熊本大学学位論文
医薬品のベネフィット/リスクを最適化 するための用法用量設定に関する研究
2019
作手 千尋
Dosing regimen selection to optimize benefit-risk balance of drugs
Chihiro Tsukude
Dosing regimen selection to optimize benefit-risk balance of drugs Department of Medical Oncology and Translational Research, Graduate School of
Pharmaceutical Sicences, Kumamoto University, Chihiro Tsukude The balance of efficacy and safety in drug development is an important factor to estimate the benefit and risk of new drugs. This balance is investigated throughout the drug development lifecycle,leading to the beneficial dosing regimens for disease treatments. In the present study, I focused on pharmacokinetics (PK) which generally have large effects on efficacy and safety, and investigated the factors which affected PK.
Firstly, in order to avoid the efficacy reduction by expression level change of drug metabolism enzymes, a new risk assessment method and an application of other disease information to a rare disease at early development stages were investigated. Then, aiming to improve safety profile of a drug, I analyzed genetic polymorphisms in a post marketing clinical study and explored possible dose adjustment.
(1) Risk assessment of CYP2C9 induction by a relative factor (RF) approach By using the RF method—a method that can simply assess CYP induction risk based on a maximum induction effect model—we evaluated the risk of CYP2C9 induction and examined its relationship with risk of CYP3A4 induction. In cryopreserved human hepatocytes, the magnitude of CYP2C9 induction by eight drugs known to induce CYP3A4 was lower than the magnitude of CYP3A4 induction, but the magnitudes of induction of both were correlated. The RF values determined for CYP2C9 had a one-to- one linear relationship with values determined for CYP3A4, supporting reports that the induction mechanism of both enzymes is the same. Furthermore, clinical CYP2C9 induction data of compounds reported to induce CYP2C9 clinically were shown to be lower than those of CYP3A4. The thresholds for CYP2C9 induction risk assessment by the RF approach were determined to be at higher steady-state plasma concentrations than those for CYP3A4. Based on these results, induction of CYP2C9 was correlated with that of CYP3A4, and induction risk could be evaluated by RF method using hepatocytes. The CYP2C9 induction risk of a compound was confirmed to be lower than its CYP3A4 induction risk.
(2) Simulating the impact of interleukin (IL)-6 levels on the PK of CYP substrates in patients with neuromyelitis optica (NMO) or NMO spectrum disorders (NMOSD) by a physiologically based Pharmacokinetic (PBPK) model
A PBPK model was used to simulate the impact of elevated levels of IL-6 on the exposure of several orally administered CYP probe substrates (caffeine, S-warfarin,
were predicted with a reasonable degree of accuracy.The PBPK model was then used to simulate the change in oral exposure of the probe substrates in patients with NMO or NMOSD with elevated plasma IL-6 levels. Moderate interactions [mean AUC fold change, ≤ 2.08 (midazolam) or 2.36 (simvastatin)] was predicted for CYP3A4 probe substrates and weak interactions (mean AUC fold change, ≤ 1.29–1.97) were predicted for CYP2C19, CYP2C9, and CYP2D6 substrates. No notable interaction was predicted with CYP1A2. Decreased levels of serum albumin (as reported in NMO patients) had little impact on the magnitude of the simulated IL-6–mediated drug interactions. This PBPK modelling approach allowed us to leverage knowledge from different disease and ethnic populations to make predictions of cytokine-related DDIs in a rare disease population where actual clinical studies would otherwise be difficult to conduct.
(3) Population PK and adverse events (AE) of erlotinib: Impact of genetic polymorphisms in metabolizing enzymes and transporters
Determinants of interindividual variability in erlotinib PK and AEs remain to be elucidated. This study with 50 Japanese non–small–cell lung cancer patients treated with oral erlotinib at a standard dose of 150 mg aimed to investigate whether genetic polymorphisms affect erlotinib PK and AE. Single nucleotide polymorphisms (SNPs) in genes encoding metabolizing enzymes or efflux transporters were analyzed as covariates in a population PK model. The ABCB1 1236C>T polymorphism, not ABCB1*2 haplotype (1236TT–2677TT–3455TT), was a significant covariate for the apparent clearance (CL/F), with the TT genotype showing a 29.4% decrease in CL/F as compared with the CC and the CT genotypes. A marginally higher incidence of adverse events (mainly skin rash) was observed in the TT genotype group; however, patients with high plasma erlotinib exposure did not always experience skin rash. None of the other SNPs affected PK or adverse events. The ABCB1 genotype is a potential predictor for erlotinib adverse events.
In the present study, a risk assessment method for enzyme induction atpreclinical stage and an application method of disease population data to a rare disease at clinical development stage were established. These methods give information about the dose selection when metabolism enzyme levels change in new drug developments. Revealing unknown patient characteristics which affect PK or safety at post marketing stage gives information about personalized dose setting/ AE prediction. Leveraging knowledge in each drug development and post marketing stage, and examining those by mathematical method such as RF method, PBPK or PPK, will lead efficacious and safe dosing regimen, improve drug development efficiency, and promote proper use of drugs after approval.
