セレコキシブ
臨床に関する概括評価
アステラス製薬株式会社
フ ァ イ ザ ー 株 式 会 社
目次
2.5 臨床に関する概括評価 ··· 1
2.5.1 製品開発の根拠 ··· 1
2.5.2 生物薬剤学に関する概括評価 ··· 14
2.5.3 臨床薬理に関する概括評価 ··· 17
2.5.4 有効性の概括評価 ··· 23
2.5.5 安全性の概括評価 ··· 34
2.5.6 ベネフィットとリスクに関する結論 ··· 45
2.5.7 参考文献 ··· 57
2.5 臨床に関する概括評価
2.5.1 製品開発の根拠
2.5.1.1 薬理学的分類
セレコキシブは,アラキドン酸をプロスタグランジン(PG)G/H に変換するシクロオキシゲナー ゼ(COX)酵素のうち,COX-2 を選択的に阻害するコキシブ(COXIB)系消炎鎮痛剤である.2.5.1.2 製品開発の臨床的・科学的背景
非ステロイド性抗炎症剤(NSAID)は COX を阻害し,PGE2等の炎症性メディエータ生成を
抑制することにより抗炎症作用,鎮痛作用及び解熱作用を示し,関節リウマチ(RA),変形性 関節症(OA),腰痛症等の炎症・疼痛性疾患に繁用されている1,2).しかし,同時に胃・十二指 腸等の上部消化管,血小板,腎等において生理機能を維持するために必要な PG の生合成をも 抑制し,上部消化管障害,血小板凝集阻害,腎機能障害等の副作用を誘発することが知られて いる3~7). 欧米では1980 年代から各種疫学調査により NSAID と消化管病変の頻度や実態が明らかにさ れてきた.NSAID による潰瘍は通常の消化性潰瘍に比べ発現頻度が高いだけでなく,必ずしも 自覚症状を伴わない無症候性であることが多いため,発見が遅れ,投与を継続している間に潰 瘍が進行し,出血,穿孔等の重篤な消化管障害に陥ることが指摘されている8~10).1997 年に報 告された米国の調査では,年間10 万人以上が NSAID による胃腸障害のために入院し,NSAID に関連する死亡者は16,500 人にのぼると推定されている11). 本邦においては,消化管障害等を軽減させるためにプロドラッグ化,徐放化等の製剤工夫が なされた NSAID が多数開発されてきたが,1991 年の日本リウマチ財団による消化管内視鏡を 用いた大規模な疫学調査の結果,これらの製剤を含む NSAID を 3 カ月以上服薬していた患者 1008 例中 15.5%に胃潰瘍(うち 41.3%は無症候性),1.9%に十二指腸潰瘍(うち 41.2%は無症候 性)が発現していたことが報告されている.また,この調査では消化管障害の既往のない患者 722 例中 73.0%に対し消化管障害の予防を目的として抗潰瘍剤が投与されていたが,抗潰瘍剤の 有無,種類によって潰瘍性病変の有病率に差はなかったと報告されている12,13). 近年,COX は単一の酵素ではなく,体内のほとんどの正常組織で広く産生される COX-1(構 成酵素)と炎症時に主に炎症組織で誘導されるCOX-2(誘導酵素)の 2 種類が存在することが 明らかになってきた14~17).COX-1 は胃,血小板,腎その他のほとんどの組織で常時発現してい るのに対し,COX-2 は活性化マクロファージや滑膜細胞においてサイトカイン等の炎症調節物 質により急速に誘導される18~20).
この新しい視点からみると,既存のNSAID はすべて COX-1 と COX-2 の両者に非選択的な阻 害剤である20,21).そこで,COX-2 を選択的に阻害して炎症部位における PGE2等の炎症性メディ
エータ生成のみを抑制し,既存の NSAID と同様の消炎・鎮痛効果を有しながら,COX-1 を阻 害しないことにより消化管障害等の副作用は既存の NSAID よりも少ない薬剤の開発が期待さ れている.
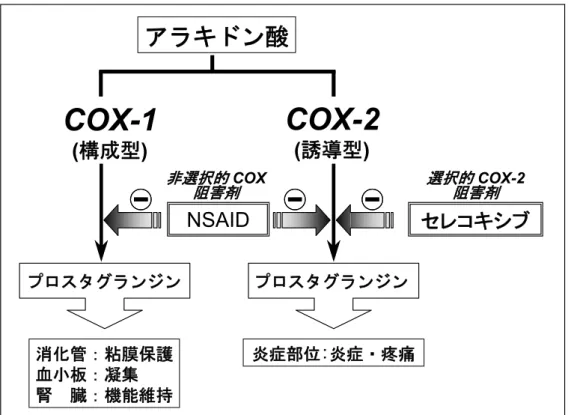
図2.5.1 に非選択的 COX 阻害剤(NSAID)及び選択的 COX-2 阻害剤(セレコキシブ)の COX-1 とCOX-2 に対する作用機序を示す.
アラキドン酸
COX-1
(構成型)
COX-2
(誘導型)
NSAID
非選択的COX 阻害剤消化管:粘膜保護
血小板:凝集
腎 臓:機能維持
炎症部位:炎症・疼痛
プロスタグランジン
プロスタグランジン
選択的COX-2 阻害剤セレコキシブ
図2.5.1 非選択的 COX 阻害剤及び選択的 COX-2 阻害剤の作用機序 セレコキシブは米国サール社(現ファイザー社)で合成されたコキシブ(COXIB)系の選択 的COX-2 阻害薬である.本薬は in vitro のヒト組換え酵素を用いた実験において,COX-2 を濃 度依存的に阻害し,その阻害活性のCOX-1 に対する比は 360 倍であった.また,COX-1 を恒常 的に発現するヒト単球様細胞株(U937 細胞)及び IL-1β刺激により COX-2 発現を誘導したヒ ト皮膚繊維芽細胞を用いた実験においてもセレコキシブは COX-2 に対して選択的な阻害作用 を示し,そのCOX-1 に対する比は 31 倍であった.in vivo では,各種炎症・疼痛モデルにおい て本薬は既存のNSAID と同等の抗炎症・鎮痛作用を示したのに対し,消化管粘膜及び血小板に 対する本薬の作用は既存のNSAID よりも弱いことが示された.単回投与毒性試験,反復投与毒 性試験,生殖発生毒性試験,抗原性試験,変異原性試験及び一般薬理試験の結果,他のNSAID と同様の所見が認められたが,臨床試験へ移行する上で重大な問題となるような所見は認めら れなかった. これら前臨床試験における知見から,本薬は既存のNSAID と同等以上の消炎・鎮痛効果を有 しながら,消化管障害や血小板凝集阻害,腎機能障害等の副作用は既存のNSAID よりも少ない 薬剤となりうることが予測され,臨床試験の開始が決定された. RA は関節滑膜の炎症を特徴とする全身性の自己免疫疾患であり,慢性的な関節の疼痛が日常生活に支障を与えるとともに,病態の進行による不可逆的な軟骨・骨破壊が身体機能障害をも たらす疾患である.RA に対する薬物療法としては NSAID,疾患修飾性抗リウマチ剤(DMARD), ステロイド剤等が用いられるが,NSAID は疼痛をやわらげるとともに炎症を抑えるための第一 選択薬であり,DMARD を投与する場合においても基礎治療薬として一般的に併用されている. ステロイド剤は強力な抗炎症作用を示す一方で副作用も強いため,長期連用は困難であり,一 時的・限定的な使用に留められている.また,「鎮痛消炎剤の臨床評価方法に関するガイドライ ン」22)の中で,「RA は,筋骨格系臓器・組織炎症のよいモデルであり,自然治癒がみられず,頻 度も高く,症状の変動がかなり小さく,過去において薬剤評価の経験も豊富なので評価の基準」 とされている. OA は荷重による膝関節,股関節等の退行性変化を基盤とした慢性疼痛疾患であり,高齢者 に多く,関節の疼痛,変形,可動域異常等により,歩行,階段昇降,姿勢変更等の日常生活に 苦痛と支障をもたらす疾患である.OA に対する治療法としては,手術,理学療法及び薬物療 法があり,薬物療法では,NSAID,ステロイド剤,関節軟骨保護剤等が用いられるが,慢性的 な疼痛を抑えるための対症治療法としてNSAID が第一選択薬である. 腰痛症,肩関節周囲炎,頸肩腕症候群及び腱・腱鞘炎は,OA と同様に,関節の退行性変化 を基盤とした疼痛,変形,可動域異常等を呈する慢性疼痛疾患であり,整形外科における保存 的対症治療法,とくにNSAID の使用法に関しても OA に極めて近い疾患群である. これらの慢性疼痛疾患では NSAID を比較的長期間にわたって投与する必要がある場合が多 く,上部消化管障害や血小板凝集阻害,腎機能障害等の副作用に対する懸念がより高いため, 本剤に対するニーズが最も高いと考えられるこれら慢性疼痛疾患を中心に本剤の臨床開発を 行った.
