アズマネックスツイストヘラー
100 μg60 吸入
アズマネックスツイストヘラー
200 μg60 吸入
第
2 部 CTD の概要
(
7)臨床概要
⑥個々の試験のまとめ
シェリング・プラウ株式会社
Section 2.7.6 Synopsis of Individual Studies
目次
略語一覧表... 1 2.7.6 個々の試験のまとめ ... 5 2.7.6.1 臨床試験一覧表 ... 5 2.7.6.2 臨床薬物動態(PK)試験 ... 8 2.7.6.2.1 第 I 相単回投与試験(JPC- -343-11) ... 8 2.7.6.2.2 第 I 相連続投与試験(JPC- -343-12) ... 322.7.6.2.3 プレドニン比較試験(C -049)(原題:Multiple-dose safety and tolerance study of mometasone furoate and lactose powder administered by dry powder inhaler in volunteers with symptoms of moderate asthma)... 56
2.7.6.3 臨床薬力学(PD)試験... 84
2.7.6.3.1 抗原誘発反応試験(I -402)(原題:Effect of inhaled mometasone furoate (SCH 32088) dry powder on allergen-induced asthmatic responses and airway inflammation (protocol I -402) )... 84 2.7.6.4 有効性及び安全性試験 ... 124 2.7.6.4.1 申請する適応症に関する比較対照試験 ... 124 2.7.6.4.1.1 第Ⅱ相試験(JPC- -343-21)... 124 2.7.6.4.1.2 BDP 効力比較試験(JPC- -343-33) ... 167 2.7.6.4.1.3 気管支喘息を対象としたフランカルボン酸モメタゾン DPI とプロピオン酸フルチカ ゾンDPI との比較試験(JPC- -343-32) ... 194 2.7.6.4.2 申請する適応症に関するオープンラベル試験 ... 234 2.7.6.4.2.1 喘息患者に対する臨床薬理試験(JPC- -343-13)... 234 2.7.6.4.2.2 高齢喘息患者に対する臨床薬理試験(JPC- -343-14) ... 258 2.7.6.4.2.3 ステロイド未使用対象試験(JPC- -343-35)... 276 2.7.6.4.2.4 高用量投与試験(JPC- -343-34)... 295 2.7.6.4.2.5 長期投与試験(JPC- -343-31)... 353 2.7.6.4.3 その他の臨床試験 ... 402 2.7.6.4.3.1 海外比較試験 ... 402
2.7.6.4.3.1.1 用量設定試験(C -134)(原題:Placebo-controlled, dose ranging study of mometasone furoate (SCH 32088) dry powder compared to beclomethasone dipropionate (VANCERIL®) in the treatment of asthma in subjects previously maintained on inhaled corticosteroids)... 402
2.7.6.4.3.1.2 FP との比較試験(I -111)(原題:Efficacy and safety of mometasone furoate (SCH 32088) dry powder and fluticasone propionate powder in the treatment of asthma)... 456
2.7.6.4.3.1.3 高用量投与試験(I -113)(原題:Efficacy and safety of three daily dose levels of mometasone furoate (SCH 32088) dry powder in the treatment of asthmatics requiring high dose inhaled corticosteroids) ... 514
Section 2.7.6 Synopsis of Individual Studies 2.7.6.4.3.2.1 長期投与試験(Cff*-135)(原題:Long-term safety study OF mometasone furoate dry
powder(SCH 32088) and beclomethasone dipropionate (VANCERIL®) in the treatment of asthma in subjects previously maintained on inhaled corticosteroids) ... 560 2.7.6.4.3.2.2 長期投与試験(C -136)(原題:Placebo-controlled efficacy and safety study with
long-term safety evaluation of mometasone furoate (SCH 32088) dry powder in the treatment of asthma in subjects previously maintained on inhaled beta-agonists) ... 617 2.7.6.4.3.2.3 長期投与試験(C -137)(原題:Placebo-controlled efficacy and safety study with
long-term safety evaluation of mometasone furoate (SCH 32088) dry powder in reducing oral steroid requirements in subjects with severe asthma)... .694 2.7.6.4.3.2.4 骨塩量の検討試験(C -210)(原題:A two-year study on the effects of mometasone
furoate(SCH 32088) dry powder inhaler (MF DPI) on bone density in adult asthmatics) ... 756 2.7.6.4.3.2.5 骨塩量の検討試験(C -302)(原題:Two-year study on the effects of mometasone
furoate dry powder inhaler (MF DPI) on bone density in young adult asthmatics) ... 799 2.7.6.5 参考文献... 838
Section 2.7.6 Synopsis of Individual Studies
略語一覧表(1 of 4)
略語 省略していない表現
A/G Albumin-globulin ratio(アルブミン-グロブリン比) ACTH Adrenocorticotropic hormone(副腎皮質刺激ホルモン) Al-p(ALP) Alkaline phosphatase(アルカリ性フォスファターゼ) ALT Alanine aminotransferase(アラニンアミノ基転移酵素) ANOVA Analysis of variance(分散分析)
AST Asparate aminotransferase(アスパラギン酸アミノ基転移酵素) ATS All treated subjects(全ての投与された患者集団)
AUC Area under the blood concentration-time curve(血中濃度曲線下面積) AUC0-12hr (0~12 時間までの血中濃度曲線下面積)
AUC0-24hr (0~24 時間までの血中濃度曲線下面積) AUC0-∞ (0~∞までの血中濃度曲線下面積) AUC0-t (0~t 時間までの血中濃度曲線下面積)
BDP Beclometasone dipropionate(プロピオン酸ベクロメタゾン)
BDP-MDI Beclometasone dipropionate metered-dose inhaler(プロピオン酸ベクロメタゾ ン定量噴霧式吸入剤)
BDP-CFC Beclometasone dipropionate chlorofluorocarbon(特定フロンをプロペラントと するプロピオン酸ベクロメタゾン定量噴霧剤)
BDP-HFA Beclometasone dipropionate hydrofluoroalkane(代替フロンをプロペラントと するプロピオン酸ベクロメタゾン定量噴霧剤)
BMD Bone mineral density(骨密度) BMI Body mass index(体容量指数) BUD Budesonide(ブデソニド)
BUD-DPI Budesonide dry powder inhaler(ブデソニド吸入用散剤) BUN Blood urea nitrogen(血中尿素窒素)
C2 Cervical vertebrae 2(第 2 頚椎)
C2hr Plasma concentration at 2 hours after inhalation(吸入 2 時間後の血漿中濃度) Cmax Maximum drug concentration(最高血漿中濃度)
CAE Clinical asthma exacerbation(臨床的喘息悪化) CBC Complete blood count(完全血球算定)
CFC Chlorofluorocarbon(クロロフルオロカーボン)
CFC MDI Chlorofluorocarbon metered dose inhaler(特定フロン使用定量噴霧式吸入剤) CI Confidence interval(信頼区間)
COPD Chronic obstructive pulmonary disease(慢性閉塞性肺疾患) CPK Creatine phospho kinase(クレアチンホスホキナーゼ) CRF Case report form(症例報告書)
Section 2.7.6 Synopsis of Individual Studies
略語一覧表(2 of 4)
略語 省略していない表現 CXX-XXX 本剤の海外臨床試験総括報告書番号(X は試験固有の数字) DD Doubling dilution(倍加希釈) DF Degree of freedom(自由度)DIP Digital image processing(デジタル画像処理) DPI Dry powder inhaler(吸入用散剤)
DEXA Dual-energy x-ray absorptiometry(二重エネルギーX 線吸収測定法) EC European Community(欧州共同体)
EES Efficacy evaluable subjects(有効性評価対象集団)
EG2 Positive eosinophils (activated eosinophils)(活性化好酸球) E-MH Extended Mantel-Haenszel test(拡張マンテル・ヘンツェル検定) EO eosinophil(好酸球)
EP End point(最終評価時)
FAS Full analysis set(最大解析対象集団)
FDA Food and Drug Administration(米国食品医薬品局)
FEF25-75% Forced expiratory flow at 25 to 75 percent of FVC(肺活量の 25~75%の努力性呼気流量) FEV1.0 Forced expiratory volume in 1 second(1 秒量)
