Corresponding author, Email: unemoto@imr.tohoku.ac.jp
J-STAGE Advance Publication date : July 1, 2016 特集「固体中の水素と材料特性」
錯体水素化物固体電解質と硫化物ガラス固体電解質の
ハイブリッド利用による室温動作可能な 4 V 級バルク型
全固体リチウム二次電池の開発
宇根本 篤
1
野 上 玄 器
2田 沢 勝
2谷 口 貢
2折 茂 慎 一
1,3 1東北大学原子分子材料科学高等研究機構 (WPIAIMR) 2三菱ガス化学株式会社新潟研究所 3東北大学金属材料研究所J. Japan Inst. Met. Mater. Vol. 80, No. 12(2016), pp. 720725 Special Issue on Hydrogen and Materials Characteristic in Solids III 2016 The Japan Institute of Metals and Materials
Development of 4 VClass BulkType AllSolidState Lithium Rechargeable Batteries by a
Combined Use of Complex Hydride and Sulfide Electrolytes for Room Temperature Operation
Atsushi Unemoto1, Genki Nogami2, Masaru Tazawa2, Mitsugu Taniguchi2and Shinichi Orimo1,3
1WPIAdvanced Institute for Materials Research, Tohoku University, Sendai 9808577 2Mitsubishi Gas Chemicals Co., Ltd., Niigata 9503112
3Institute for Materials Research, Tohoku University, Sendai 9808577
We have operated a 4 Vclass bulktype, allsolidstate LiCoO2/Li battery at room temperature. The composition consisted of a Li4(BH4)3I complex hydride electrolyte for the electrolyte layer, and a 80Li2S 20P2S5sulfide glass for an electrolyte in the positive electrode layer. The battery assembled exhibited a 92 mAh・g-1initial discharge capacity at 298 K and 0.1 C. The dis-charge capacity for the 20thcycle remained as high as 83 mAh・g-1, corresponding to a capacity retention ratio of nearly 90. [doi:10.2320/jinstmet.JD201601]
(Received March 2, 2016; Accepted May 2, 2016; Published July 1, 2016)
Keywords: complex hydride electrolyte, sulfide electrolyte, bulktype, allsolidstate battery, room temperature operation
1. 緒 言 高水素密度材料である錯体水素化物は,水素貯蔵1,2), リチウム貯蔵3,4),および高速イオン伝導57)といった, エネルギー貯蔵・変換に関連した高い機能性を有する材料群 である.典型的な錯体水素化物である LiBH4は,昇温に伴 って 390 K 付近で低温相である正方晶から高温相である六 方晶へ構造相転移する8).この高温相では,log(s/S・cm-1) =-3 を上回る高いリチウムイオン伝導率を示す5).錯体水 素化物は,その構成元素に軽元素を選ぶことができるため, 軽量材料を設計することができる.また,リチウムイオンの 輸率はほぼ 1 である.最も卑な電位を有し,高容量である 金属リチウム電極と安定な界面を形成する.結晶構造中の水 素は,錯イオンの中心元素との強固な共有結合によって安定 化されており,想定される電池の動作温度範囲では熱分解し にくく,耐熱性に優れる.また,塑性変形しやすいため,室 温での一軸加圧のみで電解質の緻密体が作製でき,電極と密 着した界面が容易に形成できるといった特長を有する57). 