2.5.1.3 臨床開発計画及び関連するガイダンス
2.5.1.3.1 本邦における臨床試験
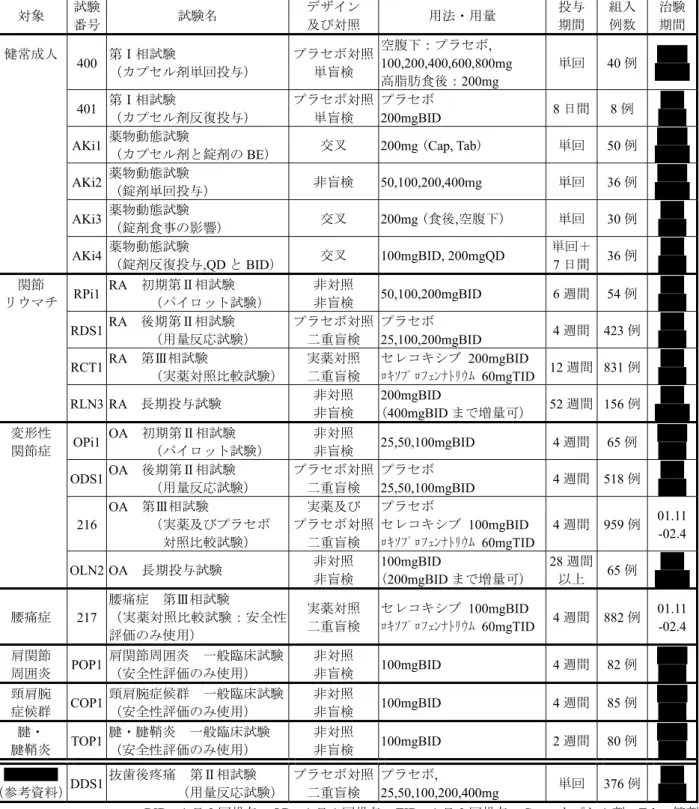
国内においては,健常成人を対象とした第I相試験を実施した後,「鎮痛消炎剤の臨床評価方 法に関するガイドライン」22)を参考に,本薬に対するニーズが最も高い慢性疼痛疾患であるRA, OA 及び類縁疾患(腰痛症,肩関節周囲炎,頸肩腕症候群及び腱・腱鞘炎)を中心に,19 試験 ( 1 試験を含む)を実施した. 国内で実施した臨床試験の一覧を表2.5.1 に示す.表2.5.1 国内で実施した臨床試験の一覧 対象 試験 番号 試験名 デザイン 及び対照 用法・用量 投与 期間 組入 例数 治験 期間 健常成人 400 第Ⅰ相試験 (カプセル剤単回投与) プラセボ対照 単盲検 空腹下:プラセボ, 100,200,400,600,800mg 高脂肪食後:200mg 単回 40 例 01.11 -02.4 401 第Ⅰ相試験 (カプセル剤反復投与) プラセボ対照 単盲検 プラセボ 200mgBID 8 日間 8 例 AKi1 薬物動態試験(カプセル剤と錠剤の
BE) 交叉 200mg (Cap, Tab) 単回 50 例 AKi2 薬物動態試験(錠剤単回投与) 非盲検 50,100,200,400mg 単回 36 例 AKi3 薬物動態試験 (錠剤食事の影響) 交叉 200mg (食後,空腹下) 単回 30 例 AKi4 薬物動態試験 (錠剤反復投与,QD と BID) 交叉 100mgBID, 200mgQD 単回+ 7 日間 36 例 関節 リウマチ RPi1 RA 初期第Ⅱ相試験 (パイロット試験) 非対照 非盲検 50,100,200mgBID 6 週間 54 例 RDS1 RA 後期第Ⅱ相試験 (用量反応試験) プラセボ対照 二重盲検 プラセボ 25,100,200mgBID 4 週間 423 例 RCT1 RA 第Ⅲ相試験 (実薬対照比較試験) 実薬対照二重盲検 セレコキシブロキソプロフェンナトリウム 200mgBID 60mgTID 12 週間 831 例 RLN3 RA 長期投与試験 非対照非盲検 (200mgBID 400mgBID まで増量可) 52 週間 156 例 変形性 関節症 OPi1 OA 初期第Ⅱ相試験 (パイロット試験) 非対照 非盲検 25,50,100mgBID 4 週間 65 例 ODS1 OA 後期第Ⅱ相試験 (用量反応試験) プラセボ対照二重盲検 プラセボ 25,50,100mgBID 4 週間 518 例 216 OA 第Ⅲ相試験 (実薬及びプラセボ 対照比較試験) 実薬及び プラセボ対照 二重盲検 プラセボ セレコキシブ 100mgBID ロキソプロフェンナトリウム 60mgTID 4 週間 959 例 OLN2 OA 長期投与試験 非対照 非盲検 100mgBID (200mgBID まで増量可) 28 週間 以上 65 例 腰痛症 217 腰痛症 第Ⅲ相試験 (実薬対照比較試験:安全性 評価のみ使用) 実薬対照 二重盲検 セレコキシブ 100mgBID ロキソプロフェンナトリウム 60mgTID 4 週間 882 例 01.11 -02.4 肩関節 周囲炎 POP1 肩関節周囲炎 一般臨床試験 (安全性評価のみ使用) 非対照 非盲検 100mgBID 4 週間 82 例 頸肩腕 症候群 COP1 頸肩腕症候群 一般臨床試験 (安全性評価のみ使用) 非対照 非盲検 100mgBID 4 週間 85 例 腱・ 腱鞘炎 TOP1 腱・腱鞘炎 一般臨床試験 (安全性評価のみ使用) 非対照 非盲検 100mgBID 2 週間 80 例 (参考資料)DDS1 抜歯後疼痛 第Ⅱ相試験 (用量反応試験) プラセボ対照 二重盲検 プラセボ, 25,50,100,200,400mg 単回 376 例
2.5.1.3.2 治験相談
本剤の臨床開発に関しては, から までの間に,医薬品 副作用被害救済・研究振興調査機構(現独立行政法人医薬品医療機器総合機構)(以下,医薬品 機構)との治験相談(本相談)を4 回実施した. 19 年 月 日及び20 年 月 日の治験相談では, について相談した.その結果,20 年 月 日の治験 相談において医薬品機構より, との結論が示された.これ を受けて, した. 20 年 月 日及び20 年 月 日の治験相談では, 方について相談し, , , , , 等について幅広く助言を得 た. は, した.2.5.1.3.3 臨床データパッケージ
本申請における臨床データパッケージ(評価資料)及び参考資料を表2.5.2 に示す.表2.5.2 本申請における臨床データパッケージ(評価資料)及び参考資料 区分 分類 地域 試験数 内容 試験番号 評価 資料 P-Ⅰ及び 薬物動態 日本 6 健常成人対象第Ⅰ相試験 及び薬物動態試験 [400][401] [AKi1][AKi2][AKi3][AKi4] (PK) - 患者対象薬物動態 (ポピュレーションPK) 国内患者対象4試験のPK併合解析 ([RDS1,ODS1,RLN3,OLN2]) 外国 9 第Ⅰ相試験及び 薬物動態試験 [001][003][032][006][037][018][044] [084][019] 3 特別な集団PK試験 [016][036][015] 14 薬物相互作用試験 [072][038][040][050][039][051][017] [095][109][114][116][117][135][171] RA 日本 4 初期第Ⅱ相試験(パイロット試験) 後期第Ⅱ相試験(プラセボ対照用量反応試験) 第Ⅲ相試験(実薬対照比較試験) 長期投与試験 [RPi1] [RDS1] [RCT1] [RLN3] OA 日本 4 初期第Ⅱ相試験(パイロット試験) 後期第Ⅱ相試験(プラセボ対照用量反応試験) 第Ⅲ相試験(実薬及びプラセボ対照比較試験) 長期投与試験 [OPi1] [ODS1] [216] [OLN2] 腰痛症 日本 1 第Ⅲ相試験(実薬対照比較試験:安全性評価 のみ使用) [217] 肩関節周囲炎 日本 1 一般臨床試験(安全性評価のみ使用) [POP1] 頸肩腕症候群 日本 1 一般臨床試験(安全性評価のみ使用) [COP1] 腱・腱鞘炎 日本 1 一般臨床試験(安全性評価のみ使用) [TOP1] 安全性 (併合解析) 日本 - 安全性(併合解析) 国内患者対象12試験の安全性併合解析 ([RPi1,RDS1,RCT1,RLN3,OPi1,ODS1, 216,OLN2, 217,POP1,COP1,TOP1]) 長期投与 安全性 外国 1 RA及びOAに対する長期投与試験
(継続長期投与) [024: Long-term Safety Study] 日本 1 抜歯後疼痛に対する第Ⅱ相試験
(プラセボ対照用量反応試験) [DDS1] 参考
資料
米国初回承認 外国 - 臨床成績概要(外国) [807: Clinical Data Summary] 申請資料 - 安全性概要(外国) [819: Integrated Summary of Safety] 外国市販後 臨床試験 外国 1 消化管安全性試験 (大規模高用量長期投与) [035-102: CLASS Study] 大腸ポリープ の再発予防 外国 1 散発性大腸腺腫再発予防効果試験 (二重盲検試験 ) [APC] 試験 外国 1 大腸腺腫性ポリープ再発予防効果試験 (二重盲検試験 ) [PreSAP] 外国 - 大腸ポリープの再発予防試験(併合解析) [APC / PreSAP] 本薬の臨床開発は,米国において先行して進められ,1998 年 12 月 31 日に「RA 及び OA の 徴候及び症状の軽減」を効能・効果として FDA より承認を取得し,1999 年 2 月から米国で販 売を開始した.米国における推奨用法・用量は,RA に対しては 100~200mg 1 日 2 回,OA に 対しては100mg 1 日 2 回又は 200mg 1 日 1 回である. 本申請における臨床データパッケージ(評価資料)は,国内で実施した健常成人を対象とし た6 試験及び慢性疼痛疾患患者を対象とした 12 試験のデータに,外国の特別な患者集団(慢性 腎障害,肝障害,高齢者)における薬物動態試験,薬物相互作用試験等を含む薬物動態試験26 試験及び外国のRA 及び OA に対する長期投与試験 1 試験のデータを加えて構成した.なお,
腰痛症に対しては, ため,本試験の有効性デー タは本申請における臨床データパッケージには含めなかった.また,肩関節周囲炎に対する一 般臨床試験[POP1](非対照試験),頸肩腕症候群に対する一般臨床試験[COP1](非対照試験) 及び腱・腱鞘炎に対する一般臨床試験[TOP1](非対照試験)の有効性データも本申請における 臨床データパッケージには含めなかった.安全性に関しては,腰痛症に対する第Ⅲ相試験[217], 肩関節周囲炎に対する一般臨床試験[POP1],頸肩腕症候群に対する一般臨床試験[COP1]及び 腱・腱鞘炎に対する一般臨床試験[TOP1]の計 4 試験も本剤の安全性を評価する上で重要なデー タと考え,本申請における安全性の臨床データパッケージに含めることとした.また,国内の 健常成人及び患者における薬物動態,並びに患者における安全性について,それぞれ併合解析 を行い,臨床データパッケージ(評価資料)に含めた. 参考資料として,本申請の効能・効果以外でこれまでに国内で実施した 抜歯後疼 痛 に対する臨床試験,米国の初回承認(RA 及び OA)申請資料である臨床データ概要及び安 全性統合概要,外国での大規模な高用量長期投与での市販後臨床試験であるCLASS 試験及び大 腸ポリープの再発予防試験(APC 試験,PreSAP 試験及び併合)のデータを添付した.