FEV1.0% Forced expiratory volume at 1 second(1 秒率,(FEV1.0/FVC)×100) Fisher Fisher’s exact test(フィッシャーの直接確率検定)
FP Fluticasone propionate(プロピオン酸フルチカゾン)
FP-DPI Fluticasone propionate dry powder inhaler(プロピオン酸フルチカゾン吸入用 散剤)
FVC Forced vital capacity(努力性肺活量)
%FVC %努力性肺活量((実測 FVC/予測 FVC)×100)
GCP Good clinical practice(医薬品の臨床試験の実施に関する基準) GH General health perceptions(全体的健康感)
GOT Glutamic oxaloacetic transaminase(グルタミン酸オキザロ酢酸トランスアミ ナーゼ)
GPT Glutamic pyruvic transaminase(グルタミン酸ピルビン酸トランスアミナー ゼ)
γ-GTP γ-Glutamyl transpeptidase(γ-グルタミルトランスペプチダーゼ) Hb Hemoglobin(血色素量)
HbA1C HemoglobinA1C(糖化ヘモグロビン)
HbsAg(HBS 抗原) Hepatitis B virus surface antigen(B 型肝炎ウイルス表面抗原) hCG Human chorionic gonadotropin(ヒト絨毛性性腺刺激ホルモン) HCV Hepatitis C virus(C 型肝炎ウイルス)
Section 2.7.6 Synopsis of Individual Studies
略語一覧表(3 of 4)
略語 省略していない表現
HPA Hypothalamic-pituitary-adrenal(視床下部-下垂体-副腎)
HPLC High performance liquid chromatography(高速液体クロマトグラフィー) HQOL Health-related quality of life(健康関連 QOL(生活の質))
HR Hour(測定時間)
Ht Hematocrit(ヘマトクリット) I-P Inorganicphosphate(無機リン) ITT intent-to-treat(包括解析)
IUD Intrauterine device(子宮内避妊器具)
IXX-XXX 本剤の海外臨床試験総括報告書番号(X は試験固有の数字) JPC-XX-343-XXX 本剤の国内臨床試験総括報告書番号(X は試験固有の数字) K-W Kruskal-Wallis test(X2分布に近似させて有意確立確率を算出) LC/MS/MS High performance liquid chromatography-tandem mass spectrometry (高速液体クロマトグラフィー・タンデム質量分析装置) LDH Lactate dehydrogenase(乳酸脱水素酵素)
LOCF Last observation carried forward(時系列データの欠測に最直前のデータを補 完すること)
log10PC20FEV1.0
Concentration of methacholine(mg/mL)required to decrease FEV1.0 20% from pre-challenge value(FEV1.0を 20%低下させるために必要なメサコリン用量 (mg/mL))の log10
LOQ Limit of quantification(定量限界) LSMean Least square mean(調整平均値)
LTD4 Leukotriene D質) 4(ロイコトリエンD4,アレルギー性炎症を起こす化学伝達物 L-trend 漸増用量に伴う非漸減用量反応(non-decreasing dose response)に関する検
定(線形対比)
MCS Mental component summary(精神的健康度) MDI Metered-dose inhaler(定量噴霧式吸入剤)
MF Mometasone furoate(フランカルボン酸モメタゾン)
MF/L-DPI Mometasone furoate and lactose powder agglomerate dry powder inhaler (フランカルボン酸モメタゾン/ラクトース粉末凝集粒吸入用散剤) MF-DPI Mometasone furoate dry powder inhaler(フランカルボン酸モメタゾン吸入用
散剤)(本剤)
MFE (フランカルボン酸モメタゾンを投与された患者No.の一部)
MF-MDI Mometasone furoate- metered dose inhaler(フランカルボン酸モメタゾン定量 噴霧式吸入剤)
MF-MDI-CFC Mometasone furoate metered dose inhaler chlorofluorocarbon(特定フロンをプロ ペラントとするフランカルボン酸モメタゾンの定量噴霧式製剤)
N.D. Not detectable(検出限界以下)
Section 2.7.6 Synopsis of Individual Studies
略語一覧表(4 of 4)
略語 省略していない表現
NOS Not otherwise specified
NSAID Nonsteroidal anti-inflammatory drug(非ステロイド性抗炎症剤) OTC Over the counter (non-prescription)(大衆薬)
P.SD Pooled standard deviation(積算した標準偏差)
PC Protocol compatible(治験実施計画書に適合した解析対象集団) PCS Phisical component summary(身体的健康度)
PEFR Peak expiratory flow rate(最大呼気速度) PFT Pulmonary function test(肺機能検査) PIFR Peak inspiratory flow rate(最大吸気速度)
PL Placebo,lactose powder(プラセボ,ラクトース散剤) pMDI Pressured metered dose inhaler(加圧式定量噴霧式吸入剤) PPS Per protocol set(治験実施計画書適合集団)
PRN pro re nata(必要に応じて) Pt Platelet(血小板)
QOL Quality of life(生活の質) RBC Red blood cell(赤血球)
RIA Radioimmunoassay(放射免疫測定法) S.D. Standard deviation(標準偏差)
S.E. Standard error(標準誤差)
SAE Serious Adverse Event(重篤な有害事象) SEM Standard error of the mean(標準誤差)
SF-36 Medical Outcomes Study 36-Item Short-Form Health Survey(MOS SF-36 健康調 査)
SOC System organ class(器官別大分類)
SPRI Schering-Plough Research Institute(シェリング・プラウ・リサーチ・インス ティテュート)
T1/2 Half-life time(血漿中濃度消失半減期)
Tmax Time to maximum concentration(最高濃度到達時間) TAA Triamcinolone acetonide(トリアムシノロンアセトニド) T-Bil Total bilirubin(総ビリルビン)
tf time of final quantifiable sample(定量可能な最終サンプルが得られるまでの 時間)
Th2 Helper T-cell 2(ヘルパーT 細胞 2 型) TP Test period(試験実施時期)
WBC White blood cell(白血球)
Section 2.7.6 Synopsis of Individual Studies
2.7.6 個々試験のまとめ
2.7.6.1 臨床試験一覧表 国内で実施した臨床試験及び参考資料とした海外臨床試験の一覧を表 2.7.6.1-1に示し た. 表 2.7.6.1-1 臨床試験一覧(1 of 3) 試験名 (試験No) 試験デザイン 対象 用法・用量 被験 者数 投与 期間 試験期間 CTD に おける 記載場所 第 I 相単回投 与試験 (JPC- -343- 11) プラセボ対照, 患者単盲検, 用量漸増, 並行群間比較 健常成人 男子志願者 本剤100 μg 単回吸入, 本剤200 μg 単回吸入, 本剤400 μg 単回吸入, 本剤800 (400×2) μg 単回吸入, 本剤1000(200×5)μg 単回吸入, 本剤1200(400×3)μg 単回吸入 33 単回 投与 年 月 ~ 年 月 5.3.3.1.1 第 I 相連続投 与試験 (JPC- -343- 12) プラセボ対照, 患者単盲検, 用量漸増, 並行群間比較 健常成人 男子志願者 本剤400 μg/日分 2 吸入, 本剤800μg/日分 2 吸入 18 14 日間 年 月 ~ 年 月 5.3.3.1.2 第II 相試験 (JPC- -343- 21) 多施設共同, 実薬対照, 非盲検, 無作為化, 並行群間比較 気管支喘息 患者 本剤200 μg/日分 2 吸入, 本剤400 μg/日分 2 吸入, 本剤800 μg/日分 2 吸入, BDP* 1-CFC* 2 400 μg/日 (分 4 又は分 2)吸入 本剤 :163 BDP-CFC :51 4 週 年 月 ~ 年 月 5.3.5.1.1 喘 息 患 者 に 対 す る 臨 床 薬 理 試験 (JPC- -343- 13) 非盲検,非対照 気管支喘息 患者 本剤800 μg/日分 2 吸入 13 4 週 年 月 ~ 年 月 5.3.3.2.1 高 齢 喘 息 患 者 に 対 す る 臨 床 薬理試験 (JPC- -343- 14) 非盲検,群間比較 気管支喘息 患者 ( 高 齢 ・ 非 高 齢) 本剤400 μg/日分 2 吸入 12 4 週 年 月 ~ 年 月 5.3.3.3.1 BDP 効力比較 試験 (JPC- -343- 33) 多施設共同, 非盲検, 前治療から切り替え BDP-CFC 吸 入 治 療 気 管 支喘息患者 本剤200 μg/日分 2 吸入 本剤400 μg/日分 2 吸入 61 BDP-CFC 4 週 + 実薬8 週 年 月 ~ 年 月 5.3.5.1.2 第III 相比較 試験 (JPC- -343- 32) 多施設共同, 実薬対照, 非盲検, 無作為化, 並行群間比較 吸 入 ス テ ロ イ ド 使 用 気 管 支 喘 息 患 者 本剤200 μg/日分 2 吸入 FP* 3-DPI* 4 200 μg/日分 2 吸入 本剤 :100 FP-DPI :104 BDP-CFC 4 週 + 実薬8 週 年 月 ~ 年 月 5.3.5.1.3 ス テ ロ イ ド 未 使用対象試験 (JPC- -343- 35) 多施設共同, 非盲検,非対照 軽症(吸入ス テ ロ イ ド 非 使用)気管支 喘息患者 本剤200 μg/日分 2 吸入 20 8 週 年 月 ~ 年 月 5.3.5.2.1 高用量投与 試験* 5 (JPC- -343- 34) 多施設共同, 非盲検,非対照 重 症 気 管 支 喘息患者 本剤400 μg/日分 2 吸入, 本剤800 μg/日分 2 吸入 75 12-24 週 年 月 ~ 年 月 5.3.5.2.2Section 2.7.6 Synopsis of Individual Studies 表 2.7.6.1-1 