近年,この LiBH4高温相をはじめとした,錯体水素化物 系固体電解質を利用したバルク型全固体リチウムイオン二次 電池の研究開発に関する報告が活発になされている.LiBH4 は還元剤としても使用されているように,還元力が強い.こ のため,金属リチウムとは安定な界面を形成する57)一方, LiCoO2といった酸化力の強い高電位正極活物質を還元分解 して電池の安定動作を阻害する.この課題に対しては, Li3PO4などを電極活物質の表面へ被覆して,錯体水素化物 固体電解質との直接接触を防ぐことにより,電池の繰り返し 動 作 が 可 能 と な る こ と が Takahashi ら か ら 報 告 さ れ て い る9,10).一方,筆者らは,LiBH 4固体電解質のみで電池作製 を行う場合,TiS27,11)や硫黄12,13)といった,低電位高容量型 正極と金属リチウムを利用した電池構成が望ましいことを提 案し,バルク型電池構成で繰り返し動作を実証した. LiBH4固体電解質を利用する電池構成では,その可動温 度が相転移温度である 390 K 以上8)に限られている.このた め,より幅広い温度領域,とりわけ室温での繰り返し動作が 可能な固体電解質の開発や電池構成の提案が望まれている. Maekawa らはこの課題に対し,ヨウ化リチウム LiI などの ハロゲン化リチウムを LiBH4へ固溶することにより,高温 相を低温領域へ安定化させる方法を提案している.例えば,
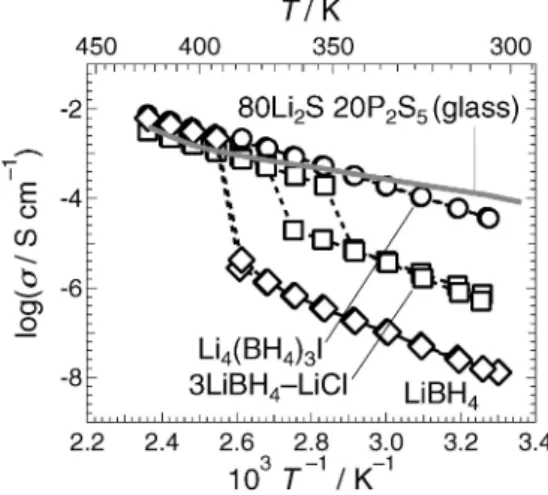
Fig. 1 Lithiumionic conductivities as a function of inverse temperature: LiBH45), Li4(BH4)3I14), 3LiBH4LiCl14) and 80Li2S 20P2S5(glass).
Li4( BH4)3I は Fig. 1 に 示 し た よ う に , 300 K に お い て
log(s/S・cm-1)=-4.7 と高いリチウムイオン伝導率を示
す14).Sveinbj äornsson らは,Li
4Ti5O12の多孔質薄膜電極と Li13/16(BH4)13/16I3/16固体電解質,金属リチウム電極を利用 した全固体電池を作製し,333 K において繰り返し動作する ことを報告している15).Yoshida らは最近,Li 4(BH4)3I 固 体電解質を電極層内部で Li4Ti5O12と混合し,電極層内部の 電極担持量を増やしたバルク型の電池構成において,室温 (296 K)だけでなく,423 K といった高温においても繰り返 し動作が可能であることを報告している16).このように, 還元力の強い錯体水素化物固体電解質であっても,その特長 に基づいた適切な電池設計により,室温での動作が可能であ り,高電位正極を備え,繰り返し動作が可能なバルク型全固 体リチウム電池が開発できる可能性がある. 代表的な硫化物固体電解質である Li2Sと P2S5からなるガ ラス固体電解質は,高いリチウムイオン伝導率を有する(例 えば,80Li2S 20P2S5ガラス固体電解質の室温でのイオン伝 導率および活性化エネルギーはそれぞれ log(s/S・cm-1)= -3.8 および 34 kJ・mol-1である17)).一連の材料は,塑性 変形性に優れるため,室温での加圧のみで電池作製が可能で ある.4 V 級正極 LiCoO2を備えるバルク型全固体リチウム 電池が室温において繰り返し動作することが報告されてい る18).硫化物系固体電解質と LiCoO 2の界面では,リチウム イオンが欠乏した空間電荷層が形成されて界面抵抗が高くな ること1922),電池の繰り返し動作に伴って,構成元素の相 互 拡散によ り抵抗層 が形成さ れる23)ことが 報告され てい る . こ の 課 題 は , 例 え ば LiNbO3な ど の 固 体 電 解 質 を LiCoO2表面へコーティングすることにより解決できる. 以上の内容を踏まえ,本研究では,錯体水素化物固体電解 質と硫化物固体電解質をハイブリッド利用し,室温での動作 が可能な 4 V 級バルク型全固体リチウム二次電池の開発を 目的とした.4 V 級正極 LiCoO2表面へ LiNbO3をおよそ 20 nmの厚みで被覆した正極活物質と,金属リチウム負極を利 用し,正極層内部の固体電解質には 80Li2S 20P2S5を,電解 質層には Li4(BH4)3I をそれぞれ利用したバルク型全固体リ チウム電池を作製し,室温において電池性能を評価した. 2. 実 験 方 法 Li4(BH4)3I 固体電解質はメカニカルミリングおよび熱処 理 を 施 す こ と に よ り 合 成 し た14). 出 発 原 料 に は LiBH 4 (90,シグマアルドリッチ社製)および LiI(99.999, シグマアルドリッチ社製)を使用した.