2.5.1.3.3.1 薬物動態に関する臨床データパッケージ
本申請における薬物動態に関する臨床データパッケージを表2.5.2.1 に示す. 本剤の健常成人における薬物動態については,国内においてカプセル剤を用いた第Ⅰ相試験 2 試験及び申請製剤(100mg 錠)を用いた薬物動態試験 4 試験を実施した.本申請においては, これら6 試験における日本人の薬物動態データに,特別な集団(慢性腎障害,肝障害,高齢者) における薬物動態,薬物相互作用等を含む外国での薬物動態試験26 試験のデータを加え,臨床 データパッケージを構成した. また,RA 及び OA 患者における薬物動態について,国内の RA,OA に対する後期第Ⅱ相試 験2 試験([RDS1][ODS1])及び長期投与試験 2 試験([RLN3][OLN2])の計 4 試験における 薬物動態のデータを併合して検討した(ポピュレーションPK). したがって,本申請における薬物動態に関する臨床データパッケージは,マスバランス試験, 肝障害及び腎障害の影響,薬物相互作用試験並びに他の疾患の影響の検討以外は,日本人を対 象に申請製剤と同一処方の100mg 錠を用いたデータで構成されている.表2.5.2.1 本申請における薬物動態に関する臨床データパッケージ
試験項目 日本 外国
試験番号 [AKi3] [AKi1] [400] [401][AKi2] [AKi4] [ODS1] [RDS1] [OLN1] [RLN1] [006] 試験BE 1) [001] [003] [015] [016] [036] [072]
相互 作用 試験2) [824] 3) マスバランス ○ 製剤BE ○ ○ 食事の影響 ○ ○ ○ 単回投与時の薬物 動態 ○ ○ ○ ○ 用量依存性 ○ ○ ○ 反復投与時の薬物 動態 ○ ○ ○ 日周変動の影響 ○ AM vs PM ○ 加齢の影響 ○ ○ ○ ○ ○ 性差 ○ ○ ○ ○ ○ ○ 肝障害患者 ○ 腎障害患者 ○ 薬物相互作用 ○ ○ CYP2C9の遺伝多型 の影響 ○ ○ ○ ○ 疾患の影響(糖尿 病、他) ○4) 患者PPK ○ ○ ○ ○ ○ 1):[037],[018],[044],[084] 2):[072],[017],[038],[039],[040],[050],[051],[095],[109],[117],[135],[114],[116],[171] 3):参考資料 4):[017],[039] 8 - -
2.5.1.3.3.2 慢性疼痛疾患患者に対する有効性及び安全性の臨床データパッケージ
本申請における慢性疼痛疾患患者に対する有効性及び安全性の臨床データパッケージを表 2.5.2.2 に示す. 本申請における有効性の臨床データパッケージは,用量反応,プラセボに対する優越性,標 準薬に対する非劣性,長期投与時の有効性等のすべてについて,国内で実施した臨床試験 8 試 験のデータから構成した. 一方,本剤の安全性については,国内で実施した短期投与10 試験及び長期投与 2 試験のデー タに,外国で実施されたRA 及び OA に対する長期投与試験[024]のデータを加え,臨床データ パッケージを構成した.2.5.1.3.3.2.1 RA に対する有効性の臨床データパッケージ
RA を対象とした臨床試験は,カプセル剤を用いた初期第Ⅱ相試験[RPi1](非対照パイロッ ト試験),申請製剤(100mg 錠及び 200mg 錠)を用いた後期第Ⅱ相試験[RDS1](プラセボ対照 用量反応試験),第Ⅲ相試験[RCT1](実薬対照比較試験)及び長期投与試験[RLN3](非対照試 験)の計4 試験を,国内で実施した.本剤の RA に対する有効性の臨床データパッケージは, これら4 試験のデータから構成した. RA に対する初期第Ⅱ相試験[RPi1]では,国内での大規模な用量設定試験(後期第Ⅱ相試験) の実施に先立ち,国内の少数例の患者を対象に,3 用量[50mg,100mg 及び 200mg 1 日 2 回(以 下,BID)]を用いて非対照非盲検で検討し,有効性の感触を得るとともに 200mgBID までの用 量で忍容性に問題がないことを確認した. RA に対する後期第Ⅱ相試験[RDS1]では,RA に対する本剤の用量反応性の確認及び至適用 量の設定を目的として,プラセボを対照に本剤3 用量(25mgBID,100mgBID 及び 200mgBID) を用いて,無作為化二重盲検並行群間比較により検討した.その結果から,本剤は RA に対し て100mgBID 及び 200mgBID の両用量において有効性が認められ,このうち 200mgBID が最も 有効性に優れ,忍容性に問題がない用量であると判断し,第Ⅲ相試験の用法・用量に設定した. RA に対する第Ⅲ相試験[RCT1]は,本剤 200mgBID の RA に対する有効性について国内の標 準薬に対する非劣性を検証し,また,安全性について標準薬と比較することを目的として,実 薬対照無作為化二重盲検並行群間比較により検討した.対照薬は,国内において RA に対する 臨床的有効性が確立され,消化管障害が少ないとされるプロドラッグであり,NSAID 市場にお ける占有率も高いロキソプロフェンナトリウム60mg 1 日 3 回(以下,TID)を選択した. RA に対する長期投与試験[RLN3]は,本剤(200mg~400mgBID)を長期投与したときの安全 性及び有効性を検討することを目的として,非対照非盲検で実施した.本試験では 200mgBID を一定期間投与した後,有効性,安全性を確認しながら増量可能なデザインとした. RA に対する臨床試験のデザイン,対象,投与期間,評価項目等については,「鎮痛消炎剤の 臨床評価方法に関するガイドライン」22),日本リウマチ協会薬効検定委員会の「委員会が基準 としている慢性関節リウマチに対する抗炎症・鎮痛剤の臨床試験実施基準方法」23)及び既存の NSAID の臨床試験成績に関する文献24,25)等を参考とし,後期第Ⅱ相試験以降の評価項目に関しては,米国リウマチ学会の「ACR コアセット」26)及び「ACR 改善基準」27)を含む米国FDA の 「RA 治療のための薬剤,医療機器及び生物製剤の臨床開発プログラム」ガイダンス 28)等の新 しい知見を取り入れて評価を行った.また,
については,医薬品機構との治験相談において助言を得て, 実施
表2.5.2.2 本申請における慢性疼痛疾患患者に対する有効性及び安全性の臨床データパッケージ 日本 外国 関節リウマチ(RA) 変形性関節症(OA) 腰痛症 肩関節 周囲炎 頸肩腕 症候群 腱・ 腱鞘炎 RA OA 位置付け・検討内容 初期 第Ⅱ相 試験 [RPi1] 後期 第Ⅱ相 試験 [RDS1] 第Ⅲ相 試験 [RCT1] 長期 投与 試験 [RLN3] 初期 第Ⅱ相 試験 [OPi1] 後期 第Ⅱ相 試験 [ODS1] 第Ⅲ相 試験 [216] 長期 投与 試験 [OLN2] 第Ⅲ相 試験 [217] 一般 臨床 試験 [POP1] 一般 臨床 試験 [COP1] 一般 臨床 試験 [TOP1] 長期 投与 試験 [024] 有効性 ・パイロット試験(瀬踏み) ○ ○ ・用量反応 (無作為化二重盲検) ○ ○ ・プラセボに対する優越性 (無作為化二重盲検) ○ ○ ○ ・標準薬に対する非劣性 (無作為化二重盲検) ○ ○ ・長期投与時の有効性 ○ ○ 安全性 ・短期投与時の安全性 ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ・長期投与時の安全性 ○ ○ ○ 11 - -
2.5.1.3.3.2.2 OA に対する有効性の臨床データパッケージ