臨床試験一覧(2 of 3) 試験名 (試験No) 試験デザイン 対象 用法・用量 被験 者数 投与 期間 試験期間 CTD に おける 記載場所 長期投与試験*6 (JPC- -343- 31) 多施設共同, 非盲検,非対照 BDP-CFC , FP-DPI , 又 は ブ テ ソ ニ ド 吸 入 気 管 支喘息患者 (本剤200 μg/日分 2 吸入) 本剤400 μg/日分 2 吸入 204 52 週 年 月 ~ 年 月 5.3.5.2.3 フ ゚ レ ト ゙ ニ ン 比 較 試験 (C -049) プラセボ・実薬対照, 評価者盲検, 複数回投与, 無作為化, 並行群間比較 軽 症 か ら 中 等 症 の 喘 息 患者 本剤 800 μg/日分 2 吸入 本剤 1600μg/日分 2 吸入 プレドニン 10 mg/日分 1 経口 プラセボラクトース DPI 0μg/日分 2 64 29 日間 年 月 ~ 年 月 5.3.3.2.3 抗原誘発反応 試験 (I -402) プラセボ対照, 二重盲検, 無作為化, 4 期クロスオーバー試験 軽 度 ア レ ル ギ ー 性 喘 息 患者 本剤 100 μg/日分 2 吸入 本剤 200 μg/日分 2 吸入 本剤 800 μg/日分 2 吸入 プラセボ 12 6 日間×4 年 月 ~ 年 月 5.3.4.1 用量設定試験 (C -134) 多施設共同, プラセボ・実薬対照, 二重盲検, 無作為化, 並行群間比較 吸 入 ス テ ロ イ ド 使 用 の 喘息患者 本剤200 μg/日分 2 吸入 本剤400 μg/日分 2 吸入 本剤800 μg/日分 2 吸入 BDP-CFC 336 μg/日分 2 吸入 プラセボ 365 12 週 年 月 ~ 年 月 5.3.5.1.4 FP 比較試験 (I -111) 国際多施設共同, 実薬対照, 二重盲検, 無作為化, 並行群間比較 吸 入 ス テ ロ イ ド 使 用 の 中 等 度 の 喘 息患者 本剤 200 μg/日分 2 吸入 本剤 400 μg/日分 2 吸入 本剤 800 μg/日分 2 吸入 FP-DPI 500 μg/日分 2 吸入 733 3 ヶ月 年 月 ~ 年 月 5.3.5.1.5 高用量投与 試験 (I -113) 国際多施設共同, 非対照, 二重盲検, 無作為化, 並行群間比較 中 等 度 か ら 重 度 の 喘 息 患者 本剤 400 μg/日分 2 吸入 本剤 800 μg/日分 2 吸入 本剤 1200 μg/日分 2 吸入 507 3 ヶ月 年 月 ~ 年 月 5.3.5.2.4 長期投与試験 (Cff*-135) 多施設共同, 実薬対照, 評価者盲検, 無作為化 吸 入 ス テ ロ イ ド 使 用 の 喘息患者 本剤 400 μg/日分 2 吸入 本剤 800 μg/日分 2 吸入 本剤800 μg/日分 1 吸入 BDP-CFC 336 μg/日分 2 吸入 239 52 週 年 月 ~ 年 月 5.3.5.1.6 長期投与試験 (C -136) 多施設共同, プラセボ対照(3 ヶ月), 二重盲検(3 ヶ月), 非盲検(9 ヶ月), 無作為化, 並行群間比較 吸入β 刺激薬 単 独 使 用 の 喘息患者 (3 ヶ月投与期間) 本剤 200 μg/日分 1(AM)吸入 本剤 400 μg/日分 1(AM)吸入 プラセボ (9 ヶ月投与期間) 本剤 200 μg/日分 1(AM)吸入 本剤 200 μg/日分 1(PM)吸入 本剤 400 μg/日分 1(AM)吸入 本剤 400 μg/日分 1(PM)吸入 3 ヶ月 236 9 ヶ月 166 3 ヶ月+ 9 ヶ月 年 月 ~ 年 月 5.3.5.1.7 長期投与試験 (C -137) 多施設共同, プラセボ対照(3 ヶ月), 二重盲検(3 ヶ月), 非盲検(9 ヶ月), 無作為化, 並行群間比較 経 口 プ レ ド ニ ン 使 用 の 重 度 の 喘 息 患者 (3 ヶ月投与期間) 本剤 800 μg/日分 2 吸入 本剤 1600 μg/日分 2 吸入 プラセボ (9 ヶ月投与期間) 本剤の各用量にて投与* 7 3 ヶ月 132 9 ヶ月 128 3 ヶ月+ 9 ヶ月 3 ヶ月: 年 月 ~ 年 月 9 ヶ月: 年 月 ~ 年 月 5.3.5.1.8
Section 2.7.6 Synopsis of Individual Studies 表 2.7.6.1-1 臨床試験一覧(3 of 3) 試験名 (試験No) 試験デザイン 対象 用法・用量 被験 者数 投与 期間 試 験 期 間 CTD に おける 記載場所 骨塩量の検討 試験 (C -210) 多施設共同, 安全性, プラセボ対照, 二重盲検, 無作為化, 並行群間比較 吸入β 刺激薬 単 独 使 用 又 は 非 ス テ ロ イ ド 性 抗 炎 症 薬 と の 併 用 使 用 の 成 人喘息患者 本剤 800 μg/日分 2 吸入 本剤 1200 μg/日分 2 吸入* 8 プラセボ 87 104 週間 年 月 ~ 年 月 5.3.5.1.9 骨塩量の検討 試験 (C -302) 多施設共同, 安全性, プラセボ対照, 二重盲検, 無作為化, 並行群間比較 吸入β 刺激薬 単 独 又 は 非 ス テ ロ イ ド 性 抗 炎 症 薬 と の 併 用 治 療 を 受 け た 若 年 成 人 喘 息患者 本剤 400 μg/日分 2 吸入 プラセボ 103 104 週間 年 月 ~ 年 月 5.3.5.1.10 *1 BDP: Beclometasone dipropionate *2 CFC: Chlorofluorocarbon *3 FP: Fluticasone propionate *4 DPI: Dry powder inhaler
*5:高用量投与試験は400μg/日及び800μg/日で投与を開始し,投与2,4,8,12,16,及び20週時に用量見直しを行い, 投与量の増量・維持・減量を決定した. *6:長期投与試験では初回投与量は400μg/日で開始するが試験中に200μg/日に減量することができる. *7:長期試験(C -137)では,3ヶ月の投与期間全体を完了した患者,もしくは有害事象又は非遵守以外の理由で早期 中止した患者は,9ヶ月間の非盲検的な本剤治療の対象となった.初回の本剤用量を1600μg/日分2とし,経口プレ ドニゾンを中止すると,用量を最低800μg/日分2まで漸減した. *8:本剤1200μg/日分2群は,治験実施計画書修正によって廃止された(19 年 月 日改訂).
Section 2.7.6 Synopsis of Individual Studie 2.7.6.2 臨床薬物動態(PK)試験 2.7.6.2.1 第 I 相単回投与試験(JPC- -343-11)〔CTD における記載箇所:5.3.3.1.1〕 1.試験の目的及び方法 (1)試験の概略 健常成人男子を対象として,本剤の単回投与時の安全性,HPA 系機能に及ぼす影響及び 薬物動態を検討した.試験方法の概略を表 2.7.6.2.1-1に示した. 表 2.7.6.2.1-1 試験方法の概略(1 of 3) 項 目 内 容 試 験 の 目 的 本邦の健常成人男子を対象として,本剤の単回投与時の安全性,HPA 系機能に及ぼす 影響及び薬物動態を検討した. 試 験 の 種 類 プラセボ対照,被験者単盲検,用量漸増,単回投与,並行群間比較試験 対 象 健常成人男子 使 用 薬 剤 1 吸入につき MF 100 μg を含有する吸入用散剤(ロット番号:35923-040) 1 吸入につき MF 200 μg を含有する吸入用散剤(ロット番号:36809-038) 1 吸入につき MF 400 μg を含有する吸入用散剤(ロット番号:36809-039) MF を含まない吸入用散剤(MF プラセボ):100 μg 用(ロット番号:35923-037) 200 μg 及び 400 μg 用(ロット番号:36809-036) 投 与 群 A 群:本剤 100 μg/被験者,プラセボ(ステップ I)+ 本剤 800 μg/被験者,プラセボ (ステップIⅤ) B 群:本剤 200 μg/被験者,プラセボ(ステップ II)+ 本剤 1000 μg/被験者,プラセボ (ステップⅤ) C 群:本剤 400 μg/被験者,プラセボ(ステップ III)+ 本剤 1200 μg/被験者,プラセボ (ステップⅤI) 投 与 方 法 ステップI: MF 100 μg/吸入又は MF プラセボを 1 回経口吸入 ステップII: MF 200 μg/吸入又は MF プラセボを 1 回経口吸入 ステップIII: MF 400 μg/吸入又は MF プラセボを 1 回経口吸入 ステップIⅤ: MF 400 μg/吸入又は MF プラセボを 2 回経口吸入 ステップⅤ: MF 200 μg/吸入又は MF プラセボを 5 回経口吸入 ステップⅤI: MF 400 μg/吸入又は MF プラセボを 3 回経口吸入 投 与 期 間 単回投与 診 断 及 び 組 み 入 れ 基 準 1.選択基準 (1) 健常成人男子 (2) 年齢:20 歳以上 40 歳以下 (3) 体重:80 kg 以下で,BMI 法による標準体重[身長(m)2 × 22]の± 20%以内にあ る者 (4) 非喫煙者 (5) 事前検診の結果,治験担当医師が被験者として適当と認めた者
Section 2.7.6 Synopsis of Individual Studie 表2.7.6.2.1-1 試験方法の概略(2 of 3) 項 目 内 容 診 断 及 び 組 み 入 れ 基 準 2.除外基準 (1) 事前検診の結果,治験担当医師が被験者として参加不可とした者 (2) 肝臓,腎臓,循環器,消化器,呼吸器疾患等の重篤な既往歴のある者 (3) アレルギー疾患の既往,特異体質及びアルコール・薬物の中毒症のある者 (4) 本試験の開始 6 ヶ月以内に 400 mL を超える採血を受けた者 (5) 本試験の開始 4 ヶ月以内に他の薬剤の試験に参加した者 (6) 本試験の開始 2 週間以内に試験の結果に影響を与える薬剤を服用していた者 (7) 本試験の開始 3 日以内にアルコール飲料を摂取した者 (8) その他,治験担当医師が被験者として不適当と判断した者 併 用 薬 剤 及 び 併 用 療 法 本試験は健常志願者を対象としたため,投与 2 週間前から投与 1 週間後(事後検診実施 日)までは原則として他の薬剤の使用を禁止した.ただし,緊急の場合を除き,被験者 からの連絡により治験担当医師が必要と認めた場合は使用できることとした. 観 察 項 目 及 び ス ケ ジ ュ ー ル 試験スケジュール 投与 前々日 投与前日 投与日 (第1日目) 時刻 検査項目 18:00 9:00 以前 9:00 投与前 9:00 9:30 10:00 10:30 11:00 本剤吸入後時間 0 0.5 1 1.5 2 入院▲/退院▽ ▲ 本剤吸入 ● 食事 問診・聴打診 ○ ○ ○ ○ 自覚症状・有害事象 体重 ○ バイタルサイン ○ ○ ○ ○ 心電図 ○ ○ 血液学的検査 ○ 血液生化学検査 ○ 尿検査 ○ 血中コルチゾール ○ ○ 尿中遊離コルチゾール 24時間蓄尿 24時間蓄尿 血漿中MF濃度測定 ○ ○ ○ ○ ○ 投与日 (第1日目) 第2日目 第8日目 時刻 検査項目 12:00 13:00 15:00 17:00 19:00 21:00 9:00 9:00 本剤吸入後時間 3 4 6 8 10 12 24 168 入院▲/退院▽ ▽ 本剤吸入 食事 ○ ○ 問診・聴打診 ○ ○ ○ ○ ○ ○ ○ 自覚症状・有害事象 体重 ○ ○ バイタルサイン ○ ○ ○ ○ ○ ○ ○ 心電図 ○ ○ 血液学的検査 ○ ○ 血液生化学検査 ○ ○ 尿検査 ○ ○ 血中コルチゾール ○ ○ 尿中遊離コルチゾール 24時間蓄尿 血漿中MF濃度測定 ○ ○ ○ ○ ○ ○ 投与前々日及び投与前日の食事は,それぞれ19:00 及び 9:00,12:00,19:00 とし,投与前日の 22:00 より投与 4 時間後までは絶食とした.