これらの出発原料を あらかじめめのう乳鉢とめのう乳棒により混合した後,内容 量 45 mL のポットへq7 mm のボール 20 個とともに封入 し,回転速度 400 rpm にて 5 時間ミリング処理(P7,フリ ッチュ社製)を施した.得られた粉末の X 線回折測定(XRD, X'pert PRO,パナリティカル社製)およびラマン分光測定 (Nicolet Almega XR,サーモフィッシャーサイエンティフ ィック社製)の結果から,合成した試料からは LiBH4高温 相8)以外のピークは現れなかった. Li4(BH4)3Iの酸化側での安定性を評価するため,TiS2正 極と金属リチウム負極を利用したバルク型全固体リチウム電 池を作製した.筆者らはこれまで,TiS2正極,金属リチウ ム負極および LiBH4を備えるバルク型全固体リチウムイオ ン電池が,393 K において 300 回以上繰り返し動作するこ とを報告している11).この結果を踏まえ,ヨウ素を含有す る固体電解質が電池特性に及ぼす影響について検討するにあ たり,類似の電池構成で性能を比較するのが適切であると考 えた.これに加え,比較のために正極内で混合する固体電解 質として LiBH4を用いた電池も作製した. TiS2と Li4( BH4)3I あ る い は LiBH4が そ れ ぞ れ 重 量 比 4060 となるよう秤量して,めのう乳ばちとめのう乳棒に より混合して混合正極とした.Li4(BH4)3I あるいは LiBH4 固体電解質粉末をそれぞれ 25 mg あるいは 20 mg 秤量して 直径 8 mm のダイスにセッティングし,60 MPa で一軸加圧 した.この後,前記混合正極を 6 mg 秤量して固体電解質上 に移動した後,正極層と固体電解質層を密着させるため, 240 MPa でさらに一軸加圧することで,正極層と固体電解 質層の二層からなる緻密体を得た.正極の反対側に金属リチ ウム箔を配置して負極とした.測定温度は 393 K で,放充 電レートを 0.05 C(電流密度 57 mA cm-2)として電池性能を
評価した(580 Battery Test System, Scribner Associates 社 製).作製した全固体電池の外観や電気化学測定の構成につ いては文献を参考にされたい1113). 電池測定前後の正極層について,Ga イオンビーム(FIB, FB2200,日立ハイテクノロジーズ社製)により断面を加工 してから,電解放射型走査電子顕微鏡(FESEM, SU9000, 日立ハイテクノロジーズ社製)およびエネルギー分散型 X 線 分析装置(EDX, Apollo XLT,アメテック社製)にてそれぞ れ微細構造観察と元素分布測定を行った. 80Li2S 20P2S5ガラス固体電解質は既報23)に倣ってメカニ カルミリング法により合成した.すなわち,Li2S(98,シ グマアルドリッチ社製)および P2S5(99,シグマアルド リッチ社製)の粉末を,めのう乳鉢とめのう乳棒を使用して あらかじめ混合しておき,内容量 45 mL のジルコニアポッ トへ q5 mm のジルコニアボール 160 個とともに封入し,回
Fig. 2 Microstructure and element distributions of the composite positive electrode layer, TiS2/Li4(BH4)3I: Before the battery test; (a) crosssectional FESEM image, and distributions of (b) I, (c) Ti and (d) S, and after the battery test; (e) crosssectional FE SEM image, and distributions of (f) I, (g) Ti and (h) S.
Fig. 3 Performance of the bulktype allsolidstate batteries operated at 393 K and 0.05 C: (a) TiS2/Li4(BH4)3I|Li4(BH4)3I |Li, and (b) TiS2/LiBH4|Li4(BH4)3I|Li.
転速度 510 rpm にて 10 時間メカニカルミリング処理を施し た.得られた試料の XRD 測定では,非晶質由来のハローパ ターンのみが現れ,目的のガラス固体電解質が得られたこと を確認した.作製したガラス固体電解質のイオン伝導率を交 流二端子法(353280 ケミカルインピーダンスメータ,日置 電機社製)により,298 から 423 K の温度範囲で評価した. LiCoO2表面へ,転動流動コーティング装置(MP01,パ ウ レ ッ ク 社 製 ) を 利 用 し て , LiNbO3を コ ー テ ィ ン グ し た19,20)( 以 下 , LiNbO 3coated LiCoO2 と 表 記 す る ) .