OA を対象とした臨床試験は,カプセル剤を用いた初期第Ⅱ相試験[OPi1](非対照パイロッ ト試験),申請製剤(100mg 錠)を用いた後期第Ⅱ相試験[ODS1](プラセボ対照用量反応試験), 第Ⅲ相試験[216](実薬及びプラセボ対照比較試験)及び長期投与試験[OLN2](非対照試験) の計4 試験を,国内で実施した. OA に対する初期第Ⅱ相試験[OPi1]では,国内での大規模な用量設定試験(後期第Ⅱ相試験) の実施に先立ち,国内の少数例の患者を対象に,3 用量(25mgBID,50mgBID 及び 100mgBID) を用いて非対照非盲検で検討し,有効性の感触を得るとともに 100mgBID までの用量で忍容性 に問題がないことを確認した. OA に対する後期第Ⅱ相試験[ODS1]では,OA に対する本剤の用量反応性の確認及び至適用 量の設定を目的として,プラセボを対照に本剤3 用量(25mgBID,50mgBID 及び 100mgBID) を用いて,無作為化二重盲検並行群間比較により検討した.その結果から,100mgBID が OA に対して最も有効性に優れ,忍容性に問題がない用量であると判断し,第Ⅲ相試験の用法・用 量に設定した. OA に対する第Ⅲ相試験[216]は,本剤 100mgBID の OA に対する有効性について,プラセボ に対する優越性を再確認した上で国内の標準薬に対する非劣性を検証し,また,安全性につい て標準薬及びプラセボと比較することを目的として,プラセボ及び実薬対照無作為化二重盲検 並行群間比較試験により検討した.対照薬は,RA と同様に,国内において OA に対する臨床的 な評価が確立されているロキソプロフェンナトリウム60mgTID を選択した. OA に対する長期投与試験[OLN2]は,本剤(100mg~200mgBID)を長期投与したときの安全 性及び有効性を検討することを目的として,非対照非盲検で実施した.本試験では 100mgBID を一定期間投与した後,有効性,安全性を確認しながら増量可能なデザインとした. OA に対する臨床試験のデザイン,対象,投与期間,評価項目等については,「鎮痛消炎剤の 臨床評価方法に関するガイドライン」22)及び既存のNSAID の臨床試験成績に関する文献 29,30)等 を参考とし,後期第Ⅱ相試験以降の評価項目等に関しては,米国FDA の「OA 治療のための薬 剤,医療機器及び生物製剤の臨床開発プログラム」ガイダンス(案)31)等の新しい知見を取り入 れて評価を行った.また, については, 医薬品機構との治験相談において助言を得て, 実施した(添付資料 5.4-3,4 医薬品機構 治験相談記録).2.5.1.3.3.2.3 安全性の臨床データパッケージ
本剤の安全性に関しては,国内で実施したRA を対象とした 4 試験([RPi1][RDS1][RCT1] [RLN3]),OA を対象とした 4 試験([OPi1][ODS1][216][OLN2])及び OA 類縁疾患を対象 とした4 試験([217][POP1][COP1][TOP1])の計 12 試験のデータに,外国で実施された RA 及びOA に対する長期投与試験[024]のデータを加え,臨床データパッケージを構成した.2.5.1.3.3.2.3.1 長期投与における安全性の臨床データパッケージ
本剤の長期投与安全性に関しては,「致命的でない疾患に対し長期間の投与が想定される新医 薬品の治験段階において安全性を評価するために必要な症例数と投与期間」のガイドラインに 基づき,国内で実施した RA に対する長期投与試験[RLN3]及び OA に対する長期投与試験 [OLN2]のデータに,外国で実施された RA 及び OA に対する長期投与試験[024]のデータを加 え,臨床データパッケージを構成した.2.5.1.3.3.2.3.2 高齢者における投与
各疾患における後期第Ⅱ相以後のすべての試験では,「高齢者に使用される医薬品の臨床評価 法に関するガイドライン」に基づき,対象患者の年齢上限を設定せず,高齢者における使用経 験例を集積し,層別解析による検討を行った.2.5.1.3.4 その他の関連するガイダンス
第 I 相試験及び初期第Ⅱ相試験は,厚生省薬務局通知「医薬品の臨床試験の実施に関する基 準(GCP)」(薬発第 874 号,平成元年 10 月 2 日付)に従い,また,後期第Ⅱ相以後の試験(後 期第Ⅱ相試験,第Ⅲ相試験,長期投与試験,一般臨床試験及び薬物動態試験)は,「医薬品の臨 床試験の実施の基準に関する省令」(平成9 年 4 月 1 日施行)に従い実施した. 各試験の有効性及び安全性の統計解析に関しては,「臨床試験のための統計的原則」及び「臨床 試験における対照群の選択とそれに関連する諸問題」のガイドラインを考慮して解析を行った. 以上の臨床試験の成績から,本申請の効能・効果における推奨用法・用量,使用上の注意等 の根拠となる有効性と安全性に関する情報が集積され,承認要件が満たされるものと判断し, 今般,原薬の輸入及び製剤の製造承認を申請する. なお,本申請では初回申請効能・効果として,本剤に対するニーズが最も高い慢性疼痛疾患 を対象とした.今回,申請効能・効果から除いた「腰痛症」,「肩関節周囲炎」,「頸肩腕症候群」 及び「腱・腱鞘炎」については である.また, に対しては,これまでに国内で抜歯後疼痛に対する第Ⅱ相 試験[DDS1](参考資料)を実施しており である.2.5.2 生物薬剤学に関する概括評価
2.5.2.1 製剤開発過程,並びに溶出試験及び比較バイオアベイラビリティ試験結果
セレコキシブは米国で先行して開発され,米国を初めとする既承認国ではカプセル剤が市販 されている.国内では錠剤に剤形を変更し,100 mg 錠及び 200 mg 錠を申請製剤として開発し た.臨床試験に使用した製剤及び試験番号一覧は表2.7.1.4.2.1~3 に示した. セレコキシブの製剤間の比例性については,国内で実施した溶出試験1 試験,臨床試験 1 試 験及び各製剤の規格試験,並びに海外で実施した臨床試験4 試験及び溶出試験から評価した. バイオアベイラビリティ及び溶出特性の相互関係を表2.5.3 に示す. 表2.5.3 製剤間のバイオアベイラビリティ及び溶出特性の相互関係(その 1) 100 mg 海外P-Ⅰ カプセル* 25 mg 海外P-Ⅲ カプセル* 50 mg 海外P-Ⅲ カプセル* 100 mg 海外P-Ⅲ カプセル* 200 mg 海外P-Ⅲ カプセル* 100 mg 海外市販 カプセル* 200 mg 海外P-Ⅱ カプセル 200 mg 海外市販 カプセル 100 mg 海外P-Ⅰ カプセル* - - - - - - [037] に よ り AUC は同等 25 mg 海外P-Ⅲ カプセル* 溶出性は 同様1) 溶出性は 同様1) 溶出性は 同様1) 溶出性は 同様1) 溶出性は 同様1) 溶出性は 同様1) 50 mg 海外P-Ⅲ カプセル* 溶出性は 同様1) 溶出性は 同様1) 溶出性は 同様1) 溶出性は 同様1) 溶出性は 同様1) 100 mg 海外P-Ⅲ カプセル* 溶出性は 同様1) [084] に よ り AUC は同等 溶出性は 同様1) [084] に よ り AUC は同等 200 mg 海外P-Ⅲ カプセル* 溶出性は 同様1) [018] に よ り 生 物 学 的 に 同等 [044] に よ り 生 物 学 的 に 同等 100 mg 海外市販 カプセル* 溶出性は 同様1) [084] に よ り 生 物 学 的 に 同等 200 mg 海外P-Ⅱ カプセル 溶出性は 同様1) 200 mg 海外市販 カプセル *:国内臨床試験で使用,-:検討なし 1):溶出条件 B( )に よる規格試験で溶出性は同様表2.5.3 製剤間の溶出特性及びバイオアベイラビリティの相互関係(その 2) 100 mg 海外市販 カプセル 25 mg 錠 50 mg 錠 100 mg 錠 200 mg 錠 100 mg 海外市販 カプセル - - [AKi1]によ りAUC は同 等 - 25 mg 錠 溶出性は 同様2) 溶出性は 同様2) 溶出性は 同様2) 50 mg 錠 溶出性は 同様2) 溶出性は 同様2) 100 mg 錠 溶出試験に より生物学 的に同等 200 mg 錠 すべて国内臨床試験で使用,-:検討なし 2):溶出条件 C( )による規格試験で溶出性は同様 セレコキシブの海外カプセル剤の処方間でバイオアベイラビリティを比較した結果,最高血 漿中濃度(Cmax)は 2 剤 2 群 4 時期クロスオーバーの繰り返しデザインで実施された試験にお いて同等であり,血漿中濃度-時間曲線下面積(AUC)はすべての試験で同等であった.また, 海外カプセル剤と申請製剤と同一処方の錠剤との製剤間の比較において,Cmax については錠剤 / カプセル剤の比(90%信頼区間)は 1.20(1.09~1.33)であり,AUC は同等であった. 初期第Ⅱ相試験で使用した含量25,50,100 及び 200 mg のカプセル剤は各規格試験結果の比 較から同様の溶出性が認められ,後期第Ⅱ相以降の試験で使用した含量25,50,100 及び 200 mg の錠剤間でも同様の溶出性が認められた.また,溶出試験の結果,申請製剤と同一処方の100 mg 錠と200 mg 錠は生物学的に同等であった(表 2.5.3).