医薬品のベネフィット/リスクを最適化するための用法用量設定に関する研究
創薬・生命薬科学専攻 ドラッグデリバリーコース 腫瘍治療・トランスレーショナルリサーチ学分野 作手 千尋
新効能医薬品の開発において有効性と安全性のバランスは、市販後における ベネフィットとリスクを予測する上での重要な要因である。このバランスは医 薬品の研究開発段階から市販後段階の長期に渡り検討され、疾患の治療に有益 な用法・用量が情報提供されている。本研究では特に、医薬品の有効性・安全 性に大きな影響を及ぼす薬物動態(PK)に着目し、PK が変動する要因につい て検討した。まず、有効性の減弱につながる代謝酵素の発現量変化に関する薬 物相互作用について、研究開発早期段階でのリスク評価方法、他疾患のデータ を希少疾患へと応用する方法を検討した。次に、安全性に影響を与える因子と して市販後臨床試験にて遺伝子多型を解析し、用量調節の必要性を検討した。
以下に本研究で得られた知見を纏める。
(1) Relative Factor(RF)法を用いたCYP2C9誘導のリスク評価
Emaxモデルに基づき簡便に誘導リスクを評価することができる RF 法を用い、
CYP2C9 の誘導のリスクと CYP3A4 誘導リスクの関係を検討した。凍結ヒト 肝細胞における、CYP3A4誘導剤 8 種による CYP2C9の誘導倍率は CYP3A4 の誘導倍率よりも低く、両者に正の相関がみられた。RF 値は、CYP2C9 と CYP3A4 で 1:1 の正の相関を示し、両酵素の誘導メカニズムが同一であるとの 報告を裏付けた。更に、臨床でCYP2C9誘導が報告されている化合物の誘導比 はCYP3A4の誘導比より低値を示した。RF法によるCYP2C9のリスク判定カ ットオフ値は、CYP2C9 にて CYP3A4 よりも高い定常状態血中濃度にて設定 された。以上より、CYP2C9の誘導は CYP3A4と相関し、ヒト肝細胞から RF 法を用いて CYP2C9 の誘導リスク評価可能であることが示された。CYP2C9 の誘導リスクはCYP3A4の誘導リスクより低いことが確認された。
(2) 視神経脊髄炎(NMO)又は視神経脊髄炎関連疾患(NMOSD)患者に おける血中インターロイキン(IL)-6 濃度が各種 CYP 分子種基質の PK に与 える影響の生理学的薬物速度論(PBPK)モデルによる予測
PBPK モデルを用い、IL-6 濃度の上昇が各種 CYP 活性指標薬(caffeine、 s-warfarin 、 omeprazole 、 dextromethorphan 、 midazolam 及 び simvastatin)の曝露量に与える影響を予測した。これら指標薬の関節リウマ
NMOSD 患者における IL-6 濃度がこれら指標薬の曝露量に与える影響を予測 した。CYP3A4指標薬は中程度の相互作用[平均AUCは2.08倍(midazolam)
又は 2.36 倍(simvastatin)以下]が、CYP2C19、CYP2C9、 CYP2D6 指標 薬は弱い相互作用(平均 AUC は 1.29-1.97 倍)が、CYP1A2 指標薬では相互 作用が無いと予測された。民俗差及び NMO 患者の血清中アルブミン濃度変動 による相互作用への影響は認められなかった。異なる疾患や民族で得られた知 識を活用しサイトカインによる相互作用を予測する本方法は、臨床試験を行う ことが困難な希少疾患での相互作用を検討できる点で有用である。
(3) エルロチニブの母集団薬物動態(PPK)及び有害事象(AE)の解析 遺伝子多型がエルロチニブの PK 及び AE 発現に与える影響を検討するため に、日本人非小細胞肺がん患者を対象に承認用量である一日一回 150mg を投 与する医師主導試験のデータをレトロスペクティブに解析した。遺伝子情報は、
エルロチニブの PK に関わることが報告されている、又は AE 発現に関与する 可能性のある代謝酵素(CYP1A1、CYP1A2、CYP2D6、CYP3A4、CYP3A5、 UGT1A1、UGT2B7、GSTM1 及び GSTT1)並びに排泄型トランスポーター
(ABCB1及び ABCG2)の一塩基多型(SNPs)を用いた。PPK解析を実施し 遺伝子多型が PK に及ぼす影響を探索し、統計学的な検討により遺伝子多型と AE との関連性を検討した。ABCB1 の 1236C>T(rs1128503)変異が経口ク リアランス(CL/F)の有意な共変量であり、TT 遺伝子型を有する患者では有 さない患者と比較してCL/Fが29.4%低く推定された。また、TT遺伝子型にお いて主に皮疹の AE の発現頻度が高い傾向が示されたが、必ずしも血漿中エル ロチニブ濃度が高い全ての患者で皮疹が発現しているわけではなかった。他の SNPsは、PK や AE発現に影響を与えなかった。ABCB1 遺伝子型はエルロチ ニブの PK に影響を及ぼすとともに AE の予測因子となりうる可能性が示され た。
本研究で行った早期研究段階における酵素誘導評価法の確立及び治験段階に おいて他疾患のデータを希少疾患へと応用する方法の確立は、新有効医薬品の 開発において、代謝酵素の酵素発現量の変動を考慮した用量の設定に関する情 報を提供する。市販後において、治験で未確認の要因と PK 及び AE 発現との 関係を明らかにしたことは、患者ごとのより詳細な用量調整や、AE 発生に関 する情報を提供する。早期研究、治験、市販後の各段階において、得られる情 報を有効活用しRF法、PBPK解析、PPK解析などの数学的手法を用いて検討 を行った本研究は、安全でかつ有効性の高い用法・用量設定へと貢献し、予測 による研究開発の効率化や市販後の適正使用に繋がることを期待する。
目次
第1章 緒言 ... 1
第2章 Relative Factor(RF)法を用いたCYP2C9誘導のリスク評価 ... 4
第1節 序説 ... 4
第2節 結果 ... 6
第 1項 ヒト凍結肝細胞を用いた in vitro における CYP2C9及び CYP3A4の誘導 . 6 第 2 項 RF 法による CYP2C9 及び CYP3A4 の臨床における誘導リスクの評価 ... 10
第 3項 臨床において CYP2C9 誘導が報告されている薬物による、臨床における CYP3A4の誘導 ... 16
第3節 考察 ... 19
第4節 小括 ... 22
第3章 視神経脊髄炎(NMO)又は視神経脊髄炎関連疾患(NMOSD)患者における 血中インターロイキン(IL)-6濃度が各種 CYP分子種基質の PKに与える影 響の生理学的薬物速度論(PBPK)モデルによる予測 ... 24
第1節 序説 ... 24
第2節 結果 ... 26
第1項 NMO又はNMOSDの患者背景を反映した母集団の作成 ... 26
第2項 IL-6のモデル開発及び妥当性確認 ... 28
第3項 仮想NMO患者でのIL-6を介した各 CYP分子種の発現量低下の影響の予 測 ... 40
第4項 IL-6を介したDDIに対する、血中アルブミン濃度の影響の検討 ... 52
第3節 考察 ... 54
第4節 小括 ... 56
第4章 エルロチニブの母集団薬物動態(PPK)及び有害事象(AE)の解析 ... 59
第1節 序説 ... 59
第2節 結果 ... 60
第1項 患者背景及び選択した遺伝子の遺伝子型頻度 ... 60
第2項 PPKモデル ... 65
