可能性が考えられる。一方、in vitro での各種ヒト悪性軟部腫瘍細胞株の増殖に対する抑制作用の検討において、本薬のSW-872 ヒト脂肪肉腫細胞株の増殖に対する抑制作用は弱いことが示されていることから (2.6.2.2.1.2.2)、SW-872 細胞株を用いたマウス異種移植モデルにおいて認められた腫瘍増 殖抑制作用は、作用機序としておもに血管新生阻害作用を介して発現すると考えられる。 さらに、本薬はヒト腎細胞癌株(CAKI-2、ACHN および 786-O)、卵巣癌細胞株 (OVCAR-3)、結腸癌細胞株(HT29)、頭頸部扁平上皮癌細胞株(HN5)、胃癌細胞株 (MKN-45)および肝細胞癌株(Hep3B)に対しても悪性軟部腫瘍と同程度の用量で抗腫瘍 効果を示したことから、悪性軟部腫瘍をはじめとする各種腫瘍に対する治療薬として期待さ れる。 他剤との比較(キナーゼ阻害作用) スニチニブおよびソラフェニブは腎細胞癌をはじめとした各種腫瘍に対する治療薬として 用いられている[スーテントカプセル 12.5mg, 2008; ネクサバール錠 200mg, 2008]。本薬、ス ニチニブおよびソラフェニブはc-Kit、Flt-3 および Raf に対する阻害作用の程度に差がみら れた(2.6.2.2.1.1.2)ものの、基本的には VEGFR および PDGFR を中心としたマルチキナー ゼ阻害による血管新生阻害作用を介して抗腫瘍効果を示す薬物であることから[Larkin, 2007]、 本薬はスニチニブおよびソラフェニブと同様に各種腫瘍に対する治療薬として有用であると 考えられる。
![図 2.6.2-1 血管新生の機序 Data source: [Ferrara, 2005]の Figure 1](https://thumb-ap.123doks.com/thumbv2/123deta/6392017.636968/10.918.209.714.87.616/図2621血管新生の機序DatasourceFerrara25のFigure1.webp)