2.5.2.2 バイオアベイラビリティ試験結果
セレコキシブの水溶解度は25 ℃,pH 7 で μg/mL,pKa は 11.1,オクタノール/水の分配 係数は>103であり,セレコキシブは と判断された.このため で あり,絶対バイオアベイラビリティ試験は実施していない. 14C-セレコキシブを用いたマスバランス試験の結果,放射能は速やかに吸収されて血漿中には 主として未変化体として存在した.また,血漿,尿及び糞中にシクロオキシゲナーゼ阻害活性 を示さない代謝物が合計3 種類検出された.投与された放射能の約 58%は糞中に,約 27%は尿 中に排泄され,尿中に未変化体は検出されず,糞中未変化体排泄率は約3%であった. 申請製剤と同一処方の100 mg 錠を用いた食事の影響試験の結果,食後投与時で空腹下投与時 に比べてセレコキシブのCmax は約 1.5 倍と高値を示し,AUC は同等であった.本剤の健常成人におけるバイオアベイラビリティの個体間変動(変動係数:CV)は Cmax で 33%以上,AUC で 29%以上,並びに最高血漿中濃度到達時間(Tmax)で 43%以上であった. また,個体内変動(CV)は Cmax で %以上,AUC で %以上であった.Cmax の個体内変動 (CV)が %を超えることから,セレコキシブは highly variable drug1)に分類された.
2.5.2.3 生物薬剤学に関する概括評価
セレコキシブの処方間あるいは製剤間の比例性については国内及び海外で実施した溶出試験, 各製剤の規格試験又は臨床試験のいずれかにより評価した. 溶出試験の結果,申請製剤と同一処方の100 mg 錠及び 200 mg 錠は生物学的に同等であるこ とが確認された.また,各製剤の規格試験の結果において,初期第Ⅱ相試験で使用した含量25, 50,100 及び 200 mg のカプセル剤の各含量間で同様の溶出性を示し,後期第Ⅱ相以降の試験で 使用した含量25,50,100 及び 200 mg の錠剤の各含量間でも同様の溶出性を示した. 臨床試験によりセレコキシブの海外カプセル剤の処方間,あるいは海外カプセル剤と申請製 剤と同一処方の錠剤との製剤間でバイオアベイラビリティを比較した結果,Cmax は 2 剤 2 群 4 時期クロスオーバーの繰り返しデザインで実施した試験において同等であり,AUC はすべての 試験で同等であった.また,繰り返しのないデザインで同等性基準を逸脱した場合も Cmax の 比の90%信頼区間は 0.7~1.43 の範囲内であった.本剤は Cmax の個体内変動(CV)が %を 超えるhighly variable drug1)であること,溶出は処方又は含量にかかわらず同様であること,及 び繰り返しデザインの試験では Cmax は同等であったことから,繰り返しのないデザインで Cmax について同等性判定基準を逸脱したのは,製剤又は処方変更の影響よりむしろ個体内又は 個体間変動によるものと推察された.本剤の安全域は比較的広いこと,及び同等性基準を逸脱 した場合もCmax の比の 90%信頼区間は 0.7~1.43 の範囲内であったことから製剤又は処方変更 の生物学的同等性に及ぼす影響は生物学的同等の許容域2)内であると推察された.したがって, 本剤のバイオアベイラビリティはカプセル剤の処方間,あるいはカプセル剤と錠剤の製剤間で 生物学的に同等であるとみなされた. 食後投与時で空腹下投与時に比べて,Cmax は約 1.5 倍と高値を示し,AUC は高値を示す傾 向はあるものの同等であった.海外においてカプセル剤と懸濁液のバイオアベイラビリティ比 較も行っており,その結果,懸濁液投与時でカプセル剤投与時に比べて Cmax は高値を示した ことから,食事の影響はセレコキシブが難溶性であることに起因すると推察された.2.5.3 臨床薬理に関する概括評価
セレコキシブの用量-反応及び濃度-応答(PK/PD)に関する検討は実施していない.本項 ではセレコキシブの薬物動態(PK)に関する知見を記載する.2.5.3.1 健常成人・患者・特別な患者集団における比較 PK の結果
2.5.3.1.1 健常成人における基本的な薬物動態
健常成人において,単回投与時の血漿中セレコキシブ濃度は投与後2~3 時間に最高値に達し た後,二相性を示して低下し,消失半減期(t1/2)は5~10 時間であった.見かけの分布容積(Vd/F) 及び経口クリアランス(CL/F)の平均値はそれぞれ 244~649 L 及び 32~41 L/h であった. 14C-セレコキシブを用いたマスバランス試験の結果,放射能は速やかに吸収され,血漿中には 主として未変化体として存在すること,血球への選択的移行性は低いことが明らかとなった. また,血漿,尿及び糞中にシクロオキシゲナーゼ-1 又は 2 の阻害作用を示さない 3 種類の代謝 物が検出され,大部分が代謝された後に主に糞中に排泄された.なお,本剤は であり,絶対バイオアベイラビリティ試験は実施していない.2.5.3.1.2 患者における薬物動態
OA 患者及び RA 患者 609 例に本剤 25,50,100,200 又は 300 mg を 1 日 2 回反復投与した ときの定常状態における血漿中未変化体濃度測定値 1160 点を用いて非線形混合効果モデルに よる母集団薬物動態(PPK)解析を行った. OA 患者及び RA 患者に本剤 25~300 mgBID を投与したときの定常状態における CL/F,Vd/F 及び吸収速度定数(ka)の母集団平均(±標準誤差)は,年齢 65 歳,体重 54 kg 並びに血清ア ルブミン濃度 4.1 g/dL と仮定したとき,それぞれ 21.2 L/h(±0.551),335 L(±33.5),及び 1.62 /h (±0.275)と推定された.また,個体間変動(CV)は CL/F で約 42%,Vd/F で約 77%,並びに ka で約 121%,個体内変動(CV)は約 39%とそれぞれ推定された.被験者背景等を用いた探索 的な共変量調整の結果,患者において年齢及び体重は本剤のCL/F に,血清アルブミン濃度は本 剤のVd/F にそれぞれ有意な共変量であることが示され,年齢が 1 歳増すと CL/F は約 1%低下 し,体重が1 kg 増すと CL/F は約 1%上昇すると推定された.また,血清アルブミン濃度が 0.1 g/dL 上昇するとVd/F は約 10%低下すると推定された.2.5.3.1.3 特別な患者集団における薬物動態
2.5.3.1.3.1 インスリン非依存性糖尿病患者
グリベンクラミド(5 mg を 1 日 1 回,又は 10 mg を 1 日 2 回)を服用しているインスリン非 依存性糖尿病患者21 名におけるセレコキシブ(200 mg,1 日 2 回 7 日間)の薬物動態パラメー タは他の試験の健常成人における値と大差なかった.2.5.3.1.3.2 RA 患者
メトトレキサート(5~20 mg/週)を服用している RA 患者 7 名におけるセレコキシブ(200 mg, 1 日 2 回 7 日間)の薬物動態パラメータは他の試験の健常成人における値と大差なかった.2.5.3.2 内因性要因
2.5.3.2.1 加齢の影響
健常高齢者群で健常非高齢者群に比べて最高血漿中濃度(Cmax)及び血漿中濃度-時間曲線 下面積(AUC)は単回投与時及び反復投与後においてそれぞれ 1.7~1.8 倍及び 1.5~1.7 倍(背 景因子で未調整の幾何平均値比)と高値を示し,加齢はセレコキシブの薬物動態の変動要因で あることが示された.また,反復投与後のCmax 及び AUCτの値は高齢者群で非高齢者群に比べ て,男性で約1.2 倍及び約 1.3 倍,女性でいずれも約 2.2 倍と高値を示し(背景因子で未調整の 幾何平均値比),加齢の影響は女性で顕著に認められた.健常高齢者で健常非高齢者に比べて高 い血漿中濃度を示すことから添付文書中では慎重投与として扱った.2.5.3.2.2 性差の影響
国内及び海外で実施されたいずれの試験においても健常非高齢者群で薬物動態に性差は認め られなかった.健常高齢者における性差の検討は海外でのみ実施されており,健常高齢者群で は女性におけるCmax 及び AUCτは男性に比べてそれぞれ約1.5 及び約 1.4 倍(背景因子で未調 整の幾何平均値比)と高値を示した.2.5.3.2.3 民族・人種の影響