Section 2.7.6 Synopsis of Individual Studie 表2.7.6.2.1-1 試験方法の概略(3 of 3) 項 目 内 容 評 価 項 目 薬物動態:Cm a x,Tm a x,AUC0 - t,AUC0 - ∞,t1 / 2 HPA 系に対する影響:血中コルチゾール濃度,尿中遊離コルチゾール排泄量 安全性:自他覚的有害事象,臨床検査値異常変動,バイタルサイン 解 析 方 法 統計手法: Paired-t-test:臨床検査の測定値(投与前と投与 24 時間後の群内比較)
Repeated Measurement ANOVA:バイタルサイン・体重及び臨床検査の追跡調査を除く 測定値(時間的な反応パターンの検討)
単回帰分析:尿中遊離コルチゾール排泄量の投与24 時間後の測定値(用量相関性の検討) Dunnett’s two side test:尿中遊離コルチゾール排泄量の投与 24 時間後の測定値(プラ
セボ群と各投与量群との比較)
Cochran-Armitage-Linear Trend test:有害事象の発現率(用量相関性の検討) 薬物動態解析プログラム「PAG-CP」:血漿中薬物濃度の薬物速度論的解析 目 標 症 例 数 各実薬群 6 例,各プラセボ群 3 例 延べ 54 例 症 例 数 の 設 定 根 拠 過去の第 I 相試験を参考に,薬物速度論的パラメータの算出が可能な被験者数として, 各ステップにそれぞれ実薬 6 名,プラセボ 3 名の症例数を設定した.これは,各種パラ メータ(臨床検査値,尿中遊離コルチゾール排泄量など)の実薬群とプラセボ群の平均 値の差が標準偏差の 2 倍であった場合に,平均値の差を検出力 80%,有意水準両側 5% で検出できる症例数である. 治 験 総 括 医 師 解 析 担 当 者 シェリング・プラウ株式会社 臨床統計解析&DM 部 部長 実 施 医 療 機 関 試 験 期 間 最初の被験者の同意取得日:19 年 月 日 最後の被験者の検査・観察終了日:19 年 月 日 (2)試験デザインの設定根拠 本試験は,健常成人男子を対象として治験薬の安全性並びに薬物動態を検討する目的で実 施した第 I 相単回投与試験である.先に実施された米国の第 I 相単回投与試験(I -130)〔セ クション 5.3.3.1.3〕,(Cee*-135)〔セクション 5.3.3.2.2〕では 100~800 μg の用量範囲 で忍容性が確認されたことから,本試験の初回投与量を100 μg とし,忍容性を確認しながら 800 μg まで段階的に増量する計画とした.そして,臨床的に問題がなければ,さらに 1200 μg まで増量することとした.各ステップとも実薬群 6 名,プラセボ群 3 名としたが,これは薬 物動態及び安全性の検討に必要な被験者数として設定したもので,通常の臨床第 I 相試験と ほぼ同様の被験者数である.実薬及びプラセボは無作為に割付けた.盲検の水準に関しては, 本試験が本剤を本邦で初めてヒトに投与する試験であることから,被験者のみ盲検とし,担 当医師に対しては盲検としなかった.測定項目には,血中薬物濃度及び一般臨床検査の他に, 本剤が合成副腎皮質ステロイド薬であることから HPA 系機能に及ぼす影響を検討する目的 で,血中コルチゾール濃度及び尿中遊離コルチゾール排泄量を追加した.
Section 2.7.6 Synopsis of Individual Studie (3)試験における用量の選択 1)用量 下記の6 用量をそれぞれ単回投与した. ステップI : 100 μg(本剤 100 μg 又はプラセボを 1 回吸入) ステップII : 200 μg(本剤 200 μg 又はプラセボを 1 回吸入) ステップIII : 400 μg(本剤 400 μg 又はプラセボを 1 回吸入) ステップIV : 800 μg(本剤 400 μg 又はプラセボを 2 回吸入) ステップV : 1000 μg(本剤 200 μg 又はプラセボを 5 回吸入) ステップVI: 1200 μg(本剤 400 μg 又はプラセボを 3 回吸入) 2)用量選択の根拠 本剤は既に米国において臨床第 I 相単回投与試験(100,200,400,800 μg)が実施さ れており,その成績の概要は以下の通りであった. ① 血中薬物動態 表 2.7.6.2.1-2 血中薬物動態 投与量(μg) 薬物速度論的 パラメータ 100 200 400 800 Cm a x(pg/mL) N.D. N.D. 28 80 AUC(tf)(pg・hr/mL) - - 64 318 (各群 12 名の平均値を示した.N.D.:検出限界以下,-:算出不能) ② 血中コルチゾール値 [AUC0-24(μg・hr/dL)] 表 2.7.6.2.1-3 血中コルチゾール値 投与量(μg) 投与群 100 200 400 800 本剤 165(-3%) 168(-7%) 157(-9%) 173(-12%) プラセボ 170 181 172 196 (各群 12 名の平均値を示した.(%)はそれぞれのプラセボ群に対する変化率を示した.いずれの投与量においても 本剤群とプラセボ群との間に有意差は認められなかった.) ③ 薬剤投与に起因すると考えられた有害事象 本剤100 μg 群に頭痛,ざ瘡(各 1 名:いずれも軽度)及びプラセボ群に頭痛(1 名: 軽度)が認められたのみであった. 以上の結果から,血中に薬物が検出されず,かつ副腎皮質機能に影響を及ぼさないと 考えられる用量(100 μg)を初回投与量とし,各投与量での安全性,副腎皮質機能への 影響及び薬物動態を確認しながら800 μg まで漸次増量し,安全性に問題がなければさら に 1200 μg まで投与することとした.なお,本試験の実施に並行して米国でも追加第 I 相試験を実施しており,本邦で1000 μg を投与する時点までには米国の 1200 μg 投与時 の成績が得られる予定であった.(注:実際には,米国で 1200 μg を投与した時の安全性 及 び 薬 物動態 に 関 する中 間 報 告書は 19 年 月 日に入手した.本試験の開始日は 19 年 月 日,1000 μg を投与したのは 月 日である.)
Section 2.7.6 Synopsis of Individual Studie (4)測定項目の適切性 標準的な臨床第 I 相試験に準じて,血漿中薬物濃度の測定及び安全性評価項目(臨床観 察,臨床検査)を設定した.この他に,本試験では血中コルチゾール濃度及び尿中遊離コ ルチゾール排泄量を測定することとしたが,それは以下の理由による. 本剤は局所に投与するステロイド薬であるが,肺内に吸入された際に全身循環血中に吸 収される可能性がある.一般に,ステロイドホルモンを長期投与すると副腎皮質機能の低 下をもたらし,血中コルチゾール濃度が低下することが知られていることから 1),本試験 でもHPA 系に本剤が及ぼす影響を検討することとした.通常,海外での同種同効薬の臨床 試験では HPA 系機能への影響を評価する目的で血中コルチゾール濃度を 24 時間測定し, その AUC の変化を評価指標とするが,AUC を算出するためには 24 時間を通しての採血 が必要で,本邦では倫理的に実施困難と判断した.このため,代替として,① 投与前及び 投与後24 時間の血中コルチゾール濃度,並びに,② 投与前 24 時間及び投与後 24 時間の 尿中遊離コルチゾール排泄量,をそれぞれ比較することとした 2~4). (5)有効性の評価項目 本試験は第 I 相臨床試験であることから,有効性の主要評価項目は設定せず,安全性,副 腎皮質機能への影響及び薬物動態をそれぞれ独立して評価した. (6)症例数の設定根拠 過去の第 I 相試験を参考に,薬物速度論的パラメータの算出が可能な被験者数として,各 ステップごとに実薬6 名,プラセボ 3 名という症例数を設定した.同時に,これは,各種パ ラメータ(臨床検査値,尿中遊離コルチゾール排泄量など)の実薬群とプラセボ群の平均値 の差が標準偏差の2 倍であった場合に,平均値の差を検出力 80%,有意水準両側 5%で検出 できる症例数でもある. 2.被験者の内訳 被験者の内訳を表 2.7.6.2.1-4 に示した.被験者数は各ステップ 9 名(実薬群 6 名,プ ラセボ群 3 名),延べ 54 名(実薬群 36 名,プラセボ群 18 名)であった. 表 2.7.6.2.1-4 被験者の内訳 (単回投与) 被験者数 ステップ 投与量(μg) 製剤(μg/吸入) 実薬 プラセボ 合計 I 100 100 × 1 6 3 9 II 200 200 × 1 6 3 9 III 400 400 × 1 6 3 9 IV 800 400 × 2 6 3 9 Ⅴ 1000 200 × 5 6 3 9 VI 1200 400 × 3 6 3 9 合 計 36 18 54
Section 2.7.6 Synopsis of Individual Studie