第3項 有効性と曝露量、EGFR遺伝子型又は患者背景の関係 ... 67
第4項 安全性と曝露量、患者背景の関係 ... 72
第5項 SNPs と AE の発現の関係 ... 73
第6項 安全性と有効性の関係 ... 76
第7項 ABCB1遺伝子型ごとの最適な用法用量の検討 ... 77 第 3 節 考察 ... 78
第 4節 小括 ... 82 第 5 章 総括 ... 84
実験及びデータ解析の部 ... 88 謝辞 ... 104 参考文献 ... 105
略語及び用語の定義
略語 英名 和名/定義
17-OHCS 17-hydroxycorticosteroid 17-ヒドロキシコルチコステ
ロイド
6-OHC 6β-hydroxycortisol 6β-ヒドロキシコルチゾール
6-OHCS 6β-hydroxycorticosteroid 6β-ヒドロキシコルチコステ
ロイド
ABC ATP-binding cassette —
AP Point above 1.25 臨床誘導比が1.25を越え、
最も低いRF × Css,uを示す点 AUC Area under the concentration-time curve 血漿中濃度-時間曲線下面積
AUC0-t AUC from time 0 to time t 時間0からtまでのAUC
BP Point below 1.25
臨床誘導比が1.25を下回 り、最も高いRF × Css,uを示
す点
CL/F 経口クリアランス
CLint Intrinsic clearance 固有クリアランス
Cmax Maximum concentration 最大血漿中濃度
CRB Carbamazepine カルバマゼピン
Css,u
Average steady-state unbound plasma concentration
定常状態平均血漿中非結合型 濃度
CTD Common Technical Document コモン・テクニカル・ドキュ
メント
DDI Drug-drug interaction 薬物相互作用
DEX Dexamethazone デキサメタゾン
DLT Dose limiting toxicity 用量制限毒性
EC50 Half-maximal effective concentration 最大の半分の効果を示す濃度
EGFR Epidermal growth factor receptor 上皮増殖因子受容体
EM Extensive metaboliser 通常の代謝活性を有する被験
者
EMA European Medicines Agency 欧州医薬品庁
Emax Maximum induction effect 最大誘導効果
FDA U.S. Food and Drug Administration 米国食品医薬品局
fp Plasma unbound fraction 血漿中非結合型分率
GST Glutathione-S-transferase グルタチオン S-転写酵素
ICH
International Conference on Harmonization of Technical Requirements
for Registration of Pharmaceuticals for Human Use
医薬品規制調和国際会議
IDL Induction detection limit 誘導検出下限値
略語 英名 和名/定義
IL-6 Interleukin-6 インターロイキン-6
IV Intravenous 静脈内投与
MHLW Ministry of Health, Labour and Welfare 厚生労働省
NC Not calculated 計算せず
ND Not determined 算出できず
NI No information 情報なし
NIF Nifedipine ニフェジピン
NMO Neuromyelitis optica 視神経脊髄炎
NMOSD Neuromyelitis optica spectrum disorders 視神経脊髄炎関連疾患
NSCLC Non-small-cell Lung Cancer 非小細胞肺がん
OMP Omeprazole オメプラゾール
OS Overall Survival 全生存期間
PB Phenobarbital フェノバルビタール
PBPK Physiologically based pharmacokinetics 生理学的薬物速度論
PD-L1 Programmed cell Death ligand 1 —
PFS Progression-free survival 無増悪生存期間
PHE Phenytoin フェニトイン
PhRMA The Pharmaceutical Research and
Manufacturers of America 米国研究製薬工業協会
PK Pharmacokinetics 薬物動態
PM Poor metaboliser 代謝活性が欠損又は著しく低
い被験者 PMDA Pharmaceuticals and Medical Devices
Agency 医薬品医療機器総合機構
PPK Population Pharmacokineitcs 母集団薬物動態
PS Performance status パフォーマンスステータス
Rb Blood-to-plasma concentration ratio 血液血漿濃度比
RF Relative Factor —
RIF Rifampicin リファンピシン
SD Standard deviation 標準偏差
SNPs Single nucleotide polymorphisms 一塩基多型
SUL Sulfinpyrazone スルフィンピラゾン
TDM Terapeutic drug monitoring 治療薬物モニタリング
UDP Uridine 5’-diphosphate —
UGT UDP glucuronosyltransferase UDPグルクロン酸転移酵素
UWDIDB University of Washington Drug
Interaction Database —
Vd Distribution volume 分布容積
本論文は、学術雑誌に掲載された次の論文を基礎とするものである。
(1) Risk of CYP2C9 induction analyzed by a relative factor approach with human hepatocytes
Drug Metab Pharmacokinet., In press
Chihiro Endo-Tsukude, Motohiro Kato, Akihisa Kaneko, Satofumi Iida, Shino Kuramoto, Masaki Ishigai, Akinobu Hamada
(2) Simulating the Impact of Elevated Levels of Interleukin-6 on the Pharmacokinetics of Various CYP450 Substrates in Patients with Neuromyelitis Optica or Neuromyelitis Optica Spectrum Disorders in Different Ethnic Populations
The AAPS Journal, 21, 42 (2019).