健常成人における本剤の薬物動態を日本人と外国人の民族間で比較することを目的として, 各試験結果の比較,及び線形混合モデルによる比較の 2 種類の併合解析を行った.解析には健 常成人を対象として実施した国内6 試験及び海外 18 試験の計 24 試験を使用した. 各試験結果の比較において,両地域で検討された項目である個体間変動,用量依存性,反復 投与時の蓄積性,日周変動の影響,健常非高齢者における性差並びに食事の影響について各試 験報告書の結果を国内と海外とで比較したところ,これらの項目において日本人と外国人とで 本剤の薬物動態における質的な差は認められなかった. 線形混合モデルによる比較において,日本人 / 外国人の比(95%信頼区間)は,国内及び海 外で共通して検討された50~600 mg の用量範囲で,Cmax で 1.11(1.04~1.19),AUCssで0.79 (0.75~0.84)と,Cmax は日本人と外国人で同程度の値を示し AUCssは日本人で低値を示した. また,人種別に比較したときにも日本人における Cmax は他の人種(Caucasian,Black 又は Hispanic)と同程度の値を示し,AUCssは日本人で低値を示した.同一試験内で日本人及び外国 人をともに対象とした試験はないため内因性民族間差を試験間差から分離して評価することは できなかったが,日本人健常成人におけるCmax は外国人と同程度であり,AUCssは外国人に比 べて低いことが示された.2.5.3.2.4 肝障害の影響
肝障害患者における血漿中セレコキシブ濃度は健常成人に比べて高値を示した.単回投与時 におけるCmax 及び AUC72hの値を肝障害患者群と健常成人群とで比較したとき,軽度肝障害患 者群ではそれぞれ約1.5 倍及び約 1.2 倍と高値を示したもののいずれの PK パラメータにも有意差はなかった一方で,中等度肝障害患者群ではそれぞれ約1.5 倍及び約 2.3 倍と高値を示し,CL/F, AUC12h及びAUC72hに有意差が認められた.反復投与後では,軽度肝障害患者群におけるCmax
及びAUCτはそれぞれ約1.4 倍及び約 1.3 倍と高値を示して Cmax に有意差が認められ,中等度
肝障害患者群のCmax 及び AUCτはそれぞれ約2.2 倍及び約 2.7 倍と高値を示し,CL/F,Cmax,
AUCτ及び AUC72hに有意差が認められた.単回投与時及び反復投与後のいずれも最高血漿中濃 度到達時間(Tmax)は軽度及び中等度肝障害患者群ともに健常成人群と同程度の値を示した. また,クレアチニンクリアランス,SGOT,SGPT 及び総ビリルビンのいずれにもすべての群に おいて投与前後で臨床的意義のある変動は認められなかった.以上のことから,肝障害はセレ コキシブの薬物動態の変動要因であることが示された.肝障害患者で健常成人に比べて高い血 漿中濃度を示すことから添付文書中では慎重投与として扱った.なお,重度肝障害患者におけ る本剤の薬物動態については臨床試験では検討していない.
2.5.3.2.5 慢性腎障害の影響
慢性腎障害患者におけるセレコキシブの薬物動態パラメータは他の試験の健常成人における 値と大差なかった.また,OA 患者及び RA 患者を対象とした PPK 解析の結果,クレアチニン, BUN,β2-マイクログロブリン及び NAG はすべて CL/F 及び Vd/F のいずれに対しても有意な共 変量とはならなかった.2.5.3.3 外因性要因
2.5.3.3.1 喫煙
セレコキシブの薬物動態に及ぼす喫煙の影響を明らかにするための試験は実施していない.2.5.3.3.2 併用薬
In vitro 代謝試験の結果,ヒトにおけるセレコキシブの代謝に主として関与するチトクロム P450(CYP)分子種は CYP2C9 であることが示された.この他に,CYP2C9 より寄与は小さい もののCYP3A4 の関与する可能性も示された.また,セレコキシブは主要な 5 種類のヒト CYP 分子種の中ではCYP2D6 を比較的強く阻害することが明らかとなった.以上のことを考慮して, セレコキシブと,臨床上併用頻度が高いと予想される薬剤,安全域の狭い薬剤,あるいは各種 CYP 分子種の基質又は阻害薬との薬物相互作用試験を実施した. 薬物相互作用試験の結果,CYP2C9 の阻害剤として知られるフルコナゾール又はフルバスタ チンとの併用によりセレコキシブの血漿中濃度は上昇し,パロキセチン又は制酸剤との併用に より本剤の血漿中濃度は低下した.本剤との併用によりCYP2D6 の基質として知られるパロキ セチン及びデキストロメトルファンの血漿中濃度は上昇した.併用薬の影響の詳細は 2.5.3.10 他の医薬品又は物質との臨床的薬物相互作用に記載した.2.5.3.3.3 食事
食後投与時において空腹下投与時に比べて,Cmax は高値を示し,AUC は高値を示す傾向は あるものの同等であり,この傾向は食事内容にかかわらず同じであった.したがって,セレコ キシブの吸収速度及び吸収量は食事内容にかかわらず食後投与時で空腹下投与時より上昇し, 吸収量における食事の影響の程度は吸収速度に比べて小さいことが示された.2.5.3.4 吸収速度及び吸収量
14C-セレコキシブを用いたマスバランス試験の結果,血漿中放射能の Tmax は 1.75 時間であ り,吸収は速やかであった.2.5.3.5 血漿たん白結合率
In vitro 及び ex vivo のいずれにおいてもセレコキシブのヒト血漿蛋白結合率は約 97%であり, in vitro で添加濃度 0.1~10 μg/mL,ex vivo で血漿中総薬物濃度 12.3~4020 ng/mL において濃度 依存性は認められなかった. ヒト血漿蛋白標準品に対する 14C-セレコキシブの in vitro 血漿蛋白結合率はアルブミンで 99.8%~100%,α1-酸性糖蛋白質で 78.6%~92.4%と,両蛋白質はいずれもセレコキシブの結合蛋 白質であることが示された.2.5.3.6 代謝経路,主代謝酵素及び遺伝多型の影響
2.5.3.6.1 代謝経路及び主代謝酵素
In vitro 試験及び14C-セレコキシブを用いたマスバランス試験の結果,セレコキシブを投与後 の血漿,尿及び糞中に代謝物として芳香環メチル基が酸化されたベンジル水酸化体のSC-60613, 更に酸化されたカルボン酸体のSC-62807,及び SC-62807 の 1-o-グルクロン酸抱合体が検出さ れた.SC-60613 及び SC-62807 はシクロオキシゲナーゼ-1 及び 2 の阻害作用を示さなかった. In vitro 代謝試験及び薬物相互作用試験の結果,セレコキシブの代謝に主として関与するヒト CYP 分子種は CYP2C9 であることが示された.2.5.3.6.2 遺伝多型の影響
国 内 で 実 施 し た 薬 物 動 態 試 験 4 試 験 の 併 合 解 析 の 結 果 , CYP2C9 の ヘ テ ロ 接 合 体 (CYP2C9*1/*3)群(15 例)では野生型(CYP2C9*1/*1)群(137 例)に比べて CL/F は低値を, 血漿中濃度は高値を示すことが明らかとなった.また,吸収速度に遺伝多型群間で差はなかっ た.CYP2C9*1/*3 群における AUCssはCYP2C9*1/*1 群に比べて約 1.6 倍(95%信頼区間; 1.4~1.9 倍)高く,これは外国人(Caucasian)において CYP2C9*1/*3 及び CYP2C9*3/*3 をあわせて CYP2C9*1/*1 と比較した結果(約 2.2 倍)と同程度であった 1).さらに,外国人(Caucasian) 健常成人(21 例)に本剤 100 mg を単回経口投与したとき,CYP2C9*1/*1(4 例)に比べて CYP2C9*3/*3(3 例)において CL/F は約 1/3 に低下し,また,t1/2は約1.5 倍に延長したこと,
並びにCmax における CYP2C9*3 変異の影響は有意でなかったことが報告されている2).日本
人のCYP2C9*3/*3 における成績はないが,外国人の CYP2C9*3/*3 でクリアランスの低下がみ られていることから,日本人のCYP2C9*3/*3 においてもクリアランスが低下し,血漿中濃度が 高値を示す可能性がある.