なお,ステップ I の 2 名,II の 1 名及び III の 3 名が,次ステップ(I,II,III の次ステ ップはそれぞれIV,V,VI)へ移行できなかったため,新たな被験者を選定して欠員を補 充した.中止した被験者の一覧を表2.7.6.2.1-5 に示した. 表 2.7.6.2.1-5 試験を中止した被験者の一覧 被験者番号 (ステップ) 中止理由 補充した被験者番号 (ステップ) 3 (I) 投与直前のγ-GTP がスクリーニング時よりさらに高値(161 IU/L) であったことがステップ I の投与終了後に判明したため,治験担 当医師が対象として不適格と判断し,ステップ IV への参加を不 可とした. 28 (IV) 6 (I) 事後検診後の追跡調査時の本人への連絡が困難で,管理上問題が あるため,治験担当医師が対象として不適格と判断し,ステップ IV への参加を不可とした. 29 (IV) 18 (II) 事後検診後の追跡調査時の本人への連絡が困難で,管理上問題が あるため,治験担当医師が対象として不適格と判断し,ステップ V への参加を不可とした. 30 (V) 20 (III) 試験参加日の都合が悪くなったため,ステップ VI に参加できな くなった. 31 (VI) 23 (III) 同上 32 (VI) 26 (III) ステップ VI 実施前日に感冒様症状発現し,被験者より辞退の申 し出があった. 33 (VI) 3.解析対象集団 解析対象とした被験者数は各ステップ9 名(実薬群 6 名,プラセボ群 3 名),延べ 54 名 で,解析から除外した被験者はなかった. 4.被験者背景 各ステップごとの被験者背景(年齢,身長,体重)の要約統計量及び分散分析の結果を表 2.7.6.2.1-6 に示した. 表 2.7.6.2.1-6 被験者背景 ステップ 投与量 (μg) 被験者数 年齢(歳) Mean ± S.D.(範囲) 身長(cm) Mean ± S.D.(範囲) 体重(kg) Mean ± S.D.(範囲) I 100 6 24.3 ± 2.7 (21~28) 166.3 ± 5.3 (160.0~175.0) 61.3 ± 6.3 (54.5~69.0) II 200 6 (24.5 ± 2.5 20~27) 169.8 ± 5.4 (161.5~178.0) 66.3 ± 9.7 (52.0~80.0) III 400 6 (25.3 ± 7.0 20~38) 173.4 ± 3.1 (169.0~178.0) 63.8 ± 8.3 (52.5~67.0) IV 800 6 (24.8 ± 2.6 21~28) 168.0 ± 7.1 (160.0~177.5) 59.8 ± 3.9 (54.5~64.0) V 1000 6 24.7 ± 2.7 (20~27) 169.8 ± 5.4 (161.5~178.0) 66.8 ± 9.8 (52.0~80.0) VI 1200 6 26.2 ± 6.5 (20~38) 174.3 ± 3.4 (169.0~177.5) 67.0 ± 6.7 (56.0~75.0) プラセボ合計 18 (23.3 ± 4.3 20~34) 171.3 ± 3.8 (165.5~177.0) 64.4 ± 5.1 (56.5~70.5) 分散分析 p = 0.8769 p = 0.0505 p = 0.4406
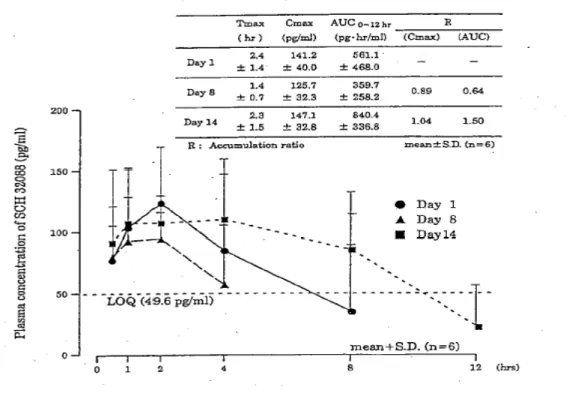
Section 2.7.6 Synopsis of Individual Studie 5.薬物動態及び副腎皮質機能への影響 (1)薬物動態 本剤100 μg 投与時の血漿中未変化体濃度は,全被験者の全測定時点で定量限界以下(< 49.8 pg/mL)であった.200 μg 投与時には定量限界をわずかに上回る濃度値が散見された だけで,400 μg 投与時でも定量限界を上回る血漿中濃度が持続したのは 6 名中 2 名のみで あった.このため,400 μg 以下の用量では薬物速度論的解析を行わなかった.いずれの用 量でも個体間変動が大きかった. 800~1200 μg 投与時の各用量での薬物速度論的パラメータの解析結果を表 2.7.6.2.1-7 に,平均血漿中未変化体濃度推移並びにCmax及びAUC0-tと用量との相関を図 2.7.6.2.1-1 に示した.800~1200 μg 投与時の血漿中未変化体濃度は,いずれの用量でも投与後 2~3 時間に最高値を示し,800,1000,1200 μg 投与時の Cmaxはそれぞれ 178.3 ± 61.1,356.2 ± 170.2 及び 342.4 ± 80.0 pg/mL で,Cmaxの平均値は1000~1200 μg で頭打ちとなった. モデルによらない方法で算出した消失半減期は,800,1000,1200 μg 投与時にそれぞれ 5.58 ± 1.95,5.70 ± 4.62 及び 9.79 ± 7.15 時間であり,投与後 24 時間の血漿中未変化体濃 度は,800 及び 1000 μg 投与時には全被験者,1200 μg 投与時には 6 名中 3 名で定量限界 以下(< 49.8 pg/mL)となった. 最終観察時点までのAUC(AUC0-t)は,800,1000,1200 μg 投与時にそれぞれ 1018 ± 493,1993 ± 961 及び 2905 ± 1183 pg・hr/mL であり,また,無限大時間までの AUC(AUC 0-∞)はそれぞれ 1657 ± 741,2673 ± 477 及び 3892 ± 1713 pg・hr/mL であった.AUC の増 加率は用量の増加をはるかに上回るものであった.
Section 2.7.6 Synopsis of Individual Studie 表 2.7.6.2.1-7 健常成人男子(n = 6)に本剤の 800,1000,又は 1200μg を 経口吸入させた際の薬物速度論的パラメータ AUC (pg・hr/mL) 投与量 (μg) Tm a x ( hr ) Cm a x (pg/mL) T1 / 2 ( hr ) 0-t 0-∞ 800 2.5±1.0 178.3±61.1 5.58±1.95 1018±493 1657±741 1000 2.3±0.5 356.2±170.2 5.70±4.62 1993±961 2673±477 1200 2.9±0.8 342.4±80.0 9.79±7.15 2905±1183 3892±1713 Non-compartment model 解析 各値は6 例の平均値± S.D. 図 2.7.6.2.1-1 健常成人男子(n=6)に本剤の 800,1000,又は 1200 μg を経口吸入 させた際の血漿中MF 未変化体濃度推移,並びに Cmax及びAUC0-tの用量相関性 Linear P lot 0 100 200 300 400 500 600 0 6 12 18 24 Time ( hr ) P las m a c onc . of M F ( pg /m L) 1200 μg 1000 μg 800 μg m e a n ± S D (N =6 ) Log-Linear P lot 10 100 1000 0 6 12 18 24 Time ( hr ) P las m a c onc . of M F ( pg /m L) 1200 μg 1000 μg 800 μg m e a n ± S D (N =6 ) Cmax 0 100 200 300 400 500 600 700 0 200 400 600 800 1000 1200 Dos e ( μg ) Cma x (p g /m L) A UC0-t 0 1000 2000 3000 4000 5000 0 200 400 600 800 1000 1200 Dos e ( μg ) AU C 0-t (p g ・hr /m L)
Section 2.7.6 Synopsis of Individual Studie (2)副腎皮質機能への影響 1)血中コルチゾール濃度 第8 日(投与後 7 日)までの血中コルチゾール濃度推移を表 2.7.6.2.1-8,投与直前と 投与24 時間後との平均値の差の検定結果を表 2.7.6.2.1-9 に示した. 400 μg までの用量では,投与後 24 時間の血中コルチゾール濃度に低下は認められなか った.一方,800 μg では投与直前 14.2 ± 4.9,投与後 24 時間 12.2 ± 2.6 μg/dL,1000 μg では投与直前13.0 ± 3.9,投与後 24 時間 11.6 ± 6.1 μg/dL と,投与後 24 時間の平均値は 投与直前値に比べて低下し,1200 μg 投与時には投与直前 13.2 ± 3.7 から投与後 24 時間 11.0 ± 4.7 μg/dL へと有意に低下した(p = 0.0103,対応のある t 検定). 次に,ステップ,薬剤,時間,時間とステップとの交互作用,時間と薬剤との交互作 用による分散分析を実施した結果,薬剤による効果は有意であったが(p = 0.0221),時 間 と ス テ ッ プ 及 び 時 間 と 薬 剤 と の 交 互 作 用 は い ず れ も 有 意 で は な く ( そ れ ぞ れ p = 0.0952,p = 0.1847),治験薬服薬による血中コルチゾール値の変化パターンはステップ 間(投与時期)及び薬剤間(実薬とプラセボ)で変わらない可能性が示唆された.薬剤, 時間,時間と薬剤との交互作用による分散分析をステップごとに実施した結果でも,時 間と薬剤との交互作用は有意ではなかった.さらに,実薬投与時の血中コルチゾール値 のみを用いて,投与量,時間,時間と投与量との交互作用による分散分析を実施したが, 時間と投与量との交互作用は有意ではなく(p = 0.5463),投与量の差による血中コルチ ゾール値の変化パターンの差は明らかではなかった. 表 2.7.6.2.1-8 血中コルチゾール濃度の推移 血中コルチゾール濃度(μg/dL)Mean ± S.D. ステップ 投与量 (μg) 被験者数 投与前日 投与直前 投与後24 時間 第8 日目 I 100 6 11.8 ± 4.9 13.0 ± 4.6 13.2 ± 3.7 12.3 ± 4.1 II 200 6 13.1 ± 3.2 15.9 ± 2.5 16.1 ± 3.6 12.1 ± 4.8 III 400 6 11.4 ± 1.7 13.0 ± 2.1 12.8 ± 2.9 10.4 ± 3.3 IV 800 6 11.3 ± 4.5 14.2 ± 4.9 12.2 ± 2.6 13.5 ± 3.2 V 1000 6 14.3 ± 4.8 13.0 ± 3.9 11.6 ± 6.1 11.3 ± 4.4 VI 1200 6 12.9 ± 3.5 13.2 ± 3.7 11.0 ± 4.7 12.6 ± 3.6 プラセボ合計 18 13.4 ± 4.2 15.6 ± 3.4 15.9 ± 3.3 16.5 ± 3.7