Krishna K Machavaram, Chihiro Endo-Tsukude, Kimio Terao, Katherine L Gill1, Oliver J Hatley, Iain Gardner, Neil Parrott, Patricia Sanwald Ducray
(3) Population Pharmacokinetics and Adverse Events of Erlotinib in Japanese Patients with Non-small-cell Lung Cancer: Impact of Genetic Polymorphisms in Metabolizing Enzymes and Transporters
Biol. Pharm. Bull., 41, 47-56 (2018).
Chihiro Endo-Tsukude, Ji-ichiro Sasaki, Sho Saeki, Norihiro Iwamoto, Megumi Inaba, Sunao Ushijima, Hiroto Kishi, Shinji Fujii, Hiroshi Semba, Kosuke Kashiwabara, Yukari Tsubata, Mitsuhiro Hayashi, Yuki Kai, Hideyuki Saito, Takeshi Isobe, Hirotsugu Kohrogi, Akinobu Hamada
第1章 緒言
新効能医薬品の開発において有効性と安全性のバランスは、市販後におけるベネフ ィットとリスクを予測する上での重要な要因である。これらのバランスは医薬品の研 究開発段階から市販後段階の長期に渡り検討され、疾患の治療に有益な用法用量が情 報提供されている。ベネフィット/リスク評価はとりわけ新しい考え方ではないが、
近年その評価プロセスについて様々な提案がなされてきた。2010 年に欧州医薬品庁
(EMA)は Benefit-Risk Methodology プロジェクトの報告書 1)を作成した。米国では 2011 年~2013 年に、米国研究製薬工業協会(PhRMA)と米国食品医薬品局(FDA)が、
評価フレームワークを提案した 2)3)。これらを受けて、医薬品規制調和国際会議(ICH) M4E(R2)ガイドラインが 2016 年にステップ 4 の合意となった 4)。本邦においても 2017年に[薬生薬審発0202第1号「新医薬品の製造販売承認申請に際し申請書に添付 すべき資料の作成要領について」の一部改正について]の厚生労働省医薬・生活衛生 局医薬品審査管理課長通知が発出され、コモン・テクニカル・ドキュメント(CTD) のセクション 2.5.6「ベネフィットとリスクに関する結論」の記載方法が全面的に見直 された。
新効能医薬品の承認申請時には明確にベネフィットがリスクを上回ることが必要と なるが、新効能医薬品の開発における状況は厳しさを増している。医薬品の研究開発 の効率は年々低下しており、1950年時点では 10億ドルの投資あたり 30程度の医薬品 がFDAに承認されていたが、2010年には1つも承認されていない 5)。これらの原因と して、新規技術の登場による開発コストの増加と、失敗コストの増加つまり失敗確率 の上昇が考えられる。製薬企業にとって後期開発段階での開発中止は、それまでかけ てきたコストが多額となるために大きな痛手である。2013 年から 2015 年までの第 II 相臨床試験又は第III相臨床試験の失敗の原因を集計した報告 6)によると、第II相試験 の失敗要因のうち約 48%が不十分な有効性によるものであり、約 25%が安全性上の理 由によるものであった。第 III 相試験の失敗要因も同様であり、約 55%が不十分な有 効性によるものであり、約 14%が安全性上の理由によるものであった。また、動物や
in vitro 実験系を用いた非臨床試験では有効性及び安全性が認められているものの、臨
床試験では想定される有効性や安全性が認められないケースも多いと考えられている。
これは新薬開発の死の谷とも呼ばれているもので、外部に情報が公開されずに失敗し
ている場合もあり、実際はどれほどの候補化合物が開発中止に至っているかさえ把握 しきれていない。このように非臨床研究段階から臨床開発段階における有効性又は安 全性の欠如による失敗は、新効能医薬品開発のコスト増加の大きな要因である。従っ て、開発候補化合物の有効性及び安全性を可能な限り早期の段階で適切に見極めるこ とは、製薬企業の経営の健全性を保つために必須である。