LiNbO3coated LiCoO2と 導 電 助 剤 の ケ チ ェ ン ブ ラ ッ ク
(KB)および 80Li2S 20P2S5ガラス固体電解質を重量比 40 606 となるよう混合して正極合剤とした.Li4(BH4)3I 固 体電解質粉末を 101.1 mg 秤量して直径 10 mm のダイスに セッティングし,20 MPa でプレスした.この後,正極合剤 を 11.5 mg 秤量し,先にプレスした固体電解質上に移動し, 240 MPa にて一軸成型した.正極の反対側に金属リチウム 箔を配置して負極とした.電池測定は 297 K において,充 放電レート 0.1 C(電流密度 73 mA cm-2)にて行った(VMP3,
BioLogic 社製).LiNbO3coated LiCoO2正極と KB および
Li4(BH4)3I からなる混合物緻密体を作製し,Ga イオンビー ムにて断面加工した試料ついて,FESEM および EDX を 利用して微細構造観察と元素分布測定を行った. 3. 結果および考察 電池測定前の正極層断面の FESEM 像およびヨウ素,チ タンおよび硫黄の分布をそれぞれ Fig. 2(a)(d)に示した. ここで見られるように,室温での一軸加圧のみで成型したに もかかわらず,正極活物質 TiS2と固体電解質 Li4(BH4)3I が 密着しており,スムーズな電荷移動反応を促す界面が形成さ れていることがわかった.このような微細構造は,TiS2と LiBH4からなる混合正極での界面と同様であり7,11),LiBH4 と LiI の固溶体 Li4(BH4)3I であっても,錯体水素化物が本 来持つ優れた加工性(塑性変形性)が失われないことを示して いる. TiS2は,以下の電気化学反応に従って電池反応が進行す る24,25).
TiS2+xLi++xe- Li
xTiS2 ( 1 )
右への反応は放電反応を,左への反応は充電反応をそれぞれ 表す.LixTiS2のリチウム濃度 x が 0 から 1 の範囲で変化す
る場合,理論容量は 239 mAh・g-1となる.Fig. 3(a)に,バ
ルク型全固体電池 TiS2/Li4(BH4)3I|Li4(BH4)3I|Li の放充電
プロファイルを示した.初回放電容量は 49 mAh・g-1であ ったのに対し,2 回目の放電容量は 141 mAh・g-1であっ た.この差の要因として,電池の動作温度 393 K において, LixTiS2を形成する TiS2と Li4(BH4)3I の固相反応,すなわ ち自己放電反応が考えられる7,11).一方,15 回目の放電容量 は 76 mAh・g-1と,繰り返し動作に伴って徐々に容量劣化
Fig. 4 Microstructure of LiNbO3coated LiCoO2 in the Li4(BH4)3I, KB and LiNbO3coated LiCoO3compact: (a) and (b) crosssectional FESEM image, and distributions of (c) Co, (d) Nb, (e) C and (f) I.
した.サイクル数の増加に伴って電圧降下が大きくなってい ることから,繰り返し電池動作中に電池内部の抵抗が大きく なり,この結果として,放電容量が小さくなったことが考え られる. 正極層で混合する固体電解質を LiBH4としたバルク型全 固体電池では,電池の動作温度である 393 K において TiS2 と LiBH4との固相反応により TiS2へ Li がドープされるた め(自己放電反応),初回放電容量こそ 61 mAh・g-1と低か ったが,2 回目の放電容量は 218 mAh・g-1と理論容量に近 い高い値が得られた.初回充電時のみ,容量は 419 mAh・ g-1と過充電となった.これは TiS 2と LiBH4の固相反応の 進行が不十分であるために,TiS2表面で未反応の LiBH4が 酸化分解したことが要因である11).15 回目の放電容量は 220 mAh・g-1と繰り返し動作に伴う顕著な容量劣化は見ら れなかった.このことは,Fig. 3(a)で見られたバルク型全 固体 TiS2/Li4(BH4)3I|Li4(BH4)3I|Li 電池のサイクル動作に