Section 2.7.6 Synopsis of Individual Studie 表 2.7.6.2.1-9 血中コルチゾール濃度推移に関する検定結果 (1)対応のある t 検定,p 値 ステップ 投与量 (μg) 投与直前vs.投与 24 時間後 I 100 0.7784 II 200 0.9015 III 400 0.8799 IV 800 0.1910 V 1000 0.4073 VI 1200 0.0103* プラセボ 0.7726 *:p<0.05 (2)分散分析,p値 ステップ 薬剤 時間 時間×ステップ 時間×薬剤 0.3187 0.0221* 0.4211 0.0952 0.1847 *:p<0.05 (3)ステップごとの分散分析,p 値 ステップ 薬剤 時間 時間×薬剤 I 0.3615 0.8965 0.8965 II 0.7756 0.0740 0.0904 III 0.0089 0.8360 0.9928 IV 0.2338 0.1026 0.6924 V 0.8491 0.7342 0.4736 VI 0.5258 0.0652 0.1598 (4)実薬投与群での分散分析,p 値 投与群(実薬のみ) 時間 時間×投与群 0.6046 0.0768 0.5463 2)尿中遊離コルチゾール排泄量 尿中クレアチニン排泄量で補正した治験薬投与前24 時間及び投与後 24 時間の尿中遊 離コルチゾール排泄量を投与量ごとにまとめて表2.7.6.2.1-10 に示した.また,尿中遊 離コルチゾール排泄量に関する検定結果を表2.7.6.2.1-11 に示した. 投与日の 24 時間に尿中に排泄された遊離コルチゾール量は,プラセボを含むいずれ の用量でも投与前 24 時間値よりも減少し,400 μg 以上の実薬群及びプラセボ群では有 意な減少を示した(p<0.05,対応のある t 検定).投与後 24 時間の尿中遊離コルチゾー ル排泄量の平均値は,400 μg 群 53.8 ± 13.0,800 μg 群 38.8 ± 22.8,1000 μg 群 34.2 ± 14.1, 1200 μg 群 29.8 ± 13.3 μg/g クレアチニンであり,これに対してプラセボ群の投与後 24 時間の尿中遊離コルチゾール排泄量の平均値は53.2 ± 11.9 μg/g クレアチニンであった. 次に,治験薬投与による尿中遊離コルチゾール排泄量の変化パターンがステップ間あ るいは薬剤間(実薬又はプラセボ)で異なるかどうかを検討する目的で,ステップ,薬 剤,時間,時間とステップとの交互作用,時間と薬剤との交互作用による分散分析を実 施した結果,時間と薬剤との交互作用が有意となり(p = 0.0198),実薬群とプラセボ群
Section 2.7.6 Synopsis of Individual Studie とで尿中遊離コルチゾール排泄量の変化パターンが異なることが示唆された.そこで, いずれの投与量群でプラセボ群との間に変化パターンの差が認められるのかを検討する 目的で,ステップごとに薬剤,時間,時間と薬剤との交互作用による分散分析を実施し た結果,ステップIV(800 μg)及び V(1000 μg)で有意差が認められた(それぞれ p = 0.0024,p = 0.0461).さらに,実薬群の尿中遊離コルチゾール排泄量のみを用い,投与 量の違いによって尿中遊離コルチゾール値の変化パターンに差が認められるかどうかを 検討する目的で,投与量,時間,時間と投与量との交互作用による分散分析を実施した ところ,時間と投与量との交互作用が有意となり,投与量の違いによって尿中遊離コル チゾール排泄量の変化パターンが異なることが示唆された(p = 0.0344). なお,プラセボ群を含む 7 群間の投与前値の一様性を一元配置分散分析により検討し たが,有意差は認められなかった.そこで,投与後値のみを用いて尿中遊離コルチゾー ル排泄量と投与量(プラセボ群を含む7 群)との相関性を単回帰分析により検討した結 果,尿中遊離コルチゾール排泄量は投与量に相関して減少した(y = 54.9182-0.019868x, p=0.0001).そこでさらに,プラセボ群と各実薬群の投与後値を Dunnett’s two side test を用いて比較したところ,プラセボ群と1200 μg 群との間に有意差が認められた(p< 0.05, 1200 μg 群-プラセボ群の平均の差= -23.483,95%信頼区間= -43.063~-3.903). 表 2.7.6.2.1-10 尿中遊離コルチゾール排泄量(μg/g クレアチニン) 尿中遊離コルチゾール排泄量:Mean ± S.D. ステップ 投与量 被験者数 投与前24 時間 投与後24 時間 I 100 6 63.8 ± 18.1 55.2 ± 11.4 II 200 6 63.9 ± 21.2 49.3 ± 14.4 III 400 6 66.4 ± 21.4 53.8 ± 13.0 IV 800 6 73.2 ± 26.6 38.8 ± 22.8 V 1000 6 58.3 ± 12.1 34.2 ± 14.1 VI 1200 6 58.2 ± 24.5 29.8 ± 13.3 プラセボ合計 18 62.3 ± 14.1 53.2 ± 11.9 表 2.7.6.2.1-11 尿中遊離コルチゾール排泄量に関する検定結果 (1)対応のある t 検定,p 値 尿中遊離コルチゾール (μg/g クレアチニン) ステップ 投与量 (μg) 投与前日 投与日 I 100 - 0.1773 II 200 - 0.0690 III 400 - 0.0288* IV 800 - 0.0009* V 1000 - 0.0094* VI 1200 - 0.0208* プラセボ - 0.0270* *:p<0.05 (2)分散分析,p 値 ステップ 薬剤 時間 時間×ステップ 時間×薬剤 0.4989 0.4046 0.0001* 0.4969 0.0198*
Section 2.7.6 Synopsis of Individual Studie (3)ステップごとの分散分析,p 値 ステップ 薬剤 時間 時間×薬剤 I 0.8446 0.0193* 0.3075 II 0.7261 0.2167 0.4060 III 0.6671 0.0052* 0.3473 IV 0.9157 0.0196* 0.0024* V 0.3670 0.0178* 0.0461* VI 0.2566 0.0096* 0.3637 *:p<0.05 (4)実薬投与群での分散分析,p 値 投与群(実薬のみ) 時間 時間×投与群 0.5524 0.0001* 0.0344* *:p<0.05 (3)薬物動態及び副腎皮質機能への影響についての結論 健常成人男子志願者に 100~1200 μg の本剤を単回経口吸入投与した際の薬物動態並び に血中コルチゾール濃度及び尿中遊離コルチゾール排泄量に及ぼす影響を検討し,以下の 結果を得た. 1)薬物動態 400 μg までの投与量では血中 MF 濃度が定量限界(49.8 pg/mL)以下もしくはこれを わずかに超える程度で,薬物速度論的パラメータを算出することができなかった.投与 量が 800 μg 以上になると全被験者で投与後の血漿中に MF が検出された.800,1000, 1200 μg 投与時の血漿中未変化体濃度はいずれも投与後 2~3 時間に最高値を示し,各投 与量のCmaxはそれぞれ 178.3 ± 61.1,356.2 ± 170.2 及び 342.4 ± 80.0 pg/mL と,Cmax の平均値は1000~1200 μg で頭打ちとなった.いずれの投与量でも被験者間の変動は大 であった.消失半減期はそれぞれ5.58 ± 1.95,5.70 ± 4.62 及び 9.79 ± 7.15 時間で,AUC0-t はそれぞれ1018 ± 493, 1993 ± 961 及び 2905 ± 1183 pg・hr/mL であった. AUC0-tでは高い用量相関性(r = 0.9998;p = 0.0123)が認められたが,y 切片の 95% 信頼区間は原点を含まず,AUC の増加率は用量の増加率をはるかに上回るものであった. しかし,低用量での解析ができなかったこともあり,この現象が体内動態の非線形性を 示唆するものかどうかを判断することは困難であった. 今回,血漿中未変化体濃度の測定に際しては,この種の薬物の濃度を測定する方法と して現在最も適していると考えられるLC-MS/MS 法を用いたが,測定可能な範囲で設定 した定量限界値でも高用量吸入時の Cmaxとの比はわずか 6~7 倍程度しかなく,このた めAUC が低用量域では過小評価された可能性がある. なお,測定感度を上げるためにはより多くの血漿量が必要であり,被験者の負担の増 大を考慮すると,測定感度を上げることは困難と考えられる. 2)血中コルチゾール濃度 投与直前と投与後24 時間との平均値の差の検定では,1200 μg 群において有意な低下 が認められた(p = 0.0103,対応のある t 検定).
Section 2.7.6 Synopsis of Individual Studie しかし,分散分析の結果からは,治験薬投与後の血中コルチゾールの変化パターンは ステップ間(投与時期)及び薬剤間(実薬とプラセボ)で変わらない可能性が示唆され た.ステップごとに実施した分散分析でも,時間と薬剤との交互作用は有意ではなかっ た.さらに,実薬投与時の血中コルチゾール値のみを用いた分散分析でも,時間と投与 量との交互作用は有意ではなく,投与量の差による血中コルチゾール値の変化パターン の差は明らかではなかった. 3)尿中遊離コルチゾール排泄量 投与後 24 時間の尿中遊離コルチゾール排泄量は,プラセボを含むすべての群で投与 前 24 時間値よりも減少し,400,800,1000,1200 μg 群及びプラセボ群では有意な減 少を示した(p<0.05,対応のある t 検定).投与後 24 時間の尿中遊離コルチゾール排泄 量の平均値は,400 μg 群 53.8,800 μg 群 38.8,1000 μg 群 34.2,1200 μg 群 29.8 μg/g クレアチニンであり,これに対してプラセボ群の投与後 24 時間の尿中遊離コルチゾー ル排泄量の平均値は53.2 μg/g クレアチニンであった.投与後 24 時間の尿中遊離コルチ ゾ ー ル 排 泄 量 と 投 与 量 と の 間 に は 有 意 な 相 関 が 認 め ら れ , プ ラ セ ボ 群 と の 多 重 比 較 (Dunnett’s two side test)でも,プラセボ群と 1200 μg 群の間に有意差が認められた.