開発候補化合物の失敗確率を低減する方策はいくつか考えられる。薬剤のコンセプ トとして、対象疾患に関連しない臓器・組織には作用が及ばないような作用機序を採 用することにより、安全性のリスク低減と有効性の増加が同時に見込まれる。また、
バイオマーカーによる患者選別によって、有効性が見込まれる患者だけに投与するこ とによって、高い効果を得られたり、従来と比較して有効性が高く安全な用法用量を 見い出すことによって失敗を避けることができたりする。以下にその背景の詳細につ いて記載する。
創薬の着想段階において開発候補化合物の有効性・安全性を高めるための方策とし て、個別化医療が注目されて久しい。特にがん領域では2002年の慢性骨髄性白血病治 療薬のイマチニブの発売を皮切りに近年分子標的薬の開発が進んできた。これらは従 来の殺細胞作用を持つ薬物と異なり、がんのドライバーとなる遺伝子に変異が入るこ とでがん化した細胞のみに作用することで、高い有効性と安全性を達成できることが 特徴である。これらは標的遺伝子変異をもつがんのみに効果を発揮するために、その 遺伝子変異の有無を確認する診断薬も同時に開発され、バイオマーカーによる患者選 別を行い、投与する患者を絞ることで明確にベネフィットがリスクを上回ることが可 能となった。
臨床開発の段階において対象患者を絞らずに有効性・安全性を高めるための方策と しては、用法用量の最適化が挙げられる。FDA は 2009年に white paper を発出し、よ り効率的な用法用量の最適化のために第 II 相試験終了後に製薬企業と相談を実施し、
モデリング&シミュレーションを用いた議論を行うと発表した 7)。本邦においても医 薬品医療機器総合機構(PMDA)が医薬品の審査にモデリング&シミュレーションを
8)
薬効の発揮、そして臨床アウトカムへと順次計算式を作成して推定することが可能と なる。また、これらの各段階において、個体内及び個体間変動の要因を明らかにする ことで、患者個人に応じた用法用量設定が可能となる場合もある。
以上のような背景を受け、医薬品の用法用量設定の効率化へ貢献することを目的と し、本研究では特に有効性・安全性に大きな影響を及ぼす薬物動態(PK)に着目し、
これらが変動する要因について評価し、医薬品開発の早期から後期までのそれぞれの 段階において新たな用法用量の設定方法を検討した。まず、研究開発早期段階で、有 効性の減弱につながる代謝酵素の発現量変化に関する薬物相互作用について、in vitro 試験及び文献調査を用いた効率的なリスク評価方法を検討した。次に、臨床開発段階 において、モデリング&シミュレーションを用いることで、治験を行わずに他疾患の データを希少疾患へと応用する方法を検討した。最後に、市販後段階において、承認 用量と比較してより適切な個別化医療(患者ごとに最適な投与量)を探索すべく、抗 がん剤の分子標的薬について、安全性に影響を与える因子として遺伝子多型を解析し、
用量調節の必要性を検討した。加えて、これら各医薬品開発の段階における用法用量 の評価方法について、その有用性を論じた。
第2章 Relative Factor(RF)法を用いた CYP2C9誘導のリスク 評価
第1節 序説
近年、FDA、EMA及び厚生労働省(MHLW)は薬物相互作用(DDI)の検討に関す るガイダンス/ガイドラインの修正版を公表した。これらは、in vitro試験実施の一般的 なフレームワークと試験結果の解釈、必要に応じた臨床試験の実施について提言して いる。FDA の draft guidance によると、CYP2C と CYP3A4/5 は同じ核内受容体、PXR の活性化により共に誘導されることから、化合物が CYP3A4 の誘導を有する場合には、
CYP2Cのリスク評価も行うことが推奨されている 9)。EMAの draft guidelineにおいて も、PXR/CARの活性化がin vitroで確認された際はin vivoにおいてCYP3Aの誘導を確 認すべきであり、CYP3Aを阻害する化合物については CYP2C9 や CYP2C19の誘導を 検討すべきだと記載されている10)。これらの記載は主に CYP3Aと CYP2Cが同じ誘導 メカニズムを有するために、CYP3A の誘導作用を持たない化合物は CYP2C の誘導作 用も持たないという考えに基づいている。
CYP2C 分子種のうち、CYP2C9 は、ヒト肝臓中のミクロソーム P450 含量のうち上
位3つに入る分子種であり、白人成人男性の肝臓におけるP450発現量の約16%を占め