伴う容量劣化が,TiS2正極と Li4(BH4)3I固体電解質界面の 安定性に起因することを示唆している. 電池動作が TiS2と Li4(BH4)3I との界面安定性に及ぼす影 響について,電池動作後の正極層の微細構造および元素分布 の観点から検討した.Fig. 2(e)(h)にそれぞれ,電池試験 後の正極層の断面 FESEM 像と,ヨウ素,チタンおよび硫 黄 の 元 素 分 布 を 示 し た . 電 池 試 験 後 に は , TiS2 と Li4(BH4)3I 界面にヨウ素が濃集していることがわかった. Fig. 3(a)に示した放充電プロファイルから,サイクル数の 増 加 に 伴 っ て 電 圧 降 下 が 大 き く な っ て い る こ と か ら , Li4(BH4)3I が TiS2との固相反応により分解して界面近傍の ヨウ素濃度が高まり,この結果として,ヨウ素を含む化合物 が析出して高抵抗につながったことが考えられる.これを抑 制して電池の安定動作を実現するためには,このような固相 反応を抑制する固体電解質の利用が有効であることがわかっ た. 硫化物固体電解質を利用したこれまでの全固体電池に関す る報告では,4 V 級正極である LiCoO2表面へ固体電解質を コーティングして緩衝層とすることにより,低抵抗かつ繰り 返し動作可能なバルク型全固体電池が作製できることが報告 されている1923).転動流動コーティング装置により作製し
た LiNbO3coated LiCoO2粒子の FESEM 像を Fig. 4(a)お
よび(b)に,コバルト,ニオブ,炭素およびヨウ素の元素マ ッピングをそれぞれ Fig. 4(c)(f)に示した.これらの結果 から,LiCoO2表面へ,厚みがおよそ 20 nm 程度の LiNbO3 が均一にコーティングされていることがわかった.本研究で 作製した 80Li2S 20P2S5ガラス固体電解質のリチウムイオン 伝導率を Fig. 1 に示した.298 K におけるリチウムイオン 伝導率と活性化エネルギーはそれぞれ log(s/S・cm-1)= -4.0 と 29 kJ・mol-1であり,既報17)とよく一致していた. LiCoO2は,以下の電気化学反応に従って電池反応が進行 する26).
LiCoO2 Li1-xCoO2+xLi++xe- ( 2 )
右への反応は充電反応を,左への反応は放電反応をそれぞれ 表す.充電上限電圧を 4.2 V とした場合,x は 0 から 0.5 に 変化することが知られており,この時の比容量は 137 mAh・ g-1に相当する.作製した 4 V 級バルク型全固体リチウム電 池の 298 K での充放電プロファイルを Fig. 5(a)に,放電容 量とクーロン効率のサイクル依存性を Fig. 5(b)にそれぞれ 示した.当初の期待通り,本研究で作製したバルク型電池は 4 V 付近で電池動作した.LiCoO2の Li 量変化に由来する充 電および放電のプラトーがそれぞれ 4.05 V および 3.8 V 付 近に現れた.初回と 20 回目の放電容量はそれぞれ 92 mAh・ g-1および 83 mAh・g-1であり,放電容量維持率はおよそ 90と顕著な容量劣化がなく繰り返し動作した.初回の クーロン効率(=放電容量/充電容量)は 75であったが,2 サイクル目以降は 97 から 100の範囲で推移しており,繰 り返し動作に対して顕著な副反応が伴うことなく動作したこ とがわかった. 以上のように,電気化学的安定性の異なる固体電解質をハ イブリッド利用することにより,それぞれ単独の材料群だけ では動作困難な電池構成であっても繰り返し動作が可能であ ることがわかった.川治らは最近,錯体水素化物固体電解質 と酸化物固体電解質を併用することで,耐熱性に優れる硫黄 フリーの 4 V 級バルク型全固体リチウムイオン電池の動作 を実証した27). 最近,Li2SP2S5ガラスへ LiBH4を分散させた固体電解質 は,298 K において log(s/S・cm-1)=-2.8 と高いイオン伝 導率を有することが報告された28).加えて,見かけの組成 90LiBH410P2S5の結晶相は,300 K において,log(s/S・ cm-1)=-3.0 のリチウムイオン伝導率を有することが報告 された29).これらの材料では,少なくとも室温以上では構
Fig. 5 Performance of the bulktype allsolidstate battery, LiNbO3coated LiCoO2/KB/80Li2S 20P2S5|Li4(BH4)3I|Li, operated at 298 K and 0.05 C: (a) Typical chargedischarge profiles, and (b) cycle performance.