こうした尿中遊離コルチゾール排泄量の変化と上記の血中コルチゾール濃度の変化を 総合的に考慮すると,本剤はコルチゾール分泌に影響を及ぼすものと考えられ,しかも コルチゾール分泌の変化は投与量に相関する可能性が示唆された.本剤は合成グルココ ルチコイドであり,HPA 系機能に対するネガティヴフィードバックによってコルチゾー ルの分泌を抑制する可能性があり,今後実施する予定の健常人を対象とした反復投与試 験でも副腎皮質機能に及ぼす影響をさらに検討する必要があると考えられた. 6.安全性 (1)有害事象 すべての有害事象について重症度別分類一覧を付表1-1 及び 1-2,因果関係別分類一覧を 付表2-1 及び 2-2 に示した.さらに,臨床検査値異常のみを抜き出し,同様に分類した一覧 を付表3-1 及び 3-2,並びに付表 4-1 及び 4-2 にそれぞれ示した. 自他覚的有害事象及び臨床検査値異常変動の要約を表2.7.6.2.1-12 及び表 2.7.6.2.1-13 に示した.自他覚的有害事象は実薬群36 名中 8 名(22.2%)に 14 件,プラセボ群 18 名中 2 名(11.1%)に 6 件発現した.これらはすべて「治験薬との関連なし」と判定された. 実薬群で認められた主な症状は,下痢(血性),出血(直腸),軟便,下腹部痛,嘔吐, 口渇,頭痛(3 件)などであり,このうち重症度が中等度と判定されたのは下痢(血性) 及び出血(直腸)であり,他はいずれも軽度と判定された.下痢(血性)及び出血(直腸) は同一被験者(被験者番号 25,投与量 400 μg)で発現した症状であった.すべての症状 は無処置で消失した. プラセボ群で認められた症状は,頭痛(2 件),全身倦怠感,咽頭痛,鼻汁,声のかすれ であり,いずれも軽度で,無処置にて消失した. 臨床検査値異常変動は,実薬群 36 名中 12 名(33.3%)に 14 件,プラセボ群 18 名中 5
Section 2.7.6 Synopsis of Individual Studie 血中コルチゾール濃度の低下 2 件及び尿中遊離コルチゾール排泄量の低下 8 件は,「治験 薬と多分関連あり」と判定されたが,残る異常変動はすべて「関連なし」と判定された. 実薬群で認められた異常変動は,血中コルチゾール濃度の低下 3 件,尿中コルチゾール 排泄量の低下8 件,GPT 上昇,CPK 上昇,無機リン上昇であった.血中コルチゾール濃度の 低下及び尿中コルチゾール排泄量の低下は,200 μg で認められた血中コルチゾール濃度の低 下1 件を除いて,すべて 800 μg 以上の投与量で認められた.いずれも無処置にて消失した. プラセボ群で認められた異常変動は,GOT 上昇,GPT 上昇,γ-GTP 上昇,中性脂肪上 昇2 件,総コレステロール上昇,尿酸上昇,A/G の低下,CPK 上昇 2 件であった.いずれ も処置を必要とせず,被験者番号 18 で認められた GPT 上昇,γ-GTP 上昇,総コレステロ ール上昇,尿酸上昇が不変であった他は,すべて消失した. 表 2.7.6.2.1-12 自他覚的有害事象の要約表(すべての事象) 本剤(μg) 項 目 プラセボ 100 200 400 800 1000 1200 検討症例数 18 6 6 6 6 6 6 発現症例数 (%) 2 (11.1)* 1 (16.7)* 1 (16.7)* 1 (16.7)* 0 3 (50.0)* 2 (33.3)* 発現件数 6 2 1 4 0 3 4 下痢(血性) 1 出血(直腸) 1 軟便 1 下腹部痛 1 嘔吐 1 口渇 1 頭痛 2 1 1 1 全身倦怠感 1 両膝関節の重い感じ 1 咽頭痛 1 鼻汁 1 1 声のかすれ 1 眼のかゆみ 1 眼の充血 1 脈拍数の減少 1 *:Cochran-Armitage-Linear-Trend-Test を実施した(p = 0.3239).
Section 2.7.6 Synopsis of Individual Studie 表 2.7.6.2.1-13 臨床検査値異常変動の要約表 本剤(μg) 項 目 プラセボ 100 200 400 800 1000 1200 検討症例数 18 6 6 6 6 6 6 発現症例数 (%) 5 (27.8)*1 2 (33.3)*1 2 (33.3)*1 0 2 (33.3)*1 3 (50.0)*1 3 (50.0)*1 発現件数 10 2 2 0 2 4 4 GOT 上昇 1 GPT 上昇 1 1 γ-GTP の上昇 1 中性脂肪上昇 2 総コレステロールの上昇 1 尿酸の上昇 1 A/G の低下 1 CPK 上昇 2 1 I-P 上昇 1 血 中 コ ル チ ゾ ー ル 濃 度 の 低下 1 1* 2 1* 2 尿中遊離コルチゾール 排泄量の低下 2* 2 3* 2 3* 2 *1:Cochran-Armitage-Linear-Trend Test を実施した(p = 0.4920). *2:治験薬との因果関係が否定できないもの (2)安全性の結論 本試験の安全性の結論を以下に示した. 自他覚的有害事象は実薬群36 名中 8 名(22.2%)に 14 件,プラセボ群 18 名中 2 名(11.1%) に 6 件発現したが,いずれも「治験薬との関連なし」と判定された.投与量別に実薬群の 自他覚的有害事象発現率をみると,100~400 μg では発現率が 16.7%(1/6 例),800 μg で は0%であったのに対して,1000 μg では 50.0%(3/6 例),1200 μg では 33.3%(2/6 例) と発現率が増加した. 実薬群の自他覚的有害事象のうち,発現頻度の多かったのは下痢(血性),出血(直腸), 軟便,下腹部痛,嘔吐の消化管障害(計 5 件)であったが,5 件のうち 3 件(下痢,出血, 下腹部痛)は 400 μg 群の同一被験者で認められたもので,これらは退院後に大量のアルコ ールを摂取したことが原因で発現したものと推定された.これを除けば,特定の器官に症 状が偏ることはなかった.また,14 件の症状のうち中等度と判定されたのは上記の下痢(血 性)及び出血(直腸)の 2 件のみで,他の症状はいずれも軽度であり,中等度の下痢及び 出血も含めて,すべての自他覚的有害事象は無処置にて消失した. 臨床検査値異常変動は実薬群 36 名中 12 名(33.3%)に 14 件,プラセボ群 18 名中 5 名 (27.8%)に 10 件みられ,これらはすべて有害事象として扱った. 実薬群で認められた異常変動は,血中コルチゾール濃度の低下 3 件,尿中コルチゾール 排泄量の減少 8 件,GPT 上昇,CPK 上昇,無機リン上昇であった.このうち,GPT の上 昇,CPK の上昇,無機リンの上昇は,いずれも治験薬との因果関係は「関連なし」と判定 された.これらは100 又は 200 μg 投与時に認められ,変化の程度も軽度であった. 次に,血中コルチゾール濃度の低下 3 件及び尿中遊離コルチゾール排泄量の低下 8 件の うち,200 μg 群の蜂に刺されたことが原因と考えられる血中コルチゾール濃度の低下 1 件 のみは「関連なし」と判定されたが,残る 10 件は「治験薬と多分関連あり」と判定され
Section 2.7.6 Synopsis of Individual Studie かった血中コルチゾール濃度又は尿中遊離コルチゾール排泄量の低下を発現した被験者の 割合は,800,1000,1200 μg 群それぞれ 33.3%(2/6 例),50.0%(3/6 例),50.0%(3/6 例)であり,有意な用量相関性が認められた(p = 0.0045,Cochran-Armitage-Linear Trend Test).なお,血中コルチゾール濃度及び尿中遊離コルチゾール排泄量の異常値はすべて追 跡検査までには正常値に復していた. バイタルサインでは,脈拍数の軽度な低下(投与前 54 回/分→投与後 2 時間 46 回/分) が投与2 時間後に認められた 1000 μg 群の名を除いて異常が認められなかった. 以上から,健常人を対象に本剤を 100~1200 μg の用量範囲で単回吸入投与した場合の 安全性はおおむね良好と考えられた.ただし,血中コルチゾール濃度の低下及び尿中遊離 コルチゾール排泄量の低下には今後も注意が必要と考えられた. 7.結論 健常成人男子に本剤を100 から 1200 μg までの用量範囲で単回吸入投与したときの忍容 性はおおむね良好と考えられた.ただし,血中コルチゾール濃度及び尿中遊離コルチゾー ル排泄量の低下は本剤投与によるものと考えられ,今回の低下の程度は臨床上問題となる ものではなかったものの,1000 μg 以上では発現頻度が 50%を示したことから,標準的な 臨床使用量は 800 μg 以下が望ましいと思われた.したがって,次の段階では,800 μg 以 下の用量を用いて健常成人男子に本剤を反復投与したときの安全性及び薬物動態を検討す るとともに,引き続いてコルチゾールに及ぼす影響を検討することが必要と判断した.