ている 11)。CYP2C9 は第一相代謝を受ける薬物のうち 15%近くの薬物の代謝を行うと
推測されている。CYP2C9 を介した DDI は、CYP2C9 の基質である薬物の血漿中濃度 に影響を与え、特にワルファリンのような治療域の狭い薬物については、治療管理上 の問題となり得る12)。
これまで、CYP2C9と CYP3A4 の誘導の比較についていくつかの報告がされている。
Hariparsad 他は、ヒト肝細胞を誘導剤と共に培養した後の mRNA 発現量及び酵素活性
における誘導倍率が CYP2C9 と CYP3A4 で相関し、CYP2C9 の誘導反応は低いと報告 した13)。彼らは更に、臨床において CYP2C9の誘導作用により DDIを起こすと報告さ れている化合物は、CYP3A4の誘導作用によるDDIも起こすと報告した。また、Nagai 他は、凍結ヒト肝細胞の mRNA 発現量における CYP2C9 の誘導倍率と CYP3A4 及び
CYP2B6 の誘導倍率との関係を検討し、CYP2C9 の誘導倍率を CYP3A4 の誘導倍率と
も、臨床における CYP2C9と CYP3A4の誘導倍率の関係は 5化合物で検討されている のみであり,網羅的に検討はされていない。
近年我々は、臨床における CYP3A4 誘導リスクを評価する方法として Relative Factor(RF)法を開発し、その CYP3A4 誘導リスク評価における有用性を示した 15, 16)。
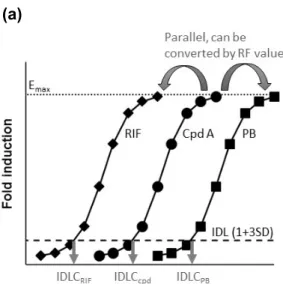
RF法は、in vitro酵素誘導試験における対数軸で示した誘導剤濃度-誘導倍率曲線の形
状がいずれの化合物においても同様であり、各化合物の最大の半分の効果を示す濃度
(EC50)値に応じて並行移動していると仮定した方法である(図 2.1)。RF法では、in
vitro 誘導試験より各化合物の有意な誘導を示す最低濃度(induction detection limit
concentration, IDLC)を算出する。標準誘導薬として規定した rifampicin(RIF)又は phenobarbital(PB)の IDLCも算出し、標準誘導薬の IDLCを各化合物の IDLCで除す る RF 値を算出する。各化合物と標準誘導薬の誘導曲線の初期の傾きを比較する値で ある RF 値を用いることにより、各化合物の臨床における血漿中非結合型濃度-誘導 比曲線を標準誘導薬の誘導比曲線へと変換できる。そのため、化合物の毒性や溶解度 の制約から誘導反応の EC50が算出できない場合においても、各化合物の IDLC 値を求 め、その臨床用量における定常状態の血漿中非結合型濃度で補正することにより、臨 床用量における誘導リスクの評価が可能となる。
(a) (b)
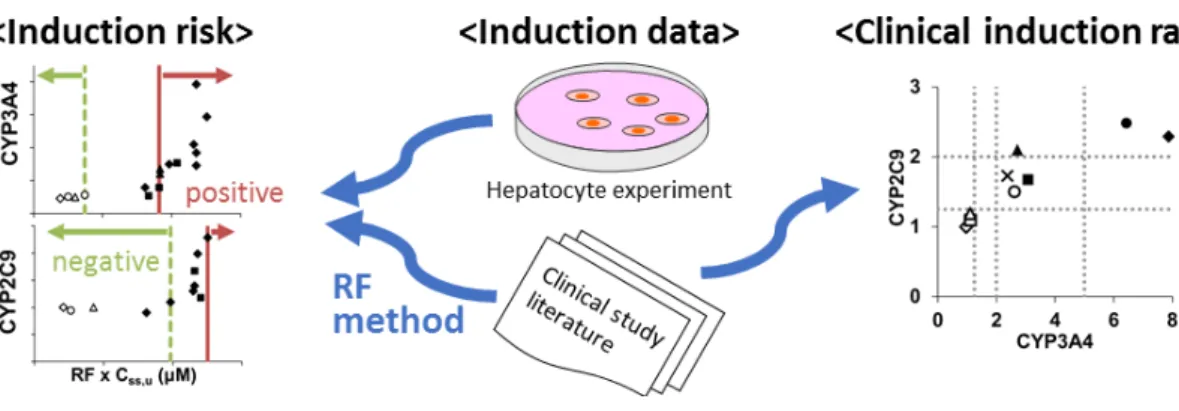
図2.1 Relative Factor(RF)法の概念
(a)in vitroの誘導における対数軸で示した濃度-反応曲線。曲線の形は化合物によらず同じ
と仮定する。標準誘導薬として rifampicin(RIF)及び phenobarbital(PB)を使用する。(b)
in vitro の誘導における濃度-反応曲線。Icpd, 化合物 A の P点における誘導倍率;P,IDL
よりも高い誘導反応を示す最低濃度における観測値;IDL(induction detection limit),溶媒 コントロールによる誘導の程度の平均値を 3 標準偏差分上回る値を誘導検出下限値とす る;IDLC(induction detection limit concentration),誘導検出下限値を上回る誘導を示す最 低濃度;Emax, 最大誘導効果。
そこで本研究では、ヒト凍結肝細胞を用い、CYP3A4 を誘導すると知られている 8 化合物についてCYP2C9及びCYP3A4の酵素誘導リスクを、酵素活性を指標としてRF 法により評価した。更に、臨床における 2C9 と 3A4 の誘導の大小関係を、臨床で
CYP2C9 誘導が報告されている化合物について調査した。本研究の目的は、化合物に
よるCYP2C9とCYP3A4両酵素の臨床における誘導リスクを比較することである。
第2節 結果
第1項 ヒト凍結肝細胞を用いた in vitro における CYP2C9及び CYP3A4の誘導 各化合物によるCYP2C9及びCYP3A4の誘導濃度依存性及び最大誘導倍率は3ロッ トで同様であり、最大誘導倍率はCYP2C9がCYP3A4より低値を示した(図2.2)。
(
(
(
図2.