2.5.3.7 排泄
14C-セレコキシブを用いたマスバランス試験の結果,総放射能の尿及び糞中排泄率は投与され た放射能のそれぞれ約27%及び約 58%であり,主として糞中に排泄されることが示された.尿 中に未変化体は検出されず,糞中未変化体排泄率は約3%であり,また,主代謝物である SC-62807 の尿及び糞中排泄率は投与された放射能のそれぞれ約19%及び約 54%であった.したがって, セレコキシブは大部分が代謝された後に糞中に排泄されることが示された.2.5.3.8 日周変動
夕投与時で朝投与時に比べてTmax は遅延し,Cmax 及び投与前値はいずれも約 0.7 倍と低値 を示したが,AUCτは同等であった.セレコキシブの薬物動態に認められた日周変動の原因は明 らかではないが消化管血流の日周変動あるいは起床時及び就寝時での体位の差が一因となって いる可能性がある.2.5.3.9 立体化学的問題
セレコキシブには光学異性体は存在しない.2.5.3.10 他の医薬品又は物質との臨床的薬物相互作用
薬物動態上の薬物相互作用の有無は各試験で Cmax 及び AUC について推定された併用時 / 非併用時の比の95%信頼区間又は 90%信頼区間がいずれも 0.8~1.25 の範囲内にあるとき薬物動 態学的な相互作用はないと判定した. セレコキシブの薬物動態に及ぼす併用薬の影響を検討した結果,セレコキシブの Cmax 及び AUC はフルコナゾール併用によりそれぞれ約 1.7 倍及び約 2.3 倍,フルバスタチン併用により いずれも約1.3 倍に上昇したことから添付文書中では併用注意として扱った. リチウム併用時の本剤の Cmax,オメプラゾール併用時の本剤の AUC,ケトコナゾール併用 時の本剤のCmax は薬物相互作用の判定基準を逸脱したがいずれも信頼区間は 0.7~1.43 の範囲 内にあり,かつ 1 を含んでいた.また,制酸剤又はパロキセチン併用により本剤の Cmax は低下 した.本剤はhighly variable drug 3)に分類されること及び安全域が広いことから,リチウム,オメ プラゾール,ケトコナゾール,制酸剤又はパロキセチンとの併用により本剤の薬物動態に認め られた薬物相互作用の臨床的意義は低い. 他の薬剤の薬物動態に及ぼすセレコキシブの影響を検討した結果,フルバスタチンの Cmax は薬物相互作用の判定基準を逸脱したが信頼区間は0.7~1.43 の範囲内であり,かつ 1 を含んで いた.オメプラゾールの Cmax は薬物相互作用の判定基準を逸脱したが信頼区間は 1 を含んで いた.以上のことから,セレコキシブ併用によりオメプラゾール及びフルバスタチンの薬物動態に認められた薬物相互作用の臨床的意義は低い.
リチウムは安全域の狭い薬剤として知られておりセレコキシブ併用により血漿中リチウム濃度 は試験期間を通じてリチウムの安全域上限である1.5 mEq/mL を超えなかったものの,Cmax 及び AUC のセレコキシブ併用時 / 非併用時の比の点推定値はそれぞれ約 16%及び約 17%高値を示し たことから添付文書中では併用注意として扱った.セレコキシブ併用により,パロキセチンの Cmax 及び AUC はそれぞれ約 1.5 倍及び約 1.8 倍,デキストロメトルファンの Cmax 及び AUC はそれぞれ約2.4 倍及び約 2.6 倍に上昇したことから添付文書中では併用注意として扱った. 肝ミクロソームを用いた In vitro 相互作用試験の結果,セレコキシブは濃度依存的に S-ワル ファリンの消失を阻害した.一方,健常成人を対象としたセレコキシブとワルファリンの薬物 相互作用試験の結果,セレコキシブはワルファリンの薬物動態及びプロトロンビン時間に影響 を及ぼさなかった.しかしながら,海外ではセレコキシブとワルファリンを併用している高齢 患者で出血時間の延長が報告されていることから,臨床上注意が必要であり,添付文書中では 併用注意として扱った. セレコキシブ併用によりメトトレキサートの薬物動態が変動しなかったことは RA 患者に臨 床上重要な利益となり得ると推察された.
2.5.4 有効性の概括評価
本有効性の概括評価では,申請効能・効果の各疾患(関節リウマチ,変形性関節症)に対す る有効性を,「2.5.4A 関節リウマチ(RA)に対する有効性」及び「2.5.4B 変形性関節症(OA) に対する有効性」の2 つのセクションに分けて記述する. なお,個々の臨床試験の試験方法及び結果の詳細は「2.7.6 個々の試験のまとめ」に記載した.
2.5.4A 関節リウマチ(RA)に対する有効性
RA を対象とした臨床試験は,カプセル剤を用いた初期第Ⅱ相試験[RPi1](非対照パイロッ ト試験),申請製剤(100mg 錠及び 200mg 錠)を用いた後期第Ⅱ相試験[RDS1](プラセボ対照 用量反応試験),第Ⅲ相試験[RCT1](実薬対照比較試験)及び長期投与試験[RLN3](非対照試 験)の計4 試験を,国内で実施した.本剤の RA に対する有効性の臨床データパッケージは, これら4 試験のデータから構成した. これら4 試験において被験薬が 1 回以上投与された症例の総数は 1359 例であった. 4 試験における人口統計学的特性は,いずれの項目においても比較的近似した値又は分布で あり,試験間における著しい差異はみられなかった.2.5.4A.1 RA に対する比較対照試験
RA に対する後期第Ⅱ相試験[RDS1]では,RA に対する本剤の用量反応性の確認及び至適用 量の設定を目的として,プラセボを対照に本剤3 用量(25mgBID,100mgBID 及び 200mgBID) を用いて,無作為化二重盲検並行群間比較により検討した.有効性の主要評価項目は最終全般 改善度とし,1~2 週間の観察期間の後,被験薬を 4 週間投与した. 本試験に組み入れられた患者は423 例であり,うち被験薬が 1 回以上投与された症例は 376 例であった.PPS(Per Protocol Set)において検討した人口統計学的特性及び投与前基準値で,群間に不均 衡が認められた項目はなかった.
PPS において,主要評価項目とした最終全般改善度による改善率(「中等度改善」以上)は, プラセボ群,25mgBID 群,100mgBID 群及び 200mgBID 群でそれぞれ 23.3%,19.2%,31.9%及 び 31.6%であった.最終全般改善度の 2 値評価(「中等度改善」以上,「軽度改善」以下)に対 するShirley-Williams 検定(有意水準片側 2.5%)では,本剤群とプラセボ群の間に統計学的に有 意な差は認められなかったものの(p=0.1496),7 段階判定(「著明改善」から「著明悪化」の 7 段階)に対する同検定では,200mgBID 群でプラセボ群に比し統計学的に有意な差が認められ た(p=0.0038).また,後に第Ⅲ相試験の主要評価項目とした ACR 改善基準(変法)による最 終評価時の改善率は,プラセボ群,25mgBID 群,100mgBID 群及び 200mgBID 群でそれぞれ 7.5%, 9.3%,16.7%及び 24.1%と,本剤の用量の増加とともに上昇しており,200mgBID 群でプラセボ 群に比して統計学的に有意に優れていた(p=0.0027).患者の疼痛評価(VAS),患者の疾患活動 性全般評価(VAS),医師の疾患活動性全般評価(VAS),患者の身体機能評価(mHAQ)等の副 次的評価項目においても用量依存的な改善が認められ,100mgBID 群及び 200mgBID 群では,
多くの評価項目においてプラセボ群に比して統計学的に有意な差が認められた.このうち 200mgBID 群では,100mgBID 群に比して,より高い有効性が認められた.