Section 2.7.6 Synopsis of Individual Studie 2.7.6.2.1 付表 付表1-1 第 I 相単回投与試験(JPC- -343-11)で発現した有害事象の重症度別分類(実薬群) 件 数 例数 (%) 件 数 例数 (%) 件 数 例数 (%) 件 数 例数 (%) 件 数 例数 (%) 件 数 例数 (%) 件 数 例数 (%) 件 数 例数 (%) 件 数 例数 (%) 件 数 例数 (%) すべての有害事象 4 2(33.3) 4 2(33.3) I-P上昇 1 1(16.7) 1 1(16.7) GPT上昇 1 1(16.7) 1 1(16.7) 軟便 1 1(16.7) 1 1(16.7) 頭痛 1 1(16.7) 1 1(16.7) すべての有害事象 3 2(33.3) 3 2(33.3) 両膝関節の重い感じ 1 1(16.7) 1 1(16.7) CPK上昇 1 1(16.7) 1 1(16.7) 血漿中コルチゾール濃度の低下 1 1(16.7) 1 1(16.7) すべての有害事象 2 1(16.7) 2 1(16.7) 4 1(16.7) 口渇 1 1(16.7) 1 1(16.7) 下腹部痛 1 1(16.7) 1 1(16.7) 出血(直腸) 1 1(16.7) 1 1(16.7) 下痢(血性) 1 1(16.7) 1 1(16.7) すべての有害事象 2 2(33.3) 2 2(33.3) 2 2(33.3) 2 2(33.3) 尿中遊離コルチゾール排泄量の低下 2 2(33.3) 2 2(33.3) 2 2(33.3) 2 2(33.3) すべての有害事象 7 4(66.7) 7 4(66.7) 4 3(50.0) 4 3(50.0) 脈拍数の減少 1 1(16.7) 1 1(16.7) 頭痛 1 1(16.7) 1 1(16.7) 鼻汁 1 1(16.7) 1 1(16.7) 血漿中コルチゾール濃度の低下 1 1(16.7) 1 1(16.7) 1 1(16.7) 1 1(16.7) 尿中遊離コルチゾール排泄量の低下 3 3(50.0) 3 3(50.0) 3 3(50.0) 3 3(50.0) すべての有害事象 8 3(50.0) 8 3(50.0) 4 3(50.0) 4 3(50.0) 嘔吐 1 1(16.7) 1 1(16.7) 眼の充血 1 1(16.7) 1 1(16.7) 頭痛 1 1(16.7) 1 1(16.7) 眼のかゆみ 1 1(16.7) 1 1(16.7) 血漿中コルチゾール濃度の低下 1 1(16.7) 1 1(16.7) 1 1(16.7) 1 1(16.7) 尿中遊離コルチゾール排泄量の低下 3 3(50.0) 3 3(50.0) 3 3(50.0) 3 3(50.0) 1000μg群 1200μg群 投与群 200μg群 400μg群 100μg群 高度 不明 中等度 800μg群 有害事象名 軽度 すべての有害事象(N=6,1例4件) 関連の否定できない有害事象(N=6,0例0件) すべての有害事象(N=6,2例2件) 関連の否定できない有害事象(N=6,2例2件) 計 すべての有害事象(N=6,2例3件) 関連の否定できない有害事象(N=6,0例0件) すべての有害事象(N=6,2例4件) 関連の否定できない有害事象(N=6,0例0件) 不明 計 軽度 中等度 高度 すべての有害事象(N=6,4例7件) 関連の否定できない有害事象(N=6,3例4件) すべての有害事象(N=6,3例8件) 関連の否定できない有害事象(N=6,3例4件)
Section 2.7.6 Synopsis of Individual Studie 付表1-2 第 I 相単回投与試験(JPC- -343-11)で発現した有害事象の重症度別分類(プラセボ群) 件 数 例数 (%) 件 数 例数 (%) 件 数 例数 (%) 件 数 例数 (%) 件 数 例数 (%) 件 数 例数 (%) 件 数 例数 (%) 件 数 例数 (%) 件 数 例数 (%) 件 数 例数 (%) すべての有害事象 16 7(38.9) 16 7(38.9) A/Gの低下 1 1(5.6) 1 1(5.6) 咽頭痛 1 1(5.6) 1 1(5.6) 声のかすれ 1 1(5.6) 1 1(5.6) CPK上昇 2 2(11.1) 2 2(11.1) GOT上昇 1 1(5.6) 1 1(5.6) GPT上昇 1 1(5.6) 1 1(5.6) 総コレステロールの上昇 1 1(5.6) 1 1(5.6) 中性脂肪上昇 2 2(11.1) 2 2(11.1) 尿酸の上昇 1 1(5.6) 1 1(5.6) 頭痛 2 1(5.6) 2 1(5.6) 鼻汁 1 1(5.6) 1 1(5.6) 全身倦怠感 1 1(5.6) 1 1(5.6) γーGTPの上昇 1 1(5.6) 1 1(5.6) 中等度 高度 不明 計 有害事象名 すべての有害事象(N=18,7例16件) 関連の否定できない有害事象(N=18,0例0件) 軽度 中等度 高度 不明 計 軽度
Section 2.7.6 Synopsis of Individual Studie 付表2-1 第 I 相単回投与試験(JPC- -343-11)で発現した有害事象の因果関係別分類(実薬群) 件数 例数(%) 件数 例数(%) 件数 例数(%) 件数 例数(%) 件数 例数(%) 件数 例数(%) すべての有害事象 4 2(33.3) 4 2(33.3) 血清無機リン上昇 1 1(16.7) 1 1(16.7) GPT上昇 1 1(16.7) 1 1(16.7) 軟便 1 1(16.7) 1 1(16.7) 頭痛 1 1(16.7) 1 1(16.7) すべての有害事象 3 2(33.3) 3 2(33.3) 両膝関節の重い感じ 1 1(16.7) 1 1(16.7) CPK上昇 1 1(16.7) 1 1(16.7) 血漿中コルチゾール濃度の低下 1 1(16.7) 1 1(16.7) すべての有害事象 4 1(16.7) 4 1(16.7) 口渇 1 1(16.7) 1 1(16.7) 下腹部痛 1 1(16.7) 1 1(16.7) 出血(直腸) 1 1(16.7) 1 1(16.7) 下痢(血性) 1 1(16.7) 1 1(16.7) すべての有害事象 2 2(33.3) 2 2(33.3) 尿中遊離コルチゾール排泄量の低下 2 2(33.3) 2 2(33.3) すべての有害事象 3 3(50.0) 4 3(50.0) 7 4(66.7) 脈拍数の減少 1 1(16.7) 1 1(16.7) 頭痛 1 1(16.7) 1 1(16.7) 鼻汁 1 1(16.7) 1 1(16.7) 血漿中コルチゾール濃度の低下 1 1(16.7) 1 1(16.7) 尿中遊離コルチゾール排泄量の低下 3 3(50.0) 3 3(50.0) すべての有害事象 4 2(33.3) 4 3(50.0) 8 3(50.0) 嘔吐 1 1(16.7) 1 1(16.7) 眼の充血 1 1(16.7) 1 1(16.7) 頭痛 1 1(16.7) 1 1(16.7) 眼のかゆみ 1 1(16.7) 1 1(16.7) 血漿中コルチゾール濃度の低下 1 1(16.7) 1 1(16.7) 尿中遊離コルチゾール排泄量の低下 3 3(50.0) 3 3(50.0) 100μg群 投与群 有害事象名 多分関連あり 明らかに関連あり 計 すべての有害事象 (N=6,2例4件) 関連なし 多分関連なし 関連の可能性あり すべての有害事象 (N=6,3例8件) 200μg群 400μg群 800μg群 1000μg群 1200μg群 すべての有害事象 (N=6,2例2件) すべての有害事象 (N=6,4例7件) すべての有害事象 (N=6,2例3件) すべての有害事象 (N=6,1例4件)
Section 2.7.6 Synopsis of Individual Studie 付表2-2 第 I 相単回投与試験(JPC- -343-11)で発現した有害事象の因果関係別分類(プラセボ群) 件数 例数(%) 件数 例数(%) 件数 例数(%) 件数 例数(%) 件数 例数(%) 件数 例数(%) すべての有害事象 16 7(38.9) 16 7(38.9) A/Gの低下 1 1(5.6) 1 1(5.6) 咽頭痛 1 1(5.6) 1 1(5.6) 嗄声 1 1(5.6) 1 1(5.6) CPK上昇 2 2(11.1) 2 2(11.1) GOT上昇 1 1(5.6) 1 1(5.6) GPT上昇 1 1(5.6) 1 1(5.6) 総コレステロールの上昇 1 1(5.6) 1 1(5.6) 中性脂肪上昇 2 2(11.1) 2 2(11.1) 尿酸の上昇 1 1(5.6) 1 1(5.6) 頭痛 2 1(5.6) 2 1(5.6) 鼻汁 1 1(5.6) 1 1(5.6) 全身倦怠感 1 1(5.6) 1 1(5.6) γ-GTP上昇 1 1(5.6) 1 1(5.6) 有害事象名 すべての有害事象 (N=18,7例16件) 関連なし 多分関連なし 関連の可能性あり 多分関連あり 明らかに関連あり 計
Section 2.7.6 Synopsis of Individual Studie 付表3-1 第 I 相単回投与試験(JPC- -343-11)で発現した臨床検査値異常の重症度別分類(実薬群) 件 数 例数 (%) 件 数 例数 (%) 件 数 例数 (%) 件 数 例数 (%) 件 数 例数 (%) 件 数 例数 (%) 件 数 例数 (%) 件 数 例数 (%) 件 数 例数 (%) 件 数 例数 (%) すべての有害事象 2 2(33.3) 2 2(33.3) I-P上昇 1 1(16.7) 1 1(16.7) GPT上昇 1 1(16.7) 1 1(16.7) すべての有害事象 2 2(33.3) 2 2(33.3) CPK上昇 1 1(16.7) 1 1(16.7) 血漿中コルチゾール濃度の低下 1 1(16.7) 1 1(16.7) すべての有害事象 すべての有害事象 2 2(33.3) 2 2(33.3) 2 2(33.3) 2 2(33.3) 尿中遊離コルチゾール排泄量の低下 2 2(33.3) 2 2(33.3) 2 2(33.3) 2 2(33.3) すべての有害事象 4 3(50.0) 4 3(50.0) 4 3(50.0) 4 3(50.0) 血漿中コルチゾール濃度の低下 1 1(16.7) 1 1(16.7) 1 1(16.7) 1 1(16.7) 尿中遊離コルチゾール排泄量の低下 3 3(50.0) 3 3(50.0) 3 3(50.0) 3 3(50.0) すべての有害事象 4 3(50.0) 4 3(50.0) 4 3(50.0) 4 3(50.0) 血漿中コルチゾール濃度の低下 1 1(16.7) 1 1(16.7) 1 1(16.7) 1 1(16.7) 尿中遊離コルチゾール排泄量の低下 3 3(50.0) 3 3(50.0) 3 3(50.0) 3 3(50.0) 有害事象名 軽度 中等度 高度 高度 不明 計 すべての有害事象(N=6,2例2件) 関連の否定できない有害事象(N=6,0例0件) 不明 計 軽度 中等度 すべての有害事象(N=6,2例2件) 関連の否定できない有害事象(N=6,0例0件) すべての有害事象(N=6,0例0件) 関連の否定できない有害事象(N=6,0例0件) すべての有害事象(N=6,2例2件) 関連の否定できない有害事象(N=6,2例2件) すべての有害事象(N=6,3例4件) 関連の否定できない有害事象(N=6,3例4件) すべての有害事象(N=6,3例4件) 関連の否定できない有害事象(N=6,3例4件) 投与群 100μg群 200μg群 400μg群 800μg群 1000μg群 1200μg群 本群において,臨床検査値異常は認められなかった.
Section 2.7.6 Synopsis of Individual Studie 付表3-2 第 I 相単回投与試験(JPC- -343-11)で発現した臨床検査値異常の重症度別分類(プラセボ群) 件 数 例数 (%) 件 数 例数 (%) 件 数 例数 (%) 件 数 例数 (%) 件 数 例数 (%) 件 数 例数 (%) 件 数 例数 (%) 件 数 例数 (%) 件 数 例数 (%) 件 数 例数 (%) すべての有害事象 10 5(27.8) 10 5(27.8) A/Gの低下 1 1(5.6) 1 1(5.6) CPK上昇 2 2(11.1) 2 2(11.1) GOT上昇 1 1(5.6) 1 1(5.6) GPT上昇 1 1(5.6) 1 1(5.6) 総コレステロールの上昇 1 1(5.6) 1 1(5.6) 中性脂肪上昇 2 2(11.1) 2 2(11.1) 尿酸の上昇 1 1(5.6) 1 1(5.6) γーGTPの上昇 1 1(5.6) 1 1(5.6) 有害事象名 すべての有害事象(N=18,5例10件) 関連の否定できない有害事象(N=18,0例0件) 軽度 中等度 高度 不明 計 軽度 中等度 高度 不明 計