造相転移に由来するイオン伝導率の不連続変化がない.この ため,これらの固体電解質を使用することにより幅広い温度 での電池駆動が可能な電池を作製できる可能性がある.一方, [B12H12]2-や[CB11H12]-に代表される籠状のクラスター 型アニオンを含有する Closoborane 系あるいは Closocar-baborane系錯体水素化物のなかで,高いリチウムイオン伝 導率3033)やナトリウムイオン伝導率30,3235)を有する材料が見 つかっており,このような固体電解質を利用した全固体電池 が動作実証されている31,32).錯体水素化物を基盤とする固体 電解質開発では,水素を含有する錯アニオンのダイナミクス がカチオン輸送速度と密接に関連している点に特徴があ る6,7,3638).この知見を踏まえ,結晶構造や組成の観点から 材料の多様化が急速に進んでおり,今後の発展が大いに期待 される. 4. 結 論 本研究では,錯体水素化物固体電解質と硫化物固体電解質 をハイブリッド利用することにより,室温での動作が可能な 4 V 級バルク型全固体リチウムイオン二次電池の開発とその 動作を実証した.金属リチウム負極を使用するため,固体電 解質層には還元力の強い Li4(BH4)3Iを利用した.LiNbO3 を LiCoO2粒子表面に 20 nm の厚みで均一コーティングし, 80Li2S 20P2S5ガラス固体電解質と導電助剤の KB を混合し て正極層に利用した.この電池は 298 K,充放電レート 0.1 Cにおいて繰り返し動作が可能であった.初回および 20 回 目の放電容量はそれぞれ 92 mAh・g-1および 83 mAh・g-1 であり,放電容量維持率はおよそ 90と高安定な電池であ った. 研究支援者の佐藤清人氏,大宮晴美氏および割舟奈穂子氏 に感謝の意を表します.本研究は,東北大学 WPIAIMR ターゲットプロジェクト 4,東北大学金属材料研究所先端エ ネルギー材料理工共創センター,科研費・基盤研究(S)(課 題番号25220911)および科学技術振興機構戦略的創造研究 推進事業・先端的低炭素化技術開発(ALCA)により実施され ました. 文 献
1) L. Schlapbach and A. Z äuttel: Nature414(2001) 353358. 2) S. Orimo, Y. Nakamori, J. R. Eliseo, A. Z äuttel and C. M. Jensen:
Chem. Rev.107(2007) 41114132.
3) W. Zaäƒdi, J.P. Bonnet, J. Zhang, F. Cuevas, M. Latroche, S. Couillaiud, J.L. Bobet, M. T. Sougrati, J.C. Jumas and L. Aymard: Int. J. Hydrogen Energ.38(2013) 47984808. 4) J. Zhang, W. Zaäƒdi, V. PaulBoncour, K. Provost, A.
Michalowicz, F. Cuevas, M. Latroche, S. Belin, J.P. Bonnet and L. Aymard: J. Mater. Chem. A1(2013) 47064717. 5) M. Matsuo, Y. Nakamori, S. Orimo, H. Maekawa and H.
Takamura: Appl. Phys. Lett.91(2007) 224103.
6) M. Matsuo and S. Orimo: Adv. Energ. Mater.1(2011) 161172. 7) A. Unemoto, M. Matsuo and S. Orimo: Adv. Funct. Mater.24
(2014) 22672279.
8) JPh. Soulie, G. Renaudin, R. Cerny and K. Yvon: J. Alloy Compd.346(2002) 200205.
9) K. Takahashi, K. Hattori, T. Yamazaki, K. Takada, M. Matsuo, S. Orimo, H. Maekawa and J. Takamura: J. Power Sources226 (2013) 6164.
10) K. Takahashi, H. Maekawa and H. Takamura: Solid State Ionics262(2014) 179182.
11) A. Unemoto, T. Ikeshoji, S. Yasaku, M. Matsuo, V. Stavila, T. J. Udovic and S. Orimo: Chem. Mater.27(2015) 54075416. 12) A. Unemoto, S. Yasaku, G. Nogami, M. Tazawa, M. Taniguchi,
M. Matsuo, T. Ikeshoji and S. Orimo: Appl. Phys. Lett. 105 (2014) 083901.
13) A. Unemoto, C. Chen, Z. Wang, M. Matsuo, T. Ikeshoji and S. Orimo: Nanotechnology26(2015) 254001.
14) H. Makekawa, M. Matsuo, H. Takamura, M. Ando, Y. Noda, T. Karahashi and S. Orimo: J. Am. Chem. Soc.131(2009) 894 895.