一方,安全性について,概括安全度による安全率(「安全である」と判定された症例の割合) は,プラセボ群,25mgBID 群,100mgBID 群及び 200mgBID 群でそれぞれ 83.2%,76.5%,82.0% 及び79.1%であった.有害事象(症状及び身体徴候)の発現率は,プラセボ群,25mgBID 群, 100mgBID 群及び 200mgBID 群でそれぞれ 21.1%,27.6%,28.1%及び 20.7%であり,本剤の用量 の増加による発現率の上昇はみられなかった.消化管潰瘍及び出血性事象は,プラセボ群で 1 例(1.1%)にみられたのみで,本剤群ではいずれの用量においても認められなかった.臨床検 査値異常変動の発現率は,プラセボ群,25mgBID 群,100mgBID 群及び 200mgBID 群でそれぞ れ 24.7%,32.3%,33.0%及び 32.6%であり,本剤の用量の増加による発現率の上昇はみられな かった. なお,本試験では200mgBID を超える用量での検討は行っていないが,外国の臨床試験にお いて400mgBID 群で 200mgBID 群を上回る有効性は認められなかった[添付資料 5.3.5.4-2(参 考)臨床成績概要(外国)]. 以上の結果から,本剤はRA に対して 100mgBID 及び 200mgBID の両用量において有効性が 認められ,このうち200mgBID が最も有効性に優れ,忍容性に問題がない用法・用量であると 判断した. 後期第Ⅱ相試験の主要評価項目とした最終全般改善度に対しては,明確な判定基準がなく, 客観性,統一性に欠けるとの指摘もあることから,次相でのより客観的な評価基準を設定する ために,探索的な検討を行った.RA に対する治療効果の客観的総合評価指標として世界的に用 いられている ACR 改善基準が候補として考えられたが,ACR 改善基準の判定項目の中には NSAID の評価には適さないとされる急性期反応物質(CRP 又は赤沈)が含まれているため,こ れを除いたACR 改善基準(変法)を考案した.後期第Ⅱ相試験のデータを用いて解析を行った 結果,ACR 改善基準(変法)による改善率では,ACR 改善基準と同様に本剤の用量反応性及び プラセボ群に対する優越性が認められただけでなく,プラセボ群の改善率が低くなるとともに 統計学的検定のp 値が小さくなり,より感度が高くなることが示された.そこで,ACR 改善基 準(変法)による改善率を第Ⅲ相試験での主要評価項目として採用した.なお,第Ⅲ相試験の 主要評価項目として ACR 改善基準(変法)による改善率を用いることについては,20 年 月 日及び同年 月 日に行った医薬品機構との治験相談において との助言を得て, した(添付資料5.4-3,4 医薬品機構 治験相談記録). RA に対する第Ⅲ相試験[RCT1]は,本剤 200mgBID の RA に対する有効性について国内の標準 薬に対する非劣性を検証し,また,安全性について標準薬と比較することを目的として,実薬対 照無作為化二重盲検並行群間比較により検討した.対照薬については,医薬品機構からの (添付資料5.4-2~4 医薬品機構 治験相談記録),国内において RA に対する臨床的有効性 が確立され,消化管障害が少ないとされるプロドラッグであり,NSAID 市場における占有率も高
いロキソプロフェンナトリウム60mg1 日 3 回を選択した.本試験では 1~2 週間の観察期間の後, 被験薬を12 週間投与した.有効性について非劣性を検証するための主要な解析として,主要評価 項目であるACR 改善基準(変法)による改善率に関し,最終観察時の 2 群の改善率の差(セレ コキシブ群の改善率-ロキソプロフェンナトリウム群の改善率)の両側 95%信頼区間の下限が, 事前に定めた非劣性の限界値( -10%)を超える場合に,本剤(200mgBID)のロキソプロフェン ナトリウム(60mgTID)に対する非劣性が検証されたと判断することとした. 非劣性の限界値に関して,第Ⅲ相試験の対照薬としたロキソプロフェンナトリウムをプラセ ボと比較して効果の大きさ(Effect Size)を検討した報告はなく,また,ロキソプロフェンナト リウムを含め国内の既存の NSAID で,ACR 改善基準(変法)を用いて非劣性を検証した報告 もない.本試験では比較的長期間(12 週間)投与での安全性を対照薬と比較することも目的の 一つであったため,倫理的な観点から内部標準としてのプラセボ群は設けられず,試験内でロ キソプロフェンナトリウムの効果の大きさを測ることもできなかった.そこで,国内の既存の NSAID の最終全般改善度を用いて非劣性(同等性)を検証した報告では両側 90%信頼区間の下 限の限界値として -10%が用いられていることを参考に,本剤(200mgBID)とロキソプロフェ ンナトリウム(60mgTID)の効果の大きさを同一と仮定し,本剤の後期第Ⅱ相試験における最 終評価時のACR 改善基準(変法)による改善率を検討した結果,効果の大きさ(200mgBID 群 の改善率-プラセボ群の改善率)は 16.6%であったことから,第Ⅲ相試験における非劣性の限 界値を -10%とすることが妥当と考えた. 第Ⅲ相試験に組み入れられた患者は831 例であり,うち被験薬が 1 回以上投与された症例は 773 例であった. PPS において検討した人口統計学的特性及び投与前基準値の群間比較では,一部の項目(罹 病期間,合併症の有無,患者の疼痛評価(VAS)及び患者の疾患活動性全般評価(VAS)等) で統計学的には有意(p<15%)であったが,これらの因子について調整した ACR 改善基準(変 法)による改善率の差の両側 95%信頼区間を算出したところ,調整を行わない場合と同様の分 布であった.したがって,これらの因子による調整を行わなくとも有効性の評価に影響はない と判断した. PPS において,主要評価項目である ACR 改善基準(変法)による最終評価時の改善率は,本 剤 200mgBID 群及びロキソプロフェンナトリウム 60mgTID 群でそれぞれ 21.4%及び 18.9%で あった.両者の差(セレコキシブ群-ロキソプロフェンナトリウム群)は2.52%,差の両側 95% 信頼区間は -4.03%~9.06%であり,その下限が,事前に治験実施計画書で規定した非劣性の限 界値 -10%を超えたことから,セレコキシブ(200mgBID)のロキソプロフェンナトリウム (60mgTID)に対する非劣性が検証されたと判断した.また,ACR 改善基準による改善率,患 者の疼痛評価(VAS),患者の疾患活動性全般評価(VAS),患者の身体機能評価(mHAQ)等ほ とんどの副次的評価項目において,本剤200mgBID 群はロキソプロフェンナトリウム 60mgTID 群と同程度の改善を示した. 一方,安全性について,概括安全度による安全率(「安全である」と判定された症例の割合) は,本剤200mgBID 群及びロキソプロフェンナトリウム 60mgTID 群でそれぞれ 77.2%及び 70.2%
であった.安全率(「安全である」,「ほぼ安全である」以下の2 値)に対する Fisher の直接確率 法(有意水準両側 5%)による検定の結果では,両群間で統計学的に有意な差が認められたが (p=0.027),概括安全度判定(「安全である」から「問題がある」の 4 段階)に対する U 検定(有 意水準両側5%)の結果では,両群間で統計学的に有意な差はみられなかった(p=0.057).有害 事象(症状及び身体徴候)の発現率は,本剤 200mgBID 群及びロキソプロフェンナトリウム 60mgTID 群でそれぞれ 50.5%及び 51.9%であった.消化管潰瘍及び出血性事象は,本剤 200mgBID 群で3 例(0.8%),ロキソプロフェンナトリウム 60mgTID 群でも 3 例(0.8%)にみられたが, そのうち重篤な消化管潰瘍及び出血性事象は,本剤 200mgBID 群では認められず,ロキソプロ フェンナトリウム60mgTID 群のみで 2 例(0.5%)にみられた.臨床検査値異常変動の発現率は, 本剤200mgBID 群及びロキソプロフェンナトリウム 60mgTID 群でそれぞれ 41.5%及び 48.7%で あった. 以上の結果より,本剤はRA に対して 100mgBID 及び 200mgBID の両用量において有効性が 認められ,このうち 200mgBID が最も優れていることが確認された.200mgBID を用いて実施 したRA に対する第Ⅲ相試験[RCT1]において,標準薬に対する非劣性が検証されるとともに, 本剤の安全性は標準薬と同程度であることが示されたことから,RA に対する推奨用法・用量は 100~200mgBID とすることが妥当であると判断した. なお,RA に対する第Ⅲ相試験[RCT1]における最終評価時の ACR 改善基準(変法)による 改善率の人口統計学的特性による層別解析の結果,性別,年齢,体重,重症度,罹病期間,合 併症,NSAID 前治療,併用薬,DMARD 併用,メトトレキサート併用の有無等において,各層 で著しく効果の異なる因子は見当たらなかった.
![表 2.5.2 本申請における臨床データパッケージ(評価資料)及び参考資料 区分 分類 地域 試験数 内容 試験番号 評価 資料 P- Ⅰ及び 薬物動態 日本 6 健常成人対象第Ⅰ相試験 及び薬物動態試験 [400][401] [AKi1][AKi2][AKi3][AKi4] ( PK ) - 患者対象薬物動態 (ポピュレーション PK ) 国内患者対象 4 試験の PK 併合解析([RDS1,ODS1,RLN3,OLN2]) 外国 9 第Ⅰ相試験及び 薬物動態試験 [001][](https://thumb-ap.123doks.com/thumbv2/123deta/8045021.844151/8.892.78.823.146.877/本申請おけるデータパッケージ薬物動態ポピュレーション国内患.webp)
![表 2.5.2.2 本申請における慢性疼痛疾患患者に対する有効性及び安全性の臨床データパッケージ 日本 外国 関節リウマチ( RA ) 変形性関節症( OA ) 腰痛症 肩関節 周囲炎 頸肩腕症候群 腱・ 腱鞘炎 RA OA 位置付け・検討内容 初期 第Ⅱ相 試験 [RPi1] 後期 第Ⅱ相試験[RDS1] 第Ⅲ相試験 [RCT1] 長期投与試験 [RLN3] 初期 第Ⅱ相試験[OPi1] 後期 第Ⅱ相試験 [ODS1] 第Ⅲ相試験[216] 長期投与試験 [OLN2] 第Ⅲ相試験[217]](https://thumb-ap.123doks.com/thumbv2/123deta/8045021.844151/13.1263.137.1152.132.647/本申請おけるに対するデータパッケージリウマチ腰痛症肩関節.webp)
![表 2.5.3 製剤間の溶出特性及びバイオアベイラビリティの相互関係(その 2 ) 100 mg 海外市販 カプセル 25 mg 錠 50 mg 錠 100 mg 錠 200 mg 錠 100 mg 海外市販 カプセル - - [ AKi1 ]によりAUCは同 等 - 25 mg 錠 溶出性は 同様 2) 溶出性は 同様2) 溶出性は 同様2) 50 mg 錠 溶出性は 同様 2) 溶出性は 同様2) 100 mg 錠 溶出試験に より生物学 的に同等 200](https://thumb-ap.123doks.com/thumbv2/123deta/8045021.844151/17.892.167.724.134.546/製剤間バイオアベイラビリティカプセルカプセルにより溶出性.webp)
![表 2.5.7 標準薬との比較試験における有効性及び安全性の結果 RA 第Ⅲ相試験[RCT1] OA 第Ⅲ相試験[216] 腰痛症第Ⅲ相試験[217] セレコキシブ 200mgBID ロキソプロフェンナトリウム 60mgTID プラセボ セレコキシブ 100mgBID ロキソプロフェンナトリウム60mgTID セレコキシブ 100mgBID ロキソプロフェンナトリウム60mgTID 68/318 ( 21.4% ) 60/318 (18.9% ) 74/151 (49.0% ) 2](https://thumb-ap.123doks.com/thumbv2/123deta/8045021.844151/48.892.95.808.144.821/ロフェンナトリウムロフェンナトリウムロフェンナトリウム.webp)