誘導試 におい (b)、
(CR
(SU (a)
(c)
(e)
2 CYP2C 試験は各ドナ いて n = 3で (d)及び(f)は RB)、●は UL)、△はn
C9及びCY ナー[ドナー で実施し、そ は CYP2C9活
は phenytoin nifedipine(N
P3A4の濃度 ー1は(a)及び その平均値を 活性の誘導倍
(PHE)、
NIF)、○は
(b
(d
(f
度-反応曲線 び(b)、ドナー を示す。(a)、
倍率である。
◇は dexam はomeprazole
b)
d)
f)
線
ー2 は(c)及び (c)及び(e)は
◆は RIF、■
methasone(D e(OMP)を
び(d)、ドナー はCYP3A4活
■は PB、▲
DEX) 、□
をそれぞれ表
ー3 は(e)及び 活性の誘導倍
▲は carbamaz は sulfinpyr す。
び(f)]
倍率を、
zepine razone
RIF、PB、CRB及びDEXの 4化合物では、全肝細胞ドナーにおいて両酵素の濃度依 存的な誘導が認められた。NIF及びOMPは CYP2C9については全ドナーで濃度依存的 な誘導が認められたが、CYP3A4についてはNIFが全ドナーで、OMPは 2ドナーで明 確な濃度依存性が認められなかった。一方、PHE及び SULは、CYP3A4については全 ドナーで濃度依存的な誘導が認められたが、CYP2C9についてはPHE及びSULで1ド ナーずつにおいて誘導の明確な濃度依存性が認めらなかった。
CYP2C9 及び CYP3A4 の誘導倍率の相関は、全化合物を用いた際の相関係数 r は
0.377–0.607であるが、他の化合物と異なる傾向を示した NIF及び OMPを除いた 6化
合物を用いた際のrは0.686–0.882であり有意な相関を示した(図2.3)。
(a)
(b)
(c)
図2.3 各ドナーにおけるCYP2C9誘導倍率とCYP3A4誘導倍率の相関
(a)、(b)及び(c)はそれぞれドナー1、ドナー2及びドナー3の結果を示し、各データは n = 3 の平均値を表す。全 8化合物について並びに NIF及びOMPを除いた 6化合物について、
ピアソンの相関係数rを算出し相関を確認した。◆はRIF、■はPB、▲はCRB、●はPHE、
◇はDEX、□はSUL、△はNIF、○はOMPをそれぞれ表す。
0 1 2 3 4 5 6
0 2 4 6 8 10
2C9 fold induction
3A4 fold induction
0 1 2 3 4 5 6
0 2 4 6 8 10
2C9 fold induction
3A4 fold induction
0 1 2 3 4 5 6
0 2 4 6 8 10
2C9 fold induction
3A4 fold induction
All 8 compounds (r = 0.453) 6 compounds (r = 0.686)
All 8 compounds (r = 0.377) 6 compounds (r = 0.716) All 8 compounds (r = 0.607) 6 compounds (r = 0.882)
第2
In ドナ ー1が めら IDLC ー1及
各 RFRIF
れぞ おり (
図2.
(a) R CYP2
◇は
In 合 物
2項 RF法に
vitro試験か ー1が 1.59、 が 1.49、ドナ れなかった Cの算出が可 及びドナー3
各化合物の I
F値とRFPB値
れ比較した
、RFPB値に (a)
4 CYP2C RFRIF値;(b) 2C9のRF値
DEX、□は
vitro試験に 物 併 用 時 の
によるCYP2
から算出した
、ドナー2が ナー2が 1.6 た場合でも、
可能であり、
3を除き算出 IDLC 値を用 値をそれぞれ た(図 2.4)。
についても同
C9のRF値 ) RFPB値。各 値=CYP3A4
SUL、△は
に用いた8化 の 酵 素 活 性
2C9及びC た IDL値は が 1.55、ド 60、ドナー3
いずれかの
、IDLCは、
出できた。
用い、RIF れ算出し、
。CYP2C9及 同様であった
値とCYP3A4 各点は 3 ド の RF値、
はNIF、○は
化合物の、C 性 / 基 質 単
CYP3A4の臨
は肝細胞ドナ ナー3が 1.8 3が 2.17で の濃度におい
CYP3A4の
と PB を標 CYP2C9及び 及び CYP3A た。
(b
4のRF値の ドナーの幾何
を示す。◆は OMPをそれ
CYP2C9及び 単 剤 投 与 時
臨床における
ナーごとに異
86であり、
であった。誘 いて IDL 値 の NIFのドナ
標準誘導剤と び CYP3A4 A4の RFRIF
b)
の相関 何平均値 ± 幾
はRIF、■は れぞれ表す。
び CYP3A4 時 の 酵 素 活
る誘導リス 異なり、CYP
CYP3A4に 誘導の明確な 値を超える誘 ナー3、並び
として各化 の RFRIF値及
F値は、ほぼ
幾何変動係数 は PB、▲は
の臨床にお 活 性 ) を 表
クの評価 P2C9につい については、
な濃度依存性 誘導を示した びに OMPの
合物の両酵 及びRFPB値 ぼ 1:1に対応
数を示す。点 CRB、●は
おける誘導比 表 2.1 に 示
いては、
ドナ 性が認 た結果 のドナ
酵素の 値をそ 応して
点線は はPHE、
比(化 す 。
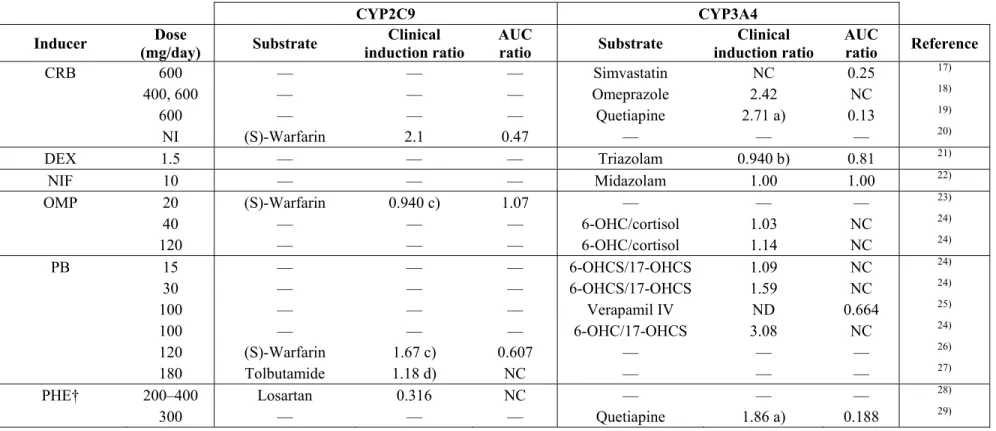
表2.1 In vitro試験に用いた化合物による、臨床におけるCYP2C9及びCYP3A4の誘導
CYP2C9 CYP3A4 Inducer Dose
(mg/day) Substrate Clinical induction ratio
AUC
ratio Substrate Clinical induction ratio
AUC
ratio Reference CRB 600 — — — Simvastatin NC 0.25 17)