15) D. Sveinbj äornsson, A. S. Christiansen, R. Viskinde, P. Norby and T. Vegge: J. Electrochem. Soc.161(2014) A1432A1439. 16) K. Yoshida, S. Suzuki, J. Kawaji, A. Unemoto and S. Orimo:
Solid State Ionics285 (2016) 96100.
17) A. Hayashi, S. Hama, H. Morimoto, M. Tatsumisago and T. Minami: J. Am. Chem. Soc.84(2001) 477479.
18) A. Sakuda, A. Hayashi and M. Tatsumisago: Chem. Mater.22 (2010) 949956.
19) N. Ohta, K. Takada, L. Zhang, R. Ma, M. Osada and T. Sasaki: Adv. Mater.18(2006) 22262229.
20) N. Ohta, K. Takada, I. Sakaguchi, L. Zhang, R. Ma, K. Fukuda, M. Osada and T. Sasaki: Electrochem. Commun.9(2007) 1486 1490.
21) K. Takada: Acta Mater.61(2013) 759770.
22) J. Haruyama, K. Sodeyama, L. Han, K. Takada and Y. Tateyama: Chem. Mater.26(2014) 42484255.
23) A. Sakuda, A. Hayashi and M. Tatsumisago: Sci. Rep.3(2013) 2261.
24) M. S. Whittingham: Science192(1976) 11261127.
25) M. S. Whittingham: Prog. Solid State Chem.12(1978) 4199. 26) T. Ohzuku and A. Ueda: J. Electrochem. Soc. 141(1994) 2972
2977.
27) J. Kawaji, S. Suzuki, K. Yoshida, A. Unemoto and S. Orimo: Extended Abstracts of the 56th Battery Symposium in Japan (2015) pp. 463.
28) A. Yamauchi, A. Sakuda, A. Hayashi and M. Tatsumisago: J. Power Sources244(2013) 707710.
29) A. Unemoto, H. Wu, T. J. Udovic, M. Matsuo, T. Ikeshoji and S. Orimo: Chem. Commun.52(2016) 564566.
30) L. He, H.W. Li, H. Nakajima, N. Tumanov, Y. Filinchuk, S.J. Hwang, M. Sharma, H. Hageman and E. Akiba: Chem. Mater. 27(2015) 54835486.
31) J. A. Teprovich Jr., H. ColonMercado, A. L. Washington II, P. A. Ward, S. Greenway, D. M. Missimer, H. Hartman, J. Velten, J. H. Christian and R. Zidan: J. Mater. Chem. A3(2015) 22853 22859.
32) W. S. Tang, A. Unemoto, W. Zhou, V. Stavila, M. Matsuo, H. Wu, S. Orimo and T. J. Udovic: Energ. Environ. Sci.8(2015) 36373645.
33) W. S. Tang, M. Matsuo, H. Wu, V. Stavila, W. Zhou, A. A. Talin, A. V. Soloninin, R. V. Skoryunov, O. A. Babanova, A. V. Skripov, A. Unemoto, S. Orimo and T. J. Udovic: Adv. Energ. Mater. (In press), DOI: 10.1002/aenm.201502237.
34) T. J. Udovic, M. Matsuo, A. Unemoto, N. Verdal, V. Stavila, A. V. Skripov, J. J. Rush, H. Takamura and S. Orimo: Chem. Commun.50(2014) 37503752.
35) T. J. Udovic, M. Matsuo, W. S. Tang, H. Wu, V. Stavila, A. V. Soloninin, R. V. Skryunov, O. A. Babanova, A. V. Skripov, J. J. Rush, A. Unemoto, H. Takamura and S. Orimo: Adv. Mater.26 (2014) 76227626.
36) T. Ikeshoji, E. Tsuchida, K. Ikeda, M. Matsuo, H.W. Li, Y. Kawazoe and S. Orimo: Appl. Phys. Lett.95(2009) 221901. 37) T. Ikeshoji, E. Tsuchida, T. Morishita, K. Ikeda, M. Matsuo, Y.
Kawazoe and S. Orimo: Phys. Rev. B83(2011) 144301. 38) P. Martelli, A. Remhof, A. Borgschulte, R. Ackermann, T.
Str äassle, J. P. Embs, M. Ernst, M. Matsuo, S. Orimo and A. Z äuttel: J. Phys. Chem. A115(2011) 53295334.