400, 600 — — — Omeprazole 2.42 NC 18) 600 — — — Quetiapine 2.71 a) 0.13 19) NI (S)-Warfarin 2.1 0.47 — — — 20) DEX 1.5 — — — Triazolam 0.940 b) 0.81 21)
NIF 10 — — — Midazolam 1.00 1.00 22) OMP 20 (S)-Warfarin 0.940 c) 1.07 — — — 23)
40 — — — 6-OHC/cortisol 1.03 NC 24) 120 — — — 6-OHC/cortisol 1.14 NC 24) PB 15 — — — 6-OHCS/17-OHCS 1.09 NC 24)
30 — — — 6-OHCS/17-OHCS 1.59 NC 24) 100 — — — Verapamil IV ND 0.664 25) 100 — — — 6-OHC/17-OHCS 3.08 NC 24) 120 (S)-Warfarin 1.67 c) 0.607 — — — 26) 180 Tolbutamide 1.18 d) NC — — — 27) PHE† 200–400 Losartan 0.316 NC — — — 28)
300 — — — Quetiapine 1.86 a) 0.188 29)
CYP2C9 CYP3A4 Inducer Dose
(mg/day) Substrate Clinical induction ratio
AUC
ratio Substrate Clinical induction ratio
AUC
ratio Reference RIF 20 Losartan 0.9 NC Quinine 1.6 NC 30)
100 Losartan 1.1 NC Quinine 3.0 NC 30) 450 Losartan 1.30 NC Midazolam ND 0.16 31) 500 Losartan 1.4 NC Quinine 4.2 NC 30) 600 (S)-Warfarin 1.99 0.256 — — — 32) 600 — — — Midazolam 7.87 0.041 33) 600 — — — Triazolam 2.93 0.051 34) 600 — — — 6-OHC/cortisol 3.71 NC 24) 1200 — — — 6-OHC/cortisol 5.90 NC 24) 1200 Tolbutamide IV 2.29 0.45 — — — 35) SUL† 400 (S)-Warfarin 0.502 1.70 — — — 36)
800 Tolbutamide IV 0.60 1.7 — — — 37) 800 — — — Verapamil IV ND 0.875 38) 800 — — — Verapamil ND 0.310 38) AUC, 血中濃度-時間曲線下面積;6-OHC/cortisol, 6β-hydroxycortisol/cortisol比;6-OHCS/17-OHCS, 6β-hydroxycorticosteroid/17-
hydoxycorticosteroid比;IV, 静脈内投与;NC, 計算せず;NI,情報なし;ND,血流律速のため算出できず。
言及のない限り基質は経口投与された。
†,PHE及びSULはCYP2C9の阻害と誘導を同時に起こすが、反復投与後の結果としてはCYP2C9の阻害であった。
a) 血液血漿濃度比(Rb)は39)より入手した。
b) 分布容積(Vd)は40)より、Rb及びfpは41)より入手した。
c) Rbは41)より入手した。
d) 血漿中非結合型分率(fp)は41)より入手した。
臨床における誘導比、その際の誘導剤の Css,u、及び算出した RFRIF値若しくは RFPB
値を用い、両酵素における誘導比と RFRIF× Css,u又は RFPB× Css,uの関係を検討した(図 2.5)。酵素誘導陽性又は酵素誘導陰性を決定する RFRIF× Css,u又は RFPB× Css,uのカット オフ値は、CYP2C9 のカットオフ値が CYP3A4 のカットオフ値より常に高値を示して いた。また、CYP2C9の最大誘導比はCYP3A4の最大誘導比の1/3以下であった。なお、
in vitro試験を行った8化合物のうち、DEX及びNIFの CYP2C9誘導比は報告されてい
なかったため、図 2.5 では誘導が無いと仮定してこれらの誘導比は 1 とした。なお、
PHEと SULは臨床では阻害のDDIが観察されているため、図 2.5の検討から除外した。
A. (a) (b) (c)
(d) (e) (f)
Negative Positive Positive Negative Positive
Negative Positive Negative Positive Negative Positive
Negative
図 2.5 RF法を用いたCYP2C9及びCYP3A4のリスク評価
凍結肝細胞3ドナー[ドナー1(a, d, g及びj)、ドナー2(b, e, h及びk)並びにドナー3(c, f, i及びl)]を用いて算出したRFRIF × Css,u (A)又は
RFPB × Css,u (B)とCYP2C9(a, b, c, g, h及びi)又はCYP3A4 (d, e, f, j, k及びl)の臨床における誘導比との関係。垂直実線は、これより右側
が化合物の臨床における用法用量が CYP誘導リスク有りと分類されるカットオフ値を示す。垂直点線は、これより左側が化合物の臨床にお ける用法用量がCYP誘導リスク無しと分類されるカットオフ値を示す。◆はRIF、■はPB、▲はCRB、●はPHE、◇はDEX、□はSUL、
△はNIF、○はOMPをそれぞれ表す。AP, 臨床誘導比が1.25を越え、最も低いRF × Css,uを示す点; BP, 臨床誘導比が1.25を下回り、最も
高いRF × Css,uを示す点。
B. (g) (h) (i)
(j) (k) (l)
Negative Positive Negative Positive
Negative Positive
Negative Positive Negative Positive Negative Positive